Metodo di determinazione della copeptina

Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento marzo 2023)
La scelta di misurare la copeptina al posto di AVP è dovuta alla sua maggiore praticabilità in termini di stabilità e praticità operativa, oltre alla comprovata affidabilità come complemento alla diagnostica delle alterazioni del bilancio dell’acqua.
I metodi più diffusi sono quelli in immuno-fluorescenza omogenea automatizzata, che utilizzano un doppio tracciante fluorescente. L’intensità del segnale è direttamente proporzionale alla concentrazione di copeptina nel campione.
Possibili interferenze:
- sepsi, shock settico, infezioni bronchiali, BPCO e cardio-vasculopatie intercorrenti possono indurre aumenti aspecifici di concentrazione ematica della copeptina;
- terapie con antagonisti della vasopressina, altre patologie che coinvolgano la secrezione di vasopressina, inducono un aumento di concentrazione ematica di copeptina;
- sono stati riportati casi di aumenti di copeptina ectopica prodotta da carcinomi bronchiali;
- l’interpretazione del dato in caso di diabete insipido centrale o periferico può essere complicata dall’incompleta espressione clinica della patologia;
- anticorpi eterofili (specialmente anti-topo) possono produrre risultati inaccurati per sovrastima o sottostima. Il laboratorio dovrebbe essere avvertito in caso di discrepanza del risultato con il quadro clinico.
Campione: plasma in EDTA (tappo lilla). Campioni emolizzati non sono accettabili.
Preparazione paziente: è indicato il digiuno e l’astensione da liquidi, inclusa acqua, nelle 8 ore precedenti il prelievo.
Conservazione: stabile 7 giorni a 4-8°C, dopo va congelato.
Riferimenti
Introduzione alla metodologia epidemiologica e statistica
La variabilità degli esami: precisione e accuratezza
La valutazione degli esami diagnostici: sensibilità e specificità
Incidenza e prevalenza di malattia
La variabilità degli esami: precisione e accuratezza
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
Il medico di laboratorio ha cura di scegliere la strumentazione e i metodi più validi, fa in modo che le fasi pre-analitiche (di preparazione del paziente e raccolta e gestione del campione) e di refertazione siano adeguate, e ottimizza l’accuratezza e la precisione dei risultati.
Un metodo si definisce “accurato” quando il risultato è prossimo al cosiddetto “valore vero”, “preciso” quando produce risultati riproducibili.
I risultati analitici sono tuttavia soggetti a errore, indipendentemente dalla qualità del laboratorio e dall’abilità di chi vi lavora. Gli aspetti quantitativi della mancanza di precisione meritano di essere chiariti. Ad esempio, se misuriamo molte volte la concentrazione di ACTH in un campione di plasma con due metodi diversi, uno molto preciso ed uno poco preciso, l’istogramma della distribuzione dei risultati avrà in entrambi i casi un aspetto (normale) gaussiano, ma nel caso del metodo preciso i risultati saranno più concentrati intorno alla concentrazione media. La deviazione standard (DS) è la misura di questa distribuzione intorno al valore medio: la DS è grande se la distribuzione è ampia ed è piccola se la distribuzione è stretta. Per i dati che hanno distribuzione gaussiana, come avviene nel caso degli errori analitici, l’aspetto della curva è completamente definito dalla media e dalla DS. In particolare, si verifica che:
- circa il 67% dei risultati è compreso nell’ambito definito da media ± 1 DS
- circa il 95% dei risultati è compreso nell’ambito definito da media ± 2 DS
- circa il 99% dei risultati è compreso nell’ambito definito da media ± 3 DS.
La figura mostra (in alto a sinistra) un esame accurato, che si avvicina cioè al valore vero, ma è scarsamente riproducibile (i risultati cioè si disperdono intorno al risultato vero), e (i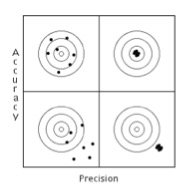 n basso a destra) un esame preciso, in cui i punti si disperdono in un’area molto stretta, ma molto distante dal valore vero (l’esame è poco accurato); in alto a destra un esame accurato e preciso e in basso a sinistra un esame inaccurato e impreciso.
n basso a destra) un esame preciso, in cui i punti si disperdono in un’area molto stretta, ma molto distante dal valore vero (l’esame è poco accurato); in alto a destra un esame accurato e preciso e in basso a sinistra un esame inaccurato e impreciso.
Poiché spesso gli esami ormonali non possiedono entrambe le caratteristiche ideali di accuratezza e precisione, è opportuno impiegare esami accurati per la diagnosi ed esami precisi per il monitoraggio (un esempio potrebbe essere rappresentato dal dosaggio del paratormone nella diagnosi di iperparatiroidismo primario e dalla determinazione della calcemia nel follow-up del paziente dopo paratiroidectomia).
È importante che il medico di laboratorio comunichi ai clinici i dati relativi all’imprecisione analitica dei diversi esami che sono a loro disposizione. Infatti, la tecnologia e l’elettronica hanno consentito progressi nell’automazione, rapidità e riproducibilità degli esami di laboratorio (soprattutto per gli esami immunometrici) ma non sempre nella loro accuratezza.
Mentre le fasi analitiche sono automatizzate, facilmente controllabili e riproducibili (la quota degli errori analitici si aggira consistentemente intorno al 10% di tutti gli errori), le fasi a carico dell’operatore umano sono suscettibili di molte possibilità di errore ed è in questa direzione che devono indirizzarsi gli sforzi per il miglioramento della qualità degli esami endocrinologici.
Sempre maggiore importanza viene data alle cause biologiche di variazione dei risultati. Infatti, la concentrazione di ogni analita, indipendentemente dalla variabilità analitica, non rimane assolutamente costante nel tempo, nello stato di salute e di malattia, ma oscilla intorno ad un punto omeostatico che è specifico del soggetto o generale. Non tutte le cause di questa variabilità sono note ed in alcuni casi tale variabilità può essere molto rilevante.
Tra le cause di variabilità intra-individuale possono essere ricordate:
- l’età, che può influenzare la concentrazione di un costituente del sangue nei primi giorni di vita, durante l’adolescenza e nell’età avanzata;
- la dieta, che influenza molti esami (tra i più classici la curva da carico di glucosio, l’escrezione urinaria di idrossiprolina e di calcio). Vi sono alimenti comuni che hanno effetti particolari e rilevanti. La caffeina, per esempio, aumenta la concentrazione di glucosio e altera la tolleranza al carico di glucosio, riduce il ritmo circadiano del cortisolo, aumenta la concentrazione di sodio, potassio, calcio e magnesio nelle urine;
- le caratteristiche secretorie specifiche dei diversi ormoni, quali il ritmo circadiano che determina concentrazioni diverse nel corso della giornata particolarmente rilevanti per ACTH e cortisolo, e i picchi secretori per il GH;
- la postura: passare dalla posizione supina a quella eretta porta ad una riduzione del volume di sangue del 10%, con conseguente aumento della concentrazione di proteine, enzimi ed ormoni proteici, poiché solo il fluido privo di proteine passa nei tessuti;
- il fumo, attraverso l’azione della nicotina, può influenzare molti esami di laboratorio in modo proporzionato al numero di sigarette fumate. La concentrazione di glucosio plasmatico può aumentare di 10 mg/dL (0.56 mmol/L) entro 10 minuti dal momento in cui si è fumato; tale effetto può protrarsi per un’ora. I fumatori tendono avere una concentrazione di glucosio più alta rispetto ai non fumatori, con un’alterata tolleranza glicemica. Il GH, inoltre, può aumentare anche 10 volte nella mezz’ora successiva al momento in cui si è fumata una sigaretta.
La valutazione degli esami diagnostici: sensibilità e specificità
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
Gli esami di laboratorio non dovrebbero esistere isolati dal contesto clinico. Infatti, nel momento in cui essi sono richiesti, il clinico ha già fatto una ipotesi provvisoria o ha approntato una lista di ipotesi diagnostiche basate sui sintomi e sui segni di ogni paziente.
Per valutare e interpretare un esame diagnostico è necessario conoscere come si comporta nel “malato” e nel “sano”. L’esame ideale dovrebbe essere anomalo nel 100% dei malati e non essere anomalo nel 100% dei soggetti sani. Questo ideale è raggiunto molto di rado; generalmente si verifica una sovrapposizione tra i due gruppi di soggetti. È pertanto utile ricordare alcune definizione di carattere epidemiologico che sono utili nel definire le capacità diagnostiche di un test.
- Sensibilità (percentuale dei risultati veri positivi) è la percentuale di risultati positivi di un esame nei pazienti affetti da una malattia. Un esame che è sempre anomalo (o positivo) nei pazienti malati ha una sensibilità del 100%.
- Specificità (percentuale dei risultati veri negativi) è la percentuale dei risultati negativi di un esame nei pazienti non affetti da malattia. Un esame che è sempre negativo nei soggetti sani ha una specificità del 100%.
- Percentuale di falsi negativi è la percentuale di risultati negativi nei pazienti affetti da malattia.
- Percentuale di falsi positivi è la percentuale di risultati positivi di un esame nei pazienti non affetti da una particolare malattia.
Incidenza e prevalenza di malattia
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
Prima di considerare l’effetto della prevalenza della malattia sui processi decisionali diagnostici, è necessario considerare il valore predittivo di un risultato positivo e di un risultato negativo. In pratica si esprime in tal modo la probabilità che un risultato anomalo sia interpretato dal clinico come proprio di un soggetto malato.
- Valore predittivo positivo di un test (VPP) esprime la probabilità che un soggetto con risultato positivo al test sia realmente affetto da malattia. Esso dipende dalla specificità e dalla prevalenza della malattia nella popolazione a cui viene applicato un test: tanto più alte sono la prevalenza e la specificità e tanto maggiore sarà il valore predittivo positivo di un test.
- Valore predittivo negativo di un test (VPN) esprime la probabilità che un soggetto con risultato negativo al test sia realmente sano. Esso dipende dalla sensibilità e dalla prevalenza della malattia nella popolazione a cui viene applicato un test: tanto più alta è la sensibilità e tanto minore è la prevalenza e tanto maggiore sarà il valore predittivo negativo di un test.
Statisticamente la probabilità di essere affetto da malattia o di non esserlo, a seconda che il test sia risultato positivo o negativo, è espressa dal teorema di Bayes. Tale teorema, pur non essendo particolarmente complicato da un punto di visto matematico, può essere applicato in modo intuitivo. Si consideri un test destinato a rivelare la presenza nel siero di anticorpi anti-HIV. Si assuma che questo test abbia una sensibilità del 100% (il test è quindi positivo nel 100% dei malati). Si assuma che il test abbia una specificità del 99.7% (il test, quindi, è negativo nel 99.7% dei soggetti sani). Si sa che la prevalenza della positività agli anticorpi anti-HIV è del 3 per mille nella popolazione generale. Qualora noi applicassimo il test a 1000 soggetti presi a caso, si avranno i seguenti risultati: 3 soggetti con positività del test, perché la prevalenza dei soggetti positivi è del 3 su mille (veri positivi), e 3 soggetti falsamente positivi, poiché, essendo la specificità del 99.7%, ci dovremo aspettare 3 positivi nei 997 soggetti sani del campione scelto a caso. In totale i soggetti positivi saranno 6, di cui solo 3 veramente positivi, con un VPP del 50%, nonostante i valori di sensibilità e di specificità siano particolarmente elevati. Per cercare di esplicitare l’influenza determinante della prevalenza della malattia nel campione di soggetti analizzato, immaginiamo ora le categorie a rischio per positività degli Ab anti-HIV. Tra gli omosessuali con partner occasionali la prevalenza di positività agli Ab anti-HIV è di circa il 10%. Questo significa che testando 1000 soggetti omosessuali promiscui avremo circa 103 pazienti con positività al test (100 dovuti alla prevalenza del 10% e 3 dovuti alla specificità del 99.7%), con un valore predittivo positivo del 97%. L’esempio consente di sottolineare come anche test considerati affidabili, per fornire un risultato significativo, devono essere effettuati dopo un attento ragionamento clinico.
L’intervallo di riferimento
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
I risultati degli esami di laboratorio sono accompagnati dai cosiddetti "intervalli di riferimento" (IR), che sono limiti con cui il clinico confronta il valore misurato nel singolo soggetto.
L’intervallo di riferimento è ottenuto misurando un particolare analita in un campione di individui estratto da una popolazione di riferimento. Gli individui di riferimento non sono necessariamente sani, ma semplicemente individui selezionati utilizzando criteri ben definiti. La valutazione della variabile nel campione di riferimento fornisce valori numerici denominati limiti di riferimento. Dal punto di vista pratico si devono selezionare in modo casuale gli individui dalla popolazione generale utilizzando dei criteri predefiniti che possono tenere conto, a seconda dei casi, di sesso, età, assunzione o meno di farmaci ed, eventualmente, dei risultati di accertamenti diagnostici di tipo generale.
Quando i risultati ottenuti nel gruppo di riferimento sono analizzati, essi si distribuiscono intorno a un valore centrale con una distribuzione che può essere simmetrica (spesso gaussiana) o più frequentemente asimmetrica (con coda a destra). Il metodo più utilizzato per scegliere l’intervallo di riferimento da questi risultati è quello di calcolare il 95% centrale della distribuzione. L’intervallo comprende cioè la popolazione compresa tra il percentile 2.5 e il 97.5. Per le variabili in cui si ha un eccesso di asimmetria con una coda importante a destra, la definizione dell’intervallo di riferimento prevede la trasformazione logaritmica dei risultati, al fine di rendere più simmetrica (gaussiana) la distribuzione dei valori.
Le caratteristiche analitiche di accuratezza e di precisione influenzano l’intervallo di riferimento: se viene usato un metodo non accurato, l’intervallo di riferimento rifletterà lo scostamento del metodo, mentre se un metodo non è preciso, l’intervallo di riferimento risulterà allargato (aumenterà la DS della distribuzione dei risultati).
L’intervallo andrebbe accompagnato sempre da un indice della variabilità campionaria della stima, il cosiddetto intervallo di confidenza, espresso di solito al 90% (vengono indicati i due valori entro cui vi è una probabilità del 90% che vi sia il limite di riferimento superiore ed inferiore).
