Metodo di determinazione del progesterone

Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
La stragrande maggioranza dei laboratori utilizza immuno-dosaggi diretti (senza estrazione) per la misura della concentrazione ematica del progesterone. Si tratta quasi sempre di metodi non isotopici. Per la preparazione degli anti-sieri sono stati utilizzati vari antigeni. La maggiore reattività crociata si verifica con il 5α-pregnandiolo (6-11%).
Con lo sviluppo della tecnologia, molti metodi di dosaggio per il progesterone sono oggi consolidati su piattaforme automatizzate a elevata produttività. Si tratta di metodi eterogenei, con dosaggi competitivi che prevedono la separazione delle frazioni libere da quelle legate all’anticorpo. Come traccianti sono adoperati enzimi, fluorocromi, sostanze chemi-luminescenti ed elettrochemi-luminescenti. Sono stati descritti anche metodi diretti a risoluzione temporale di fluorescenza.
I grandi vantaggi degli immuno-dosaggi automatizzati sono la relativa semplicità d’uso, i costi ridotti e la tempestività delle risposte, specialmente importante quando il dosaggio del progesterone viene impiegato nei protocolli di fertilizzazione in vitro per valutare la stimolazione ovarica e monitorare l’ovulazione, oltre all’abolizione di tutte le precauzioni necessarie quando si utilizzano radio-isotopi. Le criticità sono legate alla sensibilità funzionale relativamente scarsa, che rende problematica la determinazione delle basse concentrazioni, come tipicamente si verifica nel maschio, nelle donne in post-menopausa, nei bambini. Le sensibilità funzionali riportate, infatti, (0.32–1.43 nmol/L) sono da due a quattro volte più alte dei limiti di rilevazione dichiarati (0.19–0.48 nmol/L). Come metodo di riferimento è stata indicata la gascromatografia seguita da spettrometria di massa.
Come campioni si utilizzano indifferentemente siero e plasma in eparina Na/Li o EDTA. Stabilità 5 giorni a 2-8° e almeno 6 mesi a –20°C.
Bibliografia
- Kakabakos SE, Khosravi MJ. Direct time-resolved fluorescence immunoassay of progesterone in serum involving the biotin-streptavidin system and the immobilized-antibody approach. Clin Chem 1992, 38: 725-30.
- Taieb J, Benattar C, Birr AS, Lindenbaum A. Limitations of steroid determination by direct immunoassay. Clin Chem 2002, 48: 583-5.
- Stanczyk FZ, Lee JS, Santen RJ. Standardization of steroid hormone assays: why, how, and when? Cancer Epidemiol Biomarkers Prev 2007, 16: 1713–9.
- Stancyzk FZ, Clarke NJ. Advantages and challenges of mass spectrometry for assays for steroid hormones. J Steroid Biochem Mol Biol 2010, 121: 491–5.
- Patton PE, Lim JY, Hickok LR, et al. Precision of progesterone measurements with the use of automated immunoassay analyzers and the impact on clinical decisions for in vitro fertilization. Fertil Steril 2014, 101: 1629-36.
Metodo di determinazione del testosterone
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento novembre 2021)
Il testosterone circola nel sangue periferico in diverse forme:
- legato alla SHBG (circa 40-50%);
- legato (meno fortemente) all’albumina (40-50%);
- libero da legami proteici (1-2%).
La prima forma è praticamente inattiva dal punto di vista fisiologico e rappresenta una specie di deposito circolante che viene rilasciato molto lentamente per trasformarsi in una delle altre due forme, che sono prontamente disponibili per i tessuti (testosterone biodisponibile).
In determinate condizioni fisiopatologiche si verificano variazioni significative della concentrazione di SHBG. In questo caso aumenta anche la concentrazione di testosterone totale, anche se in realtà questo aumento non è in grado di causare variazioni fisiologiche significative. Al contrario, in caso di riduzione della concentrazione di SHBG, diminuirà anche la concentrazione del testosterone totale, nonostante un incremento di testosterone biodisponibile. Esempi comuni di queste condizioni sono l’obesità (SHBG ridotta, ridotto il testosterone totale ma non il biodisponibile) e l’invecchiamento (aumenta SHBG e testosterone totale ma non il biodisponibile). Comunque, nei grandi obesi il testosterone diminuisce in tutte le sue quote.
I metodi maggiormente utilizzati oggi sono quelli immunometrici, con rilevazione in chemi-luminescenza. Tipicamente, nei dosaggi competitivi il testosterone nel campione compete con un aptene marcato (es. con acridinio) per legarsi con un anticorpo monoclonale anti-testosterone. Un successivo passaggio con un reagente libera la molecola legata con le proteine di trasporto. Esiste una relazione inversa tra la quantità di testosterone presente nel campione del paziente e la quantità di RLU (Relative Light Units, unità di luce relative) rilevata dal sistema.
La diffusione dei metodi diretti è legata ai loro vantaggi: velocità, necessità di volume ridotto di campione, eliminazione di reagenti isotopici. Alcuni di questi metodi hanno sufficiente precisione e buona correlazione con i metodi LC-MS nei campioni provenienti da maschi adulti, ma spesso non hanno sensibilità sufficiente per i campioni provenienti dalle femmine e dai soggetti pre-puberi e hanno un’accuratezza non soddisfacente.
Tutti gli immuno-dosaggi per la determinazione del testosterone hanno reazione crociata per il diidro-testosterone (DHT), ma reazioni crociate trascurabili per gli altri androgeni. Nella maggior parte delle situazioni cliniche è possibile stimare la concentrazione del testosterone anche se la sua misurazione non è preceduta dalla separazione del DHT, poiché la concentrazione di questo non supera il 10-20% della concentrazione di testosterone.
Il metodo di riferimento per la determinazione del testosterone libero è la dialisi all’equilibrio, di difficile esecuzione e quindi raramente impiegato. Gli altri metodi utilizzati, come la precipitazione del testosterone legato, il calcolo degli indici androgenici e, soprattutto, quello diretto dell’analogo, non sono affidabili, come concluso dall’Expert Panel dell’Endocrine Society, che raccomanda di interpretare i risultati solo conoscendo il metodo utilizzato:
- l’intervallo di riferimento deve essere specifico per quel metodo;
- i referti devono essere corredati dalle informazioni relative al metodo utilizzato;
- i metodi diretti NON devono essere usati nella donna, nei bambini e nei pazienti ipogonadici;
- la comparabilità dei metodi NON è la stessa in tutti i campioni;
- la fase follicolare è il momento del ciclo più adatto per studiare un sospetto iperandrogenismo.
Nel caso i livelli di testosterone totale non siano chiaramente dirimenti per la diagnosi di ipogonadismo (zona grigia), potranno essere eventualmente integrati con il calcolo della frazione libera, impiegando formule che utilizzano i dosaggi di testosterone totale, albumina e SHBG (come quella utilizzabile presso ISSAM).
Dal 2010, il CDC ha lanciato un programma di standardizzazione per il testosterone con il coinvolgimento di società scientifiche e aziende produttrici, che ha consentito significativi progressi nell’affidabilità degli immuno-dosaggi.
Negli anni recenti è aumentato l’interesse nel testosterone salivare: poiché il testosterone salivare è separato completamente dalle proteine, la sua determinazione riflette la vera concentrazione dell’ormone attivo. Ma al momento il test non trova impiego nella routine corrente.
Bibliografia
- Dobs AS. The role of accurate testosterone testing in the treatment and management of male hypogonadism. Steroids 2008, 73: 1305–10.
- Albrecht L, Styne D. Laboratory testing of gonadal steroids in children. Pediatr Endocrinol Rev 2007, 5 suppl 1: 599–607.
- Taieb J, et al. Testosterone measured by 10 immunoassays and by isotope-dilution gas chromatography-mass spectrometry in sera from 116 men, women, and children. Clin Chem 2003, 49: 1381-95.
- Herold DA, et al. Immunoassays for testosterone in women: better than a guess? Clin Chem 2003, 49: 1250-1.
- Vesper HW, Botelho JC. Standardization of testosterone measurements in humans. J Steroid Biochem Mol Biol 2010, 121: 513–9.
Metodo di determinazione del 17-OH-progesterone
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento settembre 2021)
Il metodo di riferimento è la cromatografia liquida (LC) seguita da doppia spettrometria di massa (Tandem MS), con limiti di sensibilità < 1 nmol/L. Gli immuno-dosaggi diretti (senza estrazione), sia nella versione RIA che EIA, hanno trovato larghissima diffusione grazie alla loro relativa semplicità d'uso, ma sono indubbiamente limitati nella loro attendibilità. Le versioni più utilizzate prevedono anticorpi immobilizzati sulla provetta o sul fondo della micro-piastra nel caso di metodiche enzimatiche; sono stati proposti e vengono largamente utilizzati anche traccianti fotometrici, fluorescenti e chemi-luminescenti, disponibili su piattaforme automatizzate. D'altra parte, anche la LC-Tandem MS, che ha certamente sensibilità e specificità analitiche superiori agli immuno-dosaggi, non è esente da limitazioni: pur se ad elevata specificità, il metodo è esposto all'azione di interferenze che possono essere tuttavia controllate (se non eliminate) con l'uso dei rapporti ionici ("ion ratios" e "ion traps"). Ma i maggiori limiti del metodo sono legati soprattutto alla modesta armonizzazione delle differenti applicazioni, che porta a un’elevata variabilità inter-laboratorio e alla necessità di fissare intervalli di riferimento istituzione-specifici. Ecco perché è estremamente importante che nel follow-up si ricorra allo stesso metodo e, possibilmente, allo stesso laboratorio se si vuole avere la sicurezza che eventuali variazioni siano indipendenti da bias metodologici.
Raccolta e conservazione dei campioni: sono utilizzabili sia siero che plasma. I campioni sono stabili, se separati dalla parte corpuscolata, per una settimana a temperatura ambiente, fino a 14 giorni a 4°C e fino a un mese a -20°C. Campioni emolizzati e/o fortemente lipemici non possono essere accettati. Per lo screening neonatale si utilizzano capillari o cartoncini di carta da filtro, facilmente trasportabili per dosaggi centralizzati. È stato sperimentato anche il dosaggio su campioni di saliva, con il principale vantaggio della non invasività della raccolta.
Bibliografia
- Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010, 95: 4133–60.
- Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016, 101: 364-89.
- Annesley T. Mass spectrometry in the clinical laboratory: how have we done, and where do we need to be? Clin Chem 2009, 55: 1236-8.
- Bertholf RL, Cooper J, Winter WE. The adrenal cortex. In Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 6th Edition 2017, Chapter 66.
- Onuma S, Fukuoka T, Miyoshi Y, et al. Two girls with a neonatal screening-negative 21-hydroxylase deficiency requiring treatment with hydrocortisone for virilization in late childhood. Clin Pediatr Endocrinol 2021, 30: 143–8.
