Clinica e diagnostica del craniofaringioma

Marco Losa, Filippo Gagliardi, Pietro Mortini
Reparto di Neurochirurgia, Istituto Scientifico San Raffaele, Università Vita-Salute, Milano
Definizione ed epidemiologia
Il craniofaringioma è un tumore benigno di derivazione epiteliale che si sviluppa lungo il dotto cranio-faringeo ed è classificato dalla World Health Organization come un tumore di grado I, anche se sono stati riportati rari casi di trasformazione maligna.
L’incidenza annuale del craniofaringioma è stimata a 1.3 casi/milione di abitanti (1).
Vi è una chiara distribuzione bimodale dell’età di insorgenza, con un primo picco di incidenza fra i 5 e i 14 anni ed un secondo picco fra i 50 e i 74 anni (1).
Il craniofaringioma rappresenta circa il 5-15% dei tumori intracranici nell’età pediatrica (2).
Eziologia e patologia
Il craniofaringioma è un tumore monoclonale (3), per la cui origine sono principalmente accreditate due teorie: per la prima, il tumore deriva dalla trasformazione neoplastica di cellule squamose embrionali non involute del dotto cranio-faringeo (4), mentre la seconda teoria implica una metaplasia di cellule adeno-ipofisarie nel peduncolo o nella ghiandola ipofisaria (5).
Da un punto di vista anatomo-patologico esistono due tipi principali: la variante adamantinomatosa e quella papillare, anche se sono state descritte alcune forme miste (6).
Il tipo adamantinomatoso è il più comune ed è diagnosticato prevalentemente nei pazienti giovani. Macroscopicamente è caratterizzato dalla contemporanea presenza di aree cistiche e solide. Le parti cistiche contengono un materiale liquido viscoso molto caratteristico, che è stato accomunato per la sua somiglianza all’olio dei motori. La parte epiteliale è invece costituita da una palizzata di piccole cellule che somigliano alle cellule basali dell’epidermide. Al di sopra è appoggiato uno strato intermedio di variabile spessore, composto da cellule stellate e da uno strato interno formato da cellule squamose positive per la citocheratina.
La varietà papillare è quasi esclusiva degli adulti (7). E’ solitamente a struttura solida o mista con aree cistiche. E’ composta da cellule squamose epiteliali mature che formano delle pseudopapille.
L'importanza prognostica delle due varianti istologiche è ancora oggetto di discussione, ma non è stata ancora dimostrata una significativa differenza in termini di prognosi chirurgica, sensibilità alle terapie radianti e prognosi a lungo termine.
Clinica
La localizzazione del craniofaringioma è soprattutto intra- e sovrasellare, con frequente coinvolgimento del peduncolo ipofisario; in circa il 72-96% dei casi le sue dimensioni sono > 2 cm (8, 9).
La sintomatologia del craniofaringioma è diretta consequenza delle compressioni sulle vie ottiche, sul parenchima cerebrale, sul sistema ventricolare e sul sistema ipotalamo-ipofisario. I principali sintomi all’esordio sono la cefalea, presente in quasi il 60-80% dei casi, che è nel 20-40% dei casi accompagnata anche da nausea, vomito e papilledema, come manifestazione di ipertensione endocranica (10). Nel 40-70% dei casi sono rilevabili deficit dell’acuità visiva e/o difetti di tipo campimetrico, mentre nel 5-20% dei casi è rilevabile diplopia da paresi/paralisi dei nervi cranici (10). Sono meno frequenti (circa il 5-30% dei casi) sintomi da compromissione ipotalamica, quali disfunzioni neurocognitive, stato soporoso, alterazione dello stato di veglia e alterazioni del peso corporeo. Il coinvolgimento delle funzioni ipofisarie è molto variabile, a seconda della popolazione studiata, mentre il diabete insipido è presente fra il 10% e il 30% dei casi (10). Nella nostra casistica (11), l’ipogonadismo è presente nel 77% dei casi, l’ipotiroidismo nel 48%, l’iposurrenalismo nel 50% e l’ipostaturismo in età pediatrica nel 90%, mentre il diabete insipido ha una frequenza del 37%.
In età pediatrica, oltre ai comuni sintomi riferibili all’effetto massa, in quasi l’80% dei bambini è diagnosticata la presenza di deficit ormonali. Nei pazienti in età prepubere il ritardo staturale secondario al deficit di ormone della crescita tende a essere un sintomo precoce se non addirittura il primo (12).
Diagnosi
La diagnostica del craniofaringioma si basa essenzialmente sugli esami neuroradiologici.
La tomografia computerizzata (TC) mantiene la sua utilità per la valutazione dell’anatomia ossea e per la presenza di componenti solide calcifiche, che sono presenti in più della metà dei pazienti e addirittura quasi sempre evidenziabili nei bambini.
L’esame neuroradiologico più importante rimane la risonanza magnetica nucleare (RMN) con e senza contrasto, che è in grado di ben delineare le caratteristiche strutturali del tumore e i suoi rapporti con le strutture circostanti (13). L’aspetto del craniofaringioma alla RMN dipende dalla proporzione delle aree cistiche e solide del tumore, dal tipo di contenuto delle cisti (cristalli di colesterina, cheratina, sangue) e dalla quantità di calcificazioni presenti. Pur esistendo alcune differenze nelle caratteristiche radiologiche, non è possible distinguere con certezza la variante adamantinomatosa da quella papillare (14).
Infine, l’angiografia cerebrale (in parte sostituita dall’angioTAC e dall’angioRMN) può essere utile nella pianificazione degli interventi chirurgici per via trans-cranica, in quanto evidenzia i rapporti anatomici fra il tumore e i vasi arteriosi.
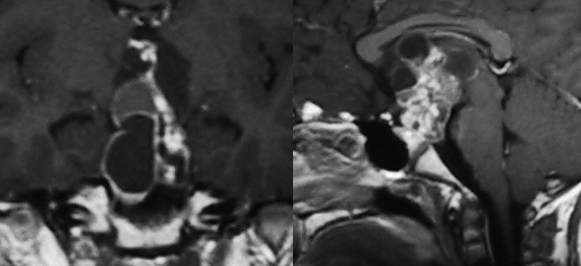
Immagine coronale (a sinistra) e sagittale (a destra) T1-pesata, dopo somministrazione di mezzo di contrasto, di craniofaringioma adamantinomatoso.
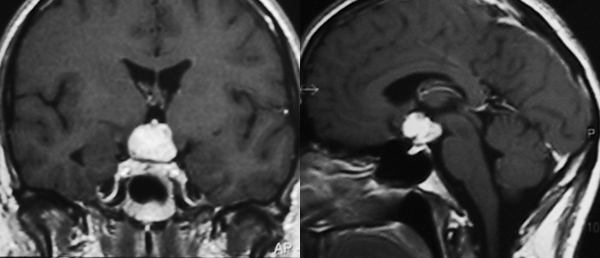
Immagine coronale (a sinistra) e sagittale (a destra) T1-pesata, dopo somministrazione di mezzo di contrasto, di craniofaringioma papillare.
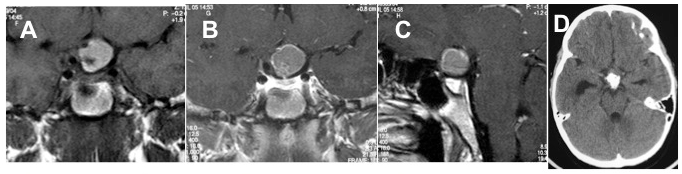
Craniofaringioma sovra-sellare nodulare. RM, sequenze SE, immagini T1, sezioni coronali senza (A) e con mdc (B) e sagittale con mdc (C) di bambino con lesione espansiva sovra-sellare, rotondeggiante, a contorni definiti, prevalentemente iperintensain T1 basale, con spot ipointenso al polo inferiore per la presenza di grossolana calcificazione intra-lesionale, ben documentata dalla TC (D). Le immagini contrastografiche documentano il fisiologico blush dell’ipofisi e solo un sottile rinforzo contrastografico periferico, attribuibile alla capsula della lesione.
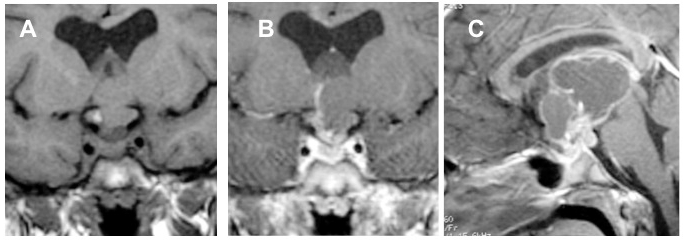
Craniofaringioma multicistico intra- e sovra-sellare. RM, sequenze SE, immagini T1, sezioni coronali senza (A) e con mdc (B) e sagittale con mdc (C) di voluminosa lesione espansiva, a prevalente sviluppo sovra-sellare, ma con minime componenti anche endo-sellare e intra-cisternale posteriore pre-pontina. Le prevalenti componenti cistiche in particolare presentano segnale disomogeneo per la presenza di cristalli di colesterina, glicoproteine, albumina e prodotti di degradazione dell’emoglobina.
La successione degli esami diagnostici in caso di sospetto clinico di un craniofaringioma dipende molto dal tipo di presentazione sintomatologica del paziente. In presenza di deficit di crescita nei bambini e sintomi da ipogonadismo o deficit tiroideo e surrenalico, l'inquadramento diagnostico iniziale prevede una valutazione ormonale approfondita seguita dagli esami neuroradiologici e oftalmologici. E' da sottolineare che la presenza di diabete insipido centrale, soprattutto in età giovanile, aumenta notevolmente la probabilità di diagnosticare un craniofaringioma. Se il paziente presenta invece inizialmente un deficit visivo di tipo campimetrico, accompagnato o meno da calo dell'acuità visiva, la valutazione oftalmologica iniziale deve comprendere necessariamente un esame del campo visivo e lo studio dell'acuità visiva e del fundus oculare. La conferma di un'alterazione compatibile con un processo espansivo intracranico rende necessaria la diagnostica radiologica e, successivamente, lo studio ormonale completo della funzione ipofisaria se si evidenzia un processo espansivo della regione ipotalamo-ipofisaria. Infine, alcuni pazienti presentano all'esordio sintomi da ipertensione endocranica (cefalea, nausea, vomito) e la diagnostica radiologica è normalmente il primo passo, seguita poi dagli studi endocrinologici e visivi.
Bibliografia
- Bunin GR, Surawicz TS, Witman PA, et al. The descriptive epidemiology of craniopharyngioma. J Neurosurg 1998, 89: 547-51.
- Kuratsu J, Ushio Y. Epidemiological study of primary intracranial tumours in childhood. A population-based survey in Kumamoto Prefecture, Japan. Pediatr Neurosurg 1996, 25: 240-6.
- Sarubi JC, Bei H, Adams FF, et al. Clonal composition of human adamantinomatous craniopharyngiomas and somatic mutation analyses of the patched (PTCH), Gsα and Gi2α genes. Neurosci Lett 2001, 310: 5-8.
- Goldberg GM, Eshbaught DE. Squamous cell nests of the pituitary gland as related to the origin of craniopharyngiomas: a study of their presence in the newborn and infants up to age four. Arch Pathol 1960, 70: 293-9.
- Hunter IJ. Squamous metaplasia of cells of the anterior pituitary gland. J Pathol Bacteriol 1955, 69: 141-5.
- Eldevik OP, Blaivas M, Gabrielsen TO, et al. Craniopharyngioma: radiologic and histologic findings and recurrence. Am J Neuroradiol 1996, 17: 1427-39.
- Crotty TB, Scheithauer BW, Young WF, et al. Papillary craniopharyngioma: a clinico-pathological study of 48 cases. J Neurosurg 1995, 83: 206-14.
- Weiner HL, Wisoff JH, Rosenberg ME, et al. Craniopharyngiomas: a clinicopathological analysis of factors predictive of recurrence and functional outcome. Neurosurgery 1994, 35: 1001-11.
- Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg 1999, 90: 237-50.
- Karavitaki N, Cudlip S, Adams CB, et al. Craniopharyngiomas. Endocr Rev 2006, 27: 371–97.
- Mortini P, Losa M, Pozzobon G, et al. Neurosurgical treatment of craniopharyngioma in adults and children: early and long-term results in a large case series. J Neurosurg 2011, 114: 1350-9.
- Garrè ML, Cama A. Craniopharyngioma: modern concepts in pathogenesis and treatment. Curr Opin Pediatr 2007, 19: 471-9.
- Hald JK, Eldevik OP, Brunberg JA, et al. Craniopharyngiomas – the utility of contrast medium enhancement for MR imaging at 1.5 T. Acta Radiol 1994, 35: 520-5.
- Sartoretti-Schefer S, Wichmann W, Aguzzi A, et al. MR differentiation of adamantinous and squamous-papillary craniopharyngiomas. Am J Neuroradiol 1997, 18: 77-87.
Terapia del craniofaringioma
Marco Losa, Filippo Gagliardi, Pietro Mortini
Reparto di Neurochirurgia, Istituto Scientifico San Raffaele, Università Vita-Salute, Milano
Nell’algoritmo terapeutico deve essere accuratamente considerato da un lato l’obiettivo di rimuovere completamente il tumore e dall’altro il rischio degli effetti collaterali. Negli ultimi anni il dibattito sulla strategia terapeutica si è spostato dalla mera considerazione del controllo completo o parziale della crescita neoplastica alle valutazioni riguardanti anche la quality of life (QoL) dei pazienti nel lungo termine (1). Così, accanto ad alcuni autori che consigliano una strategia chirurgica aggressiva con lo scopo di aumentare la probabilità di asportazione completa del tumore (2), altri autori ritengono che l’esito migliore sia ottenibile con una rimozione parziale del tumore, onde evitare manovre chirurgiche aggressive, seguita da altre terapie e, principalmente, da quella radiante (3, 4). Tuttavia, al di fuori delle estremizzazioni, la strategia chirurgica con maggiori probabilità di controllo a lungo termine del craniofaringioma dovrebbe essere quella di ricercare l’asportazione completa della lesione tutte le volte che questa sia possibile senza rischiare danni alle strutture nervose circostanti (5, 6).
Bibliografia
- Kawamata T, Amano K, Aihara Y, et al. Optimal treatment strategy for craniopharyngiomas based on long–term functional outcomes of recent and past treatment modalities. Neurosurg Rev 2010, 33: 71-81.
- Yasargil MG, Curcic M, Kis M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg 1990, 73: 3-11.
- De Vile CJ, Grant DB, Kendall BE, et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg 1996, 85: 73-81.
- Duff JM, Meyer FB, Ilstrup DM, et al. Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery 2000, 46: 291-305.
- Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg 1999, 90: 237-50.
- Mortini P, Losa M, Pozzobon G, et al. Neurosurgical treatment of craniopharyngioma in adults and children: early and long-term results in a large case series. J Neurosurg 2011, 114: 1350-9.
Terapia neurochirurgica del craniofaringioma
Marco Losa, Filippo Gagliardi, Pietro Mortini
Reparto di Neurochirurgia, Istituto Scientifico San Raffaele, Università Vita-Salute, Milano
La chirurgia è attualmente la modalità di trattamento di prima scelta. Nonostante vari ammodernamenti della tecnica chirurgica, l’intervento di asportazione del craniofaringioma rimane uno dei più difficili dal punto di vista tecnico. Ciò è dovuto principalmente alla localizzazione profonda del tumore, ai suoi margini spesso irregolari e alla sua forte tendenza ad aderire alle strutture neurovascolari circostanti.
Gli approcci chirurgici sono suddivisibili in due gruppi principali: la via trans-nasosfenoidale (TNS), meno traumatica, è riservata ai tumori ad estensione intra-sellare o anche sovra-sellari ma sotto-diaframmatici (1,2), mentre per i tumori con prevalente crescita sovra-diaframmatica è necessario utilizzare la via trans-cranica (TC). In alcuni tumori di notevoli dimensioni può essere necessario ricorrere ad entrambe le vie chirurgiche: spesso si inizia con la meno traumatica via TNS per ottenere una decompressione del tumore, seguita in un secondo tempo dall’intervento TC per togliere la parte più craniale del tumore (2, 3).
I risultati immediati dell’intervento chirurgico in alcune recenti serie chirurgiche sono riassunti nella tabella 1 (4, 5, 6-23). Si può notare come la radicalità dell’intervento chirurgico sia assai variabile fra le varie casistiche passando dal 18% al 90%.
| Tabella 1 Risultati immediati della terapia chirurgica in pazienti operati per craniofaringioma |
||||
| Autore, anno | No. Pazienti | Intervento TC | Radicalità chirurgica | Tipo di esame post-op. |
| Yasargil, 1990 | 144 | 90 | 90 | TAC/RMN |
| Hoffman, 1992 | 50 | 100 | 90 | TAC/RMN |
| De Vile, 1996 | 75 | 100 | 40 | TAC/RMN |
| Fahblusch, 1999 | 148 | 71 | 49 | TAC/RMN |
| Duff, 2000 | 121 | 68 | 57 | N/D |
| Van Effenterre, 2002 | 122 | 92 | 59 | TAC/RMN |
| Chen, 2003 | 36 | N/D | 19 | RMN |
| Maira, 2004 | 57 | 0 | 56 | RMN |
| Stripp, 2004 | 76 | 100 | 62 | TAC/RMN |
| Gonc, 2004 | 66 | 100 | 31 | TAC/RMN |
| Karavitaki, 2005 | 103 | 72 | 18 | TAC/RMN |
| Lena, 2005 | 47 | 98 | 66 | RMN |
| Minamida, 2005 | 37 | 92 | 70 | RMN |
| Shirane, 2005 | 42 | 100 | 71 | N/D |
| Sosa, 2005 | 35 | 89 | 83 | RMN |
| Thompson, 2005 | 48 | 73 | 33 | RMN |
| Tomita, 2005 | 54 | 94 | 61 | RMN |
| Zuccaro, 2005 | 153 | 99 | 69 | TAC/RMN |
| Shi, 2008 | 309 | 100 | 89 | TAC/RMN |
| Zhang, 2008 | 202 | 99 | 40 | TAC/RMN |
| Mortini, 2011 | 112 | 68 | 72 | RMN |
Buona parte di questa variabilità dipende dal tipo di popolazione studiata, ma è altrettanto ovvio che il tipo di strategia chirurgica adottato (aggressivo o conservativo) influenzi notevolmente l’esito chirurgico.
I principali fattori predittivi di persistenza di un residuo tumorale sono le dimensioni del tumore (2, 4, 5, 7, 24) e la storia di un precedente intervento chirurgico (4, 5, 9, 14). Altre caratteristiche sfavorevoli sono la localizzazione retro-chiasmatica (9) o dentro il terzo ventricolo (4).
Nella tabella 2 sono riportati invece, ove disponibili, i dati riguardanti la mortalità, la morbilità maggiore e la morbilità endocrinologica nelle stesse serie chirurgiche.
| Tabella 2 Mortalità e morbilità in pazienti operati di asportazione di craniofaringioma |
||||||||||||||
| Autore, anno | No. pazienti | Mortalità | Morbilità maggiore | Nuovo deficit visivo | Deficit ormonale post-op | Diabete insipido post-op | ||||||||
| Yasargil, 1990 | 144 | 16.7% | N/D | 13% | 79% | 90% | ||||||||
| Hoffman, 1992 | 50 | 2% | 6% | 41% | N/D | 93% | ||||||||
| De Vile, 1996 | 75 | 0 | 13% | N/D | 99% | 80% | ||||||||
| Fahlbusch, 1999 | 168 | 1.2% | 11.3% | 9.7% | N/D | N/D | ||||||||
| Duff, 2000 | 121 | 1.7% | 18.4% | N/D | 21% | 21% | ||||||||
| Van Effenterre, 2002 | 122 | 2.5% | 8% | 11% | 76% | 57% | ||||||||
| Chen, 2003 | 36 | 5.6% | 14% | 31% | 81% | 67% | ||||||||
| Maira, 2004 | 57 | 0 | 0 | 0 | 32% | 14% | ||||||||
| Stripp, 2004 | 76 | 1% | N/D | 15% | N/D | 80% | ||||||||
| Gonc, 2004 | 66 | 2% | 10.6% | N/D | 100% | 52% | ||||||||
| Karavitaki, 2005 | 103 | 1.9% | 20.4% | 5.8% | N/D | N/D | ||||||||
| Lena, 2005 | 47 | 2.4% | N/D | 16% | 89% | 86% | ||||||||
| Minamida, 2005 | 37 | 0 | 5.4% | 2.7% | 97% | N/D | ||||||||
| Shirane, 2005 | 42 | 0 | 6.7% | N/D | 81% | 52% | ||||||||
| Sosa, 2005 | 35 | 0 | 20% | 17% | 100% | 91% | ||||||||
| Thompson, 2005 | 48 | 0 | 15% | N/D | 96% | 84% | ||||||||
| Tomita, 2005 | 54 | 0 | 9% | 13% | 93% | 87% | ||||||||
| Zuccaro, 2005 | 153 | 3% | 10% | 8.5% | 85% | 50% | ||||||||
| Lee, 2008 | 66 | 0 | 6% | N/D | N/D | 67% | ||||||||
| Shi, 2008 | 309 | 3.9% | 6% | 5.5% | N/D | 53% | ||||||||
| Zhang, 2008 | 202 | 1% | 5.4% | 5% | N/D | 81% | ||||||||
| Mortini, 2011 | 112 | 2.7% | 26.6% | 13.8% | 92 | 78 | ||||||||
Con un’unica eccezione (6), probabilmente secondaria alla dichiarata intenzione di procedere aggressivamente alla rimozione del tumore, la mortalità peri-operatoria si aggira fra lo 0% e il 5%.
La morbilità maggiore varia molto fra le varie casistiche, presumibilmente per una notevole variabilità delle definizioni di complicanza chirurgica e per la diversa percentuale di pazienti operati per via TC o per via TNS. E’ evidente che quest’ultimo tipo di approccio è associato ad un rischio operatorio decisamente minore rispetto all’approccio TC. Lo stesso discorso vale per il rischio di peggioramento della funzionalità visiva. I dati nella tabella 2 confermano che la funzionalità ipofisaria è gravemente compromessa nei pazienti con craniofaringioma, sia per l’elevata frequenza di deficit endocrini già alla presentazione che per il rischio di nuovi deficit post-operatori. Poichè buona parte dei craniofaringiomi originano o sono strettamente connessi al peduncolo ipofisario, l’asportazione radicale della lesione può causare il danno di tale struttura con conseguente ipopituitarismo. D’altra parte, il sacrificio del peduncolo ipofisario non rappresenta un motivo valido per rinunciare alla radicalità chirurgica (4, 5).
La recidiva rimane uno dei problemi maggiori nella terapia del craniofaringioma. Nella tabella 3 sono riassunti i dati di recidiva e sopravvivenza libera da recidiva (PFS) a 5 e 10 anni come riportato in alcune casistiche chirurgiche.
| Tabella 3 Recidive e sopravvivenza in pazienti operati per craniofaringioma |
||||||||||||||
| Autore, anno | Follow-up (aa) | Radioterapia post-op | Recidiva | PFS a 5 aa | PFS a 10 aa | Sopravvivenza | ||||||||
| Yasargil, 1990 | N/D | 4% | 7% | N/D | N/D | 80% | ||||||||
| Hoffmann, 1992 | 4.9 | 0 | 34% | N/D | N/D | 98% | ||||||||
| De Vile, 1992 | 6.4 | 51% | 41% | N/D | N/D | 80% | ||||||||
| Fahlbusch, 1999 | 5.4 | 13% | 29% | N/D | N/D | 90% | ||||||||
| Duff, 2000 | 10 | 21% | 24% | 77% | N/D | 88% | ||||||||
| Van Effenterre, 2002 | 7 | 21% | 24% | 78% | 60% | 89% | ||||||||
| Chen, 2003 | 10.2 | N/D | 42% | N/D | N/D | N/D | ||||||||
| Maira, 2004 | 6 | 4% | 14% | N/D | N/D | 96% | ||||||||
| Stripp, 2004 | 7.6 | 56% | N/D | 63% | 53% | 89% | ||||||||
| Gonc, 2004 | 5.1 | 55% | 56% | N/D | N/D | 80% | ||||||||
| Karavitaki, 2005 | 7.8 | 32% | 36% | N/D | N/D | N/D | ||||||||
| Lena, 2005 | 9.5 | 19% | 34% | N/D | N/D | 94% | ||||||||
| Minamida, 2005 | 11.1 | 0 | 30% | 80% | 75% | 94% | ||||||||
| Shirane, 2005 | 5 | N/D | 38% | N/D | N/D | 93% | ||||||||
| Sosa, 2005 | 4.6 | 26% | 34% | N/D | N/D | 97% | ||||||||
| Thompson,2005 | 5.6 | 73% | 39% | N/D | N/D | 96% | ||||||||
| Tomita, 2005 | N/D | 15% | 44% | 62% | 49% | 90% | ||||||||
| Shi, 2008 | 2.1 | N/D | 19% | 75% | N/D | 94% | ||||||||
| Zhang, 2008 | N/D | N/D | 15% | N/D | N/D | 68% | ||||||||
| Mortini, 2011 | 6.9 | 9% | 24% | 76% | 65% | 95% | ||||||||
L’interpretazione dei dati è complicata dalla notevole variabilità nell’uso della radioterapia post-operatoria, che costituisce il fattore più importante nel modificare il rischio di recidiva (5, 8, 14). Pochi studi riportano la sopravvivenza libera da malattia a 5 e 10 anni, ma quando sono riportati entrambi, si nota solo un leggero aumento del rischio di recidiva a 10 anni, a conferma del fatto che la maggior parte delle recidive avviene nei primi 3-5 anni dall’intervento chirurgico (5, 9, 12, 16). Il fattore più importante che determina il rischio di recidiva è la radicalità della rimozione: in caso di asportazione apparentemente completa, la recidiva del tumore è chiaramente più bassa che nel gruppo di pazienti con residuo tumorale post-chirurgico (4, 5, 17).