Gli IR sono spesso utilizzati sia dal clinico che dal paziente come limiti in grado di discriminare il soggetto sano da quello malato. Si sta affermando, tuttavia, il concetto che, nonostante possano aiutare a individuare lo stato di benessere, siano spesso poco efficienti, o addirittura fuorvianti, per diagnosticare una condizione di malattia. È importante distinguere valori di riferimento cumulativi, valori di riferimento individuali e limiti decisionali (1,2).
Valori di riferimento cumulativi: il lavoro che il Committee on Reference Values della International Federation of Clinical Chemistry ha compiuto tra il 1978 ed il 1988 ha portato alla sistematizzazione della "Teoria degli Intervalli di riferimento", che ha codificato in modo preciso come calcolare gli IR (compresi in genere tra i percentili 2.5 e 97.5). Si tratta di un lavoro fondamentale dal punto di vista teorico, ma che presenta difficoltà di applicazione pressoché insormontabili nel mondo reale. Il clinico (l'endocrinologo in particolare) si trova quotidianamente di fronte alla dicotomia tra quanto è descritto in letteratura e quanto è riportato sui referti e si trova in difficoltà nell'interpretare risultati provenienti da laboratori distanti pochi chilometri che impiegano diverse nomenclature, IR e unità di misura. È fondamentale osservare come molti degli esami di laboratorio abbiano perduto la loro natura di essere diagnostici per acquisire quella di escludere, quando negativi, lo stato di malattia.
Valori di riferimento individuali: spesso l'IR calcolato nella popolazione generale è molto più ampio di quello proprio del singolo individuo, per esempio, per gli ormoni tiroidei e per il TSH (3). Variazioni nel tempo di tali ormoni possono essere clinicamente rilevanti, pur non superando i limiti di riferimento. Purtroppo, i valori di riferimento individuali non sono utilizzabili nella pratica clinica.
Limiti decisionali (o soglie decisionali, cut-off): si tratta di un concetto proposto nel 1968, che indica valori soglia, sopra o sotto ai quali il clinico prende una determinata decisione o tiene un determinato comportamento clinico. Si tratta di limiti empirici, che dipendono fortemente dal tipo di ormone e dal contesto in cui opera l'endocrinologo e sono più vicini alla diagnosi rispetto all'IR. Hanno finalità pratiche: a seconda che la concentrazione dell’analita misurata nel paziente sia sopra o sotto questi livelli, il clinico prende decisioni cliniche importanti (iniziare o sospendere una terapia, fare altre indagini diagnostiche, disporre il ricovero, programmare un intervento, dimettere il paziente). Richiedono una stretta collaborazione tra clinico e laboratorio: se il laboratorio cambia il metodo analitico senza comunicarlo tempestivamente al clinico, vi possono essere gravi conseguenze.
In Endocrinologia è particolarmente importante che nel referto sia indicato il metodo impiegato per determinare un ormone e che sia evitato l'uso di intervalli ricavati dalla letteratura. Queste considerazioni sono valide per tutti gli ormoni e i casi di calcitonina, tireoglobulina e testosterone sono solo esempi paradigmatici.
- Calcitonina (CT): il limite decisionale di 10 ng/L della CT risulta abbastanza valido con molti metodi nel maschio, mentre nella femmina produce numerosi errori di classificazione. Gli effetti di sesso, fumo (che aumenta i valori ottenuti con alcuni metodi) e massa corporea sulla concentrazione della CT dovrebbero essere, quindi, comunicati dal laboratorio (4).
- Tireoglobulina (Tg): Spencer et al. hanno misurato la Tg con 14 metodi diversi e gli anticorpi anti-Tg con 12 metodi diversi in 110 campioni e solo nel 10% dei 42 campioni studiati tutti i metodi hanno rilevato gli anticorpi anti-Tg (5,6). In molti campioni positivi per anticorpi anti-Tg tutti i metodi automatici in chemiluminescenza presentavano concentrazioni indosabili di Tg, suggerendo la presenza di interferenza, mentre tutti i metodi RIA fornivano risultati meno influenzati dalla presenza di anticorpi anti-Tg. Questo rappresenta uno dei casi più clamorosi in cui il clinico non può interpretare correttamente i risultati se non riceve da parte del laboratorio informazioni circa il metodo analitico usato sia per la Tg che per gli anticorpi anti-Tg.
- Testosterone (TT): anche se esiste la tecnologia per misurare in modo preciso, accurato e riproducibile il testosterone, la gascromatografia accoppiata a spettrometria di massa, i laboratori misurano testosterone totale e libero impiegando tecniche molto più inaccurate anche se semplici (7,8). L'intervallo nei maschi adulti è l'unico definito con una certa affidabilità: concentrazioni > 320 ng/dL sono compatibili con eugonadismo, mentre valori < 200 ng/dL con ipogonadismo. Gli intervalli nella donna adulta risentono non solo del metodo usato, ma anche della fase del ciclo mestruale, dell'età e del BMI. La determinazione del testosterone nel maschio in età avanzata e nel bambino è attendibile solo se l'analisi è eseguita in spettrometria di massa o in RIA dopo estrazione in cromatografia. In realtà, gli IR sono stati ottenuti in molti casi in popolazioni limitate e inadeguate (poche decine di soggetti di una fascia di età molto ampia, senza tener conto di ciclo mestruale, etnia e BMI). Nel 2010 è partito il programma HoST (Hormone Standardization), a cura del CDC di Atlanta e con la partecipazione tra gli altri della Endocrine Society e della American Association for Clinical Chemistry. L’obiettivo di questo progetto pluriennale (9) era quello di ottenere una standardizzazione effettiva dei metodi di dosaggio ormonale “critici”, in particolare quelli per testosterone totale nella donna e nel bambino e per l’estradiolo nell’uomo. I primi risultati sono oltremodo interessanti e lasciano sperare che i metodi immunometrici diretti possano essere affidabilmente utilizzati nella pratica clinica evitando il ricorso obbligato ai metodi estrattivi.
I clinici devono comunque sempre tenere ben presenti e conoscere le insidie presenti nella interpretazione degli IR usati nei referti degli ormoni.
Bibliografia
- Henny J. Interpretation of laboratory results: the Reference Intervals, a necessary evil? Clin Chem Lab Med 2007, 45: 939-41.
- Giavarina D. Gli intervalli di riferimento. RIMeL/IJLaM 2006, 2: 50-6.
- Andersen S, Bruun NH, Pedersen KM, Laurberg P. Biologic variation is important for interpretation of thyroid function tests. Thyroid 2003, 13: 1069-78.
- D'Herbomez M, Caron P, Bauters C, et al. Reference range of serum calcitonin levels in humans: influence of calcitonin assays, sex, age, and cigarette smoking. Eur J Endocrinol 2007, 157: 749-55.
- Spencer CA, Bergoglio LM, Kazarosyan M, et al. Clinical impact of thyroglobulin (Tg) and Tg autoantibody method differences on the management of patients with differentiated thyroid carcinomas. J Clin Endocrinol Metab 2005, 90: 5566-75.
- Schlumberger M, Hitzel A, Toubert ME, et al. Comparison of seven serum thyroglobulin assays in the follow-up of papillary and follicular thyroid cancer patients. J Clin Endocrinol Metab 2007, 92: 2487-95.
- Rosner W, Auchus RJ, Azziz R, et al. Utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007, 92: 405-13.
- Fritz KS, McKean AJS, Nelson JC, Wilcox RB. Analog-based free testosterone test results linked to total testosterone concentrations, not free testosterone concentrations. Clin Chem 2008, 54: 512-6.
- Centers for Disease Control and Prevention: Laboratory Quality Assurance and Standardization Programs. (consultato 22/03/2015)
Unità di misura
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
Ogni misurazione, per avere un significato, deve essere espressa da un’unità di misura e da un numero. L’unità di misura indica la dimensione (massa, volume o concentrazione), mentre il numero indica quante unità sono presenti nel campione. Nel corso dell’evoluzione della medicina di laboratorio sono entrate in uso nei diversi paesi modalità diverse per l’espressione della concentrazione dei costituenti di interesse medico. L’applicazione delle unità SI nel laboratorio clinico, introdotta per ovviare a questo problema, è risultata particolarmente complessa, poiché realizza il punto d’incontro di ambiti di conoscenza diversi come chimica, biochimica, fisica, tecnologia e medicina. Nel laboratorio clinico sono di solito misurate le concentrazioni di un costituente in un fluido biologico e tradizionalmente queste erano riportate in termini di massa per unità di volume. Poiché non vi erano regole o una logica precise, veniva usata una grande varietà di unità per refertare i valori di concentrazione, impiegando di solito le concentrazioni per 100 mL (o decilitro). A partire dagli anni ‘60 si è cominciato a proporre che le determinazioni fossero espresse in termini molecolari per litro invece che in termini di massa per 100 mL (o decilitro). Le principali motivazioni erano:
- i processi metabolici che avvengono nel nostro organismo seguono leggi chimiche che si svolgono in termini di reazione di un determinato numero di atomi, molecole o ioni (e non di peso);
- la mole è appropriata per molte delle tecniche analitiche classiche (spettrofotometria, fluorimetria, ecc.);
- la concentrazione dei calibratori è definita senza ambiguità, indipendentemente dalla forma chimica del materiale usato. Per esempio il glucosio può essere anidro o monoidrato: mentre 10 mmol/L di glucosio contengono la stessa quantità di glucosio nelle due forme, 180 mg/dL contengono quantità diverse;
- l’adozione delle unità SI nei paesi scandinavi e nei paesi anglosassoni (con la rilevante eccezione degli Stati Uniti) è andata avanti piuttosto rapidamente, mentre in altri paesi i referti continuano a contenere le unità convenzionali o le due unità (la scelta più frequente ma meno corretta).
Appropriatezza degli esami di laboratorio
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
Si sta estendendo anche alla Medicina di Laboratorio la classica definizione della Evidence Based Medicine (EBM) "l’esplicito e prudente impiego delle migliori prove di efficacia nel prendere decisioni circa la cura di un singolo paziente", tenendo conto delle problematiche particolari legate alle intrinseche peculiarità di questa disciplina. L’EBM può insegnarci a tenere conto dei limiti che possono influenzare i risultati, ma l’applicabilità dei principi dell’EBM nella diagnostica di laboratorio è ostacolata da aspetti come la frequente mancanza di un "gold standard" assoluto con cui confrontare i nuovi esami e la grande diversità tra i valori numerici forniti da metodi diversi per la misurazione di uno stesso analita (basti pensare al GH).
Come valutare la letteratura relativa agli esami diagnostici di laboratorio
Secondo Sackett l’EBM deve aiutare a rispondere a 3 domande:
- le prove di accuratezza di un esame diagnostico sono valide?
- questo esame è in grado di distinguere in modo accurato i pazienti con una determinata malattia da quelli che non la presentano?
- posso usare questo esame nel particolare paziente che ho di fronte in questo momento?
Come valutare la capacità di un esame di diagnosticare una malattia: Sensibilità, Specificità e Quoziente di probabilità
L’EBM si concentra sui classici concetti di sensibilità e specificità come mezzo per calcolare il Quoziente di Probabilità (Likelihood Ratio: LR), lo strumento che consente di calcolare come la probabilità di una diagnosi sia modificata dal risultato di un esame (ovvero, come si calcola la probabilità post-test a partire dalla probabilità pre-test). Concettualmente l’LR positivo e negativo indicano il rapporto (quoziente) tra la probabilità di trovare un particolare risultato (rispettivamente positivo e negativo) in un paziente affetto da una malattia rispetto a quello di trovare lo stesso risultato in un soggetto che non è affetto dalla stessa malattia. Questo è importante dal punto di vista pratico, perché consente di valutare se un esame diagnostico può essere utile alla diagnosi e quale esame è da preferire quando ve ne siano numerosi disponibili.
L’LR consente anche di tenere conto di come la concentrazione più o meno alta del risultato dal punto di vista statistico possa influenzare la probabilità di un risultato. Mentre impiegando i concetti di sensibilità e specificità il risultato può essere solo positivo o negativo, l’LR ci consente di attribuire un diverso peso a seconda del risultato numerico ottenuto. Purtroppo fino ad oggi sono stati calcolati gli LR a concentrazioni diverse solo per pochi esami diagnostici e per pochi contesti clinici. Sono stati proposti dei criteri per classificare l’utilità degli esami diagnostici in relazione al loro LR che, anche se grossolani, possono risultare molto utili nella pratica:
- LR positivi > 10 e negativi < 0.1 modificano in modo spesso conclusivo la probabilità di malattia;
- LR positivi 5 ÷ 10 e negativi 0.1 ÷ 0.2 modificano in modo discreto la probabilità di malattia;
- LR positivi 2 ÷ 5 e negativi 0.2 ÷ 0.5 modificano in modo limitato (solo raramente importante) la probabilità di malattia;
- LR positivi 1 ÷ 2 e negativi 0.5 ÷ 1 modificano scarsamente la probabilità di malattia e solo di rado sono importanti.
Uno strumento ulteriore per valutare un esame diagnostico è l’NND (Number Needed to Diagnose = numero di test necessari per ottenere una diagnosi positiva), che si calcola conoscendo la sensibilità e specificità di un esame. L’NND è il meno conosciuto ed usato strumento dell’EBM, probabilmente a causa della non immediatezza del suo calcolo:
NND = (1/sensibilità)-(1-specificità)
L’NND potrebbe tuttavia risultare utile nella pratica per confrontare il rapporto costo/beneficio degli esami di laboratorio, soprattutto nei casi in cui in un determinato contesto clinico possono essere impiegati più esami.
Uno degli aspetti più rilevanti della EBM è l’approccio pratico che introduce nell’impiego dei risultati di laboratorio per la gestione del singolo paziente. Due esempi sono gli acronimi SnNout e SpPin e il Nomogramma di Fagan. I due acronimi rendono più facile ricordare qual’è l’impiego ottimale di un esame diagnostico molto sensibile o molto specifico:
- SnNout è un acronimo inglese che deriva da ‘When a test has a very high Sensitivity, a Negative result rules out the diagnosis’ (quando un esame ha una sensibilità molto elevata, un risultato negativo esclude virtualmente la diagnosi);
- SpPin deriva invece da ‘When a test has a very high Specificity, a Positive result rules in the diagnosis’ (quando un esame ha una specificità molto elevata, un risultato positivo virtualmente conferma la diagnosi).