Metodo di determinazione degli androgeni minori
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento novembre 2021)
Androstenedione è un ormone steroideo a 19 atomi di carbonio, che serve come precursore di testosterone ed estrone. Viene sintetizzato principalmente dal deidroepiandrosterone (DHEA), mediante la 3ß-idrossisteroido deidrogenasi nell’ovaio, nel testicolo e nel surrene. Come tutti gli steroidi, viene dosato idealmente con metodi basati sulla separazione cromatografica, anche se oggi i metodi immunometrici hanno trovato una vasta applicazione per la loro semplicità, praticità ed economicità. Tipicamente, questi dosaggi automatizzati si basano su un immunodosaggio competitivo a una fase, con rivelazione in chemi-luminescenza. L’androstenedione nel campione del paziente compete con aptene marcato (ad esempio con estere di acridinio) per legarsi con un anticorpo anti-androstenedione monoclonale attaccato a una fase solida. Esiste una relazione inversa tra la quantità di androstenedione presente nel campione del paziente e la quantità di RLU (Relative Light Units, unità di luce relative) rilevata dal sistema. Sono riportate interferenze farmacologiche con alcune molecole (es. exemestano e formestano), che possono dare valori falsamente elevati. Anche la presenza di biotina può contribuire a generare risultati falsamente elevati. La presenza di anticorpi eterofili nel campione può provocare risultati falsamente ridotti o falsamente elevati. Ovviamente i risultati devono sempre essere valutati alla luce del quadro clinico.
Il DHEAS è secreto nel flusso ematico a una velocità appena maggiore rispetto al DHEA, ma ha un turn-over molto più lento e un'emivita di quasi un giorno intero. Diversamente dal cortisolo, il DHEAS non mostra importanti variazioni diurne e, diversamente dal testosterone, non circola legato alla SHBG. La sua abbondanza, insieme alla sua stabilità nelle 24 ore e giorno dopo giorno, fa del DHEAS un eccellente indicatore di produzione androgena surrenalica. La determinazione del DHEA-S è eseguita routinariamente con metodi immunometrici diretti senza estrazione o cromatografia. Sono comunque oggi disponibili metodi non isotopici in fase omogenea, che possono essere automatizzati. Di converso, poiché la concentrazione del DHEA ha un ordine di grandezza 1000 volte inferiore a quella del DHEA-S, può essere misurata solo dopo una procedura di estrazione (con diclorometano ed etil-acetato) o di separazione cromatografica. Molto usati sono gli immunodosaggi competitivi che utilizzano una tecnologia in chemi-luminescenza diretta. Si impiegano particelle magnetiche di streptavidina come fase solida, anticorpi anti-DHEAS marcati con biotina come coniugato di cattura e un DHEAS coniugato marcato con estere di acridinio. L'anticorpo anti-DHEAS e il complesso DHEAS marcato con estere di acridinio sono catturati dalla fase solida, con sviluppo del segnale. Esiste una relazione inversa tra la quantità di DHEAS presente nel campione del paziente e la quantità di RLU rilevata dal sistema. I campioni dei pazienti che contengono anticorpi eterofili possono reagire nei metodi immunologici producendo risultati falsamente elevati o ridotti. I pazienti in trattamento con biotina ad alte dosi possono avere risultati falsamente sovrastimati.
Nell’adulto, le concentrazioni sieriche di DHT sono oggi misurate accuratamente con la cromatografia liquida abbinata alla doppia spettrometria di massa (LC-MS/MS) e sono disponibili anche intervalli di riferimento coerenti.
Bibliografia
- Clinical and Laboratory Standards Institute. Defining, establishing, and verifying reference intervals in the clinical laboratory; Approved Guideline—Third Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2010. CLSI Document EP28‑A3c.
- Grimsey P, Frey N, Bendig G, et al. Population pharmacokinetics of exogenous biotin and the relationship between biotin serum levels and in vitro immunoassay interference. Int J Pharmacokinet 2017, 2: 247–56.
- Keski-Rahkonen P, Huhtinen K, Poutanen M, et al. Fast and sensitive liquid chromatography–mass spectrometry assay for seven androgenic and progestagenic steroids in human serum. J Steroid Biochem Mol Biol 2011, 127: 396–404.
- Swerdloff RS, Dudley RE, Page ST. Dihydrotestosterone: biochemistry, physiology, and clinical implications of elevated blood levels. Endocr Rev 2017, 38: 220–54.
- Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev 2004, 25: 947-70.
- Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010, 95: 4133–60.
- Rodin A, Thakkar H, Taylor N, et al. Hyperandrogenism in polycystic ovarian syndrome. Evidence of dysregulation of 11ß-hydroxysteroid dehydrogenase. N Engl J Med 1994, 330: 460–5.
- Georgopoulos NA, Papadakis E, Armeni AK, et al. Elevated serum androstenedione is associated with a more severe phenotype in women with polycystic ovarian syndrome (PCOS). Hormones (Athens) 2014, 13: 213–21.
Metodo di determinazione di inibine e AMH
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
(aggiornato al 12 settembre 2016)
Inibine
L’inibina B deriva principalmente dalle cellule della granulosa del follicolo, mentre l’inibina A è prodotta principalmente dal corpo luteo e dalla placenta.
Fino alla metà degli anni ‘90 del secolo scorso la non disponibilità di metodi di misura ha impedito di conoscere il pattern di secrezione delle due inibine e il loro ruolo. I primi metodi sono stati messi a punto in Australia nel 1994 e nel 1996 è stato misurato per la prima volta il pattern di secrezione dell’inibina B nel ciclo mestruale e dimostrata l’esistenza di una correlazione negativa tra inibina B e FSH. La determinazione nella donna ha consentito di comprendere la causa dell’aumento del FSH nella fase follicolare nella donna che si avvicina alla menopausa: la diminuzione dell’inibina B e della riserva ovarica libera l’ipofisi dall’inibizione negativa.
In ambito di ricerca sono ancora impiegati metodi immunometrici, che impiegano tracciante triziato. Nei laboratori clinici sono impiegati metodi immunoenzimatici per la determinazione di Inibina A e Inibina B, mentre sono commercializzati metodi automatici in chemiluminescenza per l’Inibina A.
I primi metodi commerciali hanno cominciato a diventare disponibili dopo il trasferimento del brevetto a DSL (acquisita poi da Beckman Coulter), che fino al 2010 è rimasto l’unico produttore e che ha messo a disposizione un metodo automatico su una piattaforma automatica per l’inibina A.
Oltre agli impieghi nella valutazione dell’infertilità maschile e femminile, l’inibina B è impiegata per rilevare recidive del cancro delle cellule granulose dell’ovaio e nella diagnosi di anomalie di sviluppo sessuale in età pediatrica e l’inibina A nella diagnosi di tumori epiteliali dell’ovaio e nello screening “integrato” per la sindrome di Down, che ha interessato nel 2011 tre milioni di donne negli Stati Uniti.
Bibliografia
- Robertson DM, Cahir N, Findlay JK, et al. The biological and immunological characterization of inhibin A and B forms in human follicular fluid and plasma. J Clin Endocrinol Metab 1997, 82: 889-96.
- Robertson DM, Stephenson T, Pruysers E, et al. Characterization of inhibin forms and their measurement by an inhibin alpha-subunit ELISA in serum from postmenopausal women with ovarian cancer. J Clin Endocrinol Metab 2002, 87: 816-24.
- Robertson DM, Stephenson T, Cahir N, et al. Development of an inhibin alpha subunit ELISA with broad specificity. Mol Cell Endocrinol 2001, 180: 79-86.
- Baly DL, Allison DE, Krummen LA, et al. Development of a specific and sensitive two-site enzyme-linked immunosorbent assay for measurement of inhibin-A in serum. Endocrinology 1993, 132: 2099-108.
- Ludlow H, Phillips DJ, Myers M, et al. A new 'total' activin B enzyme-linked immunosorbent assay (ELISA): development and validation for human samples. Clin Endocrinol (Oxf) 2009, 71: 867-73.
- Groome NP, Tsigou A, Cranfield M, et al. Enzyme immunoassays for inhibins, activins and follistatins. Mol Cell Endocrinol 2001, 180: 73-7.
- Groome NP, Evans LW. Does measurement of inhibin have a clinical role? Ann Clin Biochem 2000, 37: 419-31.
- Rawlins ML, La'ulu SL, Erickson JA, Roberts WL. Performance characteristics of the Access Inhibin A assay. Clin Chim Acta 2008, 397: 32-5.
- Groome NP, Illingworth PJ, O'Brien M, et al. Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab 1996, 81: 1401-5.
Ormone antimulleriano (AMH)
I primi metodi di misura dell’AMH sono stati messi a punto negli anni ‘90; si trattava di ELISA con una sensibilità di 0.5 mg/L, che fu seguito dal metodo sviluppato da Immunotech-Beckman Coulter e da un metodo sviluppato da DSL basato su due anticorpi monoclonali che riconoscevano epitopi nella pro-regione (F2B/7A) e delle regioni mature (F2B/12H). Questa situazione ha causato per anni molti problemi: i due metodi producevano risultati correlati (r = 0.9), ma le concentrazioni misurate dai reagenti DSL erano più basse di quelle misurate con il metodo Beckman-Coulter (4-5 volte secondo alcuni autori, del 40% secondo altri), con grossi rischi di classificazione errata nel caso di variazione di metodiche da parte del laboratorio o quando lo specialista doveva interpretare risultati prodotti da laboratori che usavano metodi diversi (all’epoca i due metodi erano egualmente diffusi nei laboratori clinici). Quando DSL fu acquistata da Beckman Coulter, i due metodi furono sostituiti da un nuovo metodo ELISA (BCI AMH Gen II), con una sensibilità di 0.08 mg/L. La svolta dal punto di vista pratico si è realizzata quando Beckman ha trasferito il metodo su una piattaforma automatica (Access), molto diffusa nei laboratori, che assicurava prestazioni di precisione e rapidità tali da essere adottata rapidamente nei laboratori clinici. Roche ha messo a punto sulla propria piattaforma automatica una metodica che usa la stessa coppia di anticorpi usati da Beckman, con risultati sovrapponibili e con precisione anche superiore. È quindi verosimile che si possa sviluppare l’impiego dell’AMH anche al di fuori di centri di terzo livello: la disponibilità di metodi automatici che impiegano gli stessi anticorpi consentirà, infatti, precisione ed omogeneità molto maggiori di quella possibile fino ad oggi.
Il coefficiente di variazione misurato da autori diversi sulle piattaforme Roche alle diverse concentrazioni è risultato < 1.7-3.7% e la determinazione non è interferita da ittero, emolisi, fattore reumatoide, activina A, inibina A, LH e FSH, mentre l’AMH non deve essere misurato prima che siano trascorse almeno 8 ore dall’assunzione di 5 mg di biotina.
La letteratura sulla stabilità dell’AMH è molto contraddittoria e risente del percorso di messa a punto di analizzatori automatizzati affidabili compiuto nell’ultimo decennio. Facendo riferimento solo ai dati relativi ai metodi automatizzati, l’AMH è stabile a temperatura ambiente per 2 giorni, a 2–8°C per 7 giorni e a temperature < -20°C per 15 mesi.
Sostanzialmente risolto il problema della standardizzazione analitica, le aziende produttrici hanno oggi bisogno della disponibilità di materiale di riferimento, che deve essere messo a disposizione al più presto dagli organismi regolatori.
Bibliografia
- La Marca A, Broekmans FJ, Volpe A, et al. Anti-Mullerian hormone (AMH): what do we still need to know? Hum Reprod 2009, 24: 2264–75.
- Nelson SM, Pastuszek E, Kloss G, et al. Two new automated, compared with two enzyme-linked immunosorbent, antimullerian hormone assays. Fertil Steril 2015, 104: 1016-21.
- Leader B, Baker VL. Maximizing the clinical utility of antimullerian hormone testing in women’s health. Curr Opin Obstet Gynecol 2014, 26: 226–36.