Bibliografia
- Honegger J, Buchfelder M, Fahlbusch R, et al. Transsphenoidal microsurgery for craniopharyngiomas. Surg Neurol 1992, 37: 189-96.
- Maira G, Anile C, Rossi GF, et al. Surgical treatment of craniopharyngiomas: an evaluation of the transsphenoidal and pterional approaches. Neurosurgery 1995, 36: 715-24.
- Duff JM, Meyer FB, Ilstrup DM, et al. Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery 2000, 46: 291-305.
- Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg 1999, 90: 237-50.
- Mortini P, Losa M, Pozzobon G, et al. Neurosurgical treatment of craniopharyngioma in adults and children: early and long-term results in a large case series. J Neurosurg 2011, 114: 1350-9.
- Yasargil MG, Curcic M, Kis M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg 1990, 73: 3-11.
- De Vile CJ, Grant DB, Kendall BE, et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg 1996, 85: 73-81.
- Hoffman HJ, De Silva M, Humphreys RP, et al. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg 1992, 76: 47-52.
- Van Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg 2002, 97: 3-11.
- Chen C, Okera S, Davies PE, et al. Craniopharyngioma: a review of long-term visual outcome. Clin Experiment Ophthalmol 2003, 31: 220-8.
- Maira G, Anile C, Albanese A, et al. The role of transsphenoidal surgery in the treatment of craniopharyngiomas. J Neurosurg 2004, 100: 445-51.
- Stripp DC, Maity A, Janss AJ, et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int J Radias Oncol Biol Phys 2004, 58: 714-20.
- Gonc EN, Yordam N, Ozon A, et al. Endocrinological outcome of different treatment options in children with craniopharyngioma: a retrospective analysis of 66 cases. Pediatr Neurosurg 2004, 40: 112-9.
- Karavitaki N, Brufani C, Warner JT, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf) 2005, 62: 397-409.
- Lena G, Paz Paredes A, Scavarda D, et al. Craniopharyngioma in children: Marseille experience. Childs Nerv Syst 2005, 21: 778-84.
- Minamida Y, Mikami T, Hashi K, et al. Surgical management of the recurrence and regrowth of craniopharyngiomas. J Neurosurg 2005, 103: 224-32.
- Shirane R, Hayashi T, Tominaga T. Fronto-basal interhemispheric approach for craniopharyngiomas extending outside the suprasellar cistern. Childs Nerv Syst 2005, 21: 669-78.
- Sosa IJ, Krieger MD, McComb JG. Craniopharyngiomas of childhood: the CHLA experience. Childs Nerv Syst 2005, 21: 785-9.
- Thompson D, Phipps K, Hayward R. Craniopharyngioma in childhood: our evidence-based approach to management. Childs Nerv Syst 2005, 21: 660-8.
- Tomita T, Bowman RM. Craniopharyngiomas in children: surgical experience at Children’s Memorial Hospital. Childs Nerv Syst 2005, 21: 729-46.
- Zuccaro G. Radical resection of craniopharyngioma. Childs Nerv Syst 2005, 21: 679-90.
- Shi XE, Wu B, Fan T, et al. Craniopharyngioma: surgical experience of 309 cases in China. Clin Neurol Neurosurg 2008, 110: 151-9.
- Zhang YQ, Ma Zy, Wu ZB, et al. Radical resection of 202 pediatric craniopharyngiomas with special reference to the surgical approaches and hypothalamic protection. Pediatr Neurosurg 2008, 44: 435-43.
- Weiner HL, Wisoff JH, Rosenberg ME, et al. Craniopharyngiomas: a clinicopathological analysis of factors predictive of recurrence and functional outcome. Neurosurgery 1994, 35: 1001-11.
Terapia radiante del craniofaringioma
Marco Losa, Filippo Gagliardi, Pietro Mortini
Reparto di Neurochirurgia, Istituto Scientifico San Raffaele, Università Vita-Salute, Milano
La terapia radiante del craniofaringioma comprende due modalità principali.
L’irradiazione intra-cistica (brachiterapia) si avvantaggia della frequente presenza di una voluminosa parte cistica del tumore. Con metodica stereotassica si immette nella cisti del tumore un isotopo ß-emittente (in genere 90Yttrio o 32Fosforo). Nella maggior parte dei casi si ha una riduzione del volume della cisti, anche se alcuni pazienti mostrano o la formazione di nuove cisti o la crescita della parte solida del tumore (1, 2). In generale la brachiterapia è raramente utilizzata al giorno d’oggi.
La radioterapia frazionata (in genere alla dose di 50-56 Gy in 25-28 sedute) è utilizzabile come terapia adiuvante dopo asportazione incompleta del tumore. In questa situazione la radioterapia abbassa notevolmente il rischio di ricrescita del tumore (3, 4, 5, 6, 7). La dose radiante sembra influire sulla probabilità di controllo della crescita tumorale, poichè dosi totali < 50 Gy sono state associate ad una più alta probabilità di ricrescita tumorale (8).
La radiochirurgia stereotassica mediante Gamma Knife ha meno applicazioni nel caso del craniofaringioma rispetto ad altre patologie della regione sellare, in quanto i residui tumorali tendono a essere più vicini o addirittura aderenti alle strutture visive. Nel caso di tumori di piccole dimensioni (in genere residuati da precedenti interventi chirurgici o recidivanti), la Gamma Knife causa una riduzione del tumore nella maggior parte dei casi, mentre circa il 10-20% dei pazienti non ottiene il controllo locale della malattia (9, 10).
Bibliografia
- Van den Berg JH, Blauuw G, Breeman WAP, et al. Intracavitary brachitherapy of cystic craniopharyngiomas. J Neurosurg 1992, 77: 545-50.
- Pollock BE, Lunsford LD, Kondziolka D, et al. Phosphorus-32 intracavitary irradiation of cystic craniopharyngiomas: current technique and long-term results. Int J Radiat Oncol Biol Phys 1995, 33: 437-46.
- Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg 1999, 90: 237-50.
- De Vile CJ, Grant DB, Kendall BE, et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg 1996, 85: 73-81.
- Duff JM, Meyer FB, Ilstrup DM, et al. Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery 2000, 46: 291-305.
- Van Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg 2002, 97: 3-11.
- Shirane R, Hayashi T, Tominaga T. Fronto-basal interhemispheric approach for craniopharyngiomas extending outside the suprasellar cistern. Childs Nerv Syst 2005, 21: 669-78.
- Sung DI, Chang CH, Harisiadis L, et al. Treatment results of craniopharyngiomas. Cancer 1981, 47: 847-52.
- Chung WY, Pan DHC, Shiau CY, et al. Gamma knife radiosurgery for craniopharyngiomas. J Neurosurg 2000, 93 (suppl 3): 47-56.
- Kobayashi T, Kida Y, Mori Y, et al. Long-term results of gamma knife surgery for the treatment of craniopharyngiomas in 98 consecutive cases. J Neurosurg 2005, 103 (Suppl. 6): 482-8.
Terapia farmacologica del craniofaringioma
Marco Faustini Fustini
Scuola AME Malattie Ipotalamo-Ipofisarie, Bologna
(aggiornamento 4/5/2026)
Introduzione
L’impiego di farmaci nella terapia del craniofaringioma è stato a lungo limitato all’impiego di iniezioni intra-cistiche di bleomicina o alfa-interferone nei pazienti pediatrici, volte principalmente a ritardare la progressione di malattia e permettere un successivo trattamento chirurgico e radiante (1,2).
L’identificazione di mutazioni somatiche, potenzialmente in grado di aprire nuovi scenari nella terapia farmacologica sistemica del craniofaringioma, è storia relativamente recente. Fin dall’inizio degli anni 2000, nel 65-70% di pazienti con la variante adamantinomatosa – che comprende complessivamente il 60-80% dei casi di craniofaringioma - furono identificate mutazioni nell’esone 3 del gene CTNNB1, inattivanti l’attività enzimatica GSK-3b (glycogen synthase kinase-3b) all’interno della via WNT, con conseguente ridotta degradazione e accumulo di ß-catenina e sua traslocazione nel nucleo (3,4). Come è noto, ß-catenina induce la trascrizione di geni e fattori di crescita che stimolano la proliferazione cellulare. Al contrario, fino a non molti anni fa non erano state ancora identificate mutazioni responsabili dello sviluppo della variante papillare.
Il punto di svolta nella possibile terapia farmacologica del craniofaringioma ci fu nel 2014, con il sequenziamento dell’intero esoma di 12 craniofaringiomi adamantinomatosi e di 3 craniofaringiomi papillari (di cui uno in un bambino di 9 anni), mediante una nuova tecnica altamente specifica che permetteva di identificare mutazioni somatiche anche con una frazione allelica molto bassa - assai comune in popolazioni cellulari eterogenee come si reperta spesso nei craniofaringiomi, dove la contaminazione con cellule stromali e tessuto reattivo è la regola (5). A conferma di studi precedenti, il gene più frequentemente mutato nei craniofaringiomi adamantinomatosi fu CTNNB1, esclusivamente nell’esone 3, in 11/12 casi (92%). Tutti e tre i craniofaringiomi papillari (100%) mostrarono mutazioni (c.1799T>A) dell’oncogene BRAFV600E, attivanti in maniera costitutiva la serin-treonin-chinasi, che regola la via MAPK/ERK coinvolta nella divisione e nella differenziazione cellulare. Questi dati furono validati nello stesso studio mediante analisi di genotipizzazione indirizzate sui bersagli individuati dal sequenziamento dell’esoma su 98 campioni di tessuti estratti da 95 pazienti (36 con variante papillare e 59 con variante adamantinomatosa): mutazioni di CTNNB1 furono individuate nel 96% dei campioni di craniofaringioma adamantinomatoso, ma in nessuno di questi fu individuata la mutazione BRAFV600E, che, viceversa, fu evidenziata nel 95% dei craniofaringiomi papillari, senza che questi mostrassero mutazioni di CTNNB1. La conclusione fu che i craniofaringiomi adamantinomatosi e i craniofaringiomi papillari mostravano mutazioni clonali mutuamente esclusive (5).
Terapia farmacologica del craniofaringioma papillare
La scoperta della mutazione BRAFV600E nei craniofaringiomi papillari suscitò subito un certo interesse anche in campo diagnostico, in particolare sui campioni tissutali scarsi ottenuti da lesioni cistiche, in cui la distinzione con la cisti della tasca di Rathke non è sempre agevole con le comuni metodiche di immuno-istochimica (6,7). Non vi è dubbio, tuttavia, che l’impatto clinico prevalente si ebbe successivamente, allorchè si aprirono nuove prospettive nella terapia farmacologica dei craniofaringiomi papillari. Mutuando l’esperienza maturata in altre neoplasie con mutazioni BRAFV600E (8-14), nel 2016 fu pubblicato il risultato della terapia con la doppia inibizione di BRAF (dabrafenib) e MEK (trametinib) in un paziente con craniofaringioma papillare BRAFV600E-mutato, in precedenza sottoposto a numerosi interventi neurochirurgici (15). Il paziente mostrò una risposta eccellente in termini di riduzione volumetrica sia della componente solida (85%) sia di quella cistica (81%). Altri autori proposero l’impiego del solo inibitore di BRAF verumafenib, che però risultava molto efficace durante il trattamento, ma gravato da rapida ricrescita a breve termine dopo la sospensione del farmaco rispetto al trattamento con la doppia inibizione BRAF/MEK (16). La possibile spiegazione è che in altre neoplasie BRAF-mutate, in cui la maggior parte dei fenomeni di resistenza al singolo inibitore di BRAF avviene per la riattivazione della via MAPK (RAF-MEK-ERK), l’aggiunta di un inibitore MEK ritarda la comparsa di cloni resistenti, oltre a ridurre lo sviluppo di carcinomi cutanei squamo-cellulari, complicanza più frequente con l’impiego del solo inibitore di BRAF (9).
Dopo le iniziali segnalazioni come singoli case report e piccole serie (15-17), furono disegnati studi in popolazioni più vaste di pazienti con craniofaringioma papillare BRAF-mutato. Uno studio prospettico (18) valutò i risultati della terapia combinata con inibitore di BRAF (vemurafenib) e inibitore di MEK (cobimetinib) a distanza di almeno tre settimane dall’intervento chirurgico in 16 adulti di nuova diagnosi, che non erano stati preventivamente trattati con radioterapia o terapie sistemiche e che avevano un residuo di neoplasia di almeno 10 mm di diametro (volume mediano 2.75 cm3). Ogni ciclo terapeutico di 28 giorni prevedeva la somministrazione di vemurafenib per tutta la durata del ciclo e di cobimetinib per 21 giorni. Dopo una mediana di 8 cicli, 15/16 pazienti (94%) mostrarono una significativa riduzione volumetrica della neoplasia (mediana 91%). L’unico paziente che non aveva raggiunto una risposta volumetrica, in realtà non era riuscito a completare neppure il primo ciclo, per la comparsa di seri effetti collaterali. La mediana del follow-up fu di 22 mesi e la percentuale stimata di pazienti che continuavano ad avere una risposta volumetrica a 12 mesi era del 93%. Tre dei 15 pazienti che avevano ottenuto una buona riduzione volumetrica avevano sviluppato progressione di malattia dopo che la terapia era stata interrotta. Complessivamente, 8 pazienti ricevettero altri trattamenti (radioterapia, secondo approccio chirurgico, inibitore di BRAF: dabrafenib) dopo l’interruzione dei cicli di terapia combinata con inibitori BRAF-MEK e 7 pazienti non ricevettero alcun trattamento: dopo follow-up mediano di 23 mesi, sei non mostrarono progressione. Ci furono 12 eventi avversi di grado 3 (eruzione cutanea, ipertensione arteriosa, disidratazione, prolungamento QT, reazioni allergiche, aumento di fosfatasi alcalina, iponatriemia) e due eventi avversi di grado 4 (aumento di CPK e iperglicemia), con necessità di interruzione del trattamento in tre casi.
Un successivo studio multicentrico di coorte reclutò retrospettivamente 16 pazienti con craniofaringioma papillare BRAF-mutato, trattati tra il 2019 e il 2023 con la doppia inibizione BRAF-MEK con l’associazione dabrafenib-trametinib (19). La popolazione era eterogenea: terapia neo-adiuvante (in 6 pazienti di nuova diagnosi con tumori voluminosi, in cui non era proponibile l’intervento neurochirurgico), terapia adiuvante (in 8 pazienti già sottoposti a procedure chirurgiche singole o multiple prima della terapia radiante) o terapia palliativa a lungo termine (in due pazienti che avevano sviluppato recidive o con residui tumorali rilevanti nonostante precedenti trattamenti multi-modali). Anche in questo studio il 94% dei pazienti (15/16) mostrò evidente riduzione volumetrica (subtotale o parziale): in media 81.4 ± 18.3% all’ultimo controllo. La terapia fu ben tollerata in 10 pazienti (62.5%); gli eventi avversi (pneumopatia ipossiemica, febbre, incremento degli indici di citolisi epatica, cefalea, vomito, astenia, eruzione cutanea, edemi periferici, diarrea, mialgie) portarono all’interruzione temporanea del trattamento in 5 pazienti (grado 1 e 3) e definitiva in tre pazienti (grado 3).
Se da un lato il trattamento mediante la doppia inibizione BRAF-MEK nei craniofaringiomi papillari ha indubbiamente aperto nuove prospettive nella gestione di questi tumori, dall’altro rimangono ancora da chiarire alcuni punti cruciali: la durata del trattamento farmacologico, il momento ideale in cui eventualmente proporlo al paziente durante il suo percorso terapeutico, l’eventuale modificazione del work-up diagnostico-terapeutico. Al momento, non siamo in grado di formulare una diagnosi pre-operatoria di craniofaringioma papillare solamente sulla base della storia clinica, dell’età del paziente e dell’imaging. Pertanto, il team multi-disciplinare (comprendente il patologo, l’endocrinologo, il neurochirurgo, il neuroradiologo, il radioterapista e l’oncologo) rimane fondamentale nel definire il work-up diagnostico e la migliore strategia terapeutica.
Terapia farmacologica del craniofaringioma adamantinomatoso
Il coinvolgimento della via WNT/ß-catenina nella tumorigenesi della variante adamantinomatosa potrebbe rappresentare un primo passo verso l’identificazione di una terapia mirata su un preciso bersaglio molecolare (3,4). Tuttavia, la complessità della via e l’incompleta conoscenza dei meccanismi fisiologici che la regolano rendono attualmente ancora prematura l’identificazione di una terapia mirata su questo bersaglio. Comunque, è meritevole di attenzione una recente scoperta che potrebbe fornire ulteriori elementi di speculazione in termini di prospettive terapeutiche, in particolare per i casi gravati da frequenti recidive e ripresa di malattia (20,21). Va aggiunto, peraltro, che le rare forme di craniofaringioma maligno descritte in letteratura derivano dalla variante adamantinomatosa in oltre i due terzi dei casi (22).
A differenza della variante papillare, in cui la via MAPK/ERK è attivata come conseguenza della mutazione somatica dell’oncogene BRAFV600E, quella adamantinomatosa non mostra generalmente l’attivazione di questa via. Tuttavia, alcuni dati mostrano che l’attivazione della via MAPK/ERK può avvenire in maniera paracrina in alcune aree di craniofaringioma adamantinomatoso, soprattutto nelle forme gravate da maggiore aggressività, in seguito all’espressione locale di fattori multipli, quali alcuni membri della famiglia FGF e EGF, come pure di ligandi e proteine trans-membrana (ad esempio CD47) (20,23). Sulla scorta di questi dati, un recente studio pre-clinico ha mostrato che in 6/14 casi di craniofaringioma adamantinomatoso recidivante era evidente l’attivazione della via MAPK/ERK nelle aree prospicienti il fronte di aggressività locale, fornendo così il presupposto per futuri studi clinici per valutare l’eventuale impiego di inibitori della via MAPK nei craniofaringiomi adamantinomatosi aggressivi e recidivanti (21).
Bibliografia
- Zheng S, Fang Y, Cai BW, et al. Intracystic bleomycin for cystic craniopharyngiomas in children. Cochrane Database Syst Rev 2016, 7: CD008890.
- Kilday J-P, Caldarelli M, Massimi L, et al. Intracystic interferon-alpha in pediatric craniopharyngioma patients: an international multicenter assessment on behalf of SIOPE and ISPN. Neuro-Oncology 2017, 19: 1398-407.
- Hofmann BM, Kreutzer J, Saeger W, et al. Nuclear ß-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and Rathke cleft cysts: a clinico-pathologic approach. Am J Surg Pathol 2006, 30: 1595-603.
- Oikonomou E, Barreto DC, Soares B, et al. ß-catenin mutations in craniopharyngiomas and pituitary adenomas. J Neurooncol 2005, 73: 205-9.
- Brastianos PK, Taylor-Weiner A, Manley PE, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet 2014, 46: 161-5.
- Marucci G, de Biase D, Zoli M, et al. Targeted BRAF and CTNNB1 next-generation sequencing allows proper classification of nonadenomatous lesions of the sellar region in samples with limiting amounts of lesional cells. Pituitary 2015, 18: 905-11.
- Kim JH, , Pailus W, Heim S. BRAF V600E mutation is a useful marker for differentiating Rathke’s cleft cyst with squamous metaplasia from papillary craniopharyngioma. J Neurooncol 2015, 123: 189-91.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010, 363: 809-19.
- Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhinition in melanoma with BRAF V600 mutations. N Engl J Med 2012, 367: 1694-703.
- Hyman DM, PuzanovI, Subbiah V, et al. Vemurafenib in multiple nonmelanoma. cancers with BRAF V600 mutations. N Engl J Med 2015, 373: 726-36.
- Dietrich S, Glimm H, Andrulis M, et al. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 2012, 366: 2038-40.
- Munoiz J, Schlette E, Kurzrock R. Rapid response to vemurafenib in a heavely pretreated patient with hairy cell leukemia and BRAF mutation. J Clin Oncol 2013, 31: e351-2.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAFV600E-mutated Erdheim-Chester disease. J Clin Oncol 2014, 33: 411-8.
- Kaye FJ, Ivey AM, Drane WE, et al. Clinical and radiographic response with combined BRAF-targeted therapy in stage 4 ameloblastoma. J Natl Cancer Inst 2015, 107: 378.
- Brastianos PK, Shankar GM, Gill CM, et al. Dramatic response of BRAF V600E mutant papillary craniopharyngioma to targeted therapy. J Natl Cancer Inst 2016, 188: djv310.
- Aylwin SJ, Bodi I, Beaney R. Pronounced responce of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary 2016, 19: 544-6.
- Bernstein A, Mrowczynski OD, Greene A, et al. Dual BRAF/MEK therapy in BRAF V600E-mutated primary brain tumors: a case series showing dramatic clinical and radiographic responses and a reduction in cutaneous toxicity. J Neurosurg 2019, 133: 1704-9.
- Brastianos PK, Twohy E, Geyer S, et al. BRAF-MEK inhibition in newly diagnosed papillary craniopharyngiomas. N Engl J Med 2023, 389: 118-26.
- De Alcubierre D, Gkasdaris G, Mordrel M, et al. BRAF and MEK inhibitor targeted therapy in papillary craniopharyngiomas: a cohort study. Eur J Endocrinol 2024, 191: 251-61.
- Apps JR, Carreno G, Gonzales-Meljem JM, et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol 2018, 135: 757-77.
- Apps JR, Gonzales-Meljem JM, Guiho R, et al. Recurrent adamantinomatous craniopharyngiomas show MAPK pathway activation, clonal evolution and rare TP53-loss-mediated malignant progression. Acta Neuropathol Commun 2024, 12: 127.
- Wang F, He Y, Wang Y, et al. Malignant craniopharingioma: a report of seven cases and review of the literature. World Neurosurg 2020, 135: e194-201.
- Zhang H, Wang C, Fan J, et al. CD47 promotes the proliferation and migration of adamantinomatous craniopharyngioma cells by activating the MAPK/ERK pathway, and CD47 blockade facilitates microglia-mediated phagocytosis. Neuropathol Appl Neurobiol 2022, 48: e12795.
Classificazione dell'ipopituitarismo
Marco Bonomi, Giovanni Goggi
Dipartimento di Medicina Endocrino-Metabolica, Ospedale San Luca – IRCCS Istituto Auxologico Italiano – Milano
Dipartimento di Scienze Cliniche e di Comunità – Università degli Studi di Milano - Milano
(aggiornamento al 14 novembre 2019)
Definizione
L’ipopituitarismo è una condizione clinica caratterizzata dalla ridotta secrezione di uno o più ormoni prodotti dall’ipofisi, che porta a un conseguente deficit funzionale delle relative ghiandole bersaglio (1).