In questa maniera è possibile assegnare in modo "automatico" un significato clinico concreto ad un esame molto sensibile o molto specifico.
Il Nomogramma di Fagan consente di semplificare la conversione della probabilità pre -test in quella post-test. Il nomogramma si usa (figura) ancorando una linea retta in corrispondenza della probabilità pre-test indicata sull’asse di sinistra e ruotandola finché raggiunge l’LR relativo all’esame che si sta considerando, indicato nell’asse al centro. È sufficiente proseguire la retta e si troverà sull’asse di destra la probabilità post-test. Il nomogramma può essere tenuto nella tasca del clinico e del laboratorista per un impiego immediato.
-test in quella post-test. Il nomogramma si usa (figura) ancorando una linea retta in corrispondenza della probabilità pre-test indicata sull’asse di sinistra e ruotandola finché raggiunge l’LR relativo all’esame che si sta considerando, indicato nell’asse al centro. È sufficiente proseguire la retta e si troverà sull’asse di destra la probabilità post-test. Il nomogramma può essere tenuto nella tasca del clinico e del laboratorista per un impiego immediato.
I vantaggi dell’impiego dei LR e del nomogramma di Fagan sono quindi numerosi:
- sono più comprensibili e più facili da usare rispetto a sensibilità e specificità;
- possono essere calcolati a livelli diversi di risultato;
- possono essere usati in modo sequenziale, in modo che la probabilità post-test che si ottiene da un esame costituisca la probabilità pre-test per il successivo;
- consentono di embricare laboratorio e clinica in modo efficace;
- forniscono gli strumenti per gli audit clinici, perchè consentono di valutare l’efficacia diagnostica degli esami richiesti dal clinico ed eseguiti dal laboratorio.
Il clinico, di fronte ad un esame, deve quindi chiedersi se questo sarà in grado di aiutarlo a risolvere un problema diagnostico, clinico o terapeutico e, per fare questo, dovrà considerare sia l’esame stesso sia il contesto clinico in cui esso sarà usato. Il clinico, per esempio, dovrebbe conoscere:
- se l’esame è disponibile, accurato, riproducibile e accessibile nel contesto in cui opera;
- qual è la probabilità pre-test;
- se la probabilità post-test che si ottiene è in grado di modificare la gestione del paziente;
- se le conseguenze mediche di un particolare esame sono accettabili per il paziente;
- se il consumo di risorse sanitarie ad esso connesse ed il rapporto costo-benefici sono accettabili.
Il ragionamento probabilistico
Marco Caputo
Dipartimento dei Servizi di Diagnosi e Cura, Ospedale G. Fracastoro, Azienda ULSS20, Verona
(aggiornato al 18 ottobre 2016)
La parte preponderante della vita professionale di un medico, qualsiasi sia la sua specializzazione, è dedicata alla raccolta e alla valutazione del dato clinico. Le decisioni che guideranno ogni strategia diagnostica e ogni scelta terapeutica dipendono in modo assoluto dalla capacità di esercitare al meglio questa attività. Si è discusso a lungo se questa capacità sia frutto dell’applicazione di un rigoroso metodo scientifico o non debba anche necessariamente avvalersi di una componente “artistica”, svincolata da dati oggettivi e valutazioni quantitative e legata invece a un’ineliminabile dote di intuito, quello che una volta veniva chiamato “occhio clinico”. Si tratta di un lavoro complicato, che solo raramente può giovarsi di certezze incontrovertibili; il ricorso a esami diagnostici è praticamente inevitabile, continuo e -naturalmente- non privo di rischi. Alcuni di questi rischi sono evidenti; altri sono più sottili ma ugualmente onerosi, come la componente di stress, paura e insicurezza che il paziente sperimenta nel suo viaggio all’interno dell’universo sanitario e che non può non ribaltare sul proprio Curante. Qui interviene un’ulteriore esigenza: quella di saper comunicare al paziente l’esito della propria indagine, in funzione delle scelte del percorso assistenziale, scelte che oggi devono essere necessariamente condivise.
Ci sono poi i “costi” legati all’acquisizione delle informazioni: costi diretti delle tecnologie utilizzate e costi indiretti dovuti alle ricadute delle decisioni assunte sulla base dei risultati ottenuti. Le evidenze ci dicono senza margini di incertezza che la variabilità di questo processo è elevatissima, non solo in paesi e organizzazioni sanitarie lontane tra loro, ma spesso anche nello stesso ambito territoriale. Gli scenari che già oggi vediamo concretizzarsi attestano che la classe medica ha sempre meno autorevolezza nel guidare questi processi complessi, spesso per oggettivi vincoli esterni, ma talvolta anche per la propria incapacità a orientarsi in un universo sempre più complesso.
L’Institute of Medicine (IOM), nell’ultimo contributo aggiunto alla fine dello scorso 2015 alla serie dedicata al “Quality Chasm”, raccomanda fortemente di curare la formazione continua e le competenze in ambito di ragionamento clinico di tutte le professioni sanitarie, come lo strumento più efficace per migliorare le prestazioni diagnostiche e di conseguenza contribuire a ridurre le conseguenze degli errori legati alle prestazioni assistenziali.
Il teorema di Bayes
Il ragionamento clinico si fonda largamente sull’inferenza bayesiana. Al reverendo Thomas Bayes (1702-1761) si deve la formulazione dell’equazione che spiega come dovrebbe ragionare la mente del medico di fronte al quesito clinico e ai test che di volta in volta vengono pensati e attuati per ridurre il margine di incertezza sull’attribuzione di determinati segni e sintomi a una determinata patologia. Ancora oggi il dilemma che grava sui medici e i loro pazienti può rappresentare un ostacolo insormontabile.
Per cercare di rendere più agevole la comprensione del modo in cui la mente umana affronta questi temi, un contributo comparso su Science offre una chiave di lettura più abbordabile. Gli autori partono da quanto le scienze psicologiche ci hanno insegnato sull’errore diagnostico e di quanto facilmente le impressioni immediate portino a sbagliare anche grossolanamente, a causa dell’approccio euristico e dei bias cognitivi che ci inducono a invitanti scorciatoie mentali, a scapito della riflessione mediata dal pacato ma lungo e a volte noioso ragionamento statistico. Ma a questa via obbligata si può sfuggire. Basta elaborare il problema in “formati”, per i quali la mente è più veloce a elaborare risposte.
Di fronte al paziente ci si trova a operare dovendo valutare un “evento singolo”, per es. una probabilità di malattia dello 0.1%. Difficile da cogliere per la nostra mente, ma se si trasforma il dato in frequenza relativa a una popolazione di riferimento, ad esempio una frequenza di 1 su 1000, è possibile facilitare la corretta comprensione del rischio e dell’inevitabile incertezza connessa al processo decisionale medico. Il rischio di errore rimane se il procedimento viene applicato ad una popolazione di riferimento non appropriata. È quindi indispensabile selezionare la giusta popolazione, come pure la prevalenza e gli esiti di malattia all’interno di quella popolazione di riferimento: senza questi elementi è impossibile prevedere realisticamente gli eventi e di conseguenza elaborare la corretta strategia.
Gli elementi della equazione di Bayes:
- Sensibilità. Immaginiamo di dovere utilizzare un test diagnostico in una popolazione di 100 pazienti, dei quali 30 siano affetti da una patologia. L’esame ideale - quello che riconosce tutti e 30 i malati e viene sempre negativo nei restanti 70 - non esiste. Nel nostro caso il test identifica correttamente 24 pazienti su 30. 24/30 = 0.80 = 80%. La sensibilità è la qualità di un test di riconoscere i colpiti da malattia. Un test “sensibile”, applicato in routine, dà forza a un risultato negativo, in altri termini aiuta a escludere la diagnosi che sospettavamo. Può essere utile l’acronimo memonico SNOUT: “high Sensitivity and Negative results rule OUT the diagnosis”.
- Specificità. Il nostro test risulta negativo in 56 delle 70 persone sane nella nostra popolazione. 56/70 = 0.80 = 80%. La specificità è la qualità di un test di riconoscere correttamente tutti gli esenti da quella malattia. Un test “specifico”, applicato in routine, dà forza a un risultato positivo, in altri termini accresce la probabilità che la malattia sia presente. Può essere utile l’acronimo mnemonico SPIN: “high Specificity and Positive results rule IN the diagnosis”.
- Prevalenza o Probabilità pre-test. È la probabilità della presenza di malattia nella nostra popolazione prima di eseguire il test.
L’equazione che esprime il teorema è la seguente:
Probabilità = (prevalenza*sensibilità)/[(prevalenza*sensibilità)+(1+prevalenza)*(1-specificità)]
Questa relazione può essere espressa in modo semplificato ricorrendo al concetto di Quoziente di Probabilità (Likelihood Ratio, LR), che non è altro che la probabilità che si abbia un determinato risultato nel soggetto malato, diviso la probabilità di ottenere lo stesso risultato in un soggetto sano. Questo ci permette di semplificare l’espressione precedente in questo modo:
LR = sensibilità/(1-specificità)
- Se LR = 1, il nostro test è perfettamente inutile ai fini dell’informazione clinica; equivale a lanciare una moneta.
- Quanto maggiore di 1 sarà LR, tanto maggiore la probabilità che la malattia che cerchiamo sia presente (LR+ per risultati positivi).
- Più LR è inferiore a 1, tanto diminuisce la probabilità di presenza di malattia (LR- per risultati negativi).
Abbiamo ora tutti gli elementi per valutare il nostro esempio e ricavarne elementi per un ragionamento clinico. Disponiamo di un esame con una sensibilità e una specificità pari a 80%, da applicare su una popolazione in cui la prevalenza di malattia è pari a 30%. In queste condizioni, il Valore Predittivo di un risultato positivo (VPP) sarà pari a 24/38 (rapporto tra i malati correttamente identificati come positivi che erano 24/30 e tutti i risultati positivi, cioè i 24 + i 12 di 70, che erano positivi pur in assenza di malattia). 24/38 = 63%.
Ma cosa succederebbe se la prevalenza di malattia nella nostra popolazione invece del 30% fosse più bassa, diciamo il 10%? Allora il VPP sarebbe 8/26 (cioè l’80% di 10, diviso 8 + 18), cioè 31% scarso. E se la prevalenza fosse ancora più bassa? Allora VPP = 1/21 = 5%.
Esiste da molti anni uno strumento relativamente semplice per calcolare i risultati delle relazioni bayesiane applicate al singolo concreto caso clinico. Si tratta del nomogramma di Fagan, un calcolatore grafico applicabile alla routine clinica (fig 1).

Figura 1: nomogramma di Fagan
Se è nota la prevalenza di malattia (colonna di sinistra) e il LR (colonna al centro), è possibile ricavare automaticamente la probabilità post-test (colonna di destra) unendo semplicemente i 2 punti precedenti. A titolo di esempio viene riportato il caso del dosaggio del TSH nell’ipotiroidismo (fig 2).
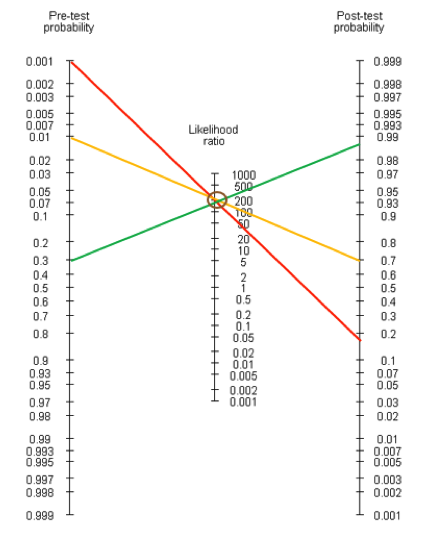
Figura 2: nomogramma di Fagan applicato al dosaggio di TSH nell’ipotiroidismo. La prevalenza di ipotiroidismo in uno screening nella popolazione generale è 0.1% (linea rossa), nell’ambulatorio di medicina generale è 1% (linea gialla), in un ambulatorio di endocrinologia è 30% (linea verde).
L’esame è dotato di prestazioni diagnostiche eccellenti assolute, in termini di sensibilità, specificità e LR. Tuttavia, come si può vedere, il valore informativo del risultato dipende in modo fondamentale dalla prevalenza della malattia, che è molto diversa a seconda del contesto in cui ci si trova.
Per cercare di rendere ancora più pratico l’utilizzo del nomogramma, è stata creata una variante (fig 3), definita “a 2 fasi” (2-step). Al formato originale sono state aggiunte due colonne, da impiegare per calcolare LR qualora non fosse noto.

Figura 3: nomogramma di Fagan 2-step
In pratica, dopo aver ottenuto sensibilità e specificità del test (dalla letteratura o dal produttore) e il risultato dell’esame (positivo o negativo), si collegano gli assi di sensibilità e specificità e si legge il corrispondente LR sull’asse centrale. Da qui si procede come con il nomogramma standard, unendo la prevalenza sulla colonna di sinistra con l’LR sulla colonna centrale e prolungando fino a incontrare la colonna di destra.
Infine, per cercare di rendere questo strumento ancora più pratico, sono da poco disponibili delle versioni app per smartphone (fig 4).
