Terapia NFPA

Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
La strategia terapeutica per gli NFPA non può prescindere dalla conoscenza sulla storia naturale della patologia. È esperienza clinica comune che si tratti di patologia estremamente eterogenea dal punto di vista biologico e clinico, che comprende lesioni di riscontro incidentale assolutamente quiescenti e forme estremamente aggressive nonostante trattamenti multimodali anche aggressivi. Uno studio tedesco (1) ha dimostrato con complessi modelli matematici che il tempo di raddoppiamento del volume tumorale varia da 8 mesi a 27 anni e diversi studi clinici che una progressione avviene in circa metà dei casi, soprattutto nei macroadenomi, mentre i micro rimangono stabili in gran parte dei casi. Sarebbe quindi utile avere a disposizione qualche strumento non invasivo che consenta di distinguere le forme evolutive da quelle stabili, per poter trattare selettivamente le prime. Purtroppo questo non esiste a tutt'oggi.
Scopo del trattamento è il controllo della crescita tumorale con le sue complicanze compressive locali (cefalea e disturbi visivi) e sistemiche (ipopituitarismo). È da notare che, anche dopo un trattamento adeguato, i pazienti affetti da NFPA hanno una riduzione della qualità della vita in confronto ai controlli sani (2), seppur non così accentuata come i pazienti con adenomi ipofisari secernenti (3).
Trattandosi di patologia eterogenea, è fondamentale un approccio individualizzato, che tenga conto delle modalità di presentazione, del volume tumorale, della compromissione funzionale e della disponibilità di presidii terapeutici adeguati. Non tutti i pazienti necessitano quindi di trattamento neurochirurgico, ma questo va senz’altro indicato (inviando il paziente ad un operatore esperto) quando la massa sia in accrescimento o vi sia compressione sulle vie ottiche.
Bibliografia
- Honegger J, et al. Growth modelling of non-functioning pituitary adenomas in patients referred for surgery. Eur J Endocrinol 2008, 158, 287–94.
- Dekkers OM, van der Klaauw AA, Pereira AM, et al. Quality of life is decreased after treatment for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab 2006, 91: 3364–9.
- van der Klaauw AA, Kars M, Biermasz NR, et al. Disease specific impairments in quality of life during long-term follow-up of patients with different pituitary adenomas. Clin Endocrinol 2008, 69: 775-8.
Terapia neurochirurgica NFPA
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
L’asportazione neurochirurgica costituisce la terapia di scelta per gli NFPA. Viene eseguita quasi sempre per via TNS e in mani esperte i risultati delle metodiche microscopica ed endoscopica non sono sostanzialmente diversi.
Quando la lesione ipofisaria viene riscontrata dopo l’esecuzione di RNM dell’encefalo per motivi clinici vari (cefalea, capogiri, follow up per altre malattie), in assenza di contesto clinico per adenoma ipofisario, e non determina né compressione delle vie ottiche né altri sintomi neurologici compressivi, bisogna considerare la presenza di incidentaloma ipofisario: nei casi in cui la lesione è intrasellare, si deve eseguire un monitoraggio seriato (6-12 mesi) della RMN, nei casi in cui la lesione è più voluminosa, la decisione terapeutica dipende dalle dimensioni della lesione e dal contesto clinico (età, comorbilità).
L’efficacia della neurochirurgia può essere valutata in base a diversi parametri: la radicalità dell'asportazione o la presenza di residuo adenomatoso, la remissione dei sintomi (miglioramento dei disturbi visivi), il ripristino della funzione ipofisaria compromessa.
Per quanto riguarda la funzione visiva, il miglioramento avviene in tre fasi (1,2):
- una fase immediata, entro una settimana;
- una fase precoce lenta, entro 6 mesi;
- una fase tardiva, più rara e solo parziale, entro 3 anni.
Nelle migliori casistiche neurochirurgiche si osserva una normalizzazione visiva nel 35-39%, un miglioramento in un altro 50-60% e un peggioramento solo nello 0.5-2.5% (2). Va sottolineato che il recupero del danno visivo può dipendere dalla condizione del n. ottico precedente l'intervento (atrofia).
Gli effetti della chirurgia sulla funzione ipofisaria negli NFPA sono contrastanti: dopo il trattamento chirurgico trans-sfenoidale alcuni studi riportano un miglioramento di grado variabile (3-6), altri non dimostrano miglioramenti significativi (7-9) ed altri ancora mostrano un peggioramento (10). La variabilità nei risultati dipende da diversi fattori: via di accesso chirurgico (la via trans-cranica, impiegata più raramente, non porta sicuramente a miglioramento dell’ipopituitarismo, 5), esperienza del neurochirurgo, criteri di valutazione dell’ipopituitarismo e, non ultimo, grado e durata dell’ipopituitarismo (un deficit completo e di più lunga durata ha meno probabilità di miglioramento di un deficit parziale di breve durata).
L’intervento trans-sfenoidale porta ad un controllo a lungo termine in circa l’80% dei pazienti (fino al 90% in alcune casistiche selezionate), ma non è chiaro, in assenza di dati attendibili sulla storia naturale degli NFPA (poiché la maggior parte dei pazienti è stata operata), se vi sia indicazione all’intervento anche in assenza di danni visivi, considerando che questa patologia viene riscontrata spesso in una popolazione anziana, portatrice di pluri-patologia che costituisce una contro-indicazione, almeno relativa, all’intervento o aumenta il rischio anestesiologico.
La presenza di danni al campo visivo costituisce a tutt’oggi l’indicazione principale per l’intervento chirurgico. In assenza di questi, l’approccio terapeutico iniziale è valutare la dimensione della lesione, la sua crescita e la funzione visiva con controlli regolari. In pazienti selezionati (età avanzata, presenza di comorbilità, ischemia cerebrale) con minime alterazioni visive, l’intervento può essere rimandato.
Dopo l’intervento neurochirurgico, il follow-up viene effettuato mediante RMN dopo 4 e 12 mesi, poi annuale per un numero arbitrario di anni (di solito 5) e poi più distanziato, in relazione alla presenza di residuo/recidiva post-operatorio e alla sua velocità di crescita. I risultati dell’esame istologico (atipia cellulare, mitosi, MIB-1, p53, immuno-istochimica (IIC) sul pezzo operatorio) possono consigliare un più stretto follow up, ma non sono elementi predittivi sicuri dell'evoluzione della malattia .
Il trattamento ottimale di un residuo tumorale è ancora controverso. Gli studi che hanno valutato il tasso di recidiva degli NFPA dopo chirurgia sono nella maggior parte dei casi retrospettivi, comprendono casistiche differenti (pazienti con piccolo residuo tumorale per i quali si è avuto un approccio conservativo, pazienti irradiati per la presenza di tumori più aggressivi o con masse tumorali residue più voluminose) e presentano fattori confondenti, quali differenti definizioni di residuo. Con queste premesse, il tasso di recidiva è stimato intorno al 17% per i pazienti con residuo minimo, mentre è del 43% nel caso di persistenza di un grosso residuo. La progressione o recidiva del tumore sembra associarsi a giovane età, invasione del seno cavernoso, estensione soprasellare del residuo tumorale, durata del follow-up (11,12). Alcuni studi indicano una maggiore aggressività per gli adenomi con immuno-istochimica positiva per ACTH, sebbene un aumento delle recidive non sia stato mostrato da tutti gli autori. Per i casi a maggior rischio (residuo di adenoma vicino alle vie ottiche, Ki-67 elevato (> 3%) o IIC positiva per ACTH o GH senza corrispettivo clinico, cosiddetti adenomi silenti), è necessario un follow-up clinico e neuroradiologico più stretto.
Bibliografia
- Dekkers OM, de Keizer RJ, Roelfsema F, et al. Progressive improvement of impaired visual acuity during the first year after transsphenoidal surgery for non-functioning pituitary macroadenoma. Pituitary 2007, 10: 61–5.
- Greenman Y, et al. How should a nonfunctioning pituitary macroadenoma be monitored after debulking surgery? Clin Endocrinol (Oxf) 2009, 70: 829-32.
- Arafah BM, Kailani SH, Nekl KE, et al. Immediate recovery of pituitary function after transsphenoidal resection of pituitary macroadenomas. J Clin Endocrinol Metab 1994, 79: 348–54.
- Marazuela M, Astigarraga B, Vicente A, et al. Recovery of visual and endocrine function following transsphenoidal surgery of large nonfunctioning pituitary adenomas. J Endocrinol Invest 1994, 17: 703–7.
- Nomikos P, Ladar C, Fahlbusch R, et al. Impact of primary surgery on pituitary function in patients with non-functioning pituitary adenomas – a study on 721 patients. Acta Neurochir 2004, 146: 27-35.
- Fatemi N, Dusick JR, Mattozo C, et al. Pituitary hormone loss and recovery after transphenoidal adenoma removal. Neurosurgery 2008, 63: 709-19.
- Comtois R, Beauregard H, Somma M, et al. The clinical and endocrine outcome to trans-sphenoidal microsurgery of nonsecreting pituitary adenomas. Cancer 1991, 68: 860–6.
- Wichers-Rother M, Hoven S, Kristof RA, et al. Non-functioning pituitary adenomas: endocrinological and clinical outcome after transsphenoidal and transcranial surgery. Exp Clin Endocrinol Diabetes 2004, 112: 323–7.
- Alameda C, Lucas T, Pineda E, et al. Experience in management of 51 non-functioning pituitary adenomas: indications for post-operative radiotherapy. J Endocrinol Invest 2005, 28: 18–22.
- Dekkers OM, Pereira AM, Roelfsema F, et al. Observation alone after transsphenoidal surgery for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab 2006, 91: 1796–801.
- Greenman Y, Ouaknine G, Veshchev I, et al. Postoperative surveillance of clinically nonfunctioning pituitary macroadenomas: markers of tumour quiescence and regrowth. Clin Endocrinol 2003, 58: 763-9.
- Losa M, et al. Early results of surgery in patients with nonfunctioning pituitary adenoma and analysis of the risk of tumor recurrence. J Neurosurg 2008, 108: 525-32.
- Dekkers M, Hammer S, de Keizer RJW, et al. The natural course of non-functioning pituitary macroadenomas. Eur J Endocrinol 2007, 156: 217–24.
- Dekkers OM, Pereira A, Romijn JA. Treatment and follow-up of clinically non functioning pituitary macroadenomas. J Clin Endocrinol Metab 2008, 93: 3717–26.
- Greenman Y, Stern N. Non-functioning pituitary adenomas. Best Pract Res Clin Endocrinol Metab 2009, 23: 625–38.
- Karavitaki N, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol (Oxf) 2007, 67: 938-43.
Terapia radiante NFPA
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
In caso di masse tumorali voluminose può essere indicata la radioterapia stereotassica (in alternativa al reintervento), mentre per piccole lesioni lontane almeno 5 mm dai nervi ottici può trovare applicazione la radiochirurgia.
È tuttora molto discussa, sempre per la relativa assenza di dati sulla storia naturale della malattia, l’utilità di una radioterapia (RT) complementare, con tecnica frazionata o radiochirurgica, dopo l’intervento chirurgico con persistenza di residuo tumorale. Gli studi focalizzati sulla ricrescita del residuo post-chirurgico senza e con RT (1-10) hanno dato risultati estremamente variabili, perché i criteri per definire la ricrescita tumorale sono molto eterogenei e per i diversi criteri di inclusione, soprattutto riguardanti la porzione di pazienti in ogni casistica avviata direttamente a RT perché ritenuta portatrice di neoplasia più aggressiva (3,5-7,11,12).
Uno studio longitudinale inglese (6) su una casistica di 73 pazienti ha dimostrato che la sopravvivenza libera da recidive era dell’82% a 5 anni, ma scendeva al 56% a 10 anni. Uno studio israeliano (8) evidenziava che le caratteristiche cliniche e neuroradiologiche del paziente prima e dopo l’intervento (in particolare l’invasione del seno cavernoso, la presenza e l’estensione sovra-sellare del residuo) sono in grado di predire la maggiore probabilità di ricrescita tumorale post-chirurgica. I fautori della RT (7) sottolineano l’alta percentuale di recidive post-chirurgiche, che viene drasticamente abbattuta con l’irradiazione del residuo tumorale. Altri (4,13) sostengono che un accurato follow-up permette di evidenziare per tempo quella frazione di pazienti il cui residuo tende a ricrescere, prima che tale crescita possa provocare danni, in maniera da avviarli alla terapia complementare più adeguata (reintervento, oppure irradiazione), risparmiando così a una buona parte di pazienti l’irradiazione.
Un recentissimo studio irlandese (14) ha valutato retrospettivamente 159 pazienti con NFPA seguiti da oltre 20 anni in un centro dove viene abitualmente applicata una politica omogenea, senza RT nella fase post-chirurgica fino a dimostrazione di ricrescita/recidiva: nel 33.5% dei casi veniva osservata ricrescita/recidiva. L’analisi multivariata dimostrava che i principali predittori negativi erano la lunghezza del follow-up (con un notevole aumento fra 5 e 10 anni, ad indicare la necessità di un follow-up prolungato) e le dimensioni del residuo (con un OR di 3.7 nei pazienti con residuo extra-sellare e la totale assenza di recidiva in quelli senza residuo). Gli autori suggerivano quindi di sottoporre a RT di routine tutti i pazienti con residuo post-operatorio extra-sellare, lasciando alla valutazione individuale la decisione sull’opportunità di RT in quelli con residuo intra-sellare.
La terapia radiante, sia frazionata convenzionale, che stereotassica, che radiochirurgica con gamma-knife, è sicuramente efficace nel ridurre il rischio di ricrescita tumorale e ha una buona efficacia nel controllo locale di malattia (4,11-13,15-17). La tossicità correlata alla RT è ben nota, dall’ipopituitarismo, alla possibilità di danno dei nervi ottici, al rischio di un secondo tumore a distanza, all’aumentata mortalità per cause cerebro-vascolari e probabilmente a deficit cognitivo (18-21). Essa dipende anche dalla dose impiegata. Va ricordato che anche la radioterapia va eseguita in Centri esperti in patologia ipofisaria.
La tabella illustra i principali risultati della letteratura.
| Evoluzione post-NCH degli NFPA (modificata da 14) | |||||||
| # | anno | FU | n (% non irradiati) | Recidiva a 5 anni | Recidiva a 10 anni | ||
| senza RT | con RT | senza RT | con RT | ||||
| 5 | 1994 | nd | 73 (100%) | 10% | - | - | - |
| 16 | 1998 | 7.5 | 26 (50%) | 32% | 7% | 52% | 7% |
| 6 | 1999 | 6.3 | 65 (100%) | 18% | - | 44% | - |
| 7 | 2000 | 5.3 | 72 (31%) | 46% | 26% | - | - |
| 22 | 2002 | 5.6 | 51 (100%) | 39% | - | - | - |
| 8 | 2003 | 4.3 | 122 (88%) | 52% | - | - | - |
| 11 | 2004 | 3.8-5.7 | 176 (75%) | 15.2% | 2.3% | 50.5% | 2.3% |
| 4 | 2006 | 5 | 97 (94%) | 6% | 19% | ||
| 23 | 2006 | 5.7 | 295 (51%) |
19.2 (senza residuo) |
18.4% | ||
| 12 | 2007 | 7.8-5.9 | 122 (23%) | 51% | 5% | 78% | 5% |
L’uso routinario della radioterapia dopo chirurgia può associarsi ad una sopravvivenza libera da recidive nel 90% dei casi dopo un’osservazione di 10 anni. Comunque nuovi studi hanno dimostrato che la radioterapia non previene la ricrescita in tutti i pazienti. La radioterapia dovrebbe dunque essere riservata a:
- tumori più aggressivi;
- tumori che crescono durante il follow-up;
- pazienti con elevato rischio operatorio in caso di reintervento.
Bibliografia
- Comtois R, Beauregard H, Somma M, et al. The clinical and endocrine outcome to trans-sphenoidal microsurgery of nonsecreting pituitary adenomas. Cancer 1991, 68: 860–6.
- Wichers-Rother M, Hoven S, Kristof RA, et al. Non-functioning pituitary adenomas: endocrinological and clinical outcome after transsphenoidal and transcranial surgery. Exp Clin Endocrinol Diabetes 2004, 112: 323–7.
- Alameda C, Lucas T, Pineda E, et al. Experience in management of 51 non-functioning pituitary adenomas: indications for post-operative radiotherapy. J Endocrinol Invest 2005, 28: 18–22.
- Dekkers OM, Pereira AM, Roelfsema F, et al. Observation alone after transsphenoidal surgery for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab 2006, 91: 1796–801.
- Bradley KM, Adams CB, Potter CP, et al. An audit of selected patients with non-functioning pituitary adenoma treated by transsphenoidal surgery without irradiation. Clin Endocrinol 1994, 41: 655-9.
- Turner HE, Stratton IM, Byrne JV, et al. Audit of selected patients with nonfunctioning pituitary adenomas treated without irradiation—a follow-up study. Clin Endocrinol 1999, 51: 281–4.
- Woollons AC, Hunn MK, Rajapakse YR, et al. Non-functioning pituitary adenomas: indications for postoperative radiotherapy. Clin Endocrinol 2000, 53: 713-7.
- Greenman Y, Ouaknine G, Veshchev I, et al. Postoperative surveillance of clinically nonfunctioning pituitary macroadenomas: markers of tumour quiescence and regrowth. Clin Endocrinol 2003, 58: 763-9.
- Losa M, et al. Early results of surgery in patients with nonfunctioning pituitary adenoma and analysis of the risk of tumor recurrence. J Neurosurg 2008, 108: 525-32.
- Chang EF, Zada G, Kim S, et al. Long-term recurrence and mortality after surgery and adjuvant radiotherapy for non-functional pituitary adenoma. J Neurosurg 2008, 108, 736-45.
- Park P, Chandler WF, Barkan AL, et al. The role of radiation therapy after surgical resection of non functional pituitary macroadenomas. Neurosurgery 2004, 55: 100-6; discussion 106-107.
- van den Bergh AC, van den Berg G, Schoorl MA, et al. Immediate postoperative radiotherapy in residual non functioning pituitary adenoma: beneficial effect on local control without additional negative impact on pituitary function and life expectancy. Int J Radiat Oncol Biol Phys 2007, 67: 863-9.
- Dekkers OM, Pereira A, Romijn JA. Treatment and follow-up of clinically non functioning pituitary macroadenomas. J Clin Endocrinol Metab 2008, 93: 3717–26.
- O’Sullivan EP, Woods C, Glynn N, et al. The natural history of surgically treated but radiotherapy naïve non-functioning pituitary adenomas. Clin Endocrinol 2009, 71: 709–14.
- Jaffrain-Rea ML, Derome P, Bataini JP, et al. Influence of radiotherapy on long-term relapse in clinically non-secreting pituitary adenomas. A retrospective study (1970-1988). Eur J Med 1993, 2: 398-403.
- Gittoes NJ, Bates AS, Tse W, et al. Radiotherapy for non-functioning pituitary tumours. Clin Endocrinol 1998, 48: 331-7.
- Höybye C, Rähn T. Adjuvant Gamma Knife radiosurgery in non-functioning pituitary adenomas; low risk of long-term complications in selected patients. Pituitary 2009, 12: 211–6.
- Brada M, Ashley S, Ford D, et al. Cerebrovascular mortality in patients with pituitary adenoma. Clin Endocrinol 2002, 57: 713-7.
- Minniti G, Traish D, Ashley S, et al. Risk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma: update after an additional 10 years. J Clin Endocrinol Metab 2005, 90: 800-4.
- Van der Klaauw AA, Biermasz NR, et al. Previous radiotherapy negatively influences quality of life during 4 years of follow-up in patients cured from acromegaly. Clin Endocrinol 2008, 69: 123-8.
- Ayuk J, Stewart PM. Mortality following pituitary radiotherapy. Pituitary 2009, 12: 35-9.
- Soto-Ares G, Cortet-Rudelli C, Assaker R, et al. MRI protocol technique in the optimal therapeutic strategy of non-functioning pituitary adenomas. Eur J Endocrinol 2002, 146: 179-86.
- Ferrante E, Ferraroni M, Castrignanò T, et al. Non-functioning pituitary adenoma database: a useful resource to improve the clinical management of pituitary tumors. Eur J Endocrinol 2006, 155: 823-9.
- Dekkers M, Hammer S, de Keizer RJW, et al. The natural course of non-functioning pituitary macroadenomas. Eur J Endocrinol 2007, 156: 217–24.
- Greenman Y, et al. How should a nonfunctioning pituitary macroadenoma be monitored after debulking surgery? Clin Endocrinol 2009, 70: 829-32.
- Greenman Y, Stern N. Non-functioning pituitary adenomas. Best Pract Res Clin Endocrinol Metab 2009, 23: 625–38.
- Karavitaki N, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol 2007, 67: 938-43.
- Losa M, Picozzi P, Motta M, et al. The role of radiation therapy in the managementof non functioning pituitary adenomas. J Endocrinol Invest 2011, 34: 623-9.
Terapia farmacologica NFPA
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Il ruolo della terapia farmacologica con i farmaci attualmente disponibili sembra assolutamente secondario, quanto meno in fase pre-operatoria (1).
I farmaci più impiegati sono stati i dopaminergici: i risultati con le varie molecole, in particolare la cabergolina, dimostrano un controllo della crescita tumorale (cioè stabilità o miglioramento, valutato come campimetria e RM) in oltre l’80% dei casi (tabella). È però stato fallimentare il tentativo di utilizzare tecniche non invasive, quali la scintigrafia recettoriale (che comunque sarebbe stata gravata da alti costi e scarsa diffusione), per predire l’efficacia del trattamento (2).