L’ipopituitarismo può interessare sia la porzione anteriore (adeno-ipofisi) che la porzione posteriore (neuro-ipofisi) dell’ipofisi e può pertanto interessare la secrezione dei seguenti ormoni: GH, PRL, ACTH, TSH, LH, FSH, ADH.
L’ipopituitarismo può essere distinto in totale (panipopituitarismo), parziale oppure unitropico, a seconda che sia rispettivamente compromessa la secrezione di tutti, alcuni o solamente uno degli ormoni ipofisari (2).
Eziologia
L’ipopituitarismo può riconoscere diverse origini.
- Patologie espansive: i tumori ipofisari (sia secernenti che non-secernenti), rappresentano da soli la causa in assoluto più frequente (61% dei casi). Si tratta spesso di tumori benigni, mentre meno comuni sono le metastasi di tumori primitivi (localizzati soprattutto a livello di mammella, colon e prostata). Un’altra possibile causa è rappresentata da tumori ipotalamici e para-ipofisari come meningiomi, gliomi e craniofaringiomi (3).
- Patologie infettive: infiammazioni purulente del seno sfenoidale possono coinvolgere la sella turcica, la capsula e il parenchima ipofisario. L’ipofisite infettiva è oggi una condizione assai rara, dato l’ampio ventaglio di terapie anti-microbiche disponibili. Alcune patologie infettive che possono esitare in tale complicanza sono la tubercolosi, l’istoplasmosi, la meningite, la sifilide e l’AIDS (2).
- Patologie autoimmuni: l’ipofisite linfocitaria è una rara affezione autoimmune, in cui il parenchima ipofisario, infiltrato da linfociti e plasmacellule, viene progressivamente distrutto, esitando in un quadro di ipopituitarismo. È una patologia che colpisce prevalentemente il sesso femminile e che solitamente insorge durante la gravidanza o il puerperio; vista la sua natura autoimmune, è responsiva al trattamento corticosteroideo. Anche le vasculiti possono interessare la regione.
- Patologie infiltrative: la sarcoidosi e l’istiocitosi X sono due patologie granulomatose sistemiche, che possono dare localizzazioni ipotalamo-ipofisarie, determinando un quadro di ipopituitarismo totale o parziale, spesso associato a diabete insipido e iperprolattinemia da deafferentazione del fascicolo dopaminergico ipotalamo-ipofisario (2).
- Patologie metaboliche: nell’emocromatosi si verifica un significativo deposito di emosiderina all’interno dell’ipofisi, in particolare nelle cellule gonadotrope, motivo per cui la più frequente manifestazione clinica che ne deriva è l’ipogonadismo ipogonadotropo. Altre patologie con localizzazioni ipofisarie sono l’amiloidosi e la mucopolisaccaridosi (2).
- Patologie vascolari: l’apoplessia ipofisaria è una rara complicanza di un adenoma ipofisario (anche se talvolta può verificarsi nel contesto di un’ipofisi normale), determinata da ischemia o emorragia ipofisaria (3). Tali eventi possono verificarsi spontaneamente oppure essere favoriti dalla terapia radiante di un adenoma, da traumi cranici o dall’esecuzione di test di stimolo con TRH o GnRH (2). La sindrome di Sheehan, invece, è l’esito di un infarto che colpisce l’ipofisi iperplastica della donna gravida in seguito ad un’importante emorragia post-partum: l’ipotensione che ne deriva, infatti, riduce l’apporto ematico all’ipofisi (che durante la gravidanza ha triplicato il suo volume e dunque anche la sua esigenza di flusso ematico), esitando in un infarto ischemico (2). L’ipopituitarismo, inoltre, può anche essere l’esito di un ictus o di un’emorragia subaracnoidea (tipicamente nell’anziano) (1). Infine, aneurismi che originano dall’arteria cerebrale comunicante anteriore possono espandersi e determinare lesioni a carico dell’ipofisi e dei nervi ottici (2).
- Patologie psicogene: in condizioni di grave disagio affettivo, violenza fisica o deprivazione di stimoli psico-sociali, può instaurarsi un deficit funzionale di GH che determina un rallentamento della crescita, potenzialmente reversibile se l’ambiente di vita viene modificato favorevolmente (2). Invece, in donne con disturbi del comportamento alimentare, o sottoposte a importante stress fisico, si riscontrano spesso disordini del ciclo mestruale dovuti a un’alterazione della secrezione pulsatile di GnRH (2).
- Iperprolattinemia: determina un quadro di ipogonadismo centrale secondario all’aumento del tono oppioide endogeno e al conseguente blocco della secrezione pulsatile di GnRH e quindi di LH e FSH (2).
- Danni iatrogeni: la radioterapia a livello cranico è in grado di determinare un danno soprattutto ipotalamico, da cui consegue più comunemente una modesta iper-PRL, un deficit di GH e/o di gonadotropine; i deficit di ACTH e TSH si manifestano in genere più tardivamente, anche 10-15 anni dopo l’irradiazione (2). Anche le forme più moderne di radioterapia, come la radiochirurgia, introdotte per avere un bersaglio più mirato con risparmio dell’ipofisi sana, si accompagnano a ipopituitarismo, seppure con minor frequenza (4). L’ipopituitarismo per danno ipotalamico e ipofisario può comparire dopo pan-irradiazione encefalica, come nelle leucosi, o dopo irradiazione di tumori della testa-collo, ad es. tumori del rinofaringe. Tale complicanza spesso si manifesta già nei soggetti trattati in età pediatrica, ma può comparire anche tardivamente. L’ipopituitarismo iatrogeno può anche essere la conseguenza di un danno riportato durante un intervento di adenomectomia selettiva (3-10% degli interventi), che in genere è precoce e permanente (2). Infine, trattamenti prolungati con glucocorticoidi sono in grado di inibire l’asse ipotalamo-ipofisi-surrene, mentre l’assunzione di farmaci oppioidi, glucocorticoidi o la sospensione di steroidi anabolizzanti può determinare un quadro di ipogonadismo centrale. Neurofarmaci e psicofarmaci ad azione anti-dopaminergica, serotoninergica e anti-istaminergica possono provocare iperprolattinemia (2). Farmaci biologici (usati come terapia di patologie oncologiche o autoimmuni) possono provocare ipofisite e ipopituitarismo.
- Lesioni traumatiche: i traumi cranici possono determinare quadri di ipopituitarismo per effetto di lesioni a carico del peduncolo o della vascolarizzazione ipofisaria. Se il difetto secretivo si instaura acutamente, prevarrà un quadro clinico di iposurrenalismo acuto, mentre i deficit delle altre tropine tendono a manifestarsi più gradualmente. Nella maggior parte dei casi si verifica un deficit di gonadotropine, mentre il 5-10% dei pazienti mostra deficit isolati o combinati di ACTH, TSH e GH. È frequentemente associato il diabete insipido (2).
- Empty sella: è il reperto radiologico di una sella turcica allargata o deformata, parzialmente o completamente riempita da liquido cerebro-spinale, come conseguenza di un’erniazione dello spazio subaracnoideo sovra-sellare, che va a determinare stiramento e compressione dell’ipofisi (1).
- Patologie congenite: difetti nella formazione della tasca di Rathke possono risultare nell’agenesia o nella grave ipoplasia dell’adeno-ipofisi, condizione che normalmente è incompatibile con la vita, ma se il neonato riesce a sopravvivere, il quadro clinico è quello di un panipopituitarismo (2). Responsabili di questo quadro possono essere mutazioni a carico di geni coinvolti nell’embriogenesi ipofisaria e ipotalamica (HESX1, LHX3/4, PROP1, PUO1F1, SOX2, SOX4, OTX2, PTX2), nella differenziazione cellulare, nella trascrizione genica, oppure che codificano per mediatori ipotalamici, per ormoni ipofisari, per recettori o per effettori post-recettoriali (vedi anche capitolo dedicato a genetica ipopituitarismi). Il quadro clinico può essere un deficit isolato (ad esempio ipogonadismo con o senza anosmia) o un deficit multiplo (MPHD, multiple pituitary hormone deficiency, ad esempio deficit di GH-PRL-TSH) (5). È importante osservare che, sebbene ereditari, i quadri clinici possono rendersi talora evidenti tardivamente, ad esempio in età giovane adulta. La diagnosi eziologica di forma ereditaria consente lo screening dei familiari. Alcune di queste mutazioni sono responsabili di espressioni fenotipiche particolari, come il panipopituitarismo associato a displasia setto-ottica (HESX1) o rigidità cervicale (LHX3), malformazioni oculari (OTX2), microftalmia (SOX2) (5). La sindrome di Kallmann, invece, è una condizione di ipogonadismo ipogonadotropo associato a ipo/anosmia (2). Infine, l’assenza o un alterato sviluppo del diaframma sellare può consentire allo spazio subaracnoideo di estendersi all’interno della sella turcica, riempiendola di liquor cefalo-rachidiano e determinando così un appiattimento dell’ipofisi (sella vuota primaria) (2).
- Idiopatica. Esiste un sottogruppo di pazienti in cui la causa dell’ipopituitarismo rimane sconosciuta. Spesso si tratta di deficit isolati e si ipotizzano traumi misconosciuti, patogenesi immunologiche, alterazioni neuro-regolatorie transitorie (vedi Deficit GH nel bambino).
Accanto alla classificazione eziologica, può essere utile considerare altri criteri di classificazione:
- cronologico: accanto alle forme permanenti, si possono osservare forme di ipopituitarismo:
- transitorie o reversibili, come nei pazienti con adenoma ipofisario sottoposti ad asportazione dell'adenoma, oppure nei pazienti affetti da prolattinoma trattati con dopaminergici, in cui si può verificare, talvolta, non solo il ripristino della funzione gonadica ma anche la correzione di altri deficit;
- funzionali:
- pazienti in terapia con glucocorticoidi o L-tiroxina a dosi farmacologiche;
- durante malattie intercorrenti severe: funzione tiroidea e gonadica;
- disturbi dell'alimentazione;
- per gravità del singolo deficit, distinguendo:
- deficit conclamato;
- deficit "latente", che può essere evidenziato solo da test provocativi, ad esempio l’insufficienza surrenalica;
- per età di comparsa:
- forme pediatriche, talora evidenti già alla nascita (forme congenite);
- forme dell'adulto;
- forme dell'anziano.
Tale classificazione risulta particolarmente utile nell’approccio iniziale del paziente ipopituitarico, indirizzando verso eziologie specifiche. Nel soggetto anziano può risultare difficile distinguere tra quella che è la normale riduzione fisiologica della produzione ormonale che si manifesta con l'età (per esempio, per il testosterone) e il risultato patologico vero dipendente dalla condizione patologica.
Bibliografia
- Curtò L, Trimarchi F. Hypopituitarism in the elderly: a narrative review on clinical management of hypothalamic-pituary-gonadal, hypothalamic-pituitary-thyroid and hypothalamic-pituitary-adrenal axis dysfunction. J Endocrinol Invest 2016, 39: 1115-24.
- Faglia G, Beck-Peccoz P, Spada S. Malattie del sistema endocrino e del metabolismo, 5° edizione. McGraw-Hill 2013.
- Gounden V, Jialal I. Hypopituitarism (panhypopituitarism). StatPearls [Internet]. Treasure Island (FL). 2019.
- Sherlock M, Ayuk J, Tomlinson JW, et al. Mortality in patients with pituitary disease. Endocr Rev 2010, 31: 301–42.
- Prabhakar VKB, Shalet SM. Aetiology, diagnosis, and management of hypopituitarism in adult life. Postgrad Med J 2006, 82: 259–66.
- Castinetti F, Reynaud R, Saveanu A, et al. An update in the genetic aetiologies of combined pituitary hormone deficiency. Eur J Endocrinol 2016, 174: R239-47.
Clinica, diagnostica e terapia dell'ipopituitarismo
Generalità di clinica, diagnostica e terapia dell'ipopituitarismo
Marco Bonomi, Giovanni Goggi
Dipartimento di Medicina Endocrino-Metabolica, Ospedale San Luca – IRCCS Istituto Auxologico Italiano – Milano
Dipartimento di Scienze Cliniche e di Comunità – Università degli Studi di Milano - Milano
(aggiornato al 14 novembre 2019)
Epidemiologia
L’incidenza oscilla tra i 12 e i 42 nuovi casi per milione di abitanti all’anno, mentre la prevalenza è di circa 300-455 casi per milione di abitanti, senza significative differenze di genere. È probabile che tali valori siano sotto-stimati, dal momento che le manifestazioni cliniche di questa condizione sono spesso, e per lungo tempo, ambigue (1).
Manifestazioni cliniche
L’ipopituitarismo si accompagna ad aumento di morbilità e mortalità. La morbilità è evidenziata anche dal persistere di una ridotta qualità di vita nonostante le terapie ormonali sostitutive. Diversi studi retrospettivi hanno dimostrato un eccesso di mortalità, principalmente cardio-vascolare, le cui cause sono controverse: una non corretta terapia sostitutiva, in particolare mancata terapia sostitutiva del deficit di GH, inadeguata terapia sostitutiva dell’ipogonadismo, sovradosaggio della terapia sostitutiva dell’iposurrenalismo; altri fattori possono contribuire, in particolare l’utilizzo della radioterapia nei tumori ipofisari (2).
I segni e i sintomi di ipopituitarismo sono molto variabili: dipendono in primo luogo dal numero e dal tipo di deficit ormonali coinvolti, ma allo stesso tempo anche dall’età di comparsa, dalla causa e dalla velocità con cui l’ipopituitarismo si verifica (2). In genere, in presenza di un danno cronico (es. lesioni ipofisarie progressive) la compromissione secretoria segue la progressione GH -> FSH/LH -> TSH -> ACTH, il che dimostra una maggior sensibilità al danno da parte delle cellule somatotrope e gonadotrope, mentre le cellule tireotrope e corticotrope sono più resistenti (3); viceversa, in caso di un insulto acuto (es. apoplessia ipofisaria) prevale l’aspetto dell’insufficienza cortico-surrenalica acuta (3).
In generale, i difetti unitropici danno luogo a quadri clinici analoghi, ma più sfumati, a quelli conseguenti ai deficit delle rispettive ghiandole bersaglio (3). I pazienti panipopituitarici invece, parallelamente alla presenza di segni e sintomi riconducibili ai deficit ormonali ipofisari, si presentano con un quadro ben più complesso: sono pazienti generalmente sovrappeso, la cui cute è sottile, pallida, depigmentata e con fini rughe al volto, tale da conferire un aspetto vecchieggiante (3). I capelli sono sottili, le unghie fragili e si osserva una caduta dei peli pubici e ascellari (3). In entrambi i sessi sarà presente un quadro di ipogonadismo centrale che, se si instaura durante l’infanzia, non consentirà lo sviluppo puberale e darà luogo a un difetto di crescita (3). La forza muscolare è ridotta e i pazienti lamentano astenia e affaticabilità: tali manifestazioni sono l’effetto di anemia secondaria al deficit di ormoni tiroidei e androgeni, del venir meno degli effetti metabolici del GH e dell’ipotensione arteriosa conseguente all’iposurrenalismo (3). Vi è inoltre tendenza all’ipoglicemia a digiuno, dovuta al difetto di GH e glucocorticoidi. I pazienti panipopituitarici, infine, non presentano generalmente deficit cognitivi, ma mostrano spesso tendenza all’isolamento, abulia, insicurezza, depressione e facile permalosità (3).
Nel caso di una lesione occupante spazio, inoltre, i sintomi dell’ipopituitarismo potranno associarsi anche a quelli dati dall’effetto massa: cefalea, nausea e vomito (sintomi riconducibili a ipertensione endocranica) e difetti del campo visivo dovuti alla compressione del chiasma ottico da parte della massa, ovvero l’emianopsia bitemporale (che talvolta, però, può essere solo monolaterale) (4).
Diagnosi
La diagnosi deve essere presa in considerazione nei pazienti con un quadro clinico compatibile o con ipoglicemia e/o iposodiemia agli esami biochimici di routine, e deve essere confermata dagli appropriati specifici esami ormonali.
Indagini di laboratorio: innanzitutto vanno eseguiti esami ematochimici basali per tutti gli ormoni ipofisari e/o i loro bersagli:
- ACTH e cortisolo
- TSH e fT4
- IGF-I
- LH, FSH, testosterone totale/estradiolo
Non sono necessarie concentrazioni francamente basse degli ormoni ipofisari per porre diagnosi di ipopituitarismo, ma è sufficiente rilevare valori inappropriatamente normali a fronte di ormoni bersaglio significativamente ridotti (3).
In considerazione della variabilità legata all’ora del giorno e alla secrezione pulsatile di alcuni ormoni ipofisari, gli ormoni basali non sono sempre utili ai fini diagnostici: per tale ragione, possono essere effettuati test dinamici per confermare il difetto di alcuni specifici ormoni ipofisari: ad esempio l’ACTH test per cortisolo o il test di ipoglicemia insulinica per GH e cortisolo (4).
Indagini radiologiche: una volta stabilita la diagnosi biochimica di ipopituitarismo, la risonanza magnetica con Gadolinio è l’esame più utile per ricercarne la causa sottostante. Tale esame è in grado di visualizzare l’ipofisi e di rilevare la presenza di eventuali lesioni espansive, infiltrative o infiammatorie nel suo contesto (4).
Campo visivo: è importante valutarlo in presenza di una massa ipofisaria che rischia di comprimere il chiasma ottico (4).
Terapia
La terapia dell’ipopituitarismo deve affrontare due differenti aspetti:
- identificare e trattare la causa sottostante (ad esempio, una massa ipofisaria può essere rimossa chirurgicamente, ecc);
- avviare un’adeguata terapia ormonale sostitutiva per supplire agli ormoni ipofisari carenti (4).
Bibliografia
- Curtò L, Trimarchi F. Hypopituitarism in the elderly: a narrative review on clinical management of hypothalamic-pituary-gonadal, hypothalamic-pituitary-thyroid and hypothalamic-pituitary-adrenal axis dysfunction. J Endocrinol Invest 2016, 39: 1115-24.
- Sherlock M, Ayuk J, Tomlinson JW, et al. Mortality in patients with pituitary disease. Endocr Rev 2010, 31: 301–42.
- Faglia G, Beck-Peccoz P, Spada S. Malattie del sistema endocrino e del metabolismo, 5° edizione. McGraw-Hill 2013.
- Gounden V, Jialal I. Hypopituitarism (panhypopituitarism). StatPearls [Internet]. Treasure Island (FL). 2019.
Ipotiroidismo centrale
Marco Bonomi, Silvia Federici
Dipartimento di Medicina Endocrino-Metabolica, Ospedale San Luca – IRCCS Istituto Auxologico Italiano – Milano
Dipartimento di Scienze Cliniche e di Comunità – Università degli Studi di Milano - Milano
(aggiornato al 14 novembre 2019)
Definizione, classificazione, eziofisiopatologia
Per ipotiroidismo centrale (ICe) si intende un difetto di produzione degli ormoni tiroidei dovuto a un’insufficiente stimolazione da parte del TSH di una ghiandola tiroidea altrimenti normale, come conseguenza di un’alterazione organica o funzionale dell’ipofisi e/o dell’ipotalamo.
È una condizione rara, che può interessare pazienti di ogni età, più frequentemente in forma sporadica, con una prevalenza stimata tra 1:16.000 e 1:100.000 casi (1,2) (circa 1 su 1000 dei pazienti ipotiroidei) (3). Questo dato è probabilmente sottostimato, in quanto la sola misurazione del TSH perde la sua accuratezza diagnostica in questa condizione, che risulta, così, spesso misconosciuta (1,3).
La patogenesi dell’ICe è eterogenea e in parte ancora indeterminata, ma possono essere individuati tre possibili meccanismi, spesso combinati tra loro (1,2,3):
- un difetto di stimolazione ipotalamica o un alterato feed-back agli ormoni tiroidei;
- una riduzione nella riserva di TSH (difetto quantitativo);
- una ridotta bioattività intrinseca del TSH prodotto (difetto qualitativo).
L’ICe può essere congenito o acquisito (di natura tumorale, traumatica, infiltrativa, infiammatoria). In entrambe le forme il difetto dell’asse tireotropo è nella maggior parte dei casi associato ad altri deficit ipofisari.
Le forme congenite (ereditarie o malformative) si manifestano generalmente nell’infanzia, ma in alcuni casi possono essere diagnosticate e/o esordire in età adulta. Il numero dei geni candidati per l’ICe è recentemente molto aumentato. Le loro mutazioni possono essere causa di forme isolate (TSHβ, TRH-R, TBL1X, IRS4) o combinate a deficit ipofisari multipli (i geni più frequentemente responsabili sono IGSF1, recentemente identificato, e PROP1), quindi potenzialmente associate a deficit di crescita, ritardo puberale, iposurrenalismo o difetti neurologici. Alcune forme genetiche si associano inoltre a caratteristiche cliniche peculiari, come il macrorchidismo per IGSF1 e i difetti uditivi per TBL1X (1,2,3). L’analisi genetica è raccomandata nei casi di ICe congeniti a esordio infantile o in qualsiasi età quando l’ICe rimane non spiegato. L’analisi genetica può essere eseguita con sequenziamento diretto in presenza di un fenotipo suggestivo oppure mediante pannello NGS (Next Generation Sequencing) dei geni candidati (2).
I tumori voluminosi della regione sellare e i loro trattamenti rappresentano la causa più frequente di ICe acquisito nell’adulto. Le cellule tireotrope, assieme alle cellule corticotrope, sono relativamente resistenti agli insulti rispetto alle altre cellule ipofisarie (somatotrope e gonadotrope), per cui l’ipotiroidismo in genere si associa o compare successivamente ai deficit di GH o di gonadotropine, mentre è infrequente che sia isolato.
Clinica
La sintomatologia clinica dell’ipotiroidismo centrale è sovrapponibile a quella dell’ipotiroidismo primitivo, anche se il quadro risulta generalmente più sfumato ed eventualmente accompagnato o mascherato da segni di altri deficit ipofisari (un deficit di GH o di ormoni sessuali può mascherare un ICe, mentre il loro trattamento lo slatentizza) o da sintomi collegati alla patologia di base (es. disturbi visivi nelle masse ipofisarie) (1,2,3).
Diagnosi
L’ipotiroidismo centrale deve essere sospettato in presenza di bassi valori sierici di FT4 associati a valori di TSH bassi o inappropriatamente normali. Va ricordato che i livelli di TSH nell’ICe possono essere normali o perfino leggermente aumentati (anche fino a 8-10 mU/L), in quanto il TSH prodotto in queste forme centrali risulta dotato di una ridotta bioattività per alterazioni nella glicosilazione (3). L’ICe deve, inoltre, essere indagato in tutti i casi di storia personale o familiare di disordini ipotalamo-ipofisari o con manifestazioni suggestive di una lesione sellare.
La diagnosi di ICe (vedi flow chart) si pone se, in almeno 2 determinazioni, si conferma la contemporanea presenza di bassi valori di FT4 (con intervallo di riferimento specifico per età) associati a valori bassi/inappropriatamente normali di TSH (1,2,3,4), dopo aver escluso tutte le condizioni potenzialmente confondenti, prima tra tutte una possibile interferenza nella misurazione ormonale (l’interferenza nel dosaggio del TSH può essere indagata per mezzo dei test di diluizione e recupero della immuno-reattività, mentre l’utilizzo di metodi di dosaggio diretti “two steps” consente una misura di FT4 e FT3 generalmente accurata)(1). Occorre, quindi, fare una diagnosi differenziale con le condizioni che possono presentare lo stesso quadro biochimico: forme severe di non-thyroidal illness, assunzione di farmaci o droghe inibenti il TSH (es. glucocorticoidi, cocaina), fase di recupero post-tireotossicosi o sindrome da sospensione della levo-tiroxina, ipotiroxinemia isolata della gravida, nascita pretermine o altre condizioni genetiche (1,2).
Rimane ancora controversa la diagnosi dell’ICe lieve, con FT4 nel range basso di norma. In questi casi la diagnosi può essere supportata da esami o dati aggiuntivi, come la presenza di familiarità, di manifestazioni cliniche associate all’ICe o di mutazioni in geni candidati, la presenza di altri deficit ipofisari, una risposta ridotta (TSH stimolato < 4 mU/L) o ritardata (picco dopo 60’) al test con TRH, l’alterazione di parametri indiretti di disfunzione tiroidea, la progressiva riduzione dell’FT4 (> 20% del valore iniziale) in un paziente in follow-up biochimico. In casi selezionati con sospetta forma di ICe lieve (FT4 borderline), è possibile tentare un trial terapeutico con levo-tiroxina per 3 mesi, che può essere proseguito in caso di miglioramento delle manifestazioni riconducibili a ipotiroidismo (1,2).