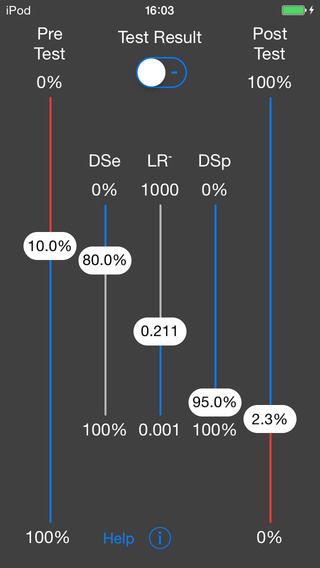
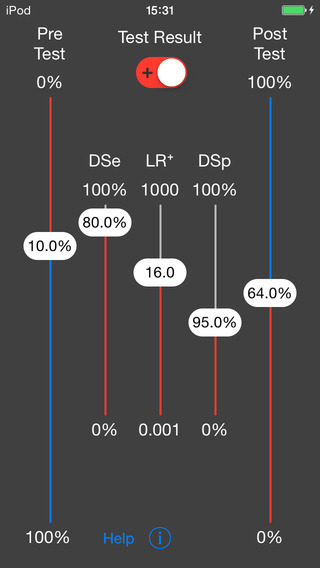
Figura 4: nomogramma di Fagan su App
In conclusione, è destinata a proseguire la lotta contro l’incertezza che condiziona il comportamento delle professioni sanitarie nel momento di applicare il risultato di un esame alla diagnosi o alla stratificazione del rischio del proprio paziente. Tuttavia, abbiamo la possibilità di utilizzare meglio le informazioni che ci arrivano da una tecnologia sempre più complessa e sofisticata. Al momento di entrare nell’era della Medicina di Precisione, in cui il nostro bagaglio di dati e conoscenze non potrà essere più interpretato come medie tra gruppi, ma andrà applicato al singolo individuo, non avremo più certezze da consegnare su ciò che è bene e ciò che è male fare per il nostro paziente, ma dovremo essere capaci di far assumere a lui le decisioni importanti per la sua salute. Per questo obiettivo, nobile ma complicatissimo da perseguire, abbiamo ancora bisogno di strumenti affidabili e pratici da utilizzare nel lavoro quotidiano, come quelli derivati dall’intuizione del reverendo Thomas Bayes.
Per approfondire
- Balogh EP, Miller BT, Ball JR, ed. Improving diagnosis in health care. Committee on Diagnostic Error in Health Care; Board on Health Care Services; Institute of Medicine; National Academies of Sciences, Engineering, and Medicine. Washington (DC): The National Academies Press; 2015.
- Operskalski JT, Barbey AK. Risk literacy in medical decision-making: how can we better represent the statistical structure of risk? Science 2016, 352: 413–4.
- Henriquez RR, Korpi-Steiner R. Bayesian inference dilemma in clinical decision making: a need for user-friendly probabilistic reasoning tools. Clin Chem 2016, 62: 1285-6.
- Caraguel CGB, Vanderstichel R. The two-step Fagan’s nomogram: ad hoc intepretation of a diagnostic test result without calculation. Evid Based Med 2013, 18: 125-8.
- Nomographs with Python, Version 0.2.2. 2010. (accesso 14/10/2016).
- Università di Adelaide. Nomogramma di Fagan a 2-fasi (scaricabile qui).
Introduzione alle tecniche di laboratorio
Romolo Dorizzi1 & Marco Caputo2
1Laboratorio, UO Corelab-Laboratorio Unico di Area Vasta Romagna, Pievesestina di Cesena (FC)
2Laboratorio Chimica Clinica ed Ematologia, Ospedale G. Fracastoro, Azienda USL 20, Verona
Molte determinazioni di laboratorio si basano su misure di energia radiante assorbita, trasmessa, emessa, diffusa, riflessa. L’uomo è capace di percepire solo una piccola frazione (luce “visibile”: da 380 a 750 nm) dell’estesa gamma di lunghezze d’onda presenti nell’energia radiante dell’universo. Gli strumenti analitici sono in grado di estendere la rilevazione a lunghezze d’onda inferiori (luce “ultravioletta”, UV) o superiori (luce “infrarossa”, IR) e questa proprietà è stata sfruttata per scopi analitici.
Il termine fotometria si riferisce alla misura della luce. Il termine spettrofotometria si riferisce alla misura dell’intensità di luce a una lunghezza d’onda selezionata. Quest’ultima è la metodologia utilizzata in Chimica Clinica per un ampio ventaglio di determinazioni qualitative e quantitative. Il principio sfrutta le proprietà di assorbimento della luce possedute dalla sostanza da analizzare o da un suo diretto derivato. L’intensità di una luce che attraversi una soluzione in cui è disciolta una sostanza assorbente (detta “cromogeno”) diminuisce in maniera proporzionale alla frazione assorbita. La rilevazione e la misura di questa frazione assorbita può essere usata per stabilire una relazione tra luce trasmessa (o assorbita) e la concentrazione della sostanza da analizzare. La cosiddetta legge di Lambert-Beer, che stabilisce la proporzionalità diretta della concentrazione di una sostanza dalla quantità di luce assorbita e la proporzionalità inversa rispetto al logaritmo della luce trasmessa è il principio su cui si è sviluppata tutta la tecnologia che adotta la spettrofotometria come principio operativo, dai più semplici strumenti monocromatici ai più complessi e potenti analizzatori in dotazione alle grandi piattaforme automatizzate.
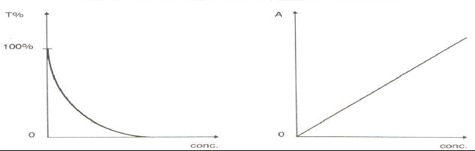
Figura 1: rappresentazione grafica della legge di Lambert-Beer. A sinistra relazione inversa fra concentrazione della sostanza in esame nel liquido biologico e luce trasmessa; a destra proporzione diretta tra concentrazione e luce assorbita.
La spettrometria in assorbimento atomico è una tecnica usata per la determinazione di oligo-elementi (rame, zinco, alluminio, piombo, litio, magnesio, ecc) in soluzione. In pratica, il metallo da ricercare viene ridotto da una fiamma o da una fonte di calore allo stato gassoso e in forma energeticamente neutra. In queste condizioni, l’atomo assorbe solo radiazioni con ampiezza di banda molto stretta, come quella emessa dalla luce di una lampada (detta “a catodo vuoto”) rivestita dallo stesso metallo che si vuole determinare. Quando la luce attraversa la camera di misurazione, subisce una netta diminuzione di intensità in presenza della sostanza corrispondente: a questo fenomeno si deve il nome di assorbimento atomico. Ci sono lampade selettive (un solo metallo) e altre composite (rivestite da più metalli) adatte alla determinazione di più analiti. Esistono strumentazioni evolute che hanno sostituito la fiamma con barre di carbonio (“fornetto di grafite”).
Si intende per fluorescenza la condizione in cui una molecola assorbe luce a una determinata lunghezza d’onda e la riemette a una lunghezza d’onda maggiore. Un atomo o molecola che possegga questa proprietà è chiamato fluoroforo. La fluorimetria è la misura della luce fluorescente emessa, e su questo principio si basa un’ampia varietà di analisi quantitative, molto accurate e sensibili, condotte attraverso l’uso di una vasta gamma di diverse strumentazioni. Esistono fluorimetri e spettrofluorimetri di varie tipologie e dimensioni. I fluorimetri a risoluzione temporale (time-resolved) ovviano a uno dei principali difetti della fluorescenza e cioè la rapidità del decadimento del segnale attraverso le proprietà di fluorofori a lunga decadenza (1,2). Esistono sistemi basati su questo principio (o una delle sue numerose varianti), per misurare gli ormoni di principale utilizzo, dalla tiroide al PTH al cortisolo. Il sistema DELFIA della Perkin Elmer è l’esempio più noto.
La citometria a flusso è una particolare applicazione della fluorimetria, dedicata all’analisi di cellule e altre particelle biologiche (3,4). I citofluorimetri sono in grado di analizzare contemporaneamente parametri multipli, quali le dimensioni cellulari, la presenza e quantità di granuli, il contenuto in DNA e RNA, i rapporti nucleotidici {(A + T)/(G + C)}, la struttura cromatinica, i recettori cellulari e altro ancora. Le applicazioni della citofluorimetria sono oggi vastissime, dall’immunologia all’ematologia, dall’oncologia alla microbiologia e alla genetica, fino alla possibile applicazione a immuno-dosaggi anche di interesse endocrinologico (per esempio il riconoscimento attraverso anticorpi monoclonali di cellule e molecole da ago-aspirati di lesioni ghiandolari), grazie all’impiego dei test basati su particelle e a fluorocromi molto potenti e a emissione prolungata come la ficoeritrina.
La chemiluminescenza è l’emissione di luce che si ha quando un elettrone ritorna da un livello energetico eccitato a un livello energetico inferiore. Se la sorgente di eccitazione non è una sostanza chimica (es. luminolo, isoluminolo, esteri di acridinio, luciferina) ma un componente di sistemi biologici si parla di bioluminescenza. I metodi in chemiluminescenza hanno trovato vastissima diffusione nei laboratori di immunometria e sono stati ampiamente applicati per il dosaggio in automazione di moltissimi ormoni, ma anche nei sistemi di rilevazione di sonde di DNA per studi di biologia molecolare. L’elettro-chemiluminescenza si diversifica dalla chemiluminescenza per il fatto che i reattivi che producono la reazione chemiluminescente sono generati elettrochimicamente sulla superficie di un elettrodo a partire da un precursore stabile (5). Il più utilizzato di questi marcatori è un chelato del Rutenio Ru2+. È un reagente molto stabile e relativamente piccolo ed è stato utilizzato per marcare apteni e molecole più grandi come oligonucleotidi e proteine. I principali vantaggi di questo metodo sono una maggiore stabilità dei reagenti e una loro preparazione semplificata e una maggiore sensibilità, con limiti di rilevazione da 200 fmol/L e un range dinamico che arriva a coprire fino a sei ordini di grandezza. La principale limitazione dei test in chemiluminescenza risiede proprio nell’estrema sensibilità analitica, che impone di operare controlli stringenti sull’integrità dei reagenti e le parti del sistema.
La misura della luce diffusa è invece realizzata attraverso due metodi e viene largamente impiegata nella quantificazione delle siero-proteine. La turbidimetria consiste nel misurare la diminuzione dell’intensità di un raggio incidente che attraversa un contenitore di particelle in soluzione. È un principio che si applica con estrema facilità agli spettrofotometri e alle piattaforme di grande automazione, anche grazie alla stabilità e al potere di risoluzione degli strumenti guidati dai moderni microprocessori. Si parla di nefelometria quando la rilevazione dell’energia luminosa diffusa o riflessa è indirizzata verso un rilevatore che non si trova sulla traiettoria diretta del raggio di luce trasmessa. Di solito i comuni nefelometri misurano la luce diffusa ad angolo retto rispetto alla luce incidente. La scelta tra nefelometria e turbidimetria dipende dalle applicazioni e dalla strumentazione disponibile. La nefelometria offre qualche vantaggio in termini di sensibilità a basse concentrazioni, ma è meno consolidabile rispetto alla turbidimetria sulle grandi piattaforme automatizzate (6).
Il termine cromatografia si riferisce alla separazione dei componenti di una miscela, in base alla loro differente distribuzione tra una fase mobile e una fase fissa o stazionaria. I componenti maggiormente presenti nella fase stazionaria sono trattenuti e si spostano più lentamente nel sistema. La tecnica è stata messa a punto all’inizio del secolo scorso per separare dei pigmenti vegetali che si evidenziavano come bande colorate (“croma-”, greco per “colore”) su una colonnina di carbonato di calcio. Le separazioni cromatografiche sono classificate in base ai meccanismi fisici o chimici utilizzati per separare i componenti: a scambio ionico, partizione, affinità, assorbimento, esclusione sterica. Tra i vari supporti che si possono utilizzare, la cromatografia su colonna è la più largamente diffusa nei laboratori clinici, nelle due varietà di gascromatografia (GC) e cromatografia liquida (LC), a seconda che la fase mobile sia gassosa o liquida (7,8). Con questa tecnica si possono misurare tutti gli ormoni steroidei, i metaboliti di metanefrine e serotonina, farmaci vari. Se la fase stazionaria in LC è costituita da particelle di piccolo diametro, si parla di “cromatografia liquida a elevate prestazioni”, o HPLC.
Il cromatografo liquido o gassoso può essere connesso a uno spettrometro di massa (MS), dando origine a una potente tecnica analitica sempre più utilizzata anche negli studi endocrinologici, la cromatografia liquida/spettrometria di massa (LC-MS) e la gascromatografia/spettrometria di massa (GC-MS). Il termine “spettrometria di massa” è in realtà un misnomero: questi strumenti non misurano la massa molecolare ma il rapporto massa/carica. Bisogna aver presente questo concetto per poter apprezzare a pieno l’architettura e la funzionalità strumentale e, fatto ancora più importante, l’interpretazione dei risultati.
La doppia spettrometria di massa, MS/MS o Tandem-MS, quando utilizzata in sinergia con l’elevatissima selettività di una GC o LC (GC/Tandem-MS, LC/Tandem-MS), accoppia la bassa incidenza di interferenze al basso costo dei reagenti e alla possibilità di analizzare molti campioni in poco tempo; non sorprende che questa tecnologia si stia rapidamente diffondendo nei laboratori clinici e sia diventata tra l’altro gold standard per la quantificazione di molte sostanze di interesse endocrinologico (per esempio gli steroidi tutti, da cortisolo a testosterone, ma anche le frazioni libere degli ormoni tiroidei, la vitamina D, ecc). I limiti alla sua diffusione universale restano al momento l’elevato costo dell’hardware e la necessità di un accurato know-how da parte di personale dedicato (9-11).
Il dosaggio “ideale” per la misurazione degli ormoni dovrebbe:
- riflettere l'attività biologica dell'ormone;
- essere abbastanza sensibile da richiedere volumi contenuti di campione;
- consentire l'automazione e una produttività molto elevata;
- essere molto accurato e riproducibile.
Gli ormoni sono misurati nella pratica corrente dei laboratori clinici con metodi immuno-metrici, radio-immunologici, immuno-radiometrici, immuno-enzimatici e in chemi-luminescenza. Tutte queste tecnologie derivano da quella radio-immunologica (RIA), un metodo analitico che unisce la specificità di una reazione immunologica (con formazione di un complesso antigene-anticorpo) con la sensibilità di un metodo radio-chimico, dato che è utilizzato un radio-isotopo.
