Scheda metirapone

Alessandro Mondin & Chiara Sabbadin
UOC di Endocrinologia, Azienda Ospedale Università di Padova
Meccanismo d’azione
Inibisce la sintesi di cortisolo e corticosterone bloccando principalmente la 11ß-idrossilasi surrenalica. Ne conseguono da un lato un accumulo di precursori, tra cui 11-desossicortisolo e desossicorticosterone, e dall’altro un calo della concentrazione plasmatica di cortisolo in grado di stimolare la secrezione di ACTH, che accelera la biosintesi di steroidi, inclusa quella di androgeni.
Indicazioni
Trattamento dell’ipercortisolismo endogeno
Test diagnostico per la carenza di ACTH
Test diagnosi differenziale Cushing ACTH-dipendente
Contro-indicazioni
Insufficienza corticosurrenalica primaria
Preparazioni, via di somministrazione, posologia
Il metirapone autorizzato in Italia è commercializzato per via orale in capsule molli da 250 mg, in confezioni da 50 capsule (Cormeto®).
La dose iniziale può variare da 250 a 1000 mg/die, a seconda della gravità e della causa dell’ipercortisolismo; la titolazione può essere rapida (entro una settimana) in base alla clinica e ai dati biochimici (cortisolemia/uria), con un periodo di aggiustamento solitamente di 1–4 settimane.
Al raggiungimento di valori normali, può essere necessario aggiungere terapia sostitutiva surrenalica (block and replace) per le forme severe di CS.
Farmaco-cinetica
È assorbito rapidamente, con picco di concentrazioni plasmatiche un’ora dopo la somministrazione orale. È eliminato rapidamente dal plasma via glucuronidazione epatica (emivita plasmatica di circa 2 ore), seppur una quota venga convertita a metabolita attivo (metirapol).
Effetti collaterali
Insufficienza surrenalica per riduzione rapida dei livelli di cortisolemia. Fondamentale riconoscerne i segni e sintomi (debolezza, astenia, anoressia, nausea, vomito, ipotensione, iperkaliemia, iponatremia, ipoglicemia) in modo da ridurre o sospendere temporaneamente la terapia e/o introdurre una terapia steroidea sostitutiva.
Ipertensione (per accumulo di DOC).
Capogiri, sedazione, cefalea, raramente anche irsutismo e dermatite allergica.
Ipopotassiemia.
Può potenziare tossicità del paracetamolo per interferenza sulla glucuronidazione (raccomandare la riduzione della dose massima giornaliera di paracetamolo).
Rischio di infezioni opportunistiche (polmonite da Pneumocystis).
Avvertenze per l’utilizzo
- I dati relativi all’uso in gravidanza sono limitati, per cui non è raccomandato in gravidanza (salvo in caso di chiara necessità, con monitoraggio della pressione arteriosa e sua gestione con misure appropriate) e in donne in età fertile che non usano misure contraccettive.
- Non sono riportate indicazioni specifiche per l’aggiustamento della dose in caso di insufficienza renale o epatica. I pazienti con cirrosi epatica presentano spesso ritardo nella risposta a causa del danno epatico che prolunga l’emivita di eliminazione plasmatica del cortisolo.
Limitazioni prescrittive
Classe A. Ricetta non ripetibile (RNRL). Medicinali soggetti a prescrizione medica limitativa, da rinnovare di volta in volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti.
Scheda mifepristone (RU486)
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
Meccanismo d’azione
Antagonista non selettivo del recettore del progesterone, dei glucocorticoidi e in misura minore degli androgeni.
A basso dosaggio (fino a 600 mg) blocca l’azione del progesterone e viene solitamente utilizzato come pillola abortiva in associazione sequenziale con un analogo delle prostaglandine, fino al 63° giorno di amenorrea. Ad alto dosaggio, invece, agisce come antagonista competitivo del recettore dei glucocorticoidi, con un’affinità 3-4 volte maggiore del desametasone e 18 volte maggiore del cortisolo (1).
Preparazioni, via di somministrazione, posologia
Compresse da 200 mg o 600 mg (Mifegyne).
Viene raccomandato di iniziare con 300 mg/die e di aumentare di 300 mg alla volta in 2-4 settimane, fino ad una posologia massima di 1200 mg (2). Essendo un antagonista recettoriale, non ci sono parametri biochimici specifici per monitorarne l’andamento, per cui la titolazione deve essere fatta solo sulla base della tollerabilità e della risposta clinica del paziente (in particolare, in termini di glicemia e calo ponderale) (3).
Indicazioni
Approvato da FDA nel 2012 come farmaco orfano nel trattamento della sindrome di Cushing endogena associata a iperglicemia o non candidabile a chirurgia o persistente/recidivata dopo chirurgia.
Contro-indicazioni
- Insufficienza surrenalica cronica.
- Desiderio o gravidanza in corso.
- Ipersensibilità al mifepristone o a uno degli eccipienti presenti nelle compresse.
- Asma severa non controllata dalla terapia.
- Porfiria ereditaria.
Avvertenze
- Data la difficoltà del monitoraggio biochimico, è fondamentale un corretto e periodico follow-up clinico del paziente.
- L’eventuale co-assunzione di forti inibitori del CYP3A4 (come ketoconazolo, itraconazolo, eritromicina e succo di pompelmo) può aumentare i livelli di mifepristone e richiedere una riduzione della posologia di tale farmaco.
Effetti collaterali
- Nausea, vomito, mal di testa.
- Amenorrea nelle donne e ginecomastia negli uomini.
- Pseudo-iperaldosteronismo: ritenzione idrica, aumento ponderale, ipertensione, ipokaliemia anche grave. Tale condizione è legata all’attivazione dell’asse corticotropo secondaria al mancato feed-back centrale: il conseguente ipercortisolismo satura l’attività dell’enzima 11β-idrossisteroido-deidrogenasi tipo 2 (in grado di inattivare il cortisolo a cortisone), permettendo l’attivazione del recettore mineralcorticoide da parte del cortisolo. Il trattamento può richiedere la supplementazione di potassio per os e l’uso di antagonisti del recettore mineralcorticoide (spironolattone e derivati).
- Insufficienza surrenalica: tale condizione è ricercata dalla terapia per il Cushing e sembra essere una complicanza rara nei soggetti con un asse corticotropo intatto. Tuttavia, se misconosciuta, severa o non adeguatamente monitorata durante stress intercorrenti, può diventare fatale. Risulta, pertanto, fondamentale monitorare le condizioni cliniche del paziente, in particolare se astenia profusa, ipoglicemia, ipotensione. Il trattamento prevede la sospensione del mifepristone e la somministrazione di desametasone ad alto dosaggio (con un rapporto di circa 1 mg per ogni 400 mg di mifepristone assunti), che va proseguita per almeno 4 giorni (considerando la lunga emivita del farmaco fino a 90 h).
- Iperplasia endometriale: dovuta all’antagonismo progestinico, per cui nel trattamento a lungo termine è raccomandato il monitoraggio con ecografia trans-vaginale.
- Transitorio ipotiroidismo, per cui si raccomanda monitoraggio dei valori di TSH, per valutare eventuale inizio di terapia ormonale sostitutiva con levotiroxina.
Limitazioni prescrittive
Nel 2009 il mifepristone è stato approvato dall’AIFA solo come pillola abortiva in fascia C con ricetta ospedaliera, mentre è stato escluso dall’elenco dei medicinali erogabili a totale carico nel SSN ai sensi della Legge 648/96, con cui era stato precedentemente autorizzato con l’indicazione per “Sindrome di Cushing di origine paraneoplastica”. L’utilizzo, pertanto, nella sindrome di Cushing risulta attualmente off-label in Italia. Inoltre, i trial clinici al momento disponibili sull’uso del mifepristone nella sindrome di Cushing sono relativamente di breve durata e focalizzati al miglioramento delle caratteristiche cliniche legate all’ipercortisolismo. Non sono noti gli effetti sulla sopravvivenza e sul rischio cardio-vascolare.
Bibliografia
- Johanssen S, Allolio B. Mifepristone (RU 486) in Cushing’s syndrome. Eur J Endocrinol 2007, 157: 561-9.
- Nieman LK, Biller B, Findling JW, et al; Endocrine Society. Treatment of Cushing's syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2015, 100: 2807-31.
- Fleseriu M, Biller BM, Findling JW, et al; SEISMIC Study Investigators. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocrinol Metab 2012, 97: 2039–49.
Terapia radiante del m. di Cushing
Giorgio Arnaldi
Clinica di Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria di Ancona, Ospedali Riuniti di Ancona
La radioterapia rappresenta una seconda scelta terapeutica nei casi in cui la chirurgia ha fallito. Può essere eseguita con tecnica frazionata o con stereotassi radiochirurgica. Può essere particolarmente indicata nei pazienti sottoposti a surrenectomia bilaterale che sviluppano sindrome di Nelson.
Effetti positivi possono esserci sulle dimensioni del tumore (stabilizzato o diminuito nel 80-90% dei casi) e sulla secrezione ormonale (ridotta nel 50% dei casi). Consente di ottenere la remissione in circa il 50-60% dei pazienti dopo 3-5 anni (1-4). Purtroppo, anche nei pazienti considerati responsivi ed in remissione si sono avute recidive dopo molti anni (5, 6).
Uno dei problemi legati a questa soluzione terapeutica è però il ritardo con cui si vedono gli effetti positivi ed il rischio molto elevato di sviluppare ipopituitarismo a lungo termine (fino al 70% dei casi in alcune casistiche). Il rischio di ipopituitarismo sembra simile tra le tecniche frazionate e quelle radiochirurgiche, anche se recenti studi hanno mostrato un minor rischio con queste ultime tecniche (4). La radioterapia convenzionale è stata associata ad un maggior rischio di eventi cerebrovascolari e disturbi cognitivi. Questi rischi sembrerebbero essere minori con le tecniche di radiochirurgia, ma al momento non vi sono dati su larghe casistiche e con lungo periodo di osservazione (6, 7).
Seppure non si possa affermare che i tumori ACTH-secernenti abbiano una maggiore radiosensibilità, alcuni studi hanno mostrato in questi tumori una più rapida comparsa degli effetti anti-secretivi rispetto ai tumori GH-secernenti e PRL-secernenti (8).
I pazienti radiotrattati dovranno essere seguiti per moltissimo tempo e valutati periodicamente, sia per la possibile comparsa di deficit ipofisari che per la possibile remissione/guarigione ormonale e i cambiamenti tumorali. In genere gli effetti si manifestano gradualmente ed il rischio di ipopituitarismo dipende da molti fattori (dose impiegata, localizzazione del residuo, distanza dal peduncolo, precedente chirurgia o radioterapia). Generalmente la valutazione ormonale ipofisaria deve avvenire annualmente. La RMN di controllo viene consigliata dopo 3 mesi dal trattamento e poi annualmente. Nel tempo necessario affinchè la radioterapia sia efficace, sarà necessario iniziare e/o proseguire la terapia medica per il controllo dell'ipercortisolismo.
Bibliografia
- Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab 2008, 93: 2454-62.
- Castinetti F, Nagai M, Dufour H, et al. Gamma knife radiosurgery is a successful adjunctive treatment in Cushing's disease. Eur J Endocrinol 2007, 156: 91-8.
- Petit JH, Biller BM, Yock TI, et al. Proton stereotactic radiotherapy for persistent adrenocorticotropin-producing adenomas. J Clin Endocrinol Metab 2008, 93: 393-9.
- Mortini P, Losa M, Barzaghi R, et al. Results of transsphenoidal surgery in a large series of patients with pituitary adenoma. Neurosurgery 2005, 56: 1222-33.
- Ronchi CL, Attanasio R, Verrua E, et al. Efficacy and tolerability of gamma knife radiosurgery in acromegaly: a 10-year follow-up study. Clin Endocrinol (Oxf) 2009, 71: 846-52.
- Castinetti F, Nagai M, Morange I, et al. Long-term results of stereotactic radiosurgery in secretory pituitary adenomas. J Clin Endocrinol Metab 2009, 94: 3400-7.
- Castinetti F, Brue T. Gamma Knife radiosurgery in pituitary adenomas: Why, who, and how to treat? Discov Med 2010, 10: 107-11.
- Jagannathan J, Yen CP, Pouratian N, Laws ER, Sheehan JP. Stereotactic radiosurgery for pituitary adenomas: a comprehensive review of indications, techniques and long-term results using the Gamma Knife. J Neurooncol 2009, 92: 345-56.
Cushing in gravidanza
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 17 dicembre 2018)
Nonostante l’impatto negativo della malattia sulla fertilità, non è infrequente l’insorgenza di una gravidanza in una paziente affetta e la sua gestione diventa ancora più difficile non solo per le possibili complicanze materno-fetali legate all’ipercortisolismo, ma anche per la gestione diagnostico-terapeutica, limitata dai possibili effetti teratogeni (1,2).
Epidemiologia della s. di Cushing diagnosticata in gravidanza
Si tratta di una condizione molto rara, con meno di 200 casi riportati in letteratura. A differenza dei quadri insorti al di fuori della gravidanza, la causa più frequente in gravidanza è l’adenoma surrenalico (40-60% dei casi), mentre l’adenoma ipofisario riguarda circa un terzo dei casi e il carcinoma surrenalico < 10%. Questa diversa distribuzione dipende probabilmente dal fatto che le forme ACTH-dipendenti si associano anche a iperandrogenismo, con conseguente maggior interferenza sull'asse gonadotropo.
La diagnosi di s. di Cushing in gravidanza
La diagnosi clinica è resa più difficile perché molti segni della gravidanza fisiologica, quali aumento ponderale, smagliature anche rubrae, irsutismo, acne, astenia e instabilità emotiva, mimano uno pseudo-Cushing. Anche ipertensione e iperglicemia possono essere frequenti complicanze gravidiche e l’ipokaliemia può esser legata all’iperemesi del I trimestre o alla fisiologica iperattivazione dell'asse renina-angiotensina-aldosterone (RAAS). In gravidanza solo la concomitante presenza di ipertensione, ecchimosi e debolezza muscolare o l’insorgenza di fratture da fragilità sembra fortemente predittiva di una vera s. di Cushing.
La diagnosi biochimica è resa altrettanto difficoltosa dalle modificazioni ormonali fisiologiche correlate all’instaurarsi della gravidanza, quali l’attivazione dell’asse corticotropo e del RAAS. In particolare, sin dal I trimestre aumentano i livelli di cortisolemia totale a causa dell’aumento delle proteine leganti il cortisolo (corticosteroid-binding globulin), secondario all’aumento degli estrogeni e alla crescente produzione placentare di CRH e ACTH già dalla 7° settimana gestazionale. Tale aumento della cortisolemia rende inaffidabile il test di soppressione con desametasone 1 mg, cui solo il 40% delle donne sane in gravidanza sembra rispondere con livelli di cortisolo < 50 nmol/L (3). Anche la cortisoluria delle 24 h aumenta di 1.4 e 1.6 volte nel II e III trimestre, rispettivamente, e solo un aumento che supera di 2-3 volte il limite maggiore di norma può essere considerato suggestivo di Cushing, soprattutto se eseguito con le metodiche più accurate di cromatografia e spettrometria di massa (4). Solo il ritmo circadiano del cortisolo sembra conservato, sebbene presenti livelli più alti di quelli normalmente attesi, soprattutto nel III trimestre. Pertanto, il cortisolo salivare notturno, se disponibile, è lo strumento più utile, almeno fino al II trimestre, per la diagnosi differenziale tra ipercortisolismo fisiologico e patologico in gravidanza.
Anche la diagnosi eziologica è resa difficile dalla mancanza in tale categoria di pazienti di dati sui test di II livello (stimolazione con CRH o desmopressina e soppressione con desametasone 8 mg). La produzione placentare di CRH e ACTH, infatti, si associa a livelli di ACTH non soppressi nel 50% delle donne con Cushing surrenalico (5). Infine, la diagnostica strumentale è limitata per i rischi teratogeni e può avvalersi solo dell’ecografia surrenalica o della RM senza mezzo di contrasto (5).
Diagnosi differenziale di ipertensione in gravidanza
L’ipertensione riscontrata durante il I trimestre di gravidanza è considerata pre-esistente e pertanto cronica, a differenza delle forme di ipertensione gestazionale e pre-eclampsia che compaiono dopo la 20° settimana gestazionale.
In gravidanza le forme secondarie di ipertensione sono molto rare e prevalentemente legate a malattie renali croniche, stenosi delle arterie renali, apnea ostruttiva notturna oltre che a forme endocrine.
Il feocromocitoma, che è la più comune causa endocrina di ipertensione in gravidanza, si associa ad alta mortalità materno-fetale (> 50% se non riconosciuta precocemente). Dal punto di vista clinico, rispetto alla s. di Cushing, non si associa ad aumento ponderale, ma può presentare ipokaliemia, iperglicemia e proteinuria. Per la diagnosi è raccomandato il dosaggio delle metanefrine libere urinarie (6).
Anche l’iperaldosteronismo primario può essere causa di ipertensione gestazionale secondaria, seppur con diversi gradi di severità per l’effetto antagonista degli elevati livelli di progesterone. Dal punto di vista clinico, oltre all’ipokaliemia, solitamente non si associa ad altri segni tipici di ipercortisolismo. La diagnosi è in parte resa difficile dalla fisiologica attivazione del RAAS durante la gravidanza, dovuta all’aumento degli estrogeni e della renina placentare, che determinano livelli di aldosterone plasmatico fino a 8-10 volte maggiori del range di normalità. Tuttavia, il riscontro in gravidanza di livelli di renina bassa o soppressa può considerarsi fortemente predittivo di iperaldosteronismo primitivo (7).