La diagnosi di ipotiroidismo centrale rende necessaria l'esecuzione di RM ipotalamo-ipofisaria e di uno studio ormonale completo per escludere una patologia ipotalamo-ipofisaria.
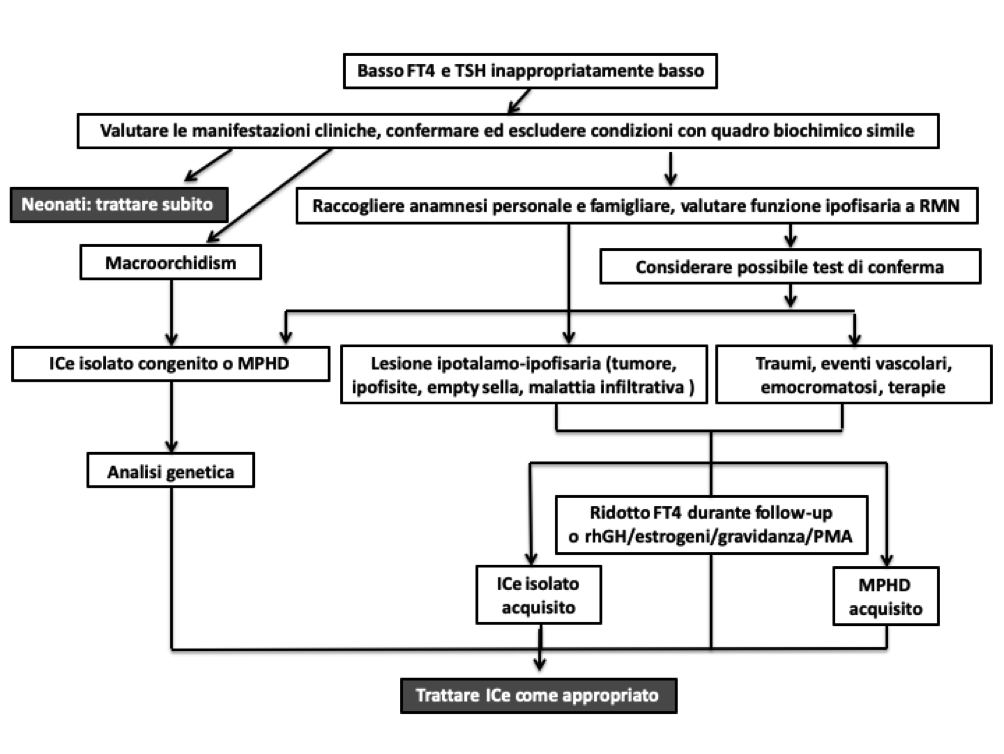
Figura 1. Flow-chart diagnostica in caso di ipotiroidismo centrale (modificata da 1)
Terapia
Prima di iniziare il trattamento deve sempre essere escluso o trattato un eventuale iposurrenalismo centrale concomitante, onde evitare di scatenare una crisi surrenalica. Il trattamento dell’ipotiroidismo centrale è basato sulla somministrazione di levo-tiroxina. Analogamente all’ipotiroidismo primitivo, per impostare la dose iniziale si devono considerare la durata e la severità dell’ipotiroidismo, l’età del paziente e la presenza di malattie concomitanti, in particolare cardiache (2,4):
- < 60 anni: 1.21–1.6 μg/kg/die;
- > 60 anni o con comorbilità cardio-vascolari: 1.0–1.2 μg/kg/die, aumentando il dosaggio gradualmente;
- > 75 anni: non è indispensabile trattare le forme lievi;
- infanzia e adolescenza: sono richieste dosi maggiori in rapporto al peso.
La dose sostitutiva finale è quindi sovrapponibile a quella utilizzata nell’ipotiroidismo primitivo, sebbene nella pratica clinica le dosi utilizzate siano spesso inferiori.
Nel monitoraggio della terapia sostitutiva dell’ICe il TSH è di scarsa rilevanza rispetto a quanto avviene nelle forme di ipotiroidismo primario in terapia (in particolare con il metodo reflex, che deve essere sempre evitato in questa condizione). In questi casi, il monitoraggio si basa sul quadro clinico e sulle concentrazioni di FT4, che devono mantenersi nella parte alta del range di riferimento. L’adeguatezza della terapia sostitutiva va verificata dopo 6–8 settimane dall’inizio, assicurandosi che il prelievo avvenga prima dell’assunzione mattutina o almeno 4 ore dopo la stessa. L’esecuzione di FT3 è utile solo per escludere il sospetto di eccesso nella terapia sostitutiva. Concentrazioni sieriche soppresse di TSH sono indicative di un’adeguata sostituzione in pazienti affetti da ICe che avevano valori di TSH pre-trattamento al di sopra del limite inferiore di riferimento, mentre un TSH > 1 mU/L suggerisce un’insufficienza della terapia sostitutiva. La determinazione del TSH è invece inutile nei pazienti che presentavano un TSH basso già prima della terapia (1,2).
Nei pazienti adulti, una volta ottenuta un’adeguata sostituzione ormonale, è raccomandato eseguire monitoraggio annuale della FT4. In caso di modifica terapeutica, è indicato rivalutare 4–6 settimane dopo la correzione. Il follow-up dei pazienti pediatrici dovrebbe essere fatto in maniera analoga ai pazienti con ipotiroidismo primario (1,2).
Bisogna infine considerare un aumento del dosaggio di levo-tiroxina in tutte le condizioni associate ad aumentato fabbisogno di ormone tiroideo (1,2):
- gravidanza;
- aumento di peso;
- sviluppo puberale;
- introduzione di terapie in grado di influire sull’assorbimento di levo-tiroxina o sul metabolismo degli ormoni tiroidei;
- introduzione di terapia sostitutiva estrogenica/contraccettivi orali;
- introduzione di terapia sostitutiva con GH (il GH promuove la conversione di T4 in T3. Il trattamento con GH del deficit di GH riduce talvolta la secrezione di TSH per incremento della secrezione somatostatinergica. Pertanto, nelle malattie ipotalamo-ipofisarie l’inizio della terapia sostitutiva con rhGH può smascherare un ipotiroidismo latente, con riduzione di TSH e soprattutto di FT4, oppure richiedere un incremento della dose nei pazienti che sono già in terapia sostitutiva con L-tiroxina) (3);
- stimolazione ovarica controllata.
Durante la gravidanza è necessario un incremento del 25-50% della dose iniziale, al fine di mantenere i livelli di fT4 nel quartile superiore del range di riferimento specifico per trimestre (2).
Una riduzione del dosaggio di L-T4 è invece indicata (1,2):
- nei pazienti anziani, soprattutto qualora siano presenti comorbilità cardio-vascolari;
- dopo il parto;
- alla menopausa;
- dopo perdita di peso;
- qualora i co-trattamenti elencati precedentemente siano interrotti.
Bibliografia
- Persani L, Cangiano B, Bonomi M. The diagnosis and management of central hypothyroidism in 2018. Endocr Connect 2019, 8: R44-54.
- Persani L, Brabant G, Dattani M, et al. 2018 European Thyroid Association (ETA) guidelines on the diagnosis and management of central hypothyroidism. Eur Thyroid J 2018, 7: 225–37.
- Beck-Peccoz P, Rodari G, Giavoli C, Lania A. Central hypothyroidism – a neglected thyroid disorder. Nature Rev Endocrinol 2017, 13: 588–98.
- Ferretti E, Persani L, Jaffrain-Rea ML, et al. Evaluation of the adequacy of levothyroxine replacement therapy in patients with central hypothyroidism. J Clin Endocrinol Metab 1999, 84: 924-9.
Iposurrenalismo centrale
Marco Bonomi, Luca Giovanelli
Dipartimento di Medicina Endocrino-Metabolica, Ospedale San Luca – IRCCS Istituto Auxologico Italiano – Milano
Dipartimento di Scienze Cliniche e di Comunità – Università degli Studi di Milano - Milano
(aggiornato al 14 novembre 2019)
Definizione, classificazione, eziofisiopatologia
Per iposurrenalismo centrale si intende un deficit della funzione corticosurrenalica secondario al deficit di ACTH.
L’ACTH viene secreto dall’adeno-ipofisi sotto lo stimolo di CRH e AVP, prodotti dall’ipotalamo, e va ad agire sul cortico-surrene, mantenendone il trofismo e stimolandone il rilascio di cortisolo e androgeni surrenalici. Pertanto, il blocco della secrezione di ACTH ha due conseguenze sul surrene:
- deficit di secrezione di glucocorticoidi (GC) e androgeni surrenalici (con conservata secrezione di mineralcorticoidi);
- ipotrofia del cortico-surrene, che si sviluppa gradualmente nel giro di alcune settimane.
L’iposurrenalismo centrale si distingue in secondario e terziario, a seconda che il deficit sia rispettivamente di origine ipofisaria o ipotalamica. Un’altra classificazione definisce secondario l’iposurrenalismo causato da una patologia ipotalamo-ipofisaria, terziario quello indotto da un eccesso di glucocorticoidi esogeni o endogeni (1,2).
La prevalenza dell’iposurrenalismo centrale, escludendo quello provocato dall’assunzione di cortico-steroidi, è stimata intorno a 150-280 per milione di abitanti.
La forma più frequente di iposurrenalismo centrale è di tipo funzionale, dovuta alla protratta inibizione dell’asse ipotalamo-ipofisi-surrene legata ad eccesso di glucocorticoidi, sia esogeni (terapie farmacologiche) che endogeni (s. di Cushing). In questo tipo di iposurrenalismo l’effetto più prolungato dei glucocorticoidi si verifica a livello ipotalamico. Resta tutt’oggi difficile predire l’insorgenza di iposurrenalismo iatrogeno sulla base di tipo di steroide utilizzato, dosaggio, via di somministrazione e durata del trattamento; il recupero della funzionalità dell’asse HPA può avvenire dopo settimane o mesi dalla sospensione o non avvenire mai (3,4).
A parte queste condizioni, il deficit di ACTH è raramente isolato, in quanto le cellule corticotrope sono le più resistenti agli insulti dannosi, mentre più spesso rientra in un quadro di deficit ipofisari multipli o di panipopituitarismo (fino a 1/3 dei pazienti ipopituitarici possono presentare insufficiente produzione di ACTH). Possibili cause sono rappresentate da tumori della regione ipotalamo-ipofisaria, in particolare craniofaringiomi (con una prevalenza di iposurrenalismo fino al 90% dopo intervento chirurgico) e adenomi ipofisari clinicamente non funzionanti (prevalenza pre-operatoria 50%, post-operatoria 75%), radioterapia per tumori cerebrali, traumi cranici e ipofisite linfocitaria. Esistono anche forme genetiche, in cui il deficit di ACTH può essere isolato (mutazioni a carico di POMC) o associato ad altri deficit ipofisari (1,2).
Clinica
È essenziale distinguere una forma di iposurrenalismo centrale che si sviluppa gradualmente, come ad esempio in corso di neoplasie ipotalamo-ipofisarie o per effetto di radioterapia della regione sellare, dall’iposurrenalismo acuto (crisi surrenalica), che si può osservare in corso di apoplessia o dopo sospensione brusca di glucocorticoidi esogeni o asportazione di adenomi ACTH-secernenti.
Il deficit di ACTH si può presentare nel neonato con ipoglicemia e crisi comiziali e nell’infanzia con ritardo di crescita. Nell’adulto, si può sospettare di fronte a ipotensione e iposodiemia euvolemica inspiegabili; si possono inoltre sovrapporre i sintomi legati ai concomitanti deficit di altre tropine ipofisarie. Tuttavia, il quadro clinico risulta spesso sfumato (astenia e ridotta performance fisica) e può precipitare in caso di stress intercorrenti. A differenza del morbo di Addison, manca l'iperpigmentazione cutanea e la funzione mineralcorticoide è preservata (quindi non c’è iperkaliemia) (2).
Diagnosi
Il sospetto di iposurrenalismo centrale si pone per sintomi o dati di laboratorio suggestivi (ipotensione cronica o acuta, iponatremia, ipoglicemia, ecc), oppure in corso di indagini per una lesione ipotalamo-ipofisaria nota.
Nel caso di esordio acuto, va prelevato un campione di sangue per il dosaggio della cortisolemia e, in attesa dei risultati, va immediatamente iniziata la terapia sostitutiva con idrocortisone (HC) ev. Negli altri casi vi è maggior tempo per il processo diagnostico.
L’iter diagnostico prevede in prima istanza la misurazione della cortisolemia basale fra le ore 8 e 9 del mattino: nei pazienti non in fase acuta (diversa è infatti l’interpretazione della cortisolemia in condizioni acute), tenendo conto di eventuali fattori interferenti (quali lo stress da prelievo e la concomitante assunzione di estrogeni orali), valori < 3 µg/dL sono diagnostici di insufficienza surrenalica, mentre valori > 15-18 µg/dL permettono di escluderla. Per valori compresi fra 3 e 15 µg/dL è indicata una successiva valutazione dinamica della riserva corticotropa (un picco di cortisolo stimolato < 18.1 µg/dL è suggestivo per iposurrenalismo). Tale zona grigia è piuttosto ampia e quindi molti soggetti con patologia ipotalamo-ipofisaria dovrebbero essere ulteriormente indagati in accordo alla letteratura. In realtà bisogna considerare anche il contesto clinico: per esempio, nei pazienti con valori di cortisolemia ai livelli alti della cosiddetta zona grigia e preservazione delle altre funzioni ipofisarie, è altamente improbabile un deficit corticotropo. Al contrario, pazienti con deficit di altre funzioni ipofisarie oppure con evoluzione progressiva del danno ipofisario (es. per effetto di radioterapia pregressa) necessitano sicuramente di test di valutazione della funzione surrenalica qualora la cortisolemia risulti in questa zona grigia. Diversa ancora risulta l’interpretazione della cortisolemia in condizioni acute, esempio malattie intercorrenti. In tali casi è importante non dilazionare la terapia sostitutiva in attesa dei test di conferma, che possono essere eseguiti una volta risolto il problema acuto.
Il dosaggio dell’ACTH plasmatico dovrebbe poi permettere la differenziazione fra forma primitiva e centrale: in realtà, sebbene elevati valori di ACTH siano diagnostici di morbo di Addison, il rilievo di livelli bassi o inappropriatamente normali potrebbe non essere altrettanto dirimente nella diagnosi di iposurrenalismo secondario (1).
Poiché i GC sopprimono l’asse HPA e provocano interferenze nel dosaggio del cortisolo, il test di stimolo andrebbe eseguito a distanza di almeno 18-24 ore (o più nel caso di GC di sintesi) dall’ultima dose di HC (5). Il test più frequentemente utilizzato è l’ACTH test. Il razionale fisiopatologico di questo test nell'insufficienza surrenalica secondaria è basato sull'atrofia del cortico-surrene, che si verifica dopo alcune settimane dall’insulto acuto ipofisario condizionante la cessazione della secrezione di ACTH, di conseguenza dovrebbe essere effettuato dopo almeno 4-6 settimane da tale evento (6). Si è molto discusso riguardo la dose di ACTH sintetico (Synacthen) da utilizzare: la dose classica di 250 μg (disponibile in commercio) determina concentrazioni sovra-fisiologiche di ACTH (60.000 pg/mL, che in soggetti normali non si raggiungono neppure in condizioni di stress), che, superando l’atrofia surrenalica, potrebbero dare falsi negativi; pertanto ha assunto maggior importanza lo stimolo con ACTH a dosi inferiori (1 μg). Anche con tale dose le concentrazioni di ACTH diventano particolarmente elevate (1000 pg/mL) e inoltre falsi positivi e falsi negativi possono derivare dalla scorretta preparazione della soluzione da iniettare. In realtà, sia il test ad alte dosi che quello a basse dosi hanno dimostrato moderata accuratezza, primariamente dovuta alla bassa sensibilità, che risulta sovrapponibile fra i due dosaggi (7). In linea generale si può affermare che il test a basse dosi probabilmente ha una maggior sensibilità ed è quindi da preferire nei centri con personale infermieristico addestrato, mentre negli altri centri è forse preferibile continuare a utilizzare il test standard, in cui la somministrazione di ACTH può essere effettuata anche per via intramuscolare.
L’altro test utilizzabile, tradizionalmente considerato come gold standard, è l’ipoglicemia insulinica (ITT): nonostante questo test permetta di indagare anche la secrezione di GH e possa essere effettuato anche subito dopo l’insorgenza dell’iposurrenalismo, è attualmente meno impiegato per via delle controindicazioni (ad esempio patologia ischemica cardio-cerebrovascolare ed epilessia) e della necessità di stretta supervisione medica continua (6). Nella pratica clinica attuale il test viene riservato a quei casi dubbi di risposta all’ACTH test, sebbene anche in questi casi la ripetizione del test all’ACTH a distanza di tempo possa chiarire la presenza di iposurrenalismo centrale.
Questi test dinamici presentano limitazioni legate principalmente alla difficoltà di definire valori cut-off di risposta, a causa dei diversi metodi di dosaggio della cortisolemia e dei fattori interferenti sui livelli di cortisolo. Per queste ragioni, ai fini di una corretta diagnosi è essenziale considerare anche la probabilità pre-test di malattia, basata sulla valutazione del quadro clinico e biochimico basale.
Terapia
La terapia dell’iposurrenalismo centrale consiste nella sostituzione del deficit di glucocorticoidi. In ragione della limitata disponibilità di dati di efficacia e sicurezza, peraltro discordanti, la terapia sostitutiva androgenica con DHEA nelle donne non è al momento indicata nella routine clinica (8). Non vi è inoltre indicazione a trattamento con fludrocortisone (1).
Per quanto riguarda la terapia di mantenimento, è raccomandato l’uso di idrocortisone (HC) o in alternativa cortisone acetato, alla dose rispettivamente di 15-20 mg/die e 18.75 - 25 mg/die, in somministrazione orale unica o frazionata. In quest’ultimo caso si possono utilizzare:
- due somministrazioni, con 2/3 della dose somministrata al risveglio ed il rimanente 1/3 nel primo pomeriggio;
- tre somministrazioni con ½ dose al risveglio, ¼ della dose a pranzo ed il rimanente ¼ nel tardo pomeriggio.
I GC a maggior durata d’azione vanno riservati a casi selezionati (1).
Le principali criticità sono rappresentate dalla mancanza di marcatori affidabili per un’ottimale titolazione della terapia e dalla difficoltà di riprodurre il fisiologico pattern di secrezione nictemerale del cortisolo, con conseguenti problemi di sovra- e sotto-dosaggio. Solitamente si inizia con un dosaggio elevato per evitare il rischio di crisi acute e successivamente si apportano aggiustamenti sulla base della risposta clinica e delle comorbilità del paziente; la dose minima sufficiente a garantire un’adeguata sostituzione della funzione surrenalica risulta associata a migliore qualità di vita e riduzione del rischio di osteoporosi e patologie metaboliche e cardio-vascolari legato al sovra-dosaggio (1). Nessuno schema è in grado al momento di simulare fedelmente il ritmo circadiano del cortisolo, nemmeno l’idrocortisone a rilascio modificato, la cui efficacia nell’iposurrenalismo centrale è ancora da dimostrare; il regime migliore, in grado di ridurre il rischio di un’eccessiva esposizione a GC, sembra essere quello in triplice somministrazione (9).
Analogamente all’iposurrenalismo primario, fondamentale è la terapia di supporto in corso di eventi stressanti (come interventi chirurgici e infezioni, specialmente gastro-intestinali) o di situazioni di mancato assorbimento (vomito, diarrea). In questi casi l’aumentato fabbisogno di cortisolo richiede un potenziamento del dosaggio secondo schemi definiti. La crisi surrenalica va trattata con immediata somministrazione parenterale di 50-100 mg di HC associata a reidratazione. La chiave per prevenire le crisi è l’educazione del paziente, che deve essere istruito dettagliatamente sulla sua condizione, in particolare deve conoscere i possibili stress intercorrenti e le manifestazioni di una crisi imminente e deve essere in grado di gestire la terapia orale e parenterale; deve inoltre portare sempre con sé carta e kit di emergenza (fornito di idrocortisone in fiale), che garantiscano l’identificazione del soggetto e un intervento tempestivo e adeguato in caso di necessità (ad esempio un incidente stradale o una sincope) (10).
Le forme di iposurrenalismo centrale secondarie al trattamento chirurgico dell’ipercortisolismo e all’eccesso di GC esogeni continuano a costituire una grossa sfida sul piano terapeutico: nel primo caso, si tende, seguendo un approccio empirico, ad utilizzare dosi più elevate nelle prime settimane dopo l’intervento, con progressivo decalage sulla base del quadro clinico; nel secondo caso, è pratica comune ridurre gradualmente le dosi di GC (tanto più lentamente quanto maggiore è la durata di terapia) e associare HC una volta raggiunti i 5 mg di prednisone (o equivalenti), rivalutando successivamente la funzionalità dell’asse HPA (4).
Monitoraggio
Il monitoraggio della terapia si basa sulla valutazione di cenestesi, peso, pressione arteriosa, elettroliti, profilo glicemico, emocromo, funzione epatica e renale. La curva giornaliera della cortisolemia ha scarso valore nella pratica clinica, ma può essere utile nei casi di sospetto malassorbimento o aumentata clearance steroidea (11). La misurazione del cortisolo libero urinario, per via dell’ampia variabilità, è di scarsa utilità (12). Inoltre, nei pazienti con deficit ipofisari multipli, bisogna prestare attenzione alle possibili interazioni fra i GC e le altre terapie sostitutive: infatti, GH e tiroxina aumentano la clearance del cortisolo e gli estrogeni orali determinano un incremento dei livelli di CBG. Altri fattori da tenere in considerazione sono la concomitante assunzione di farmaci interferenti con il metabolismo epatico ed eventuali polimorfismi del recettore per i glucocorticoidi.
Bibliografia
- Fleseriu M, Hashim IA, Karavitaki N, et al. Hormonal replacement in hypopituitarism in adults: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 3888–921.
- Crowley RK, Argese N, Tomlinson JW, Stewart PM. Central hypoadrenalism. J Clin Endocrinol Metab 2014, 99: 4027–36.
- Broersen LHA, Pereira AM, Jørgensen JOL, Dekkers OM. Adrenal Insufficiency in corticosteroids use: systematic review and meta-analysis. J Clin Endocrinol Metab 2015, 100: 2171–80.
- Dinsen S, Baslund B, Klose M, et al. Why glucocorticoid withdrawal may sometimes be as dangerous as the treatment itself. Eur J Intern Med 2013, 24: 714–20.
- Krasowski MD, Drees D, Morris CS, et al. Cross-reactivity of steroid hormone immunoassays: clinical significance and two-dimensional molecular similarity prediction. BMC Clin Pathol 2014, 14: 33.
- Petersenn S, Quabbe HJ, Schofl C, et al. The rational use of pituitary stimulation tests. Dtsch Arztebl Int 2010, 107: 437–43.
- Ospina NS, Al Nofal A, Bancos I, et al. ACTH stimulation tests for the diagnosis of adrenal insufficiency: systematic review and metaanalysis. J Clin Endocrinol Metab 2016, 101: 427–34.
- Wierman ME, Arlt W, Basson R, et al. Androgen therapy in women: a reappraisal: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014, 99: 3489–510.
- Johannsson G, Skrtic S, Lennernäs H, et al. Improving outcomes in patients with adrenal insufficiency: a review of current and future treatments. Curr Med Res Opin 2014, 30: 1833–47.
- Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 364–89.
- Bancos I, Hahner S, Tomlinson J, Arlt W. Diagnosis and management of adrenal insufficiency. Lancet Diabetes Endocrinol 2015, 3: 216–26.
- Grossman AB. The diagnosis and management of central hypoadrenalism. J Clin Endocrinol Metab 2010, 95: 4855–63.