La tecnica RIA, descritta per la prima volta nel 1959, ha soppiantato rapidamente i bio-assay, che fino a quel momento erano gli unici metodi in grado di misurare gli ormoni: per esempio, l’insulina era misurata in vivo valutando l’ipoglicemia indotta nei ratti ipofisectomizzati, surrenectomizzati e resi diabetici con alloxana e in vitro valutando la captazione di glucosio da parte del cuscinetto di tessuto adiposo epididimale di ratto. I metodi RIA classici sono basati su tre elementi:
- una molecola di ormone, che funziona da antigene, a cui è legato un tracciante radioattivo (in passato Trizio, oggi di solito 125Iodio), che emette radiazioni misurabili con un contatore beta o gamma;
- un anticorpo anti-ormone in concentrazione limitata;
- un secondo anticorpo diretto verso il primo anticorpo, che porta alla produzione di un precipitato.
Nella maggior parte dei RIA viene marcato l’antigene. La misurazione nel campione della quantità di antigene non marcato dipende della sua capacità di spiazzare l’antigene marcato (Ag*) dal legame con l’anticorpo (Ab). A questo scopo, l’anticorpo deve reagire e legarsi allo stesso modo sia con l’antigene marcato che con quello non marcato. Gli anticorpi anti-ormone possono essere policlonali, che danno risultati comparabili tra di loro, e monoclonali, che forniscono risultati più bassi, meno comparabili tra i diversi kit e con la capacità di rilevare concentrazioni basse in modo più affidabile. La produzione di anticorpi monoclonali si basa sull’immunizzazione di un topo mediante l’iniezione di un antigene (in questo caso un ormone), che produce nell’animale una risposta anticorpale. Il topo è sacrificato e le cellule che producono anticorpi sono isolate dalla sua milza. La fusione delle singole cellule che producono anticorpi con cellule di mieloma “immortalizzate” è definita “ibridoma”. Tali cellule possono essere coltivate facilmente e contengono grandi quantità di anticorpi identici, poiché derivano da un’unica cellula clonata, detti quindi monoclonali.
In radioimmunologia possono essere distinti metodi competitivi e non competitivi. Nei metodi competitivi, l’analita non legato a tracciante (non marcato) è misurato dalla sua capacità di “competere” con un analita legato a tracciante per un numero limitato (e fisso) di siti di legame di un anticorpo (fig 2): l’antigene non marcato impedisce all’antigene marcato di legarsi, perché il sito di legame è già occupato. In un metodo competitivo esiste una relazione inversa tra la concentrazione dell’antigene misurato e la concentrazione di antigene legato a tracciante misurato: più antigene “marcato” è legato, meno antigene endogeno è presente nel campione (fig 3).
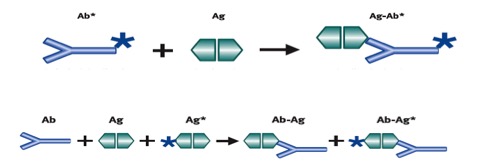
Figura 2. Metodo competitivo
(Ab = anticorpo, Ag = antigene; Ag-Ab = complesso antigene-anticorpo)
Figura 3. Curva dose-risposta per un metodo immunometrico competitivo
Nei metodi non competitivi (sandwich) (fig 4) l’analita è legato a “sandwich” tra due anticorpi ad alta specificità. La miscela di reazione comprende di norma un eccesso di anticorpo legato al tracciante: tutto l’ormone contenuto nel campione è legato ed il complesso antigene-anticorpo è misurato per determinare la concentrazione presente nel campione. La concentrazione di anticorpo legato al tracciante è, quindi, direttamente proporzionale alla concentrazione di antigene presente nel campione (fig 5).
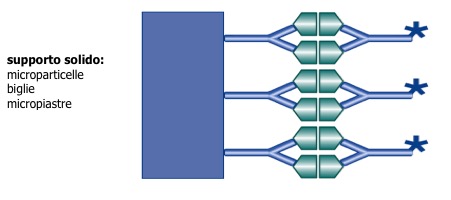
Figura 4. Metodo sandwich: gli anticorpi si legano a due epitopi sull’analita
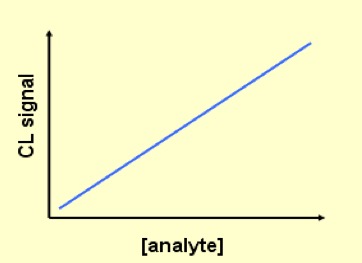
Figura 5. Curva dose-risposta per un metodo immunometrico non-competitivo
I metodi immuno-radiometrici (IRMA), in cui il tracciante è legato all’anticorpo, sono oggi poco usati, se non in qualche variante immuno-enzimatica. Attualmente sono più utilizzati i metodi a doppio anticorpo monoclonale a sandwich, che impiegano traccianti enzimatici, fluorescenti e chemi-luminescenti, e possono arrivare a sensibilità analitica (cioè alla minima concentrazione distinguibile da zero) di 0.0001 µg/L. Per esempio, i metodi a sandwich per la determinazione del TSH impiegano, di solito, un anticorpo diretto verso la subunità alfa ed uno verso la subunità beta della molecola glicoproteica eterodimerica del TSH (12).
Numerosi ormoni proteici esercitano i loro effetti legandosi a un recettore di membrana specifico, che, in rapporto con l’adenil-ciclasi, stimola la formazione intracellulare di AMP ciclico. Questi siti recettoriali della membrana cellulare sono stati isolati e utilizzati come composti leganti specifici nel dosaggio radio-recettoriale (RRA). Il principale vantaggio dato dall’uso delle proteine recettoriali, che si ottengono da tessuti differenti e non vengono completamente solubilizzate, nel RRA consiste nel fatto che il recettore misura l’attività biologica, mentre il metodo RIA misura soltanto l’attività immunologica. Tuttavia, le proteine del recettore presentano problemi tecnici considerevoli: sono presenti in piccole concentrazioni, sono instabili, e possono essere danneggiate facilmente nelle procedure di omogeneizzazione e ultra-centrifugazione necessarie per la loro preparazione. Diversi tessuti sono stati impiegati per la messa a punto di metodi RRA.
| Dosaggi RRA | |
| Tessuto | Ormone |
| Proteina del tubulo del testicolo di ratto | FSH |
| Linfociti umani e membrane di epatociti | Insulina, GH |
| Membrane di cellule della ghiandola mammaria | PRL |
| Membrane di cellule tiroidee | Immunoglobuline stimolanti il recettore del TSH |
| Cellule testicolari | hCG |
Il tracciante è sempre più frequentemente enzimatico o chemi-luminescente. Questo tipo di tracciante presenta il vantaggio di consentire l’automazione, permettendo maggiore velocità e precisione, maggiore sensibilità e ampiezza dell’intervallo di misura. I metodi immunologici che richiedono la separazione del complesso Ab-Ag sono definiti eterogenei, mentre quelli che non la richiedono sono definiti omogenei. I metodi omogenei sono in genere più semplici da mettere a punto e sono più facilmente automatizzabili.
Nei metodi competitivi, eterogenei, immuno-enzimatici (ELISA, Enzyme linked immunosorbant assay), i reagenti sono adsorbiti sulla superficie di una fase solida, di solito il pozzetto di una micro-piastra, e l’assorbanza, che è inversamente proporzionale alla concentrazione dell’analita, è misurata con un lettore di micro-piastra.
Per la messa punto dei metodi immuno-enzimatici l’enzima deve:
- essere facilmente reperibile in forma stabile e altamente purificata ad un costo ragionevole;
- essere solubile;
- contenere molti residui di lisina per la coniugazione;
- avere una struttura diversa dagli enzimi presenti nei liquidi biologici in concentrazioni significative.
Il substrato deve:
- essere voluminoso, facilmente reperibile, economico, stabile;
- non trovarsi in significative concentrazioni nei liquidi biologici.
Infine, la reazione enzima-substrato deve:
- avere un pH ottimale che non impedisca la reazione antigene-anticorpo;
- non essere inibita da una qualsiasi sostanza presente nei liquidi biologici;
- avere la possibilità di un arresto del dosaggio semplice, rapido e riproducibile.
Nessuno dei metodi a disposizione del laboratorio clinico per la misurazione degli ormoni possiede le caratteristiche proprie del dosaggio “ideale”.
Autorevoli organizzazioni hanno indicato per alcuni ormoni e test limiti decisionali che hanno validità solo nel contesto clinico e laboratoristico specificato e che possono essere tradotti nelle diverse realtà cliniche solo seguendo raccomandazioni e linee guida aggiornate, attraverso uno stretto collegamento tra l’endocrinologo, l’internista, il medico di medicina generale, il laboratorio di analisi ed il personale infermieristico ed amministrativo che ha la responsabilità delle fasi pre- e post-analitiche. È fondamentale, comunque, avere presente che non è possibile trasferire direttamente i valori di cut-off o decisionali dagli articoli scientifici o dai trattati consultati ai risultati prodotti dal laboratorio che noi utilizziamo, se non sono verificati il metodo e l’analizzatore impiegati, l’unità di misura e lo standard a cui è collegata.
Le differenze tra i risultati ottenuti con i diversi metodi dipendono da fattori diversi e il GH può rappresentare un caso esemplificativo delle problematiche comuni a molti ormoni. I risultati finali dipendono da diversi fattori (13).
- Proteine leganti: i diversi metodi valutano quantitativamente in misura diversa il GH libero, quello legato alle proteine di trasporto e quello totale.
- Isoforme: in circolo sono presenti numerose isoforme, che comprendono monomeri e oligomeri, che cross-reagiscono in modo molto diverso con i vari metodi.
- Problemi di standardizzazione: i metodi di determinazione sono calibrati con materiali di riferimento che sono aggiornati periodicamente.
CASI PARTICOLARI
Metodi basati sul principio dell’analogo. Sono disponibili molti metodi per la determinazione degli ormoni liberi ed in particolare della tiroxina (FT4) e della triiodotironina (FT3). Il metodo di riferimento per la determinazione è quello della dialisi ad equilibrio. La frazione libera del T4 è dapprima separata dalla frazione legata mediante una membrana di dialisi e poi quantificata con una metodica immunometrica tradizionale. Questa tecnica consente una misurazione molto accurata degli ormoni, ma è complessa e non risulta alla portata del laboratorio clinico. Oggi, sono impiegati universalmente metodi basati sul principio dell’”analogo” (14,15). L’FT4 legato a tracciante, che nelle metodiche immunometriche convenzionali compete con l’ormone presente nel siero del soggetto, è sostituito da una molecola “analoga” legata ad un tracciante sufficientemente simile all’ormone endogeno da consentirgli di competere con l’antigene, ma sufficientemente diverso da non legarsi con le proteine vettrici presenti nel siero. La metodica è stata automatizzata, consentendo risultati più rapidi e più precisi e favorendone la rapida diffusione nei laboratori clinici. L’accuratezza dei metodi “analoghi” commercializzati negli ultimi anni è nettamente migliorata e risulta sicuramente confrontabile a quella della dialisi ad equilibrio. È opportuno tuttavia ricordare che, a rigore di termini, la determinazione dell’FT4 mediante il metodo dell’analogo consente non la misura dell’FT4 ma la sua “stima”.
Anticorpi eterofili. Un problema importante nella determinazione del TSH è quello dell’interferenza da anticorpi eterofili, che possono comparire dopo la somministrazione di anticorpi di topo per esami immuno-scintigrafici (questi hanno indotto una reazione immune e conseguente produzione di anticorpi, ma possono essere presenti anche in soggetti che non sono stati sottoposti a trattamenti di questo tipo). Tali anticorpi interferiscono con i metodi di dosaggio basati su anticorpi monoclonali (nella maggior parte dei metodi aumentando la concentrazione del TSH) e possono essere rivelati mediante un loro dosaggio diretto o cimentando il campione con anticorpi particolari che li “bloccano”.
Bibliografia
- Diamandis E, Christopoulos TK. Europium chelate labels in time-resolved fluorescence immunoassays and DNA hybridization assays. Anal Chem 1990, 62: 1149-57.
- Harma H, Soukka T, Lovgren T. Europium nanoparticles and time-resolved fluorescence for ultrasensitive detection of prostate-specific antigen. Clin Chem 2001, 47: 561-8.
- Patrick CW. Clinical flow cytometry: milestones along the pathway of progress. MLO Med Lab Obs 2002, 34: 1-16.
- Vignali DA. Multiplexed particle-based flow cytometric assays. J Immunol Methods 2000, 243: 243-55.
- Rodríguez-Orozco AR, Ruiz-Reyes H, Medina-Serriteño N. Recent applications of chemiluminescence assays in clinical immunology. Mini Rev Med Chem 2010, 10: 1393-400.
- Sternberg J. A rate nephelometer for measuring specific proteins by immunoprecipitin reactions. Clin Chem 1977, 25: 1456-64.
- Miller JM. Chromatography: concepts and contrasts. Wiley Interscience, Malden, MA, 2009.
- Petersson P, Heaton FA, Euerby MR. Maximizing peak capacity and separation speed in liquid chromatography. J Sep Sci 2008, 31: 2346-57.
- Maurer HH. Current role of liquid chromatography–mass spectrometry in clinical and forensic toxicology. Anal Bioanal Chem 2007, 388: 1315-25.
- CLSI. Mass spectrometry in the clinical laboratory. General principles and practice: approved guideline, vol C-50A. Clinical Laboratory Standards Institute, Wayne, PA, 2007.
- Annesley T, Majzoub J, Hsing A, et al. Mass spectrometry in the clinical laboratory: how have we done, and where do we need to be? Clin Chem 2009, 55: 1236-9.
- Rawlins ML, Roberts WL. Performance characteristics of six third-generation assays for thyroid-stimulating hormone. Clin Chem 2004, 50: 2338-44.
- Dorizzi RM. Endocrinologia pediatrica e laboratorio. RIMeL/IJLaM 2007, 3: 28-44.
- Midgley JE. Direct and indirect free thyroxine assay methods: theory and practice. Clin Chem 2001, 47: 1353-63.
- Despres N, Grant AM. Antibody interference in thyroid assays: a potential for clinical misinformation. Clin Chem 1998, 44: 440-54.
Introduzione alle tecniche di patologia
Anna Crescenzi & Eleonora Perrella
UOC di Anatomia Patologica, Campus Biomedico, Roma
L’Anatomia Patologica studia le alterazioni della morfologia dei tessuti e delle cellule che li compongono attraverso l’osservazione macroscopica e microscopica. Strumento essenziale è il microscopio ottico, che permette l'osservazione diretta del materiale con ingrandimenti che consentono la valutazione delle modificazione morfologiche che sottendono alla malattia. Perché tale osservazione sia possibile, i tessuti e le cellule devono essere trattati in modo da prevenirne la decomposizione e permetterne la conservazione e la raccolta su vetrini per la successiva colorazione. Un tessuto che sia stato trattato in questo modo prende il nome di “preparato istologico o citologico”.