Le complicanze materne
La s. di Cushing florida o in non ottimale controllo terapeutico si associa ad alto tasso di complicanze materne: ipertensione (68%), diabete e pre-diabete (25%), pre-eclampsia (14%), osteoporosi e fratture (5%), disturbi psichiatrici (4%), scompenso cardiaco (3%), maggior facilità di infezioni delle ferite e morte (2%).
Le complicanze fetali
Il feto è protetto dagli effetti dei glucocorticoidi materni dall’aumentata espressione placentare dell’enzima 11ß-idrossi-steroido-deidrogenasi di tipo 2, che inattiva il cortisolo in cortisone. Tuttavia, nella s. di Cushing l’attività di tale enzima risulta saturata dagli elevati livelli di cortisolo, comportando un’elevata morbilità anche del feto, dovuta soprattutto a parto prematuro (43%), ritardo di crescita intra-uterina (21%), aborto o morte intra-uterina (5%) e insufficienza surrenalica post-partum (2%). Il tempestivo trattamento medico o chirurgico della madre affetta diventa, pertanto, fondamentale per ridurre il rischio di complicanze sia materne sia fetali.
Quando sospettare la presenza di recettori aberranti
In letteratura sono riportati alcuni casi di donne con s. di Cushing surrenalica indotta dalla gravidanza, per la presenza di recettori aberranti per LH e hCG, con un quadro clinico e biochimico che sfumava spontaneamente dopo il parto. Tali report sono molto intriganti per le possibili implicazioni terapeutiche, che sono però limitate dalla rarità e dal particolare stato della paziente affetta.
Il trattamento medico-chirurgico durante la gravidanza
Mancano dati solidi in letteratura sul miglior approccio finalizzato a ridurre l'ipercortisolismo in gravidanza.
La chirurgia, sia ipofisaria sia surrenalica, rimane la terapia di prima linea, da programmare durante il II trimestre, entro la 24° settimana gestazionale, per limitare il possibile rischio di parto prematuro e ritardo di crescita intra-uterina. Dopo l’intervento, dovrà essere sempre instaurata un’adeguata terapia sostitutiva con glucocorticoidi, da modulare secondo necessità, in particolare al momento del travaglio e del parto, per il possibile rischio di crisi surrenalica.
Nel caso in cui la chirurgia sia contro-indicata, tra i farmaci ad azione surrenalica, il metirapone sembra il più sicuro, sebbene possa peggiorare l'ipertensione e il rischio di pre-eclampsia e possa passare la barriera placentare, alterando la steroidogenesi fetale. Classicamente contro-indicati per il rischio teratogeno risultano, invece, mitotane e chetoconazolo, sebbene per quest'ultimo siano stati riportati alcuni casi di buona efficacia e tollerabilità materno-fetale. Tra i farmaci ad azione centrale, invece, attualmente solo la cabergolina potrebbe rappresentare un'opzione terapeutica efficace e sicura. Ad ogni modo, soprattutto nei casi diagnosticati tardivamente, può essere anche preso in considerazione solo un approccio conservativo mirato a trattare e ridurre le comorbilità, quali ipertensione e diabete, impostando una terapia mirata solo dopo il parto.
Il follow-up della paziente dopo il parto
Dopo il parto, la paziente operata andrà controllata periodicamente per valutare l'eventuale ripresa dell'asse corticotropo, ma anche la possibile recidiva di malattia nelle forme ipofisarie. Queste ultime, se non trattate durante la gravidanza, andranno anche rivalutate radiologicamente con RM ipofisi a 3-6 mesi dal parto. Questa è indicata anche nelle pazienti precedentemente sottoposte a bisurrenectomia per m. di Cushing, per quanto la gravidanza non sembri aumentare il rischio di s. di Nelson.
Problemi diagnostico-terapeutici del carcinoma surrenalico in gravidanza
Il carcinoma surrenalico rappresenta meno del 10% delle cause di s. di Cushing in gravidanza, con prognosi ancora più infausta rispetto alle forme diagnosticate al di fuori della gravidanza, a causa di un maggior ritardo diagnostico, di un possibile stimolo proliferativo sostenuto dagli elevati livelli di ormoni sessuali e di un minor tasso di radicalità chirurgica. La diagnosi clinica si basa solitamente sull'insorgenza di segni clinici da ipersecrezione ormonale e da compressione delle strutture adiacenti (dolore, sensazione di pienezza e gonfiore addominale). La diagnosi radiologica si basa solo su ecografia o RM diretta. La terapia è chirurgica e può esser preceduta da un breve periodo di trattamento farmacologico per ridurre l’ipercortisolismo, sempre se possibile evitando l’uso del mitotane per il suo effetto teratogeno.
La gestione della terapia contraccettiva nella donna con s. di Cushing
Considerati tutti i possibili rischi di una gravidanza, seppur rara, in corso di sindrome di Cushing, è raccomandato l’utilizzo di metodi contraccettivi a tutte le pazienti giovani ad alto rischio, soprattutto se in buon controllo farmacologico o nel post-intervento. Dato lo stato trombofilico, che può persistere anche a un anno dalla chirurgia, sono contro-indicate le classiche pillole estro-progestiniche, a favore di pillole con minor impatto sul profilo coagulativo (estrogeni naturali, solo progestinici) o di metodi barriera.
Conclusioni
La diagnosi de novo di Cushing in gravidanza è resa difficile dalle modifiche ormonali correlate alla gravidanza, che falsano gli abituali test diagnostici di primo livello, con la sola eccezione del cortisolo salivare notturno, che sembra il più affidabile. Anche la gestione della malattia durante la gravidanza è gravata da molteplici complicanze materno-fetali e contro-indicazioni (principalmente per rischio teratogeno) alla maggior parte degli abituali approcci farmacologici e chirurgici dell’ipercortisolismo. Per tali motivi, andrebbe sconsigliata la gravidanza nelle pazienti con malattia attiva, in buon controllo farmacologico o in remissione da meno di un anno.
Bibliografia
- Brue T, Castinetti F. The risks of overlooking the diagnosis of secreting pituitary adenomas. Orphanet J Rare Dis 2016, 11: 135.
- Brue T, Amodru V, Castinetti F. Management of Cushing's syndrome during pregnancy: solved and unsolved questions. Eur J Endocrinol 2018, 178: R259-66.
- Lindsay JR, Nieman LK. The hypothalamic-pituitary-adrenal axis in pregnancy: challenges in disease detection and treatment. Endocr Rev 2005, 26: 775-99.
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2008, 93: 1526-40.
- Lindsay JR, Jonklaas J, Oldfield EH, Nieman LK. Cushing’s syndrome during pregnancy: personal experience and review of the literature. J Clin Endocrinol Metab 2005, 90: 3077-83.
- Sarathi V, Lila AR, Bandgar TR, et al. Pheochromocytoma and pregnancy: a rare but dangerous combination. Endocr Pract 2010, 16: 300-9.
- Riester A, Reincke M. Progress in primary aldosteronism: mineralocorticoid receptor antagonists and management of primary aldosteronism in pregnancy. Eur J Endocrinol 2015, 172: R23-30.
Clinica e diagnostica TSHomi
Paolo Beck-Peccoz1 e Luca Persani2
Dipartimento di Scienze Cliniche e di Comunità, Università degli Studi di Milano; 1Fondazione IRCCS Cà Granda Policlinico e 2IRCCS Istituto Auxologico Italiano, Milano
Introduzione
L’ipertiroidismo secondario ad adenomi ipofisari TSH-secernenti (TSHomi) è estremamente raro, ma facilmente riconoscibile in base al quadro biochimico (1-4). Infatti, si caratterizza per il riscontro di elevati livelli di FT4 ed FT3, non dissimili da quelli reperibili in altre forme di ipertiroidismo, ma in presenza di livelli circolanti di TSH non soppressi, anzi a volte elevati. A questo particolare quadro biochimico venne dato nome di inappropriata secrezione di TSH, nel senso che rispetto ai quadri “classici” di ipertiroidismo in cui il TSH è sempre indosabile, nei TSHomi il TSH è chiaramente dosabile. Si preferisce oggi chiamare tale quadro “ipertiroidismo centrale”, dato che è decisamente appropriato per un adenoma produrre l’ormone in maniera autonoma e totalmente sganciata dal classico meccanismo di feed-back negativo (3).
È ovvio che prima di procedere nell’iter diagnostico, bisogna escludere possibili interferenze di fattori circolanti che possano dare risultati falsamente elevati sia nel dosaggio degli ormoni tiroidei che del TSH, come anticorpi anti-TSH, o quelli anti-T4 e/o anti-T3, o forme anomale di albumina o transtiretina (1, 5).
La diagnosi differenziale più impegnativa è quella tra TSHomi e sindromi da resistenza all’azione degli ormoni tiroidei (RTH), dato che questi ultimi presentano lo stesso quadro biochimico (6, 7). Il mancato riconoscimento di tali disordini tiroidei può condurre a drammatiche conseguenze, quali una improvvida tiroidectomia in pazienti con TSHoma o una inutile chirurgia ipofisaria in quelli con RTH.
La prevalenza dei TSHomi è stimata a circa 1 caso per milione (3). Non vi è dubbio che l’introduzione del dosaggio ultrasensibile del TSH e di quello diretto degli ormoni tiroidei liberi abbia comportato il riconoscimento di un notevole numero di pazienti con TSHoma, situazione che in passato è stata troppe volte confusa con la presenza di un morbo di Graves-Basedow e, di conseguenza, trattata in maniera incorretta. Dai dati della letteratura si desume che la tiroidectomia ha un effetto deleterio sulla crescita del TSHoma, con una significativa maggior frequenza di lesione macroinvasive rispetto ai pazienti non tiroidectomizzati (fig. 1).
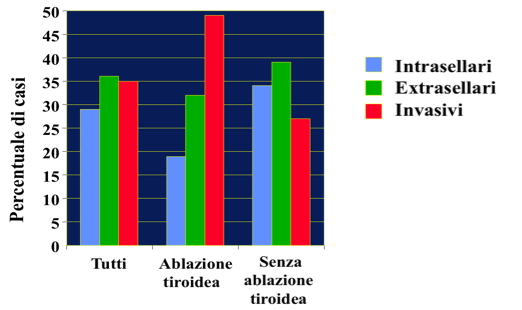
Figura 1
Effetti della tiroidectomia sul volume dei TSHomi. I dati sono stati calcolati da 264 pazienti riportati in letteratura (95 tiroidectomizzati e 169 intonsi). “Intrasellari” indica sia i micro- che i macro-adenomi intrasellari, “Extrasellari” sono i macroadenomi sovrasellari e “Invasivi” sono i macroadenomi altamente invasivi. Gli adenomi “invasivi” sono statistica-mente molto più frequenti nei pazienti tiroidectomizzati, mentre quelli “intrasellari” sono significativamente più frequenti nei pazienti intonsi (Fisher’s exact test).
Nella maggior parte dei TSHomi, l’ipersecrezione ormonale riguarda il solo TSH, i cosiddetti TSHomi “puri”. Nel 25% dei casi, i TSHomi sono tumori “misti”, potendo cosecernere GH o PRL o gonadotropine, così associando all’ipertiroidismo segni clinici di acromegalia, di amenorrea, di galattorrea, di disfunzione erettile e di infertilità.
I TSHomi sono nell’80% dei casi macroadenomi (diametro > 1 cm), anche se i recenti metodi di dosaggio ormonale e le nuove tecniche neuroradiologiche permettono oggi il riconoscimento di un numero crescente di microadenomi (2). Rarissimi sono i casi di carcinoma TSH-secernente, così come i TSHomi ectopici riscontrati in regione faringea.
Come per gli altri adenomi ipofisari, molto poco si sa sull’eziopatogenesi dei TSHomi. Gli studi più recenti hanno escluso mutazioni attivanti vari proto-oncogeni o la perdita di geni onco-soppressori (8-10), ma hanno dimostrato in qualche TSHoma la presenza di mutazioni somatiche del recettore ß degli ormoni tiroidei, che potrebbe spiegare la perdita di sensibilità al feed-back negativo degli ormoni tiroidei da parte delle cellule tumorali (11).
Clinica e diagnostica
I sintomi e i segni clinici dell’ipertiroidismo da TSHoma possono essere a volte sfumati come conseguenza di una lentissima insorgenza della malattia (1-4). A volte possono essere mascherati dalla contemporanea ipersecrezione di altri ormoni ipofisari, in particolare in caso di acromegalia (tumori misti TSH/GH), ma anche dal deficit di altri ormoni ipofisari per compressione del parenchima normale da parte di un macroadenoma TSH-secernente (1-3, 8). In tale situazione, è frequente che i segni neurologici, quali la cefalea e il restringimento del campo visivo, compaiano prima di quelli dell’ipertiroidismo.
Il gozzo è quasi sempre presente, anche in pazienti sottoposti a tiroidectomia subtotale, dato il continuo stimolo del TSH sulla replicazione delle cellule tiroidee. Può essere uni- o multinodulare, raramente pretossico o tossico (12). L’associazione tra TSHoma e carcinoma differenziato della tiroide è stata descritta in soli 5 casi. La presenza di oftalmopatia è stata descritta in pochissimi casi che avevano sviluppato un morbo di Graves-Basedow, mentre in soli 3 casi risultava secondaria ad invasione tumorale dell’orbita.
Disturbi mestruali sono stati reperiti in 1 donna su 4. Ipopituitarismo parziale è riportato nel 25% dei casi. Galattorrea e acromegalia sono stati documentati in tumori misti TSH/PRL e TSH/GH (3).
Come già accennato, la diagnosi si basa essenzialmente sul reperto di concentrazioni dosabili di TSH in presenza di livelli di FT4 e di FT3 elevati. Contrariamente a quanto descritto per altri tumori ipofisari (GH e PRL), non esiste per i TSHomi una correlazione tra volume del tumore ed ipersecrezione dell’ormone ipofisario (1). Ciò è in parte dovuto al fatto che l’attività biologica del TSH secreto è variabile, potendo essere aumentata, normale o ridotta. Tale variabilità è causata da modificazioni dei processi di glicosilazione delle molecole di TSH (13).
Utile alla diagnosi di TSHoma (ma disponibile solo in alcuni centri) è anche la misurazione della subunità alfa delle glicoproteine (alfa-GSU) e del rapporto molare alfa-GSU/TSH. Questo si ottiene dividendo l’alfa-GSU in µg/L per il TSH in mU/L, e moltiplicando poi per 10 (3, 4, 14). I valori di normalità di questi due parametri variano a seconda del sesso, dell’età e dei livelli circolanti dei vari ormoni glicoproteici (TSH, LH, FSH e hCG) (15). Elevati livelli di alfa-GSU sono presenti in circa il 60% dei casi, anche se nei microadenomi tale frequenza si riduce a circa il 10%. Il rapporto molare risulta elevato in circa il 70% dei casi. Va ricordato, comunque, che l’alfa-GSU può essere ipersecreta da quasi tutti gli adenomi ipofisari funzionanti e non funzionanti, con l’esclusione degli ACTHomi.
Nella diagnostica dei TSHomi, soprattutto per la diagnosi differenziale con l’RTH, un posto rilevante assume la misurazione dei parametri che valutano l’azione degli ormoni tiroidei a livello periferico. Tra questi, i più utili si sono dimostrati l’SHBG (sex hormone-binding globulin), l’ACE (angiotensin-converting enzyme), l’ICTP (carboxy-terminal telopeptide of type 1 collagen), l’osteocalcina ed il sIL-2R (soluble interleukin-2 receptor). Nella maggior parte dei casi di TSHoma, i valori di tali parametri risultano nel range dell’ipertiroidismo, mentre nella RTH sono nel range normale (16, 17).
Le prove dinamiche di stimolazione o inibizione della secrezione di TSH possono aumentare notevolmente la sensibilità e la specificità della diagnosi di TSHoma. Il test con TRH (200 µg ev) dimostra una assenza o insufficiente risposta del TSH in circa il 90% dei casi. Il test di inibizione della secrezione di TSH con T3 (80-100 µg/die per 8-10 giorni, T3 da somministrare ogni 8 ore) non ha mai causato una totale inibizione dei livelli circolanti di TSH nei pazienti con TSHoma sottoposti a tale test.
Recentemente, noi abbiamo sperimentato una nuova via per documentare la presenza di un TSHoma e per distinguere il TSHoma da una sindrome da resistenza all’azione degli ormoni tiroidei (18). Si tratta il paziente per 2-3 mesi con analoghi della somatostatina a lunga durata d’azione (octreotide LAR 20 mg ogni 28 giorni; lanreotide Autogel 90 mg ogni 28 giorni): in caso di TSHoma si assisterà nel 95% dei casi ad una riduzione o addirittura ad una normalizzazione dei livelli circolanti di FT4 ed FT3, mentre nell’RTH il quadro biochimico rimarrà invariato.
Infine, l’esecuzione di una risonanza magnetica nucleare o di una tomografia assiale computerizzata della regione ipotalamo-ipofisaria è obbligatoria nel sospetto di un TSHoma. È utile ricordare che in 3 differenti pazienti sono state descritte lesioni ectopiche nella sede della cosiddetta “ipofisi faringea”.
Del tutto inutile appare lo studio scintigrafico con octreotide marcata, data la poca sensibilità di tale metodologia che non distingue l’ipofisi normale da quasi tutti gli adenomi ipofisari funzionanti o non funzionanti.
Diagnostica differenziale
La diagnosi differenziale con le forme “classiche” di ipertiroidismo si basa sul reperto biochimico di TSH dosabile in presenza di elevate concentrazioni di FT4 e di FT3. Per quanto riguarda la diagnosi con le sindromi da resistenza all’azione degli ormoni tiroidei, si rimanda al capitolo specifico. Nella figura 2, sono, comunque, sintetizzate le maggiori differenze tra TSHomi ed RTH.