Ipogonadismo ipogonadotropo
Marco Bonomi, Giovanni Goggi, Biagio Cangiano
Dipartimento di Medicina Endocrino-Metabolica, Ospedale San Luca – IRCCS Istituto Auxologico Italiano – Milano
Dipartimento di Scienze Cliniche e di Comunità – Università degli Studi di Milano - Milano
(aggiornato al 14 novembre 2019)
DEFINIZIONE
L’ipogonadismo centrale o ipogonadotropo (hypogonadotropic hypogonadism, HH) è una condizione caratterizzata da specifici segni e sintomi clinici dovuti a ridotta produzione gonadica di steroidi sessuali (testosterone nel maschio ed estrogeni nella femmina) secondaria a un difetto di stimolazione gonadica da parte di ipotalamo e/o ipofisi (1).
EZIOLOGIA
Distinguiamo due principali forme di HH, correlate a forme organiche, congenite o acquisite, e forme funzionali (1) (tabella 1). Mentre la maggior parte delle forme acquisite, indipendentemente dalla loro eziologia, è responsabile di difetti secretivi ipofisari multipli o MPHD (vedi anche ipopituitarismo acquisito), le forme congenite di HH (Congenital Hypogonadotropic Hypogonadism, CHH), sono prevalentemente caratterizzate da difetti isolati (1-3), con l’eccezione delle forme di MPHD congenite e delle forme associate a iposurrenalismo congenito (vedi oltre).
| Tabella 1 Cause di ipogonadismo ipogonadotropo |
||
| Organiche | Congenite | HH (CHH normosmico, s. di Kallmann) S. di CHARGE CHH con iposurrenalismo centrale congenito MPHD congenito Altre forme sindromiche (es. s. di Prader-Willi, s. di Bardet-Biedl, s. di Noonan) |
| Acquisite | Neoplasie (es. adenomi ipofisari, craniofaringiomi, ecc) Altre malattie endocrine (es. iperPRL, ipotiroidismo, ecc) Malattie metaboliche (es. emocromatosi) Traumi cranici o eventi ischemici/emorragici Malattie infiammatorie/immunitarie (es. istiocitosi, ipofisiti, ecc) Malattie granulomatose (es. sarcoidosi) Iatrogene (es. pregressa neurochirurgia e/o radioterapia della regione ipotalamo-ipofisaria) |
|
| Funzionali | Malattie croniche (es. insufficienza renale, asma, …) Malnutrizione (es. disturbi del comportamento alimentare) Obesità (es. Late Onset Hypogonadism maschile) Eccessiva attività fisica Stress Terapie interferenti |
|
Forme organiche congenite
L’ipogonadismo ipogonadotropo congenito (CHH) è una malattia rara ed eterogenea, caratterizzata da un difetto di secrezione e/o d’azione del GnRH, che risulta 3-5 volte più frequente nei maschi (1-5). Il paziente con CHH può avere olfatto normale (CHH normosmico, nCHH) o difetti olfattivi (ipo o anosmia) che caratterizzano la sindrome di Kallmann (KS). La possibile associazione del difetto di secrezione di GnRH con il difetto olfattivo è da ricondurre alla comune origine embrionaria dei neuroni GnRH-secernenti e dei neuroni olfattivi e alla possibile implicazione di geni importanti per lo sviluppo e/o la migrazione di queste due popolazioni neuronali. Infatti, benchè la sua patogenesi non sia ancora completamente nota, è dimostrata una forte componente genetica (1,2,5-7). In passato si pensava fosse una malattia monogenica, ma è ormai noto il coinvolgimento di uno o più alleli di uno o più geni diversi (5-9). I geni implicati sono quelli responsabili dello sviluppo e/o della migrazione e/o attivazione dei neuroni GnRH-secernenti, piuttosto che il gene che codifica per il GnRH stesso o per il suo recettore (5-7). La trasmissione può essere X-linked oppure autosomica dominante o recessiva.
La sindrome CHARGE è una sindrome genetica complessa e rara, caratterizzata, oltre che da CHH e anosmia, da crescita e pubertà ritardata, associata a malformazioni oculari (coloboma), cardiopatie, atresia delle coane, anomalie renali e/o sordità. Questa condizione è dovuta a varianti alleliche del gene CHD7 (5-7). Più recentemente, sono state identificate varianti alleliche del gene CHD7 anche in pazienti che presentano solo CHH in assenza delle altre caratteristiche sopra-elencate della sindrome CHARGE (5,7).
L’iposurrenalismo centrale congenito è un’altra forma molto rara e particolare di CHH X-linked, in cui c’è un difetto combinato della secrezione gonadica e surrenalica di origine centrale, dovuta a mutazioni a carico del gene NR0B1 (prima noto come DAX1). L’associazione di CHH a difetti della secrezione surrenalica può anche essere legata a varianti alleliche dei geni POMC e NR5A1 (prima noto come SF-1) (5,8).
Altre sindromi possono presentarsi con CHH in presenza di altre caratteristiche, come la s. di Prader-Willi, la s. di Bardet-Biedl, la s. di Noonan.
Forme funzionali
In questi casi fattori esterni (tabella 1) interferiscono con la regolare attivazione dell’asse ipotalamo-ipofisi-gonadi nel suo compartimento centrale. Appartiene a questa categoria, il cosiddetto Late Onset Hypogonadism (LOH) maschile, forma di HH che si instaura progressivamente nel tempo con ridotta funzione dei neuroni GnRH. Ha una prevalenza stimata del 2.1% della popolazione generale e solitamente non è associato ad altri difetti ipofisari ma spesso a sindrome metabolica (10,11). Si ipotizza che i meccanismi all’origine di questa disfunzione ipotalamica siano diversi:
- aumento dell’attività aromatasica nel tessuto adiposo dei soggetti obesi, che convertirebbe androstenedione e testosterone rispettivamente in estrone ed estradiolo, con potente effetto di feed-back negativo a livello ipotalamo-ipofisario (12);
- infiammazione sistemica (nel contesto di sindrome metabolica, diabete e obesità) che danneggia l’ipotalamo con minor produzione di GnRH (13);
- desensibilizzazione dei neuroni GnRH con minor produzione ipotalamica di GnRH, indotta dagli alti livelli di insulina e di leptina tipici dei pazienti obesi, con sindrome metabolica (14).
È d’altra parte possibile che alcuni di questi casi siano forme di CHH a insorgenza in età adulta, come dimostrato dalla presenza anche in questa classe di pazienti di varianti tipiche delle forme a insorgenza pre-puberale (15).
MANIFESTAZIONI CLINICHE
Segni e sintomi dell’ipogonadismo variano a seconda dell’epoca di esordio.
Maschi
- Se l’ipogonadismo si instaura durante la vita fetale, i neonati possono presentare micro-pene, ipospadia e/o criptorchidismo (tipicamente bilaterale). Non troveremo mai, tuttavia, un sex-reversal: infatti, la produzione testicolare di testosterone inizia attorno all’8° settimana di gestazione per stimolo iniziale da parte della β-hCG placentare (è dunque gonadotropino-indipendente). Viceversa, la piena maturazione dell’asse GnRH-Gn-testicolo, e dunque la sua influenza sulla produzione di testosterone, si avrà solamente a partire dal II trimestre: HH non influirà, quindi, sulla determinazione del sesso fenotipico (che sarà maschile), ma solo sulla sua completa maturazione, con difetti di virilizzazione (2).
- L’ipogonadismo in età pre-pubere determina un ritardo dello sviluppo puberale: i pazienti avranno ridotto volume testicolare (< 4 mL), assenza di caratteri sessuali secondari (ridotto sviluppo pilifero, voce acuta, pene e prostata di piccole dimensioni) e potrà anche essere presente ginecomastia. Inoltre, questi soggetti tendono ad avere proporzioni eunucoidi (apertura delle braccia ≥ 5 cm rispetto all’altezza), frutto della ritardata chiusura delle epifisi delle ossa lunghe. Non andando incontro allo spurt puberale, saranno tipicamente più bassi dei loro coetanei con normale sviluppo puberale, tuttavia l’altezza finale risulta raramente compromessa. Dal punto di vista della composizione corporea, hanno ridotta massa muscolare e habitus corporeo femminile, con distribuzione del grasso più tipicamente ginoide e alterazione della maturazione ossea. Infine, sono presenti anche ridotta libido e disfunzione erettile (2).
- Negli adulti i sintomi più specifici dell’ipogonadismo sono riconducibili alla sfera sessuale: calo della libido, riduzione delle erezioni spontanee mattutine e disfunzione erettile. Il pene è di dimensioni normali, mentre i testicoli possono essere lievemente ridotti, così come anche la prostata. L’habitus è normale, tuttavia vi può essere riduzione del tono muscolare e della crescita pilifera (es. barba). Vanno ricercate inoltre la presenza di osteoporosi e di possibili alterazioni metaboliche associate, in particolare diabete, dislipidemie e obesità. Infine, questi pazienti possono riferire disturbi dell’umore, astenia, facile affaticabilità e anemia (dovuta a ridotta produzione renale di eritropoietina). Ovviamente, saranno infertili, venendo a mancare lo stimolo di FSH sulle cellule di Sertoli e la produzione di testosterone intra-testicolare indotta da LH (entrambi elementi fondamentali per una normale spermatogenesi) (1-3).
Femmine
- Un HH che si instaura già durante la vita intra-uterina non darà luogo a fenotipo patologico alla nascita: i genitali saranno quelli di una normale neonata. Infatti, la differenziazione in senso femminile dei genitali interni ed esterni avviene per assenza del gene SRY, indipendentemente dal funzionamento dell’asse GnRH-Gn-ovaio (2).
- Come nei maschi, anche nelle femmine in età pre-pubere l’ipogonadismo darà luogo a pubertà ritardata, con assenza di telarca e inevitabilmente amenorrea primaria. Il pubarca invece mostra maggior variabilità clinica, essendo influenzato sia dal grado di ipogonadismo, sia dal contributo degli androgeni surrenalici. Come per i maschi, anche nelle femmine la statura finale è simile a quella della popolazione generale (2).
- Nelle donne adulte, la manifestazione cardine è sicuramente l’amenorrea secondaria, che si potrà accompagnare a calo della libido, infertilità, osteoporosi e alterazioni del metabolismo glucidico e lipidico (2).
- In età post-menopausale la diagnosi è difficile e si pone solo a posteriori di fronte a livelli di FSH inappropriatamente basso e a RM encefalo con evidenza di lesione espansiva ipotalamo-ipofisaria.
Nelle forme congenite di HH sia maschile che femminile, inoltre, è possibile riscontrare anomalie somatiche, sensoriali o comportamentali particolari e specifiche: ad esempio, possono essere presenti anomalie neurologiche (in primis difetti dell’olfatto sotto forma di ipo/anosmia, ma anche nistagmo, sincinesie bimanuali, daltonismo o ipoacusia) o della linea mediana, quali labio-palatoschisi, agenesie/disgenesie dentali, agenesia renale unilaterale, sindattilie/polidattilie (1-3).
DIAGNOSTICA
Biochimica
Di fronte ad un quadro clinico suggestivo per ipogonadismo, dopo un’adeguata e quanto più completa raccolta anamnestica e un accurato esame obiettivo (ponendo particolare attenzione anche alla valutazione dei fenotipi associati all’HH congenito), la conferma diagnostica di HH avviene tramite specifici esami ematici.
Maschio. Il test iniziale per porre diagnosi di ipogonadismo è la determinazione del testosterone totale (TT). I livelli sierici di testosterone subiscono variazioni significative correlate ai ritmi circadiani e circannuali, alla secrezione episodica e alla variabilità nei dosaggi. Le concentrazioni dell’ormone possono essere influenzate da patologie concomitanti e da alcuni farmaci (es. glucocorticoidi), oltre che dalle alterazioni della concentrazione di SHBG. Nell’adulto il ritmo circadiano del testosterone è caratterizzato da un picco secretorio al mattino, che tende a ridursi con l’età (16). Si raccomanda pertanto di eseguire il dosaggio del TT nelle prime ore del mattino e a digiuno (16): se basso, è necessario confermare il dosaggio eseguendo una nuova determinazione. Il prelievo non deve essere eseguito in presenza di una patologia acuta o subacuta.
- Livelli di TT > 12 nmol/L (3.4 ng/mL): la diagnosi di ipogonadismo è esclusa, in assenza di alterazioni delle proteine di trasporto e di sintomi specifici.
- Livelli di TT < 8 nmol/L (2.3 ng/mL): altamente suggestivi di ipogonadismo.
- Livelli di TT compresi tra 8 e 12 nmol/L: sono considerati borderline e delineano un’area grigia di fronte alla quale è opportuno, soprattutto in presenza di sintomi specifici, procedere alla determinazione del testosterone libero calcolato (17).
Il TT sierico rappresenta la somma dell’ormone legato alle proteine (albumina e SHBG) e non legato o libero (pari allo 0.5-3%). Il termine di testosterone biologicamente attivo (biodisponibile) si riferisce alla somma della quota di testosterone legato all’albumina e di quello libero. In presenza di livelli di TT borderline (o se si sospettano alterati livelli della SHBG, come ad esempio in pazienti di età avanzata o affetti da obesità, diabete mellito, patologie croniche o tiroidee), bisognerebbe misurare le concentrazioni di testosterone libero e biologicamente attivo; il gold standard per queste determinazioni è rappresentato dalla dialisi all’equilibrio, metodo non facilmente applicabile, con costi elevati e operatore-dipendente. In alternativa, i valori calcolati con apposite formule (ad esempio la formula di Vermeulen) risultano ben correlati al gold standard (10,11,17). Il valore di testosterone libero calcolato risulta patologico e dunque compatibile con ipogonadismo se < 225 pmol.
Nell’ipogonadismo congenito maschile l’inibina B (prodotta dalle cellule di Sertoli sotto il controllo dell’FSH) è tipicamente ridotta, a indicare una scarsa popolazione di cellule di Sertoli: tale reperto è spiegato dall’assenza di stimolazione dei tubuli seminiferi, ad opera dell’FSH, durante la vita fetale e la mini-pubertà. Viceversa, i pazienti affetti da ipogonadismo acquisito, a causa di un asse GnRH-Gn-testicolo attivo durante la mini-pubertà, tendono ad avere valori di inibina B più alti. I livelli di inibina B sono correlati al volume testicolare e bassi livelli sono un fattore predittivo negativo per la fertilità (1,2).
Femmina. Nella femmina, la diagnosi di ipogonadismo è avvalorata dal riscontro di livelli bassi o indosabili di estrogeni (E2).
Per quanto riguarda il pannello degli androgeni, donne affette da HH tenderanno ad avere bassi livelli di testosterone e androstenedione, e valori normali di DHEA-S: tali valori confermano il surrene come principale fonte di androgeni circolanti, dal momento che i livelli di LH circolanti sono troppo bassi per stimolare adeguatamente le cellule della teca ovarica (2).
Donne affette da HH tendono ad avere livelli di AMH significativamente inferiori rispetto a donne sane (conseguenza di una scarsa stimolazione ovarica da parte delle gonadotropine). Tuttavia, tali valori non devono essere considerati un fattore prognostico negativo per la fertilità, poiché sia un’eventuale ripresa spontanea della secrezione pulsatile di GnRH, sia la somministrazione di gonadotropine esogene sono in grado di consentire un recupero della fertilità, portando anche ad aumento dei livelli di AMH (2).
Entrambi i sessi. Una volta posta diagnosi di ipogonadismo, occorre localizzarne l’origine andando a dosare le gonadotropine ipofisarie (LH e FSH), che nell’HH saranno basse o inappropriatamente normali e sicuramente con alterata pulsatilità. Particolare importanza riveste anche il dosaggio delle gonadotropine e degli steroidi sessuali in neonati in cui vi sia sospetto o familiarità per CHH. È infatti noto che nei primi 3-6 mesi di vita avviene la mini-pubertà, durante la quale l’asse ipotalamo-ipofisi-gonadi è ancora attivo prima di essere spento fino alla pubertà. Tale fase offre pertanto una finestra di opportunità per diagnosticare un HH: infatti, il riscontro di bassi livelli di gonadotropine ipofisarie (soprattutto se nel maschio si ha una concomitante clinica suggestiva) consente di riconoscerlo precocemente (2).
La principale sfida diagnostica riguardante il CHH si presenta al momento della pubertà, dove risulta particolarmente complesso distinguere le forme di vero CHH dalle forme transitorie di ritardo costituzionale di crescita e pubertà (RCCP). La loro differenziazione è auspicabile, dato che il RCCP non richiede terapia (se non in casi selezionati e per un tempo limitato), mentre il CHH è permanente e va sicuramente trattato. Se si considerano i livelli basali di ormoni, il quadro ormonale è sovrapponibile, perchè entrambe queste forme presentano bassi livelli di steroidi sessuali in presenza di livelli di gonadotropine bassi o inappropriatamente normali. L’uso del test di stimolo con GnRH si è dimostrato di scarsa utilità in questa diagnosi differenziale, in quanto la risposta al test è scarsa e ampiamente sovrapponibile tra i due gruppi di pazienti (1,2,19).
Nei quadri con sospetto deficit ipofisario multiplo, sia esso congenito o acquisito, sarà necessario eseguire una valutazione basale della funzione ipofisaria come descritto nel management dell’ipopituitarismo.
Imaging
Maschi. L’ecografia scrotale ha molta utilità nei pazienti affetti da HH, perchè consente di localizzare testicoli, epididimi e dotti deferenti, misurandone le dimensioni (in base alle quali è possibile determinare la severità del deficit di GnRH); inoltre, consente di monitorare nel tempo i progressi della maturazione testicolare durante la terapia con gonadotropine (2).
Femmine. L’ecografia pelvica o trans-vaginale può essere utile per indagare alcuni aspetti anatomici associati all’ipogonadismo. Ad esempio, nel CHH l’utero sarà di aspetto pre-pubere, cioè di piccole dimensioni e privo della rima endometriale. Inoltre, rispetto a donne sane di pari età, le pazienti affette da CHH hanno significativa riduzione del volume medio delle ovaie e del numero di follicoli antrali. Tuttavia, si rammenta come queste due caratteristiche non siano da considerarsi un fattore prognostico di scarsa fertilità, dal momento che entrambe rispondono bene alla terapia con gonadotropine (2).
Entrambi i sessi. È importante eseguire una RM della regione sellare, per escludere lesioni a livello ipotalamo-ipofisario, ma anche, nel sospetto di forma congenita, individuare eventuali difetti dei bulbi olfattori, del corpo calloso o della linea mediana.
Nel sospetto di forma congenita, è anche opportuno eseguire una ecografia addominale, per escludere un’eventuale agenesia renale monolaterale.
L’esecuzione della radiografia di mano e polso è di particolare importanza nel determinare l’età ossea dei soggetti pre-puberi e nel monitorare il quadro durante l’induzione della pubertà in soggetti con CHH.
L’esecuzione di densitometrie ossee regolari, infine, consente di valutare il grado di riduzione della densità minerale ossea, che è a rischio nei pazienti ipogonadici (2).
Altri esami
Test olfattometrico: per valutare la presenza di eventuali difetti olfattivi in un paziente con CHH.
Spermiogramma: non sempre possibile, poiché l’ipogonadismo si accompagna a disfunzione erettile e riduzione del volume dell’eiaculato. È tuttavia di fondamentale importanza ripeterlo in maniera seriata dopo l’inizio della terapia per la fertilità.
Analisi genetica: mediante l’NGS è possibile lo screening di un pannello che ad oggi conta oltre 50 geni noti, responsabili di CHH.
TERAPIA NEI MASCHI
Il trattamento dell’ipogonadismo deve essere effettuato considerando l’età del paziente, le condizioni psico-sessuali, l’epoca di insorgenza, la causa della patologia, l’eventuale presenza di altre affezioni e il desiderio di fertilità.
Neonati. Gli obiettivi principali a questa età sono l’aumento delle dimensioni del pene e la crescita testicolare. La terapia con testosterone può aumentare le dimensioni del pene e stimolare lo sviluppo dello scroto (senza alcun effetto sullo sviluppo testicolare). Nell’eventualità di criptorchidismo, si può optare per l’intervento chirurgico di orchidopessi o, in alternativa, per una terapia con hCG (con o senza spray nasale di GnRH), per favorire la discesa dei testicoli nella borsa scrotale. Più recentemente si è prospettata la possibilità di trattare i neonati affetti da CHH nati con micropene mediante iniezioni di gonadotropine esogene durante il primo anno di età, così da riprodurre la fisiologica mini-pubertà. Diversi studi suggeriscono, infatti, che tale approccio terapeutico durante il periodo neonatale possa avere effetti benefici sia sulla normalizzazione delle dimensioni del pene (che potrà raggiungere le dimensioni dell’adulto normale durante la terapia di induzione della pubertà), sia sulla funzione endocrina testicolare (aumenta la secrezione endogena di testosterone, INSL3, inibina B e AMH) e sullo sviluppo dei testicoli, andando a stimolare la proliferazione delle cellule di Sertoli e la crescita dei tubuli seminiferi, con incremento del volume testicolare (2).
Adolescenti. Gli obiettivi a questa età sono l’induzione della virilizzazione e della fertilità, il raggiungimento di una statura adulta ottimale, di un adeguato sviluppo osseo, di un’adeguata composizione corporea e di un normale sviluppo psico-sociale. Esistono due possibili modi per indurre la pubertà nei ragazzi affetti da HH.
- Testosterone a dosi crescenti, mediante la somministrazione di esteri, come l’enantato, iniziando con 50 mg im ogni 4 settimane, per poi incrementare progressivamente il dosaggio ogni 6 mesi circa, in base alla progressione puberale e all’accelerazione dell’età ossea, fino ad arrivare al dosaggio tipico dell’adulto. Più recentemente è stata dimostrata anche la validità dell’uso di testosterone gel 2%, partendo da un dosaggio di 10 mg a giorni alterni, con successivi incrementi ogni 6 mesi circa (20), ma tale modalità resta per ora “off-label”. Si è dimostrato che il testosterone, sia iniettivo che in gel, è in grado di portare a completamento lo sviluppo puberale, ma non permette alcuna stimolazione della crescita testicolare e della spermatogenesi.
- L’uso combinato delle gonadotropine esogene (attualmente considerato “off-label”) permette la maturazione puberale da un lato ma anche la stimolazione della crescita testicolare e la spermatogenesi dall’altro, ottenendo così un effetto psicologico positivo e una maggiore auto-stima nei giovani adolescenti (1-2). Non esistono al momento schemi terapeutici validati e internazionalmente riconosciuti. La maggior parte degli studi utilizza dosi costanti di FSH (ricombinante o estrattivo), per lo più 75 U x 3 volte/settimana, in associazione a dosi crescenti di hCG o LH ricombinante (1-2), partendo con 250 U x 3 volte/settimana, con progressivo incremento ogni 6 mesi fino alla dose tipica dell’adulto. Alcune evidenze suggeriscono l’utilità di un pre-trattamento con solo FSH della durata di 2-4 mesi prima di introdurre hCG (21).
Adulti. La terapia sostitutiva con testosterone nei maschi adulti ipogonadici è necessaria per mantenere normali livelli di testosterone, libido, funzione sessuale, densità minerale ossea e benessere generale (22,23). Possono essere impiegate diverse formulazioni disponibili: forme iniettive (come ad esempio l’enantato 250 mg ogni 2-4 settimane o l’undecanoato 1000 mg ogni 12-14 settimane) e forme trans-dermiche (gel 2%) da applicare quotidianamente. Vantaggi della terapia con gel sono la farmacocinetica più stabile e la non invasività, ma bisogna fare attenzione alla trasmissione cute-cute con bambini o donne. Tra le forme iniettive, invece, l’undecanoato garantisce concentrazioni di testosterone più stabili rispetto all’enantato nel corso delle settimane. Le controindicazioni assolute alla terapia con androgeni sono rappresentate dal carcinoma prostatico e mammario, mentre è possibile prescriverla, con opportuni monitoraggi, in caso d’ipertrofia prostatica benigna (22,23). In corso di terapia sostitutiva con testosterone andranno eseguiti controlli, inizialmente ogni 3-6 mesi, quindi annualmente, dei livelli di testosterone (da dosare 2-3 ore dopo l’applicazione del gel, 1 settimana prima dell’iniezione successiva di undecanoato o a metà tra l’ultima iniezione e la successiva di enantato), di ematocrito, PSA e naturalmente sintomi clinici di ipogonadismo (22,23).
TERAPIA NELLE FEMMINE
Adolescenti. Gli obiettivi in questa fascia di età sono sviluppo mammario soddisfacente, maturità dei genitali interni, esterni e di altri aspetti dell’aspetto femminile, normale sviluppo psico-sociale, raggiungimento di normale statura e massa ossea. Per indurre la pubertà si predilige l’impiego di estrogeni naturali trans-dermici, che garantiscono una buona efficacia e allo stesso tempo hanno un rischio cardio-vascolare ridotto rispetto alle formulazioni orali. La somministrazione di E2 viene iniziata a basse dosi, per poi aumentare gradualmente il dosaggio nell’arco di 30-36 mesi. La tabella 2 illustra un possibile schema di trattamento (24).
| Tabella 2 Schema per l'induzione della pubertà nell'ipogonadismo femminile |
|||
| Mesi | Obiettivo E2 (pg/mL) | Dose/die E2 | Note |
| 0 | 3-4 | 0.1 µg/kg | Applicare per 12 h al giorno durante la notte |
| 6 | 3-4 | 0.1 µg/kg | Applicare per tutte le 24 h, cambiando cerotto 2 volte a settimana. Dosare random E2 per verificare di essere nel range desiderato |
| 12 | 6-8 | 0.2 µg/kg | |
| 18 | 12 | 12.5 µg | Livelli di E2 inferiori potrebbero accelerare la crescita più della maturazione ossea |
| 24 | 25 | 25 µg | |
| 30 | 37 | 37.5 µg | |
| 36 | 50 | 50 µg | Aggiungere il progestinico (vedi indicazioni nel testo), anticipando l'aggiunta se compare sanguinamento |
| 42 | 75 | 75 µg | |
| 48 | 50-150 | 100 µg | Tipica dose della donna adulta |
Una volta raggiunto il massimo sviluppo mammario, e in genere al raggiungimento del dosaggio di E2 trans-dermico di 50 µg/die, si potrà aggiungere un progesterone ciclico per 12 giorni al mese, preferendo il progesterone naturale micronizzato (2).
Donne adulte. La terapia ormonale sostitutiva è necessaria per il mantenimento della salute dell’osso e dell’aspetto femminile, per migliorare la vita sessuale e promuovere un generale senso di benessere. L’E2 può essere assunto per os (1 o 2 mg/die) oppure per via trans-dermica (50 µg/die). In entrambi i casi la terapia estrogenica va associata a un progestinico ciclico (ad esempio progesterone micronizzato 200 µg/die) durante gli ultimi 14 giorni del ciclo, per prevenire l’iperplasia endometriale. Ovviamente quest’ultimo accorgimento non è necessario in donne isterectomizzate (2). Possono essere utilizzate le terapie combinate estro-progestiniche o le terapie sostitutive associate. La potenza biologica di 20 µg di etinil-estradiolo è paragonabile a quella di 1.25 mg di estrogeni coniugati e di 100 µg di 17ß-estradiolo per via trans-dermica. Il trattamento deve essere comunque individualizzato per ogni paziente. È importante ricordare che l’estrogeno somministrato per os viene metabolizzato per il 60-90% a livello epatico, determinando un sovraccarico epatico. La somministrazione non orale induce un metabolismo epatico solo per il 10-20% dell’estrogeno somministrato; i vantaggi di questa modalità di somministrazione determinano, da una parte una riduzione della sintesi di fattori pro-coagulanti e proteine della fase acuta come possibili fattori di rischio cardio-vascolare, e dall’altra una ridotta sintesi di SHBG. Gli estrogeni orali determinano una riduzione della concentrazione di IGF-I, che non si osserva con gli estrogeni trans-dermici (25). L’effetto negativo sui livelli di IGF-I indotto dalla terapia per os si osserva anche nelle pazienti ipopituitariche in trattamento sostitutivo con GH per GHD; nei casi in cui si sceglie di proseguire con la terapia estrogenica orale è necessario impiegare dosi di GH maggiori delle usuali per ottenere la stessa efficacia della terapia sostitutiva in termini di IGF-I.
Età post-menopausa. Le indicazioni per la terapia sostitutiva nelle donne normali in questa età possono ragionevolmente essere utilizzate anche per le pazienti ipogonadiche con più di 50 anni.
BIBLIOGRAFIA
- Boehm U, et al. European consensus statement on congenital hypogonadotropic hypogonadism. Pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015, 11: 547–64.
- Young J, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev 2019, 40: 669-710.
- Bonomi M, Quinton R. Congenital GnRH deficiency: a complex and genetically heterogeneous disease affecting human fertility and sexual development. Minerva Endocrinol 2016, 41: 183-7.
- Bonomi M, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol 2018, 178: 23–32.
- Cangiano B, et al. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet (submitted).
- Bonomi M, et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl 2012, 14: 49–56.
- Vezzoli V, et al. The complex genetic basis of congenital hypogonadotropic hypogonadism. Minerva Endocrinol 2016, 41: 223–39.
- Stamou MI, Georgopoulos NA. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism 2018, 86: 124–34.
- Sykiotis GP, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci 2010, 107: 15140–4.
- Wu FCW, et al. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010, 363: 123–35.
- Tajar A, et al. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab 2010, 95: 1810–8.
- Dandona P, Dhindsa S. Update: hypogonadotropic hypogonadism in type 2 diabetes and obesity. J Clin Endocrinol Metab 2011, 96: 2643–51.
- Morelli A, et al. Metabolic syndrome induces inflammation and impairs gonadotropin-releasing hormone neurons in the preoptic area of the hypothalamus in rabbits. Mol Cell Endocrinol 2014, 382: 107–19.
- George JT, et al. Exploring the pathophysiology of hypogonadism in men with type 2 diabetes: Kisspeptin-10 stimulates serum testosterone and LH secretion in men with type 2 diabetes and mild biochemical hypogonadism. Clin Endocrinol (Oxf) 2013, 79: 100–4.
- Cangiano B, et al. Evidence for a common genetic origin of classic and milder adult-onset forms of isolated hypogonadotropic hypogonadism. J Clin Med 2019, 8: 126.
- Brambilla DJ, et al. Intraindividual variation in levels of serum testosterone and other reproductive and adrenal hormones in men. Clin Endocrinol (Oxf) 2007, 67: 853–62.
- Wang C, et al. Investigation, treatment and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA and ASA recommendations. Eur J Endocrinol 2008, 159: 507-14.
- Mehta A, Hindmarsh PC, Mehta H, et al. Congenital hypopituitarism: clinical, molecular and neuroradiological correlates. Clin Endocrinol (Oxf) 2009, 71: 376-82.
- Delemarre-Van de Waal HA. Application of gonadotropin releasing hormone in hypogonadotropic hypogonadism – diagnostic and therapeutic aspects. Eur J Endocrinol 2004, 151 suppl 3: U89–94.
- Chioma L, et al. Use of testosterone gel compared to intramuscular formulation for puberty induction in males with constitutional delay of growth and puberty: a preliminary study. J Endocrinol Invest 2018, 41: 259-63.
- Dwyer AA, et al. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2013, 98: E1790-5.
- Isidori AM, et al. Outcomes of androgen replacement therapy in adult male hypogonadism: recommendations from the Italian Society of Endocrinology. J Endocrinol Invest 2015, 38: 103-12.
- Bhasin S, et al. Testosterone therapy in men with hypogonadism: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018, 103: 1715-44.
- Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab 2010, 95: 1487-95.
- Lissett CA, Shalet S. The impact of dose and route of estrogen administration on the somatotropic axis in normal women. J Clin Endocrinol Metab 2003, 88: 4668-72.
Deficit di GH nell'adulto
Ioannis Karamouzis & Gianluca Aimaretti
SCDU Endocrinologia, AOU Maggiore della Carità di Novara, Università del Piemonte Orientale
(aggiornato al 2 febbraio 2016)
Introduzione
L’ormone della crescita (GH) o ormone somatotropo, oltre alla sua attività sui processi di crescita, esercita importanti azioni metaboliche e strutturali e ha un’influenza positiva sulla qualità della vita (QOL).
La carenza di GH determina anche nell’età adulta alterazioni metaboliche, compromissioni di vari organi e sistemi e alterazioni della QOL, che caratterizzano la sindrome da deficit di GH (GHD), che comporta un aumento del rischio di morte per cause cardiovascolari.
È ampiamente dimostrato che la sindrome da GHD in età adulta può giovarsi del trattamento sostitutivo con GH biosintetico (rhGH) e tale indicazione terapeutica è stata approvata nella maggior parte delle nazioni (in Italia dal 1996).
Eziologia
Il GHD può essere espressione di un difetto isolato della funzione ipofisaria (più frequente in età pediatrica), oppure essere parte di un ipopituitarismo multiplo o totale (più frequente in età adulta). Tali difetti possono essere congeniti oppure acquisiti in età pediatrica o in età adulta.
Il GHD acquisito è tipico in età adulta in pazienti con patologie od interventi terapeutici che coinvolgono l’area ipotalamo-ipofisaria (neoplasie sellari e parasellari, processi granulomatosi o infiammatori, neurochirurgia, radioterapia). Un deficit di GH può inoltre manifestarsi in soggetti con pregresso trauma cranico o ictus (emorragia sub-aracnoidea o ischemia).
Il GHD si manifesta frequentemente come primo e talvolta unico segno di un’alterata funzione ipofisaria.
Quadro clinico
Il GHD in età adulta non è definito da un caratteristico quadro sintomatologico, poichè i segni e sintomi sono aspecifici, spesso sfumati e trascurabili o difficilmente riferibili alla carenza di GH se considerati singolarmente. Il GHD presenta inoltre una stretta analogia con la sindrome metabolica, per la presenza di alcune comuni alterazioni metaboliche e della composizione corporea, oltre all’aumento dei fattori di rischio per malattie cardiovascolari.
I segni e sintomi sono rappresentati da:
- astenia;
- aumento della massa grassa, specie addominale, con aumento del rapporto vita/fianchi;
- riduzione della massa magra;
- ipotrofia della cute e degli annessi cutanei;
- riduzione della “performance” durante esercizio fisico;
- riduzione dell’efficienza miocardica, soprattutto sotto sforzo;
- alterazioni del metabolismo lipidico (aumento di colesterolo totale e LDL, riduzione di HDL, aumento di fibrinogeno e PAI-I) associate ad aumento della resistenza periferica all’insulina;
- riduzione della densità minerale ossea;
- minore socievolezza e peggioramento della QOL.
Diagnosi
La diagnosi di GHD in età adulta si basa principalmente sulla documentazione, all’interno dell’appropriato contesto clinico, di una ridotta risposta del GH ai test di stimolo. Le recenti “consensus conference” internazionali hanno stabilito che, nell’adulto, sono parimenti affidabili i seguenti test di stimolo: 1) test con insulina (ITT); 2) test con GHRH + arginina; 3) test con GHRH + secretagoghi del GH (GHRP-6); 4) test con glucagone. Va ricordato che l’obesità e valori elevati di BMI determinano la riduzione della risposta di GH a vari stimoli provocativi e pertanto il valore soglia (cut-off) che separa la popolazione normale da quella con GHD varia in rapporto al grado di obesità e deve essere rideterminato, per ogni singolo test, in rapporto al grado di BMI.
La determinazione dell’IGF-I è un test di “screening” indicativo per la presenza di GHD, ma non permette di escludere il GHD quando il valore rientra nei limiti normali per età e sesso, specialmente nei pazienti di età più avanzata.
Test con insulina (ITT). È tuttora considerato il test di stimolo storico di riferimento per la diagnosi di GHD. Presenta limiti di riferimento ben definiti ma ha una scarsa riproducibilità. È stato documentato che il limite minimo delle risposte picco normali è 5 µg/L (3° percentile), ma una risposta picco di GH di 3 µg/L (1° percentile) è stata arbitrariamente considerata come “cut-off” per identificare con certezza i pazienti con GHD severo; tali limiti di riferimento sono stati recentemente confermati utilizzando metodologie statistiche più raffinate (curve ROC). Un paziente che mostri una risposta di GH all’ITT < 3 µg/L viene pertanto definito affetto da severo GHD e necessita di appropriata terapia sostitutiva con GH biosintetico.
Test con GHRH+arginina. Le risposte del GH a questo test sono riproducibili, non variano nel corso della vita, non presentano significative differenze nei due sessi. Il test evidenzia un’elevata specifità e sensibilità con chiari limiti di normalità; è stato documentato che il limite minimo delle risposte picco di GH nei soggetti adulti normali è 16.5 µg/L (3° percentile), ma è stato stabilito che i pazienti definibili con severo GHD debbano evidenziare un picco di GH < 9 µg/L (1° percentile). Sono stati quindi calcolati, usando la metodologia ROC, anche i “cut-off” in riferimento a tre classi di BMI:
- per soggetti in normopeso (BMI < 25 kg/m2), picco di GH = 11.5 µg/L
- per soggetti in sovrappeso (25 < BMI < 30 kg/m2), picco di GH = 8.0 µg/L
- per soggetti obesi (BMI > 30 kg/m2), picco di GH = 4.2 µg/L.
Tali valori di riferimento sono stati ora accolti dalla nota 39 AIFA nella sua ultima revisione di luglio 2014.
Sono meno utilizzati in Italia i test con GHRH + GHRP-6 (significato analogo al test con GHRH + arginina, con limite minimo delle risposte picco di GH = 15 µg/L nei soggetti adulti normali e = 5 µg/L negli obesi) e con glucagone (affidabile in età adulta, con “cut-off” di risposte picco di GH che separa i soggetti normali dai pazienti con GHD = 3 µg/L).
Risultati della terapia
Come già ricordato nel paragrafo quadro clinico precedente, il paziente adulto con GHD è un paziente con caratteristiche cliniche e rilievi obiettivi tipici della sindrome metabolica: in altre parole è un paziente a rischio di complicanze cardio-cerebrovascolari, con aumentata mortalità e morbilità. Fin dai primi studi su tale sindrome nei primi anni ’90 si è dimostrato che proprio la carenza della sostituzione con rhGH (a fronte dell’ottimizzazione e sostituzione di tutte le altre tropine ipofisarie) fosse la causa dell’aumentata mortalità cardio-cerebrovascolare dei pazienti con GHD. Pertanto, l’obiettivo principale della terapia sostitutiva con ormone somatotropo è rappresentato dal dimostrare la sua efficacia nell’attenuare tale aumentata mortalità.
Gli studi a oggi presenti non hanno ancora fornito risposte certe a tale quesito, sebbene alcune recenti metanalisi evidenzino come il trattamento sostitutivo produca un miglioramento significativo del profilo cardiovascolare globale, inducendo però un peggioramento del quadro glico-metabolico. Un lavoro recente derivante dall’analisi del Database KIMS della Pfizer (5) dimostra come già entro 2 anni dall’inizio della terapia con rhGH sia osservabile una riduzione del 50% degli indici di rischio cardiovascolare (Framingham, PROCAM, ESCscore), che permangono poi stabili e significativamente inferiori dopo altri 2 anni rispetto a una popolazione di controllo non GHD. Tale effetto è evidente, in particolare, in pazienti GHD di sesso maschile e con alti livelli di colesterolo totale e bassi di HDL. Tali dati richiedono però dimostrazioni solide e robuste e soprattutto studi su ampie casistiche che dimostrino una riduzione significativa della mortalità cardio-cerebrovascolare.
In pazienti con GHD, la QOL è generalmente compromessa. Per tale motivo la raccolta di un’accurata storia clinica e la compilazione di specifici questionari da parte del paziente sono raccomandate sia prima che in corso di terapia sostitutiva, in quanto possono essere un valido aiuto al monitoraggio della risposta alla terapia stessa. Quest’ultimo aspetto, certamente sottostimato nella pratica clinica endocrinologica italiana, riveste un ruolo fondamentale (“sine qua non”) nella pratica clinica anglosassone. Infatti, secondo le linee guida NICE la terapia con rhGH può essere prescritta solo in presenza di una documentata e chiara compromissione della “quality of life”. Il questionario più specifico, validato anche nella versione italiana (AGHDA-QOL), non è purtroppo liberamente disponibile: soggetto a copyright, con necessità di pagare una quota anche per l'utilizzo clinico nelle casistiche private.
Gli adulti con GHD presentano un rischio aumentato di sviluppare osteopenia e osteoporosi, rischio maggiore tanto più giovane è il paziente affetto da GHD, mentre nel paziente anziano tale rischio è minimo. La preservazione della massa ossea rappresenta uno degli obiettivi terapeutici durante trattamento con GH in questi pazienti. Sebbene durante il primo anno di terapia la densità minerale ossea possa ridursi per aumento del rimodellamento osseo, questo parametro tende a migliorare sensibilmente durante trattamento; viene quindi raccomandata l’esecuzione di un’indagine densitometrica (MOC/DEXA) prima di iniziare la terapia sostitutiva e periodicamente ogni 2 anni nel corso del trattamento.
INDICAZIONI PER LA PRATICA CLINICA
Quando iniziare la terapia con GH
La terapia sostitutiva con rhGH deve essere iniziata in pazienti in cui le terapie sostitutive dei deficit endocrini concomitanti siano già state impostate e ottimizzate.
Il trattamento con GH è controindicato nei pazienti affetti da neoplasia maligna in fase di attività clinica.
Come iniziare la terapia con GH, quali dosi impiegare e come monitorarne l’efficacia
La terapia con rhGH nel paziente adulto va iniziata a dosi basse (0.1-0.2 mg/die, pari a 1.25-2.5 µg/kg/die) e successivamente deve essere titolata ad intervalli di 1-2 mesi sulla base dei livelli di IGF-I (che dovranno rientrare nei limiti di norma per età, con valori ottimali compresi tra 25° e 75° centile) e sull'efficacia clinica nel determinare la remissione di sintomi e segni clinici.
Esiste una spiccata variabilità inter-individuale nella sensibilità al GH biosintetico e ciò spiega perché le dosi di mantenimento siano assai variabili. Le dosi medie di mantenimento variano nelle diverse fasi della vita:
- nel paziente giovane-adulto 5-10 µg/kg/die
- nel paziente adulto 2.5-5.0 µg/kg/die
- nel paziente anziano 1.25-2.5 µg/kg/die.
La dose di rhGH richiesta per la terapia sostitutiva nelle pazienti di sesso femminile è mediamente superiore rispetto al sesso maschile, sia se normalmente mestruate, sia soprattutto se assumono terapia sostitutiva estro-progestinica per via orale.
Cosa controllare e quando
Una volta raggiunti livelli normali di IGF-I, l'efficacia del trattamento andrà monitorata a intervalli regolari. Inoltre, è consigliato fin dall’inizio della terapia dosare FT4 e cortisolemia, per la possibile riduzione della desiodazione della T4 e della conversione di cortisone a cortisolo, che potrebbe determinare la comparsa o il peggioramento di una condizione, rispettivamente, di ipotiroidismo e/o di iposurrenalismo centrale.
Benché tale trattamento appaia relativamente sicuro, le linee guida circa la gestione del paziente con GHD raccomandano un attento e periodico monitoraggio ogni 6-12 mesi e una sorveglianza a lungo termine delle possibili complicanze. In ogni paziente è fondamentale innanzitutto l’esecuzione di un attento esame clinico, con particolare attenzione a peso, altezza, BMI e rapporto vita/fianchi, parametri antropometrici che dovranno essere rivalutati ad ogni controllo per monitorare l’efficacia della terapia sostitutiva. La comparsa di retinopatia in corso di trattamento è un evento estremamente raro.
Numerosi studi hanno dimostrato che il trattamento sostitutivo con GH nell’adulto non incrementa il rischio di recidive in pazienti con residuo post-chirurgico di tumore ipotalamo-ipofisario, né aumenta il rischio di insorgenza di nuova neoplasia. Per tale motivo, non si raccomanda un’intensificazione del follow-up nei pazienti trattati con GH, ma in caso di residuo è utile l’esecuzione periodica di RMN ipofisaria con timing sovrapponibile a quello dei pazienti non sottoposti a trattamento sostitutivo con GH, per monitorare le dimensioni della lesione, e in tutti i pazienti si suggerisce l’esecuzione di indagini di screening, come quelle applicate per la popolazione generale in base a sesso ed età. È stato pubblicato recentemente un “position statement” su trattamento con GH e sicurezza della terapia, che ha documentato che il trattamento sostitutivo con GH nell’adulto e nel bambino non aumenta il rischio di nuovi casi del tumore primario; peraltro dati disponibili nei bambini non indicano un aumento del rischio di recidive del tumore primario nei pazienti GH-trattati, mentre i dati attualmente disponibili sull’adulto per tumori maligni sono insufficienti. Infine, questo “position statement” ha indicato che i dati circa il rischio di sviluppare un secondo tumore in pazienti GH-trattati, “survivors” dei tumori pediatrici o tumori esorditi in età adulta, sono insufficienti per raggiungere una conclusione.
Inoltre in questi pazienti è raccomandato il monitoraggio periodico di pressione arteriosa, profilo lipidico, frequenza cardiaca e l’esecuzione di un elettrocardiogramma, mentre le indagini più invasive e costose, come l’ecocardiogramma o l’ecodoppler dei tronchi sovra-aortici, sono da riservare solo a casi selezionati.
Per le stesse ragioni, dovrebbero essere valutati annualmente anche i valori di glicemia a digiuno ed emoglobina glicata. Il trattamento con GH dei pazienti diabetici può richiedere un adeguamento del trattamento ipoglicemizzante in corso.
Per quanto riguarda tutti i fattori di rischio citati, i target terapeutici sono gli stessi della popolazione generale, in relazione a sesso ed età.
Bibliografia di riferimento
- Cook D, Yuen K, Biller BMK, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for growth hormone use in growth hormone-deficient adults and transition patients – 2009 update. Endocr Pract 2009, 15 Suppl 2: 1-29.
- Ho K. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol 2007, 157: 695-700.
- Molitch ME, Clemmons DR, Malozowski S, et al. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 1587-609.
- Clemmons DR. The diagnosis and treatment of growth hormone deficiency in adults. Curr Opin Endocrinol Diabetes Obes 2010, 17: 377-83.
- Schneider HJ, Klotsche J, Wittchen HU, et al. Effects of growth hormone replacement within the KIMS survey on estimated cardiovascular risk and predictors of risk reduction in patients with growth hormone deficiency. Clin Endocrinol 2011, 75: 825–30.
- Allen DB, Backeljauw P, Bidlingmaier M, et al. GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol 2015, 174: 1-9.
Deficit di GH in età transizionale
Roberto Lanzi
Unità di Endocrinologia, Istituto Scientifico Ospedale San Raffaele, Via Olgettina 60, 20132 MIlano.
Le conseguenze del deficit di ormone della crescita (GHD) variano nelle diverse età della vita. Durante l’infanzia e la pubertà il GH promuove l’accrescimento lineare e il GHD si associa a ipostaturismo armonico, mentre in età adulta comporta alterata composizione corporea e ridotta qualità di vita.
Solo una parte dei soggetti trattati con GH in età pediatrica richiede terapia sostitutiva nell’età adulta.
- Nei nati piccoli per età gestazionale (SGA), così come negli affetti da sindrome di Turner, insufficienza renale cronica o sindrome di Prader-Willi l’indicazione alla terapia cessa al raggiungimento della statura definitiva.
- Tra i soggetti con vero GHD la prosecuzione della terapia in età adulta è limitata ai casi in cui il difetto ormonale persiste al completamento dell’accrescimento lineare.
La probabilità di persistenza del GHD è particolarmente elevata in caso di deficit multiplo di almeno tre ormoni ante-ipofisari o di ridotti livelli di IGF-I (in sospensione di terapia e in assenza di condizioni cataboliche o terapia estroprogestinica), quando associati a mutazione genetica (PIT-1, PROP-1, HESX-1, LHX3/4, recettore del GHRH, ecc), anomalia strutturale congenita (ipoplasia del nervo ottico, displasia setto-ottica, ectopia della neuroipofisi, ipoplasia ipofisaria), patologie della regione ipotalamo-ipofisaria (es. craniofaringiomi), pregressa chirurgia o radioterapia encefalica (patologie sellari, cerebrali o sistemiche), oltre che in caso di GHD severo (1). E’ meno probabile nelle forme idiopatiche, isolate o nei difetti parziali. La probabilità di regressione non sembra invece influenzata dal sesso o dall’insorgenza del GHD prima o durante la pubertà (2).
La variabilità del difetto ormonale e la diversità dei criteri diagnostici di GHD nelle differenti età rendono necessario rivalutare i soggetti trattati con GH al completamento dell’accrescimento lineare. La fase di passaggio dall’infanzia all’età adulta, compresa tra il raggiungimento della statura definitiva e i 25 anni, è definita età di transizione. La nota AIFA 39 prevede il “retesting” per tutti i candidati a terapia con GH in età transizionale, con l’esclusione dei soggetti con documentata mutazione genetica o con deficit multiplo di almeno tre ormoni anteipofisari, per i quali è prevista la prosecuzione della terapia senza interruzione, adeguando il dosaggio all’età. In questi casi il riscontro di livelli circolanti di IGF-I ridotti per sesso ed età è sufficiente a confermare la persistenza del GHD, sebbene la normalità dei valori di IGF-I non ne escluda la diagnosi, suggerendo peraltro il “retesting” (1). Il “retesting” è indicato al raggiungimento della cosiddetta “near-adult height”, quando la velocità di crescita si riduce a meno di 2-2.5 cm per anno, l’altezza approccia il 98-99% di quella definitiva e corrisponde in genere una età ossea di 14-15 anni nella femmina e 16-17 nel maschio (3). Il test di stimolazione deve essere eseguito almeno un mese dopo la sospensione della terapia con GH, sebbene questa indicazione poggi, in mancanza di studi specifici, su esperienze personali. Per la diagnosi di GHD in età transizionale alcuni studi suggeriscono livelli soglia di GH più elevati rispetto all’età adulta, rispettivamente 6.1 e 19 µg/L per il test all’ipoglicemia insulinica (ITT) e arginina+GHRH (4,5); 6 e 19 µg/L sono anche i limiti di risposta ai due tests indicati dalla nota AIFA 39 per la diagnosi di GHD in età transizionale. Poiché il GHRH agisce a livello ipofisario, soggetti che sviluppano GHD di origine ipotalamica a seguito di terapia radiante possono mostrare una risposta falsamente normale al test arginina+GHRH, particolarmente nei 10 anni successivi all’irradiazione (6). Negli stessi soggetti il “retesting” in età transizionale, anche mediante stimoli diversi da arginina+GHRH, può non essere conclusivo in caso di normale risposta; possono essere indicate successive rivalutazioni della secrezione di GH per svelare casi di GHD a sviluppo tardivo. I limiti di risposta del GH allo stimolo GHRH+arginina in relazione all’indice di massa corporea (BMI) sono definiti nel soggetto adulto ma rimangono imprecisati nell’età transizionale. E' quindi richiesta cautela nell’interpretazione del test in soggetti sovrappeso o obesi. Il concomitante riscontro di ridotti valori di IGF-I può aiutare a distinguere soggetti con vero GHD da quelli con ridotta risposta del GH indotta dall’elevato BMI (1). Stimoli alternativi, come il glucagone, non sono ancora validati nel giovane adulto. Alcuni autori hanno proposto la valutazione della secrezione spontanea notturna di GH nei casi di anomalia strutturale della regione ipotalamo-ipofisaria (ectopia della neuroipofisi lungo il peduncolo ipofisario), atti a svelare casi di GHD parziale con normale risposta all’ITT (7). I criteri per la diagnosi di GHD parziale nel giovane adulto rimangono comunque tuttora imprecisati, così come rimangono aperti quesiti circa l’interpretazione di stati di discordanza tra risposta del GH ai test di stimolo e livelli circolanti di IGF-I (es. normale IGF-I con alterata risposta del GH o ridotta IGF-I con normale risposta del GH).
La terapia con GH in pazienti con GH a esordio in età pediatrica (cGHD) consente il raggiungimento di una altezza normale, sebbene circa 1 deviazione standard sotto la media della popolazione generale (8,9). A fronte di ciò, la massa magra e grassa e il contenuto minerale osseo dei pazienti con cGHD permangono significativamente ridotti rispetto ai pazienti con GHD a esordio in età adulta (aGHD) (9), evidenziando una discrepanza tra l’accrescimento mono e tridimensionale. Ciò può essere attribuito al fatto che l'incremento della massa magra (10), la variazione della distribuzione del grasso corporeo (11) e l'incremento della massa ossea fino alla fase di picco (12), processi sotto il controllo del GH, proseguono fisiologicamente oltre il termine dell’accrescimento lineare, quando tradizionalmente viene interrotta la terapia con GH. A due anni dalla sospensione della terapia, incremento della massa grassa e riduzione della massa magra sono stati documentati sia in soggetti con persistente GHD che in quelli divenuti sufficienti, ma nei primi la percentuale di massa grassa e il grasso tronculare sono risultati superiori del 50% rispetto ai controlli sani (13). Nello stesso studio i pazienti affetti da GHD hanno evidenziato il mancato fisiologico incremento della forza muscolare correlato all’età, osservato nei pazienti sufficienti per GH. Diversi studi hanno valutato gli effetti della terapia con GH nel periodo di transizione, documentando benefici effetti tanto sulla massa magra che sulla massa grassa (14, 15). A due anni dall’inizio della terapia con GH, l’utilizzo di dosi pediatriche non ha indotto significativi vantaggi rispetto all’uso di dosi per l’età adulta (15). Altri studi suggeriscono invece un effetto dose-dipendente della terapia, in particolare sulla percentuale di massa grassa corporea e sul grasso tronculare (16). Studi eseguiti su soggetti particolarmente giovani e con prevalente deficit idiopatico non hanno evidenziato significativi vantaggi della terapia (17). Incremento della densità minerale ossea è stato documentato da svariati studi (16, 18, 19), alcuni dei quali hanno evidenziato un effetto dose-dipendente del GH (16, 18) non confermato da altri (19). In uno studio recente la ripresa della terapia con GH in età transizionale ha indotto aumento dello spessore dell’osso corticale per crescita endostale (20). Benefici effetti della terapia con GH in età transizionale sono riportati sul profilo lipidico (15, 21, 22), mentre quelli sull’insulino-resistenza (14, 17), sulla morfologia e sulla funzione cardiaca rimangono controversi (14, 22). La qualità di vita nei pazienti con cGHD è in genere meno compromessa che nei soggetti con aGHD (23). Il grado di miglioramento dopo terapia è in genere proporzionale alla gravità della compromissione (24). L’insieme di questi dati supporta l'indicazione alla prosecuzione della terapia con GH in età transizionale, che al momento rimane peraltro possibile, auspicabile ma non assolutamente necessaria.
La terapia con GH deve essere iniziata a dosi di 0.2-0.3 mg/die e modificata sulla base dei livelli di IGF-I, determinati dopo 2 e 6 mesi dall’inizio della terapia e successivamente a cadenza annuale. I valori di IGF-I devono essere mantenuti nella metà superiore dell’intervallo di normalità per età e sesso, sebbene in pazienti con GHD severo insorto in età pediatrica la normalizzazione dei livelli di IGF-I non sia sempre ottenibile. E’ indicato il periodico monitoraggio dei valori di cortisolo per il noto effetto del GH sul metabolismo dei glucocorticoidi (25); anche gli indici di funzione tiroidea richiedono il periodico controllo, per il possibile smascheramento di quadri di ipotiroidismo o per adeguare la terapia sostitutiva in caso di deficit pre-esistente (26).
Bibliografia
- Molitch ME, Clemmons DR, Malozowski S, et al. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 1587-609.
- de Boer H, van der Veen EA. Why retest young adults with childhood-onset growth hormone deficiency? J Clin Endocrinol Metab 1997, 82: 2032-6.
- Molitch ME. Growth hormone treatment in adults with growth hormone deficiency: the transition. J Endocrinol Invest 2011, 34: 150-4.
- Maghnie M, Aimaretti G, Bellone S, et al. Diagnosis of GH deficiency in the transition period: accuracy of insulin tolerance test and insulin-like growth factor-I measurement. Eur J Endocrinol 2005, 152: 589-96.
- Corneli G, Di Somma C, Prodam F, et al. Cut-off limits of the GH response to GHRH plus arginine test and IGF-I levels for the diagnosis of GH deficiency in late adolescents and young adults. Eur J Endocrinol 2007, 157: 701-8.
- Darzy KH, Aimaretti G, Wieringa G, et al. The usefulness of the combined growth hormone (GH)-releasing hormone and arginine stimulation test in the diagnosis of radiation-induced GH deficiency is dependent on the post-irradiation time interval. J Clin Endocrinol Metab 2003, 88: 95-102.
- Radetti G, di Iorgi N, Paganini C, et al. The advantage of measuring spontaneous growth hormone (GH) secretion compared with the insulin tolerance test in the diagnosis of GH deficiency in young adults. Clin Endocrinol 2007, 67: 78-84.
- Guyda HJ. Four decades of growth hormone therapy for short children: what have we achieved? J Clin Endocrinol Metab 1999, 84: 4307-16.
- Attanasio AF, Howell S, Bates PC, et al. Body composition, IGF-I and IGFBP-3 concentrations as outcome measures in severely GH-deficient (GHD) patients after childhood GH treatment: a comparison with adult onset GHD patients. J Clin Endocrinol Metab 2002, 87: 3368-72.
- Barlett HL, Puhl SM, Hodgson JL, et al. Fat-free mass in relation to stature: ratios of fat-free mass to height in children, adults, and elderly subjects. Am J Clin Nutr 1991, 53: 1112-6.
- Forbes GB. Relation of lean body mass to height in children and adolescents. Pediatr Res 1972, 6: 32-7.
- Bonjour JP, Theintz G, Buchs B, et al. Critical years and stages of puberty for spinal and femoral bone mass accumulation during adolescence. J Clin Endocrinol Metab 1991, 73: 555-63.
- Johannsson G, Albertsson-Wikland K, Bengtsson BA. Discontinuation of growth hormone (GH) treatment: metabolic effects in GH-deficient and GH-sufficient adolescent patients compared with control subjects. Swedish Study Group for Growth Hormone Treatment in Children. J Clin Endocrinol Metab 1999, 84: 4516-24.
- Vahl N, Juul A, Jørgensen JO, Orskov H, et al. Continuation of growth hormone (GH) replacement in GH-deficient patients during transition from childhood to adulthood: a two-year placebo-controlled study. J Clin Endocrinol Metab 2000, 85: 1874-81.
- Attanasio AF, Shavrikova E, Blum WF, et al. Continued growth hormone (GH) treatment after final height is necessary to complete somatic development in childhood-onset GH-deficient patients. Hypopituitary Developmental Outcome Study Group. J Clin Endocrinol Metab 2004, 89: 4857-62.
- Underwood LE, Attie KM, Baptista J; Genentech Collaborative Study Group. Growth hormone (GH) dose-response in young adults with childhood-onset GH deficiency: a two-year, multicenter, multiple-dose, placebo-controlled study. J Clin Endocrinol Metab 2003, 88: 5273-80.
- Mauras N, Pescovitz OH, Allada V, et al. Limited efficacy of growth hormone (GH) during transition of GH-deficient patients from adolescence to adulthood: a phase III multicenter, double-blind, randomized two-year trial. Transition Study Group. J Clin Endocrinol Metab 2005, 90: 3946-55.
- ter Maaten JC, de Boer H, Kamp O, et al. Long-term effects of growth hormone (GH) replacement in men with childhood-onset GH deficiency. J Clin Endocrinol Metab 1999, 84: 2373-80.
- Shalet SM, Shavrikova E, Cromer M, et al. Effect of growth hormone (GH) treatment on bone in postpubertal GH-deficient patients: a 2-year randomized, controlled, dose-ranging study. J Clin Endocrinol Metab 2003, 88: 4124-9.
- Hyldstrup L, Conway GS, Racz K, et al. Growth hormone effects on cortical bone dimensions in young adults with childhood-onset growth hormone deficiency. Osteoporos Int 2012, 23: 2219-26.
- Gleeson H, Barreto ES, Salvatori R, et al. Metabolic effects of growth hormone (GH) replacement in children and adolescents with severe isolated GH deficiency due to a GHRH receptor mutation. Clin Endocrinol 2007, 66: 466-74.
- Salerno M, Esposito V, Farina V, et al. Improvement of cardiac performance and cardiovascular risk factors in children with GH deficiency after two years of GH replacement therapy: an observational, open, prospective, case-control study. J Clin Endocrinol Metab 2006, 91: 1288-95.
- Attanasio AF, Lamberts SW, Matranga AM, et al. Adult growth hormone (GH)-deficient patients demonstrate heterogeneity between childhood onset and adult onset before and during human GH treatment. Adult Growth Hormone Deficiency Study Group. J Clin Endocrinol Metab 1997, 82: 82-8.
- Murray RD, Skillicorn CJ, Howell SJ, et al. Influences on quality of life in GH deficient adults and their effect on response to treatment. Clin Endocrinol 1999, 51: 565-73.
- Filipsson H, Johannsson G. GH replacement in adults: interactions with other pituitary hormone deficiencies and replacement therapies. Eur J Endocrinol 2009, 161 Suppl 1: S85-95.
- Losa M, Scavini M, Gatti E, et al. Long-term effects of growth hormone replacement therapy on thyroid function in adults with growth hormone deficiency. Thyroid 2008, 18: 1249-5.
Scheda GH
Renato Cozzi1 & Roberto Attanasio2
Endocrinologia, 1Ospedale Niguarda & 2Istituto Galeazzi, Milano
Meccanismo d’azione
Sostituzione del deficit di GH
Indicazioni
Deficit GH nel bambino, S. di Turner, allorchè la statura cade sotto il quinto percentile, S. di Prader-Willi (in presenza di deficit dell'accrescimento), insufficienza renale cronica, Small for Gestational Age (SGA), secondo i criteri adottati in tale patologia.
GHD in età adulta, nei pazienti con patologia ipotalamo-ipofisaria (adenoma ipofisario, precedente adenomectomia ipofisaria chirurgica, lesioni ipotalamiche - craniofaringioma, lesioni granulomatose -, danno al parenchima cerebrale e/o alla regione ipotalamo-ipofisaria da radioterapia, ipopituitarismo idiopatico, post-traumatico, da neoplasie parasellari). Limitazioni: soggetti con livelli di GH < 3 µg/L dopo stimolo con ipoglicemia insulinica o, in presenza di controindicazioni al test di ipoglicemia insulinica, con picco inadeguato di GH dopo stimoli alternativi (il test con GHRH+Arginina è la prima scelta e con questo test viene utilizzato il picco di GH < 9 µg/L).
Contro-indicazioni e precauzioni
Controindicazioni assolute: neoplasia maligna in fase attiva, ipertensione endocranica benigna, retinopatia diabetica proliferativa e pre-proliferativa.
Controindicazioni relative: diabete mellito tipo 2 di difficile controllo glicometabolico.
Precauzioni: il trattamento con estrogeni orali (ma non transdermici) riduce la risposta dell’IGF-I al GH (di conseguenza va tenuto conto della sua presenza allorchè si valuta la risposta alla terapia sostitutiva, perchè non consente di valutare pienamente l'efficacia della terapia sostitutiva con GH). Al contrario quello con androgeni, normalizzando il tasso androgenico, è indispensabile per avere la risposta corretta allo stimolo, che sarebbe minore in presenza di ipogonadismo non corretto.
Monitoraggio della sicurezza
Controlli periodici (inizialmente una volta al mese, poi ogni 6 mesi) dei parametri glicemici; esame oculare nei pazienti diabetici o all'occorrenza in caso di riduzione visiva, monitoraggio neuroradiologico (inizialmente dopo 12 mesi, poi in rapporto al contesto clinico) di eventuali residui neoplastici ipofisari.
Preparazioni, via di somministrazione, posologia
Azione rapida:
- Genotropin (Miniquick 0.2 mg, 0.4 mg, 0.6 mg, 0.8 mg, 1.0 mg, 1.2 mg, 1.4 mg, 1.6 mg, 1.8 mg, 2.0 mg e Soluzione iniettabile 5.3 mg e 12 mg)
- Humatrope (6 mg, 12 mg, 24 mg)
- Norditropin (5 mg, 10 mg, 15 mg)
- Omnitrope (10 mg)
- Saizen (6 mg, 8 mg)
- Zomacton (4 mg)
(vedi scheda dispositivi di somministrazione)
Long-acting:
- lonapegsomatropin (Skytrofa 3 mg, 3.6 mg, 4.3 mg, 5.2 mg, 6.3 mg, 7.6 mg, 9.1 mg, 11 mg, 13.3 mg)
Attenzione
Conservare in frigo
Effetti collaterali
Nei casi di sovradosaggio, simili ai sintomi da eccesso di GH
Limitazioni prescrittive
Ricetta RRL, classe A, piano terapeutico, nota AIFA 39
Gli strumenti per la somministrazione di GH nel GHD
Anna Tortora
AOU San Giovanni di Dio e Ruggi d'Aragona, Università di Salerno, Scuola Medica Salernitana
(aggiornato al 9 marzo 2020)
La natura proteica del GH ne impedisce l’assorbimento per via orale e ne rende necessaria la somministrazione per via sottocutanea. Nella pratica clinica la scelta di un dispositivo per il trattamento con GH rappresenta un aspetto “tecnico” di grande importanza. Sono disponibili differenti dispositivi per la somministrazione, con alcune peculiarità circa il dosaggio in mg della soluzione, la necessità o meno di ricostituire il farmaco, la modalità di erogazione (con o senza ago), l’entità degli scatti tra un dosaggio e l’altro, le condizioni patologiche per i quali ne è approvato l’utilizzo in scheda tecnica, i dispositivi accessori che favoriscono la compliance (copri-ago) o consentono di monitorare l’aderenza.
Al momento, la somministrazione dell’ormone è giornaliera serale, con la formulazione settimanale ancora in via di approvazione.
Qui vengono schematicamente descritti i vari dispositivi in uso presentati secondo l’ordine alfabetico del farmaco, per facilitare l’individuazione del device che meglio si adatta al singolo paziente, specie in età pediatrica.
PER LA SOMMINISTRAZIONE DI GENOTROPIN (Pfizer)
GENOTROPIN GO QUICK (https://www.youtube.com/watch?v=VSzy4ac7Ib4)
Caratteristiche salienti:
- penna pre-riempita multi-dose mono-uso a due diversi dosaggi;
- 5.3 mg: range da 0.1 a 1.5 mg, scatto = 0.05 mg;
- 12 mg: range da 0.3 a 4.5 mg, scatto = 0.15 mg;
- necessita di training;
- necessita di priming per eliminare eventuale residuo d’aria (solo all’inizio di ogni penna);
- la cartuccia è mono-uso e la sua ricostituzione avviene all’inizio della nuova penna;
- l’impostazione del dosaggio va fatta all’inizio e non tutte le sere;
- dispone di finestra di “memoria” (confrontabile con dose prelevata);
- disponibile accessorio copri-ago;
- conservabile prima della ricostituzione per 1 mese fino a 25°C, per 2 anni tra +2° e +8°C;
- ogni penna può essere usata fino a 28 giorni dopo la ricostituzione, mantenendola a T tra 2° e 8°C (non congelare);
- non necessita di batteria.
Come si usa:
- avvitare l'ago sul porta-cartucce;
- ruotare il porta-cartuccia sulla penna;
- miscelare e controllare la ricostituzione;
- girare il selettore nero e impostare la dose;
- ruotare il selettore grigio per prelevare la dose;
- iniettare, tenendo premuto per alcuni secondi;
- rimuovere l’ago dalla cute;
- a esaurimento della cartuccia, cestinare tutto il dispositivo (usa e getta).
GENOTROPIN MINIQUICK
Caratteristiche salienti:
- dispositivo di iniezione pre-riempito (tubofiale a 2 comparti) mono-dose e mono-uso;
- non necessita di training;
- disponibili dosaggi già pronti da 0.2 mg, 0.4 mg, 0.6 mg, 0.8 mg, 1 mg, 1.2 mg, 1.4 mg, 1.6 mg, 1.8 mg, 2 mg (non sono disponibili i dosaggi intermedi);
- non contiene conservanti né alcool benzilico né fenolo, né (meta)cresolo;
- non necessita di priming per eliminare eventuale residuo d’aria;
- non necessita della catena del freddo dopo la consegna al paziente: la confezione integra si può conservare e/o trasportare per 6 mesi a T fino a 25°C, per 2 anni a T tra +2° e +8°C (non congelare);
- la soluzione ricostituita è stabile per 24 ore se conservata tra +2° e + 8°C.
Come si usa:
- avvitare l'ago;
- girare lo stantuffo;
- attendere e controllare la ricostituzione;
- iniettare integralmente nel sottocute, tenendo premuto per alcuni secondi;
- smaltire il dispositivo usato.
PER LA SOMMINISTRAZIONE DI HUMATROPE (Lilly)
HUMATROPEN (https://www.youtube.com/watch?v=PQgGZqF77uk)
Caratteristiche salienti:
- richiede il training pre-utilizzo (ogni cartuccia va inserita nell’apposita penna = colore e dose diversa);
- ampio range di dosaggi:
- penna da 6 mg: ogni scatto corrisponde a 0.025 mg;
- penna da 12 mg: ogni scatto corrisponde a 0.05 mg;
- penna da 24 mg: ogni scatto corrisponde a 0.1 mg;
- il priming per eliminare eventuale residuo d’aria va fatto solo all’inizio di una nuova cartuccia;
- non necessita di batterie;
- confezione integra: la cartuccia ha validità 3 anni se conservata tra +2° e + 8°C; stabile per un mese se conservata a T non > 25°C;
- il prodotto, una volta ricostituito, può essere conservato fino a un massimo di 28 giorni a temperatura compresa tra +2° e +8°C (non va congelato) al riparo dalla luce. L’esposizione a temperatura ambiente non deve superare i 30 minuti giornalieri;
- si consiglia di memorizzare la data del primo utilizzo, in quanto la penna va sostituita dopo 3 anni dal primo utilizzo.
Come si usa:
- ricostituire usando il kit Humatrope (device esterno: connettore-siringa pre-riempita con solvente);
- capovolgere delicatamente la cartuccia più volte per discioglierne il contenuto;
- inserire la cartuccia;
- attaccare l’ago (copri-ago opzionale);
- priming per eliminare eventuale residuo d’aria e controllo dell’aria rimasta;
- impostare la dose con il selettore;
- iniettare, tenendo premuto per 5 secondi;
- dopo l’iniezione, la dose nella finestra deve ritornare sullo 0.00;
- riporre il device.
PER LA SOMMINISTRAZIONE DI NORDITROPIN (Novo Nordisk)
NORDIFLEX (https://www.norditropin.com/how-to-take-it/giving-an-injection.html)
Caratteristiche salienti:
- due tipi di cartucce disponibili: ogni cartuccia va inserita nell’apposita penna (colore e dose diversa):
- 5 mg: tappo giallo arancio, dose massima 1.25 mg; ogni scatto: 0.025 mg;
- 15 mg tappo verde, dose massima 4 mg; ogni scatto: 0.075 mg;
- non memoria della dose (impostare la dose a ogni somministrazione);
- non necessita di batterie;
- liquido, senza ricostituzione (non necessita di miscelazione);
- priming, per eliminare eventuale residuo d’aria, utile ma non necessario;
- accessorio per l’inserimento automatico dell'ago e nascondi-ago (Nordi-PenMate®);
- la confezione integra ha validità di 2 anni se conservata tra +2° e +8°C;
- dopo la prima apertura: conservazione fino a 21 giorni a T ≤ 25°C e fino a 28 giorni tra +2° e +8°C (non congelare).
Come si usa:
- togliere il cappuccio;
- svitare l’alloggiamento della cartuccia;
- resettare l’asta del pistone;
- caricare la cartuccia nella penna apposita, controllando la corrispondenza dei colori (e dosaggi) diversi tra tappo della cartuccia e penna: 5 mg = giallo arancio; 15 mg = verde;
- inserire l’ago (applicare l’accessorio opzionale per nascondere l’ago, non incluso);
- fare uscire l’aria (priming utile, ma non necessario);
- impostare la dose prescritta;
- iniettare e tenere premuto per almeno 6 secondi;
- svitare l’ago e rimettere il cappuccio;
- conservare il dispositivo (fuori dal frigo fino a 25°C).
PER LA SOMMINISTRAZIONE DI NUTROPIN AQ (Ipsen)
NUTROPIN AQ PEN (http://www.digifreak.tv/video/nutropen/)

Caratteristiche salienti:
- NutropinAq è fornito come soluzione multi-dose contenente 2 mL di soluzione iniettabile: 1 cartuccia contiene 10 mg (30 UI) di somatropina e 1 mL contiene 5 mg di somatropina;
- effettuare il training pre-utilizzo;
- consente di somministrare dosi da 0.1 a 4 mg, con incrementi minimi di 0.1 mg;
- la cartuccia è già ricostituita (non è necessaria la miscelazione) e non va rimossa dal dispositivo fra una somministrazione e l’altra;
- controllo/sostituzione batteria;
- copri-ago (opzionale);
- display digitale per visualizzare la dose;
- la confezione integra ha validità di 2 anni se conservata tra +2° e +8°C;
- una volta ricostituito, si può tenere a temperatura ambiente fino a 45 minuti prima dell’uso e al massimo per un’ora al giorno; può essere conservato per un massimo di 28 giorni a temperatura compresa tra +2°C e +8°C (non va congelato) al riparo dalla luce.
Come si usa:
- rimuovere il cappuccio;
- svitare il supporto per la cartuccia;
- premere il pulsante (bianco) di reset;
- ruotare il pulsante nero fino alla posizione di start;
- caricare la cartuccia;
- attaccare l’ago;
- fare priming per eliminare eventuale residuo d’aria (pulsante nero);
- premere il pulsante bianco di reset;
- impostare la dose (i mg vengono mostrati su schermo digitale);
- iniettare (pulsante nero) e tenere premuto per 5 secondi;
- rimuovere l’ago;
- conservare il device.
PER LA SOMMINISTRAZIONE DI OMNITROPE (Sandoz)
SUREPAL (https://vimeo.com/120579331)

Caratteristiche salienti:
- Omnitrope è un medicinale biosimilare, quindi a più basso costo e strutturalmente molto simile al medicinale biologico di riferimento Genotropin;
- richiede training;
- utilizzare la cartuccia nella penna contrassegnata dal dosaggio corrispondente;
- disponibili cartucce da 5 mg, 10 mg, 15 mg pronte all'uso: nessuna ricostituzione necessaria;
- ogni scatto è pari a 0.1 mg;
- memoria della dose: registra il dosaggio del paziente;
- pulsante scorrevole: minima forza di iniezione anche per mani piccole;
- nessun priming per eliminare eventuale residuo d’aria: penna pronta per l’uso;
- copri-ago: riduce l’ansia nel paziente;
- non richiede l’uso di batterie;
- può essere conservato tra 2° e 8°C (non congelare) al riparo da luce fino a 28 giorni;
- la penna può essere utilizzata per 2 anni dal primo utilizzo.
Come si usa:
- impostare la memoria della dose (solo quando c’è un cambio di dosaggio);
- inserire la cartuccia pre-assemblata e collegare l’anello di chiusura a SurePal™;
- inserire un ago;
- richiamare la dose memorizzata, ruotando il perno della dose;
- appoggiare il device sulla cute;
- effettuare l’iniezione, mantenendo il pulsante in basso per 10 secondi dopo che il perno ha smesso di ruotare;
- rimuovere l’ago;
- riporre SurePal nella custodia senza togliere la cartuccia.
PER LA SOMMINISTRAZIONE DI SAIZEN (Merck-Serono)
ALUETTA (https://vimeo.com/342624430)

Caratteristiche salienti:
- richiede training;
- cartucce di differenti dosaggi (penna e cartuccia di differenti colori per i diversi dosaggi):
- 6 mg: dose minima somministrabile 0.15 mg, incremento minimo di dosaggio 0.05 mg;
- 12 mg: dose minima somministrabile 0.5 mg, incremento minimo di dosaggio 0.1 mg;
- 20 mg: dose minima somministrabile 0.8 mg, incremento minimo di dosaggio 0.1 mg;
- necessita di ricostituzione (fiala + solvente);
- conservare fra 10° e 40°C;
- effusore e stantuffo vanno sostituiti ogni 7 giorni, l’intero apparecchio ogni 3 anni.
Come si usa:
- accendere
- impostare la dose;
- applicare il connettore alla cartuccia ricostituita;
- inserire il connettore nel device;
- aspirare la soluzione ruotando il device;
- posizionare sulla cute;
- praticare l’iniezione premendo il bottone di rilascio (attendere alcuni secondi);
- quando l’indicatore segna 0 si può rimuovere il device;
- rimuovere il beccuccio di iniezione e riporre il device.
EASYPOD (https://manualzz.com/doc/5690720/scarica-la-guida---clickservice-online)

Caratteristiche salienti:
- penna pre-riempita multi-dose;
- necessita di training;
- cartucce di differenti dosaggi:
- 6 mg: dose minima somministrabile 0.15 mg, incremento minimo di dosaggio 0.05 mg;
- 12 mg: dose minima somministrabile 0.5 mg, incremento minimo di dosaggio 0.1 mg;
- 20 mg: dose minima somministrabile 0.8 mg, incremento minimo di dosaggio 0.1 mg;
- inserimento automatico di vari tipi di cartuccia e ago e del copri-ago;
- priming per eliminare eventuale residuo d’aria non richiesto;
- controllo elettronico di cartuccia e corretto posizionamento ago:
- si può pre-impostare (e memorizzare): tempo, velocità e profondità di iniezione e dose (in base a peso o superficie corporea);
- inibizione della somministrazione se il device non è poggiato sulla pelle grazie a un sensore cutaneo;
- registra la cronologia delle somministrazioni precedenti (compliance);
- il device indica il numero di giorni di terapia previsti con la cartuccia in corso;
- possibilità di inviare i dati a distanza grazie al trasmettitore “easypod connect” e al software dedicato;
- le cartucce integre durano 3 anni a T < 25°C;
- le cartucce, una volta utilizzate e conservate a T tra +2° e +8°C (non congelare), durano 28 giorni;
- tenere lontano da apparecchiature elettro-magnetiche;
- controllo/sostituzione batterie.
Come si usa:
- accendere;
- controllare la carica della batteria;
- impostare la dose (all’inizio e poi solo in caso di modifiche della dose);
- ricostituire (con dispositivo Click-Easy) e caricare la cartuccia;
- inserire l’ago (inserimento automatico di ago e copri-ago): il dispositivo emetterà un segnale acustico due volte;
- rimuovere il cappuccio dell’ago;
- posizionare sulla cute, attendere un beep e la luce verde e premere il pulsante di iniezione;
- la luce verde lampeggia, attendere che la luce si spenga e il device emetta un segnale acustico per due volte (tempo di iniezione: circa 5’’); allontanare il device dalla cute;
- rimuovere l’ago;
- spegnere il device.
PER LA SOMMINISTRAZIONE DI ZOMACTON (Ferring)
FERRING 33 PEN (4 mg)

Caratteristiche salienti:
- necessita di training;
- necessita di reimpostare la dose;
- permette di iniettare dosi da 0.03 mg a un massimo di 2 mg con una singola somministrazione;
- ago innovativo (sottile, 33 G/5 mm, conico all’esterno e all’interno);
- corpo esterno in alluminio anodizzato, altamente resistente;
- dopo la ricostituzione, il farmaco può essere conservato, in posizione verticale nell’astuccio, per un massimo di 14 giorni fra +2° e +8°C (non va congelato), al riparo dalla luce.
Come si usa:
- rimuovere il cappuccio protettivo della penna;
- ricostituire la cartuccia/fiala con apposito adattatore (3.3 mg/mL);
- svitare il contenitore della cartuccia e inserire la cartuccia ricostituita;
- avvitare di nuovo il contenitore della cartuccia al dispositivo;
- agganciare l’ago alla penna;
- impostare la dose;
- effettuare il priming per eliminare eventuale residuo d’aria;
- inserire l’ago nel sottocute e iniettare premendo lentamente il pulsante fino in fondo (tenere premuto per 8-10 secondi);
- scaricare il device e riporlo.
ZOMAJET (https://vimeo.com/106478539)


Caratteristiche salienti:
- è un dispositivo composto da penna, testina (3 tipi - A, B, C - di diverse dimensioni) e adattatore/flaconcino, che permette di eseguire l’iniezione senza ago (sotto pressione) nel sottocute. La testina di iniezione (senz’ago) è valida per 1 settimana;
- richiede training pre-utilizzo;
- dose: da 0.03 mg a 1.66 mg; scatto: 0.03 mg;
- non necessita di batterie;
- necessita di ricostituzione (flaconcino in polvere da 4 mg + fiala solvente da 3.5 mL);
- la confezione integra si può conservare per 2 anni tra +2° e +8°C;
- dopo la ricostituzione può essere conservato, in posizione verticale nell’astuccio, per un massimo di 14 giorni fra +2° e +8°C (non va congelato) al riparo dalla luce.
Come si usa:
- ricostituire il flaconcino/fiala;
- rimuovere il tappo del flaconcino;
- collegare l'adattatore del flaconcino alla penna;
- ruotare la penna;
- caricare la dose di priming;
- rimuovere l'adattatore e il flaconcino;
- fare priming per eliminare eventuale residuo d’aria;
- rimontare adattatore e flaconcino;
- riempire fino alla dose corretta;
- iniettare (tenendo premuto per diversi secondi);
- scaricare il device e riporlo.
| Comparazione sinottica dispositivi | ||||||||
| Device | Cartuccia (mg) | Scatto (mg) | Ago/ copri-ago | Soluzione da ricostituire | Priming | Memoria dose | Training |
Batterie |
| Aluetta | 6/12/20 | 0.05/0.1/0.1 | sì/sì | sì | no | no | sì | sì |
| Easypod | 6/12/20 | 0.05/0.1/0.1 | sì/sì | sì | no | sì | sì | sì |
| Ferring 33 Pen | 4/2 | 0.03 | sì | sì | sì | no | sì | no |
| Genotropin Go Quick | 5.3/12 | 0.05/0.15 | sì/sì | sì | inizio penna | inizio cartuccia | sì | no |
| Genotropin MiniQuick | 0.2-0.4-0.6-0.8-1.0- 1.2-1.4-1.6-1.8-2.0 |
monodose | sì | sì | no | monodose | no | no |
| Humatro-Pen | 6-12-24 | 0.025/0.05 | sì/opz | sì | inizio cartuccia | no | sì | no |
| Nordiflex | 5-15 | 0.025/0.075 | sì/opz | no | no | no | sì | no |
|
Nutropin AQ Pen |
10 | 0.1 | sì/opz | no | sì | no | sì | sì |
| SurePal | 5-10-15 | 0.1 | sì/sì | no | no | sì | sì | no |
| Zomajet |
4 | 0.03 | sì | sì | sì | no | sì | no |
INDICAZIONI TERAPEUTICHE
Bambini
Indicazioni e dosaggi:
- disturbi della crescita dovuti a GHD: 0.025-0.035 mg/kg/die oppure 0.7-1.0 mg/m2/die (autorizzati Genotropin, Humatrope, Norditropin, Nutropinaq, Omnitrope, Saizen, Zomacton);
- disturbi della crescita associati a sindrome di Turner: 0.045-0.050 mg/kg/die oppure 1.4 mg/m2/die (autorizzati Genotropin, Humatrope, Norditropin, Nutropinaq, Omnitrope, Saizen, Zomacton);
- disturbi della crescita in bambini di bassa statura nati piccoli per l'età gestazionale, che non hanno presentato recupero di crescita entro l'età di 4 anni o oltre: 0.035 mg/kg/die oppure 1 mg/m2/die (autorizzati Genotropin, Humatrope, Norditropin, Omnitrope, Saizen);
- sindrome di Prader-Willi per il miglioramento della crescita e della composizione corporea: 0.035 mg/kg/die oppure 1 mg/m2/die (non deve essere superata la dose di 2.7 mg/die) (autorizzato solo Genotropin);
- pazienti in età pediatrica con alterata funzione del gene SHOX: 0.045-0-050 mg/kg/die (autorizzato solo Humatrope).
Adulti
Trattamento sostitutivo nei pazienti adulti con GHD:
- già trattati in età pediatrica e che proseguono la terapia con GH: dosaggio 0.2-0.5 mg/die (autorizzati Genotropin, Norditropin, Nutropinaq, Omnitrope);
- con insorgenza di GHD in età adulta: la terapia va iniziata al dosaggio di 0.15-0.3 mg/die e la dose di mantenimento raramente supera 1 mg/die (autorizzati Genotropin, Humatrope, Norditropin, Nutropinaq, Omnitrope, Saizen).
CONCLUSIONI
Non esiste un device perfetto: tutti hanno pregi (molti) e qualche limite.
Il device andrebbe valutato (insieme al paziente e ai familiari) scegliendolo in base alle varie esigenze:
- la confezione mono-dose sembra più indicata nei pazienti non considerati affidabili per l’uso della penna o in quelli con problemi di intolleranza ai conservanti;
- le confezioni pluri-dose garantiscono maggiore flessibilità e precisione di dosaggio, grazie alla somministrazione con penna; esistono confezioni pluri-dose già ricostituite.
Grazie al copri-ago, si registrano raramente casi di avversione al trattamento con ago: nei pochi casi con vera e propria “agofobia”, prendere in considerazione l’uso di dispositivi senza ago.
Nella valutazione della risposta del paziente al trattamento, considerare sempre l’aspetto “tecnico” legato alla somministrazione del farmaco. Il training è un elemento importante per ottenere la compliance desiderata.
BIBLIOGRAFIA
- Molitch ME, Clemmons D R, Malozowski S, et al. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 1587-609.
- Bozzola M, Meazza C. Growth hormone deficiency: diagnosis and therapy in children. Expert Rev Endocrinol Metab 2010, 5: 273-84.
- Collett-Solberg PF, Ambler G, Backeljauw PF, et al. Diagnosis, genetics, and therapy of short stature in children: a Growth Hormone Research Society international perspective. Hormone Res Paediatr 2019, 92: 1-14.
- Agenzia italiana del farmaco. Determinazione alla nota 39 616 del 19/06/2014.
Classificazione del diabete insipido
Marco Faustini Fustini
Endocrinologia, Ospedale Bellaria, Bologna
Il diabete insipido (DI) è una condizione morbosa, nel complesso rara, caratterizzata da poliuria ipotonica. La malattia compare per effetto dell'incapacità a concentrare le urine nonostante l’aumento dell’osmolalità plasmatica.
I principali meccanismi fisiopatologici in grado di produrre DI sono due:
- l’incapacità di secernere, e spesso anche sintetizzare, l’ormone ipotalamico arginin-vasopressina (DI centrale o ipotalamico);
- l’inappropriata risposta renale all’arginin-vasopressina (DI nefrogenico).
E' stato recentemente proposto di non utilizzare più il termine di "diabete insipido", ma invece quelli di "deficit di vasopressina" per la forma centrale e "resistenza alla vasopressina" per la forma renale (2).
Esiste poi una rara e peculiare condizione morbosa, che non sarà ripresa nella successiva trattazione, il DI transitorio della gravidanza, che si manifesta allorchè i livelli di vasopressinasi sono assai più elevati di quelli riscontrati normalmente in gravidanza, con conseguente degradazione della vasopressina endogena e diabete insipido. La vasopressinasi è l’enzima (cisteina-aminopeptidasi) deputato alla degradazione dell’ormone. Nel DI transitorio della gravidanza non è rara l’associazione con altre condizioni morbose, anch’esse transitorie, tipiche della gravidanza, come la pre-eclampsia, le coagulopatie e l’epatosteatosi acuta. L’ipotesi che i prodotti di degradazione della vasopressina siano direttamente coinvolti anche nella pre-eclampsia non ha trovato unanimi consensi. Tuttavia, uno stretto legame tra queste due condizioni morbose sembra esistere, dal momento che sono stati riportati casi di donne con DI della gravidanza e pre-eclampsia che non hanno sviluppato DI in successive gravidanze non complicate da pre-eclampsia. La terapia del DI transitorio della gravidanza è la desmopressina, per contrastare l’accelerata clearance dell’arginin-vasopressina (1).
La tabella riporta la classificazione eziopatogenetica del DI.
| Classificazione eziopatogenetica del diabete insipido | ||
| Centrale (o ipotalamico) | Ereditario | autosomico dominante |
| autosomico recessivo (sindrome di Wolfram, ecc) | ||
| recessivo, legato al cromosoma X | ||
| Acquisito | tumori primitivi: craniofaringioma, germinoma, pinealoma, astrocitoma, linfoma primitivo del SNC | |
| tumori secondari: metastasi nella regione sellare, leucemie | ||
| malattie infiammatorie e granulomatose: ipofisiti, istiocitosi a cellule di Langerhans, granulomatosi di Wegener, sarcoidosi, tubercolosi, sifilide | ||
| traumi cranici | ||
| neurochirurgia (trans-sfenoidale o trans-cranica) | ||
| idiopatico | ||
| apoplessia ipofisaria (parziale o completa) | ||
| Nefrogenico | Congenito | recessivo legato al cromosoma X (mutazioni del recettore V2 di AVP) |
| autosomico recessivo (mutazioni di acquaporina-2) | ||
| Acquisito | malattie renali che alterano l’architettura renale (rene policistico, infarto renale, malattie infiltrative) | |
| perdita del gradiente midollare (eccessivo introito d’acqua, eliminazione eccessiva di sodio e acqua per uso di diuretici, …) | ||
| ridotta concentrazione di urea nella midollare (dieta molto povera di proteine) | ||
| ridotta espressione di acquaporina-2 (deplezione di potassio, ipercalcemia, dieta povera di proteine, ostruzione bilaterale delle vie escretrici renali) | ||
| farmaci (sali di litio, demeclociclina) | ||
| Transitorio della gravidanza | ||
Bibliografia
- Fenske W & Allolio B. Current state and future perspectives in the diagnosis of diabetes insipidus: a clinical review. J Clin Endocrinol Metab 2012, 97: 3426-37.
- Arima H, Cheetham T, Christ-Crain M, et al. Changing the name of diabetes insipidus: a position statement of the Working Group for Renaming Diabetes Insipidus. Eur J Endocrinol 2022, 187: P1-3.
Diagnostica del diabete insipido
Marco Faustini Fustini
Endocrinologia, Ospedale Bellaria, Bologna
La diagnosi di diabete insipido s’inserisce nell’ampio capitolo delle sindromi poliuriche/polidipsiche.
Una volta confermata la poliuria mediante la raccolta delle urine durante le 24 ore (> 2 L/m2), occorre anzitutto escludere le poliurie dovute a malattia renale e quelle di origine osmotica, di cui il diabete mellito costituisce la forma paradigmatica; entrambe sono facilmente rilevabili mediante esami di routine. Se il paziente presenta poliuria con urine ipotoniche in assenza di malattia renale, occorre poi differenziare il diabete insipido, in cui la polidipsia è secondaria alla poliuria, dalla polidipsia primaria. Come vedremo, inoltre, in alcune forme particolari di diabete insipido la polidipsia può mancare, complicando ulteriormente l’approccio diagnostico. Da qui la necessità di considerare alcuni principi fondamentali di fisiopatologia clinica, che aiuteranno alla comprensione dei test diagnostici.
In condizioni fisiologiche, l’acqua corporea totale e l’omeostasi dell’acqua sono regolate essenzialmente dal fine sistema che controlla la secrezione di AVP, incentrato sull’osmolalità plasmatica e sul volume arterioso efficace a stimolare i barocettori. Sia l’aumento dell’osmolalità plasmatica sia, in misura minore, l’ipovolemia (intesa come deplezione del volume extra-cellulare) sono in grado di stimolare la secrezione di AVP nella circolazione ematica. Raggiunto il recettore V2, posto sulla membrana baso-laterale delle cellule principali del dotto collettore, AVP induce l’inserzione dei canali dell’acqua acquaporina-2 sulla membrana apicale di tali cellule, determinando così un aumento della permeabilità all’acqua, che è così riassorbita seguendo il gradiente di concentrazione della midollare renale. La sete è indispensabile per mantenere l’omeostasi dell’acqua quando il meccanismo mediato da AVP appare insufficiente, per l’inadeguatezza intrinseca di questo sistema (diabete insipido) o per impossibilità a concentrare ulteriormente l’urina. Pertanto, se la perdita d’acqua dal rene nel paziente con diabete insipido (poliuria ipotonica) è compensata dall’introito di acqua per effetto della sensazione intatta della sete (polidipsia secondaria compensatoria), l’osmolalità plasmatica e la sodiemia potrebbero rimanere normali a lungo. Tuttavia, qualora il paziente abbia perduto il senso della sete (ad esempio per effetto di lesioni dell’ipotalamo) oppure non abbia libero accesso all’acqua (come può verificarsi, ad esempio, per un ridotto stato di vigilanza o durante la nutrizione parenterale o enterale in cui non sia previsto un adeguato apporto d’acqua in considerazione della condizione patologica di diabete insipido), l’equilibrio ben presto si altera e compare un incremento dell’osmolalità plasmatica e della sodiemia, a indicare il rischio di incipiente disidratazione intra-cellulare.
Nella polidipsia primaria l’impulso a bere non è dettato da una ridotta secrezione o attività di AVP, bensì da un difetto del meccanismo che controlla la sete o da un’aumentata sensazione della sete (diabete insipido dipsogenico) oppure, più frequentemente, da disordini psichiatrici che si accompagnano a un bisogno compulsivo di bere (polidipsia psicogena). La diagnosi differenziale tra polidipsia primaria e secondaria è essenziale per le ripercussioni terapeutiche che una diagnosi errata comporta: infatti, somministrare quotidianamente desmopressina quale presidio terapeutico a un soggetto con polidipsia primaria, erroneamente considerato affetto da DI centrale, significa aumentare notevolmente il rischio di sviluppare intossicazione d’acqua (iponatremia ipotonica severa).
Il test di disidratazione (o test di privazione d’acqua), con poche modifiche rispetto alla prima standardizzazione proposta da Miller nel 1970, è tuttora utilizzato come test diagnostico, seppure non sia dotato di elevata sensibilità e specificità (1).
- Nel DI centrale severo le urine rimangono ipotoniche (bassa osmolarità urinaria) durante il test di disidratazione, ma dopo la somministrazione di desmopressina l’osmolalità urinaria aumenta almeno del 50% (spesso oltre il 100%) e la vasopressina non è dosabile.
- Nel DI nefrogenico severo le urine possono non essere completamente diluite al termine del test, ma non aumentano in maniera rilevante dopo la somministrazione di desmopressina e le concentrazioni di AVP sono generalmente superiori a 5 pg/mL.
- Nel DI centrale parziale, la risposta al test di disidratazione può essere non molto dissimile da quella del soggetto con polidipsia primaria, e questo costituisce indubbiamente il maggiore limite del test. In entrambe queste condizioni l’osmolalità urinaria tende ad aumentare durante il test e supera spesso il valore dell’osmolalità plasmatica, senza raggiungere, tuttavia, il valore che si registra nel soggetto normale (800-1000 mOsm/kg). La risposta dell’osmolalità urinaria alla somministrazione di desmopressina è generalmente presente, seppure ridotta (10-50%), nel DI centrale parziale, mentre tende a essere pressoché nulla nella polidipsia primaria. In realtà, questa differenza può rivelarsi talora fittizia e ingannevole: soggetti con polidipsia primaria possono rispondere alla desmopressina e, d’altra parte, pazienti con DI centrale parziale possono non rispondere alla desmopressina al termine della fase di prolungata disidratazione durante la quale hanno mantenuto la capacità di secernere vasopressina, raggiungendo la massima osmolarità urinaria loro permessa e non più incrementabile dalla desmopressina esogena.
Nasce da questa indeterminatezza del test per le forme parziali di DI la necessità di individuare nuove strategie diagnostiche. Recentemente, è stato proposto di impiegare il dosaggio della copeptina – la glicoproteina C-terminale del pro-ormone AVP - come surrogato della secrezione di AVP durante il test di disidratazione con privazione d’acqua. I primi risultati sono certamente incoraggianti (2), ma attendono verifiche da studi su larga scala.
Infine, va detto che più che da un singolo test diagnostico, la diagnosi corretta del disordine che sta alla base della sindrome poliurica/polidipsia va spesso condotta sulla scorta di un insieme di dati clinici e di laboratorio. Il dato neuroradiologico (RM) può, seppure di rado, essere di aiuto, soprattutto in alcune forme pediatriche di DI centrale.
Bibliografia
- Fenske W, Allolio B. Current state and future perspectives in the diagnosis of diabetes insipidus: a clinical review. J Clin Endocrinol Metab 2012, 97: 3426-37.
- Fenske W, Quinkler M, Lorenz D, et al. Copeptin in the differential diagnosis of the polydipsia-polyuria syndrome – revisiting the direct and indirect water deprivation tests. J Clin Endocrinol Metab 2011, 96: 1506-15.