1. MATERIALI SOTTOPOSTI AD ESAME IN ANATOMIA PATOLOGICA
1.1 Campioni citologici
Il campionamento del materiale citologico si realizza attraverso 4 tecniche:
- raccolta diretta di cellule spontaneamente esfoliate da organi e cavità del corpo;
- raccolta di cellule mediante manovre meccaniche;
- citologia per apposizione;
- aspirazione di cellule mediante ago sottile.
L’esfoliazione cellulare è un processo che avviene in continuazione ed è correlato al rinnovamento dei tessuti del corpo. Cellule esfoliate spontaneamente si ritrovano nelle urine, nell’espettorato, nei versamenti (pleurico, pericardico, ascitico e sinoviale), nelle secrezioni e nel liquor. Il principale vantaggio di tale tecnica è la facilità con cui si possono ottenere i campioni in molti casi (urine, espettorato). Di contro, i campioni sono spesso costituiti da cellule mal conservate e contaminate da elementi della flogosi, microrganismi e materiale di origine estranea.
Nel campionamento attuato mediante il ricorso a manovre meccaniche (abrasione o scraping, spazzolamento o brushing, lavaggio o washing) le cellule sono rimosse direttamente dal tessuto e pertanto non mostrano modifiche legate alla degenerazione secondaria alla permanenza libera nella cavità campionata (1). L’introduzione nella diagnostica di strumenti a fibre ottiche provvisti di piccole spazzole ha rivoluzionato il campionamento citologico di organi del tratto respiratorio, gastro-intestinale e, in misura minore, del tratto urinario, consentendo l’identificazione di carcinomi iniziali dei bronchi, di carcinomi superficiali dell’esofago e dello stomaco nonché di recidive di carcinomi del colon. È inoltre possibile rimuovere cellule da lesioni non accessibili o non visibili all’endoscopista, dopo instillazione di piccole quantità di soluzioni saline che vengono successivamente aspirate e raccolte in contenitori (lavaggio peritoneale, lavaggio bronco-alveolare, lavaggio della vescica urinaria).
La tecnica della citologia per apposizione sfrutta la capacità delle cellule presenti in un tessuto di aderire a una superficie liscia quale il vetro a seguito di una leggera pressione. È una metodica facile da eseguire, economica, che consente una diagnosi rapida con una sensibilità diagnostica compresa tra 80-85%. È indicata nel caso di lesioni superficiali della cute e linfonodi e trova sempre maggiore applicazione nella diagnostica intra-operatoria (valutazione del linfonodo sentinella nel carcinoma della mammella)(2).
La tecnica di campionamento mediante aspirazione con ago sottile (FNAB) può essere eseguita su organi o lesioni superficiali o profonde (sulle sedi profonde la metodica si realizza sotto monitoraggio radiologico TAC, RMN o Eco-guidata). Sedi di prelievo più frequenti sono tiroide, mammella, linfonodi, ghiandole salivari, fegato, polmone, pancreas e reni. È una metodica facile da eseguire, relativamente economica e che consente la caratterizzazione veloce della natura della lesione, con una sensibilità diagnostica che può raggiungere il 95%. Le complicanze legate a questa procedura sono rappresentate dall’ematoma per lesioni superficiali e dal sanguinamento per lesioni profonde (la metodica non va eseguita in pazienti con difetto della coagulazione). Meno frequenti sono pneumotorace (per lesioni all’interno del torace), perforazione intestinale, pancreatite necrotizzante (per lesioni a sede nell’addome), infarto totale o parziale della lesione e impianto del tumore lungo il tragitto dell’ago (il rischio aumenta con l’utilizzo di aghi di grosso calibro e gli organi più coinvolti sono tiroide, pancreas e rene). La metodica prevede l’utilizzo di aghi non trancianti di piccolo calibro (23-27 gauge), con lunghezza variabile a seconda della profondità dell’organo bersaglio. La manovra può essere eseguita anche senza suzione, quindi senza l’impiego della siringa, con il vantaggio di un migliore controllo dell’ago e un minore traumatismo della lesione (particolarmente indicata per piccole lesioni altamente vascolarizzate, non è raccomandata per l’aspirazione di lesioni cistiche). Condizioni indispensabili per una corretta diagnosi citologica sono l’adeguatezza del campione e il suo corretto allestimento. Il corretto posizionamento dell’ago all’interno della lesione si realizza con il ricorso alle metodiche per immagini; certi di aver centrato il bersaglio, si procede al prelievo. In caso di lesioni di piccole dimensioni (diametro cm 1) è preferibile campionare la periferia, in quanto il centro può essere necrotico. Centrata e trafitta la lesione, si eseguono movimenti di “va e vieni” all’interno di essa con la siringa posta in aspirazione. Per ottenere materiale sufficiente per almeno 2 preparati, sono necessari dai 15 ai 20 movimenti (se l’ago non viene mosso all’interno della lesione si rischia di aspirare solo sangue). Prima di rimuovere l’ago, si rilascia lentamente lo stantuffo onde evitare l’aspirazione di materiale non appartenente alla lesione. Una volta fuori dalla lesione, l’ago viene rimosso dalla siringa. Previa aspirazione di qualche cc di aria nella siringa, questa viene riconnessa all’ago, la cui punta viene posta quasi a contatto con il vetrino su cui viene viene delicatamente spruzzato il materiale (3).
1.2 Campioni bioptici
Il prelievo bioptico è una procedura attraverso la quale si preleva un frammento di tessuto per poterlo analizzare microscopicamente. Il medico pone indicazione all’esecuzione della biopsia quando un esame o una condizione clinica fanno sospettare che quel tessuto sia affetto da patologia (es. evidenza radiologica di nodulo mammario, modificazione di lesione pigmentata della cute, alterazione degli enzimi epatici). Esistono numerosi tipi di biopsia, tutti mirati al prelievo di tessuto da esaminare.
L’agobiopsia è una procedura diagnostica mirata a ottenere un frammento da una massa sita all’interno del corpo. Può essere eseguita a mano libera per neoformazioni relativamente superficiali e ben delineate, oppure sotto guida radiologica per lesioni profonde o mal definibili dal contesto in cui si trovano (es. nodulo intra-ghiandolare in tiroide e ghiandole salivari, nodulo polmonare, ecc). Esistono diversi tipi di aghi bioptici che possono essere attivati manualmente o a molla e con diversa modalità di prelievo: i due sistemi principali sono il tru-cut e il Menghini. Entrambi forniscono un cilindro di tessuto (core biopsy), che viene posto in fissativo immediatamente dopo il prelievo. Il calibro dell’ago determina il diametro del cilindro di tessuto. Un cilindro di maggiori dimensioni aumenta il rischio di sanguinamento e di effetti collaterali, ma fornisce una maggiore disponibilità di materiale per la diagnosi. La scelta del calibro deve pertanto tenere conto di fattori legati al paziente, all’organo e alle caratteristiche della lesione (es estesa necrosi).
Biopsia a pinza endoscopica, broncoscopica, cistoscopica. I prelievi bioptici a pinza endoscopica vengono eseguiti in cavità del corpo raggiungibili mediante apparecchi endoscopici. La pinza, fornita di cavo e manovrata dall’esterno, entra nella cavità attraverso il canale operativo dello strumento e sotto guida ottica viene aperta in corrispondenza della lesione che si intende campionare. Le valve della pinza sono taglienti. Quando la pinza aperta è posta a ridosso della lesione, l’operatore chiude le valve e taglia un frammento di circa 2-3 mm di diametro. La pinza chiusa viene estratta dal canale operativo dell’endoscopio. Una volta estratta, la pinza viene aperta in un contenitore con fissativo dove il frammento liberato resta flottante e inizia il processo di fissazione. Per l’accurata diagnosi della lesione possono essere necessari più passaggi con prelievo bioptico. Alcuni protocolli diagnostici (es la diagnosi di malattia celiaca) richiedono un numero minimo di prelievi bioptici come criterio di adeguatezza.
Biopsia chirurgica. Il prelievo è eseguito con bisturi in un’area accessibile naturalmente (cute, cavo orale), o tramite incisione chirurgica: biopsia a cielo aperto (es nodulo sottocutaneo, nodulo della mammella, linfonodo superficiale) o per via laparoscopica (linfonodi profondi, formazioni peritoneali). Quando l’intera area è rimossa, si parla di biopsia escissionale, quando solo una parte della lesione è campionata a scopo diagnostico per programmare l’estensione dell’intervento di parla di biopsia incisionale. La biopsia chirurgica è detta anche tumorectomia, se con l'intervento chirurgico si procede alla rimozione della lesione sospetta (es nodulo mammario).
Con l’acronimo BOM si intende la biopsia osteo-midollare. Con questa procedura si preleva un frustolo di tessuto osseo spugnoso in una sede del corpo in cui questo è ricco di midollo emopoietico, di solito le creste iliache. La BOM trova indicazione in patologie ematologiche e di solito si associa a striscio midollare che consiste in citologia aspirativa della medesima sede, i cui vetrini vengono valutati per le popolazioni midollari.
Lesioni dermatologiche possono essere campionate con la “punch biopsy”. Si tratta di una pinza bioptica che, sotto anestesia, rimuove un cilindro di cute e sottocute del diametro compreso tra 3 e 10 mm. Il diametro si determina scegliendo il calibro della pinza in funzione della sede e della patologia da esaminare. La pinza è provvista di una lama circolare che produce il taglio del cilindro. Dopo la rimozione, il frammento viene posto in fissativo, facendo attenzione a evitare artefatti da compressione.
1.3 Campioni operatori
Le resezioni chirurgiche, possono essere distinte in resezioni parziali, totali o allargate, a seconda che riguardino una parte o totalità di un organo o coinvolgano più organi. Contrariamente all’esame isto-patologico su biopsie, nelle quali deve essere inclusa ed esaminata al microscopio la totalità del materiale prelevato, su resezioni chirurgiche il patologo deve selezionare l’area da esaminare al microscopio effettuando campionamenti macroscopici (c.d. “riduzione dei pezzi operatori”) secondo precisi protocolli e linee guida che consentano di definire la natura e l’estensione delle lesioni riscontrate, nonché l’adeguatezza dell’exeresi (esame dei margini di resezione) e, nel caso di patologia oncologica, la stadiazione della malattia. Per queste operazioni, che fanno parte dell’“esame macroscopico”, il patologo esamina il pezzo operatorio nella sua totalità, descrivendo con precisione forma, dimensioni, rapporti con eventuali altri organi espiantati e tutte le alterazioni che rappresentano una deviazione dalla normalità (4). Nel riportare le dimensioni (dell’organo come delle lesioni), è importante specificare se sono misurate sul pezzo a fresco o sul pezzo fissato in formalina; la fissazione, infatti, provoca una riduzione dimensionale e il dato potrebbe differire da quello rilevato in vivo dagli esami strumentali (es ecografia). Negli organi più vascolarizzati anche la misurazione a fresco può essere inferiore a quella in vivo, per via della perdita di liquidi e del circolo ematico. Il patologo, se necessario, può avvalersi di metodi di acquisizione delle immagini che permettono di documentare in modo permanente la morfologia e le caratteristiche del pezzo operatorio e le sedi dove sono stati eseguiti i campionamenti. I campionamenti delle parti da esaminare possono variare per numero e tipologia, in dipendenza delle caratteristiche del materiale asportato e dei quesiti clinici connessi. Poiché nel sezionamento del campione operatorio la prima fase è l’orientamento dello stesso, per fornire una diagnosi accurata occorre che il tessuto resecato chirurgicamente sia conservato e inviato al laboratorio di Anatomia Patologica in modo adeguato (integro, e se necessario orientato con clip metalliche o punti di sutura). Una volta asportato dal paziente e persi i riferimenti anatomici in vivo, può, infatti, non essere più ovvia la ricostruzione spaziale (es. lobo destro e sinistro in una tiroide deformata dai noduli di un gozzo). La riduzione macroscopica richiede infine da parte del patologo conoscenza dell’anatomia degli organi, per effettuare correttamente tagli e campionamento, come richiesto dalle linee guida per ciascuna patologia di organo. Le parti prelevate dal campione operatorio sono costituite da fette di spessore non > 3-4 mm, che, analogamente ai campioni bioptici, vengono poste in appositi cestelli detti biocassette e sottoposti a processazione (generalmente automatica) per la disidratazione e l’inclusione in paraffina.
2. TECNICHE DI ESECUZIONE DEGLI ESAMI IN ANATOMIA PATOLOGICA
2.1 Esame citologico
Per poter essere esaminato, il materiale citologico deve essere adeguatamente preparato e trattato. L’allestimento del campione citologico si realizza attraverso differenti tecniche, la cui scelta è in funzione del tipo di materiale.
Campioni ottenuti mediante scraping, brushing e aspirato vengono depositati in prossimità del lato corto di un vetrino porta-oggetto e, mediante un secondo vetrino, strisciati delicatamente ma con decisione affinchè lo striscio ottenuto sia sottile ed uniforme (striscio convenzionale). Strisci spessi con sovrapposizione degli strati di cellule sono difficili e spesso impossibili da interpretare.
In alternativa, il materiale raccolto può essere disciolto in un liquido fissativo (solitamente costituito da polietilen-glicole e alcol metilico) contenuto in un recipiente e posizionato in uno strumento computerizzato che provvede a depositarlo automaticamente in un singolo strato di spessore uniforme su un’area limitata del vetrino (allestimento dei preparati in strato sottile)(5). Questa metodica consente di ottenete preparati sempre in monostrato cellulare, privati degli elementi di disturbo (sangue, muco e materiale essudativo e necrotico), consente una riduzione del numero totale di vetrini allestiti e la possibilità di ottenere altri preparati sovrapponibili all’originale per la ripetizione in caso di necessità diagnostiche e/o per l’allestimento di metodiche speciali (colorazioni speciali e immuno-istochimica)(6).