Anche gli analoghi commerciali della somatostatina sono stati impiegati con successo parziale in alcuni casi: l’apparente dissociazione fra la diminuzione del volume tumorale e il miglioramento del campo visivo (più frequente) è probabilmente dovuto all’azione diretta degli SA sulla retina.
La combinazione delle 2 classi di farmaci (SA + DA), riportata aneddoticamente, non ha dato risultati migliori.
Infine, vista la biologia di questi tumori, in passato sono stati impiegati in pochi casi i superagonisti dl GnRH, con risultati assolutamente deludenti (3): pur modificando in vivo e in vitro i livelli di gonadotropine e alfa-subunità, tale trattamento non ha indotto alcuna modificazione delle dimensioni dell’adenoma. Sono stati provati in vitro anche antagonisti del GnRH (3), senza ottenere risultati.
| Efficacia dei farmaci nel trattamento primario (pre-operatorio) dei pazienti con NFPA (modificato da 1) |
||||||
| Campo visivo | Volume adenoma | |||||
| migliorato | invariato | peggiorato | diminuito | invariato | aumentato | |
| DA | 16/107 (15%) |
77/107 (72%) |
14/107 (13%) |
36/180 (20%) |
133/180 (74%) |
11/180 (6%) |
| SA | 27/84 (32%) |
50/84 (60%) |
7/84 (8%) |
11/154 (7%) |
130/154 (84%) |
12/154 (9%) |
La temozolomide, un citostatico alchilante, si è dimostrato efficace nel ridurre il volume tumorale in un piccolo numero di casi di NFPA aggressivi (4).
Bibliografia
- Colao A, et al. Medical therapy for clinically non-functioning pituitary adenomas. Endocr Relat Cancer 2008, 15: 905–15.
- de Herder WW, Reijs AE, de Swart J, et al. Comparison of iodine-123 epidepride and iodine-123 IBZM for dopamine D2 receptor imaging in clinically non-functioning pituitary macroadenomas and macroprolactinomas. Eur J Nucl Med 1999, 26: 46-50.
- Chanson P, Lahlou N, Warnet A, et al. Responses to gonadotropin releasing hormone agonist and antagonist administration in patients with gonadotroph cell adenomas. J Endocrinol Invest 1994, 17: 91-8.
- Buchfelder M. Management of aggressive pituitary adenomas: current treatment strategies. Pituitary 2009, 12: 256–60.
Clinica e diagnostica iperprolattinemie
Enrica Ciccarelli
SSD Oncologia, Ospedale Martini, Torino
(aggiornato al 1 maggio 2017)
L'iperprolattinemia rappresenta un riscontro comune nella pratica clinica endocrinologica ed è il marcatore più frequente di patologia ipotalamo-ipofisaria. L'incremento dei valori di PRL può presentarsi come patologia singola od occasionalmente essere parte di una MEN-1, in associazione con tumori delle paratiroidi e pancreatici. Eccezionalmente l'iperprolattinemia è presente in forme familiari isolate (FIPA), in adenomi determinati da mutazioni genetiche (gene AIP).
Vista la normale secrezione pulsatile della PRL, la diagnosi di iperprolattinemia richiede conferme da multipli dosaggi plasmatici. Alcune cause di iperPRL sono fisiologiche e facilmente diagnosticabili, mentre nella maggior parte dei casi è necessario un approfondimento anamnestico, biochimico e neuroradiologico. Le cause di iperPRL sono riassunte nella tabella.
| Cause di iperprolattinemia | |
| Fisiologiche |
Età neonatale |
| Farmaci |
Bloccanti dopaminergici: metoclopramide, sulpiride, domperidone, neurolettici |
| Malattie ipotalamiche | Tumori ipotalamici: craniofaringiomi, germinomi, ependimomi, gliomi, meningiomi, metastasi Lesioni infiltrative: istiocitosi, sarcoidosi, ipofisiti, TBC Malformazioni arterovenose (aneurismi) Radiazioni al cranio Traumi del peduncolo ipofisario: traumi cranici, interventi NCH |
| Malattie ipofisarie |
PRLoma |
| Altre cause | Ipotiroidismo primario Insufficienza renale cronica Insufficienza epatica Lesioni della parete toracica PCOs Sindromi paraneoplastiche IperPRL idiopatica |
Tra le cause di iperPRL patologica, quelle iatrogene rappresentano la maggior parte e pertanto è necessario un approfondimento anamnestico prima di intraprendere un costoso iter diagnostico. Quando possibile, è opportuna la sospensione del farmaco o la sostituzione con altro ad effetto neutro sui valori di PRL (es. aripiprazolo in luogo dei comuni neurolettici), con rivalutazione successiva dei valori di PRL: nel caso di riscontro di normali valori di PRL, è possibile la sospensione dell'iter diagnostico. Nel caso in cui la sospensione non possa essere attuata (es. pazienti psichiatrici), l'iter diagnostico andrà comunque completato.
L'adenoma PRL-secernente è la lesione neoplastica più frequente e rappresenta il 40% delle lesioni tumorali della regione ipotalamo-ipofisaria; livelli elevati di PRL possono però derivare da una lesione peduncolare in presenza di un tumore non PRL-secernente di grandi dimensioni (pseudo-prolattinoma).
L'adenoma PRL-secernente colpisce maggiormente il sesso femminile, anche se nel maschio si ritrovano adenomi a maggiore invasività e aggressività.
Gli adenomi PRL-secernenti vengono distinti in rapporto a:
- dimensioni: microadenomi (< 1 cm) e macroadenomi;
- invasività nei tessuti circostanti: invasivi o non invasivi;
- aggressività in rapporto ai valori di Ki67 (e all'espressione di p53): per definizione adenomi atipici se > 3%.
L'ipersecrezione della PRL può essere isolata da parte delle cellule tumorali o, più raramente, essere accompagnata dall'ipersecrezione di altri ormoni (tumori misti), come più frequentemente accade per GH (cellula mammo-somatotropa) o ACTH e TSH.
I sintomi determinati dall'iperPRL sono collegati agli effetti dei valori elevati della PRLemia e all'effetto compressivo della lesione tumorale. Un'iperPRL cronica, indipendentemente dalla causa che la determina, induce:
- nella donna alterazioni del ciclo mestruale (amenorrea primaria o secondaria, oligomenorrea, polimenorrea) e infertilità; è frequente la presenza di galattorrea spontanea o provocata, anche se questa correla più direttamente allo stato estrogenico della paziente (meno frequente nelle pazienti con amenorrea primaria, quasi costante nelle pazienti che hanno avuto gravidanze o assunto estrogeni). Come conseguenza dell'ipoestrogenismo cronico e di lunga durata, possono essere presenti dispareunia e osteoporosi (quest'ultima forse anche determinata da alterazioni nel metabolismo della vitamina D per opera diretta dell'iperPRL o dall’iperPRL di per sè);
- nel maschio vengono riscontrati disfunzione erettile, riduzione o perdita della libido, riduzione della quantità di liquido seminale e, più raramente, oligospermia; possono essere presenti ginecomastia, galattorrea e osteoporosi.
L'effetto compressivo della lesione tumorale può indurre cefalea (sia ex novo che variazione nelle caratteristiche di una precedente cefalea cronica), difetti visivi (riduzione dell'acuità visiva e alterazioni campimetriche, quali quadrantopsie, emianopsie, amaurosi, sindrome chiasmatica), paralisi dei nervi cranici (sindrome del seno cavernoso, diplopia). In alcuni casi con grave estensione sopra-sellare, può manifestarsi confusione mentale fino al coma, a causa di idrocefalo: in questi casi la diagnosi di ipersecrezione va effettuata rapidamente per iniziare quanto prima il trattamento con cabergolina. Nei pazienti con estensione infra-sellare, può manifestarsi rinoliquorrea. Nei pazienti in cui le dimensioni dell'adenoma sono rilevanti, va ricercata la presenza di insufficienza secretiva dei vari ormoni ipofisari, per effetto distruttivo sull'ipofisi sana da parte della neoplasia, con necessità conseguente di appropriato trattamento sostitutivo.
La diagnosi di iperprolattinemia e di adenoma PRL-secernente si basa su esami di laboratorio e radiologici.
Dato che la PRL è un ormone pulsatile influenzabile dallo stress, la diagnosi deve essere effettuata su campionamenti ripetuti a distanza dopo un adeguato periodo di riposo. Valori di PRL > 200 ng/mL sono diagnostici di iperprolattinemia secretoria e spesso di macroadenoma; valori inferiori non possono escludere la presenza di adenomi PRL-secernente a bassa espressione ormonale o cause di iperPRL iatrogena.
In presenza di alcune metodiche immuno-radiometriche, è possibile osservare l'hook-effect (effetto gancio), con sottostima dei valori di PRL per artefatto tecnico da saturazione di anticorpi. In questi casi è necessario che il laboratorio proceda a una diluizione del campione di plasma, al fine di non mancare la diagnosi di macroadenoma PRL-secernente, con conseguenti errati comportamenti terapeutici. In presenza di elevati livelli di PRL ed assenza di sintomi correlati, occorre prendere in considerazione la macroprolattinemia, legata alla presenza in alcuni casi di elevate concentrazioni di big-big PRL (macromolecole con aggregati anticorpali con attività biologica ridotta), che possono indurre valori falsamente elevati di PRL. Anche in questo caso viene in aiuto il laboratorio: la ripetizione del dosaggio ormonale con uso di polietilenglicole (PEG), che fa precipitare gli aggregati, porta a un ridotto recupero di PRL (< 40% del valore precedente prima di PEG). La ricerca di macroprolattinemia è particolamente rilevante nei pazienti con livelli di PRL moderatamente elevati e senza cause evidenti.
L'uso dei test dinamici, utilizzati negli anni passati per la diagnosi differenziale tra iperPRL funzionale e patologica o tra adenoma PRL-secernente e pseudo-prolattinoma, è da ritenersi desueto e privo di utilità clinica.
Dopo aver confermato la presenza di iperPRL sul piano biochimico, è necessario uno studio morfologico della regione ipotalamo-ipofisaria. L'esame di elezione è la risonanza magnetica nucleare (RM) con gadolinio. La tomografia computerizzata (TC) con mdc va riservata ai casi in cui la RM non può essere effettuata (pazienti portatori di protesi metalliche o pace-maker, importante claustrofobia); la TC è preferibile anche nei casi in cui sia da esaminare con accuratezza il profilo osseo (nei casi di rinoliquorrea) o in presenza di calcificazioni. La RM evidenzia la lesione presente all'interno della regione ipotalamo-ipofisaria e i suoi rapporti con le regioni circostanti. Nel caso in cui sia richiesta una valutazione approfondita dell'albero vascolare (es. sospetto aneurisma, fase pre-operatoria in caso di lesioni voluminose), è indicata l'esecuzione di angioRM o angiografia cerebrale. Va però considerato che difetti focali di piccole dimensioni possono essere presenti nel 10-27% di soggetti normali e possono rappresentare piccole cisti, aree di necrosi o artefatti. La RM trova anche indicazione nel follow-up delle lesioni, in particolare dopo trattamento.
Nel caso di riscontro di macroadenoma ipofisario, è necessaria una valutazione neuro-oftalmologica mediante esecuzione di fundus e campimetria; in casi particolarmente gravi, può essere utile eseguire i potenziali evocati visivi.
Bibliografia
- Frieze TW, Mong DP, Koops MK. "Hook effect" in prolactinoma: case report and review of literature. Endocr Pract 2002, 8: 296-303.
- McCudden CR, Sharpless JL, Grenache DG. Comparison of multiple methods for identification of hyperprolactinemia in the presence of macroprolactin. Clin Chim Acta 2010, 411: 155-60.
- Casanueva FF, Molitch ME, Schlechte JA, et al Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol 2006, 65: 265-73.
- Camanni F, Cannavò S, Colao A, et al. Linee di consenso diagnostico-terapeutiche: le iperprolattinemie. Documento di consenso SIE 2006.
- Melmed S, Casanueva FF, Hoffman AR. Diagnosis and treatment of hyperprolactinemia: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 273-88.
- Famini P, Maya MM, Melmed S. Pituitary magnetic resonance imaging for sellar and parasellar masses: ten-year experience in 2598 patients. J Clin Endocrinol Metab 2011, 96: 1633-41.
Terapia iperprolattinemie
Terapia neurochirurgica prolattinomi
Enrica Ciccarelli
SSD Oncologia, Ospedale Martini, Torino
(aggiornato al 1 maggio 2017)
L'intervento neurochirurgico è generalmente usato come seconda scelta nella terapia del prolattinoma, ma alcune analisi costo-efficacia (di confronto con la terapia farmacologica) e l'utilizzo di nuove tecniche di microchirurgia endoscopica endonasale lo hanno recentemente riproposto come primo approccio nei pazienti con microprolattinoma.
Nei microadenomi l’intervento può essere preso in considerazione in pazienti intolleranti o resistenti alla terapia farmacologica, oppure nei pazienti con ridotta compliance che non vogliano affrontare una terapia medica a lungo termine.
Deve essere posta indicazione a intervento chirurgico nel caso di rinoliquorrea, apoplessia ipofisaria con compromissione visiva grave o in caso di gravidanza con espansione del prolattinoma non responsivo alla terapia medica.
In mani esperte, l'adenomectomia selettiva, effettuata con le tecniche più moderne, del microprolattinoma induce normalizzazione della PRL nel 71-100% dei casi. Tuttavia, è documentata la possibilità di recidiva nel 20% dei casi. Nei pazienti con macroprolattinoma, la guarigione definitiva è rara, data la frequente natura invasiva nei tessuti circostanti. Fattori prognostici negativi sono l'età, il sesso maschile e l'aggressività della malattia.
Le complicanze di un intervento per via trans-sfenoidale sono assai ridotte (mentre hanno maggiore frequenza negli interventi effettuati per via trans-cranica), con differenze fra microadenoma e macroadenoma: rispettivamente, riduzione campimetrica 0.1% vs 1.5%, paresi oculomotoria 0.1% vs 0.6%, ictus 0.2% vs 0.6%, meningite 0.1% vs 0.5%, rinoliquorrea 1.9% vs 3.3%). Un diabete insipido transitorio è frequente in tutti gli operati, ma diviene permanente solo nell'1% dei macroadenomi. Ipopituitarismi multipli possono essere riscontrati nel 50% dei macroprolattinomi e solo occasionalmente nei microprolattinomi. Nei pazienti che presentino ipopituitarismo già in precedenza, è possibile verificare un peggioramento immediatamente dopo l'intervento, ma con recupero dopo il 4° mese. Pertanto, in tutti i pazienti dopo l'intervento NCH deve essere effettuato l'esame della funzionalità ipofisaria.
Bibliografia
- Gilliam MP, Molitch ME, Lombardi G, et al. Advances in the treatment of prolactinomas. Endocr Rev 2006, 27: 485-534.
- Jan M, Dufour H, Brue T, et al. Prolactinoma surgery. Ann Endocrinol 2007, 68: 118-9.
- Primean V, Raftopaulos C, Maiter D. Outcome of transfenoidal surgery in prolactinomas: improvement of hormonal control in dopaminergic agonist resistant patients. Eur J Endocrinol 2012, 166: 779-86.
- Tampourlou M, Trifanescu R, Paluzzi A, et al. Therapy of endocrine disease: surgery in microprolactinoma. Effectiveness and risks based on contemporary literature. Eur J Endocrinol 2016, 175: R89-96.
Terapia farmacologica iperprolattinemie
Enrica Ciccarelli
SSD Oncologia, Ospedale Martini, Torino
(aggiornato al 1 maggio 2017)
I farmaci agonisti dopaminergici hanno modificato radicalmente la strategia terapeutica dei prolattinomi.
Obiettivo della terapia
Ridurre i valori di PRLemia, ridurre il volume della neoplasia (soprattutto nei pazienti con macroadenoma) e ripristinare una normale funzione gonadica.
Farmaci disponibili: bromocriptina e soprattutto cabergolina.
La cabergolina è attualmente il farmaco di prima scelta, per la maggiore efficacia ipoprolattinemizzante e la maggiore frequenza di shrinkage tumorale. La cabergolina è un derivato ergolinico con prolungata durata d'azione; oltre alla ben nota azione dopaminergica, possiede anche una modesta attività serotoninergica. La dose di farmaco necessaria per il controllo della malattia è generalmente di 0.25-3 mg/settimana, anche se alcuni pazienti assumono fino a 11 mg/settimana. Nelle situazioni più comuni il farmaco viene frazionato in 2-3 somministrazioni settimanali. La tolleranza del farmaco è generalmente buona, ma alcuni pazienti possono lamentare ipotensione, nausea, vomito. Recentemente è stata riscontrata la presenza di valvulopatia mitralica e/o tricuspidale, causata dalla componente serotoninergica del farmaco, in alcuni pazienti con m. di Parkinson trattati con alte dosi di cabergolina (> 3 mg/die). Tale effetto non sembra manifestarsi nelle casistiche fin qui pubblicate di pazienti con prolattinoma, che assumono in genere dosi nettamente inferiori di cabergolina, anche se per periodi molto più lunghi. Per questo motivo, è consigliabile effettuare prudenzialmente un ecocardiogramma in tutti i pazienti che assumano cabergolina, in particolare quelli trattati con dosi maggiori.
La bromocriptina, un alcaloide semi-sintetico ergotaminico, è stato il primo farmaco utilizzato per la terapia medica delle iperprolattinemie. Attualmente il suo uso è limitato ai pazienti che necessitino del farmaco in corso di gravidanza o che siano intolleranti alla cabergolina. La dose utilizzata varia da 2.5 a 50 mg/die. Il farmaco deve essere assunto in dosi crescenti, a stomaco pieno con il pasto serale (occasionalmente anche in 2-3 frazioni giornaliere) per evitare effetti collaterali importanti (ipotensione ortostatica, nausea, vomito, vertigini, naso chiuso). Circa il 5-10% dei pazienti non tollera il farmaco e pertanto è necessario il passaggio alla cabergolina.
Nei rarissimi casi di neoplasia maligna PRL-secernente, può essere utilizzata la temozolomide.
Risultati.
Con la terapia medica si ottiene la normalizzazione dei livelli di PRL mediamente nel 94% dei casi, mentre lo shrinkage tumorale si verifica nell'82% degli adenomi. Vi è peraltro una significativa differenza nella risposta tra i due sessi:
- nei maschi, oltre a una maggiore presenza pre-terapia di macroadenomi con carattere di invasività (aumentati indici angiogenici, di proliferazione e di atipie cellulari nel 30% vs 8% nel sesso femminile), sono necessarie dosi di farmaco maggiori e più frequentemente si riscontrano fenomeni di resistenza;
- il sesso femminile è invece maggiormente sensibile alla terapia: nel 63% delle donne contro il 36% dei maschi è riscontrabile una risposta alla terapia con dosi di cabergolina < 1 mg/settimana.
La menopausa, a causa della riduzione dei livelli estrogenici, sembra rappresentare un fattore positivo sulla risposta alla terapia, soprattutto nelle pazienti con microprolattinoma; è quindi indicata la sospensione della terapia dopo l'evento menopausale, per verificare la possibilità di remissione a lungo termine dell'iperPRL. Questo non si verifica usualmente nella paziente con macroprolattinoma.
Nel corso della terapia farmacologica è necessario monitorare i livelli di PRL (inizialmente frequentemente, ogni 1-2 mesi, fino al raggiungimento della normalizzazione ormonale, in seguito ogni 3-6 mesi) e la RM sellare (inizialmente ogni 3-6 mesi in rapporto alla gravità della condizione clinica; dopo lo shrinkage, annualmente).
Ancora controversa è la definizione di resistenza alla terapia medica: più frequentemente si tratta di resistenza primaria, mentre più raramente si tratta di un fenomeno di escape dopo iniziale risposta. Recenti studi hanno evidenziato una correlazione con la ridotta presenza del mRNA dell'isoforma lunga del recettore D2; sono state invece escluse altre cause, come la mancanza di efficacia per ridotto assorbimento del farmaco o un’alterazione dell'affinità recettoriale per la dopamina. Attualmente si tende a utilizzare come criteri di resistenza una dose di bromocriptina > 15 mg/die e di cabergolina > 2 mg/settimana; alcuni autori includono anche la mancata riduzione dell'adenoma alla RM. In caso di resistenza alla bromocriptina, è necessaria la sostituzione con cabergolina; nel caso di resistenza a dosi di cabergolina > 3.5 mg/settimana, occorre prendere in considerazione la neurochirurgia e/o la radioterapia.
Un tentativo farmacologico con cabergolina può avere anche un significato diagnostico: infatti, una risposta positiva con shrinkage dell'adenoma è osservabile solo nei pazienti con adenoma PRL-secernente e non negli pseudoprolattinomi (NFPA, che dovranno quindi essere operati).
Nei casi di iperPRL funzionale in assenza di sintomi clinici, è indicato solo follow-up, senza terapia. Nelle pazienti con microadenoma e amenorrea, in cui non sia presente aspettativa di gravidanza, è possibile impiegare gli estroprogestinici.
La terapia farmacologica nei pazienti con macroprolattinoma deve essere proseguita spesso indefinitamente, fino al raggiungimento di tutti gli obiettivi terapeutici (normalizzazione della PRL, remissione dell'adenoma). La sospensione del farmaco è spesso seguita dalla ricomparsa di iperprolattinemia. In alcuni casi particolari si è osservata la remissione (nel 20-30% dei casi), con necessità di ripresa della cura nel caso di recidiva.
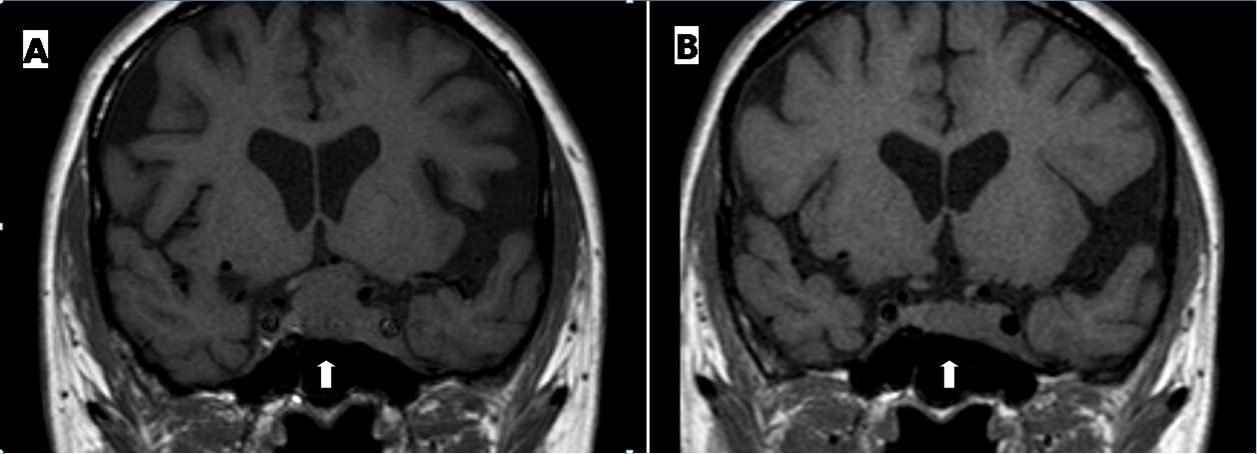
Macroprolattinoma alla diagnosi (a) e dopo terapia dopaminergica (b). La riduzione di volume dell'adenoma porta a liberazione delle vie ottiche, mentre persiste l'invasione del seno cavernoso sinistro
Bibliografia
- Ciccarelli E, Camanni F. Diagnosis and drug therapy of prolactinoma. Drugs 1996, 51: 954-65.
- Webster J, Piscitelli G, Polli A, et al The efficacy and tolerability of long term cabergoline therapy in hyperprolactinemic disorders: an open, uncontrolled multicenter study. European Multicenter Study Group. Clin Endocrinol 1993, 39: 323-9.
- Delgrange E, Daems T, Verhelst J, et al Characterization of resistance to the prolactin-lowering effects of cabergoline in macroprolactinomas: a study in 122 patiens. Eur J Endocrinol 2009, 160: 747-52.
- Vasilev V, Daly AF, Vroonen L, et al. Resistant prolactinomas. J Endocrinol Invest 2011, 34: 312-6.
Scheda farmaci dopaminergici
Renato Cozzi & Roberto Attanasio
Meccanismo d’azione
Legame a specifico recettore di membrana (D2) presente in tutte le cellule ipofisarie, ma soprattutto nelle cellule PRL- e GH-secernenti, dove inibisce la secrezione ormonale e la crescita tumorale.
Farmaco-cinetica
Cabergolina è rapidamente assorbito a livello intestinale e presenta emivita di qualche giorno (fino a 5 gg in pazienti iperprolattinemici).
Cabergolina è metabolizzata a livello epatico. In caso di compromissione della funzione renale non sembra esserci modifica della cinetica del farmaco, mentre in caso di compromissione severa della funzione epatica (Chil-Pugh-C) va considerata una riduzione della dose.
Indicazioni
- Terapia d’elezione per iperprolattinemia patologica.
- Acromegalia: forme lievi o con iperprolattinemia associata o in associazione a SA.
- Malattia di Cushing.
- Adenomi clinicamente non funzionanti (NFPA): tentativo di controllare la crescita tumorale nelle forme multirecidive.
Tranne la prima, le altre indicazioni sono attualmente off-label (modulo consenso).
Contro-indicazioni
- Psicosi in atto.
- Raynaud.
Farmaci disponibili, via di somministrazione e posologia
- Bromocriptina:
- cp 2.5 mg: Parlodel
- cp 5 mg: Parlodel
- cp 10 mg: Parlodel
capostipite dei dopaminergici, oggi largamente abbandonato per la sua minore efficacia e per gli effetti collaterali. Indicazioni attuali: iperprolattinemia in gravidanza (quando occorre)
iniziare la somministrazione a bassa dose (1.25 mg alla sera durante la cena); la titolazione (fino alla dose massima di 20-40 mg/die) va eseguita in base all’efficacia e alla tollerabilità (che spesso ne limita l’uso).
- Cabergolina:
- cp 0.5 mg: Actualene, cabergolina Aurobindo, cabergolina Ratiopharm, Dostinex
- cp 1 mg: cabergolina Ratiopharm, Cabaser
- cp 2 mg: cabergolina Ratiopharm, Cabaser
cominciare con 0.25 mg 1-3 volte alla settimana; la dose iniziale e la titolazione vanno scelte in base al quadro clinico e all’efficacia. Usualmente non si superano 3.5 mg/settimana, anche se non esiste una dose massima (nei pazienti resistenti). È stata dimostrata maggiore efficacia e tollerabilità rispetto alla bromocriptina. Se esiste rischio di rinoliquorrea (adenoma invasivo nel seno sfenoidale, erosione del pavimento sellare, è prudente iniziare con basse dosi per evitare la troppo rapida riduzione del volume tumorale
- Lisuride, metergolina (Liserdol cp 4 mg), quinagolide (Norprolac cp 25 µg, 50 µg, 75 µg): sono scarsamente impiegati (o non disponibili in Italia).
Effetti collaterali
Frequenti con bromocriptina, scarsi o assenti con cabergolina.
- Gastroenterici: nausea, vomito, anoressia
- Raynaud
- Ipotensione ortostatica (per cui è suggerita l’assunzione serale)
- Cefalea
- Senso di chiusura nasale
- Rino-liquorrea nei pazienti con riduzione volumetrica rapida di un adenoma erosivo del pavimento sellare (vedi sopra)
- Recente segnalazione di aumento di valvulopatie nei pazienti con m di Parkinson, solo per dosi molto alte (> 3 mg/die) di cabergolina)
Avvertenze per l’utilizzo
- Dopo un uso prolungato di derivati ergotaminici con proprietà agoniste per i recettori serotoninergici, come cabergolina, si sono osservati processi infiammatori delle sierose, fibrosi polmonare, fibrosi retroperitoneale e valvulopatie cardiache. Cabergolina è pertanto controindicata in pazienti con storia o sospetto di fibrosi polmonare, pericardica o retroperitoneale.
- In relazione alle valvulopatie cardiache, queste sembrano correlare all’esposizione cumulativa al farmaco. I dosaggi impiegati nella patologia ipofisaria sono usualmente inferiori a quelli usati per altre indicazioni, ma resta opportuno un ecocardiogramma basale e un monitoraggio ecografico periodico nei trattamenti a posologia e durata maggiori. Una precedente metanalisi sull’uso di cabergolina in pazienti iperprolattinemici indicava un aumento del rischio di rigurgito tricuspidalico, ma le conseguenze cliniche di tale riscontro non erano chiare.
- Cabergolina può associarsi a disturbi psichiatrici da perdita del controllo sugli impulsi (ludopatia, ipersessualità, spese eccessive o compulsive, bulimia), anche dopo vari mesi dall’avvio della terapia, ed esacerbazione di disturbi psichiatrici pre-esistenti. Il farmaco va utilizzato con cautela in pazienti affetti da patologia psichiatrica pregressa e sospeso in caso di suddetti effetti collaterali.
- Ci sono dati limitati sull’utilizzo di cabergolina in gravidanza per pazienti con malattia di Cushing (più estesi e rassicuranti per i prolattinomi).
Limitazioni prescrittive
No (tranne che per le formulazioni ad alte dosi di cabergolina, da 1 mg in su, che richiedono piano terapeutico del neurologo)
Terapia radiante prolattinomi
Enrica Ciccarelli
SSD Oncologia, Ospedale Martini, Torino
(aggiornato al 1 maggio 2017)
La radioterapia viene utilizzata solo nei casi di insuccesso della neurochirurgia e di resistenza alla terapia farmacologica, in presenza di tumori il cui volume non viene controllato dalla terapia medica per la loro aggressività locale o malignità. Non deve essere utilizzata per prevenire la recidiva tumorale post-intervento, in quanto in questi casi il controllo dell’iperprolattinemia con la cabergolina è l'indicazione terapeutica appropriata. Obiettivo della terapia radiante è l'arresto della crescita tumorale nei pazienti resistenti alla terapia farmacologica e già sottoposti a intervento chirurgico e, se possibile, la normalizzazione dell'iperprolattinemia, anche se questo viene raggiunto solo in pochi casi. Particolare attenzione va posta al trattamento di lesioni situate in stretta vicinanza al nervo ottico; in questi casi sembra preferibile un trattamento con approccio stereotassico ipofrazionato.
Per tutte le informazioni tecniche sulla radioterapia e i suoi effetti collaterali, vedi capitolo specifico.
Bibliografia
- Plowman PN, Grossman A. Radiotherapy in the treatment of pituitary tumours. Int J Rad Oncol Biol Phys 1990, 19: 229-30.
- Landolt AM, Lomax N. Gamma knife radiosurgery for prolactinomas. J Neurosurg 2000, 93 (suppl 3): 14-18.
- Puataweepong P, Dhanacai M, Hansasuta A, et al. The clinical outcome of hypofractionated stereotactic radiotherapy with cyberknife robotic radiosurgery for perioptic pituitary adenoma. Technical Cancer Res Treat 2015, 15: NP10-5.
- Cohen-Inbar O, Schlesinger D, Vance ML, Sheehan JP. Gamma knife radiosurgery for medically and surgically refractory prolactinomas: long-term results. Pituitary 2015, 18: 820-30.
- Wilson PJ, Williams JR, Smee RI. Single center experience of stereotactic radiosurgery and fractionated sterotactic radiotherapy for prolactinoma with the linear accelerator. J Med Imag Radiat Oncol 2015, 59: 371-8.
Prolattinoma in gravidanza
Enrica Ciccarelli
SSD Oncologia, Ospedale Martini, Torino
(aggiornato al 1 maggio 2017)
È ben documentato l'effetto stimolatorio degli estrogeni sulla sintesi di PRL e sulla proliferazione delle cellule lattotrope. Durante la gravidanza fisiologica l'ipofisi normale aumenta progressivamente di volume già dalle prime settimane di gestazione e i valori di PRL aumentano progressivamente, specialmente nell'ultimo trimestre quando i valori di estrogeni sono molto elevati.
L'adenoma ipofisario PRL-secernente può aumentare durante la gravidanza per la presenza di recettori per gli estrogeni anche sulle cellule tumorali. L'aumento volumetrico è proporzionale al volume iniziale del tumore: nei microadenomi il rischio di aumento è nel 3% dei pazienti, mentre sale al 30% nei pazienti con macroadenoma. Pertanto, la gravidanza deve essere attentamente pianificata, in particolare nelle pazienti con macroadenoma, nelle quali si deve ottenere uno shrinkage della neoplasia prima del concepimento.
La terapia farmacologica del prolattinoma può essere effettuata sia con cabergolina che con bromocriptina (farmaco di più lungo impiego e con esperienza più consolidata); essa deve essere sospesa al momento dell'accertamento di gravidanza. Tuttavia, nei casi di macroadenoma in cui le dimensioni dell'adenoma siano ancora voluminose o non si siano ridotte durante il trattamento farmacologico, può essere indicato proseguire la terapia durante tutta la gravidanza. In pazienti resistenti o intolleranti alla terapia farmacologica, può essere preso in considerazione l'intervento chirurgico.
Nel corso della gravidanza
Nelle pazienti con microprolattinoma, generalmente è sufficiente un controllo clinico periodico (trimestrale) per valutare eventuali sintomi da espansione tumorale (cefalea, disturbi visivi o neurologici); nelle pazienti con macroprolattinoma invece, il controllo clinico dovrà essere più stretto, specialmente nell'ultimo trimestre, con controllo campimetrico ogni 2-3 mesi, quando maggiore è l'aumento degli estrogeni e il rischio di danni neurologici da ri-espansione dell'adenoma. Non sono indicati controlli di PRL nel corso della gravidanza, in quanto anche nelle gravidanze fisiologiche si osserva un incremento ormonale. In caso di evidenza clinica di espansione tumorale, può essere eseguita la RM della regione ipotalamo-ipofisaria (senza mezzo di contrasto) a partire dal 4° mese di gravidanza.
Il parto può essere espletato per via naturale nelle pazienti con microprolattinoma, mentre in quelle con macroprolattinoma la scelta deve essere valutata in base alle caratteristiche cliniche neuroradiologiche pre-gravidanza.
Nelle pazienti con microadenoma che non abbiano dimostrato complicanze nel corso della gravidanza, l'allattamento al seno viene lasciato libero, a discrezione della paziente, mentre nelle pazienti con macroadenoma andrà valutato caso per caso (è comunque sconsigliato nelle pazienti con ri-espansione tumorale nel corso della gravidanza).
Dopo la gravidanza.
Il controllo della PRL plasmatica e di RM ipotalamo-ipofisaria andrà eseguito 2-3 mesi dopo il parto o dopo il termine dell'allattamento al seno.
Dopo la gravidanza, circa il 25% delle pazienti con microprolattinoma ha ottenuto una persistente normalizzazione dei livelli di PRL plasmatici.
Bibliografia
- Molitch ME. Pregnancy and the hyperprolactinemia women. N Engl J Med 1985, 312: 1364-70.
- Bronstein MD. Prolactinoma and pregnancy. Pituitary 2005, 8: 31-8.
- Ono M, Miki N, Amono K, et al. Individualized high-dose cabergoline therapy for hyperprolactinemic infertility in women with micro and macroprolactinomas. J Clin Endocrinol Metab 2010, 95: 2672-9.
- Zaroca Z, Tanriverdi F, Unluhisarci K, et al. Pregnancy and pituitary disorders. Eur J Endocrinol 2010, 162: 453-75.
Clinica e diagnostica acromegalia e gigantismo
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
(aggiornato al 12 aprile 2020)
DEFINIZIONE ED EZIOLOGIA
L’acromegalia è una malattia cronica sistemica (1).
Nella stragrande maggioranza dei casi è causata da un adenoma ipofisario GH-secernente (macro nel 75% e micro nel 25%) (2). In rari casi (< 1%) può essere causata dalla secrezione eutopica (gangliocitoma ipotalamico) o ectopica (carcinoide polmonare o pancreatico, microcitoma polmonare) di GHRH o ancor più raramente dalla secrezione ectopica di GH (linfomi). La diagnosi di sede può anche risultare negativa (1-2%).
Nel 25% dei casi l’adenoma ipofisario può avere secrezione mista (con PRL o più raramente TSH o ACTH) (3).
L’acromegalia può essere in qualche caso (3-5%) di origine genetica (4), anche nel quadro di sindromi più complesse: MEN-1, acromegalia familiare, FIPA, complesso di Carney, s. di McCune-Albright.
Dal punto di vista morfo-strutturale si possono distinguere (sul pezzo operatorio) due tipi principali di adenoma GH-secernente, densamente granulato e sparsamente granulato, con il secondo tipo associato a maggiore aggressività biologica (5). La distinzione però non è eseguita di routine.
EPIDEMIOLOGIA
L’acromegalia è una malattia rara (circa il 10% di tutti gli adenomi ipofisari) (6), con una prevalenza totale fra 2.8 e 13.7 casi per 100mila abitanti e un’incidenza annuale tra 0.2 e 1.1 casi/100mila abitanti (7).
La prevalenza è omogenea fra i sessi e l’età mediana di diagnosi è nella quinta decade (3,7,8).
In passato l’acromegalia è stata associata a un aumento della mortalità (9,10), ma di recente in questi pazienti è stato dimostrato un miglioramento dell’aspettativa di vita. Questo risultato è probabilmente dovuto al migliorato controllo di malattia in seguito ai progressi delle tecniche di neurochirurgia trans-sfenoidale (TNS), all’impiego di terapie farmacologiche più efficaci e all’utilizzo di criteri più rigorosi per definire la remissione di malattia. Questo andamento è stato confermato da due meta-analisi: la prima ha dimostrato una mortalità inferiore negli studi pubblicati dopo il 1995 rispetto ai precedenti (tasso standardizzato di mortalità – SMR – 1.62 vs. 2.11, rispettivamente) (11) e la seconda ha confermato che il SMR degli acromegalici non è differente da quello della popolazione generale nei 9 studi pubblicati dopo il 2008 (12), specialmente nei pazienti controllati dal punto di vista biochimico o in quelli che utilizzano analoghi della somatostatina (SSA) come trattamento adiuvante.
La causa principale di morte negli acromegalici si sta spostando progressivamente, come nella popolazione generale, dalle malattie cardio-cerebrovascolari alle neoplasie (soprattutto del polmone, colon e mammella) (10-14).
QUADRO CLINICO
Lo sviluppo della malattia dipende dall’ipersecrezione di GH (sia della secrezione basale, non dosabile nel soggetto normale, che del numero dei picchi secretori giornalieri) e di IGF-I (fattore di crescita prodotto soprattutto a livello epatico ma anche ubiquitariamente a livello tissutale). Il quadro clinico, tipico quando la malattia è conclamata, comprende:
- alterazione della fisionomia;
- aumento delle dimensioni di mani e piedi;
- macroglossia;
- abbassamento della voce;
- cefalea (indipendentemente dalle dimensioni dell’adenoma);
- ipersudorazione;
- poli-artrosi;
- astenia;
- galattorrea;
- gigantismo in epoca pre-puberale.
Nella realtà l’insorgenza delle alterazioni è così lenta che il paziente e i suoi familiari spesso non se ne accorgono. È quindi frequente un ritardo diagnostico (8-12 anni in media, più breve nei casi aggressivi), che facilita lo sviluppo di comorbilità (1,15).
COMPLICANZE
Ci possono essere danni variamente combinati (1,15,16) a carico di diversi organi e apparati.
Complicanze cardio-vascolari
Nell’acromegalia è frequente il coinvolgimento cardio-vascolare (CV) (17).
Esiste una cardiomiopatia ipertrofica specifica, che parte dall’ipertrofia bi-ventricolare concentrica con sindrome ipercinetica e poi evolve prima a disfunzione diastolica, poi a insufficienza sistolica da sforzo fino alla cardiomiopatia dilatativa con scompenso (anche se non si arriva più come in passato alla rottura di cuore). La cardiopatia è presente già alla diagnosi fino al 50% dei casi (11,17), anche se recentemente è stata riportata una frequenza inferiore (18).
Alla cardiomiopatia si può associare un’aritmia (extra-sistolia, blocchi di branca, malattia del nodo del seno, FA parossistica, TPSV e tachicardia ventricolare), soprattutto da sforzo, e i suoi effetti possono essere peggiorati dalla contemporanea presenza di ipertensione arteriosa (11,13,17).
L’ipertensione è più frequente nell’acromegalia rispetto alla popolazione generale (prevalenza media 35%) e compare in età più precoce, anche nelle prime fasi di malattia (11,19).
La prevalenza di valvulopatia (soprattutto aortica e mitralica) è molto variabile nelle diverse casistiche, dall’11% al 78% (11, 19-21).
Non sono frequenti vasculopatia periferica e cardiopatia ischemica (18).
Aterosclerosi carotidea e disfunzione endoteliale si associano ai classici fattori di rischio cardio-vascolare (18,22).
È aumentata la frequenza di eventi cerebro-vascolari e aneurismi intra-cranici (per gli aneurismi OR 4.40 rispetto al gruppo di controllo) (23). Una nuova diagnosi di aneurisma intra-cranico è stata riportata nel 17.3% dei pazienti acromegalici di una casistica italiana (24).
Sono obbligatori diagnosi precoce, monitoraggio e trattamento dei fattori di rischio e delle malattie cardio-vascolari (25).
Il position statement AME (26) dà queste indicazioni:
- raccomanda lo screening delle malattie CV alla diagnosi mediante la misurazione della pressione arteriosa e l’esecuzione di ECG ed ecocardiogramma (questo può essere posposto nei pazienti giovani senza evidenza di malattia CV);
- suggerisce l’esecuzione di angio-RM alla ricerca di aneurismi intra-cranici asintomatici;
- raccomanda il monitoraggio CV, con modalità dipendenti dalla presenza di fattori di rischio, condizioni cliniche, attività di malattia e risultati della valutazione basale.
Complicanze respiratorie
In relazione alle alterazioni anatomiche e della meccanica respiratoria, nell’acromegalia è frequente la sindrome delle apnee del sonno, soprattutto del tipo ostruttivo (OSAS), che aumenta ulteriormente il rischio CV: è riportata in una media del 69% dei casi, con un’ampia variabilità (i.e. 27-100%) (27). Di conseguenza, tutti i pazienti acromegalici alla diagnosi dovrebbero essere sottoposti a una valutazione del sonno, da ripetere nel corso del follow-up in caso di risultati patologici (25,28). Questa valutazione deve essere eseguita rapidamente soprattutto nei pazienti sintomatici, con obesità o gravi comorbilità CV, e in quelli con attività lavorativa che li pone a rischio di incidenti sul lavoro.