- Welsh P, Smith K, Nelson SM. A single-centre evaluation of two new anti-Mullerian hormone assays and comparison with the current clinical standard assay. Hum Reprod 2014, 29: 1035–41.
- van Helden J, Weiskirchen R. Performance of the two new fully automated anti-Mullerian hormone immunoassays compared with the clinical standard assay. Hum Reprod 2015, 30: 1918–26.
- Demirdjian G, Bord S, Lejeune C, et al. Performance characteristics of the Access AMH assay for the quantitative determination of anti-Müllerian hormone (AMH) levels on the Access family of automated immunoassay systems. Clin Biochem 2016, doi.org/10.1016/j.clinbiochem.2016.08.005.
- Anckaert E, Öktem M, Thies A, et al. Multicenter analytical performance evaluation of a fully automated anti-Müllerian hormone assay and reference interval determination. Clin Biochem 2016, 49: 260–7.
- De Laveleye M, Gruson D. Anti-Müllerian hormone testing: evaluation of a novel method allowing more automation. Scand J Clin Lab Invest 2015, 75: 681–5.
- Anderson RA, Anckaert E, Bosch, et al. Prospective study into the value of the automated Elecsys antimullerian hormone assay for the assessment of the ovarian growing follicle pool. Fertil Steril 2015, 103: 1074-80.
- Deeks ED. Elecsys AMH assay: a review in Anti-Mullerian Hormone quantification and assessment of ovarian reserve. Mol Diagn Ther 2015, 19: 245–9.
- Gassner D, Jung R. First fully automated immunoassay for anti-Mullerian hormone. Clin Chem Lab Med 2014, 52: 1143–52.
- Hyldgaard J, Bor P, Ingerslev HJ, Tørring N. Comparison of two different methods for measuring anti-mullerian hormone in a clinical series. Reprod Biol Endocrinol 2015, 13: 107.
Metodo di determinazione del glucosio
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
GLICEMIA
Metodi di determinazione
Il glucosio è misurato quasi esclusivamente con metodi enzimatici. L'analisi del Programma di Verifica Esterna di Qualità (VEQ) gestito dal College of American Pathologists (CAP) ha rivelato che negli Stati Uniti virtualmente tutti i laboratori misurano la concentrazione di glucosio nel sangue con metodiche basate su esochinasi o glucosio-ossidasi e solo l’1% utilizza metodi basati sulla glucosio-deidrogenasi. Il glucosio, una volta fosforilato dall’esochinasi, è ossidato dalla glucosio-6-fosfato deidrogenasi in presenza di NADP. Il NADPH generato è direttamente proporzionale alla concentrazione di glucosio. L'imprecisione (CV) nella determinazione del glucosio nel materiale di controllo e nei campioni dei pazienti è < 4%.
Raccolta dei campioni
Il campione deve essere raccolto al mattino dopo una notte di digiuno (durante questo tempo il soggetto può assumere acqua liberamente). I valori medi di glicemia a digiuno (FPG) sono più alti al mattino che al pomeriggio, per cui molti casi di diabete non sarebbero stati diagnosticati valutando valori di glicemia misurati nel pomeriggio.
Stabilità
La concentrazione del glucosio nel sangue intero diminuisce con il tempo a causa della glicolisi. La velocità della glicolisi è in media del 5-7% per ora, portando a una diminuzione di concentrazione di circa 0.6 mmol/L (10 mg/dL) per ogni ora trascorsa dal prelievo. Tale velocità dipende anche da fattori come temperatura, concentrazione del glucosio e numero di eritrociti, leucociti e piastrine (che la aumentano, portando a una più rapida diminuzione dei valori misurati).
Il processo della glicolisi può essere rallentato (migliorando quindi la stabilità delle concentrazioni misurate) da inibitori dell'enolasi, come il fluoruro di sodio (2.5 mg di fluoruro/mL di sangue) o, meno comunemente, lo iodo-acetato di litio (0.5 mg/mL di sangue). Questi reagenti si possono usare da soli o, più comunemente, con anti-coagulanti come ossalato di potassio, EDTA, citrato o litio-eparina. Anche se il fluoruro mantiene stabile a lungo la concentrazione del glucosio, la velocità di diminuzione della glicemia nella prima ora successiva alla raccolta del campione è virtualmente identica in provette con e senza fluoruro (anche se un numero di globuli bianchi molto alto può aumentare la glicolisi anche in presenza di fluoruro). Nel sangue intero a temperatura ambiente, dopo le prime 4 ore la concentrazione del glucosio è stabile per 72 ore in presenza di fluoruro. Nel siero separato, non emolizzato, sterile, senza fluoruro, la concentrazione del glucosio è stabile per 8 ore a 25°C e per 72 ore a 4°C.
Variabilità
Il glucosio a digiuno può presentare variazioni del 14% in due mattine consecutive: una glicemia di 7 mmol/L (126 mg/dL) una mattina, può calare nella successiva a 5 mmol/L (90 mg/dL) o salire a 8 mmol/L (144 mg/dL) nel 95% dei casi.
Il glucosio può essere misurato nel sangue intero, nel siero o nel plasma, ma per la diagnosi è raccomandato il plasma.
La molalità del glucosio (vale a dire la quantità di glucosio per unità di massa d'acqua) è identica nel sangue intero e nel plasma. Anche se i globuli rossi del sangue sono liberamente permeabili al glucosio (il glucosio vi entra per trasporto facilitato), il contenuto di acqua è di circa il 93% nel plasma e di circa il 73% negli eritrociti. Con un ematocrito del 45%, possiamo calcolare un contenuto di acqua medio dell’84%: infatti, 0.45*73% + 0.55*93% = 84%. Poiché il glucosio è distribuito in modo uguale nella parte acquosa del sangue, il rapporto tra il contenuto di acqua del plasma e del sangue intero è 93%/84% = 1.107. La concentrazione di glucosio nel plasma è quindi (quando l’ematocrito è del 45%) più alta dell’11% rispetto a quella nel sangue intero. ADA e WHO raccomandano di misurare la concentrazione del glucosio nel plasma (sangue raccolto in eparina). Anche se alcuni autori hanno trovato concentrazioni di glucosio più basse nel plasma che nel siero e lo hanno attribuito al passaggio dagli eritrociti al plasma causato dagli anti-coagulanti, altri hanno trovato concentrazioni più alte o uguali. Mentre il glucosio è significativamente più alto nel sangue capillare che nel sangue venoso durante OGTT, la differenza media nei campioni a digiuno è minima.
| Variabilità della glicemia | |||
| Determinazione | Plasma vs. sangue intero |
Sangue eparinato vs. siero |
Sangue capillare vs. venoso |
| random | 11% | 5%* | |
| digiuno | 2 mg/dL, 0.1 mM/L | ||
| post-OGTT | 20-25%, 30 mg/dL, 1.7 mM/L | ||
* segnalazione non unanime
| Intervalli di riferimento della concentrazione di glucosio | |||
| mg/dL | mmol/L | ||
| Plasma | Adulti* | 74-106 | 4.5-5.9 |
| Bambini | 60-100 | 3.5-5.6 | |
| Neonati a termine | 30-60 | 1.7-3.3 | |
| Neonati prematuri | 20-60 | 1.1-3.3 | |
| Sangue intero venoso | 65-100 | 3.5-5.5 | |
| Sangue intero capillare | 70-100 | 3.9-5.5 | |
| Liquor | 20-60 | 1.1-3.3 | |
*Va segnalato che alcune fonti autorevoli, come Ministero della Salute e ADA, hanno indicato come limite decisionale di euglicemia 5.5 mmol/L (100 mg/dL), mentre altre, altrettanto autorevoli come WHO, Canadian Diabetes Association, UK Diabetes Foundation, 6.1 mmol/L (110 mg/dL).
GLICOSURIA
Per la determinazione vanno preferite strisce reattive che determinano il glucosio in modo specifico, piuttosto che mediante riduzione. Non è stato dimostrato che la determinazione del glucosio sulla seconda minzione offra dei vantaggi.
Note metodologiche
In passato il glucosio era misurato con metodi in cui erano rivelate le sostanze presenti nelle urine in grado di ridurre il rame. Poiché numerosissime sostanze, oltre al glucosio, possono avere un’azione riducente di questo tipo (ad esempio: fruttosio, lattosio, galattosio, maltosio, xilosio, ribosio, acido urico, acido ascorbico, creatinina, cisteina, corpi chetonici, sulfanilamide, acido ossalico, acido ippurico, acido glucuronico, isoniazide, salicilati), oggi si impiegano metodiche basate sull’enzima glucosio-ossidasi.
In genere le strisce reattive sono impregnate dell’enzima insieme al colorante o-toluidina, che produce una colorazione blu quando la concentrazione di glucosio nelle urine supera i 100 mg/dL. Risultati falsi-positivi possono essere dati dalla contaminazione delle urine con acqua ossigenata o ipoclorito (candeggina). Risultati falsi-negativi possono essere dati dalla presenza di chetoni, acido ascorbico (contenuto come conservante in numerosi antibiotici) e salicilati.
I metodi quantitativi per la determinazione del glucosio nelle urine sono gli stessi usati nel plasma: esochinasi, glucosio-ossidasi (che non comprendono la reazione H2O2-perossidasi) e glucosio-deidrogenasi.
BIBLIOGRAFIA
- Sacks DB, Arnold M, Bakris GL, et al. Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus. Clin Chem 2011, 57: e1-e47.
- Burtis CA, Ashwood ER, Bruns DE. Tietz textbook of clinical chemistry and molecular diagnostics. Elsevier’s Saunders; St Louis 2006.
- ADA. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 S1: S81-90.
- Wu A. Tietz clinical guide to laboratory tests. Fourth Edition. WB Saunders; Philadelphia 2006.
Metodo di determinazione di emoglobina glicata
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Metodi di determinazione
Sono disponibili numerose metodiche per misurare l'emoglobina glicata. Le più usate sono basate sulla differenza di carica elettrica tra HbA1c e HbA (mini-colonnine, HPLC, elettroforesi e isoelettro-focalizzazione), sulla natura di determinanti antigenici dei primi 8 residui aminoacidici della catena ß dell’emoglobina (metodiche immunometriche) e sulla presenza di glucosio legato covalentemente all'emoglobina (cromatografia di affinità). I metodi immunometrici di più recente commercializzazione sono dotati di particolari caratteristiche di specificità per l'emoglobina A1c.
“National Glycohemoglobin Standardisation Program” (NGSP) e IFCC hanno collaborato a lungo e intensamente per ridurre significativamente, se non annullare, le differenze tra i risultati ottenuti utilizzando le diverse tecnologie disponibili commercialmente a livello internazionale (oltre un centinaio). L’IFCC assicura la tracciabilità dei metodi in commercio al metodo di riferimento attraverso un programma di standardizzazione, mentre l’NGSP definisce i limiti di accettabilità delle prestazioni dei metodi. Nel 1996 l’NGSP ha promosso un programma di standardizzazione dei risultati di HbA1c tra i laboratori, che ha consentito di renderli riferibili al DCCT. Generalmente, i risultati di tutti i metodi mostrano buone correlazioni, anche se i valori di HbA1c ottenuti sullo stesso campione di sangue possono differire, anche considerevolmente, se sono misurati con metodi diversi. Questo spiega, in parte, perchè nel corso degli anni, in mancanza di uno standard di riferimento internazionale, si siano sviluppati intervalli di riferimento metodo-dipendenti e perchè i dati ottenuti in ambiti diversi possano essere non confrontabili tra loro.