Quando il campione citologico è costituito da una quantità esigua di liquido in cui è disperso uno scarso numero di cellule (liquor), il metodo di elezione è la tecnica di centrifugazione. Questa consente, mediante il ricorso a particolari centrifughe (cito-centrifughe), la stratificazione delle cellule in monostrato, concentrandole in una superficie con diametro di 4-8 mm.
In presenza di campioni citologici liquidi con elevata dispersione cellulare (urine), è da preferire l’uso della filtrazione su membrana. Vengono utilizzate membrane filtranti di policarbonato o cellulosa, aventi pori di diametro variabile tra 5 e 8 micron. Terminata la filtrazione, la membrana viene posta sul vetrino in modo che le cellule restino adese ad esso. La modalità di filtrazione può essere manuale o automatica.
In caso di materiale difficilmente strisciabile (materiali mucosi, materiali purulenti, frustoli simil-tessutali, coaguli fibrino-ematici e liquidi ricchi di cellule fortemente ematici), si procede all’allestimento del cito-incluso (7). Tale procedura è realizzata per lo più in aggiunta ad altre preparazioni citologiche. Il materiale, dapprima fissato o con alcol etilico 95° o soluzione al 10% di formalina tamponata o altre miscele di fissativi, viene centrifugato e il sedimento cellulato viene inglobato in gel liquido (più raramente di altre sostanze) e poi lasciato solidificare. Il coagulo di gel viene poi inserito in biocassetta e processato, incluso, sezionato e colorato con ematossilina-eosina come se si trattasse di un frammento bioptico. Tale metodica incrementa la possibilità della diagnostica morfologica e ha il vantaggio di consentire ulteriori sezioni del materiale per ulteriori indagini; presenta lo svantaggio di avere tempi di allestimento più lunghi della citologia convenzionale (da 1 a 2 giorni in funzione della fissazione del materiale) e maggior carico di lavoro per il personale di laboratorio.
Come regola generale, nell’allestimento del campione citologico un passaggio cruciale è rappresentato dalla fissazione (8). La fissazione è un processo fisico-chimico che ha come obiettivo la stabilizzazione dei costituenti della cellula, al fine di garantire una morfologia cellulare inalterata, senza compromissione da processi autolitici e fenomeni putrefattivi. Ritardi o difetti legati a inadeguata fissazione si riflettono sulla qualità del preparato allestito (rigonfiamento nucleare e degenerazione citoplasmatica) e sulla buona riuscita delle immuno-colorazioni. Il fissativo di elezione in citologia è l’alcol etilico 95° (i vetrini vengono solitamente fissati per immersione in una vaschetta contenente la soluzione fissativa). È da preferire l’uso di fissativi a pellicola, quali il Cytofix (alcol 95° + 2% di cera), per materiali poco aderenti al vetrino che potrebbero staccarsi durante l’immersione nel fissativo liquido. I preparati citologici allestiti mediante la tecnica del cito-incluso vanno fissati in soluzione alcolica e in formalina neutra tamponata al 10%. Per i campioni liquidi (urine, versamenti, ecc) è opportuno l’invio in laboratorio appena eseguito il prelievo. Nel caso in cui l’allestimento del preparato non possa essere realizzato subito dopo il prelievo, è possibile attuare una prefissazione del materiale, mediante aggiunta di alcol etilico 50° in quantità uguale a quella del campione, in modo da preservarlo fino al momento dell’allestimento.
La colorazione è il procedimento che consente di rendere visibili le cellule, aumentando il contrasto dei costituenti cellulari. Le colorazioni maggiormente impiegate in citologia sono colorazioni panottiche (aspecifiche o segnaletiche che sfruttano le diverse affinità tintoriali delle strutture cellulari). La colorazione Papanicolau è quella elettiva per la citologia e viene realizzata su campioni fissati in alcol etilico o con citospray. La colorazione Ematossilina-Eosina, che è quella maggiormente utilizzata nella routine istologica per i campioni fissati in formalina, in citologia viene utilizzata per colorare i cito-inclusi. La colorazione May-Grunwald-Giemsa è quella elettiva in ematologia, ma viene utilizzata anche per colorare ago-aspirati tiroidei e linfonodali, strisci di sangue periferico e liquidi di versamento a fresco. Viene realizzata su campioni strisciati su vetrini ed essiccati all’aria dopo allestimento, senza l’utilizzo di sostanze fissative.
2.2 Esame istologico
L’esame istologico consiste nell’osservazione microscopica dei tessuti e comprende biopsie e campioni chirurgici. Il materiale da osservare deve essere trasformato in sezioni sottili, che possano essere attraversate dal fascio di luce del microscopio. Il campione deve pertanto essere portato a una consistenza che ne permetta il taglio in sezioni da 4-6 micron.
La fissazione può essere considerata l’operazione più importante della tecnica istologica, da essa dipendendo la buona riuscita di un preparato microscopico. Ha un triplice scopo:
- immobilizzare i costituenti cellulari e tissutali del campione in uno stato più vicino possibile a quello di vita;
- consentire al preparato di sopportare gli stress fisici e chimici insiti nelle successive fasi di disidratazione, inclusione e sezionamento;
- preservare i campioni dalla degradazione e dall'attacco di muffe e batteri che potrebbero proliferare.
Poiché gran parte dell’architettura cellulare è costituita da proteine, un buon agente di fissazione per l’istologia deve essere principalmente un ottimo stabilizzatore di queste macromolecole. Inoltre, più è grande il campione, maggiore deve essere la capacità di penetrazione del fissativo nel tessuto. Tutti i fissativi, utilizzati in singolo o in combinazione, devono necessariamente essere preparati come soluzioni isotoniche e a pH 7.4, per impedire fenomeni di collasso o di rigonfiamento dei campioni, legati agli stress osmotici. I composti utilizzati come fissativi in istologia possono essere suddivisi in più classi chimiche:
- aldeidi (es, formaldeide);
- alcooli (es, etanolo);
- acidi organici e minerali (es, acido acetico, tricloroacetico, picrico, cromico);
- sali di metalli pesanti (es, bicromato di potassio, cloruro mercurico).
Il fissativo più utilizzato è la formalina (soluzione di formaldeide). La fissazione si ottiene per immersione del campione in formalina in un volume di fissativo corrispondente a circa 10 volte il volume del pezzo. Per pezzi anatomici grandi, si può utilizzare minore quantità di fissativo, cambiandolo 2 o 3 volte nelle 12 ore. Gli organi cavi devono essere aperti e puliti prima dell’immersione in formalina. La fissazione ottimale richiede 12-24 ore: tempi più brevi danno fissazione incompleta specialmente nei campioni chirurgici; tempi troppo lunghi alterano l’antigenicità del tessuto. Il tempo che intercorre tra l’escissione e la fissazione del tessuto è indicato come “tempo di ischemia fredda”, viene segnalato per gli effetti deleteri sulla preservazione di antigeni e acidi nucleici. Le linee guida americane del Collegio dei Patologi elencano il tempo di ischemia fredda come campo obbligatorio nella check-list della richiesta di esame istologico, così come le linee guida dell’ASCO per l’esecuzione di analisi immunocitochimiche a scopo predittivo nel carcinoma della mammella (9). Recenti linee guida Europee per indagini molecolari su tessuti sottolineano l’importanza del tempo di ischemia fredda sull’esito dell’analisi mutazionale. Campioni di elevato volume e spessore (es isterectomie) devono essere tagliati a fresco con sezioni parallele, per permettere la migliore penetrazione del fissativo. Si possono inserire garze tra i tagli per permettere una più agevole permeazione da parte del fissativo.
Il campionamento macroscopico deve essere eseguito sotto cappa chimica che eviti la dispersione e l’inalazione della formalina. Come prima descritto, tale campionamento ha lo scopo di individuare le parti del resecato chirurgico che si intendono sottoporre all’esame microscopico. Il prelievo, che non deve essere spesso più di 3-4 mm e deve essere dimensionato alla biocassetta, va eseguito con strumenti puliti e affilati. I blocchetti di paraffina, una volta raffreddati, vengono tagliati con un microtomo in sezioni dello spessore di circa 5 micron. Le sezioni raccolte su vetrini porta-oggetto sono poi sparaffinate, idratate e colorate.
I tessuti umani sono nella maggior parte dei casi incolori e generalmente prive di pigmenti e trasparenti, tanto da risultare pressoché invisibili al microscopio ottico. La loro osservazione richiede pertanto una colorazione. Per “colorante” si intende una molecola solubile e fornita di colore proprio, capace di legarsi stabilmente a substrati cellulari e tessutali, conferendo loro colore visibile. Quasi tutti i coloranti sono soluzioni. Le cellule e i tessuti derivati da prelievi per patologie neoplastiche, infiammatorie, degenerative ecc. rappresentano campioni biologici sovente irriproducibili, pertanto esiste una responsabilità etica e legale nella corretta esecuzione di tutte le fasi, dal prelievo alla consegna al laboratorio e alla lavorazione del materiale.
2.3 Esame intra-operatorio o estemporaneo
È una procedura diagnostica che viene eseguita in corso di intervento chirurgico, al fine di stabilire la presenza e la natura della lesione e/o la sua estensione in tempi rapidi (il 90% delle procedure vengono completate entro 20 minuti, dal tempo di consegna del campione al tempo di risposta della diagnosi al chirurgo)(10). Sono richiesti al patologo esperienza, conoscenza della clinica medica, capacità di prendere una decisione sotto pressione e consapevolezza delle limitazioni dovute alla metodica (11). L’accuratezza diagnostica di questa procedura è molto alta (93.3%) e va correlata principalmente al tipo di tessuto e alla natura del processo. Estremamente rari sono i falsi positivi, da correlarsi principalmente a alterazioni iatrogene e all’assenza di informazioni cliniche; maggiori sono invece i falsi negativi, dovuti a errata interpretazione della sezione al congelatore, specialmente se coesiste intensa flogosi oscurante, o a errato campionamento (con assenza di tessuto diagnostico nel materiale congelato, presente invece nel materiale non campionato) o all’ assenza di tessuto diagnostico nelle sezioni esaminate, ma presenza di questo nelle corrispettive sezioni al definitivo. Affinché il prelievo acquisti rapidamente le caratteristiche di durezza tali da renderlo sezionabile, si ricorre al congelamento, per cui sono necessari da pochi secondi a pochi minuti. I metodi più usati sono il congelamento con azoto liquido e il congelamento con strumenti termo-elettrici che sottraggono calore (camera criostato). Per una buona conservazione della morfologia cellulare, è fondamentale che il congelamento avvenga in tempi brevi: meno rapido è il congelamento, maggiore è il rischio di formazione nei tessuti di grossi cristalli di ghiaccio che comportano rottura delle membrane cellulari e un effetto “a maglia di rete” della sezione. Il prelievo, una volta congelato, viene tagliato al criostato (microtomo situato in una camera refrigerata) in sezioni di spessore compreso tra 5 e 8 micron. Tali sezioni vengono colorate con una ematossilina-eosina rapida, montate e consegnate al patologo per essere esaminate al microscopio ottico. Terminato l’esame estemporaneo, il frammento congelato viene immerso in formalina e quindi processato secondo routine.
3. INDAGINI ANCILLARI
La routine diagnostica può avvalersi di esami speciali, che aggiungono informazioni importanti per lo specifico quesito diagnostico. Tali esami non sono eseguiti in modo routinario ma vengono richiesti dal patologo dopo la valutazione iniziale del caso.
3.1 Cito-istochimica
Si tratta di colorazioni che permettono la visualizzazione di alcuni componenti in base alla loro composizione chimica.
- Colorazione tricromica per visualizzazione contemporanea di nucleo, citoplasma e connettivo. Differenzia i vari costituenti mesenchimali. La più diffusa è la tricromica di Masson: nuclei viola, citoplasmi e tessuto muscolare rosso, collagene blu.
- Colorazione PAS per il glicogeno e i glico-coniugati, colora anche la maggior parte dei microorganismi.
- Le fibre reticolari sono delicate fibre argirofile, organizzate in reti tridimensionali che si colorano con impregnazione di argento ammoniacale (metodo di Manuel, di Wilder, ecc).
- Rosso Congo per l’amiloide.
- PASM per mucopolisaccaridi della membrana di funghi e batteri.
- Metodo di Perls per il ferro.
Esistono poi colorazioni isto-enzimatiche mirate a identificare attività di singoli enzimi nei tessuti. Tale metodica richiede spesso l’uso di sezioni di tessuto congelato, perchè l’attività enzimatica si perde con la fissazione.
3.2 Immunocitochimica-immunoistochimica
È una tecnica diagnostica che utilizza le proprietà delle reazioni immunologiche, per identificare ed evidenziare morfologicamente la localizzazione di determinati antigeni, propri di cellule e tessuti, attraverso il riconoscimento da parte di specifici anticorpi. Gli antigeni bersaglio sono rappresentati da proteine o molecole presenti nei tessuti, contro le quali è preparato in vitro l’anticorpo specifico.
La finalità della metodica è il miglioramento dell’accuratezza delle diagnosi citologiche e istologiche (marcatori diagnostici), la previsione del comportamento clinico di una neoplasia (marcatori prognostici) e la risposta alla terapia (marcatori predittivi).
L’utilità diagnostica consiste nella caratterizzazione di neoplasie scarsamente differenziate, nella differenziazione dei tumori primitivi da quelli metastatici e nella determinazione del sito di origine delle lesioni metastatiche (12,13). A tale scopo si utilizzano marcatori di specificità cellulare (es: tireoglobulina, calcitonina e paratormone, specifici rispettivamente per l’identificazione delle cellule follicolari della tiroide, delle cellule C della tiroide e del tessuto paratiroideo) e marcatori di malignità, che usano anticorpi contro antigeni associati alla malignità (es. Galectina3, CK 19, HBM-1: associazione di marcatori utilizzati per la diagnosi di malignità di lesioni tiroidee) (14,15). La certezza di una corretta interpretazione diagnostica dei risultati è strettamente legata alla qualità del preparato citologico o istologico. Fattore predominante per la realizzazione di un’immuno-colorazione ottimale è la fissazione adeguata dei campioni, che si traduce nella scelta appropriata del fissativo e della modalità di fissazione. Una fissazione inappropriata può distruggere parzialmente i determinanti antigenici o alterarne la struttura fino a renderli irriconoscibili per l’anticorpo:
- un’ipofissazione può conservare inalterato l’antigene ma può causarne la delocalizzazione (es diffusione citoplasmatica di un antigene nucleare) e può danneggiare la morfologia tissutale con conseguente difficoltà di interpretazione;
- un’iperfissazione mantiene la morfologia cellulare ma distrugge, denatura o maschera un gran numero di antigeni.