Il controllo di malattia può migliorare gli indici di gravità dell’OSAS (29). Nonostante la normalizzazione dell’ipersecrezione di GH e IGF-I porti a miglioramento dell’OSAS, in parecchi pazienti è però necessario mantenere la terapia con ventilazione a pressione positiva continua (CPAP), probabilmente per la presenza di alterazioni anatomiche irreversibili. Gli effetti positivi della CPAP sono strettamente dipendenti dalla compliance, motivo per cui è obbligatorio il follow-up regolare da parte di uno specialista.
Il position statement AME (26) raccomanda lo screening dell’OSAS attraverso la valutazione clinica e del punteggio di Epworth, da confermare preferibilmente con un esame polisonnografico formale.
Complicanze metaboliche
Intolleranza glucidica e diabete sono più frequenti negli acromegalici rispetto alla popolazione generale (30), con una prevalenza del 12-37%. L’ampia variabilità dipende dall’età e dalla familiarità, ma anche dalla durata e dal controllo della malattia acromegalica.
In tutti i pazienti acromegalici bisogna valutare la presenza di diabete mellito alla diagnosi e durante il follow-up. Il trattamento efficace dell’acromegalia può migliorare e anche far regredire le alterazioni del metabolismo glucidico, ma anche i farmaci impiegati nel trattamento possono talvolta influenzare tale metabolismo, in particolare pegvisomant (PegV) può migliorarlo e gli SSA possono peggiorarlo (31).
Nel 30-40% degli acromegalici sono riportate alterazioni del metabolismo lipidico, in particolare aumento di lipoproteina (a) e trigliceridi e diminuzione di colesterolo-HDL (32). Vista l’associazione di queste alterazioni con il rischio di complicanze CV e cerebro-vascolari, tutti i pazienti devono essere valutati da questo punto di vista alla diagnosi e durante il follow-up.
Il position statement AME (26):
- raccomanda il dosaggio di glicemia a digiuno e HbA1c alla diagnosi;
- suggerisce l’esecuzione di OGTT alla diagnosi in casi selezionati;
- raccomanda la valutazione dell’effetto sul metabolismo glucidico dei diversi farmaci impiegati nell’acromegalia;
- suggerisce la valutazione del profilo lipidico nei pazienti con fattori di rischio CV.
Rischio neoplastico
Negli studi epidemiologi recenti, in cui la mortalità degli acromegalici si è normalizzata e l’aspettativa di vita è aumentata, le cause principali di morte si sono spostate dalle complicanze CV e cerebro-vascolari alle patologie neoplastiche, come nella popolazione generale (11,12).
L’acromegalia è associata a moderato aumento del rischio oncologico (33). Una meta-analisi di 23 studi (34) ha indicato un tasso di incidenza standardizzato (SIR) di 1.5 per la totalità dei tumori, con intervallo di confidenza (IC) al 95% pari a 1.2-1.8. I dati epidemiologici e i registri oncologici di popolazione possono variare da paese a paese (35). In un recente studio italiano con una casistica di 1512 acromegalici (36), il SIR per tutte le neoplasie era significativamente aumentato in confronto alla popolazione generale (SIR 1.41, IC 95% 1.18-1.68, p < 0.001), con aumento in particolare del cancro a livello colo-rettale (SIR 1.67), renale (SIR 2.87) e tiroideo (SIR 3.99).
Le lesioni del colon associate all’acromegalia hanno alcuni aspetti particolari: rispetto alla popolazione non acromegalica, i polipi adenomatosi sono più grandi, multipli e più displasici (37). Né i livelli di GH né quelli di IGF-I sembrano correlarsi alla comparsa di cancro del colon, mentre quelli di IGF-I si correlano alla presenza di polipi. Non è ancora chiaro se, in assenza di altri fattori di rischio, lo screening tramite colonscopia debba essere effettuato in tutti i pazienti acromegalici o se sia meglio partire dopo i 40 anni. Sono disponibili solo risultati preliminari con l’utilizzo della colonografia computerizzata virtuale al posto della colonscopia in questa tipologia di pazienti.
L’apparente aumento della prevalenza di cancro della tiroide negli acromegalici può essere ascritto all’uso diffuso delle moderne tecniche diagnostiche, come nella popolazione generale (38). Rispetto a questa, nei pazienti acromegalici l’andamento clinico non è differente e la frequenza delle mutazioni di BRAF non è aumentata nel carcinoma papillare tiroideo (39).
Il position statement AME (26):
- raccomanda l’esecuzione di pancolonscopia alla diagnosi nei pazienti acromegalici dopo i 40 anni, da anticipare in presenza di altri fattori di rischio come la familiarità, e suggerisce di ripeterla in relazione all’attività di malattia e alle linee guida della popolazione generale;
- raccomanda l’esecuzione di ecografia tiroidea alla diagnosi nei pazienti con reperti sospetti all’esame obiettivo cervicale o con altri fattori di rischio.
Complicanze scheletriche
GH e IGF-I regolano l’omeostasi ossea. Anche se questi ormoni hanno effetto anabolico, che porta alla stimolazione del turnover e specialmente alla neoformazione ossea, i pazienti acromegalici hanno un’osteoporosi secondaria con aumentato rischio di frattura, anche in relazione ai differenti valori di BMD nei diversi siti scheletrici (40). Grado e durata della malattia attiva e ipogonadismo associato sono stati considerati i principali determinanti delle fratture vertebrali negli acromegalici (41). Nella prevenzione di tali fratture è quindi di importanza cruciale il controllo biochimico di malattia (42).
Negli acromegalici con fattori di rischio, quali ipogonadismo, iperparatiroidismo e trattamento sovra-fisiologico dei deficit di TSH e ACTH, è stato suggerito di valutare i livelli di vitamina D, i marcatori di turnover osseo, la densitometria con DXA, la morfologia ossea con radiografia del rachide in doppia proiezione e di correggere i fattori di rischio per malattia ossea e fratture (41,43).
La s. del tunnel carpale può colpire fino a 2/3 dei pazienti.
L’artropatia è una complicanza comune dell’acromegalia, presente già alla diagnosi nella maggior parte dei casi. È scarsamente passibile di miglioramento anche con trattamenti efficaci della malattia acromegalica e quindi costituisce uno dei principali determinanti di disabilità e peggioramento della qualità di vita, con frequente necessità di protesizzazione (44,45).
Il position statement AME (26):
- raccomanda di eseguire una raccolta anamnestica e un esame obiettivo accurati, con la ricerca attiva dei fattori di rischio per osteoporosi e fratture patologiche;
- raccomanda l’esecuzione di radiografia del rachide per una valutazione morfometrica e di densitometria alla diagnosi in casi selezionati, con follow-up a intervalli biennali, da eseguire in relazione ai dati strumentali iniziali, all’età e alle linee guida per la popolazione generale;
- nei pazienti con artropatia, raccomanda una collaborazione precoce con gli altri specialisti (ortopedici, fisiatri, reumatologi).
QUANDO SOSPETTARE LA MALATTIA?
Oltre alle situazioni di alterazioni fisionomiche (utile controllare vecchie fotografie per valutare modificazioni fisionomiche nel tempo), sono da considerare (soprattutto se presenti in combinazione o in gruppi di età più precoci rispetto all’insorgenza nella popolazione generale) la presenza di cefalea intrattabile, ipertensione resistente, artrosi in giovane età, malocclusione, diabete mellito 2 non obeso, iperidrosi, roncopatia, sindrome del tunnel carpale.
L’acromegalia va specificamente cercata con un dosaggio di IGF-I in tutti i pazienti che presentano iperprolattinemia patologica o con adenoma ipofisario anche di riscontro incidentale.
DIAGNOSI
Livelli ormonali
La secrezione di GH del soggetto normale (ma anche dell’acromegalico) può essere variabile nei diversi giorni e nella stessa giornata (picchi secretori). La sua secrezione è normalmente modificabile dai neuro-ormoni ipotalamici e dal glucosio. Il prelievo singolo è quindi poco utile, a meno che non sia molto basso o molto alto (se > 40 ng/mL non necessita di conferma). Va comunque ricordato che, quando il sospetto clinico è fondato, l’acromegalia si può accompagnare anche a valori di GH non particolarmente elevati (2-3 ng/mL). Il termine “micromegalia” definisce un’acromegalia a bassa attività secretoria, in cui i livelli di IGF-I sono modicamente aumentati e quelli medi di GH sono bassi (intorno a 1 ng/mL), con la mancanza delle fisiologiche fluttuazioni nell’ambito indosabile.
Allo scopo di facilitare confronto e interpretazione, è attualmente raccomandato l’uso di metodi di dosaggio del GH calibrati con la preparazione di riferimento internazionale 98/574 per hGH ricombinante 22 kDA (46).
Esistono alcune condizioni fisiologiche e patologiche in cui i livelli di GH sono elevati senza che vi sia acromegalia (vedi tabella 1), ma in tutte queste (tranne l’ipertiroidismo) i livelli di IGF-I sono bassi.
| Tabella 1 Aumento dei livelli di GH |
|
| Condizioni fisiologiche | Picchi secretori Digiuno Esercizio fisico Stress Ragazzi alti Sonno |
| Stati patologici | Diabete mellito tipo 1 Epatopatie Insufficienza renale cronica Depressione Malnutrizione Disturbi del comportamento alimentare Ipertiroidismo |
La secrezione di IGF-I è più costante nella giornata e dipendente da GH, età, sesso, stato nutrizionale, funzione epatica, ambiente estrogenico. Il dosaggio di IGF-I è il singolo dosaggio più utile nella diagnosi, con una correlazione semi-lineare con i valori medi di GH (arriva a plateau per valori di GH intorno a 20 ng/mL). Bisogna però fare attenzione al laboratorio (non tutti dispongono di un dosaggio adeguato), alla necessità di paragonare i valori a quelli di un gruppo di soggetti normali di pari età (nel soggetto normale i valori diminuiscono con l’avanzare dell’età, mentre le differenze di sesso sono clinicamente rilevanti solo in epoca peri-puberale) e ai possibili fattori interferenti (vedi tabella 2).
| Tabella 2 Interferenze sui livelli di IGF-I |
|
| Livelli aumentati | Pubertà Periodo post-puberale Ragazzi/e alti/e Gravidanza |
| Livelli diminuiti | Malattie acute Malattie sistemiche Insufficienza epatica Insufficienza renale Diabete mellito tipo 1 Digiuno Malnutrizione Estrogeni esogeni (o SERM) |
Come fare la diagnosi di malattia
Livelli di GH e IGF-I spiccatamente elevati sono sufficienti a porre la diagnosi di acromegalia. Nel caso in cui i valori di IGF-I non siano dirimenti, è consigliabile ripetere il dosaggio nello stesso laboratorio o in un altro laboratorio che usi un metodo di dosaggio differente. Altrimenti, la diagnosi deve essere confermata con OGTT.
L’OGTT per GH è un test di II livello, che diventa inutile per la diagnosi di malattia (mantiene l’utilità solo per valutare la tolleranza glucidica) allorchè, in presenza di contesto clinico, sono già patologici IGF-I e GH basali. Anche nei casi in cui è utile farlo, si osservano falsi positivi, cioè mancata soppressione (tabella 3) (47).
Sono considerati diagnostici per acromegalia la combinazione di mancata soppressione del GH (< 1 ng/mL o anche inferiore, fino a < 0.2 ng/mL, se si utilizzano dosaggi ultra-sensibili) (48,49) e aumento dei livelli di IGF-I rispetto ai limiti per età e sesso (50,51).
| Tabella 3 Mancata soppressione del GH in corso di OGTT |
|
| Ragazzi alti Adolescenza Diabete mellito Insufficienza epatica Insufficienza renale Malnutrizione Anoressia nervosa Depressione Tossicodipendenza da eroina |
Come monitorare l’attività di malattia
Nella pratica clinica corrente l’IGF-I è l’unico parametro utilizzabile per monitorare l’efficacia della terapia nei pazienti trattati con PegV.
Non è ancora definitivamente accertato se livelli di IGF-I lievemente al di sopra dell’intervallo di riferimento rappresentino un reale fattore di rischio sulla mortalità e per la morbilità correlata alle complicanze sistemiche della malattia (50,51); valori < 120-130% ULNR possono essere considerati accettabili, data la variabilità individuale spontanea o in casi specifici come il paziente anziano asintomatico. La diminuzione di IGF-I dopo intervento neurochirurgico può essere lenta, con stabilizzazione raggiunta di solito entro 3 mesi.
Il dosaggio di GH può essere utilizzato nel follow-up per definire il controllo di malattia del paziente acromegalico, tranne che in corso di terapia con PegV: la malattia può essere definita in remissione o controllata in presenza di livelli puntiformi di GH < 1.0 ng/mL (o inferiori, fino a < 0.2 ng/mL, con metodiche ultra-sensibili) (50).
È stato suggerito di impiegare lo stesso metodo di dosaggio per GH e IGF-I nel corso del follow-up del singolo paziente acromegalico (51).
L’uso dell’OGTT è raccomandato per valutare i risultati neurochirurgici (con gli stessi cut-off utilizzati in fase diagnostica) (28,50), mentre non ha nessun ruolo nel follow-up dei pazienti in trattamento farmacologico con SSA (52). Dopo l’intervento neurochirurgico, i risultati dell’OGTT sono attendibili già a una settimana nei pazienti che non erano stati trattati con terapia GH-soppressiva in fase pre-operatoria.
Con che frequenza ripetere gli esami?
Gli esami ormonali vanno ripetuti in rapporto all'attività dell’ipersecrezione ormonale o alla sua remissione:
- quando il paziente è in remissione post-chirurgica, la cadenza del follow-up deve essere annuale, mediante il dosaggio di IGF-I;
- quando il paziente è in trattamento farmacologico, la frequenza può essere molto più ravvicinata, in rapporto alla titolazione del dosaggio dei farmaci (vedi paragrafi specifici) oppure più allungata, nei casi di controllo stabile della malattia.
Interpretazione dei casi con valori discrepanti di GH e IGF-I
Al di là dei problemi analitici correlati ai dosaggi, in alcune situazioni si possono trovare valori discordanti di GH e IGF-I: gravidanza, fase post-operatoria precoce dopo adenomectomia, diabete mellito, insufficienza renale o epatica, malnutrizione, terapia estrogenica orale (53). Tale discrepanza è riportata in circa il 25% dei casi (54) ed è attribuita soprattutto all’uso di metodi ultra-sensibili di determinazione del GH e al trattamento con SSA.
Il position statement AME (26):
- raccomanda di utilizzare sia GH che IGF-I nella diagnosi e nel follow-up dei pazienti acromegalici (solo IGF-I nei trattati con PegV);
- raccomanda la normalizzazione dei livelli di IGF-I per età come obiettivo di ogni trattamento e suggerisce di considerare accettabili in alcuni pazienti valori di IGF-I < 120-130% ULNR;
- nel caso di discrepanza fra i livelli di IGF-I e GH, suggerisce di considerare più attendibile il valore di IGF-I;
- suggerisce che tutti gli specialisti che seguono pazienti acromegalici abbiano ben presenti le caratteristiche e i limiti dei metodi impiegati per il dosaggio di GH e IGF-I;
- durante il follow-up del singolo paziente, suggerisce che GH e IGF-I vengano misurati con lo stesso metodo e possibilmente nello stesso laboratorio (questa sarebbe stata una raccomandazione forte ma è stata diminuita di grado per le limitazioni della vita reale).
Diagnostica strumentale
La RM ipofisaria deve essere eseguita con apparecchiature ad alto campo, almeno 1.5 T, prima e dopo iniezione di Gadolinio, utilizzando piani coronali e sagittali, con sequenze T1- e T2-pesate. Lo spessore degli strati ipofisari deve essere di 2-3 mm.
Anche se l’invasione del seno cavernoso può essere valutata con buona accuratezza, sensibilità e specificità sono inversamente correlate e dipendono dai diversi criteri radiologici: i parametri più specifici sono la carotide interna intra-cavernosa completamente circondata da tessuto tumorale e l’obliterazione del compartimento venoso inferiore (55).
È stato riportato che durante terapia con SSA gli adenomi ipointensi in T2 hanno migliore soppressione dei valori di GH e IGF-I e più accentuata riduzione del volume tumorale rispetto a quelli iperintensi (56).
Nei pazienti acromegalici con contenuto sellare aumentato omogeneamente, senza chiara evidenza di adenoma, bisogna sospettare e ricercare una secrezione ectopica di GHRH, inizialmente con TC toraco-addominale, seguita poi da diagnostica per immagini medico-nucleare. La secrezione ectopica di GHRH è dimostrata definitivamente con il dosaggio dei livelli circolanti di GHRH, che però non è ampiamente disponibile.
La tempistica per il follow-up neuroradiologico post-neurochirurgia deve essere individualizzata in relazione ai risultati ormonali:
- pazienti con risposta patologica di GH a OGTT: la RM può essere eseguita a 3-4 mesi dall’intervento (per permettere l’eliminazione della componente edematosa);
- in quelli con normalizzazione ormonale: si può tranquillamente eseguire a 6 mesi.
Il follow-up neuroradiologico in corso di terapia farmacologica deve prevedere sempre il confronto dell’immagine attuale non solo con la precedente ma anche con la prima post-chirurgica, per valutare piccole variazioni progressive del volume tumorale.
Nei pazienti con remissione ormonale persistente dopo neurochirurgia e in quelli con malattia stabilmente controllata dalla terapia farmacologica, la tempistica dei controlli RM può essere progressivamente allungata con sicurezza, anche fino alla sospensione, dato che la dissociazione tra il controllo ormonale e quello tumorale è una possibilità molto remota. Visto il dibattito sugli effetti a lungo termine dell’esposizione a Gadolinio, si può ipotizzare l’uso di RM senza contrasto nel follow-up di pazienti con malattia stabilizzata, in cui sia ben chiara la posizione del residuo tumorale.
La RM non è solitamente necessaria in gravidanza e nel post-partum. Deve essere presa in considerazione (ed essere eseguita senza Gadolinio) solo in circostanze particolari, come malattia non controllata in peggioramento con grave cefalea, deficit visivo o paralisi dei nervi cranici.
È raccomandata l’esecuzione di campimetria nei casi di macroadenoma contiguo alle vie ottiche.
Per gli esami strumentali relativi alle complicanze, vedi i relativi paragrafi.
QUANDO PENSARE A UN'ORIGINE GENETICA?
Bisogna sospettare una base genetica in alcune situazioni cliniche:
- acromegalici giovani (< 30 anni), specialmente in caso di gigantismo (in questo gruppo le forme genetiche possono essere fino al 50% dei casi), particolarmente se prima dei 10 anni;
- acromegalia presente in più membri della stessa famiglia;
- altri tipi di adenoma ipofisario presenti in altri membri della famiglia;
- contemporanea presenza di clinica suggestiva per MEN-1 o altre patologie multi-organo (Carney, McCune-Albright) (tab 4).
È obbligatorio raccogliere un’anamnesi familiare e personale dettagliata, eseguire un esame obiettivo accurato e la misurazione della calcemia per la valutazione dell’iperparatiroidismo (e quindi di una potenziale MEN-1).
In questi casi, è opportuno eseguire una valutazione genetica in base al sospetto clinico, ma questa è opportuno venga eseguita nei pochi centri di riferimento in grado di dare risposte attendibili.
| Tabella 4 Forme familiari di acromegalia (modificata da 57) |
||
| Gene implicato | Prevalenza nell’acromegalia | Fenotipo (oltre all’acromegalia) |
| AIP | 50% in FIPA omogenei 4% in acromegalia apparentemente sporadica |
FIPA, pazienti più giovani, tumori invasivi, poco responsivi a SSA |
| MEN-1 | 1.2% | MEN-1: iperparatiroidismo e tumori neuroendocrini pancreatici |
| CDKN1B | Rara | MEN-4: iperparatiroidismo e tumori neuroendocrini pancreatici |
| PRKARIA | 65% dei pazienti con complesso di Carney | Complesso di Carney: mixomi cardiaci e cutanei, PPNAD e lesioni pigmentate di cute e mucose |
| SDHx | Rara | Sindrome delle 3 P: PGL/Feocromocitoma |
| GPR101 | 0-4.4% | X-LAG (gigantismo a insorgenza molto precoce) |
| GNAS | 40% | Sindrome di McCune-Albright |
Nella maggior parte dei casi familiari (50-86%) è presente un macroadenoma, con andamento aggressivo/invasivo in circa il 31-50% dei casi.
Le indagini genetiche devono essere eseguite secondo questa falsariga:
- in tutti i casi di FIPA e gigantismo ricercare le mutazioni di AIP (in questo ambito sono riportate fino al 50% dei casi);
- nel caso di accelerazione della crescita staturale nella prima infanzia sospettare X-LAG;
- nell’appropriato contesto clinico sospettare altre sindromi MEN.
Bisogna eseguire uno screening nei parenti dei portatori del gene mutato, perché gli esiti sono migliori nel caso di malattia diagnosticata a seguito di screening (58).
BIBLIOGRAFIA
- Melmed S. Medical progress: Acromegaly. N Engl J Med 2006, 355: 2558-73.
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest 2009, 119: 3189–202.
- Petrossians P, Daly AF, Natchev E, et al. Acromegaly at diagnosis in 3173 patients from the Liège Acromegaly Survey (LAS) Database. Endocr Relat Cancer 2017, 24: 505-18.
- Trivellin G, Daly AF, Faucz FR, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med 2014, 371: 2363-74.
- Cuevas-Ramos D, Carmichael JD, Cooper O, et al. A structural and functional acromegaly classification. J Clin Endocrinol Metab 2015, 100: 122-31.
- Fernandez A, Karavitaki N, Wass AH. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf) 2010, 72: 377-82.
- Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary 2017, 20: 4-9.
- Holdaway IM, Rajasoorya C. Epidemiology of acromegaly. Pituitary 1999, 2: 29-41.
- Dekkers OM, Biermasz NR, Pereira AM, et al. Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab 2008, 93: 61–7.
- Ritvonen E, Löyttyniemi E, Jaatinen P, et al. Mortality in acromegaly: a 20-year follow-up study. Endocr Relat Cancer 2016, 23: 469-80.
- Maione L, Brue T, Beckers A, et al, French Acromegaly Registry. Changes in the management and comorbidities of acromegaly over three decades: the French Acromegaly Registry. Eur J Endocrinol 2017, 176: 645-55.
- Bolfi F, Neves AF, Boguszewski CL, Nunes-Nogueira VS. Mortality in acromegaly decreased in the last decade: a systematic review and meta-analysis. Eur J Endocrinol 2018, 179: 59–71.
- Arosio M, Reimondo G, Malchiodi E, et al, Italian Study Group of Acromegaly. Predictors of morbidity and mortality in acromegaly: an Italian survey. Eur J Endocrinol 2012, 167: 189–98.
- Esposito D, Ragnarsson O, Granfeldt D, et al. Decreasing mortality and changes in treatment patterns in patients with acromegaly from a nationwide study. Eur J Endocrinol 2018, 178: 459-69.
- Nachtigall L, Delgado A, Swearingen B, et al. Changing patterns in diagnosis and therapy of acromegaly over two decades. J Clin Endocrinol Metab 2008, 93: 2035-41.
- Colao A, Ferone D, Marzullo P, et al. Systemic complications of acromegaly: epidemiology, pathogenesis and management. Endocr Rev 2004, 25: 102-52.
- Pivonello R, Auriemma RS, Grasso LF, et al. Complications of acromegaly: cardiovascular, respiratory and metabolic comorbidities. Pituitary 2017, 20: 46-62.
- Warszawski L, Kasuki L, Sá R, et al. Low frequency of cardiac arrhythmias and lack of structural heart disease in medically-naïve acromegaly patients: a prospective study at baseline and after 1 year of somatostatin analogs treatment. Pituitary 2016, 19: 582-9.
- Ramos-Leví AM, Marazuela M. Cardiovascular comorbidities in acromegaly: an update on their diagnosis and management. Endocrine 2017, 55: 346-59.
- Sharma AN, Tan M, Amsterdam EA, Singh GD. Acromegalic cardiomyopathy: Epidemiology, diagnosis, and management. Clin Cardiol 2018, 41: 419-25.
- Colao A, Spinelli L, Marzullo P, et al. High prevalence of cardiac valve disease in acromegaly: an observational, analytical, case-control study. J Clin Endocrinol Metab 2003, 88: 3196–201.
- Parolin M, Dassie F, Martini C, et al. Preclinical markers of atherosclerosis in acromegaly: a systematic review and meta-analysis. Pituitary 2018, 21: 653-62.
- Oshino S, Nishino A, Suzuki T, et al. Prevalence of cerebral aneurysm in patients with acromegaly. Pituitary 2013, 16: 195-201.
- Manara R, Maffei P, Citton V, et al. Increased rate of intracranial saccular aneurysms in acromegaly: an MR angiography study and review of the literature. J Clin Endocrinol Metab 2011, 96: 1292-300.
- Melmed S, Casanueva FF, Klibanski A, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary 2013, 16: 294–302.
- Cozzi R, et al. Italian Association of Clinical Endocrinologists (AME) and Italian AACE Chapter position statement for clinical practice: acromegaly - part 1: diagnostic and clinical issues. Endocr Metab Immune Disord Drug Targets 2020, DOI: 10.2174/1871530320666200127103320.
- Attal P, Chanson P. Endocrine aspects of obstructive sleep apnea. J Clin Endocrinol Metab 2010, 95: 483-95.
- Katznelson L, Atkinson JL, Cook DM, et al; American Association of Clinical Endocrinologists. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly—2011 update. Endocr Pract 2011, 17 (suppl 4): 1-44.
- Pagano C, Parolin M, Dassie F, et al. Obstructive sleep apnea in acromegaly and the effect of treatment: a systematic review and meta-analysis. J Clin Endocrinol Metab 2020, 105: e23-31.
- Dreval AV, Trigolosova IV, Misnikova Y, et al. Prevalence of diabetes mellitus in patients with acromegaly. Endocr Connect 2014, 3: 93–8.
- Abreu A, Tovar AP, Castellanos R, et al. Challenges in the diagnosis and management of acromegaly: a focus on comorbidities. Pituitary 2016, 19: 448-57.
- Vilar L, Vilar CF, Lyra R, et al. Acromegaly: clinical features at diagnosis. Pituitary 2017, 20: 22-32.
- Boguszewski CL, Ayuk J. Acromegaly and cancer risk: an old debate revisited. Eur J Endocrinol 2016, 175: R147-56.
- Dal J, Leisner MZ, Hermansen K, et al. Cancer incidence in patients with acromegaly: a cohort study and meta-analysis of the literature. J Clin Endocrinol Metab 2018, 103: 2182–8.
- Petroff D, Tönjes A, Grussendorf M, et al. The incidence of cancer among acromegaly patients: results from the German Acromegaly Registry. J Clin Endocrinol Metab 2015, 100: 3894-902.
- Terzolo M, Reimondo G, Berchialla P, et al; Italian Study Group of Acromegaly. Acromegaly is associated with increased cancer risk: a survey in Italy. Endocr Relat Cancer 2017, 24: 495–504.
- Lois K, Bukowczan J, Perros P, et al. The role of colonoscopic screening in acromegaly revisited: review of current literature and practice guidelines. Pituitary 2015, 18: 568-74.
- Vaccarella S, Franceschi S, Bray F, et al. Worldwide thyroid-cancer epidemic? The increasing impact of overdiagnosis. N Engl J Med 2016, 375: 614-7.
- Aydin K, Aydin C, Dagdelen S, et al. Genetic alterations in differentiated thyroid cancer patients with acromegaly. Exp Clin Endocrinol Diabetes 2016, 124: 198–202.
- Bima C, Chiloiro S, Mormando M, et al. Understanding the effect of acromegaly on the human skeleton. Expert Rev Endocrinol Metab 2016, 11: 263-70.
- Mazziotti G, Biagioli E, Maffezzoni F, et al. Bone turnover, bone mineral density and fracture risk in acromegaly: a meta-analysis. J Clin Endocrinol Metab 2015 100: 384-94.
- Chiloiro S, Mazziotti G, Giampietro A, et al. Effects of pegvisomant and somatostatin receptor ligands on incidence of vertebral fractures in patients with acromegaly. Pituitary 2018, 21: 302-8.
- Mazziotti G, Lania AGA, Canalis E. Bone disorders associated with acromegaly: mechanisms and treatment. Eur J Endocrinol 2019, 181: R45–56.
- Claessen KMJA, Mazziotti G, Biermasz NR, Giustina A. Bone and joint disorders in acromegaly. Neuroendocrinology 2016, 103: 86-95.
- Fatti LM, Cangiano B, Vitale G, et al, on behalf of The Study Group on Motor Disability in Acromegaly of the Italian Society of Endocrinology. Arthropathy in acromegaly: a questionnaire-based estimation of motor disability and its relation with quality of life and work productivity. Pituitary 2019, 22: 552-60.
- Junnila RK, Strasburger CJ, Bidlingmaier M. Pitfalls of insulin-like growth factor-I and growth hormone assays. Endocrinol Metab Clin N Am 2015, 44: 27–34.
- Arafat AM, Mohlig M, Weickert MO, et al. Growth hormone response during oral glucose tolerance test: the impact of assay method on the estimation of reference values in patients with acromegaly and in healthy controls, and the role of gender, age, and body mass index. J Clin Endocrinol Metab 2008, 93: 1254–62.
- Freda PU, Post KD, Powell JS, Wardlaw SL. Evaluation of disease status with sensitive measures of growth hormone secretion in 60 postoperative patients with acromegaly. J Clin Endocrinol Metab 1998, 83: 3808-16.
- Schilbach K, Gar C, Lechner A, et al. Determinants of the growth hormone nadir during oral glucose tolerance test in adults. Eur J Endocrinol 2019, 181: 55-67.
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014, 99: 3933-51.
- Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: a consensus on the medical treatment of acromegaly. Nat Rev Endocrinol 2014, 10: 243–8.
- Reimondo G, Bondanelli M, Ambrosio MR, et al. Growth hormone values after an oral glucose load do not add clinically useful information in patients with acromegaly on long-term somatostatin receptor ligand treatment. Endocrine 2014, 45: 122-7.
- Schilbach K, Strasburger CJ, Bidlingmaier M. Biochemical investigations in diagnosis and follow up of acromegaly. Pituitary 2017, 20: 33–45.
- Kanakis GA, Chrisoulidou A, Bargiota A, et al. The ongoing challenge of discrepant growth hormone and insulin-like growth factor I results in the evaluation of treated acromegalic patients: a systematic review and meta-analysis. Clin Endocrinol 2016, 85: 681–8.
- Dhandapani S, Singh H, Negm HM, et al. Cavernous sinus invasion in pituitary adenomas: systematic review and pooled data meta-analysis of radiologic criteria and comparison of endoscopic and microscopic surgery. World Neurosurg 2016, 96: 36-46.
- Potorac I, Petrossians P, Daly AF, et al. T2-weighted MRI signal predicts hormone and tumor responses to somatostatin analogs in acromegaly. Endocr Relat Cancer 2016, 23: 871-81.
- Gadelha M, Kasuki L, Korbonits M. The genetic background of acromegaly. Pituitary 2017, 20: 10-21.
- de Laat JM, Dekkers OM, Pieterman CR, et al. Long-term natural course of pituitary tumors in patients with MEN1: results from the DutchMEN1 study group (DMSG). J Clin Endocrinol Metab 2015, 100: 3288-96.
Terapia acromegalia e gigantismo
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
(aggiornato al 12 aprile 2020)
Obiettivi
Studi epidemiologici hanno dimostrato che la maggiore mortalità che si osserva nella malattia attiva viene ricondotta a quella della popolazione di controllo se si riportano i livelli di GH sotto 2-2.5 µg/L e si normalizzano quelli di IGF-I per gruppo di età (1).
Il trattamento ideale (2-5) normalizza rapidamente l’ipersecrezione di GH e IGF-I, elimina il tumore e i sintomi e segni della malattia acromegalica, fa regredire le comorbilità, riporta la mortalità a quella della popolazione di riferimento e migliora la qualità della vita (QoL), che può essere misurata con un questionario specifico (AcroQOL). Nonostante i fenomenali progressi degli attuali trattamenti rispetto a quanto disponibile in passato, siamo ancora ben lontani dall’aver raggiunto tutti questi obiettivi.
Ogni paziente di nuova diagnosi dovrebbe essere indirizzato a un’equipe multi-disciplinare con esperienza nel trattamento degli adenomi ipofisari, che comprenda almeno endocrinologi, neuroradiologi, neurochirurghi e radioterapisti (6).
Le 3 opzioni terapeutiche disponibili, neurochirurgica, farmacologica e radiante, non sono da considerare come mutuamente esclusive, ma come integrate e complementari.
Strategia terapeutica
La gestione terapeutica del paziente acromegalico richiede una strategia personalizzata che tenga conto di (2,3,7,8):
- caratteristiche cliniche del paziente (età, comorbilità);
- aspetto neuroradiologico dell’adenoma ed entità della secrezione ormonale;
- caratteristiche della malattia (aggressività);
- aspettative del paziente (desiderio di fertilità, accettazione dell’indicazione chirurgica).
L’intervento TNS eseguito da un neurochirurgo esperto è la modalità di trattamento primaria per la maggior parte dei pazienti, con micro-o macro-adenoma, specialmente nel caso vi siano complicanze neurologiche (danno visivo, ipertensione endocranica, coinvolgimento dei nervi cranici), perché è l’unica potenzialmente in grado di guarire definitivamente l’acromegalia. Il reintervento deve essere proposto solo ai pazienti in cui il primo intervento è stato inefficace con voluminoso residuo, o nel caso molto raro di tumore aggressivo non controllabile con la terapia farmacologica.
Nei pazienti con adenoma ampiamente invasivo e prevedibile insuccesso neurochirurgico, o condizioni cliniche scadute o che rifiutano l’intervento, bisogna proporre una terapia farmacologica primaria. Gli SSA depot di prima generazione ottengono i migliori risultati e devono essere somministrati alla dose massimale per ottenere la massima inibizione di GH/IGF-I.
La cabergolina deve essere riservata ai pazienti con ipersecrezione lieve di GH/IGF-I, indipendentemente dai livelli di PRL.
Nei pazienti non sottoposti a neurochirurgia, in cui il trattamento farmacologico primario non è riuscito a ottenere livelli di IGF-I <120-130% ULNR e/o a tenere sotto controllo le dimensioni tumorali, deve essere raccomandato l’intervento TNS.
Nei pazienti con persistenza di valori patologici di GH/IGF-I dopo neurochirurgia, deve essere avviato il trattamento farmacologico con SSA depot di prima generazione o cabergolina (questa in caso di ipersecrezione lieve), in mono-terapia o in combinazione in caso di risposta parziale agli SSA. Ai pazienti che non raggiungono l’obiettivo terapeutico con questa strategia, devono essere proposte due alternative:
- pasireotide LAR, indipendentemente dalla resistenza totale o parziale agli SSA di prima generazione;
- PegV, in mono-terapia o in combinazione con SSA nel caso sia presente un voluminoso residuo tumorale.
In questa scelta bisogna prendere in considerazione diversi fattori: compenso gluco-metabolico, dimensioni e localizzazione del residuo tumorale, costo dei farmaci e preferenze individuali.
Nei pazienti con cefalea non responsiva agli SSA di 1° generazione è fortemente raccomandato l’utilizzo di pasireotide LAR.
Nei pazienti con minimo residuo chirurgico che vogliono evitare una terapia farmacologica a lungo termine, la radiochirurgia rappresenta un’opzione eccellente. I pazienti con attività di malattia persistente o crescita tumorale nonostante chirurgia e terapia farmacologica, devono essere irradiati con la tecnica appropriata in relazione a dimensioni e localizzazione del residuo tumorale.
La strategia individualizzata per la gestione terapeutica dell’acromegalia deve sempre considerare il costo complessivo di ogni scelta. Questo non si deve limitare a considerare solo il prezzo grezzo di ogni farmaco (o procedura), ma deve sempre prevedere una valutazione integrata di eventuali effetti collaterali, QoL, trattamento delle comorbilità, giorni lavorativi persi, ecc.
Riassumendo, si deve proporre:
- come prima terapia:
- la neurochirurgia nei pazienti con:
- complicanze neurologiche, con l’obiettivo di migliorare acutamente il quadro neuro-oftalmologico (anche se l’intervento non può ottenere la guarigione);
- adenoma non invasivo, sia micro- che macro-, senza comorbilità (con l’obiettivo della guarigione);
- la terapia farmacologica con SSA depot di prima generazione in caso di:
- adenoma invasivo (in cui la guarigione chirurgica è improbabile);
- condizioni cliniche scadute;
- rifiuto della chirurgia;
- mancanza di chirurgo valido (se il paziente non può essere inviato in una sede più adeguata);
- come terapia adiuvante (in caso di persistenza di malattia dopo chirurgia):
- SSA depot di prima generazione, che dopo debulking efficace (> 75%) normalizzano frequentemente l’ipersecrezione (9,10);
- cabergolina in caso di acromegalia lieve (11);
- nei casi di resistenza/intolleranza agli SSA: pegvisomant o pasireotide LAR;
- la radioterapia nei casi di inefficacia o rifiuto delle terapie precedenti o quando il paziente voglia evitare una terapia cronica.
- la neurochirurgia nei pazienti con:
Caso particolare è quello della terapia in gravidanza e degli adenomi aggressivi.
Bibliografia
- Holdaway IM, Bolland MJ, Gamble GD. A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol 2008, 159: 89-95.
- Cozzi R, Ambrosio MR, Attanasio R, et al. Italian Association of Clinical Endocrinologists (AME) and Italian AACE chapter position statement for clinical practice: acromegaly - part 2: therapeutic issues. Endocr Metab Immune Disord Drug Targets 2020, DOI: 10.2174/1871530320666200129113328.
- American Association of Clinical Endocrinologists Acromegaly Guidelines Task Force. Medical guidelines for clinical practice for the diagnosis and treatment of acromegaly. Endocr Pract 2011, 17 suppl 4: 1-44.
- Giustina A, Barkan A, Casanueva FF, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab 2000, 85: 526-9.
- Giustina A, Chanson P, Bronstein MD, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab 2010, 95: 3141–8.
- Casanueva FF, Barkan AL, Buchfelder M, et al; Pituitary Society, Expert Group on Pituitary Tumors. Criteria for the definition of Pituitary Tumor Centers of Excellence (PTCOE): A Pituitary Society Statement. Pituitary 2017, 20: 489-98.
- Colao A, Martino E, Cappabianca P, et al, and the participants of the A.L.I.C.E. Study Group. First-line therapy of acromegaly: A statement of the A.L.I.C.E. (Acromegaly primary medical treatment Learning and Improvement with Continuous Medical Education) Study Group. J Endocrinol Invest 2006, 29: 1017–20.
- Melmed S, Colao A, Barkan A, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab 2009, 94: 1509-17.
- Petrossians P, Martins LB, Espinoza C, et al. Gross total resection or debulking of pituitary adenomas improves hormonal control of acromegaly by somatostatin analogs. Eur J Endocrinol 2005, 152: 1-7.
- Colao AM, Attanasio R, Pivonello R, et al. Partial surgical removal of growth hormone-secreting pituitary tumors enhances the response to somatostatin analogs in acromegaly. J Clin Endocrinol Metab 2006, 91: 85-92.
- Sandret L, Maison P, Chanson P. Place of cabergoline in acromegaly: a meta-analysis. J Clin Endocrinol Metab 2011, 96: 1327–35.
Terapia neurochirurgica dell'acromegalia
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
(aggiornato al 12 aprile 2020)
L’intervento TNS, con tecnica microscopica o endoscopica, è la procedura di scelta ogni volta che sia praticabile, in grado di guarire definitivamente l’acromegalia con effetti immediati (1,2).
Obiettivo
Asportazione completa dell’adenoma GH-secernente, con eliminazione dell’effetto massa e conservazione della funzione ipofisaria.
Tecnica
L’intervento è praticabile per via trans-sfenoidale (TNS) con approccio microscopico o endoscopico secondo preferenza ed esperienza del chirurgo; solo in casi rari viene utilizzata la via trans-cranica.
Risultati
La percentuale di successo dipende (3-7) da:
- soprattutto invasività del tumore (c’è un progressivo peggioramento dei risultati se si passa dai microadenomi o macroadenomi completamente intra-sellari, ai tumori ad estensione extra-sellare, soprattutto nel seno cavernoso che è la localizzazione critica per la scarsa resecabilità, agli adenomi giganti);
- livelli pre-operatori di GH (le percentuali di guarigione sono inversamente proporzionali ai livelli di GH);
- esperienza del neurochirurgo (questo fattore influenza anche la prevalenza di effetti collaterali).
I lavori più recenti dei neurochirurghi con alti volumi operatori riportano percentuali di remissione post-chirurgica fino al 75% per i macroadenomi non invasivi e fino all’85% per i microadenomi (8-10). Il tasso di remissione in caso di invasione del seno cavernoso è complessivamente del 40%, che si azzera in caso di invasione di grado 4 secondo Knosp (8,11).
I tassi di remissione sono molto inferiori (dal 20% al 37%) nel mondo reale, cioè secondo quanto riportato nei registri di malattia che combinano i risultati di chirurghi con diversi gradi di esperienza (12). Questo sottolinea la necessità di inviare i pazienti presso centri di riferimento con alti volumi.
Non esistono studi prospettici di confronto dei risultati fra le tecniche microscopica ed endoscopica, ma gli studi retrospettivi non hanno dimostrato differenze (13).
Per quanto riguarda il deficit visivo, questo migliora o si normalizza nella stragrande maggioranza dei casi, se non è passato troppo tempo dalla sua insorgenza (più di qualche settimana).
La funzione ipofisaria migliora o viene normalizzata nel 35-45% dei pazienti con deficit, non si modifica in metà e può peggiorare nel 2-6% dei casi senza deficit pre-chirurgico (2,14).
Come preparare il paziente all’intervento
Se è necessaria una decompressione chirurgica urgente per gravi problemi oftalmici o neurologici, c’è solo il tempo di valutare la funzione surrenalica e tiroidea, instaurando una terapia sostitutiva in caso di dimostrato deficit. È comunque vero che in tali casi si somministreranno steroidi a dosi generose, a scopo anti-edemigeno, by-passando del tutto il problema.
È stato suggerito che un trattamento pre-chirurgico con SSA possa migliorare la radicalità della resezione chirurgica (15), ma i dati disponibili non sono a favore dell’utilizzo routinario di tale pratica (1,15-24). Il trattamento pre-chirurgico può essere preso in considerazione per ridurre la morbilità peri-operatoria, in caso di pazienti con gravi complicanze cardio-vascolari o respiratorie dipendenti dall’acromegalia (1,15), anche se i dati pubblicati in proposito sono contrastanti (25).
Nei portatori di adenomi invasivi con risposta solo parziale agli SSA, si può considerare l’opportunità di un debulking chirurgico, che può portare a una migliore risposta post-operatoria agli SSA e fornire materiale per le analisi patologiche (1,26,27).
Dove e come operare
È opportuno che il paziente in cui una valutazione inter-disciplinare abbia posto indicazione all’intervento, venga avviato ad un centro con esperienza specifica di chirurgia ipofisaria, dove un singolo neurochirurgo abbia maturato una sufficiente esperienza e casistica personale (almeno 25 interventi ipofisari/anno). Bisogna evitare sia di mandare il paziente dal chirurgo “sotto casa”, sia di inviarlo a quei centri dove si alternano parecchi chirurghi, nessuno dei quali potrà accumulare esperienza specifica (28).
Come e quando valutare il risultato dell’intervento
Attenzione: il paziente acromegalico operato con successo ha una diuresi molto attiva nei giorni successivi all’intervento, perché elimina i liquidi accumulati dalla malattia. Questa situazione non deve essere confusa con un diabete insipido post-operatorio: anche in presenza di poliuria, il paziente acromegalico operato non è polidipsico e la sodiemia si mantiene normale.
I risultati dell’intervento sono valutati dosando i livelli di IGF-I (che devono essere analizzati in relazione all’età) e la soppressione di GH dopo OGTT. Il cut-off che distingue la guarigione chirurgica è stato progressivamente abbassato, con il miglioramento delle conoscenze fisiopatologiche, l’affinamento delle tecniche NCH e l’incremento della sensibilità dei dosaggi di GH. Attualmente è stato fissato a 0.4 ng/mL (29).
Il momento giusto per la valutazione del risultato dipende da quello che è stato fatto prima dell’intervento.
- In tutti i casi, valutare l’eventuale danno della funzione ipofisaria (come negli interventi NCH per ogni tipo di adenoma) con un prelievo in 2°-3° giornata per cortisolemia e FT4.
- Nei pazienti che non hanno fatto trattamento farmacologico GH-soppressivo pre-chirurgico, la valutazione del GH dopo OGTT è attendibile già dopo una settimana dall’intervento (30).
- Nei pazienti pre-trattati, vista la coda dell’effetto farmacologico, è opportuno rimandare la valutazione a 6-12 settimane (intervallo più lungo nei pazienti trattati più a lungo).
- Il dosaggio dell’IGF-I può non essere dirimente fino a 3 mesi dopo l’intervento.
- Il controllo della RM va fatto non prima di 3-4 mesi, per evitare immagini difficilmente valutabili dovute a processi infiammatori, di cicatrizzazione e rimaneggiamento del materiale posto dal neurochirurgo per chiudere la breccia ossea e durale, ma può essere tranquillamente posposto a 6 mesi in quelli che hanno ottenuto la normalizzazione ormonale (31).
Come e per quanto tempo proseguire il follow-up dopo chirurgia
Nei pazienti in remissione al controllo post-operatorio (GH < 0.4 ng/mL, IGF-I normalizzata) è opportuno proseguire un follow-up di minima, perché sono descritte recidive fino al 15% dei casi (32), anche se in realtà sono molto rare nei pazienti che raggiungono il cut-off di 0.4 ng/mL. Potrebbe essere sufficiente il solo dosaggio annuale di IGF-I; se sono presenti difficoltà metodologiche del dosaggio, è consigliabile aggiungere il dosaggio del GH dopo OGTT almeno ogni 2 anni.
Nei pazienti in remissione clinica e biochimica e in cui la prima immagine RM post-chirurgica era negativa, non è necessario fare una valutazione neuro-radiologica seriata (31), perché in ogni caso l’eventuale ripresa di malattia verrà intercettata dagli esami ormonali prima che si possano avere problemi di massa tumorale.
Come comportarsi nei casi ambigui
Nella maggior parte dei casi è possibile dare un giudizio sulla guarigione o persistenza della malattia dopo 3-4 mesi dall’intervento. Nei casi di persistenza, bisognerà decidere con quale terapia proseguire, tenendo presente che se c’è stato un efficace debulking del tumore (di almeno ¾ della massa tumorale), la terapia farmacologica con gli analoghi della somatostatina può diventare più efficace e normalizzare i valori di IGF-I, anche se come primo trattamento non aveva raggiunto la normalizzazione dei valori ormonali (26,33).
In circa il 30% dei pazienti esiste discrepanza fra i valori di GH e IGF-I dopo l’intervento (34). I valori di IGF-I sono più legati all’attività clinica di malattia e quindi da considerare con maggiore sospetto. Al contrario, la mancata soppressione di GH con IGF-I normale, se si escludono possibili fattori interferenti sul dosaggio di IGF-I, è probabilmente da attribuire a problemi metodologici. In entrambi i casi, comunque, il paziente va tenuto sotto controllo attento, ripetendo gli esami ogni 3-6 mesi fino a chiarire l’evoluzione (35).
Re-intervento
È un’opzione praticabile nel caso di voluminosi tumori intra-sellari persistenti o recidivanti, resistenti ai trattamenti farmacologici disponibili, anche se è gravato da una percentuale di complicanze lievemente superiore (36-38).
Il position statement AME (32):
- raccomanda di inviare il paziente a un’equipe neurochirurgica esperta;
- raccomanda la neurochirurgia come prima linea se l’adenoma è totalmente resecabile ed è prevedibile la remissione ormonale o se l’adenoma provoca effetto massa, in particolare deficit campimetrico;
- suggerisce la chirurgia di debulking nei pazienti resistenti alle terapie farmacologiche.
Bibliografia
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014, 99: 3933-51.
- Li A, Liu W, Cao P, et al. Endoscopic versus microscopic transsphenoidal surgery in the treatment of pituitary adenoma: a systematic review and meta-analysis. World Neurosurg 2017, 101: 236-46.
- De P, Rees DA, Davies N, et al. Transsphenoidal surgery for acromegaly in Wales: results based on stringent criteria of remission. J Clin Endocrinol Metab 2003, 88: 3567-72.
- Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical 'cure'. Eur J Endocrinol 2005, 152: 379-87.
- Lissett C, Peacey S, Laing I, et al. The outcome of surgery for acromegaly: the need for a specialist pituitary surgeon for all types of growth hormone (GH) secreting adenoma. Clin Endocrinol (Oxford) 1998, 49: 653–7.
- Ahmed S, Elsheikh M, Stratton IM, et al. Outcome of transphenoidal surgery for acromegaly and its relationship to surgical experience. Clin Endocrinol (Oxford) 1999, 50: 561–7.
- Wang YY, Higham C, Kearney T, et al. Acromegaly surgery in Manchester revisited - The impact of reducing surgeon numbers and the 2010 consensus guidelines for disease remission. Clin Endocrinol (Oxf) 2012, 76: 399–406.
- Anik I, Cabuk B, Gokbel A, et al. Endoscopic transsphenoidal approach for acromegaly with remission rates in 401 patients: 2010 consensus criteria. World Neurosurg 2017, 108: 278-90.
- Jane JA Jr, Starke RM, Elzoghby MA, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab 2011, 96: 2732–40.
- Honegger J, Grimm F. The experience with transsphenoidal surgery and its importance to outcomes. Pituitary 2018, 21: 545-55.
- Briceno V, Zaidi HA, Doucette JA, et al. Efficacy of transsphenoidal surgery in achieving biochemical cure of growth hormone-secreting pituitary adenomas among patients with cavernous sinus invasion: a systematic review and meta-analysis. Neurol Res 2017, 39: 387-98.
- Bates PR, Carson MN, Trainer PJ, Wass JAH, and the UK National Acromegaly Register Study Group (UKAR-2). Wide variation in surgical outcomes for acromegaly in the UK. Clin Endocrinol 2008, 68: 136–42.
- Starke RM, Raper DMS, Payne SC, et al. Endoscopic vs microsurgical transsphenoidal surgery for acromegaly: outcomes in a concurrent series of patients using modern criteria for remission. J Clin Endocrinol Metab 2013, 98: 3190–8.
- Fatemi N, Dusick JR, Mattozo C, et al. Pituitary hormone loss and recovery after transsphenoidal adenoma removal. Neurosurgery 2008, 63: 709-19.
- Fleseriu M, Hoffman AR, Katznelson L; AACE Neuroendocrine and Pituitary Scientific Committee. American Association of Clinical Endocrinologists and American College of Endocrinology disease state clinical review: management of acromegaly patients: what is the role of pre-operative medical therapy? Endocr Pract 2015, 21: 668-73.
- Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: a consensus on the medical treatment of acromegaly. Nat Rev Endocrinol 2014, 10: 243–8.
- Carlsen SM, Lund-Johansen M, Schreiner T, et al, on behalf of the POTA study group. Preoperative octreotide treatment in newly diagnosed acromegalic patients with macroadenomas increases cure short-term postoperative rates: a prospective, randomized trial. J Clin Endocrinol Metab 2008, 93: 2984–90.
- Mao Z, Zhu Y, Tang H, et al. Preoperative lanreotide treatment in acromegalic patients with macroadenomas increases short-term postoperative cure rates: a prospective, randomised trial. Eur J Endocrinol 2010, 162: 661–6.
- Li ZQ, Quan Z, Tian HL, Cheng M. Preoperative lanreotide treatment improves outcome in patients with acromegaly resulting from invasive pituitary macroadenoma. J Int Med Res 2012, 40: 517–24.
- Biermasz NR, van Dulken H, Roelfsema F. Direct postoperative and follow-up results of transsphenoidal surgery in 19 acromegalic patients pretreated with octreotide compared to those in untreated matched controls. J Clin Endocrinol Metab 1999, 84: 3551–5.
- Losa M, Mortini P, Urbaz L, et al. Presurgical treatment with somatostatin analogs in patients with acromegaly: effects on the remission and complication rates. J Neurosurg 2006, 104: 899-906.
- Nunes VS, Correa JMS, Puga MES, et al. Preoperative somatostatin analogues versus direct transsphenoidal surgery for newly-diagnosed acromegaly patients: a systematic review and meta-analysis using the GRADE system. Pituitary 2015, 18: 500-8.
- Shen M, Shou X, Wang Y, et al. Effect of presurgical long-acting octreotide treatment in acromegaly patients with invasive pituitary macroadenomas: a prospective randomized study. Endocr J 2010, 57: 1035-44.
- Pita-Gutierrez F, Pertega-Diaz S, Pita-Fernandez S, et al. Place of preoperative treatment of acromegaly with somatostatin analog on surgical outcome: a systematic review and meta-analysis. Plos One 2013, 8: e61523.
- Losa M, Donofrio CA, Gemma M, et al. Pretreatment with somatostatin analogs does not affect the anesthesiologic management of patients with acromegaly. Pituitary 2019, 22: 187-94.
- Colao AM, Attanasio R, Pivonello R, et al. Partial surgical removal of growth hormone-secreting pituitary tumors enhances the response to somatostatin analogs in acromegaly. J Clin Endocrinol Metab 2006, 91: 85-92.
- Schwyzer L, Starke RM, Jane JA Jr, Oldfield EH. Percent reduction of growth hormone levels correlates closely with percent resected tumor volume in acromegaly. J Neurosurg 2015, 122: 798-802.
- Barker FG, Klibanski A, Swearingen B. Transsphenoidal surgery for pituitary tumors in the United States, 1996–2000: mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab 2003, 88: 4709–19.
- Giustina A, Chanson P, Bronstein MD, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab 2010, 95: 3141–8.
- Feelders RA, Bidlingmaier M, Strasburger CJ, et al. Postoperative evaluation of patients with acromegaly: clinical significance and timing of oral glucose tolerance testing and measurement of (free) Insulin-Like Growth Factor I, Acid-Labile Subunit, and Growth Hormone-Binding Protein Levels. J Clin Endocrinol Metab 2005, 90: 6480-9.
- Zirkzee EJM, Corssmit EPM, Biermasz NR, et al. Pituitary magnetic resonance imaging is not required in the postoperative follow-up of acromegalic patients with long-term biochemical cure after transsphenoidal surgery. J Clin Endocrinol Metab 2004, 89: 4320-4.
- Cozzi R, Ambrosio MR, Attanasio R, et al. Italian Association of Clinical Endocrinologists (AME) and Italian AACE chapter position statement for clinical practice: acromegaly - part 2: therapeutic issues. Endocr Metab Immune Disord Drug Targets 2020, DOI: 10.2174/1871530320666200129113328.
- Petrossians P, Martins LB, Espinoza C, et al. Gross total resection or debulking of pituitary adenomas improves hormonal control of acromegaly by somatostatin analogs. Eur J Endocrinol 2005, 152: 1-7.
- Kanakis GA, Chrisoulidou A, Bargiota A, et al. The ongoing challenge of discrepant growth hormone and insulin-like growth factor I results in the evaluation of treated acromegalic patients: a systematic review and meta-analysis. Clin Endocrinol 2016, 85: 681–8.
- Zeinalizadeh M, Habibi Z, Fernandez-Miranda JC, et al. Discordance between growth hormone and insulin-like growth factor-1 after pituitary surgery for acromegaly: a stepwise approach and management. Pituitary 2015, 18: 48-59.
- Hazer DB, Işık S, Berker D, et al. Treatment of acromegaly by endoscopic transsphenoidal surgery: surgical experience in 214 cases and cure rates according to current consensus criteria. J Neurosurg 2013, 119: 1467-77.
- Negm HM, Al-Mahfoudh R, Pai M, et al. Reoperative endoscopic endonasal surgery for residual or recurrent pituitary adenomas. J Neurosurg 2017, 127: 397-408.
- Esquenazi Y, Essayed WI, Singh H, et al. Endoscopic endonasal versus microscopic transsphenoidal surgery for recurrent and/or residual pituitary adenomas. World Neurosurg 2017, 101: 186-95.
Terapia farmacologica dell'acromegalia
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
(aggiornato al 12 aprile 2020)
I farmaci attualmente disponibili appartengono a 3 differenti categorie: analoghi della somatostatina (SSA) di 1° e 2° generazione, antagonisti del recettore del GH e dopaminergici.
Nel considerare i risultati di ognuno, è opportuno ricordare che l’ambiente estrogenico influenza i risultati della terapia: le donne in età fertile possono necessitare di dosi inferiori di farmaco (1), mentre il trattamento estrogenico per via orale (per es., la pillola contraccettiva) o i SERM (2) possono influenzare il risultato della terapia, abbassando ulteriormente i livelli di IGF-I.
ANALOGHI DELLA SOMATOSTATINA (SSA) DI PRIMA GENERAZIONE
Octreotide e lanreotide sono SSA di prima generazione, approvati da oltre 30 anni per il trattamento dell’acromegalia (3). Entrambe le molecole si legano ai recettori della somatostatina (SSTR) di tipo 2 (e 5) sulle cellule adenomatose (4). È stato anche dimostrato un effetto diretto di inibizione periferica della sintesi di IGF-I a livello epatico (5).
Gli SSA di prima generazione possono essere usati in tutti i pazienti (6-8), sia come terapia primaria (intesa sia primo trattamento alla diagnosi che come unico trattamento della malattia) che adiuvante (dopo neurochirurgia inefficace e/o in attesa dei risultati della radioterapia).
Efficacia
Octreotide e lanreotide inibiscono efficacemente l’ipersecrezione ormonale nella maggior parte dei pazienti, fino a ottenere livelli di IGF-I normali (o pressoché normali) in circa metà dei casi (4,9), senza tachifilassi anche dopo molti anni di trattamento (10). È opinione comune che non ci sia differenza reale di risposta alle due molecole, anche se il passaggio dall’una all’altra può essere utile in alcuni pazienti che lamentano effetti collaterali (11,12).
L’efficacia nel controllo ormonale non è differente tra i pazienti in precedenza sottoposti a intervento neurochirurgico e quelli trattati farmacologicamente in prima battuta (13).
Il miglioramento clinico e ormonale decorre in modo parallelo, con marcato miglioramento o addirittura scomparsa di sintomi e segni di malattia e comorbilità sistemiche (a livello cardiaco, respiratorio, metabolico), almeno in quei pazienti che non sono arrivati a stadi avanzati di irreversibilità (la poli-artrosi degenerativa avanzata difficilmente può migliorare) (14-17).
Gli SSA controllano la crescita tumorale in tutti i pazienti acromegalici (nel corso di questa terapia non si verifica praticamente mai la crescita di volume dell’adenoma). Nella maggior parte dei pazienti, soprattutto nei primi mesi di trattamento, si ottiene una riduzione del volume dell’adenoma (18,19), che può essere rapida e progressiva (20). La riduzione volumetrica tumorale è più frequente e quantitativamente più marcata nei pazienti in cui gli SSA sono impiegati come terapia primaria (17,18,20).
Figura 1. Paziente acromegalico di 38 anni in cui la terapia con octreotide LAR ha ottenuto una progressiva riduzione di un voluminoso macroadenoma ad espansione sovrasellare fino all'empty sella in 24 mesi.
Le riduzioni ormonali e volumetriche di solito sono concomitanti, ma in alcuni casi può esserci una netta dissociazione, cioè normalizzazione ormonale senza nessuna variazione volumetrica o, ancor più raramente, scarse modificazioni ormonali con netta riduzione di volume (21,22).
Come prevedere la risposta al trattamento
Età e sesso non influenzano la risposta al trattamento con SSA, mentre è più controverso il ruolo degli alti livelli di GH (23,24).
La risposta del GH a un test acuto con octreotide non viene più impiegata come criterio predittivo di risposta a lungo termine (25), poiché alcuni pazienti non responsivi alla somministrazione acuta si sono poi dimostrati sensibili alla terapia cronica, anche se l’assenza completa di risposta può essere utile per identificare i pochi pazienti totalmente resistenti.
I livelli di GH/IGF-I ottenuti dopo 3-6 mesi di terapia sono predittivi del successo terapeutico a lungo termine (20,23):
- se c’è stata una buona soppressione rispetto ai livelli iniziali, anche senza aver ancora raggiunto la normalizzazione, c’è un’alta probabilità che questa venga raggiunta proseguendo la terapia;
- al contrario, in quei casi in cui non si sono verificate modificazioni significative (diminuzione dei livelli di GH inferiore al 20-30%), è inutile proseguire ed è meglio perseguire strategie alternative.
È stato segnalato che i pazienti con ipointensità del tessuto adenomatoso alla RM con le sequenze T2 hanno migliore soppressione ormonale e riduzione volumetrica durante il trattamento con SSA (26-28).
Alcuni parametri saranno disponibili solo dopo intervento neurochirurgico (solo se è disponibile un laboratorio avanzato):
- l’aspetto dei granuli di secrezione all’esame ultra-strutturale (non è più necessaria a questo scopo la microscopia elettronica) permetterà di distinguere fra i pazienti con alta probabilità di rispondere positivamente agli SSA (adenomi con granuli densi) e quelli con bassa probabilità (adenomi con granuli sparsi) (29);
- l’espressione di SSTR2 a livello delle cellule adenomatose può essere utile per identificare i pazienti con maggiore probabilità di risposta agli SSA (30);
- mutazioni di gsp sono apparentemente associate a risposta positiva agli SSA (31);
- la presenza di una forma troncata di SSTR5 è associata a resistenza (32).
Molecole, dosi, monitoraggio
Octreotide e lanreotide sono disponibili in formulazioni depot a diverso dosaggio, da iniettare ogni 28 giorni:
- octreotide LAR fl im da 10, 20, 30 mg;
- lanreotide autogel fl sc da 60, 90, 120 mg.
Octreotide è disponibile anche in formulazione short-acting, da 50, 100, 200 µg/mL, da iniettare sc e da impiegare in casi particolari.
Si inizia abitualmente con la dose media (LAR 20 mg/28 gg o ATG 90 mg/28 gg), che viene aggiustata in relazione al risultato ottenuto dopo 3 mesi:
- se i livelli di IGF-I sono rientrati nel range di normalità (o vi si sono avvicinati molto), proseguire con questa dose;
- se la terapia è solo parzialmente efficace (non ha ottenuto la normalizzazione dei valori di IGF-I), aumentare la dose (a 30 mg/28 gg di octreotide LAR o a 120 mg/28 gg di lanreotide autogel);
- se i livelli di IGF-I sono scesi al di sotto del 50% del range di normalità, diminuire la dose (a 10 mg/28 gg di octreotide LAR o a 60 mg/28 gg di lanreotide autogel), oppure allungare l’intervallo di somministrazione (di una settimana ogni 3 mesi, cioè iniettare il farmaco ogni 35, 42, 49, 56 gg), in maniera da stabilizzare i livelli di IGF-I fra il 25% e il 50% del limite massimo di normalità (33).
Nei casi particolarmente attivi (livelli ormonali molto alti, quadro clinico importante, tumore con iniziale impegno delle vie ottiche in cui vi sia qualche controindicazione alla chirurgia o i tempi di attesa dell’intervento si prolunghino), può essere utilizzata da subito la dose maggiore (octreotide LAR 30 mg o lanreotide autogel 120 mg), oppure può essere utile, dopo aver praticato la prima iniezione del long-acting, che richiede una settimana circa per raggiungere livelli terapeutici efficaci, somministrare contemporaneamente per 1-2 settimane 1 fl di octreotide sc ogni 8 ore per accelerare l’effetto del trattamento.
Per ottenere un miglior controllo ormonale nei pazienti parzialmente resistenti, si può aumentare la quantità di farmaco somministrata: l’aumento di dose è efficace con entrambe le molecole (per octreotide LAR è autorizzata la dose fino a 40 mg/28 giorni), mentre l’accorciamento dell’intervallo fra le somministrazioni è efficace solo con lanreotide ATG (120 mg/21 giorni) (34,35).
Per valutare l’efficacia della terapia, si misurano i livelli plasmatici di GH e IGF-I su campioni prelevati subito prima della successiva somministrazione del farmaco, a intervalli di 3 mesi nella fase di titolazione e ogni 6-12 mesi a dosaggio stabilizzato.
Gli analoghi short-acting hanno ancora un ruolo importante nella terapia della cefalea acromegalica e spesso sono l'unico rimedio efficace in questa complicazione invalidante (anche se oggi vale la pena di provare pasireotide, vedi oltre). Talvolta è necessaria la loro somministrazione per via sc continua con un micro-infusore.
Effetti collaterali
Sono tollerati molto bene anche durante terapie molto prolungate.
Fastidio in sede di iniezione e disturbi gastro-enterici sono solitamente di intensità lieve-moderata e tendono a scomparire con le iniezioni successive (14).
La colelitiasi è frequente ma raramente è sintomatica o richiede chirurgia in urgenza (36) e obesità e dislipidemia sembrano giocare un ruolo importante. È opportuno rimarcare che la colelitiasi sintomatica è invece frequente dopo sospensione degli SSA, perché si ripristina la contrattilità della colecisti che a quel punto è piena di bile densa o calcoli (37).
Gli effetti sul metabolismo glucidico sono ampiamente variabili ma raramente di rilevanza clinica (38). La comparsa o il peggioramento del diabete sono soprattutto a carico dei pazienti resistenti agli SSA o di quelli con familiarità diabetica. Una recente meta-analisi sugli effetti degli SSA sul metabolismo glucidico (39) concludeva che il parametro maggiormente coinvolto è la glicemia post-prandiale, che dovrebbe quindi essere l’obiettivo di farmaci mirati, come quelli che agiscono sul sistema delle incretine (40). È comunque esperienza comune che non è solitamente necessaria una terapia anti-diabetica aggressiva nei pazienti con modesto peggioramento del metabolismo glucidico in corso di terapia cronica con SSA.
SSA DI SECONDA GENERAZIONE
Pasireotide è un SSA multi-ligando, in grado di attivare differenti recettori (SSTR5, SSTR2, SSTR1, SSTR3) con differenti affinità.
Dopo alcuni studi pre-clinici e aperti, uno studio multi-centrico prospettico randomizzato di confronto diretto ha dimostrato la superiorità di pasireotide LAR rispetto a octreotide LAR nel controllo dell’ipersecrezione ormonale (41). Quello studio ha suscitato molto scalpore, perché i risultati di pasireotide LAR erano sovrapponibili a quanto comunemente osservato in precedenza con gli SSA di 1° generazione, mentre i risultati di octreotide LAR erano decisamente peggiori.
Il successivo studio PAOLA (42) ha arruolato pazienti parzialmente resistenti agli SSA di 1° generazione a dose massimale, e li ha randomizzati a pasireotide LAR o al proseguimento del trattamento precedente. Pasireotide LAR è risultato decisamente migliore, con risultati persistenti fino a 24 mesi (43,44). Anche un recente studio retrospettivo “real life” (45) ha dimostrato la normalizzazione di IGF-I in 19/35 pazienti che non erano adeguatamente controllati con octreotide. Dato oltremodo interessante era la completa scomparsa della cefalea nei pazienti che ne soffrivano. Gli effetti di pasireotide sulla cefalea refrattaria sono stati riportati anche da altri autori e un recente position paper del gruppo di Rotterdam raccomanda l’uso di questo farmaco per la cefalea non responsiva agli SSA di 1° generazione (46).
Il differente assetto molecolare degli SSTR, in particolare di SSTR5, nell’adenoma potrebbe spiegare la risposta differente a pasireotide rispetto agli SSA di 1° generazione.
Gli effetti collaterali di pasireotide sono simili a quelli degli SSA di 1° generazione, con l’eccezione di alterazioni glicemiche più frequenti e più gravi, soprattutto in quei pazienti che già prima del trattamento avevano uno screzio glicemico (glicemia a digiuno > 100 mg/dL) e/o erano in trattamento con farmaci anti-diabetici. Le alterazioni glicemiche non sono correlate all’efficacia del trattamento sull’acromegalia (47). È quindi imperativo ottimizzare le terapie anti-diabetiche prima di iniziare il trattamento con pasireotide ed eseguire uno stretto monitoraggio dei livelli glicemici fin dalle prime settimane di trattamento (48).
Pasireotide LAR è autorizzato per il trattamento dell’acromegalia nel caso la neurochirurgia non sia indicata o non abbia avuto successo e nel caso le terapie farmacologiche alternative non siano state efficaci o non siano tollerate.
Pasireotide LAR è disponibile in fiale iniettabili intramuscolo ogni 28 giorni, ai dosaggi di 20, 40 e 60 mg.
ANTAGONISTA DEL RECETTORE DEL GH
Pegvisomant (PegV) è una molecola di GH modificata, in grado di legarsi al suo recettore ma non di attivare i meccanismi post-recettoriali (49). Si comporta quindi come un antagonista funzionale, bloccando la sintesi e provocando la diminuzione dei livelli circolanti di IGF-I.
PegV è autorizzato in Italia per i pazienti acromegalici con malattia ancora attiva dopo intervento neurochirurgico, se gli SSA non sono efficaci o tollerati o in attesa del risultato delle terapie radianti.
In corso di terapia con PegV l’attività di malattia può essere valutata dal punto di vista biochimico solo con il dosaggio di IGF-I (il GH tende ad aumentare specularmente alla diminuzione dell’IGF-I e con i dosaggi commerciali verrebbe dosato anche il farmaco) (50).
Efficacia
Negli studi registrativi la mono-terapia con PegV ha ottenuto la normalizzazione di IGF-I nel 63-97% dei casi (51,52), con un parallelo miglioramento di sintomi e segni della malattia e della QoL. La molecola è efficace anche nel controllo delle complicanze cardio-vascolari e scheletriche della malattia (50).
Rispetto agli studi registrativi, la percentuale di successo negli studi osservazionali è inferiore (53): nella pratica clinica la normalizzazione di IGF-I si raggiunge in meno del 65-70% dei casi, dato attribuito a inadeguata titolazione della dose.
Il trattamento con PegV si accompagna di solito a miglioramento del metabolismo glucidico (54,55). Si può quindi prendere in considerazione questo trattamento di seconda linea nei diabetici, soprattutto nel caso di scompenso metabolico in corso di trattamento con SSA.
Molecola e dosi
Pegvisomant fl da 10, 15, 20, 25, 30 mg, da iniettare sottocute tutti i giorni.
La dose iniziale è di 10 mg/die, da incrementare in relazione ai risultati, a intervalli mensili, aumentandola di 5 mg/die ogni 4-6 settimane, fino a ottenere la normalizzazione dei livelli di IGF-I.
Secondo le indicazioni del foglietto illustrativo, la dose massima autorizzata è di 30 mg/die, ma sono stati riportati casi in cui il successo terapeutico è stato ottenuto con la somministrazione off-label di dosi fino a 40-60 mg/die. Il successo terapeutico è stato talora ottenuto somministrando una o due volte alla settimana la dose cumulativa settimanale (anche in questo caso off-label).
Le dosi più alte possono essere necessarie nei maschi e nei pazienti con diabete, obesità o livelli molto alti di GH (53).
A equilibrio raggiunto, si consiglia il monitoraggio ogni 6-12 mesi.
Indicazioni e precauzioni
PegV è autorizzato in Europa solo come trattamento adiuvante, cioè nei pazienti con persistenza di attività di malattia, già sottoposti in precedenza a terapia neurochirurgica e/o radiante e resistenti/intolleranti agli SSA.
PegV può essere particolarmente indicato nei pazienti con alterazioni del metabolismo glucidico, perché migliora la sensibilità all’insulina (55).
Dal punto di vista della sicurezza, anche se PegV non ha azione a livello dell’adenoma, nel corso degli studi osservazionali a lungo termine le dimensioni tumorali sono rimaste stabili nella maggior parte dei casi (53). Raramente (circa 3% dei casi) è stato riportato un incremento clinicamente significativo del volume tumorale, attribuito alla storia naturale dell’adenoma o alla sospensione di un precedente trattamento con SSA, che aveva ottenuto una riduzione di tale volume. In alcuni casi è stata invece osservata una riduzione delle dimensioni tumorali. È sempre raccomandata la sorveglianza con RM, soprattutto nei casi con voluminoso residuo tumorale e malattia aggressiva, non precedentemente irradiati.
In meno del 3% dei pazienti è stato riportato un transitorio aumento degli enzimi epatici in corso di terapia con PegV, soprattutto nei maschi, nei diabetici, nei portatori di s. di Gilbert e nei pazienti che fanno terapia combinata con SSA. Si raccomanda uno stretto monitoraggio clinico e biochimico (anche settimanale) nei casi in cui gli esami di funzione epatica aumentano di oltre tre volte sopra il limite superiore dell’intervallo di normalità, con sospensione quando le transaminasi sono > 3-5 volte il limite massimo di normalità: una volta ottenuta la normalizzazione delle transaminasi dopo sospensione del trattamento, PegV può essere nuovamente somministrato. Negli altri casi si può proseguire la terapia, rallentando il ritmo di incremento del dosaggio o tornando per qualche settimana al dosaggio inferiore, in attesa della normalizzazione delle transaminasi, ripartendo poi con una prudente e lenta titolazione della dose.
Viene raccomandata la rotazione dei siti in cui praticare l’iniezione, per minimizzare il rischio di lipo-ipertrofia.
Efficacia
La terapia dopaminergica è spesso efficace sui sintomi, ma normalizza l’ipersecrezione ormonale solo in alcuni casi. Una metanalisi (56) ne ha confermato l’utilità in circa un terzo dei casi, se usata in mono-terapia. Può essere associata agli altri farmaci (vedi oltre).
In alcuni casi ottiene anche una riduzione del volume tumorale.
Molecole e dosi
Le molecole utilizzabili sono:
- bromocriptina cp 2.5, 5 e 10 mg
- cabergolina cp 0.5 mg.
Cabergolina è più efficace e meglio tollerata. È consigliabile partire con la somministrazione di ½ cp da 0.5 mg alla sera durante il pasto, da aumentare progressivamente (½ cp in più alla settimana) fino alle dosi massime tollerate o a 0.5 mg/die. È consigliabile eseguire il primo controllo di efficacia (se non ci sono problemi di tollerabilità) quando si è raggiunta la dose di 1.5 mg/settimana.
Indicazioni e precauzioni
Da utilizzare soprattutto come terapia adiuvante nei pazienti con ipersecrezione ormonale moderata. La presenza di iperprolattinemia associata non è pre-requisito per il suo impiego. In ogni caso si tratta di terapia off-label (serve quindi consenso informato e non è rimborsabile dal SSN).
Recentemente sono stati avanzati dubbi sulla sicurezza di questo trattamento a lungo termine, a seguito delle osservazioni nei pazienti con m. di Parkinson trattati con cabergolina ad alte dosi (> 3 mg/die), in cui è stato riportato un aumento dell'incidenza di valvulopatie, con un effetto additivo di dosi di farmaco e tempi prolungati di somministrazione (57). Le dosi impiegate nell’acromegalia sono sicuramente inferiori alle alte dosi utilizzate nel Parkinson, però va ricordato che la terapia va portata avanti per lungo tempo e i pazienti acromegalici hanno talvolta un’intrinseca predisposizione alle valvulopatie, correlata alla malattia. Anche se non è stato trovato un aumento della prevalenza di valvulopatie nei pazienti endocrinopatici trattati con cabergolina (58), è comunque opportuno valutare preliminarmente la situazione valvolare con un’ecocardiografia (che dovrebbe rientrare comunque nella stadiazione delle complicanze di malattia). Nel caso di lesioni valvolari conclamate clinicamente o emodinamicamente rilevanti, la terapia è da ritenersi controindicata. L’ecocardiografia dovrà essere ripetuta nel tempo, a intervalli congrui con il rischio di partenza (più frequentemente nei pazienti con qualche lesione di partenza) e con la dose di farmaco impiegata: dopo 6 mesi in chi assume 0.5 mg/die, ma non prima di 24 mesi in chi assume 1 mg/settimana.
INDICAZIONI DEL POSITION STATEMENT AME (7) SULLE MONO-TERAPIE FARMACOLOGICHE
Raccomandiamo di iniziare il trattamento farmacologico con un SSA di prima generazione.
Suggeriamo di trattare con pasireotide LAR i pazienti resistenti agli SSA di prima generazione, se hanno normale equilibrio gluco-metabolico.
Raccomandiamo di trattare con PegV i pazienti resistenti o intolleranti agli SSA dopo intervento neurochirurgico non risolutivo o in attesa degli effetti delle terapie radianti.
Raccomandiamo uno stretto monitoraggio precoce dei livelli glicemici in corso di terapia con pasireotide.
Suggeriamo di considerare la terapia con pasireotide negli acromegalici con cefalea resistente.
Raccomandiamo l’utilizzo del solo IGF-I per monitorare l’efficacia di PegV.
Raccomandiamo il monitoraggio regolare del volume tumorale, soprattutto nei pazienti con voluminoso residuo e malattia clinicamente aggressiva.
Raccomandiamo il monitoraggio dei test di funzione epatica all’inizio del trattamento con PegV e durante le fasi di titolazione del dosaggio.
I FARMACI POSSONO ESSERE COMBINATI?
La combinazione di due farmaci diversi, che agiscono in maniera additiva o sinergica, può migliorare l’efficacia, ridurre gli effetti collaterali delle singole molecole, diminuire la frequenza delle iniezioni e/o la dose di farmaco, migliorare la compliance a lungo termine, e perfino ridurre i costi. Non sono disponibili RCT che abbiano valutato terapie farmacologiche combinate per l’acromegalia.
SSA + Cabergolina
L’aggiunta di cabergolina agli SSA in pazienti parzialmente resistenti ha portato alla normalizzazione dei livelli di IGF-I nel 30-45% dei casi (56,59,60). È importante sottolineare che né i livelli di PRL, né la positività immuno-istochimica per PRL, né l’espressione di D2R sono predittivi dell’efficacia del trattamento con cabergolina (60,61).
SSA + PegV
Parecchi studi hanno dimostrato che in pazienti considerati parzialmente resistenti agli SSA, l’aggiunta di PegV (anche con uno schema di somministrazione una o due o tre volte a settimana) porta alla normalizzazione dei livelli di IGF-I nel 62-100% dei casi (62-68). Anche l’associazione con pasireotide LAR ha avuto successo in piccole casistiche (69,70).
Questa combinazione avrebbe il vantaggio di consentire il controllo anche della crescita tumorale in quei pazienti in cui questo può costituire un problema di rilievo (residuo voluminoso o comunque in vicinanza delle vie ottiche).
Visto il lievitare dei costi (si tratta di farmaci entrambi molto costosi), la sua rimborsabilità SSN è fonte di controversie con le autorità regolatorie, anche se a questa combinazione si dovrebbe ricorrere solo in una piccola minoranza di pazienti nell’ambito di una patologia rara ed è segnalata la possibilità di ridurre le dosi di entrambi i farmaci e/o l’allungamento dell’intervallo tra le iniezioni di PegV (a giorni alterni o anche in mono-somministrazione settimanale).
Secondo i dati disponibili, l’associazione di SSA e PegV è da prendere in considerazione nei pazienti con risposta parziale agli SSA e nei pazienti con voluminoso residuo tumorale che non hanno raggiunto il controllo ormonale.
Pegvisomant + Cabergolina
Anche se non sono ancora disponibili dati conclusivi (71), l’associazione di PegV e cabergolina può essere presa in considerazione nei pazienti con modesto aumento dei livelli di IGF-I in corso di mono-terapia con PegV o in quelli con ipersecrezione mista GH-PRL.
Il position statement AME (7):
- suggerisce un trattamento combinato per migliorare l’efficacia o ridurre gli effetti collaterali associati ai singoli farmaci, diminuire la frequenza delle iniezioni e/o la dose dei farmaci, migliorare la compliance e ridurre i costi;
- nel caso vi sia l’indicazione a una terapia farmacologica combinata, suggerisce la somministrazione di una combinazione con efficacia dimostrata: SSA + cabergolina o SSA + PegV.
QUALE FARMACO SCEGLIERE?
Vale quasi sempre la pena di partire con un farmaco in mono-terapia. Nel paziente acromegalico “tipo”, il farmaco di prima scelta è un SSA di 1° generazione. Infatti, gli altri farmaci hanno indicazioni particolari:
- cabergolina (o bromocriptina) si impiega nel paziente con ipersecrezione modesta, nella speranza di riuscire a ottenere gli obiettivi terapeutici con un farmaco impiegato per via orale (la sua prescrizione è off-label);
- pasireotide è autorizzato solo nel paziente in cui l’intervento chirurgico non è indicato o non è stato efficace, se gli SSA di 1° generazione non hanno raggiunto la normalizzazione ormonale;
- pegvisomant è autorizzato solo come terapia di 3° linea, quindi nel paziente già operato e/o irradiato, resistente o intollerante agli SSA.
Il riscontro sul pezzo operatorio dei recettori per somatostatina, in particolare il sottotipo SSTR2, sembrerebbe associato alla successiva risposta alla terapia con SSA (72). In realtà questa determinazione non è eseguita routinariamente.
Sono pochi gli studi comparativi diretti fra octreotide LAR e lanreotide autogel. La scelta si basa su esperienza personale, tollerabilità (può capitare che un paziente tolleri il secondo farmaco dopo aver avuto effetti collaterali con il primo) (11,12) e maneggevolezza: lanreotide autogel è preparato in siringhe pre-riempite da iniettare sc, quindi più facile da utilizzare senza assistenza infermieristica, mentre octreotide LAR deve essere preparato sul momento (la diluizione del farmaco richiede attenzione).
PROSPETTIVE PER NUOVI FARMACI O FORMULAZIONI
Sono in cantiere nuove modalità di trattamento che potranno essere disponibili nei prossimi anni: octreotide orale alla dose di 40-80 mg/die (73), SSA a lunga durata d’azione con iniezioni trimestrali di 84 mg (74), oligonucleotidi anti-senso (75).
COME MONITORARE GLI EFFETTI DELLA TERAPIA FARMACOLOGICA
Controlli dell’ipersecrezione ormonale e della funzione ipofisaria:
- dosare GH e IGF-I (in corso di pegvisomant solo IGF-I; in caso di ipersecrezione associata, anche gli altri ormoni coinvolti):
- in fase di titolazione del farmaco a intervalli trimestrali;
- successivamente ogni 6 mesi (o anche annualmente nei pazienti stabili, ben controllati);
- funzione ipofisaria: per i pazienti ipopituitarici vedi il capitolo ipopituitarismo, per gli irradiati vedi radioterapia nell’acromegalia. Nei pazienti con funzione ipofisaria normale, non irradiati e in cui la terapia farmacologica per il controllo dell’acromegalia ha successo, non serve ricontrollare gli esami di funzione ipofisaria annualmente, ma basta farlo ogni 2-3 anni (specialmente in quelli operati).
Controlli biochimici:
- metabolismo glucidico: glicemia a digiuno e HbA1c, a intervalli inizialmente trimestrali e successivamente semestrali;
- profilo lipidico: secondo il profilo di rischio individuale.
Controlli neuroradiologici: ripetere RM ipofisi:
- nei pazienti denovo dopo 3 e 12 mesi e in seguito in base al comportamento ormonale;
- negli operati dopo il 1° controllo post-operatorio ripetere a 1 anno e poi se in remissione ormonale non serve ripetere il controllo;
- in terapia farmacologica ogni 12-24 mesi, sempre in base al comportamento ormonale.
Controlli comorbilità:
- nei pazienti trattati con SSA, ecografia addominale a intervalli annuali (associare terapia con sali biliari in caso di fattori di rischio per colelitiasi o evidenza di fango biliare all’ecografia);
- per le altre comorbilità, vedi capitolo clinica e diagnosi dell’acromegalia.
I FARMACI POSSONO ESSERE SOSPESI?
I farmaci sono in grado di tenere sotto controllo l’attività di malattia ma non di guarirla. Al di là di sporadiche segnalazioni (76,77), quando la terapia farmacologica viene sospesa (anche dopo lungo tempo), si osserva la progressiva ripresa dapprima dell’ipersecrezione ormonale, poi dell’attività di malattia e da ultimo della ricrescita tumorale (78,79). Quindi, di regola non ha senso provare a sospendere la terapia nel paziente che non abbia subito trattamenti ablativi.
COSA FARE NEI PAZIENTI RESISTENTI
Per resistenza al trattamento si intende il mancato raggiungimento degli obiettivi terapeutici, cioè la mancata normalizzazione dell’ipersecrezione di GH/IGF-I o la crescita del tumore dopo un periodo di trattamento di alcuni mesi a dosi piene di farmaco (4,80). Le strategie terapeutiche nei pazienti resistenti possono essere diverse.
- Nei pazienti trattati con SSA cambiare analogo: in alcuni casi il passaggio da un analogo all'altro può migliorare la soppressione di GH/IGF-I.
- Nei pazienti mai operati cambiare strategia terapeutica: il paziente con parziale responsività a un primo trattamento con SSA, può normalizzare i valori ormonali con un secondo ciclo di SSA dopo intervento (81,82).
- Associare diversi farmaci: vedi sopra.
- Usare dosaggi non convenzionali: è stato riportato che l’utilizzo di dosi potenziate (fino a 60 mg di octreotide LAR ogni 28 gg) riesce a normalizzare una certa porzione di pazienti con resistenza parziale (34). Siamo comunque nell’ambito dell’off-label.
- Sospendere l'SSA impiegato e iniziare PegV.
- Nei pazienti con adenoma aggressivo/carcinoma è stato approvato l’uso della temozolomide.
- Anche per l’acromegalia (come già per altri tumori) sono stati segnalati casi di efficacia di terapia radio-recettoriale con somatostatina radio-marcata (in analogia a quanto si fa con il radioiodio nei tumori tiroidei differenziati).
BIBLIOGRAFIA
- Eden Engström B, Burman P, Karlsson FA. Men with acromegaly need higher doses of octreotide than women. Clin Endocrinol (Oxf) 2002, 56:73-77.
- Stone JC, Clark J, Cuneo R, et al. Estrogen and selective estrogen receptor modulators (SERMs) for the treatment of acromegaly: a meta-analysis of published observational studies. Pituitary 2014, 17: 284–95.
- Cozzi R, Attanasio R. Octreotide LAR for acromegaly. Expert Rev Clin Pharmacol 2012, 5: 125-43.
- Gadelha MR, Wildemberg LE, Bronstein MD, et al. Somatostatin receptor ligands in the treatment of acromegaly. Pituitary 2017, 20: 100–8.
- Murray RD, et al. Central and peripheral actions of somatostatin on the growth hormone-IGF-I axis. J Clin Invest 2004, 114: 349-56.
- Melmed S, Colao A, Barkan A, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab 2009, 94: 1509-17.
- Cozzi R, Ambrosio MR, Attanasio R, et al. Italian Association of Clinical Endocrinologists (AME) and Italian AACE chapter position statement for clinical practice: acromegaly - part 2: therapeutic issues. Endocr Metab Immune Disord Drug Targets 2020, DOI: 10.2174/1871530320666200129113328.
- Katznelson L, et al. American Association of Clinical Endocrinologists Acromegaly Guidelines Task Force. Medical guidelines for clinical practice for the diagnosis and treatment of acromegaly. Endocr Pract 2011, 17 suppl 4: 1-44.
- Freda PU, et al. Long-acting somatostatin analog therapy of acromegaly: a meta-analysis. J Clin Endocrinol Metab 2005, 90: 4465–73.
- Maiza JC, Vezzosi D, Matta M, et al. Long-term (up to 18 years) effects on GH/IGF-1 hypersecretion and tumour size of primary somatostatin analogue (SSTa) therapy in patients with GH-secreting pituitary adenoma responsive to SSTa. Clin Endocrinol 2007, 67: 282–9.
- Andries M, Glintborg D, Kvistborg A, et al. A 12-month randomised cross-over study on the effects of Lanreotide Autogel and Octreotide long-acting repeatable on GH and IGF-l in patients with acromegaly. Clin Endocrinol 2008, 68: 473-80.
- Tutuncu Y, Berker D, Isik S, et al Comparison of octreotide LAR and lanreotide autogel as post-operative medical treatment in acromegaly. Pituitary 2012, 15: 398–404.
- Alquraini H, Del Pilar Schneider M, Mirakhur B, Barkan A. Biochemical efficacy of long-acting lanreotide depot/Autogel in patients with acromegaly naïve to somatostatin-receptor ligands: analysis of three multicenter clinical trials. Pituitary 2018, 21: 283-9.
- Lancranjan I, Atkinson BA, the Sandostatin LAR Group. Results of a European multicentre study with Sandostatin LAR in acromegaly patients. Pituitary 1999, 1: 105–14.
- Colao A, Ferone D, Marzullo P, et al. Systemic complications of acromegaly: epidemiology, pathogenesis and management. Endocr Rev 2004, 25: 102-52.
- Maison P, Tropeano AI, Macquin-Mavier I, et al. Impact of somatostatin analogs on the heart in acromegaly: a metaanalysis. J Clin Endocrinol Metab 2007, 92: 1743–7.
- Caron PJ, Bevan JS, Petersenn S, et al, The PRIMARYS Investigators Group. Effects of lanreotide Autogel primary therapy on symptoms and quality-of-life in acromegaly: data from the PRIMARYS study. Pituitary 2016, 19: 149–57.
- Bevan JS. The antitumoral effects of somatostatin analog therapy in acromegaly. J Clin Endocrinol Metab 2005, 90: 1856-63.
- Melmed S, Sternberg R, Cook D, et al. A critical analysis of pituitary tumor shrinkage during primary medical therapy in acromegaly. J Clin Endocrinol Metab 2005, 90: 4405-10.
- Cozzi R, Montini M, Attanasio R, et al. Primary treatment of acromegaly with octreotide LAR: a long-term (up to 9 years) prospective study on its efficacy in the control of disease activity and on tumor shrinkage. J Clin Endocrinol Metab 2006, 91: 1397-403.
- Casarini APM, Pinto EM, Jallad RS, et al. Dissociation between tumor shrinkage and hormonal response during somatostatin analog treatment in an acromegalic patient: preferential expression of somatostatin receptor subtype 3. J Endocrinol Invest 2006, 29: 826-30.
- Resmini E, Dadati P, Ravetti J-L, et al. Rapid pituitary tumor shrinkage with dissociation between antiproliferative and antisecretory effects of a long-acting octreotide in an acromegalic patient. J Clin Endocrinol Metab 2007, 92: 1592–9.
- Cozzi R, Attanasio R, Montini M, et al. Four-year treatment with Octreotide-Long-Acting Repeatable in 110 acromegalic patients: predictive value of short-term results? J Clin Endocrinol Metab 2003, 88: 3090-8.
- Colao A, Pivonello R, Cappabianca P, et al. Effect of gender and gonadal status on the long-term response to somatostatin analogue treatment in acromegaly. Clin Endocrinol (Oxf) 2005, 63: 342-9.
- Pokrajac A, Claridge AG, Shakoor SKA, Trainer PJ. The octreotide test dose is not a reliable predictor of the subsequent response to somatostatin analogue therapy in patients with acromegaly. Eur J Endocrinol 2006, 154: 267–74.
- Potorac I, Petrossians P, Daly AF, et al. T2-weighted MRI signal predicts hormone and tumor responses to somatostatin analogs in acromegaly. Endocr Relat Cancer 2016, 23: 871-81.
- Shen M, Zhang Q, Liu W, et al. Predictive value of T2 relative signal intensity for response to somatostatin analogs in newly diagnosed acromegaly. Neuroradiology 2016, 58: 1057–65.
- Potorac I, Beckers A, Bonneville JF. T2-weighted MRI signal intensity as a predictor of hormonal and tumoral responses to somatostatin receptor ligands in acromegaly: a perspective. Pituitary 2017, 20: 116–20.
- Fougner SL, Casar-Borota O, Heck A, et al. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol 2012, 76: 96–102.
- Casarini AP, Jallad RS, Pinto EM, et al. Acromegaly: correlation between expression of somatostatin receptor subtypes and response to octreotide-LAR treatment. Pituitary 2009, 12: 297–303.
- Barlier A, Gunz G, Zamora AJ, et al. Prognostic and therapeutic consequences of Gs alpha mutations in somatotroph adenomas. J Clin Endocrinol Metab 1998, 83: 1604–10.
- Luque RM, Ibanez-Costa A, Neto LV, et al. Truncated somatostatin receptor variant sst5TMD4 confers aggressive features (proliferation, invasion and reduced octreotide response) to somatotropinomas. Cancer Lett 2015, 359: 299–306.
- Neggers SJ, Pronin V, Balcere I, et al, LEAD Study Group. Lanreotide Autogel 120 mg at extended dosing intervals in patients with acromegaly biochemically controlled with octreotide LAR: the LEAD study. Eur J Endocrinol 2015, 173: 313-23.
- Giustina A, Bonadonna S, Bugari G, et al. High-dose intramuscular octreotide in patients with acromegaly inadequately controlled on conventional somatostatin analogue therapy: a randomised controlled trial. Eur J Endocrinol 2009, 161: 331-8.
- Giustina A, Mazziotti G, Cannavò S, et al. High-dose and high-frequency lanreotide autogel in acromegaly: a randomized, multicenter study. J Clin Endocrinol Metab 2017, 102: 2454-64.
- Attanasio R, Mainolfi A, Grimaldi F, et al. Somatostatin analogs and gallstones: a retrospective survey on a large series of acromegalic patients. J Endocrinol Invest 2008, 31: 704-10.
- Paisley AN, Roberts ME, Trainer PJ. Withdrawal of somatostatin analogue therapy in patients with acromegaly is associated with an increased risk of acute biliary problems. Clin Endocrinol 2007, 66: 723–6.
- Mazziotti G, Floriani I, Bonadonna S, et al. Effects of somatostatin analogs on glucose homeostasis: a meta-analysis of acromegaly studies. J Clin Endocrinol Metab 2009, 94: 1500-8.
- Cozzolino A, Feola T, Simonelli I, et al. Somatostatin analogs and glucose metabolism in acromegaly: a meta-analysis of prospective interventional studies. J Clin Endocrinol Metab 2018, 103: 2089-99.
- Baroni MG, Giorgino F, Pezzino V, et al. Italian Society for the Study of Diabetes (SID)/Italian Endocrinological Society (SIE) guidelines on the treatment of hyperglycemia in Cushing's syndrome and acromegaly. J Endocrinol Invest 2016, 39: 235-55.
- Colao A, Bronstein MD, Freda P, et al. Pasireotide versus octreotide in acromegaly: a head-to-head superiority study. J Clin Endocrinol Metab 2014, 99: 791–9.
- Gadelha MR, Bronstein MD, Brue T, et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): a randomised, phase 3 trial. Lancet Diabetes Endocrinol 2014, 2: 875-84.
- Sheppard M, Bronstein MD, Freda P, et al. Pasireotide LAR maintains inhibition of GH and IGF-1 in patients with acromegaly for up to 25 months: results from the blinded extension phase of a randomized, double-blind, multicenter, Phase III study. Pituitary 2015, 18: 385-94.
- Bronstein MD, Fleseriu M, Neggers S, et al; Pasireotide C2305 Study Group. Switching patients with acromegaly from octreotide to pasireotide improves biochemical control: crossover extension to a randomized, double-blind, Phase III study. BMC Endocr Dis 2016, 16: 16.
- Shimon I, Adnan Z, Gorshtein A, et al. Efficacy and safety of long-acting pasireotide in patients with somatostatin-resistant acromegaly: a multicenter study. Endocrine 2018, 62: 448-55.
- Coopmans EC, Muhammad A, van der Lely AJ, et al. How to position pasireotide LAR treatment in acromegaly. J Clin Endocrinol Metab 2019, 104: 1978-88.
- Schmid HA, Brue T, Colao A, et al. Effect of pasireotide on glucose- and growth hormone-related biomarkers in patients with inadequately controlled acromegaly. Endocrine 2016, 53: 210-9.
- Fleseriu M, Rusch E, Geer EB, on behalf of the ACCESS Study Investigators. Safety and tolerability of pasireotide long-acting release in acromegaly—results from the acromegaly, open-label, multicenter, safety monitoring program for treating patients who have a need to receive medical therapy (ACCESS) study. Endocrine 2017, 55: 247–55.
- Kopchick JJ, Parkinson C, Stevens EC, Trainer PJ. Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev 2002, 23: 623-46.
- Giustina A, Ambrosio MR, Beck Peccoz P, et al. Use of pegvisomant in acromegaly. An Italian Society of Endocrinology guideline. J Endocrinol Invest 2014, 37: 1017–30.
- Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone–receptor antagonist pegvisomant. N Engl J Med 2000, 342: 1171–7.
- van der Lely AJ, Hutson TK, Trainer PJ, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001, 358: 1754–9.
- Buchfelder M, van der Lely AJ, Biller BMK, et al. Long-term treatment with pegvisomant: observations from 2090 acromegaly patients in ACROSTUDY. Eur J Endocrinol 2018, 179: 419-27.
- Drake WM, Rowles SV, Roberts ME, et al. Insulin sensitivity and glucose tolerance improve in patients with acromegaly converted from depot octreotide to pegvisomant. Eur J Endocrinol 2003, 149: 521–7.
- Feola T, Cozzolino A, Simonelli I, et al. Pegvisomant improves glucose metabolism in acromegaly: a meta-analysis of prospective interventional studies. J Clin Endocrinol Metab 2019, 104: 2892–902.
- Sandret L, Maison P, Chanson P. Place of cabergoline in acromegaly: a meta-analysis. J Clin Endocrinol Metab 2011, 96: 1327-35.
- Schade R, Andersohn F, Suissa S, et al. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med 2007, 356: 29-38.
- Valassi E, Klibanski A, Biller BM. Potential cardiac valve effects of dopamine agonists in hyperprolactinemia. J Clin Endocrinol Metab 2010, 95: 1025-33.
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014, 99: 3933-51.
- Cozzi R, Attanasio R, Lodrini S, Lasio G. Cabergoline addition to depot somatostatin analogs in resistantacromegalic patients: efficacy and lack of predictive value of prolactin status. Clin Endocrinol (Oxf) 2004, 61: 209-15.
- Suda K, Inoshita N, Iguchi G, et al. Efficacy of combined octreotide and cabergoline treatment in patients with acromegaly: a retrospective clinical study and review of the literature. Endocr J 2013, 60: 507-15.
- Feenstra J, de Herder WW, ten Have SM, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet 2005, 365: 1644-6.
- Neggers S, de Herder W, Janssen J, et al. Combined treatment for acromegaly with long-acting somatostatin analogues and pegvisomant: long-term safety up to 4.5 years (median 2.2 yrs) of follow-up in 86 patients. Eur J Endocrinol 2009, 160: 529-33.
- Trainer PJ, Ezzat S, D’Souza GA, et al. A randomized, controlled, multicentre trial comparing pegvisomant alone with combination therapy of pegvisomant and long-acting octreotide in patients with acromegaly. Clin Endocrinol 2009, 71: 549–57.
- Madsen M, Poulsen PL, Ørskov H, et al. Cotreatment with pegvisomant and a somatostatin analog (SA) in SA-responsive acromegalic patients. J Clin Endocrinol Metab 2011, 96: 2405–13.
- Neggers SJCMM, de Herder WW, Feelders RA, et al. Conversion of daily pegvisomant to weekly pegvisomant combined with long-acting somatostatin analogs, in controlled acromegaly patients. Pituitary 2011, 14: 253-8.
- van der Lely AJ, Bernabeu I, Cap J, et al. Coadministration of lanreotide Autogel and pegvisomant normalizes IGF1 levels and is well tolerated in patients with acromegaly partially controlled by somatostatin analogs alone. Eur J Endocrinol 2011, 164: 325–33.
- Neggers SJ, Franck SE, de Rooij FW, et al. Long-term efficacy and safety of pegvisomant in combination with long-acting somatostatin analogs in acromegaly. J Clin Endocrinol Metab 2014, 99: 3644–52.
- Muhammad A, van der Lely AJ, Delhanty PJD, et al. Efficacy and safety of switching to pasireotide in patients with acromegaly controlled with pegvisomant and first-generation somatostatin analogues (PAPE Study). J Clin Endocrinol Metab 2018, 103: 586-95.
- Chiloiro S, Bima C, Tartaglione T, et al. Pasireotide and pegvisomant combination treatment in acromegaly resistant to second line therapies: A longitudinal study. J Clin Endocrinol Metab 2019, 104: 5478-82.
- Higham CE, Atkinson AB, Aylwin S, et al. Effective combination treatment with cabergoline and low-dose pegvisomant in active acromegaly: a prospective clinical trial. J Clin Endocrinol Metab 2012, 97: 1187–93.
- Gatto F, Feelders RA, van der Pas R, et al. Immunoreactivity score using an anti-sst2A receptor monoclonal antibody strongly predicts the biochemical response to adjuvant treatment with somatostatin analogs in acromegaly. J Clin Endocrinol Metab 2013, 98: E66–E71.
- Melmed S, Popovic V, Bidlingmaier M. Safety and efficacy of oral octreotide in acromegaly: results of a multicenter phase III trial. J Clin Endocrinol Metab 2015, 100: 1699-708.
- Chieffo C, Cook D, Xiang Q, Frohman LA. Efficacy and safety of an octreotide implant in the treatment of patients with acromegaly. J Clin Endocrinol Metab 2013, 98: 4047–54.
- Trainer PJ, Newell-Price JDC, Ayuk J, et al. A randomised, open-label, parallel group phase 2 study of antisense oligonucleotide therapy in acromegaly. Eur J Endocrinol 2018, 179: 97–108.
- Ronchi CL, Rizzo E, Lania AG, et al. Preliminary data on biochemical remission of acromegaly after somatostatin analogs withdrawal. Eur J Endocrinol 2008, 158: 19-25.
- Auriemma RS, Galdiero M, Grasso LF, et al. Complete disappearance of a GH-secreting pituitary macroadenoma in a patient with acromegaly: effect of treatment with lanreotide Autogel and consequence of treatment withdrawal. Eur J Endocrinol 2010, 162: 993-9.
- Hatipoglu E, Bozcan S, Kadioglu P. Discontinuation of somatostatin analogs while acromegaly is in long-term remission. Pituitary 2015, 18: 554–60.
- Casagrande A, Bronstein MD, Jallad RS, et al. Long-term remission of acromegaly after octreotide withdrawal is an uncommon and frequently unsustainable event. Neuroendocrinology 2017, 104: 273–9.
- Colao A, Auriemma RS, Lombardi G, Pivonello R. Resistance to somatostatin analogs in acromegaly. Endocr Rev 2011, 32: 247-71.
- Petrossians P, Martins LB, Espinoza C, et al. Gross total resection or debulking of pituitary adenomas improves hormonal control of acromegaly by somatostatin analogs. Eur J Endocrinol 2005, 152: 1-7.
- Colao AM, Attanasio R, Pivonello R, et al. Partial surgical removal of growth hormone-secreting pituitary tumors enhances the response to somatostatin analogs in acromegaly. J Clin Endocrinol Metab 2006, 91: 85-92.
Scheda octreotide e lanreotide
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Meccanismo d’azione
Legame a specifici recettori di membrana (SSTR-2 e 5) presenti sulle cellule ipofisarie e variamente accoppiati ad inibizione della secrezione e/o della proliferazione cellulare.
Indicazioni
- terapia adiuvante o primaria dell’acromegalia e gigantismo
- terapia adiuvante o primaria del TSHoma e inappropriata secrezione di TSH
- terapia dei tumori neuroendocrini
Contro-indicazioni
Nessuna.
Porre attenzione nei pazienti diabetici (per soppressione della secrezione anche di insulina) o con litiasi biliare (per soppressione della colecistochinina e inibizione della motilità colecistica e intestinale).
Farmaci disponibili, via di somministrazione e posologia
- Octreotide
- Octreotide sc (fl 0.05 mg/mL, 0.1 mg/mL, 0.2 mg/mL, 0.5 mg/mL, 1 mg/mL): octreotide Bioindustria, octreotide Sun, Sandostatina, Siroctid, Treoject
- Octreotide capsule gastroresistenti (20 mg): Mycapssa
- Octreotide LAR im (10, 20, 30 mg): Longastatina LAR, Sandostatina LAR, octreotide Teva
- Lanreotide
- Lanreotide autogel siringa preriempita (60, 90, 120 mg): Ipstyl, Myrelez
Effetti collaterali
- Dolori addominali e diarrea
- Dolore o granuloma in sede di iniezione
- Colelitiasi (raramente colecistite acuta)
- Possibile deterioramento del metabolismo glucidico
- Alopecia
- Malassorbimento vitamine liposolubili
- Bradicardia
- Ipotiroidismo centrale
Limitazioni prescrittive
Abolita la nota AIFA 40 da novembre 2016, rimane piano terapeutico.
Scheda antagonisti del recettore del GH
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Meccanismo d’azione
Analogo modificato del GH, che agisce come antagonista a livello del suo recettore, blocca la sintesi di IGF-I nei tessuti bersaglio del GH. Questa azione si svolge a livello di tutti i tessuti, è indipendente dalla presenza di recettori specifici a livello ipofisario e si accompagna ad aumento dei livelli circolanti di GH per la riduzione del feed-back dell’IGF-I.
Preparazioni, via di somministrazione, posologia
Pegvisomant (Somavert) fl 10, 15, 20, 25, 30 mg, iniezione sottocutanea quotidiana.
Indicazioni
Acromegalia e gigantismo ancora attiva dopo intervento neurochirurgico di adenomectomia e/o radioterapia, in paziente resistente o intollerante a SA e/o DA.
Contro-indicazioni
Da impiegare con cautela nei pazienti con residuo post-chirurgico voluminoso vicino alle vie ottiche o con epatopatia.
Effetti collaterali
- Possibile incremento delle dimensioni dell’adenoma, specialmente nei pazienti con adenoma aggressivo non radio-trattato e/o in quelli con precedente riduzione del volume tumorale durante terapia con SA
- Tossicità epatica: aumento delle transaminasi (significativo: > di 3 volte il limite superiore di riferimento), indipendente dalla dose e dalla durata (effetto idiosincrasico), che talvolta regredisce senza richiedere la sospensione del trattamento
- Lipodistrofia in sede di iniezione
- Cefalea
Precauzioni
Conservare in frigorifero, al riparo dalla luce
Limitazioni prescrittive
Dal 1 dicembre 2012, in fascia A con piano terapeutico.