Oggi sia l’American Association for Clinical Chemistry (AACC) che l’ADA raccomandano che i laboratori usino solo metodi per l’emoglobina glicata certificati dall’NGSP e che partecipino a un programma di Verifica Esterna di Qualità (VEQ). I progressi registrati in venti anni sono evidenti. Nel 1993 solo il 50% dei laboratori degli Stati Uniti misurava l’emoglobina glicata come HbA1c, nel 1996 la percentuale è salita all’80%, nel 2004 al 99% e oggi è arrivata al 100%. L’NGSP ha lavorato anche con il College of American Pathologists per migliorare progressivamente le prestazioni dei laboratori anche utilizzando il Programma di VEQ che gestisce oltre 3000 laboratori (in maggioranza fuori degli Stati Uniti).
Il livello di accuratezza accettabile, che era inizialmente considerato uno scostamento del 15%, è stato progressivamente diminuito, portandolo al 12% nel 2008, al 10% nel 2009, all’8% nel 2010 e al 7% nel 2011.
Nuova posizione di consenso sulla determinazione dell'HbA1c
Minore successo ha avuto in Italia la raccomandazione dell’IFCC del 2005, basata su rigorose considerazioni metrologiche, di sostituire, contestualmente all’introduzione del nuovo sistema di riferimento IFCC, la refertazione dell’HbA1c in percentuale con la nuova unità di misura mmol di HbA1c /mol emoglobina. L’IFCC ha raccomandato di eliminare la percentuale perchè ambigua e non conforme al Sistema Internazionale (SI) e di ricorrere alla doppia refertazione solo per un breve periodo, non superiore ai due anni.
Ricavare la concentrazione di HbA1c in unità convenzionali da quella in unità SI e viceversa è più complesso che nel caso del glucosio (nel quale si passa da mg/dL a mmol/L dividendo il valore numerico per 18) e non è sufficiente applicare un fattore di divisione o di moltiplicazione. A una HbA1c NGSP del 4% corrisponde un valore IFCC cinque volte più alto (20 mmol/mol), mentre a un valore del 12% corrisponde un valore IFCC nove volte più alto (108 mmol/mol). La figura 1 mostra che, anche se esiste una relazione lineare tra i valori ottenuti in unità NGSP e quelli ottenuti in unità IFCC, la retta ha pendenza diversa da 1 e intercetta diversa da 0. Per convertire unità NGSP e IFCC occorre la cosiddetta master equation, una semplice equazione lineare (NGSP = 0.09148(IFCC) + 2.152; IFCC = 10.93(NGSP) – 23.50).
Solo un terzo dei laboratori italiani, anche se ufficialmente la conversione è totale, usano una sola unità di misura SI, come raccomandato, in qualche caso per la resistenza dei laboratori, in altri per quella dei clinici. L’adozione di un’unità di misura per un esame così critico come l’emoglobina glicata è un processo complesso e delicato che, se non gestito correttamente, può produrre confusione e inconvenienti. È necessario e urgente affrontare la questione in modo condiviso e, quando si procede alla conversione, occorre fornire in modo capillare e tempestivo a laboratoristi, clinici e, soprattutto, pazienti tutte le informazioni e gli strumenti necessari ad evitare che la nuova unità di misura produca danni al paziente (facilmente reperibili in rete, ad esempio su http://www.ngsp.org/convert1.asp).
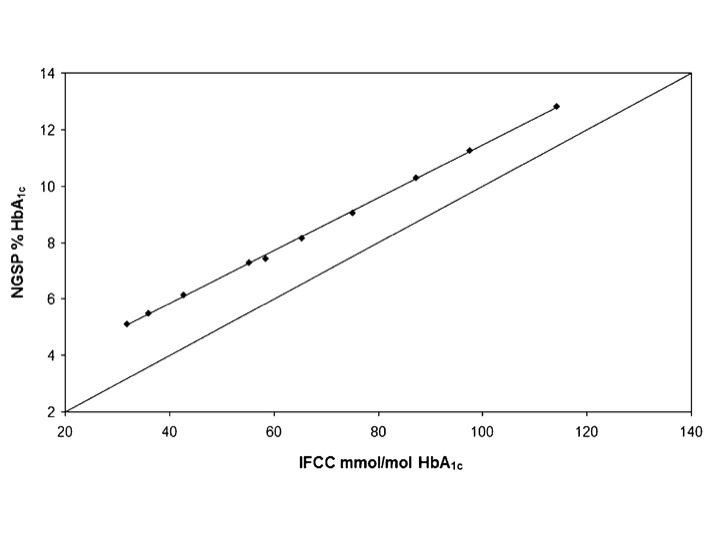
Confronto dei valori di HbA1c ottenuti dai network NGSP (media di 7 laboratori) e IFCC (media di 13 laboratori)
◆ rappresenta la retta di regressione, mentre la linea continua la retta di identità
Raccolta dei campioni
Non occorre il digiuno. I campioni possono essere raccolti in qualunque momento del giorno, non sono alterati da fattori acuti come stress ed esercizio fisico.
I campioni di sangue da utilizzare devono essere raccolti usando un anti-coagulante (di norma EDTA, ma si può usare anche ossalato o fluoruro).
Stabilità
L’analisi deve essere eseguita il più presto possibile, perché la glicazione continua in vitro, soprattutto quando la glicemia è elevata. I campioni in eparina devono essere analizzati entro due giorni e possono non essere adatti ad alcuni metodi come l’elettroforesi. Dopo 3-4 giorni, il metabolismo cellulare produce delle emoglobine modificate, come l’HbA1d e l’HbA3,che possono interferire soprattutto con i metodi cromatografici. Una conservazione impropria dei campioni, per esempio a temperature elevate, può provocare artefatti rilevanti, dipendenti dal metodo di determinazione utilizzato, che non sono sempre identificabili.
In generale, i campioni di sangue intero sono stabili per una settimana a 4°C. Con la maggior parte dei metodi, i campioni conservati a temperatura ≤ –70°C sono stabili per almeno un anno. I campioni non sono stabili a –20°C.
Variabilità
Il trial DCCT ha stimato che ogni aumento dell'1% dell’emoglobina glicata corrisponde a un aumento medio della glicemia di 1.66 mmol/L (30 mg/dL), sia pure con una notevole dispersione dei risultati che rende poco impiegabile questo calcolo nel singolo paziente. Inoltre, i pazienti si distribuirebbero in due popolazioni, in cui l'emoglobina lega il glucosio ad alta e a bassa velocità (rispettivamente high e low "glycators").
Anche la soglia renale può influenzare l'emoglobina glicata. Poiché una volta superata la soglia renale, tutto il glucosio che arriva al rene è escreto, più alta è questa soglia, più alta è la concentrazione di glucosio (e di emoglobina glicata) che può essere raggiunta prima che il glucosio sia misurabile nelle urine.
| Fattori che interferiscono nella misurazione dell’emoglobina glicata | |
| Aumento dei globuli bianchi | Elevati livelli di globuli bianchi (LLC) possono interferire positivamente con numerose metodiche (soprattutto immunochimiche) |
| Carenza di ferro | L’anemia sideropenica si associa a valori più elevati di HbA1c (è possibile che l’aumento di malon-dialdeide in carenza di ferro aumenti la glicazione dell’emoglobina |
| Emoglobinopatie | Interferenza (positiva o negativa) analitica (soprattutto HPLC a scambio ionico) e biologica (effetti sulla vita media eritrocitaria) |
| Età del soggetto | L’HbA1c aumenta leggermente con l’età (0.03% per anno) |
| Insufficienza renale | L'urea può formare, come cianato, emoglobina carbamilata, che generalmente co-eluisce con le metodiche a scambio ionico Spesso nei pazienti uremici la vita media eritrocitaria è ridotta |
| lpertrigliceridemia | Interferenza positiva nelle metodiche immuno-turbidimetriche |
| Processi emolitici | Possono causare valori falsamente diminuiti per diminuzione della vita media eritrocitaria |
| Etnia del soggetto | Differenze (≤ 0.4%) dovute a fattori quali la cinetica di ingresso del glucosio negli eritrociti e variazioni della vita media eritrocitaria |
| Variabilità stagionale | Discreto effetto (fino a circa il 7%) di tipo ciclico, con periodo semestrale |
Raccomandazioni (riguardano sia i pazienti con DM-T1 che quelli con DM-T2)
L'HbA1c deve essere misurata routinariamente in tutti i pazienti con diabete mellito per documentarne il controllo glicemico. Livello e qualità di evidenza: A, moderata
I laboratori devono utilizzare solo metodi di determinazione dell'HbA1c di cui sia stata certificata la “tracciabilità” al metodo DCCT. Infatti, solo i metodi con tale caratteristica producono risultati confrontabili con il metodo DCCT e tra di loro. Livello e qualità di evidenza: Buona pratica di laboratorio
Laboratori e clinici devono conoscere le potenziali interferenze dei metodi analitici per la misura della HbA1c comprese le emoglobinopatie. Alterazioni del turn-over eritrocitario possono causare risultati spuri. Livello e qualità di evidenza: Buona pratica di laboratorio
I laboratori devono utilizzare metodi di misura dell’emoglobina glicata con CV intra-laboratorio < 2% e inter-laboratorio < 3.5%. Livello di evidenza: B, bassa
Gli obiettivi del trattamento prevedono il mantenimento della concentrazione di HbA1c< 7% e obiettivi più stringenti in pazienti selezionati a basso rischio di ipoglicemia. Livello di evidenza e qualità: A, alta
La determinazione dell’emoglobina glicata deve essere eseguita due volte l’anno in tutti i pazienti e quattro volte l’anno nei casi in cui la terapia sia stata cambiata o non siano stati raggiunti gli obiettivi di trattamento. Livello e qualità di evidenza: B, bassa
Bibliografia
- Mosca A, Branca MT, Carta M. et al. Raccomandazioni per l'implementazione della standardizzazione internazionale della misura dell'emoglobina glicata in Italia. Biochimica Clinica 2009, 33: 258-61.
- Braga F, Panteghini M. Standardizzazione della misura e traguardi analitici per l’emoglobina glicata. Biochimica Clinica 2013, 37: 100-7.
- Little RR, Rohlfing CL, Sacks DB. Status of hemoglobin A1c measurement and goals for improvement: from chaos to order for improving diabetes care. Clin Chem 2011, 57: 205-14.
- Sacks DB. Measurement of hemoglobin A(1c): a new twist on the path to harmony. Diabetes Care 2012, 35: 2674-80.
- Sacks DB. 2011 consensus meeting on the worldwide standardization of hemoglobin A1c measurement. Clin Chem 2013, 59: 857-8.
- Sacks DB, Arnold M, Bakris GL, et al. Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus. Clin Chem 2011, 57: e1-e47.
- Sacks DB; ADA/EASD/IDF working group of the HbA1c assay. Global harmonization of hemoglobin A1c. Clin Chem 2005, 51: 681–3.
Metodo di determinazione dei chetoni
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Metodi di determinazione
Sono state descritte numerose metodiche di misura. La più usata è la reazione colorimetrica che avviene tra corpi chetonici e nitroprussiato (sodio ferro-cianuro) e che produce colore porpora. Questo metodo è ampiamente disponibile in forma di strisce reattive e di compresse ed è utilizzato per misurare chetonuria e chetonemia (nel siero e nel plasma). Sono commercializzate strisce reattive che misurano sia glucosio che chetoni.
Il metodo del nitroprussiato misura solo acido acetico (AcAc)(se il reagente contiene glicina, misura anche l’acetone, ma in modo molto meno sensibile). I metodi che si basano sulla reazione al nitroprussiato devono essere utilizzati con cautela nella diagnosi di chetoacidosi, perché non quantificano il ß-OH-butirrato (ß-HBA), il chetone predominante nella chetoacidosi, e non devono essere usati per monitorarne la terapia, poiché nel corso di trattamento efficace il ß-HBA diminuisce mentre AcAc e acetone possono aumentare. I metodi che misurano in maniera specifica il ß-HBA sono utili sia per la diagnosi che per il follow-up della chetoacidosi.
Non vi è ancora consenso se nei pazienti diabetici i corpi chetonici debbano essere misurati nel sangue o nelle urine. Nel monitoraggio del trattamento della chetoacidosi la misura semi-quantitativa dei corpi chetonici è più accurata nel sangue che nelle urine. Tuttavia, anche se i corpi chetonici non sono escreti nelle urine in modo proporzionale alla loro concentrazione nel sangue, è maggiormente impiegata la determinazione nelle urine, più pratica.
Raccolta dei campioni
I campioni per la determinazione dell’acetone, altamente volatile, devono essere conservati in contenitori chiusi. I corpi chetonici possono essere misurati nel siero o nel plasma con strisce reattive e compresse, analoghe a quelle utilizzate per la chetonuria e i campioni possono essere diluiti con soluzione fisiologica per titolarne la concentrazione (i risultati sono espressi generalmente come positivi ad una diluizione 1:x). Anche in questo caso non è misurato il ß-HBA, il corpo chetonico prevalente nella chetoacidosi.
Stabilità
L’AcAc è labile ed è convertito rapidamente in ß-HBA; può pertanto essere misurato solo in campioni addizionati con acido perclorico. Il ß-HBA è stabile nel sangue intero per 4 ore a temperatura ambiente e per 48 ore in siero/plasma a 4-8°C.
Variabilità
Gli intervalli di riferimento dei metodi di misura per il ß-HBA sono diversi, ma la concentrazione in soggetti sani a digiuno è generalmente < 0.5 mmol/L (5 mg/dL).
Pazienti con chetoacidosi ben documentata (CO2 < 17 mmo/L) hanno generalmente una concentrazione di β-HBA > 2 mmol/L (20 mg/dL).
In condizioni fisiologiche il rapporto tra ß-HBA ed AcAc è 1:1; nel digiuno prolungato può salire a 6:1, nella chetoacidosi può superare 10:1.
Raccomandazioni
- Nella diagnosi o nel monitoraggio di chetoacidosi non deve essere utilizzata la determinazione dei corpi chetonici nelle urine. Livello di evidenza: Buona pratica di laboratorio.
- I metodi per misurare i corpi chetonici nel sangue che si basano sulla reazione al nitroprussiato possono essere utilizzati per confermare la diagnosi di chetoacidosi, ma non per monitorarne il trattamento. Per la diagnosi ed il monitoraggio di chetoacidosi devono essere utilizzati metodi specifici per il ß-HBA nel sangue. Livello e qualità di evidenza: B, moderata.
Bibliografia
- Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev 1999, 15: 412–26.
Metodo di determinazione di insulina, proinsulina e peptide C
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Insulina
Poco utilizzati sono i bio-dosaggi e i metodi in spettrometria di massa che producono risultati più bassi rispetto a quelli immunometrici (maggiormente impiegati, anche se sono spesso influenzati dagli anticorpi sviluppati dai pazienti trattati con insulina). Una misura accurata dell’insulina può essere effettuata precipitando con polietilen-glicole (PEG) gli anticorpi endogeni e l’insulina ad essi legata. Anche recentemente l’Insulin Standardization Work Group, istituito dall’ADA, ha concluso che non tutti i metodi in commercio misurano l’insulina in modo preciso, accurato e con una sufficiente sensibilità. Attualmente si devono considerare alcuni aspetti fondamentali.
Numerose preparazioni di insulina umana sono usate come calibratori, espressi di solito in UI: uno dei più utilizzati è il 1st IRP 66/304 che contiene il 100% di insulina umana (43 mg di questo materiale corrisponde a 1 UI). Il workgroup ha tuttavia raccomandato che la concentrazione di insulina sia refertata in unità SI pmol/L, evitando i riferimenti alle unità tradizionali di insulina basato sulla attività biologica dell’insulina per milligrammo di preparazione. Le preparazioni attuali di insulina, preparate con tecniche ricombinanti, sono più purificate rispetto a quelle prodotte in passato e, assumendo un peso molecolare dell’insulina monomerica di 5808 Da, è possibile assegnare un numero accurato di moli ai calibratori usati in ogni metodica (12 pmol/L = 2 mU/L nelle vecchie unità).
Anticorpi sviluppati contro l’insulina hanno una certa cross-reattività con la pro-insulina (ma non con il peptide-C). Anche se l’interferenza è poco rilevante nei soggetti sani (che hanno bassi livelli di pro-insulina), l’interferenza diventa rilevante nei pazienti con insulinoma e con diabete (che hanno un’elevata concentrazione di pro-insulina), causando una sovrastima della concentrazione di insulina. Poichè l’attività biologica della pro-insulina è molto bassa, ne risulta che il paziente può essere classificato erroneamente.
Non è stata ancora raggiunta la standardizzazione dei laboratori perché mancano, secondo i requisiti dell’International Organization for Standardization, materiale primario di riferimento, metodo di misura di riferimento e materiale secondario di riferimento. Esistono, tuttavia, un metodo di misura di riferimento candidato e una certa quantità di materiale primario di riferimento certificato che sono già stati usati con risultati molto incoraggianti.
| Dosaggio insulina | |
| Metodologia adottata | RIA, IRMA, ELISA, Chemiluminescenza |
| Campione richiesto | Provetta da siero senza gel e con gel, 6 mL |
| Volume minimo | 500 µL |
| Stabilità del campione | Il siero è stabile a temperatura ambiente per 24 ore, a 2-8°C per una settimana, a – 20°C per 3 mesi |
| Intervallo di riferimento | 2-25 mU/L (24-300 pmol/L) < 9 mU/L (108 pmol/L) (metodi ad elevata specificità) < 200 mU/L (2400 pmol/L) dopo OGTT |
Pro-insulina
La determinazione della pro-insulina risulta difficile per la bassa concentrazione della molecola e per la difficoltà della produzione di anticorpi che non cross-reagiscano con l’insulina ed il peptide-C, presenti in concentrazioni molto più alte (la pro-insulina cross-reagisce con gli anticorpi anti-insulina per una percentuale che può superare il 25%).
I dati disponibili sono scarsi, per la difficoltà del reperimento di preparazioni pure di pro-insulina da usare per la calibrazione del dosaggio e perché i metodi disponibili misurano anche delle forme intermedie di clivaggio della pro-insulina. Solo recentemente è stata prodotta una molecola sintetica di pro-insulina che ha consentito di produrre anticorpi monoclonali e calibratori affidabili.
| Dosaggio pro-insulina | |
| Metodologia adottata | RIA, IRMA, ELISA |
| Campione richiesto | Provetta da siero senza gel, 6 mL |
| Volume minimo | 500 µL |
| Stabilità del campione | Il siero è stabile a – 70°C per 3 mesi |
| Intervallo di riferimento | 1.1-6.9 pmol/L |
Peptide-C
Il peptide-C subisce un metabolismo epatico molto modesto e la sua determinazione non è influenzata dalla presenza di anticorpi anti-insulina. I metodi di dosaggio sono, tuttavia, caratterizzati da una specificità diversa degli anticorpi usati, da una cross-reazione variabile con la pro-insulina e dal tipo di calibratore impiegato (inizialmente è stato utilizzato il calibratore IRR 84/510 e successivamente il 76/561).
| Dosaggio peptide-C | |
| Metodologia adottata | RIA, IRMA, ELISA, Chemiluminescenza |
| Campione richiesto | Provetta da siero senza gel, 6 mL |
| Volume minimo | 500 µL |
| Stabilità del campione | Il siero NON è stabile a – 20°C (diminuisce del 28% in 4 settimane); stabilità a – 70°C non valutata |
| Intervallo di riferimento | 0.25-0.6 nmol/L 0.9-1.9 nmol/L o 3-5 volte il valore pre-stimolo (dopo stimolo con glucosio o glucagone) |
Bibliografia
- Clark PM. Assays for insulin, proinsulin(s) and C-peptide. Ann Clin Biochem 1999, 36: 541–64.
- Marcovina S, Bowsher RR, Miller G, et al. Standardization of insulin immunoassays: report of the American Diabetes Association’s Workgroup. Clin Chem 2007, 53: 711–6.
- Miller WG, Thienpont LM, Van Uytfanghe K, et al. Toward standardization of insulin immunoassays. Clin Chem 2009, 55: 1011–8.
- Sapin R. Insulin immunoassays: fast approaching 50 years of existence and still calling for standardization. Clin Chem 2007, 53: 810-1.
- Sacks DB, Arnold M, Bakris GL, et al. Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus. Clin Chem 2011, 57: e1-e47.
- Little RR, Rohlfing CL,Tennill AL, et al. Standardization of C-peptide measurements. Clin Chem 2008, 54: 1023-6.
- Staten MA, Stern MP, Miller WG, et al. Insulin assay standardization: leading to measures of insulin sensitivity and secretion for practical clinical care. Diabetes Care 2010, 33: 205-6.
- Heinemann L. Insulin assay standardization: leading to measures of insulin sensitivity and secretion for practical clinical care: response to Staten et al. Diabetes Care 2010, 33: e83.
- Staten MA, Miller WG, Bowsher RR, et al. Insulin assay standardization: leading to measures of insulin sensitivity and secretion for practical clinical care: response to Heinemann. Diabetes Care 2010, 33: e84.
Metodo di determinazione dei lipidi
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Metodi di determinazione
I metodi enzimatici hanno virtualmente sostituito i metodi chimici di misura del colesterolo totale. Tali metodi hanno in comune i primi due passaggi, che vedono prima l'idrolisi degli esteri del colesterolo tramite colesterolo-esterasi e poi l'ossidazione dello stesso con produzione di acqua ossigenata. Quest'ultima può essere misurata amperometricamente o, più comunemente, col metodo di Trinder, che prevede la formazione di un composto chinonico colorato a partire da acqua ossigenata, amino-antipirina e fenolo tramite l'enzima perossidasi. All'end-point della reazione, il colore sarà direttamente proporzionale alla concentrazione del colesterolo (e di altri steroli di solito non significativi) presenti nel campione. Sul metodo di Trinder interferisce la presenza di sostanze riducenti: in particolare la concentrazione dell'acido ascorbico e della bilirubina. Quest'ultima può avere su questa reazione effetti molto complessi e di tipo ambivalente. Il principale effetto di sottostima può essere indicato nell'attività anti-ossidante; quello di sovrastima nella capacità di assorbire la luce alla stessa frequenza (500 nm) del composto colorato. Per la maggioranza dei metodi, comunque, si inizia ad avere una sottostima del colesterolo per bilirubinemia > 5 mg/dL.
Per questo il metodo di riferimento rimane un metodo chimico con estrazione: il metodo Abell Kendall–modificato CDC. Si tratta di un metodo in cui il colesterolo in forma di estere è rilasciato per saponificazione di un campione di siero con idrossido di potassio alcolico. Dopo estrazione in esano ed evaporazione di un'aliquota dell’estratto, il cromoforo ottenuto con il reagente di Liebermann-Burchard si legge a 620 nm. La complessità del metodo e l’impiego di reagenti corrosivi spiega perché non è eseguito nei laboratori clinici. Oggi tutti i laboratori clinici misurano il colesterolo impiegando metodi enzimatici colorimetrici automatizzati.
Storicamente la concentrazione dell'HDL colesterolo era determinata dalla misura del colesterolo sul sovranatante dei campioni dopo aver precipitato le ß-apolipoproteine tramite miscele di polianioni cationici divalenti che davano risultati non omogenei di precipitazione. Attualmente, grazie all'eliminazione del pre-trattamento del campione, sono divenute molto popolari misure omogenee. Queste prevedono un primo reagente, che si lega al colesterolo non HDL impedendogli di partecipare alle successive reazioni, e un secondo reagente che misura il colesterolo rimasto disponibile. A seconda delle ditte produttrici, il primo reagente può essere costituito da un PEG modificato (ad esempio, Roche), un'immuno-inibizione (ad esempio, Wako) o una miscela di polimeri sintetici poli-anionici e detergenti selettivi (ad esempio, Beckman).
Il modo più diffuso per il calcolo del colesterolo LDL è l’impiego dell'equazione di Friedewald:
Colesterolo-LDL calcolato (mg/dL) = (Colesterolo totale - Colesterolo-HDL) - Trigliceridi/5.
L’equazione di Friedewald parte dal presupposto di un rapporto fisso fra colesterolo VLDL e trigliceridi (dove il rapporto è uguale a 5 in caso di misure in mg/dL e a 2.22 (2.17 per alcune fonti come l’Henry's) in caso di misura in mmol/L) e dall'assunto che tutti i trigliceridi siano contenuti nelle VLDL. Entrambi i presupposti non sono perfettamente veri. Sicuramente non sono veri in assenza di un perfetto digiuno per la presenza di chilomicroni e altre particelle lipidiche come residui del lento assorbimento dei grassi da parte dell'intestino (per questo sono consigliate anche 12-14 ore di digiuno prima di un prelievo per la determinazione dell'assetto lipidico). Si deve rilevare che il rapporto è sicuramente diverso quando i trigliceridi superano la concentrazione di 400 mg/dL e in questi casi la formula non è applicabile.
Poiché questa formula produce valori di colesterolo LDL artificialmente bassi anche in presenza di piccole quantità di chilomicroni o di lipoproteine patologiche, durante gli anni ‘90 del secolo scorso alcune aziende e alcuni autori hanno proposto metodi diretti per la determinazione del colesterolo LDL che non richiedono fasi preparatorie o calcoli. In analogia con la determinazione del colesterolo HDL, questi metodi prevedono l'uso di un primo reagente, per “sequestrare” il colesterolo non legato alle lipoproteine LDL, e di un secondo per misurare il colesterolo rimasto disponibile, cioè quello LDL. Uno dei metodi disponibili in commercio (Roche) prevede l'uso di un composto zuccherino per inibire la reazione chimica di HDL, VLDL e chilomicroni e in contemporanea, l'uso di un detergente non ionico per solubilizzare e quindi misurare il colesterolo LDL. I metodi diretti, pur essendo molto precisi, non sono altrettanto accurati e presentano spesso delle discrepanze quando confrontati con il metodo di riferimento dell'ultra-centrifugazione, soprattutto quando la concentrazione di trigliceridi è molto elevata (> 400 mg/dL). I vantaggi rispetto all'equazione di Friedewald sono pertanto modesti, soprattutto considerando il costo molto elevato dei reagenti.
La determinazione enzimatica dei trigliceridi, che impiega una reazione a punto finale secondo Trinder, è stata messa a punto negli anni ‘60 e ‘70 del secolo scorso. Presenta una buona precisione (nel controllo di qualità del CAP i coefficienti di variazione si aggirano attorno al 5-6%), ma ha una tendenza alla sovrastima, in quanto la reazione di base dei diversi metodi messi a punto prevede l'idrolisi dei trigliceridi a glicerolo ed acidi grassi. La misura del glicerolo sarebbe un indicatore fedele della concentrazione dei trigliceridi se si potesse eliminare il glicerolo endogeno, i mono e i di-gliceridi presenti abitualmente in concentrazioni molto piccole. La misura del glicerolo può essere eseguita con lettura all'ultravioletto del NADH prodotto dopo fosforilazione dello stesso e deidrogenazione. Un secondo metodo prevede l'associazione di un cromogeno (formazano), che viene ossidato dal NADH e produce un colore più facilmente misurabile fra 500 e 600 nm. Un terzo applica il metodo di Trinder dopo ossidazione del composto fosforilato e produzione di acqua ossigenata.
L’iniziale reazione di saponificazione alcalina con idrossido di potassio è stata sostituita con un’idrolisi enzimatica, impiegando enzimi come lipasi, una miscela di lipasi e di proteasi, esterasi del fegato combinata con una lipasi particolarmente efficace di Rhizopus arrhizus. Il metodo di Wahlefeld utilizza una lipoprotein-lipasi di microrganismi per l’idrolisi rapida e completa dei trigliceridi a glicerolo, con la conseguente ossidazione a diidrossi-acetone fosfato e a perossido d’idrogeno, che, insieme al 4-aminofenazone e al 4-clorofenolo, provoca una reazione catalizzata dalla perossidasi, formando un colore rosso. L’intensità di colore del colorante rosso formatosi è direttamente proporzionale alla concentrazione di trigliceridi e può essere misurata fotometricamente.
Raccolta dei campioni
I campioni per i lipidi diversi dal colesterolo vanno raccolti a digiuno per evitare l’effetto della dieta. Le decisioni diagnostiche e terapeutiche non vanno prese sulla base di un singolo prelievo per la variabilità biologica relativamente elevata.
Stabilità
Il colesterolo è stabile nel sangue e non richiede precauzioni particolari. Dato che riducendo il colesterolo si riduce l’infiammazione, e uno stato di infiammazione diminuisce il colesterolo, è preferibile evitare di raccogliere campioni durante uno stato infiammatorio acuto (per esempio in caso di ricovero per dolore retro-sternale è meglio misurare il colesterolo al momento del ricovero piuttosto che il giorno successivo quando la concentrazione può essere diminuita in modo significativo e questa diminuzione può durare molti giorni).
| Stabilità | |
| Colesterolo | a 15–25°C: 1 giorno a 2–8°C: 7 giorni a -20°C: 3 mesi |
| Colesterolo HDL e LDL diretto | a 2–8°C: 7 giorni a -70°C: 30 giorni |
| Trigliceridi | a 2–8°C: 7 giorni a -20°C: 3 mesi a -60°C: alcuni anni |
Variabilità
La variabilità inter-individuale è del 22.3%, quella intra-individuale dell’8.2% e quella analitica è intorno al 3%. La differenza critica è intorno al 22%.
Il colesterolo diminuisce nella malattia acuta e cronica, soprattutto negli stati infiammatori e nei periodi di ridotta assunzione calorica. Le concentrazioni di colesterolo aumentano nel corso dell’infanzia e successivamente sono leggermente più alte nelle femmine che nei maschi.
Poiché l’associazione tra rischio cardiovascolare aumentato e colesterolo plasmatico si estende a concentrazioni trovate nei soggetti sani, si impiegano limiti decisionali piuttosto che intervalli di riferimento. Nella tabella sono indicati i limiti più comunemente utilizzati.
| Limiti decisionali (mg/dL) | |
| Colesterolo totale | 199 |
| HDL | 40 |
| LDL | 129 |
| Trigliceridi | 149 |
Bibliografia
- Nauck M, März W, Jarausch J, et al. Multicenter evaluation of a homogeneous assay for HDL-cholesterol without sample pretreatment. Clin Chem 1997, 43: 1622–9.
- Rifai N, Warnick GR, McNamara JR, et al. Measurement of low-density-lipoprotein cholesterol in serum: a status report. Clin Chem 1992, 38: 150–60.
- Nordestgaard BG, Benn M. Fasting and nonfasting LDL cholesterol: to measure or calculate? Clin Chem 2009, 55: 845–7.
- Mora S, Rifai N, Buring JE, Ridker PM. Comparison of LDL cholesterol concentrations by Friedewald calculation and direct measurement in relation to cardiovascular events in 27 331 women. Clin Chem 2009, 55: 888-94.
- Stein EA, Myers GL. National Cholesterol Education Program recommendations for triglycerides measurement: executive summary. Clin Chem 1995, 41: 1421–6.
- McPherson RA, Pincus MR. Henry's clinical diagnosis and management by laboratory methods. 22nd Edition WB Saunders; Philadelphia 2011.
- Khera AV, Mora S. Fasting for lipid testing. Is it worth the trouble? Arch Intern Med 2012, 172: 1710-1.
- Sidhu D, Naugler C. Fasting time and lipid levels in a community-based population. A cross-sectional study. Arch Intern Med 2012, 172: 1707-10.
- Cuhadar S, Atay A, Koseoglu M, et al. Stability studies of common biochemical analytes in serum separator tubes with or without gel barrier subjected to various storage conditions. Biochem Med (Zagreb) 2012, 22: 202-14.
- http://ltd.aruplab.com/tests/pub/0020468
Metodo di determinazione dell'albuminuria
Romolo Dorizzi
Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
Nel presente capitolo sono usati i termine “microalbumina” e “microalbuminuria” che è raccomandato abbandonare. La nomenclatura corretta consiste nell’indicazione dell’analita (albumina) e del sistema nel quale questo viene misurato (urine, U). Il referto deve quindi riportare “U-Albumina” o “Albuminuria” e i prescrittori devono richiedere “albumina urinaria” o “albuminuria”.
Metodi di determinazione
Sono commercializzati numerosi metodi semi-quantitativi per lo screening della microalbuminuria. Si tratta di strisce reattive che sono ottimizzate per dare una lettura “positiva” a una concentrazione pre-determinata di albumina. Per essere utili a questo scopo, devono avere un’elevata sensibilità clinica che, per l’identificazione di un’escrezione > 30 mg/24 ore, varia dal 95% al 65%, a seconda della competenza dell’operatore. La percentuale dei risultati falsi positivi arriva al 20%. I metodi disponibili, che impiegano strisce reattive per la microalbumina, non sembrano essere adatti a programmi di screening nell’ambulatorio del medico di famiglia o al domicilio del soggetto. I metodi adatti allo screening devono avere una bassa percentuale di falsi negativi, in maniera che solo i risultati positivi richiedano conferma con un metodo quantitativo. Per esempio, il metodo Micral (Roche) usa IgG anti-albumina monoclonali complessate a ß-galattosidasi. L’albumina presente nelle urine si lega all’anticorpo coniugato con l’enzima impregnato nella striscia reattiva e migra nella zona di reazione, dove reagisce con un substrato (rosso clorofenolo-galattoside) producendo un colore rosso. L’analisi richiede la semplice immersione della striscia reattiva nelle urine per 5 secondi (tempo critico) e l’intensità del colore, valutata dopo 5 minuti, è proporzionale alla concentrazione dell’albumina.
Tutti i metodi quantitativi per la determinazione della microalbuminuria sono immunometrici e impiegano anticorpi contro l’albumina. Sono disponibili in commercio metodi basati su tre tecnologie: RIA, ELISA, immuno-turbidimetria. I limiti di rivelabilità di tutti sono intorno a 20 mg/L e la maggior parte dei metodi presenta una buona concordanza. Tutte le tecnologie hanno vantaggi e svantaggi, ma quella immunometrica è la più diffusa, perché semplice, automatizzabile, rapida e poco costosa.
Raccolta dei campioni
In passato il campione ideale era considerato quello della raccolta delle 24 ore (che consentiva la possibilità di misurare contemporaneamente la clearance della creatinina). Potevano essere accettati campioni raccolti per intervalli più brevi tenendo conto della notevole variazione circadiana dell’escrezione di albumina (30-50% più bassa durante la notte). Per ovviare alle difficoltà della raccolta, soprattutto nel paziente ambulatoriale (ma non solo), era stato proposto di utilizzare un campione estemporaneo, esprimendo il risultato in mg/L. ADA, National Kidney Foundation (NKF) e JNC 7 raccomandano oggi il primo campione di urina del mattino, sul quale eseguire la misura di albumina e creatinina e l’espressione del risultato come rapporto albumina/creatinina (in questo caso il valore soglia è 30 mg/g di creatinina). Il rapporto ha, infatti, una variabilità biologica (intra-persona) più bassa rispetto a quella presentata da campioni raccolti random nel corso della giornata e non risente dell’eventuale interferenza da attività fisica o da proteinuria posturale intra-persona. Il soggetto deve essere a digiuno e, preferibilmente, non deve avere assunto cibo per almeno due ore.
Stabilità
L’albumina è stabile per 24 ore a temperatura ambiente e alcuni giorni nelle urine non trattate conservate a +4°C.
Se sono necessari periodi più lunghi di conservazione, è necessario congelare a -80°C (la concentrazione di albumina in urine non trattate/centrifugate/filtrate non dimostra alcuna diminuzione dopo sei mesi). Il congelamento a -20°C è sconsigliato, in quanto la concentrazione di albumina in urine non trattate/centrifugate/filtrate diminuisce dello 0.27% al giorno.
Variabilità
La variabilità intra-soggetto dell’escrezione urinaria di albumina (della concentrazione di albumina e del rapporto albumina/creatinina nelle 24 ore) è notevole nei soggetti senza diabete e anche maggiore in quelli con diabete.
Nei soggetti sani, la minore variabilità intra-soggetto dell’albumina (CV del 36%) e del rapporto albumina/creatinina (CV del 31%) si osservano nella prima minzione del mattino. È stato pertanto raccomandato di misurare la concentrazione dell’albumina nelle urine della prima minzione.
Nei pazienti con diabete, il CV intra-soggetto per la concentrazione di albumina nella prima minzione supera il 60% e quello del rapporto albumina/creatinina nello stesso tipo di campione si avvicina al 40%.
Secondo gli standard dell’ADA si può parlare di aumento dell’escrezione dell’albumina o di progressione dell’albuminuria quando la concentrazione è risultata alterata in due campioni su tre raccolti nel corso di 3-6 mesi. Esercizio fisico nelle 24 h precedenti, infezioni, febbre, insufficienza cardiaca, iperglicemia e ipertensione severe possono aumentare l’escrezione di albumina.
| Dosaggio albuminuria | ||||
| Metodologia adottata | Immuno-nefelometria, Immuno-turbidimetria, ELISA, RIA, Point of care Testing | |||
| Campione richiesto | Prima minzione (meno consigliati raccolta 24 ore e raccolta temporizzata) | |||
| Volume minimo | 5 mL | |||
| Stabilità del campione | 24 ore a temperatura ambiente; 3 giorni a 4-8°C; 6 mesi a – 20°C; 12 mesi a – 70°C | |||
| Intervallo di riferimento | mg/24 ore | µg/min | µg/mg creatinina | |
| Normale | < 30 | < 20 | < 30 | |
| Microalbuminuria | 30-300 | 20-200 | 30-300 | |
| Macroalbuminuria | > 300 | > 200 | > 300 | |
Raccomandazioni
- La misurazione annuale della microalbuminuria nei pazienti che non presentano proteinuria clinica deve cominciare nei bambini alla pubertà o dopo 5 anni dalla diagnosi di diabete e nei pazienti con DM-T2 al momento della diagnosi. Livello e qualità di evidenza: B, moderata
- Il CV analitico dei metodi per misurare la microalbuminuria deve essere < 15%. Livello e qualità di evidenza: B, moderata
- Sono accettabili raccolte temporizzate di urine per la determinazione dell’albumina e campioni di urine temporizzate o non temporizzate per la determinazione del rapporto albumina/creatinina. Livello e qualità di evidenza: B, moderata
Bibliografia
- Burtis CA, Ashwood ER, Bruns DE. Tietz textbook of clinical chemistry and molecular diagnostics. Elsevier’s Saunders; St Louis 2006.
- Incerti J, Zelmanovitz T, Camargo JL, et al. Evaluation of tests for microalbuminuria screening in patients with diabetes. Nephrol Dial Transplant 2005, 20: 2402-7.
- Graziani MS, Caldini AL. Indicazioni per la misura dell’albumina nelle urine per l’accertamento e il monitoraggio della nefropatia diabetica. Biochimica Clinica 2011, 35: 127-30.
- Giampietro O, Penno G, Clerico A, et al. How and how long to store urine samples before albumin radioimmunoassay: a practical response. Clin Chem 1993, 39: 533-6.
- KDOQI. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis 2007, 49: S12–154.
- KDOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Part 5. Evaluation of laboratory measurements for clinical assessment of kidney disease. Guideline 5. Assessment of proteinuria. 2000.
- Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension 2003, 42: 1206–52.
- ADA. Standards of medical care in diabetes—2014. Diabetes Care 2014, 37 (suppl 1): S14–80.
Dosaggi relativi all'endocrinologia oncologica
Metodo di determinazione della cromogranina
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
La famiglia delle granine ha tre componenti, che si trovano nelle cellule neuroendocrine di tutto l’organismo:
- cromogranina A (CgA);
- cromogranina B (CgB);
- secretogranine II, III, IV, e V.
CgA è la molecola meglio studiata e quella per la quale si dispone di metodi di dosaggio commerciali - essenzialmente immuno-dosaggi - che utilizzano sia anticorpi policlonali che monoclonali.
È estremamente importante conoscere la specificità dell’antisiero, in quanto CgA è estesamente catabolizzata dopo il rilascio e può rapidamente scomparire dal campione, dando risultati falsi negativi.
Tra i differenti metodi, forse gli IRMA danno la migliore sensibilità e specificità. Sono metodi non competitivi, con un doppio anticorpo diretto contro i frammenti 145-197 e 198-245.
Tra i fattori che possono spiegare le differenze tra metodi si possono elencare:
- la differente sorgente antigenica dell’antisiero di ogni kit;
- le diverse unità di misura in cui viene espresso la concentrazione di CgA (nmol/L, ng/mL, U/L);
- la soglia fissata come limite superiore di riferimento.
Ecco perchè è importante che il laboratorio specifichi nella risposta il metodo utilizzato e perchè è fortemente raccomandato di utilizzare lo stesso laboratorio per dosaggi seriati nel tempo.
Può essere dosata indifferentemente nel siero o nel plasma.
Ci sono diverse cause in grado di provocare falsi positivi: esercizio fisico estremo, farmaci (soprattutto inibitori di pompa protonica e anti-H2) e patologie (insufficienza renale, insufficienza cardiaca, ipertensione, iperparatiroidismo, gastrite atrofica, cirrosi, epatiti, pancreatite, artrite reumatoide).
Bibliografia
- Oberg K. Circulating biomarkers in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer 2011, 18: 17-25.
- Nehar D, Lombard-Bohas C, Olivieri S, et al. Interest of chromogranin A for diagnosis and follow-up of endocrine tumours. Clin Endocrinol (Oxf) 2004, 60: 644-52.
- Bilek R, Safarik L, Ciprová V, et al. Chromogranin A, a member of neuroendocrine secretory proteins as a selective marker for laboratory diagnosis of pheochromocytoma. Physiol Res 2008, 57 suppl 1: 171-9.
- Welin S, Stridsberg M, Cunningham J, et al. Elevated plasma chromogranin A is the first indication of recurrence in radically operated midgut carcinoid tumors. Neuroendocrinology 2009, 89: 302-7.
Metodo di determinazione della gastrina
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
L’ormone maturo è una catena di 34 amino acidi (gastrina-34), sintetizzato dalle cellule G della mucosa dell’antro come pre-pro-ormone, che viene ulteriormente clivata a una molecola più corta, gastrina-17, che è la forma biologicamente più attiva dell’ormone. In circolo si possono trovare ulteriori frammenti minori con modificazioni post-traduzionali, che sono in grado di interferire con i metodi di dosaggio comunemente in commercio. L’emivita di gastrina-17 è più breve di gastrina-34 (5’ vs 25’) e l’eliminazione è renale.
Dal punto di vista analitico è estremamente importante sapere quanto il metodo utilizzato è specifico per la forma bioattiva dell’ormone e quante reazioni crociate può esibire, ed è utile che il laboratorio che esegue il dosaggio ne specifichi la denominazione nella risposta.
In commercio esistono diversi immuno-dosaggi basati su differenti principi di rivelazione (RIA, EIA, CLIA) e con specificità anticorpale molto diversa. Il risultato di un singolo dosaggio deve essere interpretato alla luce del quadro clinico complessivo e, in particolare, del pH gastrico e delle misure di secrezione acida.
Il campione da utilizzare è il siero. Il prelievo va effettuato a digiuno al mattino (essenziale, altrimenti il dato non è interpretabile), e possibilmente in wash-out farmacologico, dato che l’azione dei farmaci soppressori di secrezione gastrica causa valori falsamente elevati dell’ormone. La molecola è instabile e il campione deve pervenire al laboratorio nel più breve tempo possibile. Essendo la gastrina eliminata dai reni, nei pazienti in insufficienza renale può risultare una pseudo iper-concentrazione. Gli intervalli di riferimento (di solito < 100 pg/mL) non differiscono in base all’età.
Bibliografia
- Rehfeld JF, Gingras MH, Bardram l, et al. The Zollinger–Ellison syndrome and mismeasurement of gastrin. Gastroenterology 2011, 140: 1444–53.
- Jais P, Mignon M. Normal serum gastrin concentration in gastrinoma. Lancet 1995, 2: 1421–2.
- Wolfe MM, Paquet RJ, Reel GM. Specificity of commercially available antibodies used for gastrin measurement. J Lab Clin Med 1985, 105: 417–21.
- Metz DC. Diagnosis of the Zollinger–Ellison syndrome. Clin Gastroenterol Hepatol 2012, 10: 126-30.
Metodo di determinazione dell'acido 5-OH-indolacetico
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
I metodi di determinazione dell’acido 5-idrossi-indolacetico (5-HIAA) sono oggi più sensibili e specifici che nel passato. La cromatografia liquida ad alta pressione (HPLC) è il metodo più diffuso nei laboratori clinici, ed è in grado di determinare contemporaneamente altre sostanze di interesse clinico sullo stesso campione. Alternative all’HPLC includono gli immuno-dosaggi e la spettrometria di massa. Quando si tratta di rilevare basse concentrazioni di HIAA si preferisce di solito la rilevazione elettrochimica con misure amperometriche. Il potenziale di ossidazione è < 0.6 V e sono poche le sostanze in grado di interferire a questi potenziali. Di solito si procede a un’estrazione con solventi organici o resine a scambio anionico per purificare il campione, e sono descritti metodi automatizzati che incorporano un’estrazione in fase solida per velocizzare le procedure. In molti sistemi è prevista l’iniezione diretta del campione di urina sulla colonna analitica. In questi casi il campione è solo diluito con tampone e centrifugato per limitare la contaminazione del sistema HPLC.
Esistono infine metodi per la determinazione simultanea di HIAA, acido vanil-mandelico e HMV. Si tratta di sistemi automatizzati, che eliminano le interferenze cromatografiche e riducono i tempi di analisi (2-6’ a campione).
Le condizioni per la raccolta e la conservazione dei campioni sono di particolare importanza. Si raccomanda la raccolta di due campioni di urine 24 h, perché la concentrazione in urine spot è estremamente variabile. In alternativa si potrebbero utilizzare raccolte temporizzate (più brevi) riferite all’escrezione di creatinina. Il contenitore deve essere opaco alla luce, perché il composto è fotosensibile. Inoltre, essendo instabile in ambiente alcalino, è necessario acidificare le urine e usare preservativi stabilizzanti. I campioni di urine acidificate possono essere conservati per 2–4 settimane a temperatura di frigorifero, e per periodi più lunghi a – 20°C.
Importante anche seguire una dieta per i giorni precedenti e nel giorno di raccolta: vanno limitati gli alimenti fonti di 5-OH-indolo (avocado, ananas, banane, prugne, pomodori). Inoltre, se possibile, vanno sospesi i farmaci che aumentano o diminuiscono la concentrazione di HIAA (fenacetina, acetaminofene, benzodiazepine, levoDOPA).
Bibliografia
- Deacon AC. The measurement of 5-hydroxyindoleacetic acid in urine. Ann Clin Biochem 1994, 31: 215-32.
- Lionetto L, Lostia AM, Stigliano A, et al. HPLC-mass spectrometry method for quantitative detection of neuroendocrine tumor markers: vanillylmandelic acid, homovanillic acid and 5-hydroxyindoleacetic acid. Clin Chim Acta 2008, 398: 53-6.
- Mills KC. Serotonin syndrome: a clinical update. Crit Care Clin 1997, 13: 763-83.
Metodo di determinazione di VIP e glucagone
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
Metodi di dosaggio del glucagone
È un polipeptide di 29 amino acidi a catena singola, secreto come pro-ormone da ipotalamo e cellule alfa delle isole pancreatiche.
La molecola in circolo è estremamente labile e il prelievo deve essere eseguito rispettando rigide precauzioni per evitarne il degrado in vitro. Sono considerazioni importanti, dal momento che le informazioni cliniche derivanti da questo dosaggio possono essere rilevanti solo in un contesto clinico sufficientemente suggestivo di ipersecrezione non fisiologica.
Si utilizzano immuno-dosaggi (RIA ed EIA). La marcata variabilità inter-assay dipende dalle differenti preparazioni di anti-siero utilizzate e dalla mancata standardizzazione del dosaggio, pertanto risultati dello stesso campione in laboratori differenti sono estremamente difficili da confrontare.
Il campione più utilizzato è plasma in EDTA, con aggiunto un inibitore della proteolisi. È raccomandato il prelievo in provetta pre-raffreddata, l’immediata centrifugazione a 2-4°C e il congelamento del plasma separato.
Come tutti gli immuno-dosaggi, anche questo può subire interferenze non sempre prevedibili o evidenziabili. È indispensabile che risultati non attesi rispetto al quadro clinico che ha indotto la richiesta vengano discussi con personale competente del laboratorio che ha eseguito l’esame.
Bibliografia
- Vinik AI, Woltering EA, O’Dorisio TM, et al. Neuroendocrine tumors: a comprehensive guide to diagnosis and management. 5th ed. InterScience Institute, Inglewood (CA), 2012.
- Sacks DB. Diabetes mellitus. In: Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 5th ed. 2012: 1227-8.
Metodi di dosaggio del VIP
Il VIP è un neuro-modulatore più che un ormone, responsabile di una ben definita sindrome clinica che configura un quadro caratteristico di una neoplasia neuroendocrina non frequentemente riscontrata.
Per il dosaggio si utilizzano immuno-assay, essenzialmente RIA.
Il campione è costituito da plasma in EDTA, eventualmente con aggiunta di aprotinina come anti-proteolitico. Il dosaggio non è effettuabile in pazienti che abbiano da poco ricevuto radionuclidi, per le interferenze che rendono non interpretabile il risultato. È indispensabile che risultati non attesi rispetto al quadro clinico che ha indotto la richiesta vengano discussi con personale competente del laboratorio che ha eseguito l’esame.
Bibliografia
- Ghaferi AA, Chojnacki KA, Long WD, et al. Pancreatic VIPomas: subject review and one institutional experience. J Gastrointest Surg 2008, 12: 382-93.
- Salwen MJ, Siddiqi HA, Gress FG, et al. Laboratory diagnosis of gastrointestinal and pancreatic disorders. In: Henry's Clinical Diagnosis and Management by Laboratory Methods, 22nd Ed, 2011: 319-20.
Metodo di determinazione del sodio
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
Anche se esistono altre tecniche di determinazione, tra cui l’assorbimento atomico o la spettrometria da emissione di fiamma, che oggi rivestono interesse storico o sono adoperate in laboratori di ricerca, oggi la maggior parte dei laboratori clinici utilizza metodi di dosaggio basati sull’impiego di elettrodi ione-selettivi (ISE).
Bisogna distinguere due tipologie di metodi ISE:
- i metodi indiretti misurano la concentrazione ionica dopo aver diluito il campione. Questi sono i metodi utilizzati di preferenza sulle piattaforme automatizzate di più frequente impiego nei laboratori generali;
- nei metodi diretti, invece, il campione è presentato all’elettrodo senza preventiva diluizione. Questa tecnologia è montata di preferenza sugli strumenti per emogasanalisi e su quelli per Point of Care Testing (POCT). Tale applicazione è stata introdotta solo dopo che si sono resi disponibili elettrodi miniaturizzati.
I metodi indiretti misurano la concentrazione elettrolitica nel volume plasmatico totale, mentre i metodi diretti lo fanno sulla sola componente plasmatica acquosa, che riflette la frazione fisiologicamente attiva. Molti laboratoristi e clinici ritengono che il metodo di scelta sia quest’ultimo, anche se nella pratica i valori ottenuti con metodi diretti vengono comunque riportati alla frazione plasmatica totale. È un bene che sia così, perchè oggi la maggior parte dei laboratori impiega metodi indiretti per la misura degli ioni, ma in condizioni di alterata concentrazione proteica o lipidica (per niente rare oggi in clinica) il ricorso a metodi diretti è la scelta più oculata.
Sono utilizzabili siero, plasma e urine. Se non testati subito, i campioni possono essere conservati in frigorifero o congelati. Contrariamente al Potassio, il Sodio è dieci volte più concentrato nel plasma che all’interno dei globuli rossi; pertanto l’emolisi non interferisce normalmente nella determinazione della sodiemia. I campioni lipemici invece andrebbero chiarificati mediante ultra-centrifugazione e si dovrebbe utilizzare l’infra-natante; in alternativa, andrebbero impiegati metodi ISE diretti.
Per dosaggi fecali vanno prese in considerazione solo le emissioni liquide, perchè solo le diarree profuse sono in grado di indurre una perdita significativa di sodio.
Bibliografia
- Apple FS, Koch DD, Graves S, et al. Relationship between direct-potentiometric and flame-photometric measurement of sodium in blood. Clin Chem 1982, 28: 1931-5.
- Dimeski G, Barnett RJ. Effects of total plasma protein concentration on plasma sodium, potassium and chloride measurements by an indirect ion selective electrode measuring system. Crit Care Resusc 2005, 7: 12-5.
- Tietz NW. Clinical guide to laboratory tests. WB Saunders, Philadelphia, PA, 2005.
Metodo di determinazione del potassio
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 24 novembre 2015)
Anche se esistono altre tecniche di determinazione, tra cui l’assorbimento atomico o la spettrometria da emissione di fiamma, che oggi rivestono interesse storico o sono adoperate in laboratori di ricerca, oggi la maggior parte dei laboratori clinici utilizza metodi di dosaggio basati sull’impiego di elettrodi ione-selettivi (ISE).
Bisogna distinguere due tipologie di metodi ISE:
- i metodi indiretti misurano la concentrazione ionica dopo aver diluito il campione. Questi sono i metodi utilizzati di preferenza sulle piattaforme automatizzate di più frequente impiego nei laboratori generali;
- nei metodi diretti, invece, il campione è presentato all’elettrodo senza preventiva diluizione. Questa tecnologia è montata di preferenza sugli strumenti per emogasanalisi e su quelli per Point of Care Testing (POCT). Tale applicazione è stata introdotta solo dopo che si sono resi disponibili elettrodi miniaturizzati.
I metodi indiretti misurano la concentrazione elettrolitica nel volume plasmatico totale, mentre i metodi diretti lo fanno sulla sola componente plasmatica acquosa, che riflette la frazione fisiologicamente attiva. Molti laboratoristi e clinici ritengono che il metodo di scelta sia quest’ultimo, anche se nella pratica i valori ottenuti con metodi diretti vengono comunque riportati alla frazione plasmatica totale. È un bene che sia così, perchè oggi la maggior parte dei laboratori impiega metodi indiretti per la misura degli ioni, ma in condizioni di alterata concentrazione proteica o lipidica (per niente rare oggi in clinica) il ricorso a metodi diretti è la scelta più oculata.
Il plasma è la matrice preferita per il dosaggio ematico del potassio, in quanto la concentrazione di questo ione nel siero risulta artificialmente aumentata a causa della liberazione di potassio dalle piastrine nel processo di coagulazione. Se non testati subito, i campioni possono essere conservati in frigorifero o congelati. Nel prelievo di campioni per la determinazione di K bisogna avere assoluta cura di evitare l’emolisi che aumenta la concentrazione di K:
- leggera emolisi (Hb ≈ 50 mg/dL) del ≈ 3%;
- emolisi marcata (Hb ≈ 200 mg/dL) del 12%;
- emolisi massiccia (Hb > 500 mg/dL) di oltre il 30%.
Nessuna correzione matematica è raccomandata come affidabile. Particolare attenzione deve essere posta nelle misure di K con strumenti per POCT o emogasanalisi: se si sospetta una pseudo-iperkaliemia, il campione di sangue intero deve essere centrifugato e ispezionato visivamente per la presenza di emolisi. Un effetto opposto di pseudo-ipokaliemia è possibile in campioni conservati a caldo e in presenza di leucocitosi. La glicolisi accelerata provoca un flusso di ioni K dentro le cellule, risultando in una falsa diminuzione della concentrazione plasmatica.
Bibliografia
- Apple FS, Koch DD, Graves S, et al. Relationship between direct-potentiometric and flame-photometric measurement of sodium in blood. Clin Chem 1982, 28: 1931-5.
- Brydon WG, Roberts LB. The effect of haemolysis on the determination of plasma constituents. Clin Chim Acta 1972, 41: 435-8.
- Dimeski G, Barnett RJ. Effects of total plasma protein concentration on plasma sodium, potassium and chloride measurements by an indirect ion selective electrode measuring system. Crit Care Resusc 2005, 7: 12-5.
- Tietz NW. Clinical guide to laboratory tests. WB Saunders, Philadelphia, PA, 2005.