In teoria andrebbe scelto un fissativo specifico per ogni campione da esaminare, in base a natura e composizione di quest’ultimo (procedura non attuabile nella diagnostica istocito-patologica di routine, soprattutto a causa dei costi elevati). Il fissativo maggiormente utilizzato nella routine istopatologica è la formalina (soluzione al 10% di formalina neutra tamponata), che determina una buona preservazione dell’antigenicità, presenta un elevato potere di penetrazione, non induce coartazione dei tessuti e ha un costo relativamente basso. Si considerano ottimali tempi di fissazioni in formalina di 2-6 ore per piccole biopsie, 24-48 ore per campioni costituiti da pezzi chirurgici. Per i preparati citologici si ricorre alla fissazione alcolica. Le variabili che incidono sui risultati delle immuno-colorazioni (rendendosi responsabili di artefatti che si traducono in false colorazioni) possono essere molteplici e legate a errori nella varie fasi attraverso cui si realizza la metodica (tempi di lavorazione del campione, fissazione, sistema di ripristino antigenico, scelta dell’anticorpo, inibizione di enzimi endogeni, sistema di rivelazione dell’antigene, formazione del complesso substrato-cromogeno, lettura ed interpretazione). Artefatti possono anche essere legati al prelievo: nei preparati citologici non in monostrato i reagenti possono venire intrappolati tra gli strati e conferire al preparato una colorazione di fondo che rende difficile l’interpretazione dei risultati; tessuti prelevati mediante elettro-bisturi o crio-bisturi presentano fenomeni di coagulazione cellulare sul versante dell’exeresi, con conseguente migrazione e diffusione interstiziale di enzimi ossidativi, prevalentemente lisosomiali, responsabili di un’immuno-colorazione diffusa non specifica. Artefatti possono essere secondari anche al non corretto campionamento del pezzo operatorio da parte del patologo: sezioni spesse rendono difficoltosa la penetrazione del fissativo, con conseguente ipofissazione della porzione centrale del campione che si traduce in una perdita di antigenicità nelle aree più interne. Campioni costituiti da tessuti necrotici o ricchi in stravasi emorragici o infiltrato infiammatorio presentano un’immuno-colorazione non specifica (intenso background), secondaria alla liberazione di enzimi ossidativi responsabili di modificazioni dell’assetto antigenico delle cellule.
Anche la lettura e l’interpretazione del preparato immuno-istochimico possono rappresentare una trappola diagnostica. In un esame immuno-istochimico è necessario individuare la localizzazione della positività (nucleare, citoplasmatica, di membrana) e la sua compatibilità con il marcatore impiegato, per accertare se il reperto ottenuto è un vero positivo o se la colorazione è artefattuale; successivamente, va analizzato il pattern di colorazione (granulare, lineare, dot-like) e va verificato se questo è espressione di condizioni normali o patologiche. Contemporaneamente va ricercato il controllo interno, ovvero vanno individuate all’interno del tessuto quelle cellule che sono sicuramente positive per l’antigene ricercato (es. proteina S100 nei tronchi nervosi). Quando non si dispone di un controllo interno, il confronto viene fatto con campioni selezionati in precedenza, che fungono da controllo esterno per gli specifici antigeni. Tali sezioni di controllo sono spesso costituite da tissue array (in un’unica inclusione vengono montati insieme cilindri di tessuto di differenti campioni scelti sulla base del contenuto di antigene)(16): in questo modo un’unica sezione fornisce controlli positivi e negativi per l’immuno-istochimica. Per ridurre al minimo il rischio di errore e ottenere la certezza di una corretta interpretazione diagnostica dei risultati, si rende indispensabile la standardizzazione dell’intera procedura immuno-istochimica.
L’immuno-istochimica può essere eseguita anche mediante l’utilizzo di anticorpi coniugati con fluorofori: in tal caso si parla di immuno-fluorescenza e deve essere osservata con microscopi dotati di lampade apposite.
3.3 Microscopia elettronica
È una risorsa diagnostica relativamente complessa e costosa, che trova il suo utilizzo quando si devono ottenere ingrandimenti microscopici superiori alla risoluzione del microscopio ottico. Il potere risolutivo cresce proporzionalmente al decrescere della lunghezza d’onda della radiazione impiegata; infatti, la scoperta che gli elettroni hanno una radiazione di bassissima lunghezza d’onda ha suggerito la possibilità di usare fasci di elettroni per ottenere poteri risolutivi assai elevati. Il microscopio elettronico è un tipo di microscopio che non sfrutta la luce come sorgente di radiazioni, ma un fascio di elettroni. Usando un fascio di elettroni, infatti, è possibile raggiungere una risoluzione parecchi ordini di grandezza superiore.
Nel microscopio elettronico a trasmissione (TEM) gli elettroni che costituiscono il fascio attraversano una sezione dove è stato creato precedentemente il vuoto, per poi passare completamente attraverso il campione. Dopo che il fascio ha attraversato il campione, viene focalizzato da una lente “obiettivo” e quindi allargato e proiettato su uno schermo fluorescente. Le zone dello schermo che appaiono scure sono dovute, appunto, a un’irregolare deviazione degli elettroni da parte delle dislocazioni della struttura del campione. L’uso diagnostico della microscopia elettronica è ristretto a poche patologie (es. glomerulopatie renali) e richiede una fissazione dedicata (gluteraldeide o miscele di gluteraldeide) e la post-fissazione in Osmio. I campioni, dovendo essere tagliati in sezioni ultrasottili, vengono inclusi in resine invece che in paraffina e tagliati con lame estremamente dure. Le sezioni ultrasottili sono raccolte su speciali retini inseribili nel microscopio.
4. PROCEDURE DI PREPARAZIONE E CONSEGNA DEL MATERIALE IN LABORATORIO
4.1 Confezionamento ed etichettatura dei campioni isto-citologici
L'équipe operatoria deve verificare, per ogni richiesta di esame, la corretta etichettatura del o dei contenitori (sulle pareti e non sul coperchio) con le seguenti informazioni:
- identificazione del paziente con almeno tre elementi (nome, cognome e data di nascita oppure nome, cognome e codice fiscale);
- identificazione del materiale, specificando data del prelievo, tipo di prelievo, localizzazione topografica e lateralità del prelievo;
- in caso di prelievi multipli differenziati, deve essere riportato il numero arabo progressivo corrispondente a quello indicato nella richiesta d’esame;
- è richiesta la notifica di rischio biologico quando sia nota ai curanti una rilevante patologia infettiva (es. HIV+).
Il Ministero del Lavoro, della Salute e delle Politiche Sociali ha prodotto nel 2009 il “Manuale per la Sicurezza in sala operatoria: Raccomandazioni e Checklist” (17), dove, nell’Obiettivo “Identificare in modo corretto i campioni chirurgici”, viene sottolineato che “La non corretta identificazione dei campioni chirurgici può causare gravi conseguenze ai pazienti e la prevenzione di tali errori è fondamentale per la sicurezza dei pazienti”.
4.2 Richiesta di esame
Ogni esame istologico o citologico deve essere accompagnato da modulo di richiesta correttamente compilato (cartaceo o informatico a seconda del livello di automazione della Struttura). L'équipe operatoria deve verificare la corretta compilazione della richiesta con le seguenti informazioni:
- identificazione del paziente (nome, cognome, data di nascita, sesso o sistemi di identificazione mediante codice a barre o similari);
- identificazione del richiedente (unità operativa, nome, cognome e firma del richiedente);
- identificazione del materiale, specificando data del prelievo, tipo di prelievo, localizzazione topografica e lateralità del prelievo (es. lobo superiore del polmone sinistro, ecc.);
- numero di contenitori; in caso di prelievi multipli differenziati deve essere riportato il numero arabo identificativo del campione, specificando quanto riportato nel punto 3;
- notizie cliniche pertinenti (es. patologie pregresse correlabili, risultati di esami di imaging, terapie in corso o pregresse);
- firma leggibile del medico richiedente (preferibilmente associata a timbro o stampa del nome);
- è richiesta la notifica di rischio biologico quando sia nota ai curanti una rilevante patologia infettiva (es. HIV+).
Le procedure descritte, sia con riferimento alla richiesta che al contenitore, devono essere considerate come obbligatorie, al fine di garantire una corretta identificazione del paziente, del campione e della provenienza e di ridurre al minimo rischi di scambio, smarrimento o incompleta valutazione del materiale (18).
4.3 invio dei campioni al laboratorio
Il trasporto dei campioni al laboratorio deve essere eseguito utilizzando un’apposita valigetta porta-campioni. I campioni devono viaggiare accompagnati dalla relativa richiesta di esame. Alla consegna del materiale il tecnico di Anatomia Patologica appone una firma per materiale ricevuto sulla lista dei campioni (lista di riscontro). Tale firma equivale alla presa in carico dell’esame. In caso di non conformità, il materiale viene restituito al camminatore con la nota per le correzioni del caso.
In ciascuna Istituzione deve essere implementata una procedura atta a garantire la sicurezza degli operatori coinvolti nelle operazioni di confezionamento, trasporto e spedizione di campioni diagnostici e di materiali biologici a potenziale rischio infettivo, impedire la dispersione nell’ambiente di potenziali agenti infettanti e far sì che il materiale giunga a destinazione nei tempi e nelle condizioni ottimali, al fine di poter essere analizzato, garantendo la sicurezza del personale di laboratorio e l’attendibilità del risultato diagnostico.
5. CONSERVAZIONE DEI CAMPIONI
La conservazione del materiale diagnostico tiene conto della distinzione tra il materiale processato ai fini dell’esame microscopico (vetrini e inclusioni in paraffina) e il materiale non processato, cosiddetta “riserva non campionata” costituita dai residui di materiale su cui è stato eseguito il campionamento macroscopico e che non è stato sottoposto a processazione (es. residuo di segmento intestinale da cui sono stati prelevati per esame istologico il tumore, i linfonodi ed i margini di resezione). Mentre per la prima categoria di materiale si pone un problema di archiviazione per lungo periodo, per la seconda categoria le esigenze sottese alla relativa conservazione sono limitate temporalmente.
5.1 Vetrini e inclusioni
La circolare n. 61 del 19 dicembre 1986 N. 900.2/ AG. 464/260, ha definito il “periodo di conservazione della documentazione sanitaria presso le istituzioni sanitarie pubbliche e private di ricovero e cura”. In analogia a quanto stabilito per le radiografie, è stato ritenuto che il materiale isto-citologico possa essere considerato parte della “restante documentazione diagnostica” e possa essere applicato lo stesso periodo di conservazione di venti anni. In data 14 ottobre 1987 la terza sezione del Consiglio Superiore della Sanità ha precisato che soltanto il materiale diagnostico istologico (costituito dal vetrino e dalla relativa inclusione), indipendentemente dalla positività o negatività del referto, debba essere considerato parte integrante della “restante documentazione diagnostica” di cui alla circolare e quindi conservato 20 anni, mentre si esprime un’indicazione alla conservazione per cinque anni per i preparati citologici.
5.2 Contenitori con residui biologici
Vengono conservati in armadi aspirati per prevenire la diffusione di vapori di formalina. Sono disposti nei ripiani in modo tracciato, per poterli recuperare in caso di necessità di ulteriori prelievi. Di solito vengono smaltiti 30 giorni dopo la refertazione del caso. Eccezioni alla conservazione del residuo possono essere circostanziate da particolari situazioni (es. patologia rara).
BIBLIOGRAFIA
- Blumenfeld W, Hashmi N, Sagerman P. Comparison of aspiration, touch and scrape preparations simultaneously obtained from surgically excised specimens. Effect of different methods of smear preparation on interpretive cytologic features. Acta Cytol 1998, 42: 1414-8.
- Shiver SA, Creager AJ, Geisinger K, et al. Intraoperative analysis of sentinel lymph nodes by imprint cytology for cancer of the breast. Am J Surg 2002, 184: 424-7.
- Abele JS, Miller TR, King EB, Lowhagen T. Smearing techniques for the concentration of particles from fine needle aspiration biopsy. Diagn Cytopathol 1985, 1: 59-65.
- Lott R, Tunnicliffe J, Sheppard E, et al. Pre-microscopic examination specimen handling. Guidelines in the surgical pathology laboratory. College of American Pathologists (CAP) and National Society for Histotechnology. Published by HISTOQIP 2014: 1-49.
- Michael CW, Hunter B. Interpretation of fine-needle aspirates processed by the ThinPrep technique: cytologic artifacts and diagnostic pitfalls. Diagn Cytopathol 2000, 23: 6-13.
- Dey P, Luthra UK, George J, et al. Comparison of ThinPrep and conventional preparations on fine needle aspiration cytology material Acta Cytol 2000, 44: 46-50.
- Karnauchow PN, Bonin RE. "Cell-block" technique for fine needle aspiration biopsy. J Clin Pathol 1982, 35: 688.
- Mygind H, Lauritzen AF. Influence of prefixation and fixation times on cellular preservation and epithelial cellularity in filter imprints. Acta Cytol 1987, 31: 950-3.
- Raccomandazioni Gruppo Italiano di studio di Patologia Mammaria (GIPaM). 2013.
- White VA, Trotter MJ. Intraoperative consultation/final diagnosis correlation: relationship to tissue type and pathologic process. Arch Pathol Lab Med 2008, 132: 29-36.
- Lechago J. The frozen section: pathology in the trenches. Arch Pathol Lab Med 2005, 129: 1529-31.
- DeYoung BR, Wick MR. Immunohistologic evaluation of metastatic carcinomas of unknown origin: an algorithmic approach. Semin Diagn Pathol 2000, 17: 184-93.
- DeLellis RA, Dayal Y. The role of immunohistochemistry in the diagnosis of poorly differentiated malignant neoplasms. Semin Oncol 1987, 14: 173-92.
- Leong AS. