Test con prelievi multipli per PRL durante fisiologica

Maria Rosaria Ambrosio1 & Romolo Dorizzi2
1Sezione di Endocrinologia, Dipartimento di Scienze Biomediche e Terapia Avanzate Università degli Studi di Ferrara
2Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
| Test con prelievi multipli per PRL durante fisiologica | |
| Indicazioni |
Valutazione della secrezione basale di PRL (sensibilità della sua secrezione in rapporto allo stress) e costituiscono la prima tappa nella diagnosi di iperprolattinemia patologica. |
| Controindicazioni | Nessuna |
| Materiale necessario per l’esecuzione | Soluzione fisiologica |
| Relazione con età, sesso, peso corporeo, gravidanza |
Marcato aumento in gravidanza (considera la gravidanza nella paziente amenorroica). |
| Precauzioni | In alcuni casi di iperprolattinemia da grave stress, in assenza di contesto clinico, sono necessari prelievi per un periodo più prolungato. |
| Esecuzione | Non esiste una metodica standardizzata ed i prelievi sono eseguiti ogni 20-30 minuti con un minimo di tre. |
| Dosaggio | PRL |
| Possibili effetti collaterali | Nessuno |
| Parametri da monitorare durante l’esecuzione | Nessuno |
| Manovre da eseguire dopo la fine del test | Nessuna |
| Valutazione risultati |
Valori patologici di PRL (> del valore superiore di riferimento) in assenza di interferenza farmacologica e di macroprolattinemia, richiedono sempre l’esecuzione di RMN sellare con gadolinio. |
| Interpretazione |
L’entità dell’ipersecrezione di PRL nei casi di macroadenoma PRL-secernente è frequentemente proporzionale alle dimensioni dell’adenoma. |
| Attendibilità e ripetibilità dei risultati |
Falsi positivi (riscontro di valori elevati in assenza di contesto clinico): presenza di macroprolattinemia e di anticorpi eterofili. |
| Giudizio complessivo costo beneficio e costo-efficacia |
Indice dell’entità della secrezione di PRL, alla portata di ogni laboratorio. |
Diagnostica ormonale dell'asse vasopressina-osmolarità
Maria Rosaria Ambrosio1 & Romolo Dorizzi2
1Sezione di Endocrinologia, Dipartimento di Scienze Biomediche e Terapia Avanzate Università degli Studi di Ferrara
2Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Indagini basali
Test dinamici
Valutazione basale dell'osmolarità
Maria Rosaria Ambrosio1 & Romolo Dorizzi2
1Sezione di Endocrinologia, Dipartimento di Scienze Biomediche e Terapia Avanzate Università degli Studi di Ferrara
2Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
L’osmolalità descrive la pressione osmotica di una soluzione, quando si aggiungono dei soluti ad un solvente puro come l’acqua. L’unità di misura per l’osmolalità, secondo il sistema SI, è la mOsm/kg H2O. Per osmolalità si intendono le concentrazioni relative alla massa di solvente (una soluzione 1 osmolale contiene 1 Osm/kg di acqua), mentre per osmolarità si intendono le concentrazioni relative al volume di soluzione (una soluzione 1 osmolare contiene 1 Osm/L di soluzione). Dal punto di vista termodinamico è più corretto usare il termine osmolalità, perché le concentrazioni espresse sulla base del peso sono indipendenti dalla temperatura, mentre quelle basate sul volume variano con la temperatura. Le principali sostanze osmoticamente attive sono sodio, cloro, glucosio ed urea.
È possibile calcolare l’osmolalità con numerose equazioni empiriche; la più usata è la seguente:
- mOsm/kg = (1.86 * sodiemia (in mmol/L)) + glicemia (in mmol/L) + urea (in mmol/L) + 9
- mOsm/kg = (1.86 * sodiemia (in mmol/L)) + (glicemia (in mg/dL)/18) + (urea (in mg/dL)/2.8) + 9
Le 9 mOsm/kg aggiunte nelle due equazioni rappresentano il contributo delle altre sostanze osmoticamente attive presenti nel plasma, come il potassio, il calcio e le proteine, mentre 1.86 rappresenta circa due volte il coefficiente osmotico del sodio, riflettendo il contributo del sodio e del cloro.
| Alterazioni dell'osmolarità | ||
| Diminuite da | Aumentate da | |
| Cause fisiopatologiche | inappropriata secrezione di ADH carico idrico (intossicazione d’acqua) |
disidratazione febbre diuresi osmotica (diabete scompensato) esercizio fisico ad alta temperatura diabete insipido centrale o nefrogenico iperaldosteronismo primario ipodipsia ipotalamica |
| Modificazioni farmaco-indotte | FANS | soluzioni ipertoniche mannitolo vaptani |
Come calcolare l’osmolalità senza osmometro
Paolo Zuppi, Francesca Rota, Maurizio Cosentino*
UOSD Endocrinologia, Ospedale San Camillo-Forlanini, Roma
*Endocrinologo Ambulatoriale Territorio ASL RM2
(aggiornato al 15 giugno 2021)
La conoscenza dei valori di osmolarità (1) plasmatica e urinaria è indispensabile per una gestione corretta di condizioni cliniche rilevanti fra cui l’iposodiemia. Purtroppo, molti laboratori, anche di grandi centri, sono sprovvisti dell’osmometro, l’apparecchio che permette la determinazione dell’osmolarità reale. Negli anni sono stati ricercati e proposti dei surrogati calcolati dell’osmolalità. Molti analiti contribuiscono all’osmolarità dei liquidi biologici: sodio, cloro, potassio, fosfati, solfati, calcio, magnesio, creatinina, glucosio, acido urico, lipidi, proteine ecc. Le formule proposte in letteratura sono semplificazioni necessariamente imprecise, nonostante, quasi sempre, contengano una costante per tentare di considerare il contributo di queste sostanze.
Osmolalità urinaria
L’osmolarità urinaria può essere, approssimativamente, dedotta o calcolata:
- dedotta dal peso specifico delle urine (2). Ogni punto di peso specifico oltre 1000 corrisponde a 35 unità di osmolarità.
| Tabella 1 Osmolarità urinaria |
|
| Peso specifico | Osmolarità |
| 1000 | 0 |
| 1001 | 35 |
| 1002 | 70 |
| 1010 | 350 |
| 1020 | 700 |
Le diverse modalità di rilevazione del peso specifico possono essere alterate dalla presenza di:
- pH, chetoni, bilirubina, urobilinogeno, glucosio e proteine, se il peso specifico è determinato con strisce reattive;
- chetoni, bilirubina ed emoglobina, se il peso specifico è determinato con rifrattometro;
- calcolata, approssimativamente, secondo le formule (3,4):
- 1.09 [(1.86 x Na) + (glucosio/18) + (urea/6)];
- [(Na + K) x 2] + (urea/6).
L’approssimazione di questi metodi aumenta in alcune condizioni cliniche in cui sono presenti nelle urine grandi molecole, come ad esempio nel diabete mellito scompensato, nella sindrome nefrosica, mezzi di contrasto, mannitolo, carbenicillina.
Osmolalità plasmatica
Sono state proposte numerose formule per calcolare, in assenza di osmometro, l’osmolarità plasmatica. Le più usate sono le 14 sotto-elencate nella tabella 2, in ordine per osmolarità crescente a parità dei dati.
| Tabella 2 Osmolarità plasmatica |
|
| Formula | Ref |
| 1.75 Na + (glu/18) + (urea/6) + 10.1 | 5 |
| 1.86 Na + (glu/18) + (urea/6) | 6 |
| 1.86 Na + (glu/18) + (urea/6) + 5 | 7 |
| 1.86 Na + (glu/18) + (urea/6) + 9 | 8 |
| [1.86 (Na + K)] + (glu/18) + (urea/6) + 9 | 9 |
| [1.86 (Na + K)] + (glu/18) + (urea/6) + 10 | 9 |
| 1.89 Na + 1.38 K + [1.08 (glu/18)] + [1.03 (urea/6)] + 7.45 | 9 |
| [1.90 (Na + K)] + (glu/18) + (urea/6) + 5 | 10 |
| 1.897 Na + (glu/18) + (urea/6) + 13.5 | 10 |
| 1.85 Na + 1.84 K + (glu/18) + (urea/6) + Ca + 1.17 Mg + 1.15 | 11 |
| 2 Na + (glu/18) + (urea/6) | 12 |
| 2 Na + [1.15 (glu/18)] + (urea/6) | 13 |
| [1.86 (Na+ K)] + [1.15 (glu/18)] + (urea/6) + 14 | 13 |
| [2 (Na + K)] + (glu/18) + (urea/6) | 14 |
Le diverse formule danno risultati fortemente discrepanti. I lavori di confronto fra osmolarità misurata e osmolarità calcolata con le diverse formule suggeriscono che la formula di Worthley (12), (2 Na + glucosio/18 + urea/6), offre una buona affidabilità (6). La presenza di sostanze non considerate nella formula applicata determina una differenza fra osmolarità reale e osmolarità calcolata: gap osmolare. Tale discrepanza deve far sospettare la presenza di sostanze quali etanolo, metanolo, acetone, glicole etilenico e numerosi farmaci (7).
Sebbene la osmolarità surrogata sia imprecisa, in mancanza di un osmometro permette comunque un orientamento al clinico.
Nel file che si può scaricare qui (Formule), inserendo i valori disponibili verrà automaticamente calcolato il valore con le diverse formule.
Breve glossario
- Azoto: elemento chimico, peso atomico 14.00, è un gas incolore, inodore, insapore e inerte, principale costituente delle molecole biologiche.
- Azotemia: valore che indica la quantità dell’azoto non proteico nel sangue. In gran parte è costituita da urea ma anche da acido urico, creatina, creatinina, purine, aminoacidi ed altri composti azotati. Valori normali 15-50 mg/dL.
- Azoto ureico = BUN (blood urea nitrogen): è la quantità dell'azoto nell’urea presente nel sangue. Poiché vi sono 28 g di azoto per 60 g di peso di una mole di urea, il BUN si calcola dividendo il valore di urea espresso in mg/dL per 2.14 (60/28). Il peso molecolare dell'azoto è 28.02, il doppio del peso atomico. Una mole di azoto (N2) corrisponde pertanto a 28 g di azoto. Nelle formule per determinare l’osmolarità plasmatica il valore di BUN è espresso in mmol/L. Per effettuare la conversione da mg/dL a mmol/dL bisogna dividere per 28. Ottenuto il risultato in mmol/dL, per convertirlo in mmol/L bisogna moltiplicare per 10. Ciò equivale a dividere il valore in mg/dL per 2.8. I valori normali sono 9–20 mg/dL, ma vi possono essere differenze fra i laboratori in base al metodo di dosaggio.
- Urea: composto chimico di formula CO(NH2)2, prodotto finale, non tossico, del catabolismo delle proteine eliminata dai reni. Ogni molecola di urea possiede due atomi di azoto, ciascuno con massa molare di 14 g/mol. Il peso molecolare dell'urea è 60.06, cioè 1 M di urea corrisponde a 60 g di urea. Nelle formule per determinare l’osmolarità plasmatica il valore di urea è espresso in mmol/L. Per effettuare la conversione da mg/dL a mmol/dL bisogna dividere per 60. Ottenuto il risultato in mmol/dL, per convertirlo in mmol/L bisogna moltiplicare per 10. Ciò equivale a dividere il valore in mg/dL per 6. I valori normali nell’uomo sono di 18-40 mg/dL.
- Uremia: valore misurato di urea nel sangue, ma è diventato nome comune dell'insufficienza renale terminale, sebbene sarebbe corretto dire iperuremia.
Bibliografia
- Rasouli M. Basic concepts and practical equations on osmolality: Biochemical approach. Clin Biochem 2016, 49: 936-41.
- Voinescu GC, Shoemaker M, Moore H, et al. The relationship between urine osmolality and specific gravity. Am J Med Sci 2002, 323: 39-42.
- Bianchi V, Bidone P, Arfini C. Siero ed urine: osmolalità calcolata o osmolalità misurata? RIMeL – IJLaM 2009, 5: 206-11.
- Youhanna S, Bankir L, Jungers P, et al. Validation of surrogates of urine osmolality in population studies. Am J Nephrol 2017, 46: 26-36.
- Edelman IS, et al. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest 1958, 37: 1236–56.
- Glasser L, et al. Serum osmolality and its applicability to drug overdose. Am J Clin Pathol 1973, 60: 695–9.
- Boyd DR, et al. Osmometry: a new bedside laboratory aid for the management of surgical patients. Surg Clin North Am 1971, 51: 241–50.
- Dorwart W, et al. Comparison of methods for calculating serum osmolality form chemical concentrations, and the prognostic value of such calculations. Clin Chem 1975, 21: 190–4.
- Bhagat CI, et al. Calculated vs measured plasma osmolalities revisited. Clin Chem 1984, 30: 1703–5.
- Rasouli M, et al. Comparison of methods for calculating serum osmolality: multivariate linear regression analysis. Clin Chem Lab Med 2005, 43: 635–40.
- Garcia-Morales EJ, et al. Osmole gap in neurologic-neurosurgical intensive care unit: Its normal value, calculation, and relationship with mannitol serum concentrations. Crit Care Med 2004, 32: 986–91.
- Worthley LI, Guerin M, Pain RW. For calculating osmolality, the simplest formula is the best. Anesth Intens Care 1987, 15: 199-202.
- Khajuria A, et al. Osmolality revisited-deriving and validating the best formula for calculated osmolality. Clin Biochem 2005, 38: 514–9.
- Gerich JE, et al. Clinical and metabolic characteristics of hyperosmolar nonketotic coma. Diabetes 1971, 20: 228–35.
- Martín-Calderón JL, Bustos F, Tuesta-Reina LR, et al. Choice of the best equation for plasma osmolality calculation: Comparison of fourteen formulae. Clin Biochem 2015, 48: 529-33.
- Kar E, Kocatürk E, Küskü Kiraz Z, et al. Comparison of measured and calculated osmolality levels. Clin Exp Nephrol 2020, 24: 444-9.
Test dell’assetamento
Maria Rosaria Ambrosio1 & Romolo Dorizzi2
1Sezione di Endocrinologia, Dipartimento di Scienze Biomediche e Terapia Avanzate Università degli Studi di Ferrara
2Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
| Test dell’assetamento | |
| Indicazioni | Test confermatorio di diabete insipido centrale, va praticato solo nei casi incerti di poliuria e polidipsia non accompagnati da ipersodiemia. Diagnosi differenziale fra diabete insipido centrale e periferico e polidipsia primaria. |
| Meccanismo d’azione | Valuta la riserva di ADH. L’assetamento (restrizione idrica) determina disidratazione e incremento dell’osmolarità plasmatica. La stimolazione degli osmocettori aumenta la secrezione di ADH che, agendo sul tubulo renale, aumenta il riassorbimento di acqua e l’osmolalità urinaria. |
| Controindicazioni | Ipersodiemia all’inizio dell’esame. Paziente con poliuria dopo neurochirurgia. Paziente con ipodipsia causata da lesione del SNC (ipotalamica). Attenzione: il paziente con ipodipsia non è cosciente della sua condizione. |
| Materiale necessario per l’esecuzione | Desmopressina (Minirin) 10 μg nasale o 4 μg sottocute o endovena. |
| Relazione con età, sesso, peso corporeo, gravidanza | Considera: diabete insipido gestazionale, causato dalla liberazione di vasopressinasi placentare (normale risposta alla desmopressina). |
| Precauzioni | Eseguire in regime di ricovero, al mattino e interrompere l’idratazione 2-3 ore prima di iniziare il test (evitare la restrizione di liquidi durante la notte, che può causare severa deplezione idrica e ipersodiemia). |
| Esecuzione | Pesare il paziente per calcolare il massimo tollerato di diuresi o perdita di peso prima dell’interruzione del test (p.e. nel paziente di 70 kg la diuresi non deve superare i 3.5 litri o il peso non deve scendere sotto i 66.5 kg). Il paziente deve essere tenuto sotto stretto controllo per evitare che introduca liquidi (come spesso accade nelle polidipsie psicogene). Dopo il prelievo basale, assetamento (6 ore circa) fino al raggiungimento di osmolalità urinaria normale (600 mOsm/kg). Considera: la diuresi totale e/o perdita di peso non devono superare il 5% del peso corporeo. Far urinare il paziente ogni ora (o quando ne sente il bisogno), misurando la quantità oraria delle urine. Allorché l’assetamento non determina più contrazione della diuresi oraria in 2-3 raccolte successive, somministrazione di Desmopressina endonasale (10 μg). |
| Dosaggio | Misurare peso, diuresi, osmolarità plasmatica e urinaria ai tempi 0, +1h, +2h, +3h, +4h, +5h, +6h, +7h. |
| Possibili effetti collaterali | Disidratazione |
| Parametri da monitorare durante l’esecuzione | Diuresi oraria e peso specifico, osmolarità plasmatica e urinaria, elettroliti, azotemia, emocromo, peso corporeo, stato di coscienza, pressione arteriosa, ritmo e frequenza cardiaca. Il test va sospeso, eseguendo prelievo ematico per elettroliti e osmolarità, allorché la diuresi complessiva è maggiore del 5% del peso corporeo e/o quando compaiono segni di deplezione di volume. |
| Manovre da eseguire dopo la fine del test | Libero accesso ai liquidi. |
| Valutazione risultati | Valori normali: osmolarità plasmatica 275-290 mOsm/kg, osmolalità urinaria: 600-900 mOsm/kg. Nel diabete insipido l’assetamento aumenta l’osmolarità plasmatica, mentre quella urinaria rimane invariata. Nei casi di diabete insipido parziale, frequente nelle forme centrali, l’osmolalità urinaria può raggiungere valori vicini alla normalità (600 mOsm/kg), con conseguente contrazione della diuresi. La somministrazione di desmopressina (che consente la diagnosi differenziale tra le forme centrali e le forme nefrogeniche) contrae la diuresi oraria e aumenta i valori di osmolarità urinaria (> 100-800% nelle forme complete; > 15-50% in quelle parziali) nelle forme centrali. |
| Interpretazione | Nel soggetto normale, quando l’assetamento è prolungato, la secrezione di ADH aumenta in maniera massimale; l’aumento dell’osmolarità plasmatica determina l’aumento massimale della osmolalità urinaria (> 800 mOsm/kg). Nel soggetto normale la somministrazione di ADH (desmopressina) non aumenta ulteriormente l’osmolalità urinaria, già stimolata al massimo dall’ADH endogeno. Polidipsia primaria (o psicogena): l’assetamento di solito aumenta l’osmolalità urinaria (500-600 mOsm/kg); nessuna risposta alla somministrazione di desmopressina in quanto la secrezione endogena di ADH è intatta (e quindi già stimolata). Nelle forme più gravi (con soppressione funzionale della secrezione di ADH) l’osmolalità urinaria aumenta dopo somministrazione di desmopressina. Diabete insipido nefrogenico (ereditario, iatrogeno): l’osmolalità urinaria rimane iso-osmotica (300 mOsm/kg), risponde poco o nulla alla somministrazione di desmopressina. |
| Attendibilità e ripetibilità dei risultati | Nelle forme gravi di polidipsia primaria la secrezione di ADH è soppressa: questi casi frequentemente si presentano con iposodiemia e rispondono al test come i pazienti con diabete insipido centrale; la somministrazione di desmopressina può quindi aggravare l’iposodiemia e determinare intossicazione d’acqua. |
| Giudizio complessivo costo beneficio e costo-efficacia | Test di difficile interpretazione nei casi di deficit parziale di ADH, che rappresentano la maggior parte della casistica (i risultati del test possono erroneamente indirizzare verso una forma di polidipsia primaria): l’osmolalità urinaria può aumentare solo dopo l’aumento a livelli sopranormali di quella plasmatica e la somministrazione di desmopressina non riesce ad aumentare ulteriormente l’osmolalità urinaria. Da tenere in considerazione: la modalità d’esordio, l’aspetto psicologico del paziente, la presenza di iposodiemia, il risultato di un trial con desmopressina, eventuale RMN sellare. |
| Bibliografia |
|
Copeptina nella diagnosi differenziale delle sindromi poliuriche-polidipsiche e delle iponatremie
Rita Indirli
SC Endocrinologia, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano. Dipartimento di Scienze Cliniche e di Comunità, Università degli Studi di Milano.
(aggiornato al 10 maggio 2023)
ARGININA-VASOPRESSINA E COPEPTINA
L’ormone antidiuretico, o arginina-vasopressina (AVP), viene sintetizzato a partire da un peptide precursore (pre-pro-AVP) nel corpo cellulare dei neuroni magno-cellulari contenuti nei nuclei sovra-ottico e para-ventricolare dell’ipotalamo. La molecola di pre-pro-AVP dà origine a tre peptidi: AVP, neurofisina II, e la molecola C-terminale copeptina. Quest’ultima viene co-secreta dalla neuro-ipofisi in quantità equimolare ad AVP, e ne viene per questo considerata un marcatore surrogato (1).
Diverse caratteristiche di AVP rendono infatti difficoltoso il suo dosaggio: basso peso molecolare, ridotta emivita in vivo (12 minuti) e instabilità ex vivo (può essere conservata a -20°C fino a massimo 24 ore), legame alle piastrine circolanti, lunghe tempistiche di dosaggio (> 48 ore) con tecnica radio-immunometrica. Al contrario, copeptina ha emivita di circa 26 minuti, può essere conservata a temperatura ambiente oltre 7 giorni ed essere dosata con tecniche immuno-luminometriche automatizzabili, che richiedono da 30 minuti a 2.5 ore (tab 1).
| Tabella 1 Confronto tra le caratteristiche di arginin-vasopressina (AVP) e copeptina (1) |
||
| AVP | Copeptina | |
| Forma molecolare | Peptide di 9 aminoacidi | Glicopeptide di 39 aminoacidi |
| Concentrazione mediana (e range) nei soggetti sani | 1 pg/mL (0.5-6.1) | 4 pmol/L (0.9-21.4) |
| Emivita | 12 minuti | 26 minuti |
| Legame alle piastrine | Sì | No |
| Dosaggio | Radio-immunometrico | Immuno-luminometrico |
| Quantità necessaria al dosaggio | 1 mL | 50 µL |
| Procedure pre-analitiche | Estrazione, inibitori delle proteasi | Non necessarie |
| Tempo necessario per il dosaggio | > 48 ore | 0.5-2.5 ore |
| Stabilità ex vivo | 24 ore a -20°C (instabile) | ≥ 7 giorni a temperatura ambiente, 14 giorni a -4°C (stabile) |
La funzione biologica di copeptina non è ancora stata identificata: si ipotizza che possa avere un ruolo nel ripiegamento e nella maturazione proteolitica di AVP, mentre non è noto se esista un recettore per questa molecola (2).
SINDROME POLIURIA-POLIDIPSIA
In presenza di una sindrome poliuria-polidipsia (diuresi > 50 mL/kg/die nell’adulto, introito idrico > 3 L/die), avendo escluso cause di diuresi osmotica (iperglicemia, ipercalcemia) e verificata l’emissione di urine ipotoniche (osmolalità urinaria, U-Osm, < 800 mOsm/kg), è necessario differenziare tra diabete insipido (DI, centrale -DIC- o nefrogenico -DIN-) e polidipsia primaria (PP) (3).
Il riscontro di sodiemia > 147 mEq/L con osmolalità plasmatica (P-Osm) > 280 mOsm/kg permette immediatamente la diagnosi di DI, mentre la sodiemia < 135 mEq/L la esclude (3). La maggior parte dei casi, tuttavia, si presenta con sodiemia e P-Osm normali, richiedendo quindi una diagnostica di approfondimento.
Misurazioni basali
La misurazione basale (senza precedente restrizione di fluidi) dei livelli di copeptina, in assenza di eventi acuti infettivi o cardio-cerebro-vascolari, è impiegata per la diagnosi di DIN: un valore ˃ 21.4 pmol/L ha infatti specificità e sensibilità del 100% (4,5).
È stato inoltre proposto il valore di copeptina basale < 2.9 pmol/L come indicativo di DIC, con sensibilità dell’82% e specificità del 78%, mentre valori < 2.6 pmol/L sarebbero diagnostici di DIC completo (4,5). Tuttavia, considerata l’accuratezza diagnostica non ottimale, questi cut-off ad oggi non rientrano nell’algoritmo diagnostico.
Test dell’assetamento
Il primo test utilizzato storicamente è stato il test dell’assetamento. L’assetamento porta a uno stimolo osmotico, che induce il rilascio di AVP, l’aumento di U-Osm e la contrazione della diuresi. La tabella 2 ne riassume le modalità di esecuzione e l’interpretazione.
Il test dell’assetamento indiretto ha accuratezza globale solo del 70-76% (4,6); in particolare, mentre è affidabile nell’identificare i casi di DIC completo, rimane insoddisfacente la sua performance nella diagnosi differenziale tra DIC parziale e PP (accuratezza diagnostica per PP 41%).
Per questi motivi, già nel 1981 era stato proposto di modificare il test classico con la misurazione diretta di AVP in corso di assetamento (7). Tuttavia, i valori di AVP misurati in corso di assetamento sono largamente sovrapponibili tra pazienti con DIC, DIN, e senza DI (8) e mostrano accuratezza diagnostica solo del 46% (4), come atteso date le difficoltà di dosaggio di questo ormone.
La misurazione di copeptina al termine del test di assetamento (prima della somministrazione di desmopressina) permette una diagnosi corretta nel 44-72% dei casi (4,6). Un rapporto tra Δ-copeptina e Δ-sodiemia (tra le ore 8 e le ore 16) ≥ 0.2 pmol/mmol differenzia tra DIC parziale e PP, con sensibilità dell’86% e specificità del 100%.
La performance diagnostica del test dell’assetamento è inficiata dal fatto che una condizione di polidipsia-poliuria cronica può portare a una serie di modifiche fisiopatologiche (down-regolazione dell’espressione di AVP nell’ipotalamo e di acquaporina-2 nell’epitelio renale, wash-out del gradiente renale midollare), che possono ridurre il rilascio di AVP in risposta all’assetamento e la capacità di concentrazione urinaria dopo la somministrazione di desmopressina (1). Inoltre, durante il test di assetamento non tutti i pazienti raggiungono P-Osm > 290 mOsm/kg e la conseguente adeguata stimolazione del sistema AVP-copeptina. Infine, il test di assetamento è un test lungo, impegnativo per pazienti e operatori sanitari e con possibili effetti avversi (es. iponatremia dopo somministrazione di desmopressina (6).
Test con soluzione salina ipertonica
Recentemente, allo scopo di superare i limiti del test dell’assetamento, è stato proposto il test con soluzione salina ipertonica al 3%, dapprima nella forma di un test combinato: i pazienti venivano sottoposti a un test di assetamento “breve” (con inizio alle ore 8.00 senza precedente restrizione di fluidi) e, nel caso di mancato raggiungimento di sodiemia > 147 mEq/L alle ore 13.00, a successiva infusione di soluzione salina al 3% fino al raggiungimento di tale valore soglia di sodiemia (5). Un valore stimolato di copeptina > 4.9 pmol/L era in grado di differenziare tra DIC e PP, con sensibilità del 94% e specificità del 96%.
Successivamente, il test con soluzione ipertonica è stato validato singolarmente (senza precedente assetamento), dimostrando accuratezza diagnostica del 96.5% (6). Il cut-off di copeptina stimolata di 4.9 pmol/L è in grado di differenziare tra PP e DIC, con sensibilità del 93.2% e specificità del 100%. È importante sottolineare che il cut-off di copeptina stimolata di 4.9 pmol/L è stato validato utilizzando per il dosaggio la piattaforma B.R.A.H.M.S Kryptor. I valori soglia di copeptina richiedono apposita validazione quando si utilizzino metodi di dosaggio differenti (1).
Confrontato con il test dell’assetamento, il test con salina ipertonica è preferito dai pazienti, pur comportando un aumento del rischio di ipersodiemia grave (˃ 155 mEq/L), comunque in assenza di eventi avversi seri. La tabella 2 ne riassume le modalità di esecuzione.
Test con arginina
Un nuovo test con arginina (tab 2), che impiega quindi uno stimolo non osmotico, ha dimostrato accuratezza diagnostica del 93%, con cut-off di copeptina di 3.8 pmol/L (sensibilità 93%, specificità 92%) (9). Lo studio di validazione di questo test (CARGOx study, NCT03572166) è stato da poco concluso e se ne attende la pubblicazione.
| Tabella 2 Test diagnostici per la diagnosi differenziale delle sindromi poliuro-polidipsiche (3) |
|||
| Test | Procedura | Durata | Interpretazione |
| Copeptina basale | Prelievo venoso basale. | - | Copeptina > 21.4 pmol/L = DIN. |
| Test di assetamento indiretto | Restrizione di fluidi dalle ore 00:00 alle ore 16:00. Somministrazione di DDAVP alle ore 16:00. Valutazione della risposta a DDAVP alle ore 17:00. Interruzione precoce se calo ponderale > 5%, P-Osm ≥ 300 mOsm/kg. |
17 ore |
Al termine dell’assetamento:
|
| Test con soluzione salina ipertonica |
Somministrare soluzione salina ipertonica al 3% ev:
Misurare il sodio su EGA venoso ogni 30 minuti. |
2-4 ore | Copeptina > 4.9 pmol/L = PP. Copeptina ≤ 4.9 pmol/L = DIC. |
| Test con arginina (non ancora validato) | Somministrare arginina cloridrato 21% al dosaggio di 0.5 g/kg (massimo 40 g) diluita in 500 mL di soluzione fisiologica 0.9%, in infusione in 30 minuti. Misurare copeptina 60 minuti dopo l’inizio dell’infusione. |
1-2 ore | Copeptina > 3.8 pmol/L = PP. Copeptina ≤ 3.8 pmol/L = DIC. |
| DI: diabete insipido. DIC: diabete insipido centrale. DIN: diabete insipido nefrogenico. PP: polidipsia primaria. DDAVP: desmopressina. EGA: emogasanalisi. | |||
La figura riporta l’algoritmo diagnostico per la diagnosi differenziale delle sindromi poliuria-polidipsia.
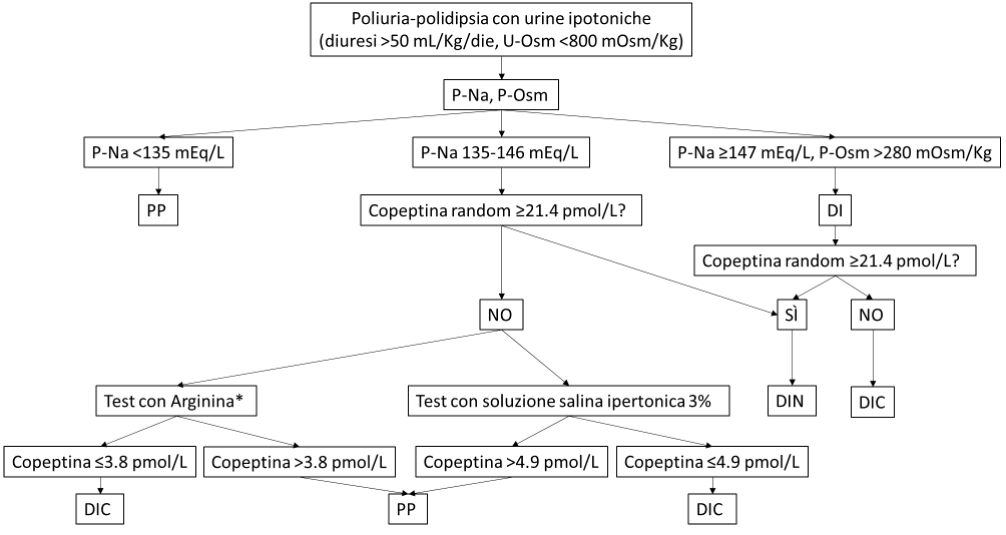
Algoritmo diagnostico per la diagnosi differenziale della poliuria-polidipsia (3)
*In attesa di validazione (studio CARGOx study, NCT03572166, concluso, in attesa di pubblicazione).
U-Osm: osmolalità urinaria. P-Na: sodiemia plasmatica. P-Osm: osmolalità plasmatica. PP: polidipsia primaria. DI: diabete insipido. DIN: diabete insipido nefrogenico. DIC: diabete insipido centrale.
PREDIZIONE DEL DIABETE INSIPIDO POST-CHIRURGICO
Diversi studi hanno valutato l’impiego di copeptina per predire lo sviluppo di DIC dopo chirurgia della regione ipotalamo-ipofisaria (1). Due studi hanno individuato un cut-off di 2.5 pmol/L in prima giornata post-intervento come diagnostico di DIC (area sotto la curva ROC 0.72) (10,11). Altri studi hanno provato a valutare i livelli di copeptina con tempistiche diverse (1 ora dopo l’estubazione, seconda giornata post-operatoria, 2-3 mesi dopo l’intervento) con risultati discordanti (1). Sono quindi necessari ulteriori studi per validare l’impiego di copeptina in questo ambito.
IPONATREMIA
Nella diagnostica differenziale delle iponatremie ipotoniche è fondamentale differenziare forme:
- euvolemiche: sindrome da inappropriata anti-diuresi (SIAD), PP, uso di diuretici;
- ipovolemiche: disidratazione da perdita renale o extra-renale;
- ipervolemiche; stati edematosi (es. scompenso cardiaco, cirrosi epatica).
I valori di copeptina sono tendenzialmente più elevati nelle forme ipo- e ipervolemiche rispetto ai pazienti con SIAD, ma con ampia sovrapposizione tra le tre condizioni (12).
La misurazione di copeptina sembrerebbe avere un modesto valore diagnostico nell’identificazione dei pazienti con PP (< 3.9 pmol/L, specificità 91% e sensibilità 58%) e delle forme ipovolemiche (> 84 pmol/L, specificità 90% e sensibilità 23%).
Oltre al valore di copeptina, un altro parametro che è stato valutato per l’identificazione della SIAD è il rapporto tra copeptina e sodio urinario: valori < 0.3 pmol/mmol hanno dimostrato sensibilità del 61% e specificità del 60% (12,13).
Ad oggi, i parametri che hanno riportato il maggiore potere diagnostico per SIAD rimangono comunque la sodiuria su urine spot, la frazione di escrezione dell’urea e la frazione di escrezione dell’acido urico (13). Nei pazienti con PP, una U-Osm < 200 mOsm/kg può aiutare nella diagnosi (specificità 87% e sensibilità 97%) (12,13).
Infine, quando applicata ai soli pazienti con SIAD, la misurazione di copeptina non sembra utile nella differenziazione tra forme paraneoplastiche e altre cause (14).
Complessivamente, allo stato attuale non c’è indicazione alla misurazione di copeptina nella diagnostica differenziale dell’iponatremia ipotonica.
BIBLIOGRAFIA
- Mu D, Ma C, Cheng J, et al. Copeptin in fluid disorders and stress. Clin Chim Acta 2022, 529: 46–60.
- Martino M, Arnaldi G. Copeptin and stress. Endocrines 2021, 2: 384–404.
- Christ-Crain M, Winzeler B, Refardt J. Diagnosis and management of diabetes insipidus for the internist: an update. J Intern Med 2021, 290: 73–87.
- Fenske W, Quinkler M, Lorenz D, et al. Copeptin in the differential diagnosis of the polydipsia-polyuria syndrome - revisiting the direct and indirect water deprivation tests. J Clin Endocrinol Metab 2011, 96: 1506–15.
- Timper K, Fenske W, Kühn F, et al. Diagnostic accuracy of copeptin in the differential diagnosis of the polyuria-polydipsia syndrome: a prospective multicenter study. J Clin Endocrinol Metab 2015, 100: 2268–74.
- Fenske W, Refardt J, Chifu I, et al. A copeptin-based approach in the diagnosis of diabetes insipidus. N Engl J Med 2018, 379: 428–39.
- Zerbe RL, Robertson GL. A comparison of plasma vasopressin measurements with a standard indirect test in the differential diagnosis of polyuria. N Engl J Med 1981, 305: 1539–46.
- de Fost M, Oussaada SM, Endert E, et al. The water deprivation test and a potential role for the arginine vasopressin precursor copeptin to differentiate diabetes insipidus from primary polydipsia. Endocr Connect 2015, 4: 86–91.
- Winzeler B, Cesana-Nigro N, Refardt J, et al. Arginine-stimulated copeptin measurements in the differential diagnosis of diabetes insipidus: a prospective diagnostic study. Lancet 2019, 394: 587–95.
- Winzeler B, Zweifel C, Nigro N, et al. Postoperative copeptin concentration predicts diabetes insipidus after pituitary surgery. J Clin Endocrinol Metab 2015, 100: 2275–82.
- Vanasuntorn A, Hansasuta A, Chailurkit LO, Sriphrapradang C. Postoperative copeptin as a biomarker for development of diabetes insipidus following hypothalamic-pituitary surgery. Endocr Pract 2021, 27: 463–70.
- Fenske W, Störk S, Blechschmidt A, et al. Copeptin in the differential diagnosis of hyponatremia. J Clin Endocrinol Metab 2009, 94: 123–9.
- Nigro N, Winzeler B, Suter-Widmer I, et al. Evaluation of copeptin and commonly used laboratory parameters for the differential diagnosis of profound hyponatraemia in hospitalized patients: ‘The Co-MED Study’. Clin Endocrinol (Oxf) 2017, 86: 456–62.
- Winzeler B, Steinmetz M, Refardt J, et al. Copeptin is not useful as a marker of malignant disease in the syndrome of inappropriate antidiuresis. Endocr Connect 2020, 9: 20–7.
Analisi genetica nelle malattie ipotalamo-ipofisarie
Maria Rosaria Ambrosio
Sezione di Endocrinologia, Dipartimento di Scienze Biomediche e Terapia Avanzate Università degli Studi di Ferrara
(aggiornato al 30 gennaio 2016)
I tumori ipofisari familiari sono rari (5% degli adenomi ipofisari) e possono rientrare in varie sindromi: neoplasie endocrine multiple di tipo 1 (MEN-1), Carney Complex (CNC), neoplasie endocrine multiple di tipo 4 (MEN-4), sindrome feocromocitoma-paraganglioma (PHEO-PGLs). Esistono inoltre gli adenomi ipofisari familiari isolati (FIPA), forme familiari di adenomi ipofisari non associate ad altre patologie endocrine. Inoltre, è stata recentemente descritta una sindrome di acrogigantismo legata al cromosoma X, con tipica insorgenza in età infantile.
| Patologia | Gene | Cromosoma | Proteina | Trasmissione | Manifestazioni |
| MEN-1 | MEN-1 | 11q13 | Menina | AD | Iperparatiroidismo primario, neoplasie neuroendocrine gastro-entero-pancreatiche |
| Carney | PRKAR1A | 17q22-24 | Subunità regolatoria 1-α della protein kinasi A | AD | Lesioni cutanee pigmentate, mixomi, schwannomi, altre manifestazioni endocrine (tra cui PPNAD) |
| MEN-4 | CDKN1B (+ altri geni codificanti per inibitori di kinasi ciclino-dipendenti) | 12p13 | p27 (+ p15, p18, p21, …) | AD | MEN-like |
| PHEO/PGLs | SDHA – SDHB- SDHC – SDHD – VHL |
vari | Subunità della succinato-deidrogenasi | AD | Feocromocitoma/ paraganglioma |
| FIPA | AIP | 11q13 | Aryl hydrocarbon receptor interacting protein | AD |
L’analisi genetica del DNA per la ricerca di mutazioni è oggi disponibile in diversi laboratori. Tale analisi ha diversi scopi:
- stabilire la diagnosi di una sindrome a trasmissione genetica nel paziente affetto da una determinata patologia;
- identificare nel soggetto malato una mutazione, così da ricercarla anche nei suoi familiari;
- identificare tra i familiari del soggetto malato chi è portatore della mutazione del gene;
- diagnosi prenatale.
Il ruolo dell’analisi genetica in queste sindromi non è ancora ben chiaro, in particolare per la scarsità di dati che mostrino come l’identificazione preclinica della MEN-1 porti ad interventi in grado di migliorarne la morbilità e la mortalità. Tuttavia, l'esame genetico per la ricerca di mutazioni:
- per MEN-1 e Carney Complex è raccomandato per identificare i portatori in cui deve essere eseguita sorveglianza clinica e biochimica;
- per MEN-4 deve essere riservato ai casi con manifestazioni endocrine multiple, ma negativi all'analisi del gene MEN-1;
- per PHEO-PGL eve essere eseguita solo nei casi di coesistenza di tumore ipofisario e feocromocitoma;
- per AIP può essere eseguita nei pazienti con adenomi familiari, se negativi per MEN-1 e Carney complex, e con adenomi ipofisari sporadici quando portatori di macroadenomi in giovane età.
Bibliografia
- Brandi ML, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001, 86: 5658-71.
- Vasilev V, et al. Familial pituitary tumor syndromes. Endocr Pract 2011, 17 Suppl 3: 41-6.
- Fukuoka H, Takahashi Y. The role of genetic and epigenetic changes in pituitary tumorigenesis. Neurol Med Chir 2014, 54: 943-57.
- Beckers A, et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer 2015, 22: 353-67.
Anticorpi anti-ipofisi
Maria Rosaria Ambrosio1 & Romolo Dorizzi2
1Sezione di Endocrinologia, Dipartimento di Scienze Biomediche e Terapia Avanzate Università degli Studi di Ferrara
2Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Il dosaggio degli anticorpi diretti contro antigeni citoplasmatici di cellule ipofisarie può essere utile nella diagnostica differenziale delle masse ipofisarie; tali anticorpi sono stati, infatti, riscontrati nel 25% dei pazienti con accertata ipofisite linfocitaria (e occasionalmente anche in donne con sindrome di Sheehan studiate a 5 anni dal parto).
La determinazione degli Ab anti-ipofisi trova ancora molteplici difficoltà metodologiche ed interpretative:
- in particolare la loro frequenza tende a decrescere con il progredire della malattia, per cui il momento della loro ricerca potrebbe condizionarne in modo critico l’identificazione
- le cellule ACTH-secernenti sono in grado di fissare in maniera aspecifica le immunoglobuline umane potendo dare vita a falsi positivi
- il modo più preciso di identificare tali auto-anticorpi è quello di usare ipofisi umane fetali o linee cellulari ipofisarie in doppia immunofluorescenza
- la difficoltà di identificare ed isolare i vari autoantigeni ha impedito sinora lo sviluppo di metodiche standardizzate e più pratiche per la loro determinazione.
In circa il 50% dei pazienti con ipofisite linfocitaria possono essere presenti altri autoanticorpi organo-specifici, indicatori del coinvolgimento autoimmune di altri distretti corporei.
Bibliografia
- Betterle C. Le malattie autoimmuni. Piccin Editore, 2001.
Imaging neuroradiologico ipotalamo-ipofisario
Alessandro Bozzao, Serena Palizzi, Valentina Frezza, Sara De Giorgi
Sapienza Università di Roma, Facoltà di Medicina e Psicologia, Dipartimento NESMOS (Neuroscienze, Salute Mentale, Organi di Senso)
UOC Neuroradiologia, AO "Sant'Andrea", Roma
(aggiornato al novembre 2022)
ANATOMIA NEURORADIOLOGICA DELLA REGIONE SELLARE
L’ipofisi è una ghiandola endocrina fondamentale per l’omeostasi del nostro organismo. È alloggiata nel basi-cranio, sul fondo della sella turcica (fig 1), all’interno di un recesso denominato fossetta ipofisaria.
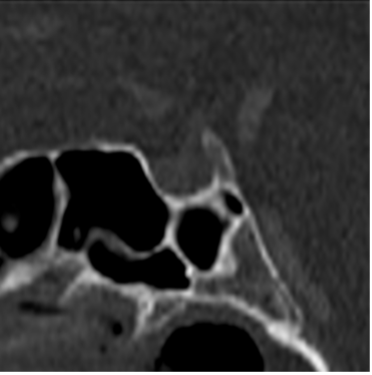
Figura 1
Sella turcica (sezione sagittale TC con MdC)
La sella turcica è una cavità ossea, che costituisce la porzione superiore del corpo dell'osso sfenoide, un osso impari e mediano del neuro-cranio. Nella sua parte anteriore, la sella è delimitata da un rilievo trasversale, detto tuberculum sellae, e latero-anteriormente dai processi clinoidei anteriori. Davanti al tubercolo della sella si trova il solco pre-chiasmatico, che accoglie l'omonima struttura nervosa. Il solco si prolunga lateralmente fino ai fori ottici, punti di passaggio dei nervi ottici e delle arterie oftalmiche. Il margine posteriore della sella turcica è segnato dalla lamina quadrilatera o dorso della sella, un rilievo di forma quadrangolare, il cui margine libero termina lateralmente con i processi clinoidei posteriori e che prosegue posteriormente unendosi alla base dell'occipitale, formando il clivus (1,2).
Anteriormente alla sella turcica si trova il seno sfenoidale, soggetto a una considerevole variabilità di dimensione, forma e pneumatizzazione. Il seno sfenoidale è attraversato da setti, assai variabili come numero, forma, direzione, spessore. La loro valutazione può essere oggetto di studio mediante tomografia computerizzata (TC) in caso di pianificazione per intervento per via trans-sfenoidale (fig 2).
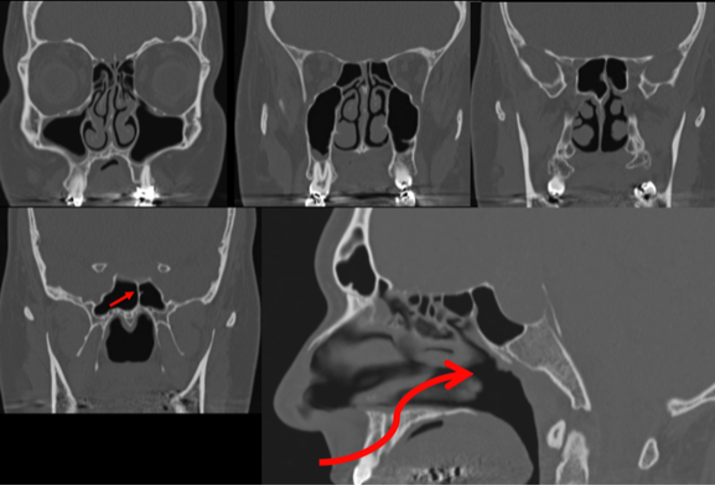
Figura 2
Sezione TC sagittale e coronale
In questi casi è importante anche valutare l’integrità della parete del seno sfenoidale, che può essere molto sottile o assente e consentire all’arteria carotide interna di aggettare direttamente verso il seno protetta da sola mucosa. Questa condizione è ovviamente estremamente pericolosa per la possibilità di lacerare l’arteria durante un intervento per via trans-sfenoidale (fig 3) (3).
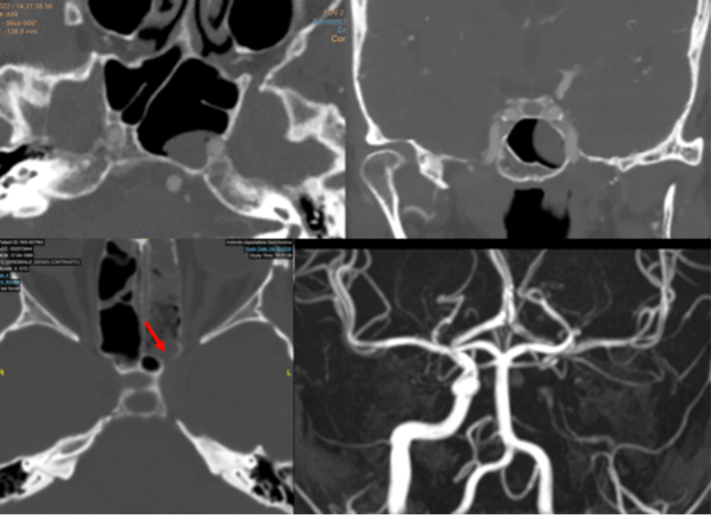
Figura 3
Variante anatomica con procidenza della carotide interna nel seno sfenoidale
In basso le immagini post-operatorie con l’occlusione dell’arteria carotide interna
I canali ottici protrudono nella porzione supero-laterale del seno sfenoidale, mentre la II branca del trigemino protrude nella sua parte infero-laterale. Il recesso ottico-carotideo è situato lateralmente e superiormente, fra canale ottico e protuberanza carotidea (4).
Lateralmente alla sella turcica si trovano i seni cavernosi (fig 4), che contengono sangue venoso. All'interno di essi decorre la porzione orizzontale o cavernosa della carotide interna e un segmento del VI nervo cranico, mentre il III, il IV e la I branca del V nervo cranico si trovano nel tetto e nella parete laterale del seno cavernoso (fig 4).
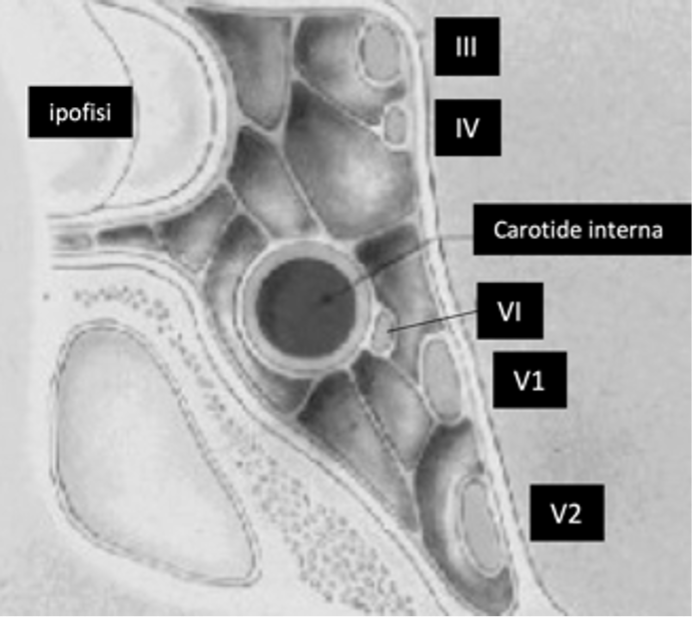
Figura 4
Anatomia del seno cavernoso in proiezione coronale
La parete mediale del seno cavernoso è costituita da dura sottile, di spessore variabile ma normalmente più sottile nella porzione posteriore del seno, sede in cui, infatti, si ritiene più probabile l’infiltrazione del seno da parte dei tumori ipofisari (5). La distanza fra arteria carotide e faccia laterale dell'ipofisi in condizioni normali varia fra 1 e 3 mm. Vista dall'alto la parete laterale del seno cavernoso si estende dalla fessura orbitaria superiore anteriormente, fino all'apice della porzione petrosa dell'osso temporale, posteriormente. Il III nervo cranico entra nel seno dal tetto, lateralmente al dorso sellare, il IV entra in posizione più posteriore e laterale, la branca oftalmica del V entra dalla parte inferiore della parete laterale e il VI entra dalla parete posteriore del seno fra carotide medialmente e III nervo lateralmente (1,2,4). Il VI nervo cranico è l’unico che decorre all’interno del seno (fig 4).
Superiormente il tetto della sella è costituito dal diaframma sellare, un setto orizzontale durale, che divide la loggia sellare dalla regione sovra-sellare. Nella sua porzione centrale è presente uno iato per il passaggio del peduncolo ipofisario, struttura che connette l’ipotalamo all’ipofisi. Il diaframma è più sottile attorno al peduncolo e la sua apertura centrale è talvolta più ampia di quanto necessario per il passaggio del peduncolo. In questi casi può essere presente un'invaginazione dell'aracnoide sovra-sellare.
La sella è internamente rivestita da periostio (2).
ANATOMIA DELLA REGIONE SOVRA-SELLARE
La regione sovra-sellare ha limiti anatomici meno precisi ed è delimitata anteriormente dal tubercolo sellare, inferiormente dal diaframma sellare, posteriormente dalla membrana di Lillequist, superiormente dai nervi ottici, dal chiasma e dal pavimento del III ventricolo e lateralmente dalle carotidi interne.
La membrana di Lillequist è una membrana aracnoidea, che decorre fra il dorso della sella e la faccia anteriore dei corpi mammillari e separa la cisterna chiasmatica dalla cisterna inter-peduncolare. La membrana non è visualizzabile all’imaging.
Il III ventricolo è una struttura impari e mediana, localizzata superiormente alla sella ed è in stretto rapporto con il poligono di Willis e con il sistema venoso profondo del cervello. A livello del pavimento del III ventricolo troviamo l'infundibolo (fig 5), una struttura imbutiforme localizzata fra il chiasma e il tuber cinereum, connessa all’ipofisi tramite gli assoni infundibolari che raggiungono il lobo posteriore dell'ipofisi. Il tuber cinereum si fonde nell'infundibolo (2).
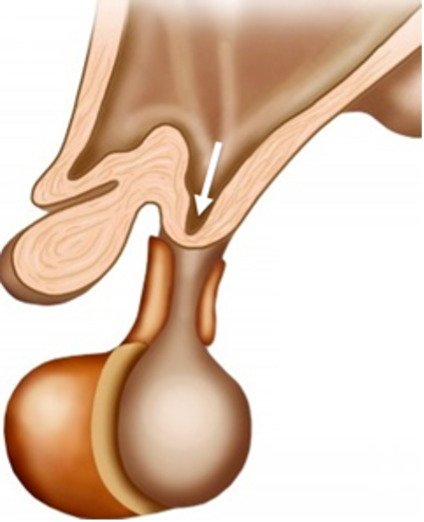
Figura 5
Infundibolo (freccia)
L’ipotalamo (fig 6) costituisce la parte ventrale del diencefalo, localizzata intorno al III ventricolo, di cui costituisce il pavimento e parte delle pareti laterali, tra il chiasma ottico e i peduncoli cerebrali, formando due metà simmetriche che si uniscono in basso per formare il tuber cinereum. L’ipotalamo è sede di diversi nuclei, di cui i principali sono i nuclei sovra-ottico e para-ventricolare, che secernono ossitocina e vasopressina (2,4).
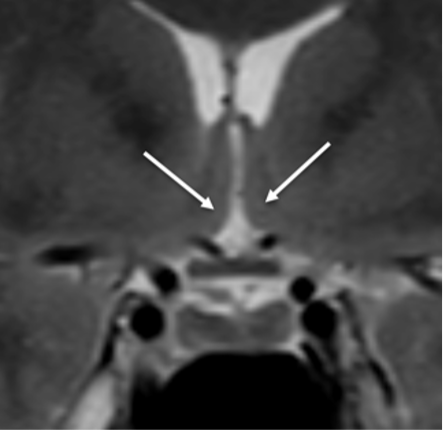
Figura 6
RM in T2 in proiezione coronale: ipotalamo (frecce).
ANATOMIA DELL’IPOFISI
L’ipofisi è una ghiandola di forma ovoidale, costituita da un lobo anteriore (adeno-ipofisi), che avvolge la parte più distale del peduncolo ipofisario, costituendo la pars tuberalis, e da un lobo posteriore (neuro-ipofisi), più aderente all'osso della sella di quanto non sia il lobo anteriore. Poiché il lobo anteriore è separato dal lobo posteriore, la pars tuberalis è più frequentemente inserita nel lobo posteriore (2).
L’ipofisi ha un volume complessivo di 420 mm3 e il peso di 1 grammo. L’adeno-ipofisi rappresenta circa il 75-80% del volume ghiandolare e avvolge antero-lateralmente la neuro-ipofisi.
L’adeno-ipofisi si sviluppa dall’estroflessione della tasca di Rathke, che origina dall’ectoderma embrionale che avvolge il tetto della cavità orale. È suddivisa in tre parti:
- la pars distalis, anteriore, che ne rappresenta la parte principale;
- la pars intermedia o lobo intermedio, formata da una stretta lamina verticale contigua alla neuro-ipofisi e separata dal lobo anteriore da una fessura, residuo della tasca di Rathke;
- la pars tuberalis o lobo tuberale o infundibolare, adesa alla superficie antero-laterale dell’infundibolo, che circonda a C e con cui concorre a formare il peduncolo ipofisario.
Nell’adeno-ipofisi sono presenti sei tipi cellulari diversi:
- le cellule tireotrope, che secernono il TSH;
- le cellule corticotrope, che secernono l’ACTH;
- le cellule lattotrope, che secernono la PRL;
- le cellule somatotrope, che secernono il GH;
- le cellule gonadotrope, che secernono le gonadotropine;
- le cellule follicolo-stellate, che potrebbero rappresentare cellule staminali ipofisarie e la cui funzione sembra importante per la secrezione di fattori di crescita e citochine e per mantenere i corretti rapporti (e quindi l’equilibrio paracrino) fra i diversi tipi cellulari.
Le cellule corticotrope e tireotrope tendono a raggrupparsi insieme nelle zone più centrali della ghiandola, mentre le cellule somatotrope si distribuiscono nelle porzioni più laterali e le cellule gonadotrope, lattotrope e follicolo-stellate sono diffusamente sparse nel parenchima adeno-ipofisario (1,2,4).
La neuro-ipofisi (fig 7) origina da un’estroflessione del pavimento del terzo ventricolo ed è quindi di derivazione neuro-ectodermica. È costituita principalmente (75%) dalla parte terminale degli assoni delle cellule che secernono vasopressina e ossitocina, i cui corpi cellulari sono contenuti nell’ipotalamo (nei nuclei sopra-ottico e para-ventricolare). La residua neuro-ipofisi (25%) è composta dai pituiciti (cellule gliali) e da vasi sanguigni (1).
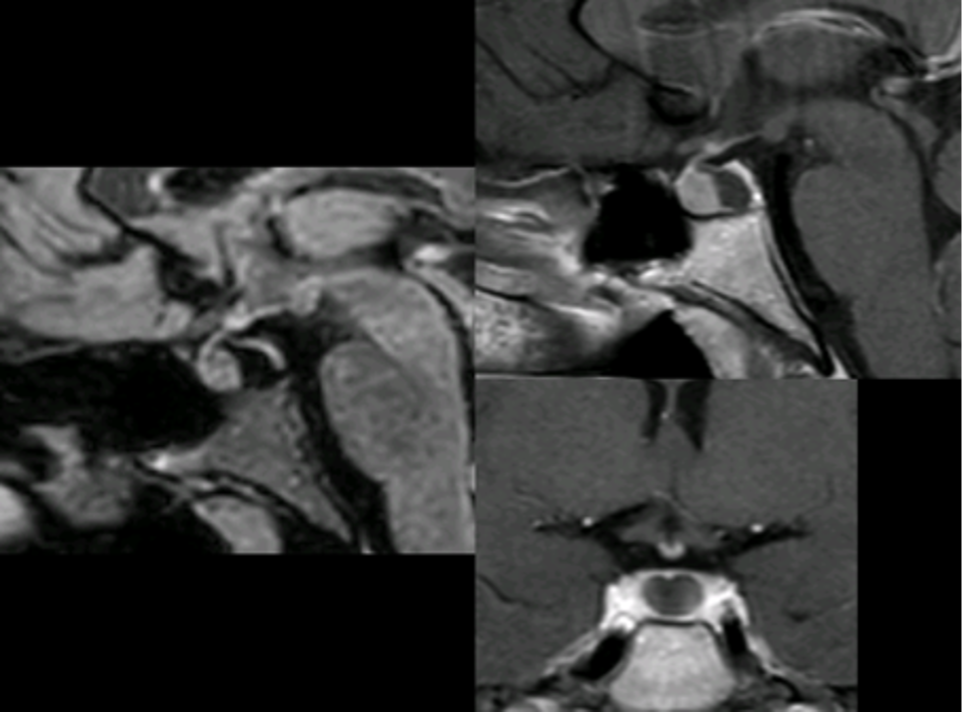
Figura 7
RM sagittali (T2 FLAIR a sinistra e T1 con gadolinio in alto a destra) e coronale T1 post gadolinio (in basso a destra): cisti della pars intermedia
Talvolta l’adeno-ipofisi e la neuro-ipofisi non si fondono correttamente e si formano cisti sulla linea mediana che divide le due parti (cisti della pars intermedia). Queste cisti sono spesso reperti occasionali nel corso degli esami diagnostici e possono entrare in diagnosi differenziale con i micro-adenomi o con le cisti della tasca di Rathke (fig 7).
Il peduncolo ipofisario origina dall'eminenza mediana ipotalamica e si porta in avanti e in basso, attraverso il diaframma sellare, fino a raggiungere l'ipofisi. È costituito da assoni neuronali e da sinusoidi del sistema portale ipotalamo-ipofisario, che ne occupano la porzione anteriore (6).
VASCOLARIZZAZIONE DELLA REGIONE SELLARE E PARA-SELLARE
Il sistema vascolare ipotalamo-ipofisario ha un significato prettamente funzionale, e permette all'ipotalamo di controllare l'ipofisi anteriore senza un collegamento diretto di tipo neuronale: le arterie del peduncolo ipofisario formano un sistema di capillari, sui quali terminano gli assoni dei neuroni neuro-secernenti dell'ipotalamo, che scaricano i propri fattori ormonali nei suddetti vasi. Dalle venule del peduncolo ipofisario, il sangue passa alle venule dell'adeno-ipofisi, le cui cellule ricevono i fattori appena immessi nel sangue. Questo decorso vascolare è ben evidente nelle RM dinamiche e appare con un “gomitolo” detto “tuft sign” alla base del peduncolo durante le prime fasi della sequenza (fig 8) (7). Il sangue infine raggiunge le vene dei seni cavernosi.
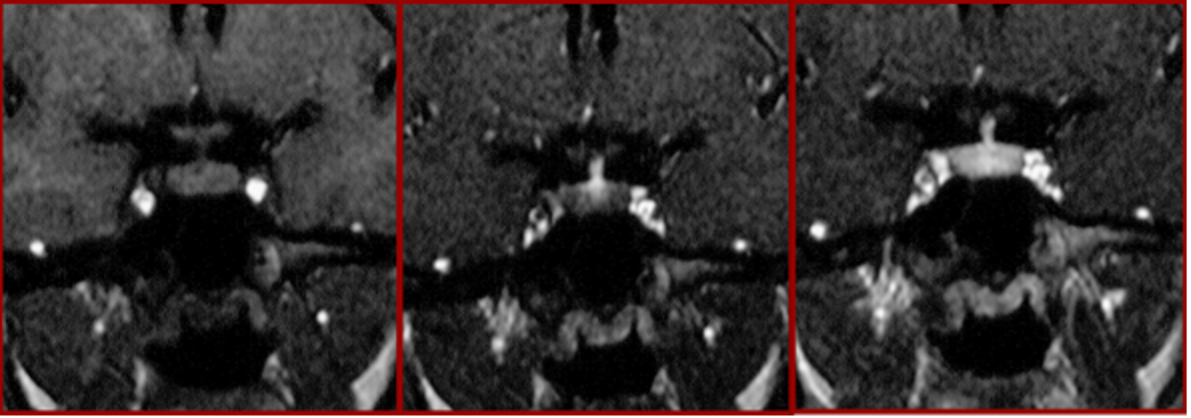
Figura 8
RM dinamica: nell’immagine centrale il segno del gomitolo
La ghiandola ipofisaria riceve sangue arterioso, prevalentemente nella sua porzione posteriore, da rami superiori e da uno inferiore, che originano, prevalentemente, dalla carotide interna (8). I rami arteriosi ipofisari superiori originano dalla porzione sopra-clinoidea (C6) della carotide interna e alcuni, più piccoli, dalle arterie cerebrali anteriore e posteriore; si portano a formare un anello intorno al peduncolo infundibolare ipofisario, e irrorano l'eminenza mediana, la pars tuberalis adeno-ipofisaria e il peduncolo stesso. Il ramo arterioso ipofisario inferiore origina dalla porzione cavernosa (C4) della carotide interna e irrora la pars nervosa della neuro-ipofisi. La pars distalis adeno-ipofisaria risulta perciò priva di vascolarizzazione diretta di sangue arterioso, ma è provvista di un sistema portale ipotalamo-ipofisario, che fornisce una comunicazione ormonale diretta tra l'ipotalamo sovrastante e l'adeno-ipofisi. Nell'eminenza mediana, le arterie ipofisarie superiori si riducono a capillari fenestrati, che vanno a formare il plesso capillare primario. Da questo plesso si formano le vene portali ipofisarie, che scendono lungo la pars tuberalis e, raggiunta la pars distalis adeno-ipofisaria, vi costituiscono il plesso capillare secondario. Questa rete vascolare trasporta i releasing hormones (RH) prodotti dall'ipotalamo e permette all'adeno-ipofisi di riceverli in breve tempo, instaurando una comunicazione diretta. È opportuno ricordare che l’arteria ipofisaria inferiore o meningo-ipofisaria può avere rami anastomotici diretti, tramite le arterie che irrorano la II e III branca trigeminale, con l’arteria meningea media, ramo della carotide esterna. Queste anastomosi, ben conosciute, possono essere estremamente pericolose durante procedure di embolizzazione di patologie vascolari (tipicamente le fistole durali) che vengano irrorate dall’arteria meningea media.
Il drenaggio venoso di neuro-ipofisi e adeno-ipofisi avviene perlopiù attraverso le vene ipofisarie inferiori, che si immettono nei seni durali venosi, soprattutto nel seno cavernoso, e consentono di portare in circolo gli ormoni liberati dalla ghiandola, e quindi ai rispettivi organi bersaglio. Esistono inoltre altre due possibilità di drenaggio della neuro-ipofisi: verso l'adeno-ipofisi e verso l'ipotalamo. Quest'ultima rappresenta una via diretta di regolazione a feed-back negativo sull'ipotalamo, vantaggiosa in termini di tempo perché inibisce la liberazione dei RH, prima che gli ormoni ipofisari compiano l'intero percorso sistemico (8,9). I seni durali venosi che drenano la ghiandola confluiscono nei seni petrosi inferiori che, a loro volta, drenano nel golfo della giugulare (fig 9).
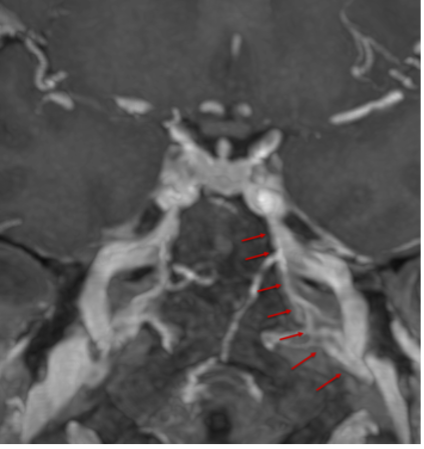
Figura 9
Ricostruzione MIP (proiezione di massima intensità) di RM con mdc per la definizione dei seni petrosi inferiori (frecce)
Esistono ampie varianti anatomiche nella conformazione dei seni petrosi inferiori, che rendono ragione della necessità di eseguire un venogramma diagnostico per definire l’anatomia dei seni prima del loro sampling. Non è infatti sempre definito che il seno petroso inferiore di un lato dreni l’emi-ghiandola corrispondente. Questo non consente quindi di definire con precisione la lateralità di un micro-adenoma secernente (più spesso ACTH). Gli spazi subaracnoidei della regione sellare e para-sellare sono poi attraversati da vasi perforanti che irrorano fra l'altro i nervi ottici, il chiasma, i tratti ottici, le pareti del III ventricolo e l'ipotalamo. Le vene della regione sellare e sovra-sellare sono di piccolo calibro e la regione sovra-sellare è quasi completamente drenata da tributarie delle vene basali (9).
ANATOMIA RADIOLOGICA DELL’IPOFISI
Morfologicamente alla nascita l’ipofisi si presenta globosa e presenta segnale iperintenso (fig 10).
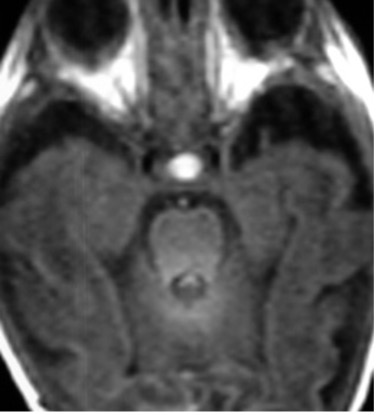
Figura 10
RM assiale in T1: ipofisi neonatale iperintensa
A 2-3 mesi di età la forma e il segnale dell’ipofisi sono simili a quelli dell’adulto. L’altezza globale dell’ipofisi viene misurata sul piano coronale nelle immagini pesate in T1 e varia in funzione di età e sesso. Nei primi 4 anni l’ipofisi misura 2-7 mm di altezza e 3-12 mm di lunghezza. In questa fase della vita può essere clinicamente utile (tipicamente nei deficit di GH) definire anche il volume della ghiandola, cui si può fare riferimento mediante tabelle auxologiche dedicate (6,10).
Nei maschi adulti e nelle donne in menopausa l'altezza dell'ipofisi risulta normale fino a 8 mm. Si registra invece un incremento nella curva dimensionale dell’ipofisi nel sesso femminile in età puberale (fig 11), dove la ghiandola può andare a misurare fino a 8-10 mm. Ovviamente queste ghiandole presentano generalmente segnale omogeneo e normale, sia in T1 che in T2.
Figura 11
RM coronale in T1: ipofisi puberale. È ben evidente la salienza della ghiandola, il cui segnale è omogeneo, nella cisterna sovra-sellare
Nelle donne in allattamento o in gravidanza la ghiandola ipofisaria può misurare fino a 14-15 mm, con profilo superiore convesso (6).
Molte condizioni, oltre a quelle già descritte, possono determinare un aumento fisiologico delle dimensioni della ghiandola. In primo luogo, dobbiamo ricordare le conformazioni anatomiche della sella. Una sella di piccole dimensioni o l’estroflessione dei sifoni carotidei all’interno della sella (kissing carotids) possono determinare una salienza della ghiandola verso la cisterna sovra-sellare (fig 12).
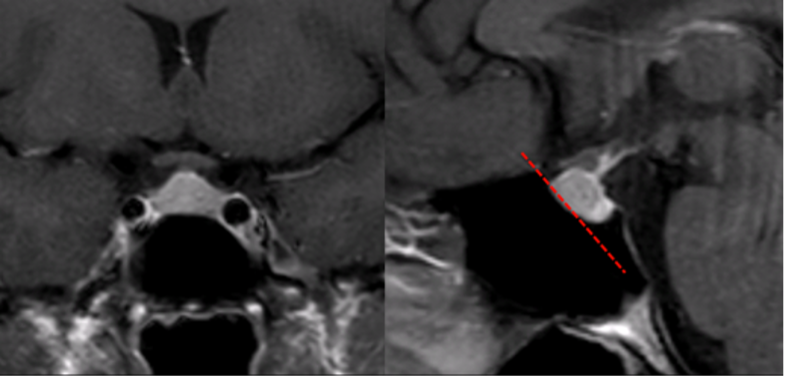
Figura 12
RM coronale (a sinistra) e sagittale (a destra) in T1 post-mdc: cavo sellare “piccolo”, con salienza della ghiandola verso la cisterna sovra-sellare
La ghiandola può aumentare di dimensioni anche nell’ipotensione liquorale, diagnosi che viene agevolmente posta in uno studio dell’encefalo, documentando il tipico scollamento dei foglietti durali. Una ghiandola di grandi dimensioni può anche essere la conseguenza di iperplasia secondaria. Esistono iperplasie tireotrope da grave ipotiroidismo primario e, più rare, nell’Addison e nella s. di Klinefelter, menopausa precoce, amenorrea secondaria iatrogena, con progestinici.
Come detto, la ghiandola ipofisaria presenta caratteristiche di segnale tipiche: con l'eccezione dei neonati, il lobo anteriore ha un'intensità di segnale simile a quella della sostanza bianca in tutte le sequenze, mentre il lobo posteriore si distingue per la sua iperintensità nelle immagini T1 (senza mdc), legata all'accumulo di vasopressina e ossitocina e alla presenza di strutture granulari neuro-secretorie prodotte nell'ipotalamo. Tali granuli non contengono lipidi, quindi non ci sarà un abbattimento del loro segnale nelle sequenze con saturazione del tessuto adiposo (fig 13).
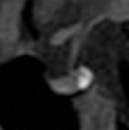
Figura 13
RM sagittale T1 con saturazione del grasso: neuro-ipofisi
È importante non confondere la neuro-ipofisi con il tessuto adiposo del clivus; questo è frequentemente presente e potrebbe mimare, in modo erroneo, la presenza di una neuro-ipofisi normale (fig 14).
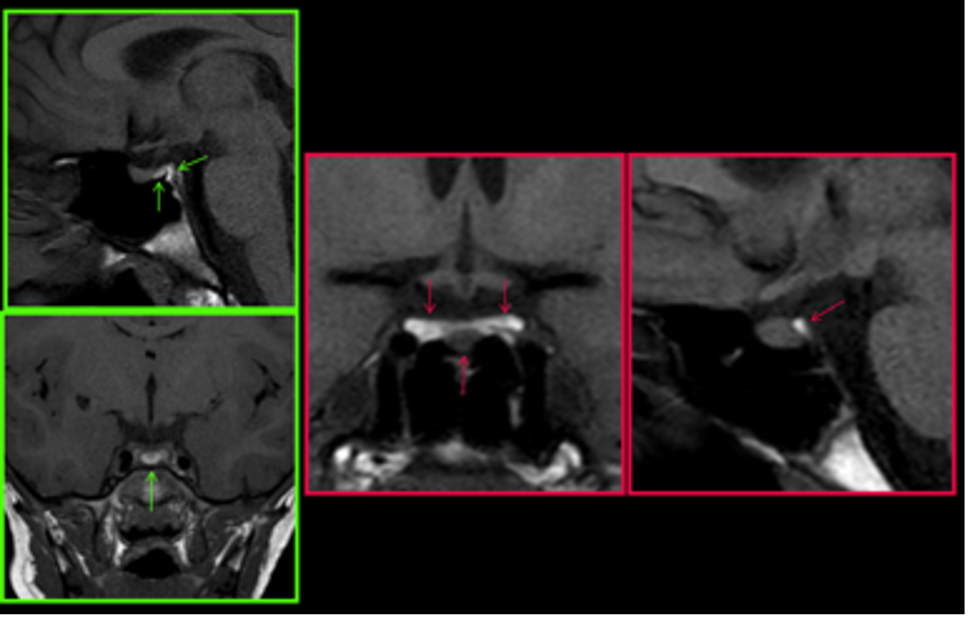
Figura 14
RM sagittali e coronali T1 non saturate
Diagnosi differenziale fra neuro-ipofisi e tessuto adiposo del clivus: con bordo verde ghiandola normale, con bordo rosso mancata visualizzazione della neuro-ipofisi (le frecce indicano il clivus)
Nel dubbio possono essere utili le sequenze con soppressione del tessuto adiposo, che satureranno il segnale del clivus, ma non quello della neuro-ipofisi. L'assenza di rilevabile iperintensità di segnale negli adulti nelle immagini T1 non è un reperto univocamente patologico; può trattarsi di una condizione funzionale rilevata nel 10-20% dei pazienti asintomatici (11).
Dopo somministrazione di mdc paramagnetico, il potenziamento avviene fisiologicamente dall’alto, coinvolgendo il peduncolo e l’infundibolo e, nei secondi successivi, si diffonde omogeneamente all’interno della ghiandola. L’adeno-ipofisi e il peduncolo ipotalamo-ipofisario presentano una marcata impregnazione, per la mancanza della barriera emato-encefalica, appena meno intensa rispetto a quello degli adiacenti seni cavernosi. L'impregnazione della neuro-ipofisi è di minore entità e ovviamente meno evidente proprio per la fisiologica iperintensità nelle immagini T1 senza mdc. Nello studio dinamico l'impregnazione inizia dal peduncolo ipofisario, procede sino al punto di inserzione sulla ghiandola e si estende con andamento centrifugo a tutta l’adeno-ipofisi; entro 60 secondi l'intera ghiandola risulta impregnata (fig 8) (6,11).
L'ipotalamo presenta la stessa intensità di segnale del tessuto encefalico e, a differenza delle strutture ipofisarie, non subisce alcuna impregnazione dopo somministrazione di mezzo di contrasto, ad esclusione dell'eminenza mediana, che rappresenta l'unica porzione ipotalamica sprovvista di barriera emato-encefalica. I diversi nuclei ipotalamici non sono riconoscibili all'esame RM (7).
Il peduncolo ipofisario negli adulti ha una morfologia triangolare, con dimensioni da 3.5 mm (eminenza mediana) a 2.9 mm (zona intermedia) fino a 1.9 mm (vicino alla ghiandola). Nei bambini qualsiasi ingrandimento > 3 mm dovrebbe essere considerato sospetto e segnalato come patologico. In questa ottica possono essere utili le acquisizioni volumetriche T2 (CIS), per misurare esattamente il diametro del peduncolo ipofisario (6,11).
BIBLIOGRAFIA
- Adams RD, Victor M. Principi di neurologia, 5ª ed, McGraw-Hill, 1994, ISBN 88-386-2033-4.
- Anastasi G, et al. Trattato di anatomia umana, 4ª ed, 2007, Edi Ermes, ISBN 88-7051-286-X.
- Liu J, Qu Q, Yang D, et al. Transnasal endoscopic anatomy and approaches to the cavernous sinus. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2013, 48: 901-7.
- Daniel PM. Anatomy of the hypothalamus and pituitary gland. J Clin Pathol Suppl (Assoc Clin Pathol) 1976, 7: 1-7.
- Yilmazlar S, Kocaeli H, Eyigor O, et al. Clinical importance of the basal cavernous sinuses and cavernous carotid arteries relative to the pituitary gland and macroadenomas: quantitative analysis of the complete anatomy. Surg Neurol 2008, 70: 165-74; discussion 174-5.
- Dal Pozzo G. Compendio di risonanza magnetica cranio e rachide, 1ª ed. 2009, UTET SpA, ISBN 978-88-02-05705-7.
- Bonneville JF, Cattin F, Moussa-Bacha K, Portha C. Dynamic computed tomography of the pituitary gland: the "tuft sign". Radiology 1983, 149: 145-8.
- Gibo H, Hokama M, Kyoshima K, Kobayashi S. Arteries to the pituitary. Nihon Rinsho 1993, 51: 2550-4.
- Daniel M, Prichard MM. Observations on the vascular anatomy of the pituitary gland and its importance in pituitary function. Amer Heart J 1966, 72: 147-52.
- Sari S, Sari E, Akgun V, et al. Measures of pituitary gland and stalk: from neonate to adolescence. J Pediatr Endocrinol Metab 2014, 27: 1071-6.
- Colosimo C. Neuroradiologia. 1ª ed, 2014, EDRA LSWR S.p.A., ISBN 978-88-214-2909-5.
DIAGNOSTICA NEURORADIOLOGICA
RISONANZA MAGNETICA
Indicazioni
La Risonanza Magnetica (RM) è l’indagine diagnostica di prima scelta per lo studio dell’ipofisi e delle regioni sellare e para-sellare. In relazione alle dimensioni della ghiandola, gli esami RM dovrebbero essere eseguiti con apparecchiature da 1.5 T o superiori. Sono sconsigliati gli esami con magneti a potenza di campo inferiore.
L’ipofisi è un organo di piccole dimensioni, pertanto la RM, metodica con elevata risoluzione spaziale e di contrasto, è in grado di riconoscere alterazioni strutturali anche molto sottili. Il compito della RM è di definire l’estensione della lesione, i suoi rapporti con le strutture vicine e fornire un’ipotesi diagnostica; è indicata pertanto per individuare la lesione responsabile dell’ipersecrezione.
Tecnica di esecuzione
Lo studio RM inizia con una sequenza rapida di centraggio sui tre piani dello spazio (scout-view). Il protocollo diagnostico standard prevede l’utilizzo di sezioni sottili (non > 3 mm), sequenze TSE pesate in T1 sagittali e coronali e sequenze TSE T2 coronali (fig 15).
| Figura 15 Protocollo diagnostico di base RM ≥ 1.5 T per lo studio dell’ipofisi |
|
| Sagittale TSE-T1 2-3 mm |  |
| Coronale TSE-T1 2-3 mm | 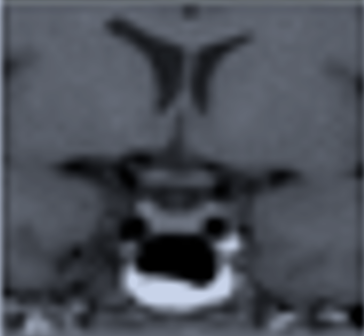 |
| Coronale TSE-T2 2-3 mm |  |
| Coronale TSE-T1 2-3 mm post Gd |  |
| Sagittale TSE-T1 2-3 mm post Gd | 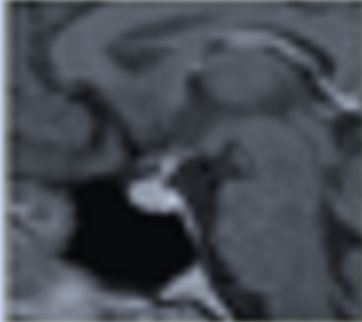 |
In particolare:
- le immagini sagittali permettono una buona valutazione dimensionale della ghiandola, la visualizzazione del peduncolo, delle cisterne sovra-sellari, del chiasma ottico, del tuber cinereum, dei corpi mamillari e del terzo ventricolo;
- le immagini coronali, con inclinazione parallela al peduncolo ipofisario, permettono una valutazione della simmetria dell’ipofisi, del peduncolo e delle strutture anatomiche para-sellari (ad esempio i seni cavernosi);
- le immagini assiali vengono utilizzate esclusivamente in casi selezionati, quando risulta necessario andare a valutare l’estensione para-sellare della lesione.
In genere, risulta necessario ripetere sequenze TSE T1 dopo somministrazione endovenosa di Gadolinio, in particolar modo nella ricerca di micro-adenomi. Il mezzo di contrasto permette una migliore definizione della lesione e della sua estensione.
Gli studi dinamici vengono utilizzati per valutare la vascolarizzazione dell’asse ipotalamo-ipofisario. Si realizzano mediante rapida acquisizione di sequenze pesate in T1 (con risoluzione temporale di 10-20 sec), durante infusione a bolo di mezzo di contrasto. Questa sequenza permette di visualizzare un eventuale ritardo di impregnazione dell’adenoma rispetto al tessuto ghiandolare sano.
Possono essere utilizzate sequenze aggiuntive, in relazione al tipo di lesione osservata:
- le sequenze GRE permettono di evidenziare depositi di calcio e di sostanze para-magnetiche;
- le sequenze FLAIR identificano lesioni a segnale simil-liquorale (dermoidi);
- le sequenze SPIR sopprimono il segnale del grasso e risultano utili nello studio di lipomi, teratomi e nei controlli post-chirurgici;
- le sequenze di diffusione possono evidenziare la presenza di lesioni ad elevata cellularità.
TOMOGRAFIA COMPUTERIZZATA
Indicazioni
La metodica risulta solitamente complementare alla RM, qualora risulti necessario studiare le modificazioni ossee primarie e secondarie indotte dalla lesione o per ricercare calcificazioni o sanguinamenti intra-lesionali. Viene inoltre utilizzata per lo studio dell’anatomia della regione etmoido-sfenoidale nella pianificazione pre-chirurgica. La TC è indicata nei pazienti che non possono sottoporsi a un esame RM (ad esempio portatori di pace-maker, protesi metalliche o soggetti con grave claustrofobia).
Tecnica di esecuzione e indicazioni
L’esame è realizzato tramite acquisizione volumetriche con ricostruzioni multi-planari sui piani ortogonali dello spazio. L'utilizzo del mezzo di contrasto organo-iodato permette una migliore definizione della patologia e anche l'esecuzione di studi dinamici.
PATOLOGIE DELLA REGIONE SELLARE E SOVRA-SELLARE
GHIANDOLA "PICCOLA" E SELLA VUOTA
La sella vuota è un reperto di riscontro molto comune durante l’esecuzione di esami RM del cranio. È legata a un’incontinenza del diaframma sellare con quello che si può definire come un micro-meningocele intra-sellare (fig 16).
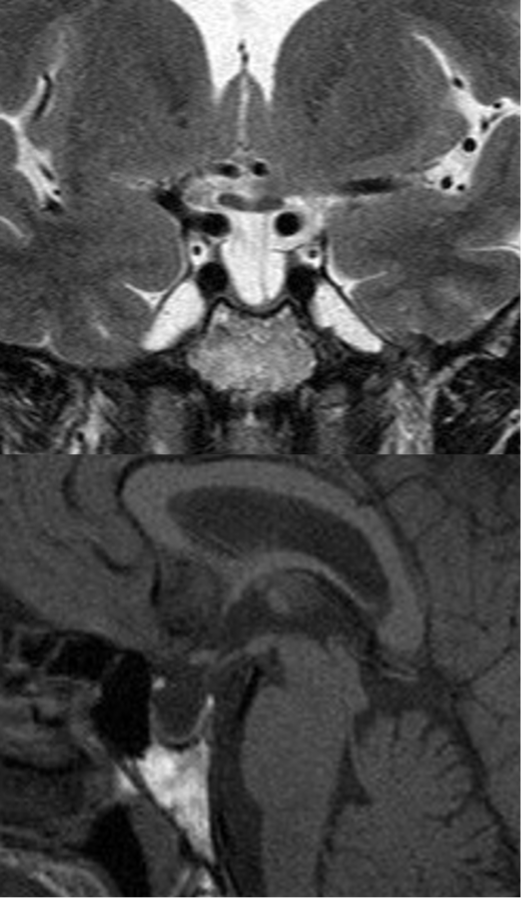
Figura 16
RM coronale T2 (in alto) e sagittale T1 (in basso): sella vuota (micro-meningocele intra-sellare)
È spesso un reperto occasionale, asintomatico, più frequente nelle donne di età > 50 anni. Non necessita di follow-up e l’unica diagnosi differenziale è con le cisti aracnoidee (vedi relativo capitolo).
Diverso è il discorso della cosiddetta sindrome della sella vuota, una condizione di ipertensione liquorale che si associa a cefalea, papilledema e iperprolattinemia per compressione del peduncolo. In questa condizione la sella vuota è espressione di una ipertensione liquorale cronica.
La sella vuota può anche essere la conseguenza di danni della ghiandola, che si possono sviluppare, ad esempio, in caso di apoplessia, nel post-chirurgico, nel post-radioterapia e, più raramente, nei trattamenti prolungati con i dopamino-agonisti.
Non bisogna confondere la sella vuota di un adulto con l’ipoplasia ghiandolare nel bambino, specie se affetto da deficit di GH isolato o da deficit ormonali multipli. Questa condizione è una dei tre possibili riscontri di deficit ormonali in età pediatrica, insieme a una ghiandola normale e alla più grave disconnessione ipotalamo-ipofisaria (fig 17). Questa condizione, nettamente più frequente nei deficit multipli, è caratterizzata dalla presenza di neuro-ipofisi ectopica infundibolare con il tipico segnale in T1. In caso di ipoplasia ghiandolare, è opportuno misurare il volume della ghiandola, che può essere predittivo di una buona risposta con terapia sostitutiva.
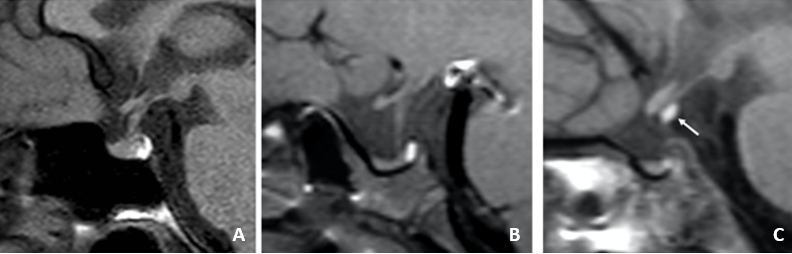
Figura 17
RM sagittali T1: A: ghiandola normale; B: ipoplasia; C: ectopia della neuro-ipofisi. Ognuno di questi tre quadri si può associare a deficit ormonale
Bibliografia
- Mehla S, Chua AL, Grosberg B, Evans RW. Primary empty sella. Headache 2020, 60: 2522-5.
- Chiloiro S, Giampietro A, Bianchi A, De Marinis L. Empty sella syndrome: multiple endocrine disorders. Handb Clin Neurol 2021, 181: 29-40.
LESIONI CISTICHE
Il quadro clinico varia a seconda della localizzazione della cisti, delle dimensioni e della sua eventuale rottura.
Le cisti epidermoidi si trovano comunemente a livello dell'angolo ponto-cerebellare; la seconda localizzazione più comune è la regione para-sellare. Alla RM le cisti epidermoidi appaiono isointense/sfumatamente iperintense nelle sequenze T1 e T2-pesate e non mostrano potenziamento dopo mezzo di contrasto (a differenza dei macro-adenomi).
Le cisti dermoidi sono meno comuni delle cisti epidermoidi. Contrariamente agli epidermoidi, che sono spesso localizzati lateralmente, le cisti dermoidi sono generalmente localizzate sulla linea mediana a livello della fossa cranica posteriore o nella cisterna para-sellare. Radiologicamente appaiono come lesioni ipodense non potenzianti alle scansioni TC. Occasionalmente possono apparire iperdense alla TC a causa della presenza di micro-emorragie (1). In RM le cisti dermoidi appaiono iperintense nelle sequenze T1-pesate e con aspetto variabile nelle sequenze T2-pesate, in assenza di potenziamento post-contrastografico.
Le cisti di Rathke sono gli incidentalomi cistici dell'ipofisi più comuni. Sono lesioni benigne, cistiche, a prevalente localizzazione intra-sellare originate dalla fessura di Rathke nella pars intermedia vestigiale dell'ipofisi. Occasionalmente, hanno una posizione sovra-sellare e possono causare deficit ipofisari o compressione del chiasma ottico o del nervo (2). La diagnosi viene effettuata mediante l'imaging e le caratteristiche cliniche.
In TC la cisti di Rathke appare come una lesione omogenea ipodensa, isodensa o sfumatamente iperdensa. Nel 10-15% dei casi può essere iso-ipodensa, e può contenere piccole calcificazioni curvilinee nella parete. Tendenzialmente non presenta potenziamenti post-contrastografici, ad eccezione della parete, che talvolta può potenziare. In RM la cisti di Rathke appare come una lesione omogenea e ben delimitata, con occasionale potenziamento della parete dopo mdc. L'intensità del segnale in RM è altamente variabile e dipende dal contenuto della cisti:
- una cisti contenente liquido trasparente appare iso-ipointensa in T1 e iperintensa in T2 (fig 18), mentre una cisti contenente materiale mucoide appare iperintensa in T1 e isointensa in T2;
- una cisti contenente sangue presenta segnale iperintenso sia in T1 che in T2 (fig 19).
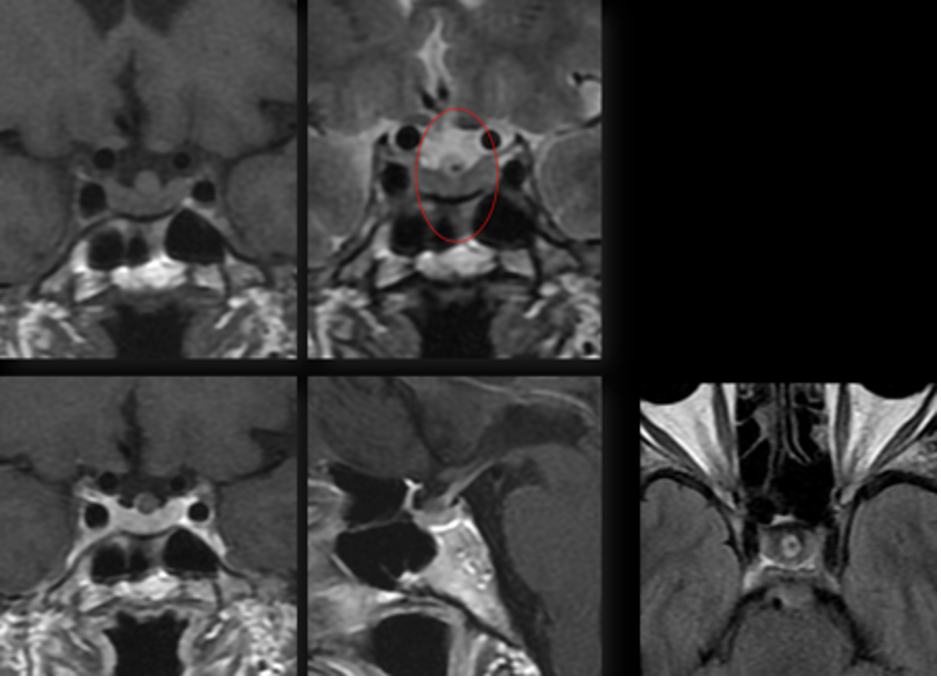
Figura 18
RM in T1 coronale (in alto e a sinistra), sagittale (al centro in basso) e assiale (a destra): cisti della tasca di Rathke con la tipica concrezione ipointensa in T1 (circolo)
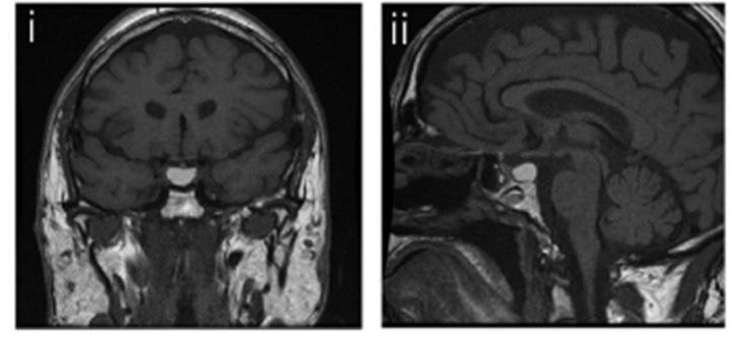
Figura 19
Immagini coronale (i) e sagittale (ii) pesate in T1, che mostrano segnale iperintenso: cisti della tasca di Rathke
Una caratteristica quasi patognomonica per la cisti di Rathke è un nodulo intra-cistico, non potenziante, che mostra bassa intensità in T2 e ipersegnale in T1 (fig 18) (3).
L’ascesso ipofisario, condizione estremamente rara, è una lesione sellare cistica, di solito ad aspetto omogeneo, con segnale ipo o isointenso in T1, iso o iperintensità in T2 e parete che potenzia dopo somministrazione di mdc.
Le cisti aracnoidee, all’esame TC e RM, sono strutture ben circoscritte, con parete scarsamente apprezzabile, che tendono a esercitare effetti compressivi sulle strutture adiacenti, specie sul peduncolo ipofisario (fig 16). Se di grandi dimensioni, nel tempo possono esercitare un effetto rimodellante sulle strutture ossee adiacenti. Se con localizzazione intra-sellare, possono entrare in diagnosi differenziale con la sella vuota (fig 20).
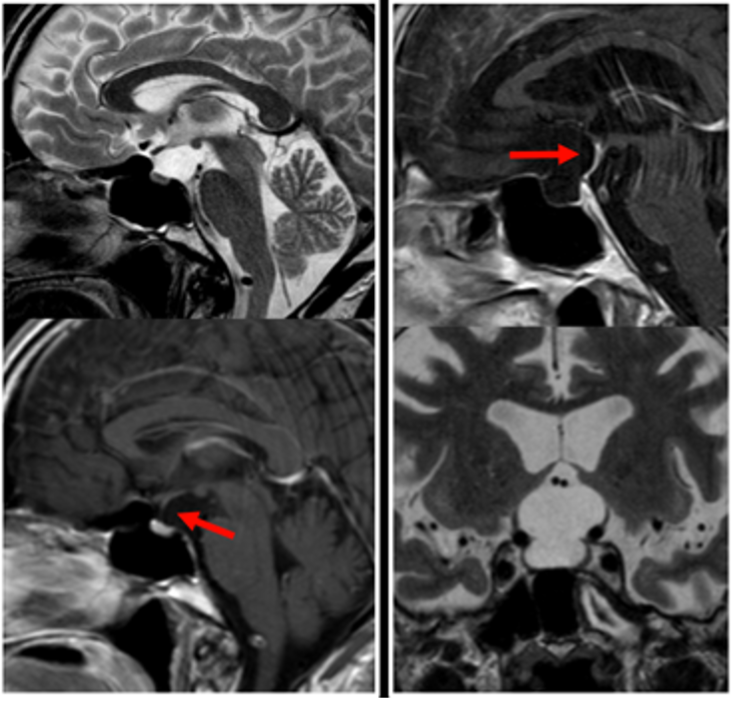
Figura 20
RM sagittali in T2 (in alto a sinistra) e T1 (in basso a sinistra e in alto a destra) e coronale in T2 (in basso a destra): cisti aracnoidee con dislocazione del peduncolo anteriore (in basso a sinistra) e posteriore (in alto a destra)
Bibliografia
- Gao PY, Osborn AG, Smirniotopoulos JG, et al. Radiologic-pathologic correlation. Epidermoid tumor of the cerebellopontine angle. AJNR Am J Neuroradiol 1992, 13: 863-72.
- Shin JL, Asa SL, Woodhouse LJ, et al. Cystic lesions of the pituitary: clinicopathological features distinguishing craniopharyngioma Rathke’s cleft cyst and arachnoid cyst. J Clin Endocrinol Metab 1999, 84: 3972–82.
- Bonneville F, Chiras J, Cattin F, et al. T2 hypointense signal of Rathke cleft cyst. AJNR Am J Neuroradiol 2007, 28: 397.
- Kornienko VN, Pronin IN. Diagnostic neuroradiology. Springer Verlag 2008: ISBN 3540756523.
- Valassi E, Biller BMK, Klibanski A, et al. Clinical features of nonpituitary sellar lesions in a large surgical series. Clin Endocrinol 2010, 73: 798–807.
- Famini P, Maya MM, Melmed S. Pituitary magnetic resonance imaging for sellar and parasellar masses: ten-year experience in 2598 patients. J Clin Endocrinol Metab 2011, 96: 1633–41.
- Spampinato MV, Castillo M. Congenital pathology of the pituitary gland and parasellar region. Top Magn Res Imaging 2005, 16: 269-76.
- Byun WM, Kim OL, Kim D. MR imaging findings of Rathke’s cleft cysts: significance of intracystic nodules. AJNR Amer J Neuroradiol 2000, 21: 485-8.
Micro-adenomi
Sono lesioni ipofisarie intra-sellari con diametro massimo < 10 mm, di gran lunga più frequenti dei macro-adenomi, verso cui progrediscono solo in rari casi (1).
I micro-adenomi si presentano spesso con alcune caratteristiche (2):
- aspetto asimmetrico della ghiandola ipofisaria, con aumento di volume a carico del lobo interessato dalla lesione;
- possibile sotto-slivellamento del pavimento sellare;
- possibile deviazione contro-laterale del peduncolo ipofisario, verso il lobo sano.
È stato osservato che i micro-adenomi presentano spesso una specifica localizzazione in funzione della diversa origine delle cellule secretorie. In particolare, i prolattinomi e i tumori GH-secernenti sono localizzati lateralmente, mentre i tumori secernenti TSH e ACTH si dispongono lungo la linea mediana.
Nelle sequenze T1-pesate, i micro-adenomi appaiono generalmente ipointensi rispetto alla ghiandola sana; tuttavia, un 25% di essi può presentarsi anche come lesione isointensa. Nelle sequenze T2-pesate, i micro-adenomi si presentano come lesioni iperintense in circa l’80% dei casi; la maggior parte dei tumori GH-secernenti appaiono, invece, ipo o isointensi (3).
La somministrazione di mdc accentua, nelle sequenze T1, la differenza di segnale fra la ghiandola sana, che si impregna più rapidamente, e l’adenoma ipofisario, che appare relativamente ipointenso (fig 21). Pertanto, è opportuno acquisire le immagini subito dopo la somministrazione di mdc o con scansioni dinamiche.
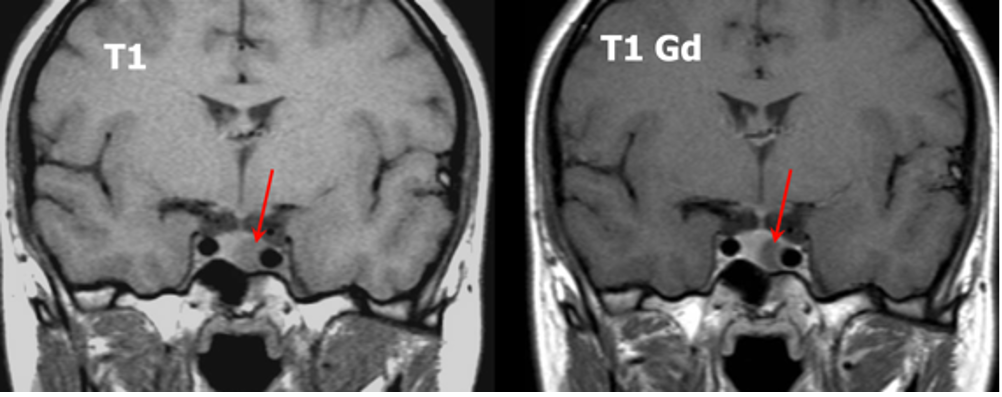
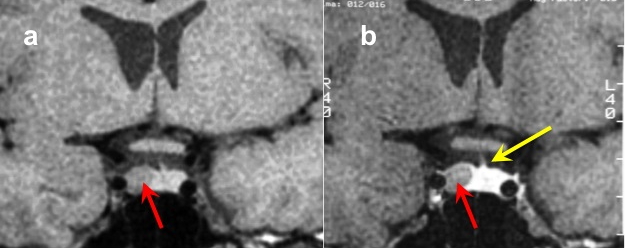
Figura 21
RM coronale T1 senza (a sinistra) e con Gd (a destra) in due diversi pazienti (sopra e sotto, grazie a Regina Barbò per questa immagine). Si evidenzia una massa intra-ipofisaria ben demarcata (freccia rossa), di diametro < 10 mm, ipointensa rispetto all'ipofisi normale. Dopo mdc l'impregnazione del microadenoma è meno rapida di quella dell'ipofisi normale circostante. Notare la deviazione controlaterale del peduncolo ipofisario nelle immagini in basso (freccia gialla)
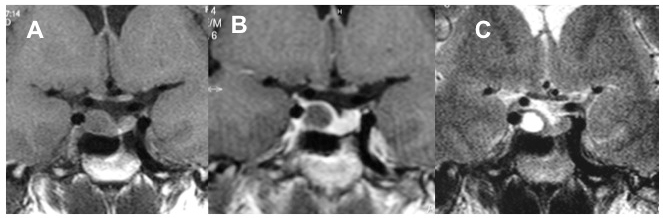
Figura 21bis. RM, sequenze SE, immagini T1, sezioni coronali senza (A) e con mdc (B) e T2 (C). Microadenoma cistico: lesione espansiva del lobo destro dell’adeno-ipofisi, ovalare a contorni definiti, modicamente, uniformemente ipointensa in T1 basale e caratterizzata da franca ipointensità nell’immagine dopo somministrazione di mdc, che condiziona un fisiologico blush del parenchima ipofisario sano. La spiccata, omogenea iperintensità di segnale nell’immagine T2 conferma la struttura cistica dell’adenoma, del tutto verosimilmente secondaria a sanguinamento intra-lesionale (grazie a Regina Barbò)
Circa il 10-30% dei micro-adenomi (specie PRL) vengono individuati esclusivamente nelle sequenze post-contrastografiche e la tecnica dinamica incrementa la sensibilità di un ulteriore 10% (4).
In alcuni casi, soprattutto a seguito di un trattamento farmacologico, nei micro-adenomi si possono sviluppare aree di necrosi o di emorragia, che si presentano come aree di alterato segnale nelle sequenze T1 e T2.
Un discorso a parte meritano i micro-adenomi ACTH-secernenti. Si tratta infatti di lesioni molto piccole, spesso < 3 mm (pico-adenomi), localizzati in sede centrale, con segnale e potenziamento spesso analogo alla ghiandola normale. Le dimensioni dell’adenoma non correlano, peraltro, con i livelli di ACTH prodotto. Se la sensibilità della RM nella diagnosi dei micro-prolattinomi è intorno all’90%, quella degli ACTHomi è del 50-60%, ma aumenta, in alcune casistiche, all’80% con l’impiego di tecniche dinamiche con bassa dose di Gadolinio (0.05 mmol/kg) e acquisizioni volumetriche. Bisogna tuttavia considerare che queste tecniche aumentano l’incidenza di falsi positivi. Alcuni studi hanno proposto l’uso di magneti a 3 T nella diagnosi di micro-adenoma ACTH-secernente, con risultati non superiori al 1.5 T (5). Altri (6) hanno ottenuto migliori risultati, con sensibilità del 72% e specificità del 100%. In particolare, la RM 3 T avrebbe identificato lesioni non visibili con l’1.5 T nel 31% (15/49 casi), ma in 2 di questi non c’è stato un riscontro chirurgico corrispondente (su 5 operati) e in 8 casi la lesione era visibile retrospettivamente anche nello studio a 1.5 T. I veri positivi del 3 T rispetto all’1.5 T erano, alla fine dell’analisi, 7/49 ovvero il 14%. Attualmente, da linee guida, vengono considerate positive solo lesioni > 6 mm in pazienti con dati di laboratorio congrui; in tutte le altre condizioni esiste l’indicazione al cateterismo dei seni petrosi inferiori. Molto recentemente (7), Bonneville ha proposto un algoritmo diagnostico (fig 22) negli ipercortisolismi ACTH-dipendenti, che considera un imaging di base, con l’impiego di sequenze post-contrasto volumetriche sub-millimetriche a 3 T. In caso di sospetti artefatti o incidentalomi, suggerisce la necessità di acquisire immagini assiali T1 con saturazione adiposa e FLAIR post-contrasto tardive, utili alla definizione di una lesione funzionante. In caso di RM normale, sottolinea la necessità dell’impiego di sequenze volumetriche 3D post-Gd e di sequenze FLAIR post-Gd. Solo in caso di ulteriore negatività, consiglia di acquisire una PET/RM e infine il cateterismo dei seni petrosi inferiori.
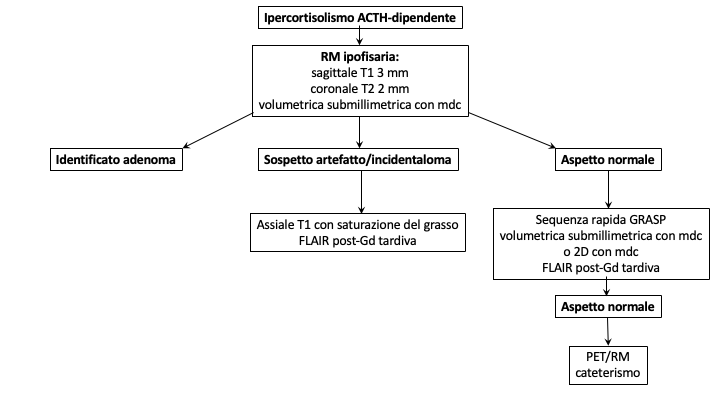 Figura 22
Figura 22
Algoritmo diagnostico micro-adenomi ACTH-secernenti
È chiaro che si tratta di protocolli non ancora applicabili in tutte le strutture, ma sottolineano comunque la necessità di un maggiore sforzo in questo tipo di diagnosi. In un articolo dello stesso anno (8) si sottolinea l’importanza sia dell’aspetto tecnico che dell’esperienza del lettore. In sostanza, i pazienti con ipercortisolismo ACTH-dipendente dovrebbero essere valutati solo in centri di riferimento, dove questi pazienti dovrebbero essere presi in carico.
| Caratteristiche RM micro-adenomi | |
| Sede | Intra-sellare |
| Caratteristiche tipiche | Asimmetria del profilo ghiandolare Slivellamento del pavimento sellare Deviazione controlaterale del peduncolo ipofisario |
| Degenerazione emorragica | Più frequente che in altre lesioni ipofisarie |
| Sequenze T1-pesate | Generalmente ipointenso (isointenso nel 25% dei casi) |
| Sequenze T2-pesate | Iperintenso nell’80%; adenoma GH-secernente ipointenso |
| Potenziamento dopo Gd | Minimo rispetto al resto del parenchima ghiandolare |
Macro-adenomi
Possono presentare un’estensione:
- sovra-sellare: si osserva un aspetto a “pupazzo di neve” per restringimento del macro-adenoma a livello del diaframma sellare; comporta la compressione delle strutture ottico-chiasmatiche (fig 23);
- infero-sellare: verso il seno sfenoidale, con possibili aspetti erosivi sul clivus della sella turcica; tale aspetto è più frequente nei macro-adenomi GH-secernenti;
- laterale o para-sellare: si presenta con il coinvolgimento dei seni cavernosi (9).
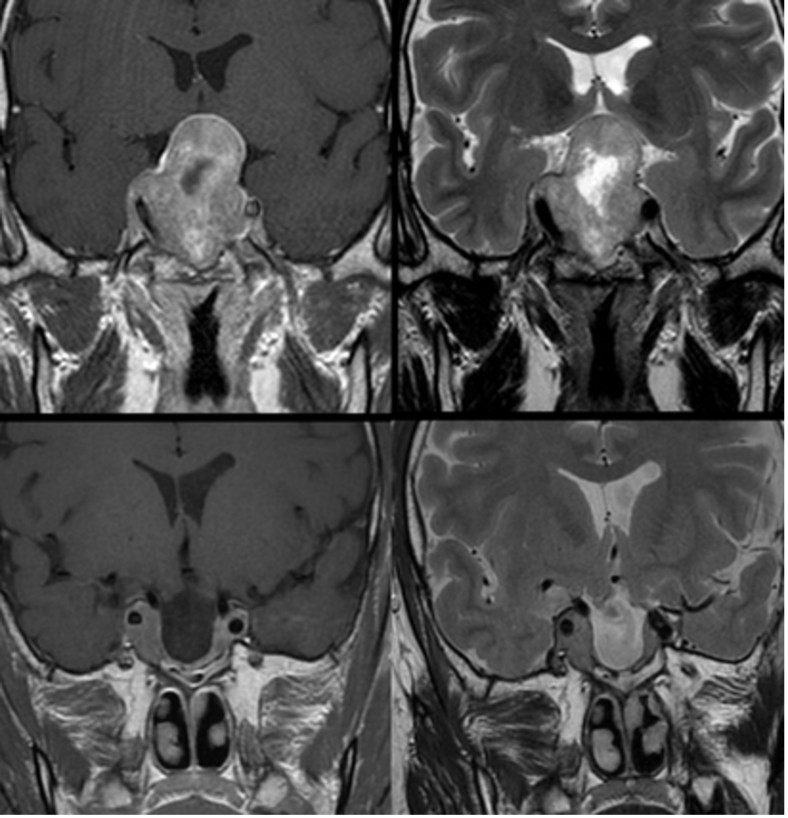
Figura 23
RM coronali in T1 (a sinistra) e T2 (a destra): macro-adenoma ipofisario con morfologia a pupazzo di neve; nelle immagini in basso il post-operatorio
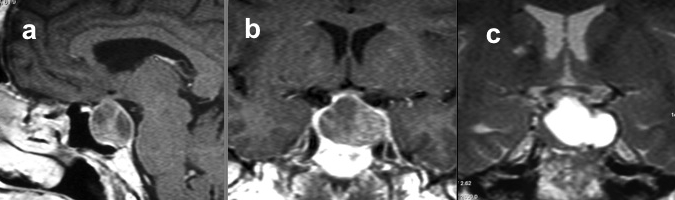
Figura 23 bis. Macroadenoma cistico. Le immagini RM sagittale (a) e coronale (b), T1 dopo mdc, evidenziano una lesione espansiva intra- e sovra-sellare, disomogeneamente ipointensa e circondata da un sottile potenziamento contrastografico periferico, più accentuato al margine laterale destro ed attribuibile al residuo parenchima ipofisario ed al peduncolo, entrambi compressi. L'immagine pesata in T2 (c) documenta la struttura cistica, secondaria a necrosi o sanguinamenti intra-lesionali, della lesione che risulta francamente iperintensa (grazie a Regina Barbò).
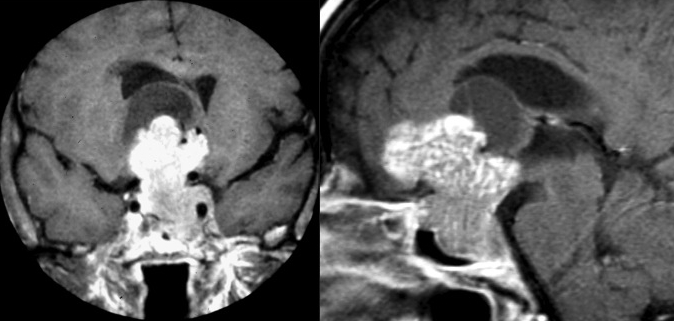
Figura 23 ter. Macroadenoma gigante. Voluminosa lesione espansiva con epicentro in corrispondenza del cavo sellare, che risulta armonicamente ampliato, con depressione del pavimento e sviluppo inferiore intra-sfenoidale, laterale intra-cavernoso e prevalente sovra-sellare. Non stenosi dei tratti intra-cavernosi dei sifoni carotidei. La componente parenchimatosa della lesione presenta un'impregnazione contrastografica moderata e finemente disomogenea, con aspetto"reticolato", mentre la componente cistica polare superiore risulta moderatamente iperintensa rispetto al liquor nelle immagini T1 contrastografiche coronale (a) e sagittale (b). Il macroadenoma gigante comprime il terzo ventricolo ed i forami di Monro, con secondario idrocefalo ostruttivo (grazie a Regina Barbò).
L’infiltrazione del seno cavernoso viene classificata secondo i criteri di Knosp, che definiscono un’invasione circonferenziale del seno fino al completo inglobamento dell’arteria carotide nel seno cavernoso (10,11). Si ritiene che il superamento della linea inter-carotidea esterna e il contatto sulla circonferenza del sifone carotideo siano segni affidabili di infiltrazione del seno (fig 24).
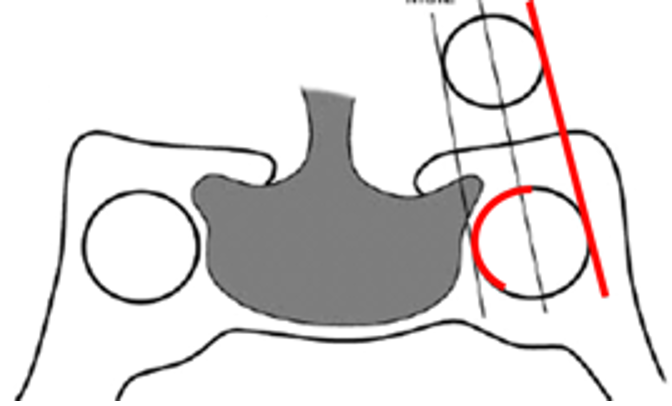
Figura 24
Schema in coronale che mette in rilievo i rapporti dell’ipofisi con il seno cavernoso e le carotidi a sinistra: segni di infiltrazione del seno cavernoso
Nella sua estensione nel seno, il macro-adenoma tende a circondare il tratto cavernoso dell’arteria carotide interna (ACI) senza comprimerlo. La stenosi del lume della ACI è indicativa della presenza di una lesione non adenomatosa, ad esempio di un meningioma (fig 25) (12).
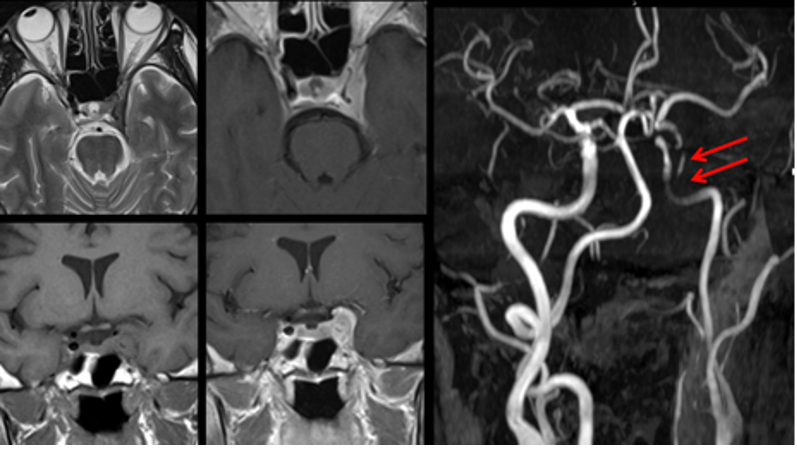
Figura 25
RM assiale T2 (in alto a sinistra), coronale T1 (in basso a sinistra) e assiale e coronale T1 con Gd (al centro): meningioma
Sequenza RM con mdc con ricostruzione MIP (a destra): coinvolgimento carotide interna con stenosi vascolare (frecce)
Del tutto recentemente sono stati proposti segni RM di infiltrazione diretta della dura localizzata tra la ghiandola e il seno, visibili mediante acquisizione di tomogrammi RM ad alta risoluzione con magneti a 3 T (7). Questi reperti devono essere confermati da studi prospettici, ma definiscono una nuova frontiera nella definizione di questa problematica.
Il segnale dei macro-adenomi è solo lievemente più elevato rispetto a quello dei micro-adenomi nelle sequenze T1 ed è generalmente disomogeneo per la presenza di aree di necrosi o emorragia, tanto più frequenti quanto maggiori sono le dimensioni della lesione (13). È importante sottolineare che il segnale dell’adenoma non è predittivo della sua solidità durante gli interventi per via trans-sfenoidale. Solo un tumore con componenti emorragiche può essere considerato soffice in previsione di un intervento chirurgico; in tali casi le aree emorragiche si presentano con un segnale iperintenso nelle sequenze T1-pesate (fig 26). In tale ottica, alcuni lavori sottolineano che un precoce potenziamento della lesione durante le fasi dinamiche contrastografiche può essere espressione di minore consistenza della lesione in virtù della sua vascolarizzazione (14).
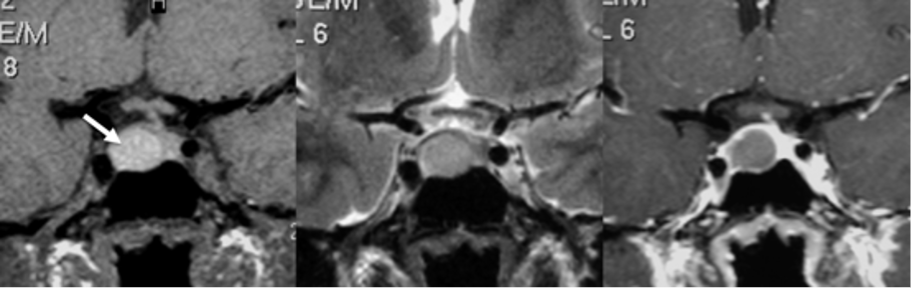
Figura 26
RM coronali T1 senza Gd (a sinistra), T2 (al centro) e T1 con Gd (a destra): macro-adenoma emorragico (freccia)
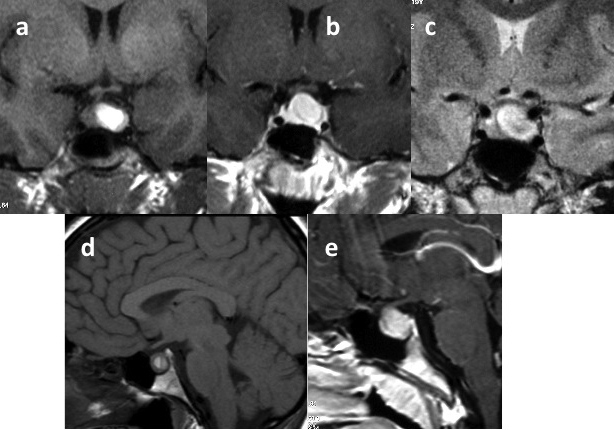
Figura 26 bis. Emorragia intra-adenoma. Adenoma ipofisario con area di elevato segnale(a,c) e "livello" tra componente fluida e corpuscolata (d) nelle immagini Ti basali e T2, in rapporto ad emorragia intra-lesionale recente. Le immagini T1 contrastografiche coronale (b) e sagittale (e) mostrano l'omogenea impregnazione della componente adenomatosa periferica (grazie a Regina Barbò).
Il segnale degli adenomi in T2 è importante nelle forme GH-secernenti. Infatti, le lesioni con segnale ipointenso presentano migliore risposta alla terapia con analoghi della somatostatina di prima generazione. All’opposto, la terapia con pasireotide si è dimostrata più efficace nelle forme con ipersegnale in T2 (15). Questi studi sottolineano la possibilità di utilizzare i dati radiologici per una terapia sempre più personalizzata di queste patologie.
I macro-adenomi presentano, in genere, un disomogeneo potenziamento post-contrastografico. La somministrazione di mdc risulta utile sia per una migliore caratterizzazione della lesione, sia per localizzare la quota di tessuto adeno-ipofisario sano riconoscibile alla periferia della lesione (16). Questa informazione può essere meglio definita con le immagini della RM dinamica (fig 27).
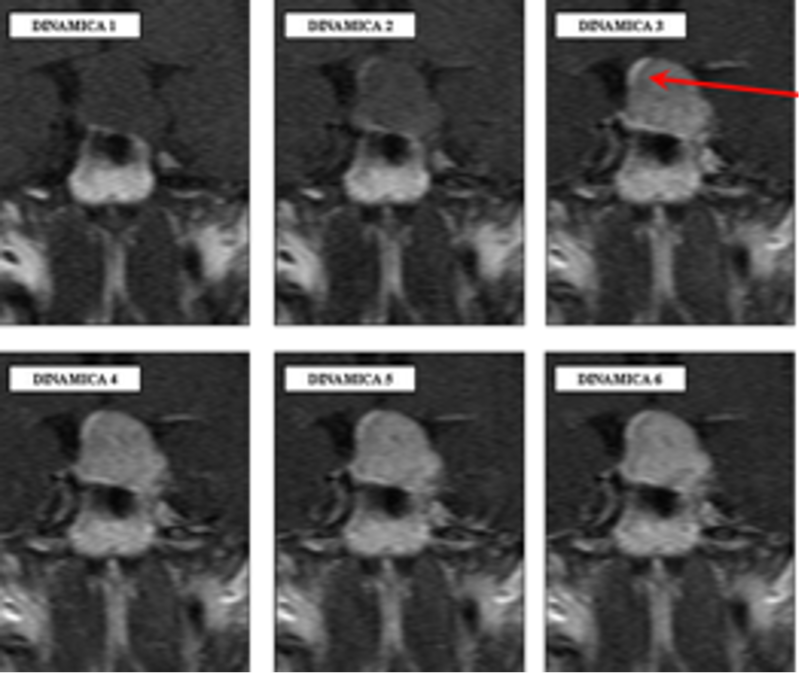
Figura 27
Sequenza dinamica in coronale nel macro-adenoma: la freccia visualizza la residua ghiandola sana e il peduncolo
| Caratteristiche RM macro-adenomi | |
| Sede | Intra- e peri-sellare |
| Sviluppo | Sovra-sellare Infero-sellare Para-sellare |
| Necrosi ed emorragia | Più frequente rispetto ai micro-adenomi |
| Segnale in T1 e T2 | Disomogeneo |
IL FOLLOW-UP
Micro-adenomi. Le linee guida dell’Endocrine Society suggeriscono di ripetere la RM di routine entro 1 anno. È stato riportato come estremamente raro l’aumento volumetrico del micro-adenoma, sia spontaneo che durante la terapia con farmaci dopaminergici. Questa osservazione ha portato alcuni esperti a raccomandare di non eseguire la RM di follow-up, a meno che la PRL non aumenti significativamente (per esempio > 250 ng/mL), o in caso di sviluppo di cefalea grave, compromissione dei campi visivi o dell'acuità visiva o sviluppo di paralisi dei nervi cranici.
Macro-adenomi. Nei non operati il controllo va previsto entro 1-6 mesi. In caso di mancata risposta alla terapia farmacologica (ormoni, nuovi disturbi visivi), si suggerisce un follow-up più ravvicinato (fig 28).
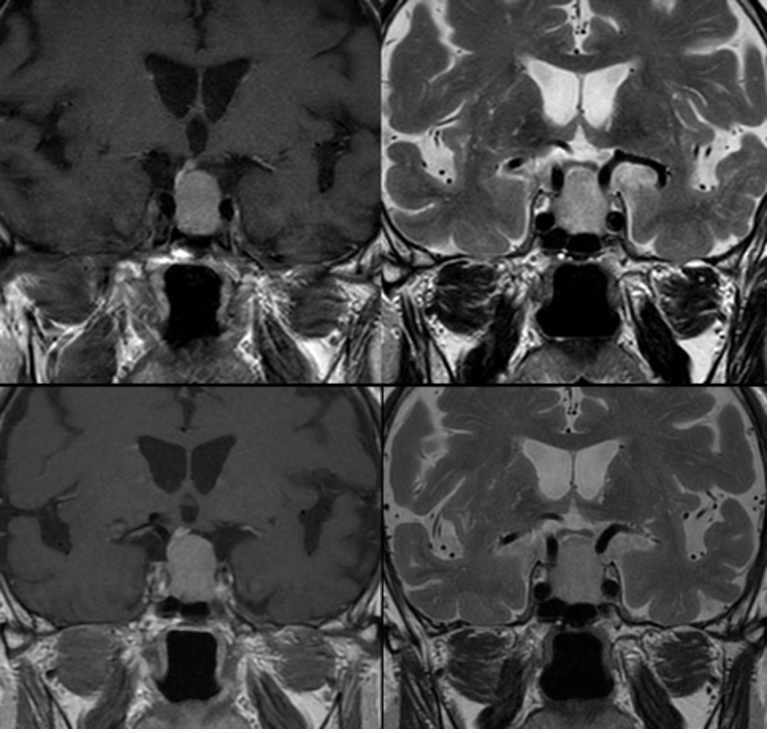
Figura 28
RM coronali T1 (a sinistra) e T2 (a destra): follow-up macro-adenoma a distanza di 3 anni
La tempistica di un ulteriore follow-up della RM dopo il primo dovrebbe basarsi sul contesto clinico individuale: cambiamenti del quadro neuro-oftalmologico ed endocrinologico, dimensioni dell'adenoma prima del trattamento, segni di invasività, chirurgia precedente, tasso di diminuzione ormonale e contrazione volumetrica del tumore durante il trattamento farmacologico, sesso, stato degli estrogeni, nonché aderenza alla terapia. A causa della ben nota deposizione della maggior parte dei chelati di Gd in molti tessuti e del dibattito riguardante gli effetti a lungo termine dell'esposizione al Gd, si suggerisce di evitare l’uso del Gd nel follow-up se non indispensabile (fig 29).
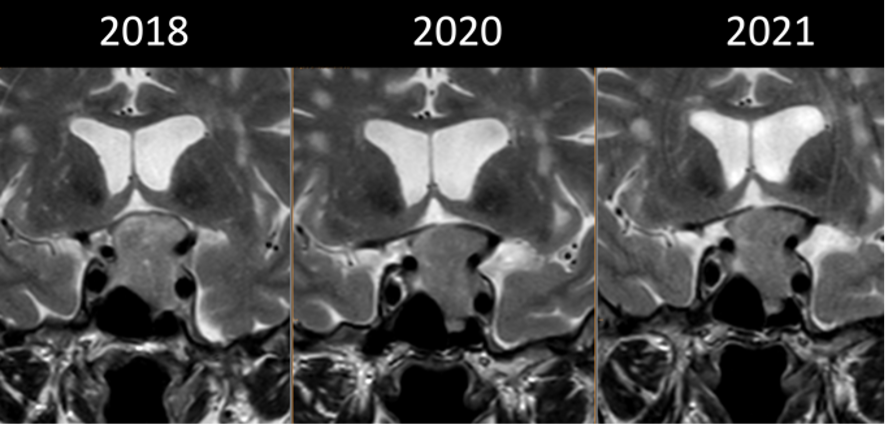
Figura 29
RM coronale in T2: follow-up del macro-adenoma senza Gd (quadro stabile)
Durante un trattamento farmacologico cronico è estremamente importante rivedere l'intera serie di immagini RM disponibili, perché cambiamenti minimi possono non essere rilevati quando il confronto si limita a due soli studi consecutivi (fig 30).
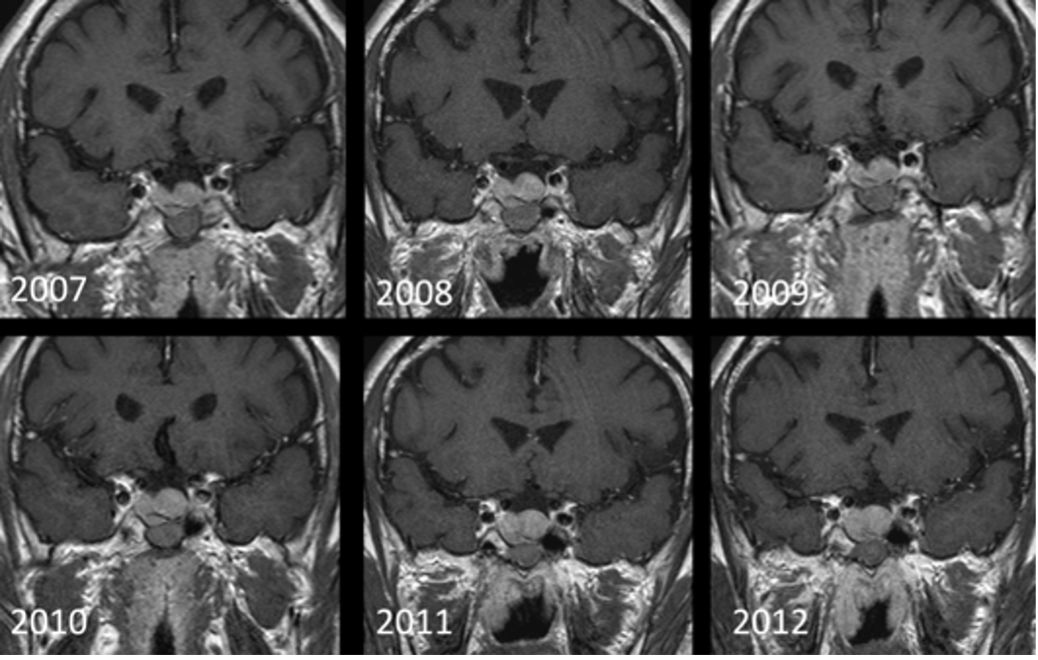
Figura 30
RM coronali T1 con Gd: controlli seriati di macro-adenoma. Il confronto tra il primo (2007) e l’ultimo esame (2012) documenta un chiaro aumento della lesione.
Nel follow-up dei macro-adenomi operati è consigliata l’esecuzione del controllo post-operatorio a 6 mesi dall’intervento, al fine di differenziare la persistenza di materiale di packing da quello necrotico-emorragico. È suggerito che le sequenze T2 siano le più sensibili, documentando un segnale dell’eventuale residuo, analogo al tumore originario (fig 31).
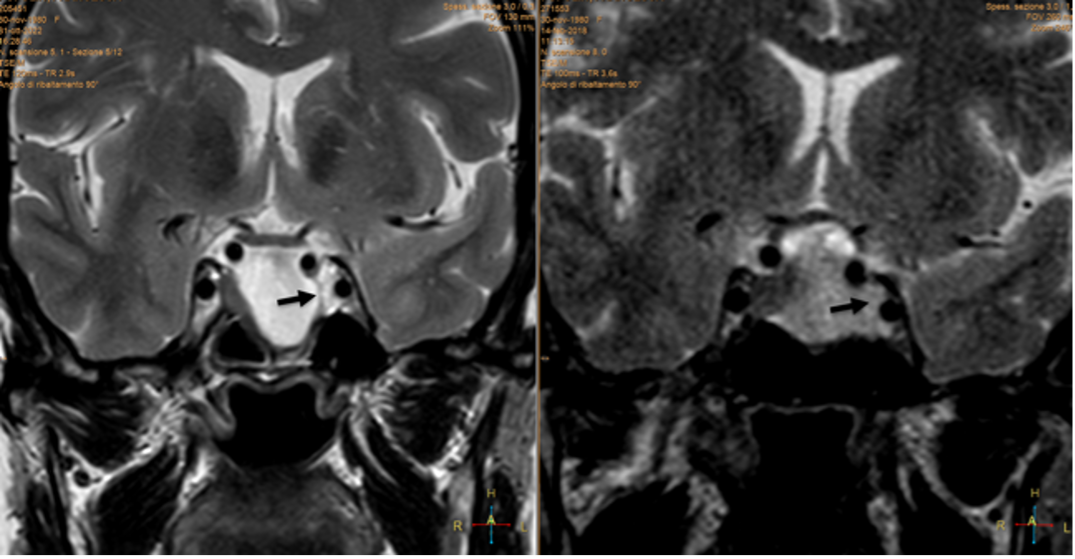
Figura 31
RM coronali T2: follow -up post-chirurgico di macro-adenoma. A sinistra il pre-operatorio con il tessuto nel seno cavernoso, a destra il residuo post-operatorio (freccia) in paziente del tutto asintomatico (ACTH).
L’IPOFISITE
Radiologicamente si osserva un ispessimento del peduncolo, con aspetto rigonfio dell’adeno-ipofisi, che presenta potenziamento intenso ed uniforme. Si associa un ispessimento durale adiacente (fig 32) e la mancata visualizzazione della neuro-ipofisi.
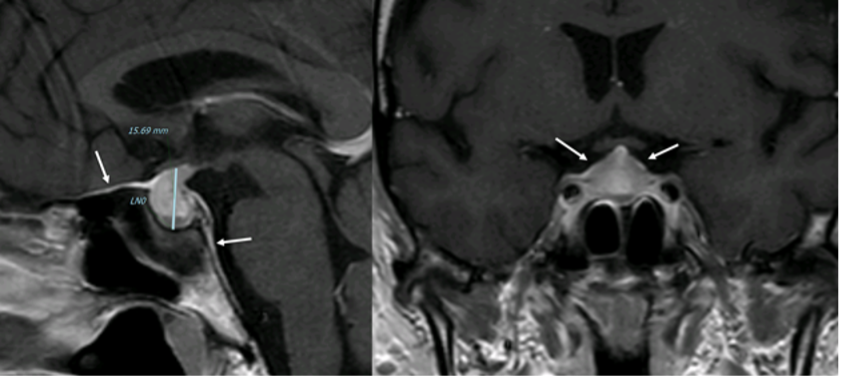
Figura 32
RM T1 con Gd sagittale (a sinistra) e coronale (a destra): ipofisite
La diagnosi differenziale con un adenoma non è semplice e deve basarsi fondamentalmente sui dati clinici, ma, nonostante tutti gli sforzi, nel 40% delle ipofisiti viene posta diagnosi di adenoma (17). Tra gli elementi a favore dell’ipofisite, oltre al profilo clinico è necessario considerare: l’età più giovane, la mancata visualizzazione della neuro-ipofisi (fig 33), la cavità sellare di dimensioni normali, l’espansione simmetrica della ghiandola, l’allargamento del peduncolo, il segnale e il potenziamento omogeneo e l’intenso potenziamento durale.
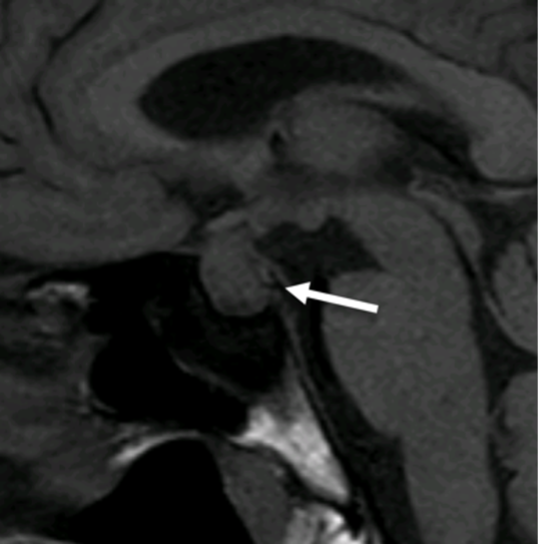
Figura 33
Ipofisite: mancata visualizzazione della neuro-ipofisi nelle sequenze T1 sagittali senza Gd
L’APOPLESSIA IPOFISARIA
Si tratta di una sindrome clinica acuta, con cefalea, difetti della vista e deficit ormonali variabili. L’apoplessia ghiandolare si sviluppa in un pre-esistente adenoma (ma anche in ghiandole normali), talvolta misconosciuto al momento della diagnosi. Deve essere differenziata dalle assai più frequenti emorragie intra-ghiandolari, principalmente asintomatiche, di riscontro per lo più occasionale (spesso durante il follow-up), riportate nel 10-15% delle casistiche chirurgiche e nel 20% di quelle radiologiche. Solo il 25% dei pazienti con emorragia da adenoma riferisce un'apoplessia clinica.
Poiché i sintomi non sono specifici, è probabile che la maggior parte dei pazienti venga sottoposto a una TC di base del cranio. Questa può essere normale, poiché il sanguinamento ipofisario può essere visualizzato solo in caso di tumore/emorragie di grandi dimensioni, con una sottostima della patologia. Per questo motivo, in un paziente con intensa cefalea “a colpo di tuono” deve sempre essere presa in considerazione l’apoplessia ipofisaria. Alla RM si riscontra più spesso una lesione intra- e sovra-sellare, con un segnale aspecifico in T1 e T2 (specie nei primi momenti) e potenziamento periferico della lesione (fig 34). Si associano segni di irritazione mucosa nel seno sfenoidale.
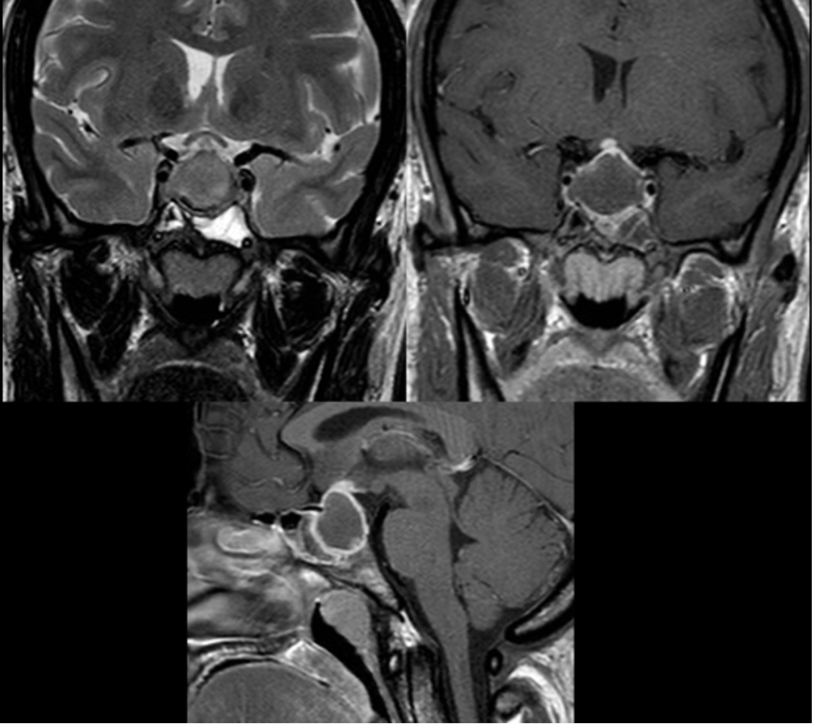
Figura 34
Apoplessia ipofisaria: RM coronale T2 (in alto a sinistra, segnale aspecifico) e coronale e sagittale (in basso) T1 post mdc, dove si apprezza potenziamento periferico
La presenza di compressione sul chiasma e il coinvolgimento dei nervi oculo-motori sono segni a favore di un rapido intervento decompressivo per via trans-sfenoidale. Il quadro evolve in atrofia ghiandolare e talvolta in sella vuota secondaria (18-19).
BIBLIOGRAFIA
- Daly AF, Beckers A. The epidemiology of pituitary adenomas. Endocrinol Metab Clin North Am 2020, 49: 347-55.
- Vasilev V, Rostomyan L, Daly AF, et al. Pituitary 'incidentaloma': neuroradiological assessment and differential diagnosis. Eur J Endocrinol 2016, 175: R171-84.
- Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am 1999, 28: 81-117.
- Buchfelder M, Schlaffer S. Imaging of pituitary pathology. Handb Clin Neurol 2014, 124: 151-66.
- Vitale G, Tortora F, Baldelli R, et al. Pituitary magnetic resonance imaging in Cushing's disease. Endocrine 2017, 55: 691-6.
- Fukuhara N, Inoshita N, Yamaguchi-Okada M, et al. Outcomes of three-Tesla magnetic resonance imaging for the identification of pituitary adenoma in patients with Cushing's disease. Endocr J 2019, 66: 259-64.
- Bonneville JF, Potorac I, Petrossians P, et al. Pituitary MRI in Cushing's disease - an update. J Neuroendocrinol 2022, 34: e13123.
- Nasi-Kordhishti I, Grimm F, Giese S, et al. The importance of MRI quality and reader's experience for detecting an adenoma in Cushing's disease. Eur J Endocrinol 2022, 187: 349-59.
- Colonnese C, Bozzao A, Pantano P, Fantozzi, Lezioni di neuroradiologia, Editore Esculapio, III Edizione 2019.
- Knosp E, Steiner E, Kitz K, Matula C. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery 1993, 33: 610-7; discussion 617-8.
- DiRisio AC, Feng R, Shuman WH, et al. The Knosp criteria revisited: 3-dimensional volumetric analysis as a predictive tool for extent of resection in complex endoscopic pituitary surgery. Neurosurgery 2022, doi: 10.1227/neu.0000000000002170.
- Gallucci M, Andreula C, Cirillo S, Scarabino T, Manuale di neuroradiologia, Editore Poletto, II Edizione 2016.
- Bonneville JF. Magnetic resonance imaging of pituitary tumors. Front Horm Res 2016, 45: 97-120.
- Romano A, Coppola V, Lombardi M, et al. Predictive role of dynamic contrast enhanced T1-weighted MR sequences in pre-surgical evaluation of macroadenomas consistency. Pituitary 2017, 20: 201-9.
- Tortora F, Negro A, Grasso LFS, et al. Pituitary magnetic resonance imaging predictive role in the therapeutic response of growth hormone-secreting pituitary adenomas. Gland Surg 2019, 8 suppl 3: S150-8.
- Faje A, Tritos NA, Swearingen B, Klibanski A. Neuroendocrine disorders: pituitary imaging. Handb Clin Neurol 2016, 136: 873-85.
- Leung GKK, Lopes MBS, Thorner MO, et al. Primary hypophysitis: a single-center experience in 16 cases. J Neurosurg 2004, 101: 262-71.
- Barkhoudarian G, Kelly DF. Pituitary apoplexy. Neurosurg Clin N Am 2019, 30: 457-63.
- Muthukumar N. Pituitary apoplexy: a comprehensive review. Neurol India 2020, 68 (supplement): S72-8.
ISPESSIMENTO DEL PEDUNCOLO IPOFISARIO
I germinomi sono neoplasie infantili maligne, rare, che originano dalle cellule germinali primitive.
Si localizzano lungo le strutture della linea mediana, a livello della ghiandola pineale, del pavimento del III ventricolo e del peduncolo ipofisario. In particolare, quest’ultima localizzazione si associa, spesso, all’insorgenza di diabete insipido (1): in un bambino con diabete insipido la presenza di peduncolo ipofisario di dimensioni > 3 mm deve essere considerata sospetta e rende necessario un controllo RM a 6 mesi (2).
Il germinoma si associa alla produzione di alfa-feto proteina e ß-hCG nel liquor e nel sangue. Presenta una tendenza infiltrante, con disseminazione per via liquorale.
All’esame RM, il germinoma si manifesta come lesione solida iso/ipointensa in T1 e iperintensa in T2. Il potenziamento dopo somministrazione di mdc è massivo e omogeneo. Non si associa alla presenza di calcificazioni (fig 35) (3).
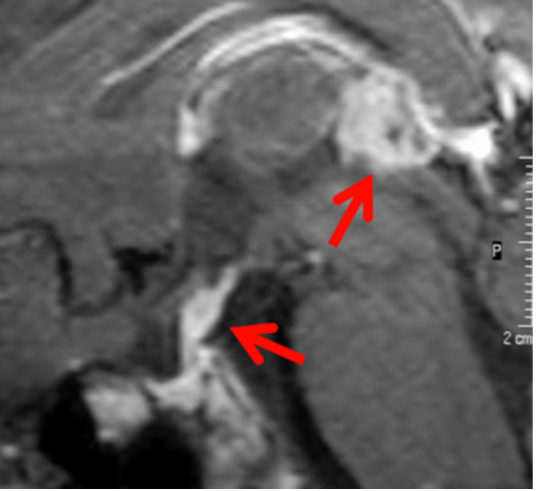
Figura 35
RM sagittale T1 con mdc: germinoma con la tipica doppia localizzazione infundibolare e pineale
L'istiocitosi a cellule di Langerhans nella localizzazione infundibolare causa diabete insipido e deficit di GH. Si associano lesioni litiche dell’osso (specie teca), esoftalmo e tipiche alterazioni in T2 nel cervelletto.
Le alterazioni ossee possono oggi essere studiate con la RM whole-body, che si è dimostrata superiore all’esame radiografico e alla scintigrafia, risultando peraltro molto meno invasiva dal punto di vista biologico (4).
Anche la sarcoidosi può avere una rara localizzazione infundibolare.
Bibliografia
- Faje A, Tritos NA, Swearingen B, Klibanski A. Neuroendocrine disorders: pituitary imaging. Handb Clin Neurol 2016, 136: 873-85.
- Colonnese C, Bozzao A, Pantano P, Fantozzi, Lezioni di neuroradiologia, Editore Esculapio, III Edizione 2019.
- Gallucci M, Andreula C, Cirillo S, Scarabino T, Manuale di neuroradiologia, Editore Poletto, II Edizione 2016.
- Kim JR, Yoon HM, Jung AY, et al. Comparison of whole-body MRI, bone scan, and radiographic skeletal survey for lesion detection and risk stratification of Langerhans cell histiocytosis. Sci Rep 2019, 9: 317.
ALTRE LESIONI
Craniofaringioma
Il craniofaringioma è un raro tumore benigno intra-cranico non gliale della regione sellare e para-sellare, derivante da malformazioni del tessuto embrionale (tasca di Rathke), a basso grado istologico (OMS I). È il più frequente tumore non gliale in età pediatrica, con crescita lenta e propensione alla recidiva post-operatoria.
La maggior parte presenta esclusivamente una componente sovra-sellare, solo raramente vi è estensione anche nella fossa anteriore (9%), media (8%) o posteriore (12%). Altre sedi rare includono il rino-faringe, l'area para-nasale, l'osso sfenoide, il seno etmoide, l'area intra-chiasmatica, il lobo temporale, la ghiandola pineale, la fossa cranica posteriore, l’angolo ponto-cerebellare, la porzione media del mesencefalo o la porzione interna del terzo ventricolo.
Il craniofaringioma è tipicamente un tumore cistico rotondeggiante, a densità eterogenea per la presenza di componenti solide che prendono mezzo di contrasto e calcificazioni curvilinee o nodulari. Spesso le lesioni sono di grandi dimensioni (oltre i 5 cm) al momento della diagnosi. Si distinguono le forme adamantinose, più comuni, spesso multi-loculate, con componenti fluide (a olio di macchina) e papillari, caratterizzate da lesioni solido-cistiche (con contenuto fluido simil-liquorale).
La RM è la modalità di imaging che permette di ottenere informazioni cruciali sulla posizione e sui rapporti con le strutture adiacenti (1). L'intensità di segnale del craniofaringioma è molto variabile, in relazione alla concentrazione proteica del liquido cistico, alle componenti solide e alla presenza di calcificazioni (2).
Nelle sequenze T1-pesate:
- nella componente solida appare isointenso, ipointenso o a intensità di segnale disomogenea;
- nella componente cistica può apparire iperintenso;
- può presentare elevata intensità di segnale in relazione a elevati livelli di proteine e colesterolo, calcificazioni o sanguinamenti.
Nelle sequenze T2-pesate:
- nella componente solida appare ipointenso o iperintenso;
- nella componente cistica appare iperintenso o ipointenso.
Dopo somministrazione endovenosa di gadolinio il craniofaringioma mostra intenso potenziamento periferico delle pareti delle cisti e nelle componenti solide (noduli intra-cistici) (figura 36).
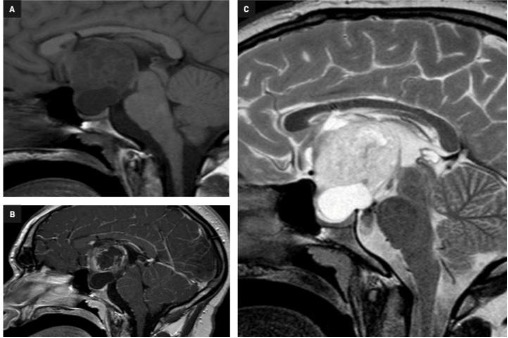
Figura 36
Craniofaringioma
(A) T1 sagittale: lesione grossolana, eterogenea, che origina a livello della sella turcica e va a comprimere e dislocare il fornice e il III ventricolo. La massa presenta una componente inferiore ipointensa a livello della sella e una componente maggiore superiore leggermente iperintensa
(B) T1 sagittale post-contrastografica: potenziamento periferico delle pareti
(C) Immagine T2 sagittale: componente inferiore cistica iperintensa e componente superiore lievemente ipointensa rispetto alla componente cistica
L’angio-RM può essere utile per valutare la vicinanza del tumore alle carotidi interne e alle porzioni adiacenti al circolo di Willis (3-5).
Nelle sequenze DWI, il craniofaringioma presenta un coefficiente di diffusione maggiore rispetto al macro-adenoma. Tale caratteristica può essere dirimente nella diagnosi differenziale con gli adenomi o altre lesioni della regione sellare, in aggiunta alle altre caratteristiche tipiche del craniofaringioma, come la presenza di calcificazioni, la localizzazione sovra-sellare e la compressione sul III ventricolo.
La TC è l’unica metodica in grado di evidenziare o escludere la presenza di calcificazioni, di frequente riscontro in questa lesione (circa il 90%) (6). In TC il tumore può avere densità eterogenea, per la presenza di componenti cistiche e solide, con impregnazione contrastografica in corrispondenza delle porzioni solide. Si possono inoltre osservare alterazioni secondarie a livello della base cranica, come l'allargamento della sella turcica o l'erosione del dorso della sella (figura 37).
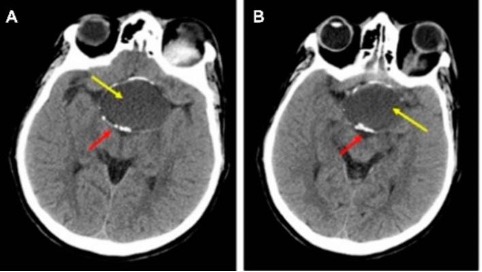
Figura 37
Craniofaringioma: TC senza mdc che mostra una grossolana lesione cistica (freccia gialla) occupante lo spazio sellare e sovra-sellare, con calcificazioni periferiche curvilinee (freccia rossa)
Cordoma
I cordomi sono rari tumori dell’osso, che si sviluppano principalmente a carico dell’osso sacro e della base cranica; quelli della base cranica sono localizzati principalmente a livello del clivus. Si pensa derivino da residui della notocorda, che permangono a livello dello scheletro assile per tutta la vita e che possono subire trasformazione in cordoma a qualsiasi età (7,8).
Il cordoma si presenta come lesione solida, con effetti osteolitici sulla corticale e coinvolgimento dei tessuti molli. Si localizza maggiormente a livello della metà superiore del clivus, ma si può estendere anche alla sua metà inferiore, al processo clinoideo posteriore, al seno cavernoso e al condilo occipitale (9).
TC e RM sono le metodiche standard per la diagnosi iniziale del tumore, la pianificazione del trattamento e il follow-up post-trattamento.
Alla TC senza mezzo di contrasto il cordoma appare come lesione eterogenea, generalmente iso-iperdensa, ben circoscritta, con associata erosione ossea (10).
Alla RM nelle sequenze T1-pesate può presentare segnale eterogeneo, generalmente iso-ipointenso con alcune aree iperintense (legate alla presenza di materiale mucoide o emorragie), setti e aree di necrosi (11). Nelle sequenze T2-pesate appare generalmente iperintenso. È comune la presenza di calcificazioni. Dopo somministrazione endovena di gadolinio, il cordoma mostra moderato/marcato potenziamento con aspetto a favo d’ape, con alcune aree lineari di non captazione (12).
Condrosarcoma
I condrosarcomi sono spesso descritti insieme ai cordomi poiché ne condividono la presentazione clinica, le caratteristiche di imaging e la localizzazione (generalmente in sede posteriore). Sono localizzati più comunemente a livello della fessura petro-occipitale.
La TC mostra una lesione solida tendenzialmente iso-iperdensa, con erosione ossea ed una matrice condroide calcifica, con il classico aspetto “ring and archs” ed alcune calcificazioni (13).
Alla RM i condrosarcomi appaiono iso-ipointensi in T1, iperintensi in T2 e mostrano disomogeneo potenziamento (14,15).
Bibliografia
- Rossi A, Cama A, Consales A, et al. Neuroimaging of pediatric craniopharyngiomas: a pictorial essay. J Pediatr Endocrinol Metab 2006, 19 suppl 1: 299-319.
- Karavitaki N, Cudlip S, Adams CB, Wass JA. Craniopharyngiomas. Endocr Rev 2006, 27: 371-97.
- Lee I, Zan E, Bell W, et al. Craniopharyngiomas: radiological differentiation of two types. J Korean Neurosurg Soc 2016, 59: 466-70.
- Pusey E, Kortman KE, Flannigan BD, et al. MR of craniopharyngiomas: tumor delineation and characterization. AJR Am J Roentgenol 1987, 149: 383-8.
- Byrne JV. Imaging of the pituitary. In Wass JAH, Shalet SM, eds. Oxford Textbook of Endocrinology and Diabetes. 1st Oxford University Press 2002: 136–45.
- Sartoretti-Schefer S, Wichmann W, Aguzzi A, Valavanis A. MR differentiation of adamantinous and squamous-papillary craniopharyngiomas. AJNR Am J Neuroradiol 1997, 18: 77-87.
- Kyriakos M. Benign notochordal lesions of the axial skeleton: a review and current appraisal. Skeletal Radiol 2011, 40: 1141-52.
- Yakkioui Y, van Overbeeke JJ, Santegoeds R, et al. Chordoma: the entity. Biochim Biophys Acta 2014, 1846: 655-69.
- Meyer JE, Oot RF, Lindfors KK. CT appearance of clival chordomas. J Comput Assist Tomogr 1986,10: 34–8.
- Brown RV, Sage MR, Brophy BP. CT and MR findings in patients with chordomas of the petrous apex. AJNR Am J Neuroradiol 1990, 11: 121–4.
- Meyers SP, Hirsch WL Jr, Curtin HD, et al. Chordomas of the skull base: MR features. AJNR Am J Neuroradiol 1992, 13: 1627–36.
- Pamir MN, Ozduman K. Analysis of radiological features relative to histopathology in 42 skull-base chordomas and chondrosarcomas. Eur J Radiol 2006, 58: 461–70.
- Lee YY, Van Tassel P. Craniofacial chondrosarcomas: imaging findings in 15 untreated cases. Am J Neuroradiol 1989, 10: 165-70.
- Almefty K, Pravdenkova S, Colli BO, et al. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer 2007, 110: 2457-67.
- Meyers SP, Hirsch WL, Curtin HD, et al. Chondrosarcomas of the skull base: MR imaging features. Radiology 1992, 184: 103-8.
Meningioma
I meningiomi sono tumori benigni a lenta crescita, che possono coinvolgere la regione sellare, ma in genere non determinano allargamento della sella turcica. Originano generalmente dalle pareti sellari, o più frequentemente da aree adiacenti del basi-cranio, quali il planum etmoido-sfenoidale, la grande e piccola ala dello sfenoide, il canale ottico. I meningiomi sellari propriamente detti originano dal tubercolo, dal diaframma, dai processi clinoidei anteriori, dalla lamina quadrilatera, dalle pareti laterali della sella turcica/seni cavernosi (1,2). Presentano segnale isointenso in T1 e T2, con impregnazione rapida, netta e omogenea dopo mdc, talvolta estesa alla dura madre con il tipico segno della “coda durale”, sebbene quest’ultimo non sia considerato patognomonico della lesione.
In alcuni casi i meningiomi possono presentare calcificazioni intra-lesionali e iperostosi adiacente alla base di impianto, reperti meglio evidenti all’esame TC. È possibile osservare stenosi della carotide intra-cavernosa (fig 25).
La diagnosi differenziale si pone principalmente con l’adenoma, che al contrario del meningioma presenta potenziamento meno intenso ed eterogeneo, con un time-to-peak più lungo. La ghiandola ipofisaria inoltre non è separata dalla lesione (1,2).
Riassumendo le principali caratteristiche del meningioma:
- epicentro: para-sellare/sovra-sellare;
- ipofisi distinguibile dalla lesione;
- sella turcica di normali dimensioni;
- segnale T1/T2 isointenso;
- potenziamento contrastografico rapido, netto, omogeneo, con “coda durale”;
- calcificazioni intra-lesionali evidenziabili in TC;
- “encasement” dell’arteria carotide interna.
Glioma ottico-diencefalico
È un glioma tipico dell’età infantile, la cui presentazione clinica dipende dalla localizzazione (1):
- se chiasmatica, determina aspetto ingrossato del chiasma e/o dei nervi ottici, mostra segnale RM isointenso in T1 e iperintenso in T2, con disomogenea impregnazione dopo MdC;
- se ipotalamica, ha un aspetto maggiormente disomogeneo.
Bibliografia
- Colosimo C. Neuroradiologia. 1ª ed, 2014, EDRA LSWR S.p.A., ISBN 978-88-214-2909-5.
- Huang BY, Castillo M. Nonadenomatous tumors of the pituitary and sella turcica. Top Magn Reson Imaging 2005, 16: 289-99.
- Smith JK. Parasellar tumors: suprasellar and cavernous sinuses. Top Magn Reson Imaging 2005, 16: 307-15.
- Rennert J, Doerfler A. Imaging of sellar and parasellar lesions. Clin Neurol Neurosurg 2007, 109: 111–24.
- Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am 1999, 28: 81-117.
GLI INCIDENTALOMI
Un incidentaloma è una condizione patologica non correlata alla motivazione di esecuzione di un esame diagnostico.
Nella regione sellare se ne stima una prevalenza (su serie autoptiche) di circa il 10%, che sale fino al 38% in quelle RM.
Nel 90% dei casi si tratta di adenomi o cisti della tasca di Rathke.
È utile distinguere gli incidentalomi solidi dai cistici. I solidi sono più frequentemente micro-adenomi non funzionanti, anche se viene riscontrata ipersecrezione subclinica in oltre il 20% dei casi (18% PRL, 4% ACTH, 3% GH).
Ma un incidentaloma è anche l’interpretazione in senso patologico di una variante anatomica o di un artefatto tecnico di un esame diagnostico. Sono questi gli incidentalomi più gravi, perché trasformano un soggetto normale in un paziente (falsi incidentalomi). Molti di questi sono legati ad artefatti da volume parziale e all’uso di sequenze dinamiche.
La gestione degli incidentalomi varia in relazione alla tipologia di reperti (1-3):
- nelle forme cistiche si consiglia, se asintomatiche, un controllo RM dopo 1 anno e quindi ogni 2 anni;
- nei micro-adenomi senza segni clinici eseguire un controllo a un anno e quindi ogni due anni (ma il follow-up può essere interrotto dopo un anno se stabile < 5 mm);
- nei macro-adenomi il controllo RM va eseguito a 6 mesi, quindi meno frequentemente, monitorando il campo visivo se prossimi al chiasma.
Bibliografia
- Constantinescu SM, Maiter D. Pituitary incidentaloma. Presse Med 2021, 50: 104081.
- Vasilev V, Rostomyan L, Daly AF, et al. Pituitary 'incidentaloma': neuroradiological assessment and differential diagnosis. Eur J Endocrinol 2016, 175: R171-84.
- Paschou SA, Vryonidou A, Goulis DG. Pituitary incidentalomas: A guide to assessment, treatment and follow-up. Maturitas 2016, 92: 143-9.
Campimetria ottica e potenziali evocati visivi
Chiara Martini & Pietro Maffei
Clinica Medica 3°, Azienda Ospedaliera di Padova
PERIMETRIA
L’analisi del campo visivo è una tecnica diagnostica di particolare utilità nell’algoritmo decisionale terapeutico delle patologie espansive ipofisarie. Esistono varie metodiche di esame e in questo capitolo tratteremo brevemente la perimetria statica, la perimetria dinamica e la perimetria automatica computerizzata. Tutte queste tecniche consentono di effettuare lo screening, la diagnosi ed il follow-up dei disturbi del campo visivo dovuti a patologie coinvolgenti le componenti principali del sistema visivo, quali ad esempio l’occhio, la retina, il nervo ottico e la corteccia cerebrale. La patologia ipofisaria può determinare un’alterazione del campo visivo patognonomica, mediante compressione del nervo ottico soprattutto nel punto di decussazione delle sue fibre a livello del chiasma. Le alterazioni del campo visivo sono tuttavia causate, con maggiore frequenza, da altre patologie a carico dell’occhio (ad esempio il glaucoma) o della retina (ad esempio retiniti).
Generalmente definiamo come campo visivo la percezione visiva globale ottenuta durante la fissazione di un oggetto statico, mantenendo la testa e il corpo in posizione fissa. Rispetto al punto di fissazione dello sguardo, nella visione mono-oculare, uno stimolo visivo può essere fisiologicamente percepito fino a circa 50° superiormente, 70° inferiormente, 50° nasalmente e 100° temporalmente. Poiché la papilla del nervo ottico, localizzata a 10-15° nasalmente, non presenta fotorecettori, essa determina fisiologicamente un’area di scotoma assoluto (area cieca) all’interno del campo visivo. Altre aree di scotoma o di riduzione dell’acuità visiva vanno invece considerate come potenzialmente patologiche.
La perimetria è la tecnica utilizzata per misurare l’estensione del campo visivo di un soggetto e per valutare la sensibilità dell’occhio nel percepire gli stimoli visivi. Quando gli stimoli sono presentati su una superficie piatta la tecnica viene anche definita campimetria. Attualmente gli stimoli vengono invece presentati su una superficie concava di tipo sferico in cui l’occhio del soggetto in esame viene posizionato al centro geometrico della stessa. Nella perimetria statica lo stimolo viene presentato in diverse zone del campo visivo, ma la sua intensità luminosa viene modificata anche in relazione alle risposte del soggetto. Nella perimetria dinamica lo stimolo mantiene intensità e forma costanti e viene progressivamente spostato dalle zone cieche verso quelle visibili del campo visivo.
L’esame non è invasivo ed è ripetibile nel tempo, ma tuttavia richiede una discreta collaborazione. Lo scopo dell’esame deve essere spiegato con attenzione e il soggetto deve essere accuratamente posizionato e preparato dal perimetrista, al fine di evitare artefatti di registrazione ed errate interpretazioni. E’ importante inoltre evitare alcune fonti comuni di errore come ad esempio: identificazione paziente, età, mancata correzione vizio di rifrazione, artefatti da lenti, allineamento, pupille miotiche o presenza di cataratta. Viene esaminato un occhio alla volta e il controlaterale viene opportunamente mascherato. In linea generale il soggetto esaminato deve premere un pulsante tutte le volte in cui percepisce la comparsa di uno stimolo, generalmente luminoso, all’interno del campo visivo. Tutte la risposte del soggetto vengono poi riportate su un tracciato o grafico perimetrico, che riproduce la mappa del campo visivo, con le risposte assolute e il loro grado di scostamento rispetto ai parametri di riferimento. La tipologia delle mappe e la rappresentazione grafica delle risposte può variare in relazione alla metodica utilizzata.
L’esame può essere effettuato sia con perimetri manuali (perimetria Goldman) che automatici.Fra i perimetri automatici più diffusi in commercio ricordiamo i perimetri di Humphrey e i perimetri Octopus di cui daremo alcune informazioni (Figura 1).
Entrambi forniscono la cosiddetta mappa delle soglie o dei valori in cui vengono riportati, per ciascun occhio, una rappresentazione dei valori soglia di segnale luminoso che, in base al grado di intensità percepita, vengono tradotti in una scala numerica su ogni punto del campo visivo esaminato. Le mappe sono analizzabili punto per punto e in caso di mancanza assoluta di segnale questo si traduce in un valore 0 (mappe di Humphrey; Fig 1a parte superiore) o in un quadretto nero (mappe Octopus; Fig 1b parte superiore). Oltre a queste rappresentazioni vengono presentate parallelamente altre mappe di più immediata interpretazione visiva che “traducono” il valore numerico in una scala di grigi (mappe di Humphrey; Fig 1a parte superiore) o in una scala di colore (mappe Octopus; Fig 1b parte superiore), la cui gradazione varia in rapporto alla perdita di segnale. Le scale di grigio o di colore rappresentano tuttavia il segnale percepito con un'approssimazione maggiore rispetto ai valori assoluti. Entrambe le strumentazioni forniscono anche le mappe dei confronti o delle probabilità. Si tratta di altre due coppie di mappe (quattro in totale; Fig 1a e b parti cerchiate in rosso) che identificano sia con valori assoluti che simbolici (scala di grigi sia per Humphrey che Octopus) gli scostamenti del soggetto rispetto a: 1) popolazione di riferimento (divisa per decadi) chiamata total deviation (Humphrey) o comparison (Octopus); 2) gradiente di sensibilità del soggetto in esame chiamato pattern deviation (Humphrey) o corrected comparison (Octopus). Nel primo caso nella mappa viene rappresentata la probabilità statistica di ciascuna deviazione rispetto alla popolazione generale, mentre nel secondo viene sottratto il segnale del danno omogeneamente distribuito nel campo visivo del soggetto al fine di far risaltare i difetti di maggiore profondità. Segnaliamo infine le curve cumulative di difetto (Octopus; Fig 1b parte con quadrato rosso), che riportano su una linea, che prende la forma nella situazione normale di una S allungata e coricata, la sensibilità globale del soggetto passando progressivamente dai punti di maggiore sensibilità (in alto a sinistra) verso quelli a minore sensibilità (in basso a destra). Al di sopra e al di sotto di questa linea vengono posizionate le linee di riferimento. Gli scostamenti rispetto alla norma forniscono un’immagine immediata della sensibilità globale o dei difetti localizzati puri. La curva non fornisce tuttavia la topografia dei difetti.
Come si diceva la presenza di un adenoma ipofisario può determinare difetti del campo visivo, individuabili con la perimetria, in zone diverse in relazione al danno del nervo ottico lungo il suo decorso (Figura 2). Generalmente l’adenoma ipofisario determina una compressione dal basso verso l’alto a livello del chiasma; tuttavia la posizione relativa del chiasma e dell’adenoma possono variare, determinando quadri compressivi con associata componente pre e post-chiasmatica. Quando il difetto sembra rispettare la linea verticale mediana è fondamentale analizzare entrambi gli occhi poiché generalmente si possono distinguere lesioni:
- pre-chiasmatiche (difetti monoculari);
- chiasmatiche (binoculari, emianopsie eteronime, ossia coinvolgenti l’emicampo destro e il sinistro controlaterale, solitamente emianopsie bitemporali e raramente binasali);
- post-chiasmatiche (binoculari, emianopsie omonime, ossia coinvolgenti lo stesso emicampo).
Per la particolare configurazione anatomica delle fibre nel punto di decussazione, le compressioni del chiasma dal basso determinano perdita di segnale prevalentemente negli emicampi superiori, mentre le compressioni dall’alto interessano maggiormente gli emicampi inferiori.
POTENZIALI EVOCATI VISIVI (PEV)
Il segnale visivo determina fisiologicamente la depolarizzazione di alcuni gruppi neuronali, che producono una variazione dell’attività elettro-encefalografica (EEG) registrabile con un elettrodo posto sulla superficie del capo (potenziali evocati visivi; PEV). La tecnica standard di registrazione dei PEV prevede l’applicazione di tre elettrodi sulla regione occipitale, che registrano la risposta EEG quando vengono presentati all’interno del campo visivo specifici segnali caratterizzati da variazioni di contrasto (reversing pattern o P-PEV) o di luminanza (flash PEV o F-PEV). Nel primo caso viene generalmente utilizzato uno schermo con luminosità costante in cui ad esempio l’occhio viene stimolato da un’immagine a scacchiera in cui si alternano, con una cadenza predefinita, elementi bianchi e neri (quadrati o barre), mentre nel secondo caso l’impulso luminoso è generato da una lampada stroboscopica, di cui si possono modulare l’intensità e la frequenza d’impulso. La tecnica P-PEV è la preferita poiché meno soggetta a variabilità inter- e intra-individuale; è inoltre più sensibile e accurata rispetto all’F-PEV. L’F-PEV viene invece riservata ai pazienti poco collaboranti che non mantengono fisso lo sguardo durante il test. Le condizioni del soggetto risultano pertanto critiche nell’interpretazione corretta dei P-PEV ed in particolare bisognerà tener conto dei seguenti fattori: vizi di rifrazione (vanno corretti con lenti), affaticamento nel mantenimento del fuoco, grado di vigilanza e attenzione, capacità di mantenere fisso lo sguardo, miosi pupillare, anisocorie.
Con la metodica P-PEV è generalmente documentabile un tracciato EEG con picco di positività a livello occipitale e con una latenza di circa 100 ms in risposta alla variazione di contrasto (pattern reversal). Le componenti della risposta registrate dall’elettrodo vengono definite in base alla loro polarità e alla latenza. Le oscillazioni negative e positive sono denominate rispettivamente N e P. In base alla latenza dopo la variazione di contrasto, si riconosce abitualmente a livello occipitale un segnale EEG trifasico con picco N75, P100 ed N145. Le latenze possono variare in relazione allo stimolo e alla velocità della variazione di contrasto applicata (la scacchiera è la tecnica più utilizzata). Durante il test il soggetto viene invitato a mantenere lo sguardo fisso in un punto che non coincide con quello in cui viene presentato lo stimolo con un’angolazione variabile. L’ampiezza del PEV viene calcolata sia sulla deflessione N75-P100 che su quella P100-N145. In sintesi le alterazioni del segnale PEV comprendono le modifiche di ampiezza, morfologia e topografia.
Per valutare la presenza di una patologia a carico del chiasma ottico il segnale EEG trifasico registrabile nell’emisfero occipitale destro e sinistro viene posto in relazione alle stimolazioni provenienti singolarmente dall’occhio destro e sinistro. L’analisi del segnale prenderà in esame la sua ampiezza, latenza e topografia. Le stimolazioni possono essere effettuate per singolo occhio a campo pieno o anche ad emicampo. Nella patologia chiasmatica si determina generalmente una asimmetria crociata (“crossed”) dei PEV; in questo caso il PEV risulta maggiormente alterato nell’emisfero controlaterale all’occhio stimolato e questa asimmetria - fra i due emisferi occipitali - si inverte dopo stimolazione dell’occhio controlaterale. Quando si è in presenza di una patologia retro-chiasmatica, si registra invece una asimmetria non crociata (“uncrossed”) del segnale PEV a livello occipitale; ossia il PEV alterato di un emisfero occipitale non si modifica in relazione all’occhio che viene stimolato. Nel caso infine di una patologia del nervo ottico prechiasmatica non si registra asimmetria dei PEV.
Nell’individuazione delle patologie compressive a carico del chiasma ottico i PEV sono risultati più sensibili rispetto alla perimetria. E’ stato infatti documentato che pazienti con adenoma ipofisario e perimetrie oculari nella norma possono presentare alterazioni dei PEV. Il risultato dei PEV potrebbe quindi in alcuni casi modificare le decisioni terapeutiche ed in particolare l’indicazione all’approccio chirurgico.
Bibliografia
- Schiefer U, Patzold J, Dannheim F. Konventionalle perimetrie. Teil I: Einfuhrung - Grundbegriffe. Ophthalmologe 2005, 102: 627-46.
- Odom JV, Bach M, Barber C, et al. Visual evoked potentials standard (2004). Doc Ophthalmol 2004, 108: 115-23.
- American Clinical Neurophysiology Society. Guideline 9B. Guidelines on visual evoked potentials. J Clin Neurophysiol 2006, 23: 138-56.
Tomografia a coerenza ottica (OCT)
Stefano Amodeo
Facoltà di Medicina e Psicologia UOC di Oftalmologia, Università di Roma Sapienza; Ospedale Sant'Andrea di Roma
(aggiornato al febbraio 2025)
Cosa è
La tomografia a coerenza ottica, comunemente nota come OCT, è una tecnologia di imaging non invasiva, che permette di ottenere immagini ad alta risoluzione delle strutture oculari. Viene utilizzata principalmente in oftalmologia per analizzare la retina, il nervo ottico e la cornea, fornendo informazioni dettagliate sulla loro morfologia e stato di salute.
Come funziona
L'OCT sfrutta la luce per acquisire immagini delle strutture oculari in sezione trasversale, in modo simile a una TC, ma senza l'uso di radiazioni ionizzanti. Questa tecnologia si basa sul principio dell'interferometria a bassa coerenza, che consente di misurare il tempo impiegato dalla luce per riflettersi dai diversi strati dei tessuti oculari. I dati raccolti vengono elaborati da un software dedicato, che genera immagini dettagliate delle strutture osservate.
Il funzionamento dell'OCT si basa sull'uso di un raggio laser a bassa coerenza, solitamente nella banda dell'infrarosso, che viene diviso in due fasci. Uno di questi viene diretto verso il tessuto oculare, mentre l'altro è inviato a uno specchio di riferimento. La luce riflessa dalle diverse strutture dell'occhio viene poi confrontata con quella dello specchio, generando un'interferenza che permette di ricostruire l'immagine dei tessuti. Questa tecnica consente di ottenere immagini con una risoluzione dell’ordine di pochi micron, rendendola uno strumento essenziale per la diagnosi precoce di molte patologie oculari.
Uno dei principali vantaggi dell'OCT è la capacità di fornire immagini in tempo reale senza necessità di contatto diretto con l'occhio, rendendola una tecnica sicura e ben tollerata. Inoltre, la rapidità di esecuzione permette di integrare facilmente l'esame nella pratica clinica quotidiana. Tuttavia, come tutte le tecnologie diagnostiche, presenta alcune limitazioni, tra cui la riduzione della qualità dell'immagine in presenza di opacità dei mezzi diottrici, come cataratta avanzata o edema corneale.
Per cosa si usa
L'OCT viene utilizzata principalmente per la diagnosi e il monitoraggio di malattie della retina, come la degenerazione maculare legata all’età, la retinopatia diabetica e il pucker maculare. Inoltre, è fondamentale nello studio del nervo ottico, permettendo di valutare i danni legati al glaucoma, permette di misurare lo spessore delle fibre nervose, evidenziando gli assottigliamenti tipici del glaucoma e permettendo di seguire nel tempo l’evoluzione della patologia nonché di valutare l’idoneità del trattamento terapeutico locale messo in atto.
L’OCT trova applicazione anche nelle patologie della cornea: utile nella valutazione di cheratocono o nel follow-up post-chirurgico di interventi refrattivi.
Prospettive
Negli ultimi anni, l’evoluzione della tecnologia ha portato allo sviluppo dell’OCT ad alta velocità e dell’Angio-OCT, che permette di visualizzare la micro-vascolarizzazione retinica senza l’uso di mezzi di contrasto. Questo ha reso l’OCT uno strumento sempre più potente per la diagnosi precoce e il monitoraggio di numerose malattie oculari, migliorando significativamente la gestione clinica dei pazienti.
In conclusione, l’OCT rappresenta uno dei più importanti, utili e necessari strumenti diagnostici in oftalmologia. La sua capacità di fornire immagini ad alta risoluzione delle strutture oculari interne ha rivoluzionato la diagnosi e il trattamento di molte patologie. Grazie ai continui progressi tecnologici, l’OCT continuerà a giocare un ruolo chiave nella pratica clinica, consentendo diagnosi sempre più precoci e precise. Le nuove tecniche di imaging permetteranno di studiare porzioni di retina sempre più vaste, fino ad arrivare ad ottenere fotografie ad alta risoluzione di tutta la retina; l’applicazione nel segmento anteriore dell’occhio permetterà di comprendere in modo ancor più specifico le alterazioni a carico degli strati profondi della cornea, consentendo diagnosi sempre più precoci e dettagliate.
Utilizzo in endocrinologia
L’OCT è particolarmente utile nello studio della retina nei pazienti diabetici, permettendo di individuare precocemente segni di retinopatia diabetica, edema maculare e alterazioni micro-vascolari. Ciò consente un monitoraggio più accurato delle complicanze oculari legate al diabete mellito, migliorando la gestione complessiva del paziente.
Inoltre, l’OCT può essere impiegata nella valutazione dei nervi ottici in pazienti con orbitopatia di Graves, per monitorare lo stato delle fibre nervose e l’eventuale compressione orbitale.
Alcuni studi sperimentali stanno anche esplorando il ruolo dell’OCT nel controllo delle complicanze neuro-oftalmologiche delle malattie ipofisarie, fornendo un supporto aggiuntivo alla diagnostica tradizionale.
Grazie alla sua rapidità, sicurezza e precisione, la tomografia a coerenza ottica rappresenta un importante strumento complementare nella pratica endocrinologica moderna, favorendo una diagnosi precoce e un follow-up più mirato delle complicanze sistemiche.
Figura 1. Esame normale
L'immagine mostra un esame OCT perfettamente nella norma, in cui sono riconoscibili i singoli strati retinici ed è mantenuta la corretta morfologia dei singoli strati e della retina in toto; è ben identificabile la fisiologica depressione foveale.
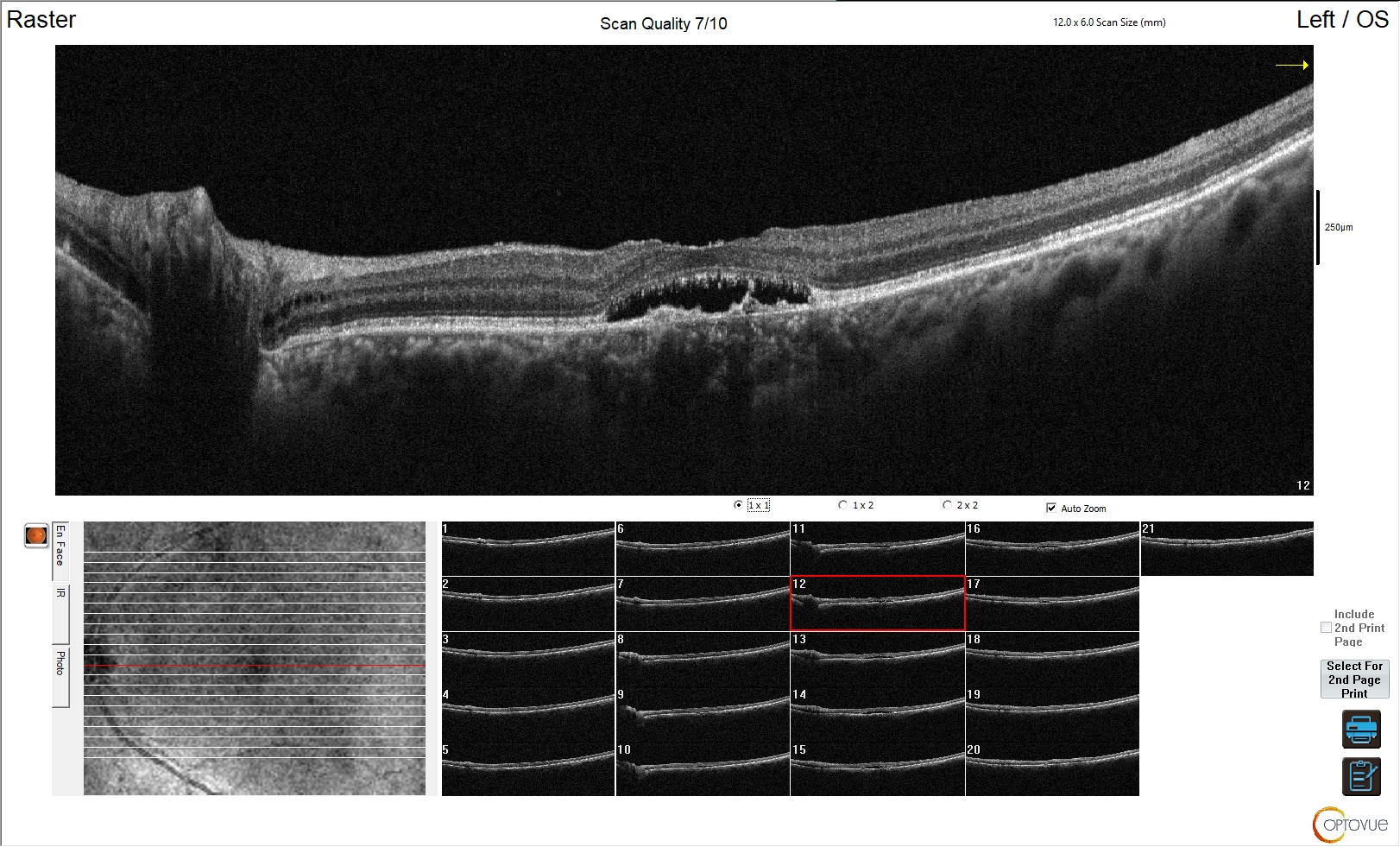
Figura 2. Maculopatia neo-vascolare nel soggetto anziano
L'esame OCT in questa immagine mostra un’alterazione contenuta della morfologia retinica. È scomparsa la fisiologica depressione foveale ed è presente negli strati interni della retina a livello dell'epitelio pigmentato un'area iporiflettente, con irregolarità di morfologia a livello del complesso epicoriocapillare. Tale quadro è riconducibile a una maculopatia senile neo-vascolare.
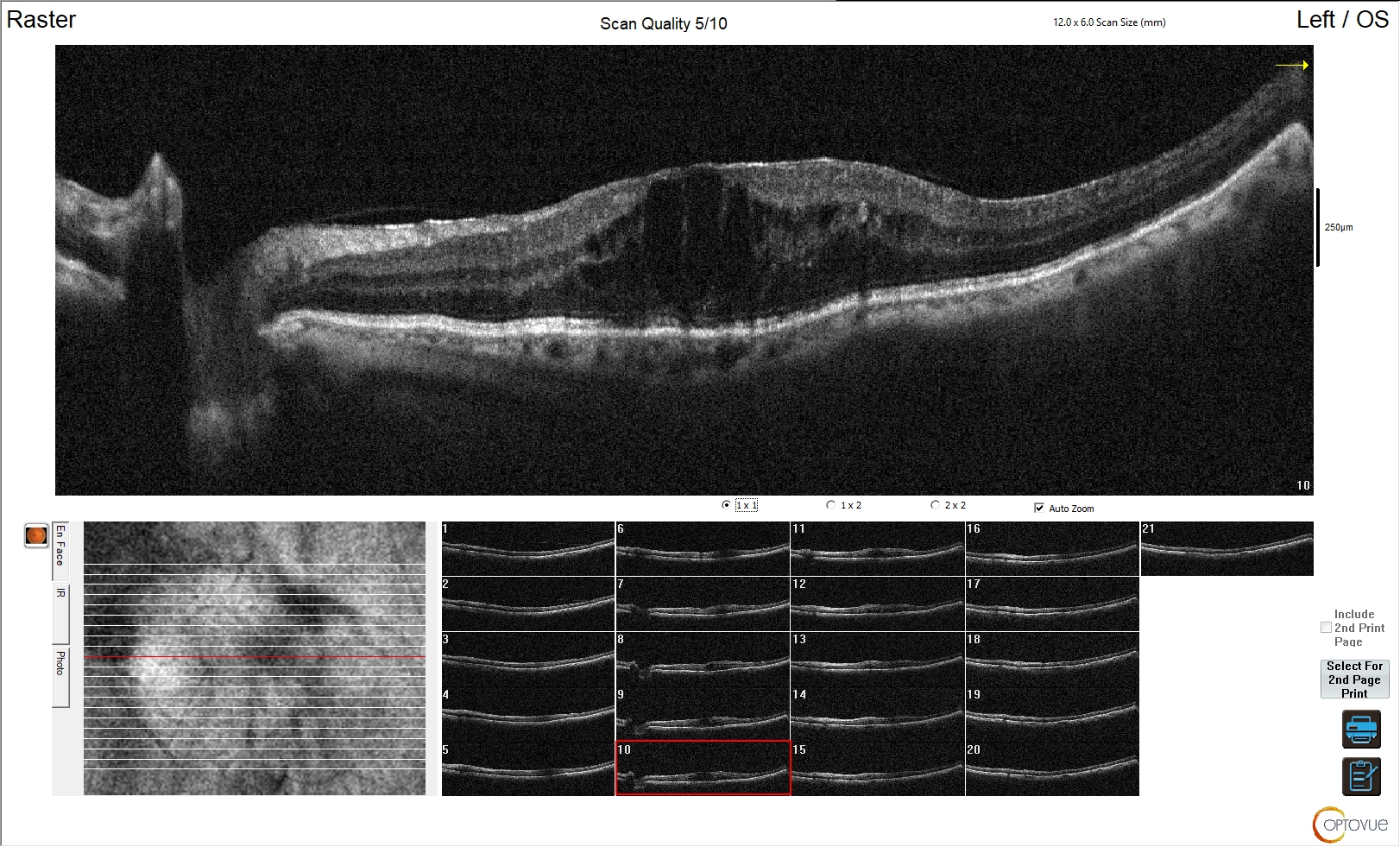
Figura 3. Edema maculare cistoide da retinopatia diabetica
L'esame OCT della regione maculare mostra marcato aumento di spessore della retina. Risulta sovvertita la normale architettura e morfologia della retina, con scomparsa della fisiologica depressione foveale. È evidente nella porzione centrale dell'esame un edema cistico da retinopatia diabetica con interessamento maculare.
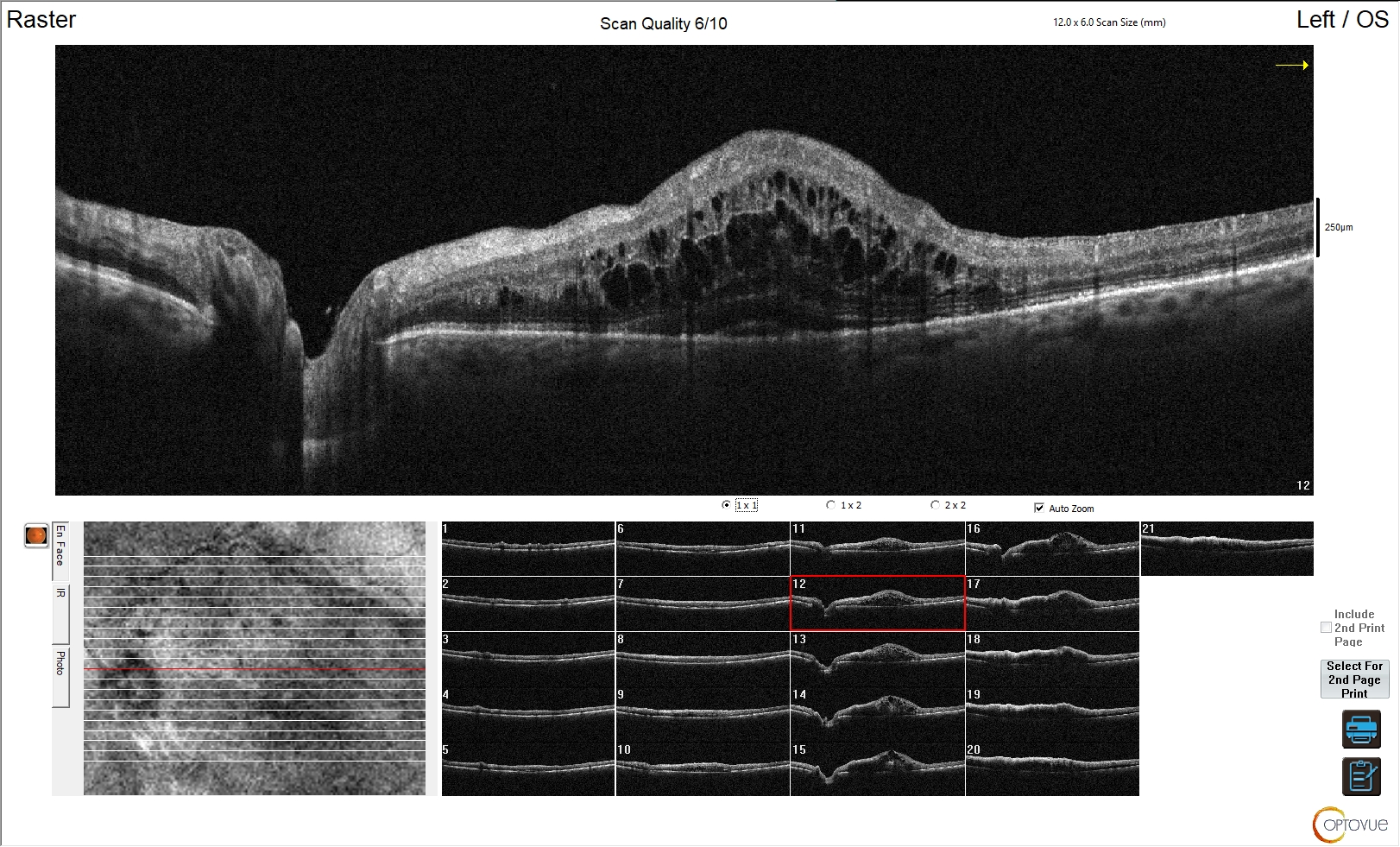
Figura 4. Edema maculare indotto da trombosi vascolare venosa
L'esame OCT in questa immagine evidenzia un’alterazione della fisiologica morfologia sia degli strati esterni che degli strati interni della retina. Non è riconoscibile la fisiologica depressione foveale ed è evidente la presenza di edema maculare ed extra-maculare, con alterazione degli strati interni della retina. Tale quadro è riconducibile a trombosi venosa retinica, con tipico edema intra-retinico diffuso nell'area ischemica.
Bibliografia
- Crincoli E, Sacconi R, Querques L, Querques G. OCT angiography 2023 update: focus on diabetic retinopathy. Acta Diabetol 2024, 61: 533-41.
- Nesper PL, Soetikno BT, Zhang HF, Fawzi AA. OCT angiography and visible-light OCT in diabetic retinopathy. Vision Res 2017, 139: 191-203.
- Reznicek L, Kolb JP, Klein T, et al. Wide-field Megahertz OCT imaging of patients with diabetic retinopathy. J Diabetes Res 2015, 2015: 305084.
Generalità di patologia, classificazione, epidemiologia delle malattie ipotalamo-ipofisarie
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Tutte le strutture della regione ipotalamo-ipofisaria possono essere coinvolte da un’ampia gamma di lesioni di diversa natura (tabella).
| Lesioni | ||
| Tumori derivanti dalle cellule adeno-ipofisarie | ||
| Tumori derivanti dalle cellule neuro-ipofisarie | Pituicitomi Tumori a cellule granulari (coristomi) |
|
| Tumori parasellari | Maligni | Gliomi Tumori a cellule germinali Linfomi primitivi Metastasi Tumori neuroectodermici primitivi sovratentoriali Ependimoblastomi |
| Potenzialmente maligni (basso-grado) |
Cordomi Condrosarcomi Emangiopericitomi Tumori fibrosi solitari Istiocitosi a cellule di Langerhans Plasmocitomi |
|
| Solitamente benigni | Craniofaringiomi Meningiomi Paragangliomi Lipomi Neurinomi/Schwannomi Gangliocitomi |
|
| Lesioni malformative | Cisti della tasca di Rathke Epidermoidi Dermoidi Amartomi Empty sella Cisti aracnoidi |
|
| Lesioni granulomatose, infettive, infiammatorie | Ipofisiti Ascessi ipofisari Pseudotumori Tuberculosi Micosi Sarcoidosi Granulomatosi di Wegener Mucocele sfenoidale Sindrome di Tolosa–Hunt |
|
| Lesioni vascolari | Aneurismi Fistola carotido-cavernosa Trombosi del seno cavernoso |
|
I dati dei registri oncologici suggeriscono che la prevalenza dei tumori primitivi del SNC sia di 130–230/100.000 (1). Le lesioni della regione sellare e parasellare siono molto comuni, fino al 10–15% delle masse intracraniche su casistiche neurochirurgiche (2), e 3–24% su autopsie non mirate (a seconda del numero di sezioni esaminate) (3).
La malignità potenziale di questi tumori può essere definita secondo la classificazione WHO dei tumori del SNC (4,5):
- tumori di grado I WHO, con basso potenziale proliferativo e buona possibilità di guarigione neurochirurgica;
- tumori di grado II WHO, infiltrativi con bassa attività mitotica, che possono recidivare e progredire a malignità maggiore;
- tumori di grado III WHO, con evidenza istologica di malignità;
- tumori di grado IV WHO, mitoticamente attivi con rapida evoluzione.
La regione parasellare può essere coinvolta da un gran numero di altre lesioni non neoplastiche, infiammatorie, granulomatose, infettive o vascolari. La tabella elenca le varie possibili lesioni secondo un criterio eziopatogenetico e anatomico. È importante ricordare come tutte le classificazioni (e questa non fa eccezione), create per ordinare in qualche modo un grande gruppo di entità diverse, da quelle tipiche e frequenti alle atipiche e rarissime, non sono mai in grado di incasellare in modo del tutto soddisfacente l’ampia varietà delle malattie umane (3).
Secondo diverse casistiche neurochirurgiche comprendenti migliata di casi (1120 casi in 18 anni in un unico centro (6), il Registro Tedesco dei tumori ipofisari con 4122 casi in 10 anni (7), 1469 casi in 10 anni in un unico centro (8), gli adenomi ipofisari comprendono circa il 90% delle lesioni di questa regione. Quindi tutto il resto delle numerosissime cause rappresenta l’~8-15% dei casi: tumori nel 4.2-5.6%, lesioni malformative nel 2.9-5.2%, lesioni infiammatorie nello 0.7-1.2% dei casi. In queste casistiche NCH esiste ovviamente una sottostima delle lesioni vascolari.
Un’osservazione dal punto di vista neuroradiologico, con la valutazione retrospettiva di 2598 RM in 11 anni (9), ha dimostrato che, dopo aver escluso tutte le ipofisi normali (47%), le lesioni non adenomatose rappresentavano il 18% delle lesioni rilevate.
Bibliografia
- Davis FG, Kupelian V, Freels S, McCarthy B, & Surawicz T. Prevalence estimates for primary brain tumors in the United States by behavior and major histology groups. Neuro-oncology 2001, 3: 152–8.
- Terada T, Kovacs K, Stefaneanu L, Horvath E. Incidence, pathology, and recurrence of pituitary adenomas: study of 647 unselected surgical cases. Endocr Pathol 1995, 6: 301–10.
- Kovacs K, Horvath E, & Vidal S. Classification of pituitary adenomas. J Neurooncol 2001, 54: 121-7.
- Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007, 114: 97–109.
- Lloyd RV, Kovacs K, Young Jr WF, et al. Pituitary tumors. In: Introduction. WHO classification of tumors of the endocrine organs: pathology and genetics of endocrine organs, Eds. DeLellis R, Lloyd RV, Heitz PV, Eng C, International Agency for Research and Cancer (IARC), Lyon, 2004: pp. 10–13.
- Freda PU, & Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin N Amer 1999, 28: 81-117.
- Saeger W, Lüdecke DM, Buchfelder M, et al. Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. Eur J Endocrinol 2007, 156: 203-16.
- Valassi E, Biller BMK, Klibanski A, & Swearingen B. Clinical features of nonpituitary sellar lesions in a large surgical series. Clin Endocrinol 2010, 73: 798–807.
- Famini P, Maya MM, & Melmed S. Pituitary Magnetic Resonance Imaging for Sellar and Parasellar Masses: Ten-Year Experience in 2598 Patients. J Clin Endocrinol Metab 2011, 96: 1633–41.
Generalità di clinica delle malattie ipotalamo-ipofisarie
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Se si escludono i casi di sindromi ipersecernenti, la presentazione clinica e radiologica delle lesioni della regione è spesso simile: riscontro incidentale con indagine neuroradiologica praticata per motivi non correlati in assenza di ogni sintomo o segno, agli effetti compressivi locali di una massa che cresce in prossimità di strutture delicate e critiche, la cui gravità dipende da localizzazione, dimensioni e velocità di crescita (1,2).
I sintomi più comuni sono i disturbi visivi per compressione delle vie ottiche a diversi livelli (con uno spettro continuo che va dal minimo difetto campimetrico alla cecità bilaterale) e la cefalea che può essere gravemente invalidante, dipendente sia dalla grandezza della massa, che dalla distorsione del diaframma sellare o dall’irritazione della dura (3,4).
Sono comuni anche ipopituitarismo e iperprolattinemia (per la mancanza della normale azione inibitoria di dopamina sulla secrezione di PRL), mentre diabete insipido e paralisi dei nervi cranici sono rari negli adenomi ma comuni in altre lesioni della regione.
La localizzazione ipotalamica può produrre una sindrome diencefalica nel bambino (deperimento fino alla cachessia, obesità, iperpiressia, diabete insipido, adipsia, deficit di crescita e maturazione sessuale) o alterazione del controllo dell’appetito nell’adulto (con grave obesità o denutrizione).
La diagnosi di queste lesioni richiede quindi un approccio multidisciplinare, con studi endocrini, oculistici e neuroradiologici. Un corretto orientamento diagnostico è preliminare a qualunque terapia, perché anche se molte lesioni sono di competenza del NCH, alcune possono o devono essere trattate con farmaci (in modo preliminare o esclusivo) e anche nel campo NCH l’approccio o la tecnica chirurgica possono essere diversi in relazione al tipo di lesione ipotizzata/diagnosticata.
Bibliografia
- Famini P, Maya MM, & Melmed S. Pituitary Magnetic Resonance Imaging for Sellar and Parasellar Masses: Ten-Year Experience in 2598 Patients. J Clin Endocrinol Metab 2011, 96: 1633–41.
- Glezer A, Belchior Paraiba D, et al. Rare sellar lesions. Endocrinol Metab Clin North Am 2008, 37: 195-211.
- Arafah BM, Prunty D, Ybarra J, et al. The dominant role of increased intrasellar pressure in the pathogenesis of hypopituitarism, hyperprolactinemia, and headaches in patients with pituitary adenomas. J Clin Endocrinol Metab 2000, 85: 1789–93.
- Levy MJ, Jäger HR, Powell M, et al. Pituitary volume and headache: size is not everything. Arch Neurol 2004, 61: 721–5.
Classificazione ed epidemiologia degli adenomi ipofisari
Sofia Asioli1,2 & Maria Vittoria Altavilla1
1Dipartimento di Scienze Biomediche e Neuromotorie (DIBINEM), Alma Mater Studiorum Università di Bologna
2Programma Neurochirurgia Ipofisi - Pituitary Unit, IRCCS Istituto delle Scienze Neurologiche di Bologna
(aggiornato al 9/12/23)
Generalità
I tumori che originano dalle cellule dell’adeno-ipofisi sono stati tradizionalmente (sec. World Health Organization 2017) definiti come “adenomi ipofisari”. Poichè queste neoplasie possono essere localmente invasive e metastatizzare, è stata introdotta la nomenclatura di “Tumori Neuroendocrini Ipofisari (PitNET)” (1-4), per meglio definirne l’entità come neoplasie neuroendocrine e soprattutto per racchiudere l’ampio spettro di andamento clinico: da lesioni piccole e indolenti, a quelle dimensionalmente più importanti, localmente invasive e non resecabili chirurgicamente. Generalmente sono localizzate nella regione della sella turcica, spesso con estensione in sede sovra-sellare, ma vi sono anche localizzazioni ectopiche che includono il seno sfenoidale e, raramente, il clivus.
Clinicamente gli adenomi/PitNETs ipofisari possono essere di piccole dimensioni (< 1 cm, micro-adenomi/PitNETs) e a lenta crescita, motivo per cui vengono diagnosticati perlopiù come riscontro occasionale; possono anche dare origine a sindromi da eccesso ormonale, tra cui iperprolattinemia, acromegalia o gigantismo, morbo di Cushing o ipertiroidismo. Gli adenomi/PitNETs di grandi dimensioni (> 1 cm, macro-adenomi/PitNETs) possono causare invece sintomi da massa intra-cranica (tra i più comuni, ad esempio, cefalea e disturbi del campo visivo) e portare a un quadro di ipopituitarismo. Alcuni possono estendersi verso le sedi inferiori e apparire come masse nasali o para-nasali, mentre in altri casi possono andare incontro a necrosi emorragica acuta, con rapida espansione e conseguente presentazione clinica definita "Apoplessia ipofisaria", caratterizzata da cefalea grave, letargia e sintomi da aumento della pressione intra-cranica (5).
Gli adenomi/PitNETs ipofisari per la maggior parte non sono invasivi e possono presentare una crescita espansiva nella regione sellare. Gli adenomi/PitNETs ipofisari invasivi (sotto-gruppo che rappresenta una percentuale variabile in letteratura, di circa il 30%) nonostante la terapia multi-modale possono comportare la presenza di residuo neoplastico e recidiva, anche precoce e multipla (1). Pertanto, dopo l’intervento neurochirurgico di prima linea, eseguito nel 56% dei PitNETs diagnosticati (6), è richiesta la sorveglianza clinica e in un numero considerevole di pazienti un trattamento adiuvante.
Nella maggior parte dei casi l’identificazione di queste neoplasie è di tipo incidentale, in corso di approfondimenti, perlopiù strumentali, per altri quadri clinici. In particolare, esami strumentali come la risonanza magnetica con mezzo di contrasto (Gadolinio) hanno una forte utilità anche nell’inquadramento e nella classificazione degli adenomi ipofisari, perché permettono di identificare la localizzazione della massa (sede intra-sellare e/o sovra-sellare e/o para-sellare e/o infra-sellare e/o retro-sellare), di caratterizzarne le dimensioni (micro- o macro-adenoma/PitNET), di verificare l’eventuale compressione del chiasma ottico e l’invasione delle strutture contigue (es. seno cavernoso o seno sfenoidale) e la presenza di emorragia o di alterazioni cistiche (1,5). L’evidenza radiologica di crescita invasiva sembra oltretutto avere un valore prognostico, identificando i tumori con potenziale aggressivo e guidandone il trattamento, in aggiunta alla valutazione dei biomarcatori tumorali (es. recettori della somatostatina-SSTR, degli estrogeni-ER) (7).
Epidemiologia
I tumori dell’adeno-ipofisi sono al terzo posto (12-15%) tra le neoplasie più frequenti del sistema nervoso centrale nell’adulto. Circa il 20% delle ghiandole ipofisarie "normali" presenta una lesione incidentale radiologicamente rilevabile (8).
Numerosi studi di popolazione degli ultimi 15 anni hanno identificato una prevalenza compresa tra 78 e 116 casi per 100.000 persone (6,9-11). I tumori lattotropi sono i più comuni PitNET funzionali (12), solitamente trattati con terapia farmacologica, per cui la maggior parte di essi non viene rilevata nelle statistiche oncologiche o chirurgiche. Il 56% dei PitNET diagnosticati clinicamente viene sottoposto a resezione chirurgica (6) e tra questi più del 40% sono masse funzionalmente subcliniche o silenti della linea SF1 (13), circa il 15% della linea TPIT, e circa il 30% di quella PIT1, dando luogo in più della metà dei casi a un eccesso di GH (6,13).
L'incidenza degli adenomi ipofisari/PitNETs aumenta con l'età. Circa il 5% dei pazienti riceve la diagnosi prima dei 20 anni (14). Gli adenomi ipofisari/PitNETs si manifestano in ugual misura in entrambi i sessi, anche se alcuni studi mostrano una predominanza femminile di alcuni sottotipi. In particolare, la malattia di Cushing e i tumori lattotropi sono più comuni nei pazienti di sesso femminile, mentre i tumori non funzionanti sono più spesso resecati chirurgicamente nei pazienti di sesso maschile (9,11). La prevalenza degli adenomi ipofisari/ PitNETs è in aumento (6) ed è probabilmente destinata ad aumentare ulteriormente con l’invecchiamento della popolazione e il miglioramento delle tecniche di imaging (9). Tali neoplasie rimangono tuttavia una patologia sotto-diagnosticata, anche quando sono clinicamente rilevanti. Questo è particolarmente vero nella popolazione anziana, perché i sintomi a lenta progressione (anche di adenomi/PitNETs funzionanti, come i tumori corticotropi) vengono confusi con disturbi dovuti all’età avanzata, come è stato ben evidenziato per l'acromegalia in uno studio in cui, con una ricerca attiva della malattia, si è trovata una prevalenza di 1000 casi per milione, 10 volte superiore a quella degli studi inglese e belga (10-12).
Classificazione
La classificazione secondo la 4a edizione della World Health Organization (WHO 2017) ha proposto la delineazione dei tipi e sotto-tipi dei tumori primitivi dell’adeno-ipofisi sulla base di criteri anatomo-patologici e dell’espressione dei fattori di trascrizione specifici per ogni linea cellulare, con l’obiettivo di stabilire un approccio univoco e riproducibile ai fini diagnostici. L’ultima modifica di nomenclatura, utilizzata attualmente e presente nella 5° edizione sia della classificazione WHO 2022 dei tumori endocrini e neuroendocrini (1) sia della classificazione WHO 2021 dei tumori del sistema nervoso centrale (5) ha portato al cambio di nomenclatura e di IC-COD (da 0 a 3) sostituendo il termine “adenoma” con “Pituitary Neuroendocrine Tumour/adenoma” (PitNET/adenoma) (15). Inoltre, ha supportato l’utilizzo dei fattori di trascrizione specifici e degli ormoni ipofisari per classificare la linea evolutiva dei PitNET. Tuttavia, nonostante venga suggerito di riportare marcatori di proliferazione (come Ki67 e/o conta mitotica) non è consigliato l’utilizzo di un grading come per le altre neoplasie endocrine. A tal proposito si suggerisce di integrare il grading francese proposto da Trouillas (16), che riporta la suddivisione di adenomi ipofisari/PitNets in 5 categorie (vedi paragrafo “Adenomi/PitNETs aggressivi e grading”).
Infine, viene suggerito l’utilizzo del report standardizzato proposto nel 2019 dall’European Pituitary Pathology Group (15).
La classificazione definisce innanzitutto tre categorie sulla base dell’espressione dei fattori di trascrizione PIT1, TPIT e SF1. All’interno di ognuna vengono poi descritti i sotto-tipi distinti a seconda del tipo di cellula ipofisaria d’origine (figura):
1. PiNETs della linea PIT1:
- somatotropo;
- mammo-somatotropo;
- lattotropo;
- tireotropo;
- pluri-ormonale maturo della linea PIT1;
- immaturo;
- a cellule staminali acidofile;
- misto somatotropo-lattotropo;
2. PitNETs della linea TPIT:
- corticotropo;
3. PitNETs della linea SF1:
- gonadotropo;
4. PitNETs senza una distinta linea di differenziazione:
- pluri-ormonali;
- “null cell”;
5. PitNETs multipli
- multipli e sincroni di distinte linee.
Inoltre, sulla base dell’espressione della CAM 5.2 (cito-cheratina a basso peso molecolare), valutata alla caratterizzazione immuno-istochimica, alcuni adenomi vengono distinti ulteriormente in densamente e sparsamente granulati (tabella).
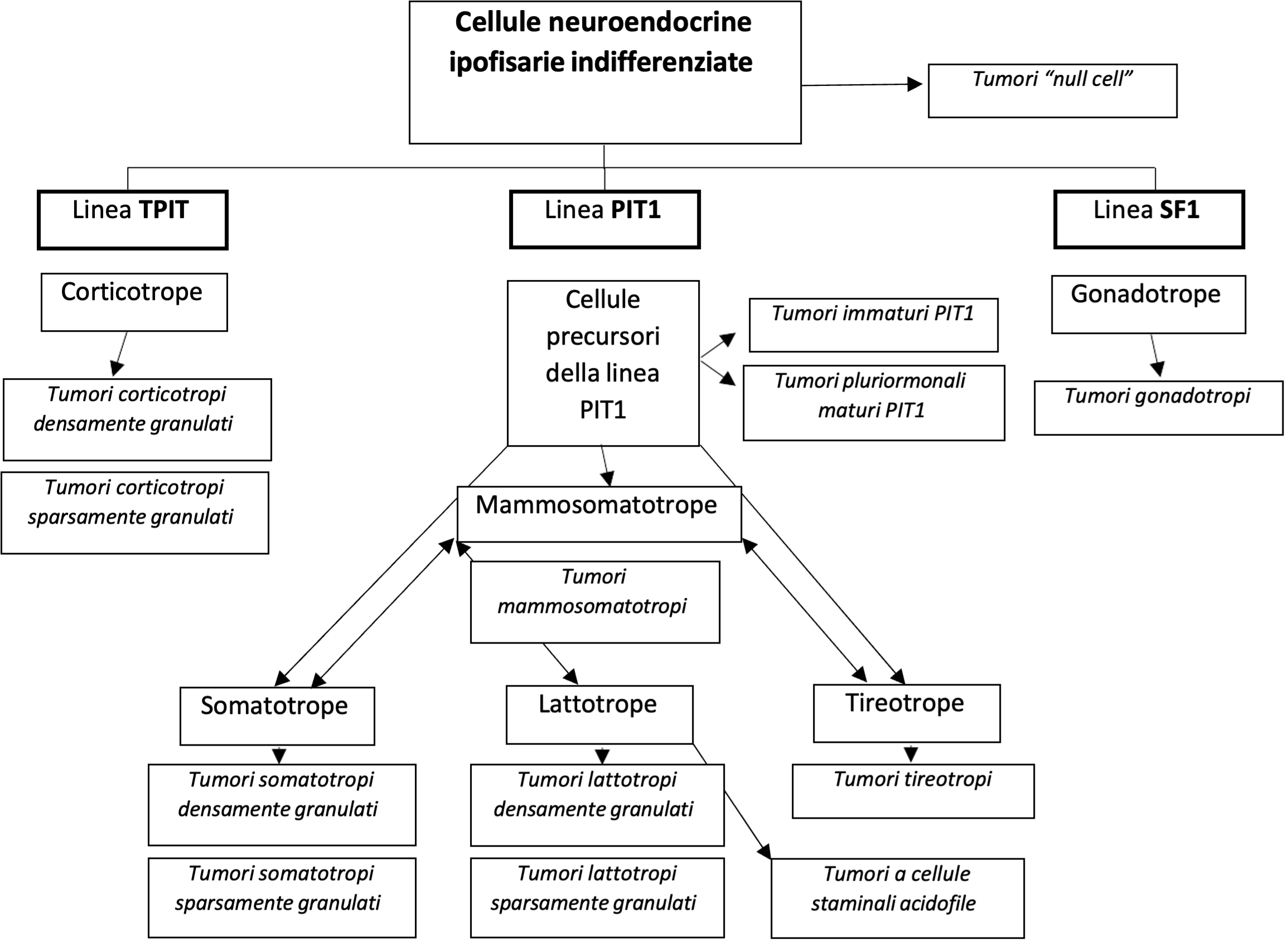
Le cellule adeno-ipofisarie secernenti ormoni si differenziano lungo tre linee per formare cellule mature. Ogni tipo di cellula matura dà origine ad almeno un tipo di adenoma/tumore ipofisario. Esistono poi adenomi senza alcuna differenziazione cellulare d’origine, detti “null cell” (modificata da 4).
| Classificazione PitNETs | |||
| Tipo di adenoma | Ormoni | Cheratina (CAM 5.2) | Sotto-tipo tumorale |
| Linea PIT-1 | |||
| Somatotropi | GH, α-subunità | Peri-nucleare | Tumori somatotropi densamente granulati |
| GH | Corpi fibrosi (>70%) | Tumori somatotropi sparsamente granulati | |
| Lattotropi | PRL (para-nucleare) | Debole/negativa | Tumori lattotropi sparsamente granulati |
| PRL (citoplasmatico) | Debole/negativa | Tumori lattotropi densamente granulati | |
| Mammo-somatotropi | GH (spesso predominante) PRL, α-subunità | Peri-nucleare | - |
| Tireotropi | α-subunità, β-TSH | Debole/negativa | - |
| Pluri-ormonali maturi della linea PIT-1 | GH (spesso predominante), PRL, α-subunità, β-TSH | Peri-nucleare | - |
| Adenomi a cellule staminali acidofile | PRL (predominante), GH (focale/ variabile) | Sparsi corpi fibrosi | - |
| Immaturi della linea PIT-1 | GH, PRL, α-subunità, β-TSH | Focale/variabile | - |
| Linea TPIT | |||
| Corticotropi | Variabile | Variabile | Tumori corticotropi densamente granulati |
| Variabile | Tumori corticotropi sparsamente granulati | ||
| Peri-nucleare intensa (ad anello) | Tumori a cellule di Crooke | ||
| Linea SF1 | |||
| Gonadotropi | α-subunità, β-FSH, β-LH | Variabile | - |
| Senza linea di differenziazione | |||
| Pluri-ormonali non classificabili | Espressioni multiple | Variabile | - |
| Null cell | - | Variabile | - |
Adenomi/PitNETs aggressivi e grading
Storicamente, in assenza di chiari segni patologici di malignità, solo i rari tumori ipofisari con metastasi confermate (0.2%) venivano considerati maligni e definiti “carcinomi ipofisari” (17). La classificazione WHO del 2014 introdusse inoltre il termine "adenoma atipico", definito da caratteristiche morfologiche (Ki-67 ≥ 3%, colorazione estesa di p53 e numerose mitosi) che di solito mancano nei tumori benigni. In assenza però di una validazione clinica, questo sistema di classificazione è stato abbandonato nella classificazione WHO del 2017.
Nonostante i vari studi precedenti (16,18-21), la nuova edizione della classificazione WHO (5° edizione, WHO 2022) non supporta alcun sistema di stratificazione prognostica e considera più aggressivi solo alcuni tipi e sotto-tipi di adenomi ipofisari/PitNETs. Tra questi rientrano i tumori lattotropi nei pazienti di sesso maschile, i tumori immaturi della linea PIT1, i tumori a cellule di Crooke, i tumori a cellule staminali acidofile e i tumori corticotropi "silenti" ovvero biochimicamente non funzionanti. Questo approccio fa dell'isto-tipo l’indicatore prognostico e predittivo più forte, ma è ancora dibattuta in letteratura l'evidenza di una prognosi peggiore per questi sotto-tipi cosiddetti "ad alto rischio di recidiva".
Tra le classificazioni, tuttavia, quella maggiormente accreditata è la stratificazione prognostica a 5 gradi proposta da Trouillas et al (16) e validata su 2565 pazienti da studi indipendenti (22-26). In tale sistema la stratificazione viene definita in base all'invasione e all’attività proliferativa. L'invasione viene definita sulla base dell’evidenza radiologica di invasione del seno cavernoso o sfenoidale e si definisce assente (= 1) o presente (= 2). I tumori sono poi considerati proliferanti (designati "b"), se due di questi tre marcatori risultano al di sopra di cut-off pre-definiti:
- Ki-67 ≥ 3%;
- conta mitotica > 2 per 10 campi ad alto ingrandimento;
- p53 positivo (> 10 nuclei fortemente positivi per 10 campi ad alto ingrandimento).
In base a questi criteri, i tumori sono classificati come segue:
- grado 1a: non invasivo e non proliferante;
- grado 1b: non invasivo e proliferante;
- grado 2a: invasivo e non proliferante;
- grado 2b: invasivo e proliferante;
- grado 3: tumore maligno con metastasi.
In conclusione, si è evidenziato come i tumori 2b, se definiti accuratamente, rappresentino circa l'8% di tutte le neoplasie resecate chirurgicamente e come mostrino effettivamente un significativo aumento del rischio di recidiva e progressione (da 4 a 8 volte), indipendentemente dal tipo e dal sotto-tipo istologico (18,20).
Altre malattie della regione ipofisaria-sellare
1. Altri tumori dell’ipofisi anteriore:
- craniofaringioma adamantinomatoso;
- craniofaringioma papillare;
- blastoma
2. Neoplasie dell’ipofisi posteriore e dell’ipotalamo:
- tumori derivanti dalle cellule ipofisarie:
- pituicitoma;
- tumore a cellule granulari della regione sellare/pituicitoma a cellule granulari;
- oncocitoma a cellule fusate/pituicitoma oncocitico;
- pituicitoma ependimale;
- tumori neuronali:
- gangliocitoma e adenoma/PitNET-gangliocitoma misto;
- neurocitoma sellare.
3. Altri tumori sellari:
- meningioma;
- cordoma.
L'adeno-ipofisi deriva dall'ectoderma orale e i tumori che la riflettono con morfologia squamosa e adamantinomatosa sono noti come craniofaringiomi. Gli studi molecolari hanno identificato due vie molecolari distinte: i cranio-faringiomi papillari ospitano mutazioni BRAF p.V600E, mentre i sotto-tipi adamantinomatosi presentano mutazioni CTTNB1, che determinano immuno-reattività nucleare della ß-catenina (27).
I blastomi ipofisari sono tumori primitivi molto rari, composti da una commistione di cellule neuroendocrine adeno-ipofisarie, cellule primitive della tasca di Rathke e cellule follicolo-stellate. Sono un segno distintivo della sindrome DICER1 (28).
I tumori dell’ipofisi posteriore sono stati a lungo riconosciuti come neoplasie delle cellule ipofisarie (29,30). Ad oggi sappiamo che la neuro-ipofisi normale contiene sotto-tipi cellulari, quali cellule fusate, cellule oncocitiche, granulari ed ependimali (31,32), da cui si ipotizza derivino le diverse neoplasie di questo gruppo, quali i pituicitomi, i tumori a cellule granulari della regione sellare/pituicitomi a cellule granulari, gli oncocitomi a cellule fusate/pituicitomi oncocitici e i pituicitomi ependimali. Tali neoplasie, tutte positive all’indagine immuno-istochimica per anticorpo anti-TTF1, sembrano rappresentare lo spettro di un'unica entità nosologica e nella clsassificazione WHO 2022 dei tumori endocrini e neuroendocrini viene suggerito di classificarle uniformemente come sotto-tipi di pituicitoma (1,30).
Da molti anni è nota anche l'esistenza dei gangliocitomi nella sella turcica, che possono avere una secrezione ormonale e/o possono essere associati agli adenomi/PitNET.
Lo spettro dei tumori neuronali è stato recentemente ampliato dal riconoscimento dei neurocitomi sellari (33).
Bibliografia
- WHO Classification of Tumours Editorial Board. Endocrine and neuroendocrine tumours. Lyon (France): International Agency for Research on Cancer; 2022. (WHO classification of tumours series, 5th ed, vol. 10).
- Asa SL, Casar-Borota O, Chanson P, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer 2017, 24: C5-8.