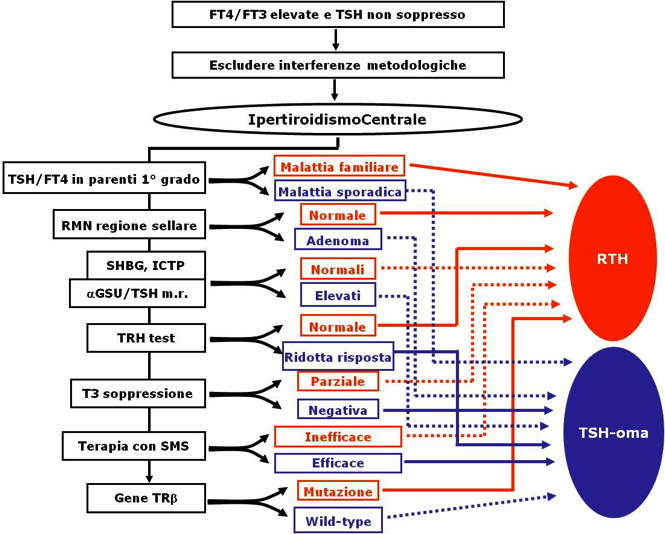
Figura 2.
Flow-chart della diagnosi differenziale tra TSHomi ed RTH. Per una accurata descrizione si rimanda al Capitolo sulla resistenza all’azione degli ormoni tiroidei.
Bibliografia
- Beck-Peccoz P, Brucker-Davis F, Persani L, et al. Thyrotropin-secreting pituitary tumors. Endocr Rev 1996, 17: 610-38 .
- Socin HV, Chanson P, Delemer B, et al. The changing spectrum of TSH-secreting pituitary adenomas: diagnosis and management in 43 patients. Eur J Endocrinol 2003, 148: 433-42.
- Beck-Peccoz P, Persani L, Lania A. Thyrotropin-Secreting Pituitary Adenomas, Chapter 20, in: Endotext, Thyroid Disease.
- Brucker-Davis F, Oldfield EH, Skarulis MC, et al. Thyrotropin-secreting pituitary tumors: diagnostic criteria, thyroid hormone sensitivity, and treatment outcome in 25 patients followed at the National Institutes of Health. J Clin Endocrinol Metab 1999, 84: 476-86.
- Gurnell M, Halsall DJ, Chatterjee VK. What should be done when thyroid function tests do not make sense? Clin Endocrinol (Oxf) 2011, 74: 673-8.
- Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev 1993, 14: 348-99.
- Gurnell M, Visser TJ, Beck-Peccoz P, Chatterjee VKK. Resistance to Thyroid Hormone. In: Endocrinology, Adult and Pediatric (6th Edition, vol. II), edited by Jameson LJ and DeGroot LJ. Saunders Elsevier, Philadelphia, PA, 2010, pp. 1745.
- Bertholon-Grégoire M, Trouillas J, Guigard MP, et al. Mono-and plurihormonal thyrotropic pituitary adenomas: pathological, hormonal and clinical studies in 12 patients. Eur J Endocrinol 1999, 140: 519-27.
- Boggild MD, Jenkinson S, Pistorello M, et al. Molecular genetics studies of sporadic pituitary tumors. J Clin Endocrinol Metab 1994, 78: 387-92.
- Dong Q, Brucker-Davis F, Weintraub BD, et al. Screening of candidate oncogenes in human thyrotroph tumors: absence of activating mutations of the Gaq, Ga11, Gas, or thyrotropin-releasing hormone receptor genes. J Clin Endocrinol Metab 1996, 81: 1134-40.
- Ando S, Sarlis NJ, Oldfield EH, et al. Somatic mutation of TR-β can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab 2001, 86: 5572-6.
- Abs R, Stevenaert A, Beckers A. Autonomously functioning thyroid nodules in a patient with a thyrotropin-secreting pituitary adenoma: possible cause-effect relationship. Eur J Endocrinol 1994, 131: 355-8.
- Beck-Peccoz P, Persani L. Variable biological activity of thyroid-stimulating hormone. Eur J Endocrinol 1994, 131: 331-40.
- Terzolo M, Orlandi F, Bassetti M, et al. Hyperthyroidism due to a pituitary adenoma composed of two different cell types, one secreting alfa-subunit alone and another cosecreting alfa-subunit and thyrotropin. J Clin Endocrinol Metab 1991, 72: 415-21.
- Beck-Peccoz P, Persani L, Faglia G. Glycoprotein hormone α-subunit in pituitary adenomas. Trends Endocrinol Metab 1992, 3: 41-5.
- Beck-Peccoz P, Roncoroni R, Mariotti S, et al. Sex hormone-binding globulin measurement in patients with inappropriate secretion of thyrotropin (IST): evidence against selective pituitary thyroid hormone resistance in nonneoplastic IST. J Clin Endocrinol Metab 1990, 71: 19-25.
- Persani L, Preziati D, Matthews CH, et al. Serum levels of carboxyterminal cross-linked telopeptide of type I collagen (ICTP) in the differential diagnosis of the syndromes of inappropriate secretion of TSH. Clin Endocrinol (Oxf) 1997, 47: 207-14.
- Mannavola D, Persani L, Vannucchi G, et al. Different response to chronic somatostatin analogues in patients with central hyperthyroidism. Clin Endocrinol (Oxf) 2005, 62: 176-81.
Terapia TSHoma
Paolo Beck-Peccoz1 e Luca Persani2
Dipartimento di Scienze Cliniche e di Comunità, Università degli Studi di Milano; 1Fondazione IRCCS Cà Granda Policlinico e 2IRCCS Istituto Auxologico Italiano, Milano
Terapia chirurgica
La terapia neurochirurgica di adenomectomia per via trans-nasosfenoidale o, più raramente, per via trans-cranica rimane il primo approccio alla terapia dei TSHomi.
L’obiettivo è quello di rimuovere la massa tumorale e di restaurare una normale funzione ipofisaria e tiroidea (1). Tale obiettivo è limitato dalla frequente presenza di macroadenomi, a volte invasivi e di consistenza dura, tanto che alcuni autori inglesi hanno parlato di “pituitary stones” (2).
Il paziente deve essere preparato all’intervento in prima istanza con analoghi della somatostatina (sia octreotide sc, 50-750 µg 2-3 volte al giorno, che analoghi a lunga durata d'azione, octreotide LAR, lanreotide autogel) e beta-bloccanti, quali il propranololo, che oltre a bloccare l’effetto delle catecolamine a livello cardiaco, bloccano la trasformazione periferica della T4 a T3, l’ormone biologicamente più attivo. I farmaci anti-tiroidei (metimazolo o propil-tiouracile) non sono indicati in questi pazienti, in quanto una riduzione dei valori degli ormoni tiroidei potrebbe aumentare i valori di TSH e comportare un aumento delle dimensioni tumorali
Il paziente operato può risultare ipopituitarico e la valutazione del parziale o completo ipopituitarismo deve essere effettuata dopo 2-3 mesi dall’intervento.
Terapia radiante
In caso di fallimento della neurochirurgia o di residuo tumorale cospicuo, così come nei pazienti in cui tale trattamento è controindicato, deve essere presa in considerazione una terapia radiante, eseguita possibilmente con gamma-knife. La dose di radiazione deve essere superioreai 45 Gy se la radioterapia viene eseguita con tecnica classica frazionata (2 Gy al giorno) e ai 25 Gy se attuata in una singola dose. Dosi superiori a quelle prima indicate devono essere valutate dal radioterapista e variano a seconda del volume e della posizione del tumore.
Troppi pochi casi di TSHoma sono stati trattati con la radioterapia per poter quantizzare in modo significativo i benefici di tale approccio terapeutico (2,3). Comunque la radioterapia da sola o insieme alla chirurgia ipofisaria non è stata dimostrata essere efficace (2).
Terapia farmacologica
Il trattamento farmacologico dei TSHomi si basa sulla somministrazione degli analoghi a lunga durata d’azione dell’octreotide o del lanreotide (octreotide: LAR 10-30 mg ogni 28 giorni; lanreotide: 60-120 mg ogni 28 giorni).
Nessun altro tumore ipofisario o neuroendocrino è così sensibile al trattamento con analoghi della somatostatina quanto i TSHomi (3-5): circa il 95% dei TSHomi normalizza la secrezione di TSH e degli ormoni tiroidei, ripristinando così lo stato di eutiroidismo nel paziente. Nel caso di adenomi misti TSH/GH, non solo viene curato l’ipertiroidismo ma anche l’acromegalia (3-5).
Tale terapia è in grado anche di ridurre la massa tumorale in circa il 50% dei casi.
Come tutti i pazienti trattati con analoghi della somatostatina, anche quelli con TSHoma devono essere monitorati per quanto riguarda la possibile induzione di colelitiasi e di alterazioni del metabolismo glicidico (2,3).
In pazienti con adenoma misto TSH/PRL possono essere fatti tentativi con farmaci dopaminergici, quali bromocriptina e cabergolina, a volte con successo e, ovviamente, con costi minori.
Criteri di cura e follow-up
Sebbene non supportati da sufficiente documentazione scientifica, tra i criteri di cura si annoverano:
- la remissione dell’ipertiroidismo
- la regressione dei segni neurologici della presenza del tumore (cefalea e riduzione del campo visivo)
- la normalizzazione dei parametri biochimici precedentemente alterati (6).
Ovviamente, molti dei criteri suddetti non sono applicabili ai soggetti precedentemente tiroidectomizzati o sottoposti a terapia ablativa con radioiodio. È utile sottolineare che anche nei pazienti con normalizzazione dei criteri clinici e biochimici, non è assicurata una cura completa. Infatti, una rilevante riduzione della massa tumorale potrebbe di per sé determinare una situazione di apparente normalizzazione. Solo il test di soppressione con T3 è in grado di confermare la completa risoluzione, qualora la secrezione del TSH fosse totalmente inibita.
La recidiva del tumore appare essere rara, almeno nei primi anni dopo la resezione chirurgica.
Buona norma è ricontrollare gli esami biochimici 2-3 volte nel primo anno dopo l’intervento chirurgico e poi annualmente. La RMN deve essere eseguita dopo 6 mesi dall’intervento e, in presenza di residui tumorali una volta all’anno. In assenza di residui e con un test di soppressione con T3 normale, dopo i controlli del primo anno, le indagini suddette dovrebbero essere effettuate ogni 3-5 anni.
Bibliografia
- Losa M, Mortini P, Franzin A, et al. Surgical management of thyrotropin-secreting pituitary adenomas. Pituitary 1999, 2: 127-31.
- Beck-Peccoz P, Brucker-Davis F, Persani L, et al. Thyrotropin-secreting pituitary tumors. Endocr Rev 1996, 17: 610-38 .
- Beck-Peccoz P, Persani L, Lania A. Thyrotropin-Secreting Pituitary Adenomas, Chapter 20, in: Endotext, Thyroid Disease.
- Kuhn JM, Arlot S, Lefebvre H, et al. Evaluation of the treatment of thyrotropin-secreting pituitary adenomas with a slow release formulation of the somatostatin analog lanreotide. J Clin Endocrinol Metab 2000, 85: 1487-91.
- Caron P, Arlot S, Bauters C, et al. Efficacy of the long-acting octreotide formulation (octreotide-LAR) in patients with thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab 2001, 86: 2849-53.
- Losa M, Giovanelli M, Persani L, et al. Criteria of cure and follow-up of central hyperthyroidism due to thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab 1996, 81: 3084-90.
Gli adenomi ipofisari familiari e sindromici
Ernesto De Menis
UO Medicina Interna, Dipartimento Medicina Clinica, Ospedale Generale, Montebelluna
(aggiornato al 5 luglio 2019)
Le forme familiari di adenomi ipofisari sono più frequenti di quanto ritenuto in passato e attualmente si stima che il 5% degli adenomi ipofisari abbia una base eredo-familiare (1). È importante riconoscere precocemente la base ereditaria, perché ciò determina prognosi/terapie specifiche e ricerca di altre manifestazioni cliniche legate ad es. a presenza di altre neoplasie, come nella MEN-1. Inoltre lo screening nei familiari permette l’identificazione di soggetti in fase precoce, verosimilmente modificando il loro esito clinico.
In ogni adenoma ipofisario, indipendentemente dall’età, devono essere valorizzati:
- anamnesi familiare e personale;
- esame obiettivo;
- calcemia alla diagnosi e nel follow-up.
Particolare attenzione deve essere rivolta agli adenomi in età pediatrica, indipendentemente da altri aspetti clinici.
Le forme di riferimento per rilevanza epidemiologica e clinica sono sicuramente la MEN-1 e poi AIP.
Si distinguono due categorie di tumori ipofisari su base ereditaria, a seconda che comprendano o meno altre manifestazioni cliniche extra-ipofisarie.
SOLO TUMORI IPOFISARI
Questo gruppo comprende 2 entità.
FIPA
L’acronimo sta per Familial Isolated Pituitary Adenoma, forma clinica definita dalla presenza in una famiglia di tumori solamente ipofisari. I membri delle famiglie possono presentare:
- adenomi dello stesso tipo (classicamente tumori GH-secernenti, acromegalia familiare), definiti in tal caso come FIPA omogenei;
- adenomi di diverso tipo, definiti come FIPA eterogenei.
Nelle famiglie FIPA prevalgono nettamente gli adenomi GH/PRL secernenti.
Dal punto di vista clinico questi tumori mostrano età alla diagnosi più precoce e maggior aggressività rispetto ai corrispettivi adenomi sporadici (2).
Due consorzi internazionali hanno dimostrato mutazioni inattivanti germinali del gene AIP nel 20% dei casi di FIPA globalmente considerati, ma nei casi di FIPA omogenei GH-secernenti la presenza di mutazioni di AIP supera il 30% (3). Tali mutazioni sono presenti inoltre nel 30-40% dei casi di gigantismo ipofisario (4). Il gene AIP codifica per Aryl hydrocarbon receptor Interacting Protein, proteina dotata di molteplici interazioni, inclusa la mediazione del segnale della somatostatina nelle cellule somatotrope.
Non vi sono linee guida su FIPA/AIP, ma si suggerisce la ricerca di mutazioni germinali di AIP in:
- tutti i casi di FIPA;
- tutti i casi di gigantismo;
- negli adenomi GH-secernenti insorti prima dei 18 anni;
- nei macroadenomi GH/PRL-secernenti insorti prima dei 30 anni, specie se presentano ridotta risposta alla terapia.
In caso di positività per mutazione di AIP, i familiari di primo grado devono essere sottoposti a screening genetico: se positivi per la mutazione, dovranno eseguire periodico follow-up endocrinologico.
X-LAG
X-LAG è l’acronimo di X-linked acrogigantism, sindrome descritta nel 2014 (5). Questi soggetti hanno un quadro clinico ben caratterizzato: precocissimo aumento staturale (fin dai primi mesi/anni di vita), stigmate acromegaliche, voluminosi adenomi GH-secernenti, aspetti istologici caratteristici, resistenza alla terapia. Il quadro è dovuto a duplicazioni/mutazioni attivanti il gene che codifica per GPR101, un recettore orfano accoppiato a una G-protein. Mentre nell’acromegalia dell’adulto non sono state trovate alterazioni di questo gene, queste sono responsabili di circa il 10% dei gigantismi non sindromici. Specie nei maschi le mutazioni insorgono in fase post-zigotica, con mosaicismo tissutale (analogamente alla sindrome di McCune-Albright) e quindi l’analisi del DNA del sangue periferico può risultare negativa (6).
TUMORI IPOFISARI ASSOCIATI AD ALTRE MANIFESTAZIONI CLINICHE
Questo gruppo comprende entità cliniche caratterizzate dalla coesistenza di adenomi ipofisari, altri tumori endocrini e non endocrini e/o manifestazioni non tumorali.
MEN-1
È sicuramente la forma identificata da più tempo e più comune e il suo riconoscimento è rilevante anche per gli aspetti relativi alla mortalità. È dovuta a mutazioni inattivanti il gene onco-soppressore per la menina.
Nel 30-40% dei soggetti affetti sono presenti adenomi ipofisari, che costituiscono la manifestazione d’esordio della malattia nel 17% dei soggetti (in questi casi solo il 27% presenta una concomitante manifestazione di MEN-1, in genere l’iperparatiroidismo). Sono stati riportati tutti i tipi di adenomi ipofisari, anche se vi è una prevalenza dei prolattinomi. Nella MEN-1 esistono anche casi di acromegalia da secrezione ectopica di GHRH da parte di NET.
Gli adenomi ipofisari nella MEN-1 sono stati considerati più aggressivi dei corrispettivi adenomi sporadici (7). Tuttavia, studi recenti hanno evidenziato che, con l’applicazione dello screening suggerito dalle linee guida, gli adenomi ipofisari vengono diagnosticati più precocemente, con un migliore esito clinico (8).
Le linee guida internazionali (9) indicano quali pazienti sottoporre a screening per MEN-1, ma non forniscono alcuna indicazione nel caso di adenomi ipofisari isolati e apparentemente non familiari: in questi pazienti appare comunque opportuno eseguire un controllo del metabolismo calcio-fosforico. Nel caso di paziente con MEN-1 le linee guida suggeriscono sia i tempi che le modalità di follow-up.
MEN-4
Nel ratto era nota una sindrome (MEN X) in cui coesistevano tumori endocrini tipici della MEN-1 e della MEN-2. Il quadro è stato riportato anche in famiglie umane e la sindrome è stata definita MEN-4. Accanto ad adenomi ipofisari, si riscontrano adenomi paratiroidei, carcinoidi, feocromocitomi, tumori testicolari, lipomi e angiomiolipomi renali.
I geni responsabili codificano per proteine regolatorie del ciclo cellulare, inibitori di CDKI, in particolare la CDKN1B – p27 (10). Si tratta di una forma nettamente più rara della MEN-1, da sospettare soprattutto nel paziente con un quadro clinico MEN-like senza mutazioni per MEN-1.
SINDROME 3P
Negli ultimi anni è stata segnalata la concomitanza di adenomi ipofisari (specie GH e PRL-secernenti) e feocromocitomi: il quadro è stato definito sindrome 3P (Pituitary/Pheo/Paraganglioma). I geni coinvolti sono quelli delle sindromi familiari feocromocitoma/paraganglioma, in particolare delle succinico-deidrogenasi. Gli adenomi hanno caratteristiche morfologiche particolari (es. la vacuolizzazione).
La rarità dell’associazione indica che nei feo/paragangliomi non è indicato lo screening per la presenza di adenomi ipofisari, ma in caso di coesistenza delle due forme si deve procedere all’analisi genetica (11).
Complesso di Carney (CNC)
In circa il 75% dei pazienti con CNC sono presenti lievi incrementi di PRL, GH e IGF-I, ma è molto più rara la frequenza di adenomi ipofisari (esclusivamente GH/PRL-secernenti). Questi tumori si caratterizzano per la frequente multi-focalità e la coesistenza di iperplasia (12). La causa nettamente più frequente è una mutazione inattivante del gene codificante una subunità regolatoria della protein-kinasi A (PRKAR1A).
Sindrome di McCune-Albright
La sindrome è dovuta a una mutazione post-zigotica attivante il gene per GNAS.
Nella sua forma classica, accanto alle lesioni ossee e cutanee, sono presenti endocrinopatie iperfunzionanti. Nel caso dell’ipofisi, questa iperfunzione autonoma si manifesta soprattutto come acromegalia, dovuta a una fase iperplastica seguita da sviluppo di adenoma (13).
La gestione di questi pazienti è complessa. La diagnosi iniziale può essere ritardata: l’iniziale aumento della velocità di crescita lineare viene attribuito alla pubertà precoce, le alterazioni morfologiche del cranio alla displasia ossea.
La terapia si presenta complessa per la difficoltà dell’approccio neurochirurgico legato alle anomalie ossee, i rischi potenziali della radioterapia (trasformazione sarcomatosa delle lesioni ossee del cranio), ridotta risposta agli analoghi della somatostatina; il pegvisomant sembra rappresentare una valida alternativa.
DICER 1
Il gene DICER codifica per una endo-ribonucleasi coinvolta nei processi differenziativi degli organi e sue mutazioni determinano tumori disontogenetici nell’infanzia. Sono stati segnalati casi eccezionali di particolari tumori ipofisari nell’infanzia (blastoma) con quadro clinico di Cushing (14).
Bibliografia
- Vasilev V, et al. Familial pituitary tumor syndrome. Endocr Pract 2011, 17 suppl 3: 41-6.
- Daly A, et al. Clinical characterization of familial isolated pituitary adenomas. J Clin Endocrinol Metab 2006, 91: 3316-23.
- Beckers A, et al. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev 2013, 34: 239-77.
- Rostomyan l, et al. Clinical and genetic characterization of pituitary gigantism: an International collaborative study in 208 patients. Endocr Relat Cancer 2015, 22: 745-57.
- Trivellin G, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med 2014, 371: 2363-74.
- Iacovazzo D, et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol Commun 2016, 4: 56.
- Verges B, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 2002, 87: 457-65.
- De Laat JM, et al. Long-term natural course of pituitary tumors in patients with MEN1: results from the Dutch MEN1 study group (DMSG). J Clin Endocrinol Metab 2015, 100: 3288-96.
- Thakker RV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012, 97: 2990-3011.
- Pellegata NS. MENX and MEN4. Clinics (Sao Paulo) 2012, 67 suppl 1: 13-8.
- Denes J, et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab 2015, 100: E531-41.
- Correa R, et al. Carney complex: an update. Eur J Endocrinol 2015, 173: M85-97.
- Salenave S, et al. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab 2014, 99: 1955-69.
- Schultz KAP, et al. DICER1 and associated conditions: identification of at-risk individuals and recommended surveillance strategies. Clin Cancer Res 2018, 24: 2251-61.
Clinica e diagnostica dell'apoplessia ipofisaria
Marco Faustini Fustini
IRCCS Istituto delle Scienze Neurologiche di Bologna (ISNB), Ospedale Bellaria, Bologna
(aggiornato all'11 aprile 2016)
L’apoplessia ipofisaria è un’evenienza rara, ma non eccezionale, con un’incidenza - riportata in casistiche neurochirurgiche - variabile tra lo 0.6 e il 9.1% (1). Colpisce pazienti con tumore ipofisario, solitamente un macroadenoma; casi sporadici sono stati eccezionalmente riportati in pazienti con altre patologie espansive sellari (cisti della tasca di Rathke, ipofisiti).
Nell’80% dei casi costituisce il sintomo d’esordio del tumore ipofisario e avviene in pieno benessere, mentre nel restante 20% compare in pazienti con patologia espansiva della regione sellare già nota (1,2).
I più importanti fattori favorenti la comparsa dell'apoplessia (1-5) sembrano essere l’ipertensione arteriosa e la terapia anti-coagulante. Altri fattori sono gli interventi chirurgici (soprattutto il by-pass aorto-coronarico), le procedure invasive (angiografia, anestesia spinale), il diabete mellito, le coagulopatie, i test di stimolo con ormoni ipotalamici (GnRH, TRH, eccezionalmente CRH) e l'ipoglicemia insulinica, la terapia anti-aggregante piastrinica, la terapia con analoghi del GnRH, i dopamino-agonisti, gli estro-progestinici, il trauma cranico, la gravidanza, la permanenza alle alte quote e il sesso maschile. Il possibile ruolo favorente degli analoghi della somatostatina è tuttora oggetto di dibattito (1,6).
L’apoplessia ipofisaria può presentarsi in tutte le età della vita, ma predilige la V-VI decade.
Nella sua forma “classica” l'apoplessia è un’emergenza medico-chirurgica conseguente a emorragia o infarto dell’ipofisi (1,5). Si caratterizza per la comparsa improvvisa di:
- cefalea intensa, solitamente frontale, retro-orbitale o fronto-temporale, spesso accompagnata da vomito;
- compromissione della funzione visiva (riduzione del visus, deficit campimetrici, oftalmoplegia con ptosi palpebrale);
- alterazione dello stato di coscienza (talora con segni di irritazione meningea e fotofobia in caso di stravaso di sangue nello spazio subaracnoideo).
Raramente può progredire rapidamente verso lo stato di coma, soprattutto quando i fenomeni compressivi coinvolgono strutture neuro-vascolari di primaria importanza, quali il sistema ventricolare, il peduncolo cerebrale, l'arteria cerebrale media. Eccezionalmente è stata descritta l’evoluzione verso l'ictus.
Non mancano, tuttavia, quadri clinici meno imponenti (apoplessia "subacuta"), in cui alcuni segni e sintomi, soprattutto quelli visivi, possono essere completamente assenti o talmente fugaci da essere interpretati come aura nell'ambito di un attacco di emicrania. La sindrome di Sheehan (necrosi ischemica post-partum) manca del caratteristico corteo sintomatologico tipico dell’apoplessia ipofisaria e si manifesta clinicamente con l’ipopituitarismo che ne consegue.
Il quadro clinico neurologico è causato dal rapido aumento della pressione all’interno della sella turcica (7), che a sua volta può ripercuotersi lateralmente sul seno cavernoso, la cui parete mediale sovente è stata in precedenza intaccata dall’invasione del tumore ipofisario; ne può conseguire oftalmoplegia (70%), per sofferenza ischemica acuta del III nervo cranico. L’espansione verso l’alto della massa necrotico-emorragica coinvolge il chiasma ottico nel 75% dei casi, causando riduzione dell’acuità visiva, che può esitare in cecità e deficit campimetrici (soprattutto emianopsia bitemporale).
In circa l'80% dei casi si sviluppa ipopituitarismo che può essere globale o parziale (1,5,7). Diversi meccanismi possono costituirne il fondamento:
- aumento acuto della pressione intra-sellare;
- compressione del peduncolo ipofisario o dei vasi portali;
- vasospasmo;
- estensione del processo necrotico-emorragico alla porzione ancora normale dell’ipofisi.
Le conseguenze cliniche dell’ipopituitarismo possono essere rilevanti, se tale condizione non è riconosciuta precocemente. Ciò che maggiormente condiziona la prognosi è l’iposurrenalismo secondario acuto (che si manifesta con ipoglicemia e iposodiemia), presente nel 70-75% dei casi (1); rappresenta la causa più frequente di mortalità, oggi inferiore al 10%, ma in passato assai elevata (fino al 75%). Causa assai più rara di mortalità, nelle forme particolarmente gravi e drammatiche, è la compressione della massa necrotico-emorragica sul peduncolo cerebrale. Il diabete insipido permanente è un’evenienza sporadica (0-8%) come pure l'ictus per compressione acuta della porzione intra-cavernosa dell’arteria carotide (1).
La diagnosi di apoplessia deve essere presa in considerazione in tutti i pazienti che manifestano cefalea intensa e severa, acuta e/o di recente insorgenza, soprattutto se associata con sintomi e segni neuro-oftalmologici. Questi sintomi associati a un primo riscontro neuroradiologico (TC) di lesione espansiva sellare suggeriscono fortemente la diagnosi di apoplessia ipofisaria, che può richiedere misure terapeutiche urgenti.
La diagnosi differenziale va posta con emorragia subaracnoidea, meningite, ictus, trombosi del seno cavernoso, emicrania, neurite ottica. La risonanza magnetica (RM) è la metodologia radiologica di prima scelta e conferma la diagnosi in oltre il 90% dei casi. Sono stati descritti quadri neuroradiologici diversi, in relazione all’intervallo trascorso dall’esordio, ma generalmente il sospetto dello stravaso emorragico nasce dal caratteristico aspetto iperintenso nelle immagini T1-pesate, nelle sue variabili sfaccettature (1).
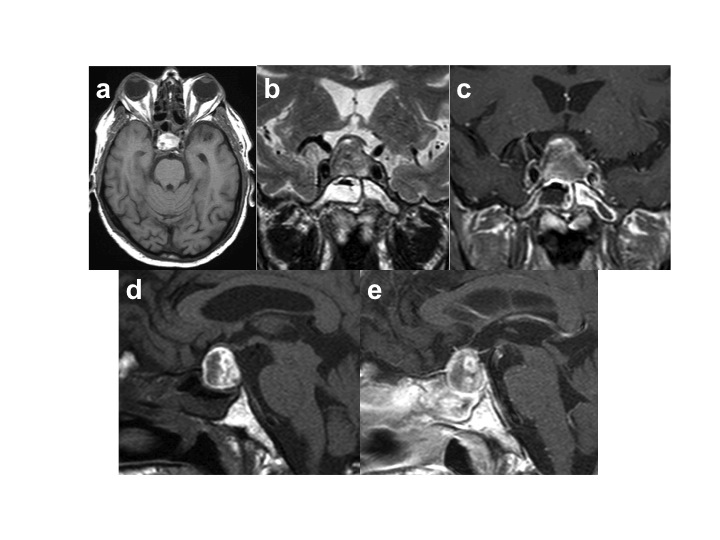
Apoplessia ipofisaria emorragica (RM eseguita a 6 ore dall'esordio clinico). Le immagini in T1 assiale (a) e e sagittale (d) mostrano iperintensità confluente nell'ambito di un'ipofisi ingrandita con moderata presa periferica di mdc. La massa intra e sovrasellare appare ipointensa nella sequenza T2 coronale (b). Il chiasma ottico è stirato (c & e).
Indagini di laboratorio. Ipoglicemia e iponatremia, quali conseguenze dell’iposurrenalismo secondario, sono fortemente suggestive. L’iponatremia isolata è stata talora descritta quale conseguenza di SIADH, che può accompagnare, seppure assai di rado, l’apoplessia ipofisaria (1). I dosaggi ormonali non sempre sono disponibili in tempi rapidi e spesso servono più a confermare il sospetto che a porre la diagnosi. È indispensabile raccogliere un campione di siero sul quale eseguire (anche in un secondo tempo) la determinazione della cortisolemia, prima di iniziare la terapia con corticosteroidi. In realtà, non è stato ancora definito con certezza un cut-off definito e condiviso di cortisolemia per la diagnosi di iposurrenalismo secondario in corso di un evento stressante, quale appunto è l’apoplessia ipofisaria.
L’evoluzione dell’ipopituitarismo dipende dall’estensione del processo necrotico-emorragico: quando coinvolge globalmente la ghiandola, l'ipopituitarismo è irreversibile; negli altri casi può risolversi, in parte o totalmente, a distanza di qualche settimana o mese dall'evento acuto (8). Per contro, negli adenomi ipofisari ipersecernenti l’apoplessia si può accompagnare al miglioramento o alla regressione completa dello stato ipersecretorio (1).
Bibliografia
- Capatina C, Inder W, Karavitaki N, et al. Pituitary tumour apoplexy. Eur J Endocrinol 2015, 172: R179-90.
- Vanderpump, M, Higgens, Wass JAH. UK guidelines for the management of pituitary apoplexy a rare but potentially fatal medical emergency. Emerg Med J 2011, 28: 550-1.
- Semple PL, Jane JA, Laws ER. Clinical relevance of precipitating factors in pituitary apoplexy. Neurosurgery 2007, 61: 956-61.
- Biousse V, Newman NJ, Oyesiku NM. Precipitating factors in pituitary apoplexy. J Neurol Neurosurg Psychiatry 2001, 71: 542-5.
- Rajasekaran S, Vanderpump M, Baldeweg S, et al. UK guidelines for the management of pituitary apoplexy. Clin Endocrinol 2011, 74: 9-20.
- Bakiri F, Herrera J, Riestra M, et al. Pituitary apoplexy after somatostatin analogue administration: coincidental or causative? Clin Endocrinol 2014, 81: 471-3.
- Zayour DH, Selman WR, Arafah BM. Extreme elevation of intrasellar pressure in patients with pituitary tumor apoplexy: relation to pituitary function. J Clin Endocrinol Metab 2004, 89: 5649-54.
- Prescott H, Ellis E, Soule S. Pituitary infarction: a potentially fatal cause of postoperative hyponatraemia and ocular palsy. BMJ 2011, 342: 704-5.
Terapia dell'apoplessia ipofisaria
Marco Faustini Fustini
IRCCS Istituto delle Scienze Neurologiche di Bologna (ISNB), Ospedale Bellaria
(aggiornato all'11 aprile 2016)
La sintomatologia neuro-oftalmologica che caratterizza l’apoplessia ipofisaria può essere di non facile e immediato inquadramento nelle strutture di pronto Soccorso e di Medicina d’Urgenza, che solitamente devono gestire pazienti con altre patologie acute. Allo scopo di ottimizzare il percorso diagnostico-terapeutico del paziente con sospetta apoplessia ipofisaria, la commissione di esperti che ha redatto le linee guida del Regno Unito (1) ha proposto l’accesso pianificato del paziente a un team specializzato multi-disciplinare (neurochirurgo dedicato alla chirurgia ipofisaria, endocrinologo, neuroradiologo, oftalmologo).
In passato l’apoplessia ipofisaria è stata considerata unicamente un’emergenza neurochirurgica. Tuttavia, numerosi dati indicano nella terapia conservativa con glucocorticoidi una possibile alternativa nei casi meno gravi, ossia in assenza di segni neuro-oftalmologici severi e progressivi o in presenza di un quadro clinico di oftalmoplegia isolata.
Inoltre, pur non essendo stati ancora ben delineati criteri certi per definire il deficit neuro-oftalmologico che richiede l’intervento neurochirurgico decompressivo, esiste accordo sulla necessità di istituire prontamente la terapia con corticosteroidi al momento della diagnosi, ancor prima di avere disponibili i risultati degli esami ormonali e indipendentemente dalla scelta di inviare poi il paziente alla chirurgia decompressiva trans-sfenoidale, in quanto l’iposurrenalismo secondario acuto non trattato è la causa più frequente di mortalità e morbilità (1-4).
In assenza di edema cerebrale evidente - condizione in cui si usa preferibilmente il desametazone ev a dosi variabili da 4 a 16 mg/die - il farmaco più impiegato è l’idrocortisone (100 mg in bolo ev seguito da 2-4 mg/h in infusione continua oppure da 50-100 mg ogni 6 ore per via im o ev). Le indicazioni per l’impiego empirico di corticosteroidi comprendono:
- instabilità emodinamica
- alterazione - anche iniziale e lieve - dello stato di coscienza
- riduzione dell’acuità visiva
- presenza di deficit campimetrici.
Nei pazienti con apoplessia ipofisaria subacuta, la cortisolemia inferiore a 550 nmol/L (19.9 µg/dL) potrebbe costituire di per sé un’indicazione al trattamento con corticosteroidi. Tuttavia, sul livello soglia sotto il quale iniziare il trattamento empirico con corticosteroidi non esiste un larghissimo consenso e alcuni autori consigliano di ridurlo almeno a 500 nmol/L (18.1 µg/dL).
L’ipopituitarismo compare in percentuale variabile tra il 70 e il 100% (1,2,5), ma può essere transitorio (15-60%). Pertanto, è consigliabile rivalutare il paziente almeno a distanza di 4-8 settimane dall’evento acuto (1). In realtà, alcuni studi indicano che la possibilità di recupero può richiedere tempi maggiori. Esistono dati discordanti sul fatto che la decompressione precoce mediante chirurgia transfenoidale possa migliorare il recupero della funzione ipofisaria (2,5,6).
I pazienti senza segni neuro-oftalmologici o con segni neuro-oftalmologici lievi e stabili possono essere trattati in maniera conservativa, mantenendo un monitoraggio stretto a causa dell’evoluzione, talvolta imprevedibile, dell’apoplessia ipofisaria. Nel caso in cui i segni neuro-oftalmologici non migliorino entro una settimana o peggiorino, deve essere presa in considerazione la decompressione chirurgica per via transfenoidale.
D’altra parte, non esiste un livello di evidenza sufficiente per raccomandare il trattamento conservativo in pazienti con deficit dell’acuità visiva o deficit campimetrici, seppure alcuni esperti suggeriscano, con cautela, il trattamento conservativo anche in questi casi, purchè il monitoraggio dei parametri neuro-oftalmologici sia giornaliero e il paziente sia affidato a un team multi-disciplinare di esperti, con la possibilità di eseguire rapidamente la decompressione chirurgica, qualora questa si rendesse necessaria.
Nella gestione clinica del paziente con apoplessia ipofisaria, l’area di maggiore incertezza riguarda proprio l’indicazione e la scelta della tempistica dell’eventuale terapia chirurgica decompressiva. Infatti, se da una parte studi retrospettivi indicano che segni neuro-oftalmologici lievi tendono a migliorare spontaneamente in molti pazienti trattati con terapia conservativa, dall’altra esistono evidenze a favore di un significativo miglioramento dell’acuità visiva e della campimetria ottica quando la decompressione chirurgica è eseguita nelle prime otto ore dall’esordio. Occorre prendere atto, comunque, che non esistono studi clinici controllati di buona qualità che confrontino l’approccio chirurgico con quello conservativo e che gli studi finora condotti sono tutti gravati da un evidente bias di selezione, poiché i pazienti più gravi sono quelli che più facilmente sono avviati alla decompressione chirurgica (2). Preso atto di questa limitazione, la decompressione chirurgica per via transfenoidale è l’opzione terapeutica da preferire nel paziente che, dopo la stabilizzazione dal punto di vista emodinamico con corticosteroidi, presenti segni neuro-oftalmologici significativi o un ridotto stato di coscienza (1-7). Secondo queste indicazioni, devono essere avviati alla terapia chirurgica decompressiva i pazienti:
- con segni neuro-oftalmologici gravi (acuità visiva severamente ridotta, deficit del campo visivo severi e persistenti o in peggioramento);
- con deterioramento del livello di coscienza;
- che, in assenza di riduzione severa dell’acuità visiva o del campo visivo, presentino la persistenza o il peggioramento dell’oftalmoplegia per interessamento dei nervi dell’oculomozione contenuti e compressi nel seno cavernoso (III, IV o VI nervo cranico) dopo 4-7 giorni dall’inizio della terapia conservativa;
- che, in corso di terapia conservativa, presentino la comparsa o il peggioramento del quadro neuro-oftalmologico o un deterioramento dello stato di coscienza.
La decompressione chirurgica deve essere preferibilmente eseguita da un’equipe neuro-chirurgica esperta, possibilmente entro una settimana dall’esordio.
In conclusione, un consiglio pratico condivisibile è quello di valutare inizialmente ogni ora (poi, allorchè le condizioni cliniche siano divenute stabili o in miglioramento, ogni 4-6 ore) il paziente acuto sintomatico, avviandolo senza indugi alla decompressione chirurgica in caso di deterioramento delle condizioni neurologiche e/o neuro-oftalmologiche, tenendo ben presente che occorre un contatto diretto e costante con l’equipe chirurgica del team multi-disciplinare che affronta l’emergenza.
Bibliografia
- Rajasekaran S, Vanderpump M, Baldeweg S, et al. UK guidelines for the management of pituitary apoplexy. Clin Endocrinol 2011, 74: 9-20.
- Capatina C, Inder W, Karavitaki N, et al. Pituitary tumour apoplexy. Eur J Endocrinol 2015, 172: R179-90.
- Bi WL, Dunn IF, Laws ER Jr. Pituitary apoplexy. Endocrine 2015, 48: 69-75.
- Briet C, Salenave S, Chanson P. Pituitary apoplexy. Endocrinol Metab Clin North Am 2015, 44: 199-209.
- Moller-Goede DL, Brandle M, Landau K, et al. Pituitary apoplexy: re-evaluation of risk factors for bleeding into pituitary adenomas and impact on outcome. Eur J Endocrinol 2011, 164: 37-43.
- Arafah BM, Harrington JF, Madhoun ZT, et al. Improvement of pituitary function after surgical decompression for pituitary tumor apoplexy. J Clin Endocrinol Metab 1990, 71: 323-8.
- Singh TD, Valizadeh N, Meyer FB, et al. Management and outcomes of pituitary apoplexy. J Neurosurg 2015, 122: 1450-7.
Adenomi ipofisari aggressivi
Roberto Attanasio
Istituto Galeazzi IRCCS, Milano
(aggiornato al 14 marzo 2020)
Definizioni
Occorre fare chiarezza su termini spesso usati inappropriatamente come equivalenti o sinonimi.
Il concetto di resistenza è tipicamente clinico: per esempio un PRLoma è definito resistente al trattamento se non si ottiene normalizzazione dei livelli di PRL e riduzione del volume tumorale con un’appropriata titolazione della terapia con cabergolina.
L’invasività viene definita in base a dati radiologici, chirurgici e istopatologici (1):
- infiltrazione attiva delle strutture perisellari: seno cavernoso e sfenoidale, osso e meno comunemente vasi sanguigni e guaine nervose (in base ai rilievi neuroradiologici o istopatologici);
- invasione di seno cavernoso e/o sfenoidale (25–55% dei tumori), mentre l’estensione sovrasellare non costituisce un criterio di invasività;
- nella classificazione di Hardy (2), gli adenomi invasivi sono quelli di grado III (erosione ossea focale) e IV (erosione ossea della base cranica e delle strutture parasellari);
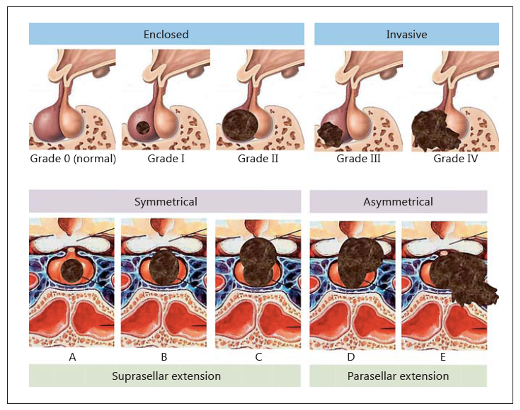
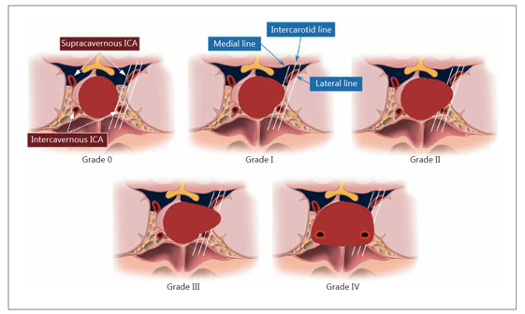
Classificazione di Hardy
- nella classificazione di Knosp (3), il grado 3 e 4 definiscono la vera invasione intra-cavernosa del tumore.
Classificazione di Knosp
Un macroadenoma può essere invasivo ma solitamente l’aspetto patologico è tipico (vedi oltre) e l’andamento clinico benigno senza aggressività clinica.
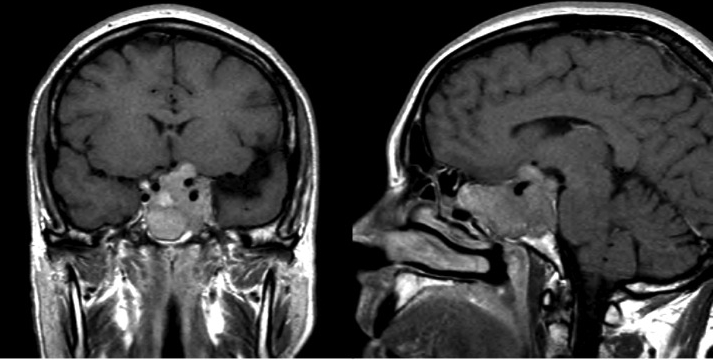
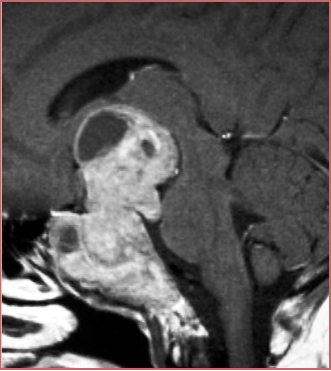
Casi di aggressività neuroradiologica: in alto ampia invasione del seno cavernoso e del clivus, in basso invasione del seno sfenoidale (grazie a Marco Losa)
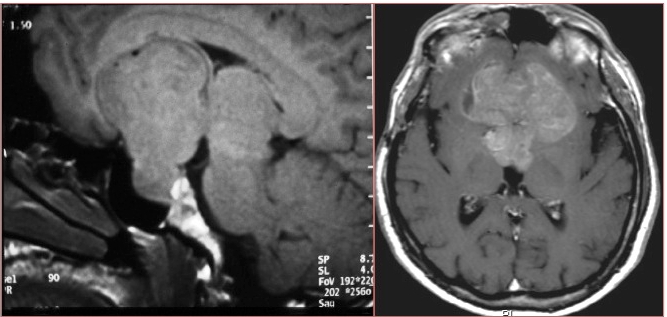
Casi di aggressività neurochirurgica: ampia estensione extra-sellare di forma irregolare ed eccentrica (grazie a Marco Losa)
L’aggressività, che riguarda un piccolo sottogruppo di tumori ipofisari, ha una definizione più sfuggente. Un tumore viene definito aggressivo sulla base di un tasso di crescita insolitamente rapido o plurime recidive con crescita tumorale rilevante nonostante un ottimale trattamento standard che include neurochirurgia, radioterapia e farmaci. In generale i tumori aggressivi sono macroadenomi o adenomi giganti, invasivi (ed è intuitivo che l’invasione del clivus rappresenta un dato più caratteristico e preoccupante di quella del seno cavernoso). Esistono comunque eccezioni, come i microadenomi ACTH-secernenti aggressivi o altri tumori localizzati, caratterizzati da alto tasso di recidiva o mancata risposta alle terapie.
Recidiva non è sinonimo di aggressività: un conto è una recidiva dopo 10 anni dal primo intervento (che può essere considerato un comportamento benigno) e un altro dopo 6 mesi.
Secondo la classificazione WHO 2004 (ora superata), i tumori ipofisari venivano classificati in 3 categorie: adenomi tipici e atipici e carcinomi. Il termine adenoma si applica a neoplasie epiteliali benigne con struttura simil-ghiandolare. Vengono considerate benigne perché gli aspetti macro e microscopici suggeriscono una crescita localizzata passibile di resezione chirurgica radicale. Il carcinoma è definito solo dalla presenza di metastasi a distanza, mentre il termine di adenoma atipico veniva attribuito a un adenoma con alcune caratteristiche microscopiche peculiari: un indice di proliferazione (valutato con Ki-67) > 3%, estesa immunoreattività per p53 e alto indice mitotico.
Quindi i termini:
- tipico o atipico si riferiscono solo ad aspetti istopatologici;
- invasivo o non invasivo ad aspetti radiologici, chirurgici o morfologici di invasione;
- aggressivo o non aggressivo al comportamento clinico. Il cosiddetto adenoma aggressivo sarebbe quindi una lesione benigna con potenzialità maligna (4).
Ma come spesso accade, i dati del mondo reale hanno la perniciosa abitudine di non adattarsi alle classificazioni teoriche. La classificazione tipico/atipico spesso non correlava con l’aspetto radiologico o il comportamento clinico: adenomi tipici possono essere invasivi con comportamento aggressivo e adenomi atipici possono essere non invasivi e non aggressivi. Questo ha portato a cambiare la classificazione e quella adottata dal WHO dal 2017 (5) usa criteri differenti. Le forme considerate clinicamente aggressive secondo questa classificazione sarebbero quindi:
- tumori somatotropi sparsamente granulati;
- tumori lattotropi densamente granulati;
- tumori mammo-somatotropi;
- tumori a cellule staminali acidofile;
- vari tipi di tumori silenti (tireotropi, corticotropi sparsamente granulati, adenomi a cellule di Crooke cell, adenomi silenti sottotipo 3, adenomi null cell).
È stato infine proposto che i tumori ipofisari aggressivi e i carcinomi sono clinicamente o istologicamente simili, tanto da suggerire per gli adenomi aggressivi il termine di tumori con potenziale maligno senza metastasi, e di rimpiazzare il termine adenomi con quello di tumori ipofisari neuroendocrini (PitNET) (6,7).
La raccomandazione della LG europea (8) è quindi di prendere in considerazione la diagnosi di tumore ipofisario aggressivo nei pazienti con tumore radiologicamente invasivo e con velocità di crescita rapida, oppure crescita clinicamente rilevante nonostante l’utilizzo di terapie standard ottimali (NCH, RT e farmaci).
Epidemiologia
È descritta invasione locale nel 25-55% degli adenomi sottoposti a NCH (1,9), che sembra correlata alle dimensioni: avviene nell’80% degli adenomi giganti (> 4 cm), nel 22% dei macroadenomi e nel 2% dei microadenomi (10).
I carcinomi sono molto rari, pari allo 0.1-0.2% di tutti i tumori ipofisari (11).
Vista la definizione elusiva, la prevalenza degli adenomi aggressivi non è nota.
Quando sospettare
Si può distinguere un tumore aggressivo fin dall’inizio? L’identificazione precoce di un tumore aggresssivo è una sfida di grande importanza clinica, perchè si tratta di patologia associata ad aumentata morbilità e mortalità (anche in assenza di metastasi). Il decorso aggressivo può essere teoricamente suggerito da aspetti clinici, morfologici o molecolari.
Molti tumori ipofisari che alla fine si dimostreranno carcinomi mostrano precocemente un comportamento aggressivo, non rispondono alle terapie standard fin dall’inizio o recidivano precocemente dopo resezione chirurgica (11,12). Nella maggior parte dei casi tali tumori non sono inizialmente distinguibili da quelli con un decorso più “classico”, con segni di compressione sui tessuti circostanti e/o di ipersecrezione ormonale. Ma la presentazione clinica più comune è quella di recidiva precoce dopo il primo intervento neurochirurgico con rapida crescita locale ed estensione tumorale (13).
Alcuni aspetti clinici possono aiutare nell’identificare precocemente un tumore ipofisario che si dimostrerà poi aggressivo (14):
- la resistenza al trattamento nei prolattinomi, con necessità di aumentare progressivamente le dosi di dopaminergico in un paziente con buona compliance;
- la resistenza secondaria al trattamento in un paziente che era stato inizialmente responsivo per periodi prolungati;
- la modifica dell’aspetto secretorio, come un ACTHoma silente che si trasforma in un Cushing florido.
Non ci sono aspetti neuroradiologici in grado di predire con attendibilità un comportamento aggressivo, ma la combinazione di invasione (con erosione del pavimento sellare o del clivus) con la presenza di marcatori di proliferazione può rappresentare un campanello d’allarme.
Aspetti istopatologici e molecolari
Gli adenomi a cellule di Crooke (in cui c’è una diffusa deposizione intra-cellulare di cheratina) e gli ACTHomi silenti hanno un andamento più aggressivo della maggior parte degli ACTHomi funzionanti (13).
Una conta mitotica > 2 per 10 campi ad alto ingrandimento (HPF) è associata ad aumentato rischio di recidiva e si trova più facilmente nei carcinomi che negli adenomi.
A livello molecolare i tumori ipofisari accumulano con il tempo anomalie nelle vie molecolari, che contribuiscono alla progressione da adenoma “benigno” a tumore ipofisario aggressivo recidivante, fino a carcinoma in casi eccezionalmente rari. I dettagli sono poco noti e nessun singolo marcatore si è dimostrato attendibile per predire questa progressione.
Nei tumori ipofisari sono stati studiati diversi marcatori biologici (13,15-20):
- alterazioni cromosomiche: instabilità genomica di 11q in associazione a perdita di 11p;
- microRNA;
- marcatori di proliferazione: Ki-67, PTTG;
- oncogeni: RAS, TP53;
- oncosoppressori: p53;
- fattori di crescita e loro recettori: EGF, VEGF, FGR-4-R;
- fattori correlati all’angio-genesi: densità micro-vascolare;
- fattori di adesione cellulare: metallo-proteinasi di matrice.
Anche se la classificazione WHO del 2017 (5) ha abbandonato il termine di adenoma atipico, un Ki-67 > 3% rimane un marcatore prognostico utile per identificare precocemente quegli adenomi con maggior potenzialità di evoluzione a comportamento aggressivo (13,15). Un valore di Ki-67 ≥ 10% è stato proposto come segno di malignità.
L’oncosoppressore p53 è un fattore di trascrizione con aumentata espressione nucleare nei tumori ipofisari aggressivi e nei carcinomi metastatici. Però, non è stato ancora validato un metodo attendibile di quantificazione, cosa che ha portato a rimuovere la sua valutazione nella classificazione WHO del 2017 (5).
Anche se i marcatori molecolari attualmente a disposizione sono scarsamente predittivi di evoluzione a comportamento aggressivo, i tumori con un profilo molecolare caratteristico di aggressività possono essere seguiti più strettamente e tale profilo può influenzare le scelte decisionali, per esempio sull’uso di una radioterapia adiuvante.
Terapia
Il trattamento degli adenomi aggressivi rappresenta una sfida per le dimensioni, l’invasivià, la rapida crescita e l’alta incidenza di recidive. Il problema iniziale è capire il momento in cui il trattamento deve diventare aggressivo con un approccio multimodale.
Se esiste una terapia farmacologica, è opportuno impiegarla per prima alle massime dosi (14):
- PRLomi: cabergolina
- GHomi: SSA/pegvisomant
- ACTHomi: pasireotide
- TSHomi: SSA.
Il ruolo della terapia farmacologica è duplice: primo, controllare l’ipersecrezione ormonale dove questa esista; secondo, tenere sotto controllo la crescita tumorale e prevenire le recidive. Nella pratica questi due aspetti si sovrappongono, dato che i farmaci più impiegati, dopaminergici e SSA, hanno sia proprietà anti-secretorie che anti-tumorali. Però, in questi tumori aggressivi le terapie farmacologiche sono inefficaci parzialmente o del tutto e la tappa successiva è tentare qualche forma di debulking neurochirurgico. Una terapia multimodale con NCH, RT e chemioterapia, anche se è non in grado nella maggior parte dei casi di ottenere la remissione totale o parziale, riuscirà almeno a mantenere il controllo della crescita/ricrescita tumorale locale.
L’approccio neurochirurgico alle lesioni aggressive offre parecchi vantaggi:
- debulking con alleviazione dell’effetto massa sulle strutture circostanti;
- creazione di uno spazio di sicurezza fra la lesione residua e le strutture circostanti, che rende più sicuro il successivo approccio radiochirurgico;
- possibile miglioramento della sensibilità alla terapia farmacologica;
- fornire materiale per gli studi patologici e molecolari (dove disponibili).
Dato che anche la NCH difficilmente porterà alla guarigione nei casi più aggressivi, l’obiettivo è quello di ottenere la massima riduzione della massa tumorale, con decompressione delle vie ottiche e riduzione del tessuto iperfunzionante, mantenendo l’equilibrio fra l’esigenza di rimuovere il più possibile il tessuto tumorale e quella di non fare danni (10).
Per definizione, i tumori aggressivi invadono e infiltrano le strutture circostanti, rendendo difficile l’asportazione radicale, particolarmente nel caso di invasione del seno cavernoso, sfenoidale e dell’osso e nel caso di re-interventi. L’approccio NCH endoscopico può dare vantaggi significativi (13) nei confronti dell’invasione para-sellare. Nel caso di estesa estensione sovra-sellare, può essere talvolta utile l’approccio trans-cranico. Interventi successivi possono portare a migliore rimozione del tessuto tumorale residuo, che può consentire una migliore efficacia delle terapie complementari farmacologiche e radianti. Molto più che nella “semplice” neurochirurgia ipofisaria, è fondamentale in questi casi l’esperienza del neurochirurgo, per decidere se valga la pena operare ancora e per ottenere i migliori risultati.
Nel caso di persistenza di crescita tumorale o di rapida ricrescita post-NCH nonostante la terapia farmacologica, se è visibile un “bersaglio”, di solito la tappa successiva è utilizzare qualche forma di radiazione (13). La radiochirurgia stereotassica con applicazione di un’alta dose di radiazioni, di solito in un’unica seduta, ha buona efficacia. In alternativa, se il residuo tumorale è troppo voluminoso o troppo vicino a strutture radio-sensibili (soprattutto le vie ottiche), il trattamento radiante può essere somministrato in piccole dosi frazionate nel corso di 4-6 settimane.
La temozolomide (TMZ) è un chemioterapico alchilante, cioè induce un danno del DNA attaccando un gruppo metilico a una guanina (14,21,22). La guanina metilata viene riconosciuta da un sistema di riparazione intra-cellulare del DNA, che porta all’attivazione di una cascata apoptotica e alla morte cellulare. Alcune caratteristiche della molecola la rendono particolarmente attraente in questo ambito: la possibilità di somministrazione orale, l’attivazione spontanea del precursore in forma attiva indipendente dall’ingestione di cibo, la capacità di attraversare la barriera emato-encefalica, la non specificità per la fase del ciclo cellulare, che la rende adatta ad agire anche nei tumori a lenta crescita.
La metilguanina-metiltransferasi (MGMT) è un enzima che rimuove la base metilata, restaurando la guanina nativa e quindi contrastando l’effetto di TMZ e salvando la cellula dall’apoptosi. L’espressione di MGMT nei tumori ipofisari può essere predittiva della risposta al trattamento con TMZ: alti livelli di MGMT si associano a resistenza a TMZ. Però questa osservazione non è stata del tutto confermata e possono essere implicati anche altri meccanismi, per cui la valutazione di MGMT non costituisce un pre-requisito per candidare un paziente a questa terapia.
La dose standard da impiegare in questo ambito è di 150-200 mg/m2/die per 5 giorni ogni 28 giorni, ma sono stati impiegati anche altri protocolli (150 mg/m2 nei giorni 1–7 e 14–21 di un ciclo di 28 giorni, oppure un protocollo metronomico con una somministrazione quotidiana continuativa di 50–75 mg/m2).
Le alterazioni morfologiche indotte da TMZ comprendono emorragia, necrosi, fibrosi focale, infiltrazione infiammatoria, riduzione delle mitosi e degli indici di proliferazione (21). Nei pazienti responsivi, entro poche settimane si osserva una rapida riduzione del volume tumorale (e dell’ipersecrezione se presente). La mancata risposta dopo 3 cicli di trattamento è quindi considerata predittiva di resistenza, mentre la risposta iniziale non è sempre associata a risposta positiva a lungo termine.
I dati combinati di diverse coorti di piccole dimensioni (12) hanno evidenziato una risposta nel 55% dei casi di carcinoma e nel 41% dei casi di adenoma aggressivo, ma le percentuali salgono al 70% in entrambi i gruppi se si includono anche i casi con stabilizzazione.
La survey dell’ESE (23) ha riportato i dati di 166 pazienti (40 con malattia metastatica) da 23 Paesi: dopo TMZ di prima linea è stata riportata risposta biochimica completa nel 19% dei casi, parziale nel 34%, stabilizzazione nel 27% e progressione nel 21%. Era più probabile una risposta positiva nei tumori funzionanti rispetto ai non funzionanti (45% vs 17%), independentemente dallo stato di MGMT.
Il trattamento concomitante con RT migliorava la risposta: la risposta era completa o parziale nel 71% vs il 34% dei trattati con sola TMZ (23).
Lo studio francese (24) è stato il primo a dimostrare un aumento della sopravvivenza nei responsivi a TMZ: mediana di 44 mesi vs 16 mesi nei non responsivi.
Un secondo ciclo di trattamento sembra meno efficace.
Data la mancanza di marcatori che possano indicare in anticipo quali tumori si comporteranno in modo aggressivo, è difficile decidere quando iniziare il trattamento con TMZ. Bisogna tenere in considerazione diversi fattori con un approccio inter-disciplinare: età, pregressi trattamenti, invasione, marcatori di proliferazione, sottotipo istologico. È necessario un bilancio fra i benefici (e rischi) di iniziare la TMZ e i rischi di reintervento e re-irradiazione. Attualmente TMZ viene ancora impiegato come “ultima spiaggia” in (4,7,25):
- carcinomi ipofisari;
- PRLomi aggressivi resistenti a dopaminergici, con dimostrata crescita dopo NCH e RT;
- ACTHomi aggressivi, soprattutto a cellule di Crooke e tipo Nelson, non controllati dopo NCH e RT (eventualmente in combinazione con pasireotide);
- GHomi aggressivi, non controllati da altre terapie (eventualmente in combinazione con cabergolina, SSA e pegvisomant);
- adenomi recidivanti con pregressi interventi multipli inefficaci per consistenza fibrosa, dove TMZ potrebbe ottenere una diminuzione di consistenza della lesione, rendendola più facilmente aggredibile da un ulteriore intervento.
Le LG europee (8) raccomandano:
- la valutazione della risposta dopo 3 cicli: in caso di progressione sospendere; in caso di risposta proseguire per almeno 6 mesi in totale;
- il monitoraggio degli effetti avversi (ematologici ed epatici in particolare);
- nel caso non sia ancora stata somministrata la massima dose di terapia radiante, la combinazione secondo il protocollo di Stupp: 75 mg/m2/die di TMZ per 6 settimane durante RT, seguito da 6–12 mesi di dose standard.
Esiste un razionale per la terapia radio-recettoriale: la somministrazione di una dose radiante tossica a cellule neoplastiche che esprimono SSTR (identificati con Octreoscan o 68Ga-DOTATATE PET/CT) con SSA radiomarcati con 90Y o 177Lu (26). I risultati preliminari non sono stati incoraggianti.
Riguardo alla target therapy, sono stati riportati risultati negativi con everolimus e altre molecole (lapatanib, sunitinib, erlotinib). C’è un singolo case report positivo con bevacizumab (27).
Con la chemioterapia classica sono stati riportati risultati in casi aneddotici con diversi agenti: CCNU/5FU, doxorubicina e CCNU, methotrexate e 5-FU; CCNU, procarbazina ed etoposide; cisplatin (carboplatin)–etoposide; ciclofosfamide, adriamicina e 5-FU; cisplatino, procarbazina, CCNU e vincristina.
L’impianto intra-tumorale di carmustina ha mostrato stabilizzazione o risposta positiva nella maggior parte dei 9 casi riportati (28).
È assolutamente indispensabile che questi pazienti complessi vengano indirizzati a centri con esperienza specifica nel trattamento multi-discipinare di casi difficili.
Bibliografia
- Di Ieva A, et al. Aggressive pituitary adenomas — diagnosis and emerging treatments. Nature Rev 2014, 10: 423-35.
- Hardy J, Vezina JL. Transsphenoidal neurosurgery of intracranial neoplasm. Adv Neurol 1976, 15: 261–73.
- Knosp E, et al. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery 1993, 33: 610-7.
- Syro LV, et al. Treatment of pituitary tumors with temozolomide: an update. Endocr Relat Cancer 2018, 25: T159-69.
- Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol 2017, 134: 521–35.
- Asa S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Related Cancer 2017, 24: C5-8.
- Trouillas J, et al. Aggressive pituitary tumours and carcinomas: two sides of the same coin? Eur J Endocrinol 2018, 178: C7-9.
- Raverot G, et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol 2018, 178: G1-24.
- Raverot G, et al. Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: a French multicenter experience. J Clin Endocrinol Metab 2010, 95: 4592-9.
- Buchfelder M. Management of aggressive pituitary adenomas: current treatment strategies. Pituitary 2009, 12: 256-60.
- Heaney A. Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab 2011, 96: 3649-60.
- Chatzellis E, et al. Aggressive pituitary tumors. Neuroendocrinology 2015, 101: 87-104.
- Heaney A. Management of aggressive pituitary adenomas and pituitary carcinomas. J Neuroncol 2014, 117: 459-68.
- Bengtson D, et al. Long-term outcome and MGMT as a predictive marker in 24 patients with atypical pituitary adenomas and pituitary carcinomas given treatment with temozolomide. J Clin Endocrinol Metab 2015, 100: 1689-98.
- Lenders N, McCormack A. Malignant transformation in non-functioning pituitary adenomas (pituitary carcinoma). Pituitary 2018, 21: 217-29.
- Righi A, et al. Galectin-3 expression in pituitary adenomas as a marker of aggressive behavior. Hum Pathol 2013, 44: 2400-9.
- Mete O, et al. Biomarkers of aggressive pituitary adenomas. J Mol Endo 2012, 49: R69-78.
- Coli A, et al. Minichromosome maintenance protein 7 as prognostic marker of tumor aggressiveness in pituitary adenoma patients. Eur J Endocrinol 2016, 174: 307-14.
- Chen C, et al. Treatment of aggressive prolactinoma with temozolomide. A case report and review of literature up to date. Medicine 2017, 96: e8733.
- Rotondo F, et al. The role of molecular pathology in the classification and clinicopathologic evaluation of pituitary neoplasms. Diagnost Histopathol 2018, 24: P95-103.
- Raverot G, et al. Pituitary carcinomas and aggressive pituitary tumours: merits and pitfalls of temozolomide treatment. Clin Endocrinol 2012, 76: 769-75.
- Hirohata T, et al. DNA mismatch repair protein (MSH6) correlated with the responses of atypical pituitary adenomas and pituitary carcinomas to temozolomide: the national cooperative study by the Japan Society for Hypothalamic and Pituitary Tumors. J Clin Endocrinol Metab 2013, 98: 1130-6.
- McCormack A, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol 2018, 178: 265-76.
- Lasolle H, et al. Temozolomide treatment can improve overall survival in aggressive pituitary tumors and pituitary carcinomas. Eur J Endocrinol 2017, 176: 769-87.
- Halevy C, Whitelaw BC. How effective is temozolomide for treating pituitary tumours and when should it be used? Pituitary 2017, 20: 261-6.
- McLean J, et al. Peptide receptor radionuclide therapy for aggressive atypical pituitary adenoma/carcinoma: variable clinical response in preliminary evaluation. Pituitary 2014, 17: 530-8.
- Ortiz LD, et al. Anti-VEGF therapy in pituitary carcinoma. Pituitary 2012, 15: 445-9.
- Laws E, et al. Gliadel for pituitary adenomas and craniopharyngiomas. Neurosurgery 2003, 53: 2555-69.
Carcinomi ipofisari
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
I carcinomi ipofisari sono una rarità (< 0.5% degli adenomi): per la diagnosi di carcinoma è necessaria la presenza di metastasi (1). L’attuale definizione richiede il contemporaneo assolvimento dei seguenti requisiti (2):
- diagnosi istologica di tumore ipofisario primitivo;
- presenza di metastasi senza alcuna continuità con il tumore ipofisario primitivo;
- stretta corrispondenza o almeno somiglianza degli aspetti istocitologici e dei marcatori biomolecolari fra il tumore ipofisario primitivo e le metastasi;
- esclusione di qualsiasi possibile tumore primitivo alternativo.
Tuttavia la definizione attuale è ben lungi dall’essere soddisfacente dal punto di vista logico e clinico perchè da un lato non si può impiegare il termine carcinoma in tumori molto aggresssivi localmente che richiedono molteplici interventi e terapie adiuvanti (radioterapia) per la mancanza di metastasi; dall’altra il termine carcinoma viene applicato a casi in cui si trovano metastasi ma che sono assolutamente indistinguibili da “normali” adenomi per proliferazione, mitosi, pleiomorfismo nucleare, necrosi, emorragie (3).
La grande maggioranza dei carcinomi ipofisari sono secernenti (soprattutto ACTH e PRL, ma sono state descritte tutte le forme) con progressiva perdita di ipersecrezione parallela alla sdifferenziazione.
Il periodo di latenza fra la diagnosi iniziale di adenoma e la comparsa di metastasi è variabile (in media 7 anni) (1) dopodichè la sopravvivenza media è < 4 anni.
Elementi che possono indurre il sospetto di trasformazione maligna (soprattutto in un PRLoma) sono la perdita secondaria di sensibilità alla terapia dopaminergica, per esempio in un paziente che nonostante dosi sempre maggiori di farmaco vede salire i livelli di PRL, oppure la crescita volumetrica del tumore prima ben controllato dalla terapia (4).
Le metastasi possono essere cerebrali e spinali (40% dei casi, per invasione dello spazio subaracnoideo, figura), e/o sistemiche (47% per disseminazione linfatica o ematica, soprattutto a fegato e osso).
RM con immagini coronali in T1 con mdc. L'ipofisi appare di dimensioni asimmetricamente aumentate, con impegno della cisterna chiasmatica, prevalente a destra, e secondaria deviazione sin del peduncolo. L'impregnazione contrastografica, disomogenea a livello ipofisario per la presenza di componenti cistico-necrotiche, evidenzia un'estensione della lesione alla porzione superiore del seno cavernoso destro, inglobante il sifone carotideo. Ulteriori potenziamenti contrastografici patologici sono presenti in sede sia intra-parenchimale in regione fronto-basale e fronto-temporale destra, che extra-parenchimale in corrispondenza del tratto ottico destro, di significato ripetitivo.
Dal punto di vista terapeutico, bisogna essere aggressivi, con interventi neurochirurgici mirati al debulking del tumore primitivo e delle metastasi, quando aggredibili, e radioterapia. Per quanto riguarda i farmaci, oltre alle dosi massimali degli anti-secretori (dopaminergici, analoghi della somatostatina e pegvisomant, da soli o in combinazione, a seconda dell'istotipo di partenza), recentemente è stata impiegata la temozolomide, in alcuni casi con marcato vantaggio, mentre la chemioterapia ha avuto scarso successo (4).
Bibliografia
- Kaltsas GA, Nomikos P, Kontogeorgos G, et al. Diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab 2005, 90: 3089–99.
- Lopes MBS. The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathol 2017, 134: 521–35.
- Kovacs K, Horvath E, Vidal S. Classification of pituitary adenomas. J Neurooncol 2001, 54: 121–7.
- Heaney AP. Pituitary carcinoma: difficult diagnosis and treatment. J Clin Endocrinol Metab 2011, 96: 3649–60.
Scheda temozolomide
Marco Losa
Reparto di Neurochirurgia, Istituto Scientifico San Raffaele, Università Vita-Salute, Milano
Meccanismo d’azione
La Temozolomide è un agente alchilante di seconda generazione che viene assunto per via orale. La Temozolomide è un pre-farmaco che spontaneamente va incontro ad una trasformazione chimica nel metabolita attivo MTIC. Il meccanismo d'azione citotossico avviene attraverso la metilazione del DNA, con accumulo di sostanze alchilate, per cui la cellula va incontro ad apoptosi. L'enzima O6-metilguanina-DNA metiltransferasi (MGMT), che è implicato nei meccanismi di riparazione del DNA, rimuove i prodotti alchilati indotti dalla Temozolamide e controbilancia il suo effetto antineoplastico. La Temozolomide penetra la barriera emato-encefalica con buona efficacia (1).