- Asa SL, Asioli S, Bozkurt S, et al. Pituitary neuroendocrine tumors (PitNETs): nomenclature evolution, not clinical revolution. Pituitary 2020, 23: 322-5.
- Asa SL, Mete O, Cusimano MD, et al. Pituitary neuroendocrine tumors: a model for neuroendocrine tumor classification. Mod Pathol 2021, 34: 1634-50.
- WHO Classification of Tumours Editorial Board. Central nervous system tumours. Lyon (France): International Agency for Research on Cancer; 2021. (WHO classification of tumours series, 5th ed, vol. 6).
- Daly AF, Rixhon M, Adam C, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab 2006, 91: 4769-75.
- Delgrange E, Vasiljevic A, Wierinckx A, et al. Expression of estrogen receptor alpha is associated with prolactin pituitary tumor prognosis and supports the sex-related difference in tumor growth. Eur J Endocrinol 2015, 172: 791-801.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer 2004, 101: 613-9.
- Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf) 2010, 72: 377-82.
- Fontana E, Gaillard R. Epidémiologie des adénomes hypophysaires: étude dans une agglomération urbaine de Suisse. Rev Med Suisse 2009, 5: 2172-4.
- Agustsson TT, Baldvinsdottir T, Jonasson JG, et al. The epidemiology of pituitary adenomas in Iceland, 1955-2012: a nationwide population-based study. Eur J Endocrinol 2015, 173: 655-64.
- Daly AF, Beckers A. The epidemiology of pituitary adenomas. Endocrinol Metab Clin North Am 2020, 49: 347-55.
- Mete O, Cintosun A, Pressman I, Asa SL. Epidemiology and biomarker profile of pituitary adenohypophysial tumors. Mod Pathol 2018, 31: 900-9.
- Kane LA, Leinung MC, Scheithauer BW, et al. Pituitary adenomas in childhood and adolescence. J Clin Endocrinol Metab 1994, 79: 1135-40.
- Villa C, Vasiljevic A, Jaffrain-Rea ML, et al. A standardised diagnostic approach to pituitary neuroendocrine tumours (PitNETs): a European Pituitary Pathology Group (EPPG) proposal. Virchows Arch 2019, 475: 687-92.
- Trouillas J, Roy P, Sturm N, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol 2013, 126: 123-35.
- Dekkers OM, Karavitaki N, Pereira AM. The epidemiology of aggressive pituitary tumors (and its challenges). Rev Endocr Metab Disord 2020, 21: 209-12.
- Trouillas J, Burman P, McCormack A, et al. Aggressive pituitary tumours and carcinomas: two sides of the same coin? Eur J Endocrinol 2018, 178: C7–9.
- McCormack A, Dekkers OM, Petersenn S, et al & ESE survey collaborators. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol 2018, 178: 265–76.
- Raverot G, Ilie MD, Lasolle H, et al. Aggressive pituitary tumours and pituitary carcinomas. Nature Rev Endocrinol 2021, 17: 671–84.
- Burman P, Trouillas J, Losa M, et al & ESE survey collaborators. Aggressive pituitary tumours and carcinomas, characteristics and management of 171 patients. Eur J Endocrinol 2022, 187: 593–605.
- Raverot G, Dantony E, Beauvy J, et al. Risk of recurrence in pituitary neuroendocrine tumors: a prospective study using a five-tiered classification. J Clin Endocrinol Metab 2017, 102: 3368–74.
- Lelotte J, Mourin A, Fomekong E, et al. Both invasiveness and proliferation criteria predict recurrence of non-functioning pituitary macroadenomas after surgery: a retrospective analysis of a monocentric cohort of 120 patients. Eur J Endocrinol 2018, 178: 237–46.
- Asioli S, Righi A, Iommi M, et al. Validation of a clinicopathological score for the prediction of post-surgical evolution of pituitary adenoma: retrospective analysis on 566 patients from a tertiary care centre. Eur J Endocrinol 2019, 180: 127–34.
- Guaraldi F, Zoli M, Righi A, et al. A practical algorithm to predict postsurgical recurrence and progression of pituitary neuroendocrine tumours (PitNET)s. Clin Endocrinol 2020, 93: 36–43.
- Sahakian N, Appay R, Resseguier N, et al. Real-life clinical impact of a five-tiered classification of pituitary tumors. Eur J Endocrinol 2022, 187: 893–904.
- Fukuhara N, Iwata T, Inoshita N, et al. Immunohistochemistry or molecular analysis: which method is better for subtyping craniopharyngioma? Endocr Pathol 2021, 32: 262-8.
- De Kock L, Sabbaghian N, Plourde F, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol 2014, 128: 111-22.
- Brat DJ, Scheithauer BW, Staugaitis SM, et al. Pituicytoma: a distinctive low-grade glioma of the neurohypophysis. Am J Surg Pathol 2000, 24: 362-8.
- Mete O, Lopes MB, Asa SL. Spindle cell oncocytomas and granular cell tumors of the pituitary are variants of pituicytoma. Am J Surg Pathol 2013, 37: 1694-9.
- Roncaroli F, Scheithauer BW, Cenacchi G, et al. 'Spindle cell oncocytoma' of the adenohypophysis: a tumor of folliculostellate cells? Am J Surg Pathol 2002, 26: 1048-55.
- Takei Y, Seyama S, Pearl GS, Tindall GT. Ultrastructural study of the human neurohypophysis. II. Cellular elements of neural parenchyma, the pituicytes. Cell Tissue Res 1980, 205: 273-87.
- Asa SL, Mete O. Hypothalamic endocrine tumors: an update. J Clin Med 2019, 8: 1741.
Clinica generale degli adenomi ipofisari
Liana Cortesi1, Chiara Carzaniga2
1 Unità di Endocrinologia, Ospedali Riuniti di Bergamo, Bergamo, Italia
2 Unità di Endocrinologia, Istituto Auxologico Italiano, Milano, Italia
Dal punto di vista clinico gli adenomi dell’ipofisi si possono manifestare con 3 problematiche:
- sintomi neurologici/visivi legati all’effetto massa della neoplasia
- sintomi legati all’eccesso di produzione di ormoni ipofisari
- sintomi legati all’ipopituitarismo dovuto all’effetto massa compressivo sull’ipofisi
I disturbi neurologici sono legati alle dimensioni della neoplasia e sono in relazione alla sede di sviluppo della stessa.
L’estrinsecazione in sede sovra-sellare si associa in genere allo stiramento della dura madre (diaframma della sella), con conseguente comparsa di cefalea gravativa in regione frontale, temporale o retro-orbitaria, che non si modifica con i movimenti posturali ed è generalmente resistente alle comuni terapie analgesiche (1). Va comunque tenuto presente che la cefalea causata dall'adenoma ipofisario non è mai caratteristica per localizzazione e modalità di presentazione, salvo che in alcuni casi (per es., invasione del seno cavernoso da parte di un adenoma GH-secernente; di forte intensità con vomito associato nei casi di apoplessia).
Lo sviluppo sovra-sellare può inoltre determinare compressione sul chiasma ottico con conseguente comparsa di disturbi del campo visivo, a carico dei quadranti temporali. L’alterazione visiva che si osserva più di frequente è l’emianopsia bitemporale, cioè la perdita della visione negli emicampi visivi bilateralmente, legata alla compressione del chiasma nella parte centrale dove avviene l’incrocio delle fibre nasali (2).
Raramente vi possono essere disturbi legati al coinvolgimento delle strutture ipotalamiche, con alterazioni della temperatura corporea, presenza di disturbi del ritmo sonno-veglia e iperfagia. Nei casi di apoplessia dell'adenoma, può manifestarsi, dopo qualche giorno, un quadro di diabete insipido, che può essere transitorio, ma anche permanente.
Se lo sviluppo dell’adenoma avviene in senso laterale, l’invasione mono o bilaterale dei seni cavernosi determina un coinvolgimento delle strutture in essi contenuti: arteria carotide interna, III, IV e VI nervo cranico con possibile comparsa di oftalmoplegia, diplopia o dolore nella sede di distribuzione del ramo oftalmico del trigemino.
Più raramente l’adenoma ha uno sviluppo verso il basso determinando erosione del pavimento sellare e possibile perdita di liquido cefalo-rachidiano (liquorrea).
Nel caso di adenomi voluminosi (adenomi giganti), si può verificare compressione delle vie di drenaggio del liquor, con conseguente ipertensione endocranica che clinicamente si manifesta con cefalea intensa accompagnata da vomito e edema della papilla (3).
Sintomi legati all’eventuale presenza di ipersecrezioni ormonali che causano sindromi tipiche:
- ipersecrezione di GH: acromegalia/gigantismo
- ipersecrezione di PRL: ipogonadismo con oligo-amenorrea nella donna, deficit della libido e della potenza nell’uomo, galattorrea
- ipersecrezione di ACTH: ipercortisolismo
- ipersecrezione di TSH: ipertiroidismo
Va ricordato che gli adenomi secernenti di solito producono un solo ormone in eccesso, ma talvolta si può verificare l’ipersecrezione di 2 o più ormoni, l’associazione più frequente è GH/PRL.
Si può inoltre verificare una condizione di iperprolattinemia (con valori di PRL di solito < 200 ng/mL) da disconnessione ipotalamica per compressione dell’adenoma sulle vie di trasporto della dopamina, fisiologico inibitore della PRL (4).
E’ fondamentale differenziare questa condizione dal macroprolattinoma per il diverso approccio terapeutico.
La compressione dell’adenoma sul tessuto ipofisario sano può dare quadri clinici di ipopituitarismo, che può interessare solo alcune tropine ipofisarie (ipopituitarismo parziale) oppure tutte le tropine ipofisarie (panipopituitarismo)(5).
Il deficit ipofisario che compare per primo è il deficit di GH, seguito dall’ipogonadismo, dall’ipotiroidismo e iposurrenalismo centrali (6). Quest’ulitma condizione va identificata e trattata con tempestività prima di iniziare qualsiasi altra terapia sostitutiva.
Di solito lo sviluppo dell’adenoma avviene lentamente e la comparsa dei sintomi è graduale. Vi sono però alcune situazioni, in particolare nei tumori di grosse dimensioni, dove la comparsa della sintomatologia è improvvisa per emorragia intra-tumorale o apoplessia ipofisaria.
Bibliografia
- Melen O. Neuro-ophtalmologic features of pituitary tumors. Endocrinol Metab Clin North Am 1987, 16: 585-608.
- Hollenhorst RW, Young BR. Ocular manifestations produced by adenomas of the pituitary gland: analysis of 1000 cases. In: Kohler PO, Ross GT, eds. Diagnosis and Treatment of Pituitary Tumors. New York American Elsevier, 1973: 53-64.
- Lania A, Ferrante E. Adenomi ipofisari non funzionanti. In: Beck-Peccoz P, et al. Ipofisi. Società Editrice Universo, 2009: 121-9.
- Attanasio R, Cozzi R. Gli adenomi ipofisari. In: Diagnosi e terapia delle malattie della regione ipotalamo-ipofisaria. AME 2007: 49-51.
- Ferrante E, Ferraroni M, Castrignanò T, et al. Non-functioning pituitary adenoma database: a useful resource to improve the clinical management of pituitary tumors. Eur J Endocrinol 2006, 155: 823-9.
- Vance ML. Hypopituitarism. N Engl J Med 1994, 330: 1651-62.
Adenomi clinicamente non funzionanti (NFPA)
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Clinica e diagnostica NFPA
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Sono il tipo più frequente di adenoma ipofisario nelle casistiche NCH.
Dal punto di vista biologico sono un gruppo eterogeneo, classificabili in adenomi:
- gonadotropi (immunoistochimica positiva per ß-FSH e/o ß-LH e/o a-subunità);
- silenti (immunoistochimica positiva per un singolo ormone: GH, PRL, ß-TSH/a-subunità, ACTH);
- null cell (immunoistochimica negativa);
- silenti sottotipo 3 (immunoistochimica positiva per più ormoni ipofisari);
- oncocitomi (citoplasma ricco di mitocondri).
Gli adenomi gonadotropi e null cell costituiscono circa l’85% dei NFPA, mentre i silenti circa il 15% (di cui la maggior parte esprime solo ACTH).
Questa classificazione presenta delle implicazioni cliniche, poiché l’adenoma silente ACTH-positivo e il sottotipo 3 sembrano avere un comportamento più aggressivo e una maggiore invasività nel seno cavernoso.
I sintomi e segni sono spesso aspecifici. Il sintomo principale è il danno visivo, che, quando non acuto, viene spesso diagnosticato tardivamente, solo dopo aver eseguito un campo visivo. I sintomi neurologici meccanici (cefalea, diplopia, ptosi palpebrale) sono molto meno frequenti. Dal punto di vista endocrinologico, l'ipopituitarismo è di difficile diagnosi: a parte il caratteristico fenotipo ipopituitarico dei pazienti con malattia di vecchia data, i sintomi lamentati più di frequente sono i disordini della sfera sessuale (disfunzione erettile, amenorrea secondaria) e l'astenia. Raramente la prima manifestazione può essere un quadro drammatico di apoplessia ipofisaria, con ipopituitarismo acuto (ipoglicemia, iposodiemia, collasso circolatorio). In un recente studio multicentrico italiano (1) su 295 casi di NFPA, al momento della diagnosi erano presenti alterazioni visive nel 67.8% dei pazienti, cefalea nel 41.4%, mentre nel 62% dei casi erano stati rilevati deficit ipofisari multipli o isolati.
L’ipopituitarismo può dipendere dall’ostruzione determinata dalla massa sul flusso portale, oppure dalla distruzione del normale tessuto dell’ipofisi anteriore. Lo sviluppo dell’ipopituitarismo è spesso lento e insidioso, con progressiva perdita delle funzioni dell’ipofisi anteriore, a partire dalla secrezione di GH (considerata l’anomalia endocrina più comune in corso di ipopituitarismo, anche se clinicamente quasi silente nell’adulto) e gonadotropine, seguite nel tempo da un deficit di TSH, e infine di ACTH e PRL.
I livelli di PRL sono invece di solito aumentati, per compressione del peduncolo ipofisario ed interruzione del trasporto di dopamina, se non in una fase molto tardiva associata alla completa distruzione del parenchima ipofisario. Si pone, quindi, il quesito diagnostico di differenziare il NFPA dal prolattinoma: livelli di prolattina < 100 ng/mL fanno generalmente pensare ad iperprolattinemia funzionale associata a NFPA. La diagnosi differenziale è importante per la scelta terapeutica, poiché nella grande maggioranza dei casi negli NFPA è di prima scelta la neurochirurgia e nei prolattinomi la terapia dopaminergica.
Gli NFPA devono essere differenziati, oltre che dagli adenomi ipofisari funzionanti, da altre masse sellari e parasellari, come cisti della tasca di Rathke, craniofaringiomi, meningiomi, cordomi, germinomi, metastasi, processi granulomatosi, ipofisiti. La presentazione clinica è di poco aiuto, in quanto i sintomi sono aspecifici, riflettendo l’effetto massa del tumore. Poiché in caso di adenoma ipofisario la secrezione dell’ipofisi posteriore è colpita solo molto raramente (si osserva solo nei casi di apoplessia parziale o completa dell'adenoma, per compressione acuta del peduncolo), il riscontro di diabete insipido dovrebbe indirizzare ad una lesione non ipofisaria (ipotalamica, granulomatosa, neoplastica).
Molti studi osservazionali hanno tentato di definire la storia naturale degli NFPA, senza alcun trattamento (2,3,4). Nel 2007 sono stati pubblicati i dati di uno studio clinico olandese su 28 pazienti con NFPA, non operati, seguiti per un periodo di più di 7 anni per studiarne il decorso naturale (5): in 14/28 (50%) si è osservato un incremento di volume dell’adenoma (con un deficit campimetrico in metà di essi), in 8/28 una riduzione spontanea di volume nel follow-up a lungo termine (solo in due dei quali, attribuibile ad apoplessia), mentre il volume tumorale è rimasto stabile negli altri sei casi. Nello stesso anno uno studio inglese (6) sull’evoluzione naturale di 40 casi di NFPA dimostrava che circa il 70% andava incontro a crescita dell’adenoma entro 6 anni. Scomponendo però i dati in relazione alle caratteristiche dell’adenoma al momento della diagnosi, si dimostrava che solo il 20% dei microadenomi aumentava di volume, mentre la crescita si verificava nel 100% dei macroadenomi. Una recente revisione sistematica sull’argomento (7) dimostrava che di 304 casi pubblicati, con un follow-up medio di 52 mesi, il 28.9% dimostrava tendenza alla crescita, l’11.2% alla diminuzione di volume, mentre tutti gli altri rimanevano stabili.
Anche se questi dati sembrano rassicuranti sulla possibile evolutività di questo tipo di adenoma in assenza di trattamento, cionondimeno bisogna ricordare che alcuni di questi tumori, seppure una minoranza, hanno un comportamento molto aggressivo e quindi è importante eseguire un attento follow-up. Inoltre, un recente lavoro su 38 casi di microadenoma clinicamente non funzionante (8) ha evidenziato come, anche in questa situazione apparentemente a minor rischio, ben il 50% dei casi aveva un deficit di GH, come dimostrato dalla scarsa risposta al test con GHRH + arginina.
Per valutare le modalità della crescita tumorale sono stati impiegati modelli matematici utilizzati per analizzare le curve di crescita di altre neoplasie. Ciò che descrive meglio la crescita di un tumore, anche se si applica soprattutto alle neoplasie maligne a rapida crescita, è una sigmoide, che ha tre fasi distinte: una fase iniziale esponenziale, una successiva lineare ed una finale che va a plateau, che è stata spiegata con il deficit di vascolarizzazione delle parti centrali che vanno incontro a necrosi. Con questi modelli, si può calcolare il tempo di raddoppiamento del volume tumorale. Questo calcolo è stato utilizzato solo raramente negli adenomi ipofisari, trovando risultati molto eterogenei, con un range da 8 mesi a 27 anni (9-11).
È appena uscita una revisione sistematica (12) di 1614 pazienti post-NCH senza radioterapia, con metanalisi di 19 studi (9 prospettici e 10 retrospettivi) per un totale di 971 pazienti, divisi in 2 gruppi, con e senza residuo. Il tasso di recidiva complessivo risultava del 12% in quelli senza residuo post-NCH (4% a 5 anni e 18% a 10 anni) e del 46% in quelli con residuo (44% a 5 anni e 60% a 10 anni). Il tempo di raddoppiamento medio del volume tumorale residuo era di 3.4 anni.
La diagnosi definitiva di NFPA si basa sul reperto neuroradiologico alla RMN di espanso a partenza dalla regione sellare. In circa due terzi dei casi viene riscontrato che il macroadenoma invade le strutture circostanti (seno sfenoidale e seno cavernoso) e può dislocare il peduncolo ipofisario e comprimere il chiasma ottico.
L’eventuale compressione del chiasma ottico viene indagata tramite studio del campo visivo, mentre lo studio dei potenziali evocati visivi permette di indagare con maggiore sensibilità l’integrità funzionale dei nervi ottici.
Come mezzo diagnostico morfo-funzionale è stato suggerito l’utilizzo della scintigrafia (13-15), ma nessuna tecnica scintigrafica a scopo diagnostico è entrata nell’uso comune in questa patologia per la scarsa attendibilità dei risultati e gli alti costi.
Bibliografia
- Ferrante E, Ferraroni M, Castrignanò T, et al. Non-functioning pituitary adenoma database: a useful resource to improve the clinical management of pituitary tumors. Eur J Endocrinol 2006, 155: 823-9.
- Reincke M, Allolio B, Saeger W, et al. The ‘incidentaloma’ of the pituitary gland. Is neurosurgery required? JAMA 1990, 263: 2772–6.
- Sanno N, Oyama K, Tahara S, et al. A survey of pituitary incidentaloma in Japan. Eur J Endocrinol 2003, 149: 123–7.
- Donovan LE, Corenblum B. The natural history of the pituitary incidentaloma. Arch Intern Med 1995, 155: 181–3.
- Dekkers OM, Hammer S, de Keizer RJ, et al. The natural course of non-functioning pituitary macroadenomas. Eur J Endocrinol 2007, 156: 217-24.
- Karavitaki N, Collison K, Halliday J, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol 2007, 67: 938-43.
- Dekkers OM, et al. Treatment and follow-up of clinically nonfunctioning pituitary macroadenomas. J Clin Endocrinol Metab 2008, 93: 3717-26.
- Yuen KCJ, Cook DM, Sahasranam P, et al. Prevalence of GH and other anterior pituitary hormone deficiencies in adults with nonsecreting pituitary microadenomas and normal serum IGF-1 levels. Clin Endocrinol 2008, 69: 292–8.
- Ekramullah SM, Saitoh Y, Arita N, et al. The correlation of Ki-67 staining indices with tumour doubling times in regrowing non-functioning pituitary adenomas. Acta Neurochir 1996, 138: 1449–55.
- Tanaka Y, Hongo K, Tada T, et al. Growth pattern and rate in residual nonfunctioning pituitary adenomas: correlations among tumor volume doubling time, patient age, and MIB-1 index. J Neurosurg 2003, 98: 359–65.
- Honegger J, Zimmermann S, Psaras T, et al. Growth modeling of non-functioning pituitary adenomas in patients referred for surgery. Eur J Endocrinol 2008, 158: 287–94.
- Chen Y, Cheng W, Su ZP, et al. Natural history of postoperative nonfunctioning pituitary adenomas: a systematic review and meta-analysis. Neuroendocrinology 2012, 96: 333-42.
- de Herder WW, Reijs AE, de Swart J, et al. Comparison of iodine-123 epidepride and iodine-123 IBZM for dopamine D2 receptor imaging in clinically non-functioning pituitary macroadenomas and macroprolactinomas. Eur J Nucl Med 1999, 26: 46-50.
- Colombo P, Siccardi AG, Paganelli G, et al. Three-step immunoscintigraphy with anti-chromogranin A monoclonal antibody in tumours of the pituitary region. Eur J Endocrinol 1996, 135: 216–21.
- Duet M, Mundler O, Ajzenberg C, et al. Somatostatin receptor imaging in non-functioning pituitary adenomas: value of an uptake index. Eur J Nucl Med 1994, 21: 647-50.
- Greenman Y, Stern N. Non-functioning pituitary adenomas. Best Pract Res Clin Endocrinol Metab 2009, 23: 625-38.
- Jaffe C. Clinically non-functioning pituitary adenoma. Pituitary 2006, 9: 317-21.
- Korbonits M, Carlsen E. Recent clinical and pathophysiological advances in non-functioning pituitary adenomas. Horm Res 2009, 71 (suppl 2): 123-30.
- Molitch M. Pituitary incidentalomas. Best Pract Res Clin Endocrinol Metab 2009, 23: 667-75.
- Sam S, Molitch M. The pituitary mass: diagnosis and management. Rev Endocr Metab Dis 2005, 6: 55-62.
Incidentaloma ipofisario
Endocrinologia, Ospedale Niguarda, Milano & 2Comitato Scientifico AME
Sapienza Università di Roma, Facoltà di Medicina e Psicologia, Dipartimento NESMOS (Neuroscienze, Salute Mentale, Organi di Senso)
UOC Neuroradiologia, AO "Sant'Andrea", Roma
L’ampio uso odierno di sofisticate metodiche diagnostiche porta al riscontro incidentale sempre più frequente di queste lesioni: nelle RM eseguite per motivi non correlabili a patologia ipofisaria (escludendo quindi manifestazioni di eccesso o deficit ormonale e disturbi visivi) si trovano microadenomi di almeno 3 mm in circa il 10% (1) e macroadenomi nello 0.2-0.3% dei casi (2-4). C’è quindi un’epidemia di incidentalomi ipofisari.
Sensibilità e specificità di RM (in confronto ad una diagnosi patologica considerata come gold standard) sono del 99% e 29%, rispettivamente (5).
Qual è quindi l’approccio corretto dal punto di vista clinico e dei costi, alla diagnosi e durante il follow-up (6-8)?
Alla diagnosi
Va fatto in tutti i casi uno screening delle forme ipersecretorie, anche in assenza di quadro clinico specifico. Questo deve comprendere:
- dosaggio PRL, dopo esclusione delle cause fisiologiche (gravidanza e stress), farmacologiche e secondarie ad altre patologie, per confermare o escludere l’iperPRL;
- dosaggio di IGF-I per escludere l’acromegalia, se è disponibile un dosaggio attendibile e sono state escluse le cause di interferenza sul dosaggio di IGF-I (vedi tabella), soprattutto epatopatie e malnutrizione;
- dosaggio FT4 e TSH per escludere l’ipertiroidismo centrale;
- test di Nugent (o cortisolo salivare notturno se disponibile) come screening del Cushing.
Se questi dati preliminari lasciano adito a dubbi, si passa a una valutazione più formale e strutturata:
- livelli di PRL > 200 ng/mL sono diagnostici of PRLoma, ma livelli inferiori nel range patologico necessitano di ulteriori indagini per distinguere tra la deconnessione ipotalamo-ipofisaria, compatibile con qualunque lesione della regione (per l’annullamento della fisiologica inibizione della dopamina sui livelli di PRL), in cui raramente PRL è > 100 ng/mL e altre situazioni. I dosaggi seriati di PRL (3-4 prelievi a distanza di 20-30’ in corso di infusione di fisiologica) elimineranno il fattore stress. La diluizione (1:10 e/o 1:100) supererà l’effetto “gancio” (dovuto a saturazione dell’anticorpo di cattura in presenza di grande eccesso di antigene, come si può osservare con certi dosaggi quando i valori di PRL sono stellari come accade in alcuni macroPRLomi molto voluminosi). La precipitazione del campione con PEG eliminerà la macroprolattinemia, situazione in cui esistono macroaggregati di PRL e anticorpi circolanti, senza corrispettivo clinico). Le cause farmacologiche (che comunque raramente causano valori di PRL > 100 ng/mL) possono essere eventualmente rimosse (se non ci sono controindicazioni): è sufficiente un wash-out di alcune ore per gli anti-emetici, di alcuni giorni per neurolettici standard e anti-depressivi, ma sono necessarie alcune settimane per i neurolettici depot.
- Il dosaggio di GH durante OGTT per perfezionare la diagnosi di acromegalia ove i livelli di IGF-I siano non disponibili, inattendibili, con interferenze o inconcludenti, perché in questa patologia si perde la fisiologica inibizione operata dal glucosio sul GH.
- Uno o più dei numerosi test disponibili per la diagnostica di II livello del Cushing.
Lo screening dell’ipopituitarismo, che può essere parziale o totale, va fatto in tutti i macroadenomi, mentre di solito i microadenomi lasciano intatta la secrezione dell’ipofisi normale, anche se un recente lavoro pone il dubbio di deficit di GH anche in questa situazione (9). Gli esami utili sono:
- dosaggio cortisolemia al mattino h 8 per escludere l’iposurrenalismo;
- dosaggio FT4 per escludere l’ipotiroidismo;
- dosaggio testosterone per escludere l’ipogonadismo maschile; per escludere l’ipogonadismo femminile è sufficiente la storia mestruale in età fertile e il dosaggio di FSH dopo la menopausa.
Non serve dosare le tropine ipofisarie (ACTH, TSH, gonadotropine in età fertile) per lo screening dell’ipopituitarismo perché la sensibilità nel range basso (quello che ci interessa) è molto scarsa. La diagnosi di deficit di GH è più complessa (in questo caso è l’IGF-I che è poco sensibile) e va perseguita solo quando si pensa possa essere utile l’eventuale trattamento sostitutivo.
Trattamento
Tutti i tumori ipersecernenti indipendentemente dalle dimensioni vanno trattati appropriatamente secondo le rispettive linee-guida: PRLomi, acromegalia, Cushing, TSHoma.
Il caso degli NFPA è diverso. C’è indicazione NCH nei macroadenomi soprattutto se vicini alle vie ottiche, ma non nei micro. Una crescita progressiva nella valutazione seriata per molti anni è stata osservata solo nel 10% dei microadenomi incidentali, mentre nel 6% dei casi c’era una diminuzione e gli altri erano stabili (10,11). Una metanalisi con un follow-up di 472 anni-persona (12) ha mostrato che ogni anno c’è crescita nell’8.2% degli incidentalomi ma solo nell’1.7% dei micro (senza nessuna comparsa di deficit visivo). Quindi nei micro è opportuno un atteggiamento attendista con valutazioni seriate (13).
Follow-up degli incidentalomi non operati
È inutile ripetere lo screening delle ipersecrezioni (a meno che il quadro clinico lo indichi).
Per quanto riguarda l’ipopituitarismo, solo nei macroadenomi ripetere una volta all’anno i dosaggi di cortisolemia, FT4 e testosterone.
Dal punto di vista neuroradiologico, nei microadenomi, controllo RM a 12 mesi e poi dopo altri 18-24 mesi: se non c’è crescita non è necessario ripetere altre RM.
Nei macroadenomi, RM ogni 6 mesi nel primo anno poi annuale. E' indispensabile che le immagini vengano sempre confrontate con la prima della serie (e non solo con l’ultima). L'esame campimetrico riveste anch'esso utilità. Se non ci sono alterazioni campimetriche né crescita tumorale, si può progressivamente allungare l’intervallo fra i controlli di RM.
Bibliografia
- Hall WA, Luciano MG, Doppman JL, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Int Med 1994, 120: 817–20.
- Nammour GM, Ybarra J, Naheedy MH, et al. Incidental pituitary macroadenoma: a population-based study. Am J Med Sci 1997, 314: 287–91.
- Vernooij MW, Ikram A, Tanghe HL, et al. Incidental findings on brain MRI in the general population. N Engl J Med 2007, 357: 1821–18.
- Yue NC, Longsteth Jr. WT, Elster AD, et al. Clinically serious abnormalities found incidentally at MR imaging of the brain: data from the Cardiovascular Health Study. Radiology 1997, 202: 41–6.
- Famini P, Maya MM, & Melmed S. Pituitary Magnetic Resonance Imaging for Sellar and Parasellar Masses: Ten-Year Experience in 2598 Patients. J Clin Endocrinol Metab 2011, 96: 1633–41.
- King Jr JT, Justice AC, & Aron DC. Management of incidental pituitary microadenomas: a cost-effectiveness analysis. J Clin Endocrinol Metab 1997, 82: 3625–32.
- Dekkers OM, Pereira AM, & Romijn JA. Treatment and follow-up of clinically non functioning pituitary macroadenomas. J Clin Endocrinol Metab 2008, 93: 3717–26.
- Randall BR, Kraus KL, Simard MF, & Couldwell WT. Cost of evaluation of patients with pituitary incidentaloma. Pituitary 2010, 13: 383–4.
- Yuen KCJ, Cook DM, Sahasranam P, et al. Prevalence of GH and other anterior pituitary hormone deficiencies in adults with nonsecreting pituitary microadenomas and normal serum IGF-1 levels. Clin Endocrinol 2008, 69: 292–8.
- Dekkers OM, Hammer S, de Keizer RJW, et al. The natural course of non-functioning pituitary macroadenomas. Eur J Endocrinol 2007, 156: 217–24.
- Karavitaki N, Collison K, Halliday J, et al. What is the natural history of nonoperated, nonfunctioning pituitary adenomas? Clin Endocrinol 2007, 67: 938–43.
- Fernandez-Balsells MMM, Barwise A, Gallegos-Orozco J, et al. The natural history of pituitary incidentalomas: a systematic review and meta-analysis. J Clin Endocrinol Metab 2011, 96: 905-12.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary Incidentaloma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2011, 96: 894–904.