Preparazioni, via di somministrazione, posologia
La Temozolomide è commercialmente disponibile sotto forma di capsule per uso orale. Sono disponibili compresse contenenti 5, 20, 100, 140, 180 e 250 mg (Temodal, Temomedac, Temozolomide Accord, Temozolomide Hexal, Temozolomide Sandoz, Temozolomide Sun, Temozolomide TEVA). Il farmaco deve essere somministrato intero a digiuno con un bicchier d'acqua.
La Temozolomide viene normalmente somministrata fino ad un massimo di 12 cicli in monoterapia. La dose del ciclo 1 è di 150 mg/m² una volta al giorno per 5 giorni consecutivi, seguito da 23 giorni senza trattamento. All’inizio del ciclo 2, il dosaggio viene aumentato a 200 mg/m² se non è presente tossicità ematologica. Nei pazienti con grave disfunzione epatica o renale non è richiesta una riduzione della dose, tuttavia in questi casi il farmaco deve essere somministrato con cautela.
Indicazioni
La temozolomide è stata utilizzata con successo nel trattamento dei gliomi ad alto grado, delle metastasi cerebrali dei melanomi (2, 3). Inoltre la Temozolomide ha dimostrato attività antineoplastica anche nei tumori neuroendocrini maligni (4).
La pubblicazione nel 2006 di due case report dimostranti l'efficacia della Temozolomide nella terapia di pazienti con carcinoma ipofisario (5, 6) ha aperto la porta all'utilizzo di questo farmaco sia in altri pazienti con carcinoma ipofisario che in pazienti con adenoma ipofisario aggressivo e resistente alle terapie standard. Da allora sono stati pubblicati vari case report e anche mini-serie di pazienti trattati con Temozolomide (7-10). Il dato consistente che si può trarre da queste pubblicazioni è l'efficacia nel 30-50% dei casi trattati, mentre la restante parte dei pazienti non mostra riduzione della massa tumorale. In alcuni pazienti si è assistito ad una stabilizzazione della crescita tumorale (8, 10), ma non è ancora chiaro se questo risultato persiste a lungo termine o è solo temporaneo. E' da sottolineare come in alcuni casi responsivi si è assistito a una remissione completa della malattia che si è protratta anche a distanza dalla sospensione del farmaco (10). La probabilità di attività clinica nei pazienti con carcinoma o adenoma ipofisario aggressivo sembra essere correlata con l'assenza o bassi livelli di espressione della MGMT nelle cellule adenomatose (9). Inoltre l'assenza di una risposta clinica o radiologica nei primi 3-6 mesi di trattamento sembra indicare i casi resistenti al trattamento (8, 10).
E' da sottolineare come l'indicazione ufficiale al trattamento con Temozolomide dei carcinomi ipofisari o degli adenomi ipofisari aggressivi sia stata recepita, caso unico in Europa e negli Stati Uniti, dall'Agenzia Italiana del Farmaco, come pubblicato sulla Gazzetta Ufficiale N. 118 del 23 Maggio 2011.
Contro-indicazioni
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti.
Ipersensibilità alla dacarbazina (DTIC).
Grave mielosoppressione.
Nei pazienti anziani sembra esservi un aumentato rischio di neutropenia e trombocitopenia.
Precauzioni d'uso
Si può riscontrare una maggiore comparsa di polmonite da pneumocisti quando la Temozolomide viene somministrata in un regime posologico prolungato. Prima della somministrazione devono essere valutati i parametri ematologici di laboratorio. Il giorno 22 (21 giorni dopo la prima somministrazione) o entro le 48 ore successive deve essere effettuato un esame emocromocitometrico completo: se la conta dei neutrofili si riduce a < 1.0 x 109/L o la conta delle piastrine è < 50 x 109/L durante un ciclo, il dosaggio del ciclo successivo deve essere ridotto di un livello. I livelli di dose sono 100 mg/m², 150 mg/m² e 200 mg/m². La dose più bassa raccomandata è di 100 mg/m².
Non c'è esperienza d'uso in bambini al di sotto dei tre anni e l'esperienza in ambito pediatrico è comunque limitata.
La temozolomide può avere effetti genotossici. Pertanto agli uomini in trattamento con la temozolomide si raccomanda di non procreare fino a 6 mesi dopo la conclusione del trattamento e di informarsi sulla crio-conservazione dello sperma prima dell’inizio del trattamento a causa della possibile, irreversibile infertilità legata alla terapia con la Temozolomide. È stata dimostrata la tossicità teratogena e/o fetale in studi preclinici, condotti su ratti e conigli. La Temozolomide pertanto non deve essere normalmente somministrato a donne gravide. Le donne in età fertile devono essere avvisate di evitare la gravidanza in corso di trattamento con Temozolomide. Non è noto se la temozolomide sia escreta nel latte umano; pertanto l’allattamento al seno deve essere interrotto durante in trattamento con Temozolomide.
Effetti collaterali
La temozolomide è un agente embriotossico, teratogeno e genotossico. La trombocitopenia e/o la neutropenia sono gli effetti collaterali dose-limitanti più comunemente osservati. Nei pazienti con tumori cerebrali si sono verificate forme gravi di mielodepressione, richiedenti ospedalizzazione o la sospensione del trattamento, quali trombocitopenia e neutropenia rispettivamente nell'8% e nel 4% dei trattati. Altre reazioni avverse sono: nausea (42%), vomito (35%) con forme severe nel 4% dei casi, affaticamento (21%), stipsi (15%), cefalea (13%), anoressia (11%) e diarrea (8%).
Limitazioni prescrittive
La Temozolomide è dispensata dalle Aziende Ospedaliere con File F.
Bibliografia
- Newlands ES, Stevens MFG, Wedge SR, et al. Temozolomide: a review of its discovery, chemical properties, preclinical development and clinical trials. Cancer Treat Rev 1997, 23: 35-61.
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomized phase III study: 5-year analysis of the EORT-NCIC trial. Lancet Oncol 2009, 10: 459-66.
- Agarwala SS, Kirkwood JM. Temozolomide a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000, 5: 144-51.
- Ekeblad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res 2007, 13: 2986-91.
- Lim S, Shahinian H, Maya MM, et al. Temozolomide: a novel treatment for pituitary carcinoma. Lancet Oncol 2006, 7: 518-20.
- Fadul CE, Kominsky AL, Meyer LP, et al. Long term response of pituitary carcinoma to temozolomide. J Neurosurg 2006, 105: 621-6.
- Hagen C, Schroeder HD, Hansen S, et al. Temozolomide treatment of a pituitary carcinoma and two pituitary macroadenomas resistant to conventional therapy. Eur J Endocrinol 2009, 161: 631-7.
- Raverot G, Sturm N, de Fraipont F, et al. Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: A French multicenter experience. J Clin Endocrinol Metab 2010, 95: 4592-9.
- Bush ZM, Longtine JA, Cunningham T, et al. Temozolomide treatment for aggressive pituitary tumors: correlation of clinical outcome with O6-methylguanine methyltransferase (MGMT) promoter methylation and expression. J Clin Endocrinol Metab 2010, 95: 280-90.
- Losa M, Mazza E, Terreni MR, et al. Salvage therapy with Temozolomide in patients with aggressive or metastatic pituitary adenomas: experience in six cases. Eur J Endocrinol 2010, 163: 843-51.
Terapie post-NCH
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
La terapia dipende da vari fattori:
- diagnosi radiologica pre-chirurgica: adenoma, craniofaringioma;
- presenza o meno di ipopituitarismo pre-chirurgico;
- via di accesso alla neoplasia: trans-cranica (TC) o trans-sfenoidale (TNS);
- visualizzazione da parte del neurochirurgo (durante intervento TNS) di tessuto ipofisario sano dopo l’asportazione della neoplasia.
GENERALITÀ E ADENOMI CLINICAMENTE NON FUNZIONANTI
Terapia steroidea (1-4)
Fase intra-operatoria: l’adenomectomia per via TNS è uno stress minore e non richiede obbligatoriamente steroidi nei casi con funzione ipofisaria normale prima dell’intervento.
Fase post-operatoria. Il trattamento sostitutivo steroideo post-chirurgico non è indispensabile nei casi in cui il neurochirurgo (esperto in chirurgia ipofisaria) ha visualizzato l’ipofisi sana residua e l’intervento non è stato complicato. Nei casi in cui queste condizioni non sono presenti, è indispensabile iniziare il giorno dopo l’intervento il trattamento sostitutivo steroideo (cortisone acetato 18.75-25 mg/die, idrocortisone 15-20 mg/die) in 2-3 somministrazioni: al risveglio e alle h 16, oppure al risveglio, a pranzo e alle h 18), da continuare a questa dose dopo la dimissione. La valutazione della funzione dell’asse ACTH-cortisolo può essere già fatta nei primi giorni dopo l’intervento, prima di assumere la dose di steroide del mattino, attraverso il dosaggio della cortisolemia alle ore 8:
- se la cortisolemia è > 10 µg/dL, lo steroide può essere sospeso con gradualità nei giorni successivi;
- se la cortisolemia è < 3 µg/dL, la terapia sostitutiva con steroidi va mantenuta e la dose va titolata in base al BMI del paziente;
- nei casi in cui la cortisolemia è compresa tra 3 e 10 µg/dL, lo steroide va mantenuto e dopo 30-45 giorni dall’intervento (intervallo ritenuto necessario per escludere interferenze legate allo stato funzionale surrenalico pre-intervento) deve essere compiuta la valutazione della funzionalità dell’asse ACTH-cortisolo.
La valutazione dell’asse ACTH-cortisolo viene eseguita mediante l’ACTH test alla dose di 1 µg (test da eseguirsi dopo almeno 12 ore di sospensione della terapia steroidea e in presenza di normale funzione tiroidea, spontanea o con sostituzione farmacologica):
- nei casi in cui il test mostra valori di cortisolemia normali (basali e dopo stimolo) la terapia cortisonica va sospesa;
- nei casi in cui l’esame è deficitario, la terapia cortisonica va mantenuta titolandola in base al peso del paziente e alla cortisolemia prima e dopo lo stimolo con ACTH (in media cortisone acetato: 12.5-25 mg/die; idrocortisone 10-20 mg/die).
Nei casi deficitari lo stimolo va ripetuto (con le stesse precauzioni e modalità) a distanza di 6 mesi dall’intervento, perché talvolta, nei casi di deficit parziale, si può osservare il recupero di una normale funzione dell’asse ACTH-cortisolo.
Nei casi in cui l’accesso è per via TC (condizione di stress chirurgico maggiore, in cui il paziente verrà trattato dal neurochirurgo con desametasone ad alta dose con finalità anti-edema), la possibilità di indurre ipopituitarismo è nettamente superiore: in questi casi alla sospensione del desametasone è indispensabile proseguire con lo steroide sostitutivo per via orale (alle dosi precedenti), fino all’esecuzione del test con ACTH 1 µg (con le modalità precedenti).
Va ricordato che i pazienti con iposurrenalismo post-chirurgico possono andare incontro a crisi di iposurrenalismo acuto nelle condizioni di stress fisico, quali febbre, caldo ambientale, interventi chirurgici, che richiedono l’impiego di dosi maggiori di steroidi.
Nei pazienti con iposurrenalismo pre-chirurgico, la terapia sostitutiva steroidea va potenziata durante l’intervento e mantenuta dopo la chirurgia, fino all’esecuzione del test di riserva surrenalica con ACTH: talvolta si può osservare la remissione dell’iposurrenalismo pre-chirurgico.
Nei casi di iposurrenalismo post-chirurgico, non c’è quasi mai indicazione a terapia sostitutiva con mineraloattivi (ad es fludrocortisone), perché la secrezione di aldosterone, indipendente dall’ACTH, viene mantenuta.
Il monitoraggio dell’adeguatezza della terapia sostitutiva cortisonica è molto difficile: possono essere utili il valore di cortisolemia prima dell’assunzione della terapia sostitutiva steroidea, oppure durante la giornata in corso di somministrazione del farmaco, tenendo comunque presente che nel soggetto normale la cortisolemia si riduce marcatamente dopo le prime ore del giorno. Molto utili sono i parametri indiretti di attività dello steroide: leucocitosi, ipopotassiemia, variazioni dei livelli glicemici (ipoglicemia in caso di dosaggio insufficiente, iperglicemia nei casi di eccesso di dose) e dei lipidi circolanti.
Bilancio idro-salino (5-8)
Costituisce l’emergenza maggiore dopo la chirurgia.
Nei casi di chirurgia TC il diabete insipido è molto frequente. Talvolta può aversi un’evoluzione in 3 fasi, con diabete insipido subito dopo l’intervento, che poi scompare per liberazione massiccia dell’ADH endogeno, per poi ripresentarsi successivamente.
In altri casi può comparire verso la quinta-settima giornata iposodiemia da inappropriata secrezione di ADH, la cui entità può essere in rapporto alla sofferenza post-chirurgica del sistema arginin-vasopressina; può peggiorare fino alla decima-dodicesima giornata, per poi generalmente regredire spontaneamente. Talvolta l’iposodiemia compare a distanza dall’intervento (10-15 giorni) e quando è marcata può determinare confusione mentale e stato di agitazione che richiedono l’ospedalizzazione; talvolta l’iposodiemia è seguita da diabete insipido permanente.
Quando è presente iposodiemia, è indispensabile la restrizione idrica.
Nei casi di diabete insipido il trattamento con desmopressina va iniziato con cautela, per evitare accumulo di acqua e conseguente iposodiemia, specialmente allorchè questa compare spontaneamente dopo l’intervento. È meglio quindi nei primi giorni post-intervento lasciare che la sodiemia sia leggermente superiore alla norma, piuttosto che nella parte medio-bassa del range di normalità.
Il controllo dell’adeguatezza del trattamento del diabete insipido viene effettuato monitorando nei primi giorni dopo la chirurgia il bilancio idrico (liquidi in entrata e in uscita nelle 24 ore) e la sodiemia: valori di sodio inferiori alla norma sono espressione di una dose elevata di desmopressina. Nei pazienti operati per via TC, quindi incoscienti nelle prime ore dopo l’intervento, è indispensabile monitorare la diuresi: allorchè questa supera il valore di 200 mL/h, va iniziato il trattamento con desmopressina sc.
Va sottolineato che nella fase post-chirurgica non va mai forzata da parte del medico e/o del personale infermieristico l’idratazione per via orale: il paziente deve bere secondo lo stimolo della sete, in quanto l’iperidratazione può precipitare una condizione di eccessiva emodiluizione, allorchè si manifesta una sindrome da inappropriata secrezione di ADH. Il quadro elettrolitico va monitorato quotidianamente nei pazienti operati per via TC dopo la ripresa di alimentazione spontanea, perché talvolta la chirurgia, specialmente nelle lesioni ipotalamiche, è gravata dalla comparsa di ipo/adipsia.
Funzione tiroidea
Il controllo va fatto dopo 7-10 giorni dall’intervento: se compare ipotiroidismo la terapia sostitutiva con L-tiroxina va iniziata subito a dose piena in base al peso corporeo del paziente.
Nei pazienti ipotiroidei prima dell’intervento, talvolta si osserva la remissione dell’ipotiroidismo, che va valutata a distanza di 15-20 giorni dalla sospensione della terapia sostitutiva.
Il monitoraggio della dose di L-tiroxina va eseguito attraverso il dosaggio della sola FT4. Il dosaggio del TSH è inutile e fuorviante e soprattutto non va eseguito il TSH Reflex.
Funzione gonadica
È spesso gravemente compromessa già prima dell’intervento nei pazienti con adenoma ipofisario non funzionante. In questi casi generalmente va mantenuto il trattamento sostitutivo con steroidi sessuali.
Nei casi in cui il deficit pre-chirurgico era modesto, è indicato valutare eventuali variazioni dello stato gonadico a distanza dall’intervento, per evitare le interferenze negative dovute allo stress chirurgico e ai farmaci (steroidi) impiegati in fase chirurgica.
ADENOMI SECERNENTI
GHomi
Nei pazienti acromegalici l’asportazione dell’adenoma GH-secernente, quando completa, è seguita da una poliuria profusa, talvolta anche di diversi litri, senza polidipsia. Tale poliuria può essere erroneamente interpretata come diabete insipido post-chirurgico e quindi talvolta viene somministrata desmopressina; in realtà si tratta dell’eliminazione dei liquidi trattenuti in maniera patologica dal paziente acromegalico a causa della sua malattia.
Il mantenimento o la ripresa della terapia GH-soppressiva dipende dal risultato della valutazione ormonale, il cui timing va programmato in base alla presenza di eventuale terapia GH-soppressiva pre-chirurgica: nel caso di paziente pretrattato con analoghi della somatostatina a lunga durata d’azione con buon controllo farmacologico della malattia, è opportuno lasciar trascorrere 2-3 mesi per avere una valutazione “pulita”.
PRLomi
I prolattinomi sottoposti a intervento chirurgico sono una rarità: a parte alcuni rari casi di microadenoma, che vogliono una guarigione definitiva, generalmente vengono operati pazienti con malattia aggressiva, che non risponde alla terapia farmacologica o che “scappa” al suo effetto terapeutico. Questi pazienti richiedono un controllo molto stretto, perchè i PRLomi resistenti al trattamento dopaminergico sono gli adenomi ipofisari più aggressivi dal punto di vista del comportamento biologico. La funzione ipofisaria potrebbe essere maggiormente compromessa rispetto agli altri tipi di adenomi per le caratteristiche neuroradiologiche e di invasività del prolattinoma. La presenza di valori dosabili di PRL nella fase post-chirurgica è un’indicazione al mantenimento della terapia soppressiva con dopaminergici, anche nei pazienti in cui l’asportazione neurochirurgica viene ritenuta completa.
Adenomi ACTH-secernenti
La gestione terapeutica post-chirurgica in questi pazienti è controversa. Alcuni autori preferiscono non somministrare terapia sostitutiva steroidea in maniera preventiva dopo l’intervento e attendere il risultato del dosaggio della cortisolemia e/o l’insorgenza dei primi sintomi di iposurrenalismo. Questa condotta richiede grande attenzione e la presenza di un team all’interno del quale sia sempre presente l’endocrinologo. Dal momento che questa condizione si realizza molto raramente, è preferibile e più sicuro iniziare dopo l’intervento in maniera sistematica la terapia sostitutiva steroidea, che può richiedere dosi maggiori rispetto a quelle che si impiegano nelle forme non funzionanti (cortisone acetato 37.5 mg/die, idrocortisone 30 mg/die, in 2-3 somministrazioni).
CRANIOFARINGIOMI
Visto il tipo di lesione e il tipo di intervento, il panipopituitarismo, se non era già presente prima dell’intervento, è comunque la regola dopo l’intervento e deve essere adeguatamente trattato con dosi piene di farmaci. Anche il diabete insipido è praticamente obbligatorio (9).
BIBLIOGRAFIA
- Inder WJ, Hunt PJ. Glucocorticoid replacement in pituitary surgery: guidelines for perioperative assessment and management. J Clin Endocrinol Metab 2002, 87: 2745-50.
- Nemergut EC, Dumont AS, Barry UT, Laws ER. Perioperative management of patients undergoing transsphenoidal pituitary surgery. Anesth Analg 2005, 101: 1170-81.
- Wentworth JM, Gao N, Sumithran KP, et al. Prospective evaluation of a protocol for reduced glucocorticoid replacement in transsphenoidal pituitary adenomectomy: prophylactic glucocorticoid replacement is seldom necessary. Clin Endocrinol 2008, 68: 29-35.
- Cozzi R, Lasio G, Cardia A, et al. Perioperative cortisol can predict hypothalamus-pituitary-adrenal status in clinically non-functioning pituitary adenomas. J Endocrinol Invest 2009, 32: 460-4.
- Fraser JF, Stieg PE. Hyponatremia in the neurosurgical patient: epidemiology, pathophysiology, diagnosis, and management. Neurosurgery 2006, 59: 222–9.
- Kristof RA, Rother M, Neuloh G, et al. Incidence, clinical manifestations, and course of water and electrolyte metabolism disturbances following transsphenoidal pituitary adenoma surgery: a prospective observational study. J Neurosurg 2009, 111: 555-62.
- Kinoshita Y, Tominaga A, Arita K. Post-operative hyponatremia in patients with pituitary adenoma: post-operative management with a uniform treatment protocol. Endocr J 2011, 58: 373-9.
- Hannon MJ,Finucane FM, Sherlock M, et al. Disorders of Water Homeostasis in Neurosurgical Patients. J Clin Endocrinol Metab 2012, 97: 1423–33.
- Smith D, Finucane F, Phillips J, et al. Abnormal regulation of thirst and vasopressin secretion following surgery for craniopharyngioma. Clin Endocrinol (Oxf) 2004, 61: 273–9.
Ipertrofie ipofisarie fisiologiche
Liana Cortesi1, Chiara Carzaniga2
1Unità di Endocrinologia, Ospedali Riuniti di Bergamo, Bergamo, Italia
2Unità di Endocrinologia, Istituto Auxologico Italiano, Milano, Italia
L’ipofisi è un organo di forma ovale, situato all’interno della sella turcica nel contesto dell’osso sfenoide. La ghiandola cresce rapidamente durante la vita fetale: da 3 mg tra la 10° e la 14° settimana di gestazione ai 50 mg alla 25° settimana, per arrivare ai 100 mg alla nascita ed aumentare fino a 600-900 mg circa nella vita adulta (1). Le dimensioni dell’ipofisi nell’adulto sono variabili da 11 a 15 mm di diametro trasverso, 7-11 mm di diametro anteroposteriore e 4-7 mm di diametro verticale. L’ipofisi nel sesso femminile è più grande che nell’uomo (2).
L’iperplasia ipofisaria è una condizione infrequente, poco studiata e spesso non diagnosticata. E’ un processo che attraversa diverse fasi, da un lieve aumento del numero di cellule che non interessa l’architettura tessutale, ad un incremento di spazio che occupa l’intera ghiandola determinando alterazioni a livello architetturale e morfologico. Esistono tre tipi differenti di iperplasia ipofisaria: la forma diffusa, quella focale e quella nodulare.
L’iperplasia diffusa indica un aumento numerico delle cellule ghiandolari senza alterazioni maggiori a carico della morfologia e dell’architettura acinare. L’iperplasia focale è un piccolo, circoscritto accumulo di un tipo specifico di cellule ipofisarie, talvolta di riscontro incidentale durante le autopsie. L’iperplasia nodulare è la forma più avanzata dell’iperplasia focale. A seconda del grado di proliferazione, gli acini sono ingranditi e popolati da un aumentato numero di cellule. La massa iperplasica non è mai monomorfica e spesso sono coinvolti altri tipi cellulari. L’iperplasia nodulare non determina mai problemi di compressione a carico delle strutture adiacenti (3).
Le cause di iperplasia ipofisaria fisiologiche sono la pubertà, la gravidanza e l’allattamento.
Le dimensioni della ghiandola variano infatti nel corso delle varie epoche di vita, aumentando notevolmente nei primi due anni di vita, crescendo progressivamente fino a raggiungere un plateau. All’età puberale si realizza un secondo picco di crescita, con una fisiologica ipertrofia che si manifesta nei maschi con incremento di volume della ghiandola, mentre nel sesso femminile si assiste anche a un cambiamento nella forma con margine superiore convesso (figura) (5). Queste variazioni anatomiche riflettono l’attivazione dell’asse ipotalamo-ipofisi-gonadi (6).
Ragazza adolescente. La RM in T1 sagittale (a) mostra un ingrandimento diffuso dell'ipofisi con margine superiore convesso. Il mdc (b) impregna omogeneamente la ghiandola. E' presente il fisiologico bright spot posteriore. (Cortesia della dott.ssa P. Doneda)
L’ipofisi è uno degli organi che si modifica maggiormente durante la gravidanza. La ghiandola aumenta di volume a causa dell’iperplasia delle cellule lattotrope. La massima altezza ed una forma maggiormente convessa si raggiunge nella prima settimana dopo il parto (7). L’incremento dimensionale può arrivare al 120 % del volume ghiandolare e si verifica per un terzo nel primo trimestre (8). L’aumento dell’altezza è correlato all’età gestazionale, con un aumento di 0.08 mm per settimana. Le dimensioni ghiandolari rientrano nei limiti entro 6 mesi dal parto.
Concludendo, è importante effettuare un’accurata definizione neuroradiologica di questi casi di ipertrofia fisiologica, al fine di differenziarle da lesioni adenomatose, ipofisiti o lesioni infiltrative sellari. Le moderne tecniche di indagine neuroradiologica (RM) permettono di distinguere con maggiore accuratezza i casi di ipertrofia fisiologica dalle patologie, evidenziando specifiche modifiche del segnale. Ad esempio, è possibile trovare un’iperintensità in T1 sia durante la gravidanza che nel post-partum. Tale segnale va differenziato da un possibile infarcimento emorragico. Al contrario il segnale iperintenso in T1 della neuroipofisi può essere meno evidente.
Bibliografia
- Wolpert SM, Molitch ME, Goldman JA, et al. Size, shape and appearance of the normal female pituitary gland. AJR Am J Roentgenol 1984: 143: 377-81.
- Lurie SN, Doraiswamy PM, Husain MM, et al. In vivo assessment of pituitary gland volume with magnetic resonance imaging: the effect of age. J Clin Endocrinol Metab 1990, 71: 505-8.
- Horvath E, Kovacs K, Scheithauer BW. Pituitary hyperplasia. Pituitary 1999, 1: 169-79.
- Peyster RG, Hoover ED, Viscarello RR, et al. CT appearance of adolescent and preadolescent pituitary gland. AJNR 1983, 4: 411-4.
- Elster AD, Chen MYM, Williams DW, et al. Pituitary gland: MR imaging of physiological hypertrophy in adolescence. Radiology 1990, 174: 681-5.
- Gonzalez JG, Elizondo G, Saldivar D, et al. Pituitary gland growth during normal pregnancy: an in vivo studio using magnetic resonance imaging. Amer J Med 1988, 85: 217-20.
- Dinç H, Esen F, Demirci A, et al. Pituitary dimension and volume measurements in pregnancy and post partum. MR assessment. Acta Radiol 1998, 39: 64-9.
- Elster AD, Sanders TG, Vines FS. Size and shape of the pituitary gland during pregnancy and post partum: measurement with MR imaging. Radiology 1991, 181: 531-5.
Adenomi ipofisari da feed-back
Chiara Carzaniga1, Liana Cortesi2
1Unità di Endocrinologia, Istituto Auxologico Italiano, Milano, Italia
2Unità di Endocrinologia, Ospedali Riuniti di Bergamo, Bergamo, Italia
Iperplasia/ipertrofia ipofisaria si osservano in stati di deficit prolungato di ghiandole bersaglio, come l’ipotiroidismo primario e l'ipogonadismo primario, mentre nell'iposurrenalismo primario questa evenienza è molto più rara. Inoltre iperplasia delle cellule GH-secernenti si osserva nei rarissimi casi di ipersecrezione eutopica o ectopica (carcinoidi) di GHRH, mentre eccezionalmente l’iperplasia delle cellule ACTH-secernenti si osserva in casi di ipersecrezione eutopica o ectopica di CRH. Spesso è difficile distinguere dal punto di vista neuroradiologico un’iperplasia/ipertrofia da un adenoma ipofisario, per cui queste alterazioni vengono anche comunemente chiamate adenomi da feed-back.
L’iperplasia delle cellule tireotrope è un frequente riscontro nei casi di ipotiroidismo primario di lunga durata. Si osserva specialmente nei pazienti giovani, in fase puberale, e regredisce nel momento in cui viene impostata un’adeguata terapia sostituiva con ormoni tiroidei (1). In alcuni casi l’ingrandimento dell’ipofisi simula una massa intra-soprasellare che può arrivare a stretto contatto delle vie ottiche, senza comunque determinare danni campimetrici, mimando un adenoma. In altri pazienti l’iperprolattinemia associata all’ipotiroidismo domina il quadro clinico, caratterizzato da amenorrea e galattorrea suggerendo una diagnosi di prolattinoma. La terapia con ormoni tiroidei è in grado di risolvere la sintomatologia (2). Raramente l’iperplasia raggiunge un grado tale da determinare una compromissione delle altre tropine ipofisarie. Importante è fare una corretta diagnosi differenziale dal punto di vista biochimico tra l’iperplasia delle cellule tireotrope (TSH elevato, FT4 ridotto) e l’adenoma ipofisario TSH-secernente (TSH e FT4 entrambi elevati). La TAC ha una bassa specificità, mentre con la RMN l’iperplasia si può chiaramente distinguere dall'adenoma per l'assenza di aree ipofisarie a diversa intensità di impregnazione da parte del gadolinio. Pertanto la diagnosi differenziale si basa soprattutto sulla storia clinica, sui dati di laboratorio e sulla sintomatologia. Alcuni studi ritengono che anche il dosaggio dell’alfa-subunità abbia un’alta sensibilità nel discriminare queste due forme, essendo alta negli adenomi TSH-secernenti e normale nell’iperplasia. Si ricorda inoltre che in età infantile sono riportati numerosi casi di iperplasia ipofisaria secondaria ad ipotiroidismo. I bambini, tuttavia, raramente presentano manifestazioni neurologiche secondarie all’effetto massa (3) (vedi figura).
Ingrandimento ipofisario secondario. Bambina di 8 anni con ipotiroidismo primario. RM alla diagnosi (a-c): ipofisi omogeneamente ingrandita e contrastata con espansione sovra-sellare. E' presente il fisiologico bright spot della neuroipofisi. Normali caratteristiche dell'ipofisi dopo terapia sostitutiva tiroidea (d).
L’iperplasia delle cellule GH-secernenti è molto rara. Gli unici casi riportati riguardano un’iperplasia secondaria alla produzione di GHRH da tumori extra-ipofisari come carcinoidi bronchiali, tumori delle isole pancreatiche, carcinomi midollari della tiroide, feocromocitoma, ecc. Si tratta per lo più di casi di iperplasia diffusa in cui le cellule sono acidofile. Nei tumori secernenti GHRH sono riportati allargamenti della sella turcica: la riduzione delle dimensioni della lesione ipofisaria fino alla scomparsa dopo asportazione del tumore GHRH-secernente fa supporre che trattasi più probabilmente di iperplasia. Studi in vitro dimostrano che una stimolazione prolungata da parte del di GHRH determina non solo rilascio di GH ma anche proliferazione delle cellule somatotrope (1,2). In letteratura sono segnalati inoltre rari casi di sindrome di McCune-Albright e di gigantismo causati da un’iperplasia delle cellule mammosomatotrope.
L’iperplasia delle cellule prolattino-secernenti è frequente e si verifica fisiologicamente durante la gravidanza e l’allattamento. E’ un processo reversibile che termina alla fine dell’allattamento. L’iperplasia delle cellule lattotrope si verifica anche in corso di terapia con estrogeni, come osservato nei casi di transessualismo maschi-femmine.
L’iperplasia delle cellule gonadotrope è rara, va comunque tenuta presente nei casi di giovani donne con amenorrea secondaria ipergonadotropa determinata da menopausa precoce che alla RM praticata per altri motivi mostrino apparentemente una lesione adenomatosa ipofisaria. Nella sindrome di Klinefelter, l’assenza di un feed-back negativo da parte degli androgeni, determina un’iperattività delle cellule gonadotrope. Pochi studi riportano casi di adenomi ipofisari, risultati immunoreattivi per FSH e alfa-subunità quando asportati chirurgicamente e altri positivi per “gonadal deficiency cell”, osservate nei casi di deficit gonadico, inclusa la castrazione. E’ ragionevole pensare che, a causa del mancato feed-back negativo, la stimolazione cronica delle cellule dell’adeno-ipofisi determini dapprima iperplasia e successivamente la formazione di un adenoma. Tale ipotesi non può essere confermata a causa dei pochi casi riportati in letteratura (4). Sono segnalati anche casi di alterazioni ipofisarie in pazienti affette da sindrome di Turner. Trattasi di adenomi ipofisari non secernenti, adenomi corticotropi, prolattinomi o costituiti da “gonadal deficiency cells”. Non è ancora chiaro se questi riscontri siano incidentali o se vi sia una relazione ma, essendo patologie rare, la possibilità di un’associazione eziologica è da considerare.
Considerando che l’incidenza degli adenomi gonadotropino-secernenti è del 10-15 % di tutti gli adenomi ipofisari e che essi si verificano esclusivamente in età avanzata quando la funzionalità gonadica è compromessa, è ragionevole pensare ad un legame tra l’ipogonadismo e l’insorgenza di adenomi. Il mancato feed-back negativo da parte degli steroidi sessuali porterebbe ad una stimolazione permanente delle gonadotropine che rappresenterebbe il passo cruciale verso lo sviluppo dell’adenoma. Questo starebbe a significare che gli incidentalomi riscontrati in età avanzata potrebbero essere dei gonadotropinomi (6).
Dati in letteratura riportano che l’iperplasia o gli adenomi delle cellule corticotrope si possono sviluppare in pazienti affetti da morbo di Addison non trattato per lungo tempo. Tuttavia, tale riscontro è segnalato anche nei pazienti trattati con terapia ormonale sostitutiva convenzionale. Infatti, dal momento che la terapia sostitutiva con idrocortisone o cortisone acetato non è in grado di inibire completamente l’ACTH, questo aspetto determina di conseguenza una proliferazione delle cellule corticotrope (7).
Anche nei rarissimi casi riportati in letteratura di sindrome di Cushing causata da secrezione ectopica di CRH viene segnalata un’iperplasia delle cellule corticotrope. L’iperplasia delle cellule corticotrope è responsabile di una piccola percentuale di casi di malattia di Cushing, causata invece da microadenomi ipofisari. In una minoranza di casi, iperplasia ed adenoma possono essere presenti simultaneamente (1,8). Anche la sindrome di Nelson può essere considerata un adenoma da feed-back.
Bibliografia
- Horvath E, Kovacs K, Scheithauer BW. Pituitary hyperplasia. Pituitary 1999, 1: 169-79.
- Sano T, Asa SL, Kovacs K. Growth Hormone-Releasing Hormone-Producing Tumors: clinical, biochemical, and morphological manifestations. Endocr Rev 1988, 9: 357-73.
- Joshi AS, Woolf PD. Pituitary hyperplasia secondary to primary hypothyroidism: a case report and review of the literature. Pituitary 2005, 8: 99-103.
- Scheithauer BW, Moschopulos M, Kovacs K, et al. The pituitary in Klinefelter syndrome. Endocr Pathol 2005, 16: 133-8.
- Scheithauer BW, Kovacs K, Horvath E, et al. The pituitary in Turner syndrome. Endocr Pathol 2005, 16: 195-200.
- Nicolis G, Shimshi M, Allen C, et al. Gonadotropin-producing pituitary adenoma in a man with long standing primary hypogonadism. J Clin Endocrinol Metab 1988, 66: 237-41.
- Zhou J, Ruan L, Li H, et al. Addison's disease with primary hyperplasia: a case report and review of the literature. Endocrine 2009, 35: 285-9.
- Schteingart DE, Lloyd RV, Akil H, et al. Cushing’s syndrome secondary to ectopic Corticotropin-Releasing Hormone-Adrenocorticotropin secretion. J Clin Endocrinol Metab 1986, 63: 770-5.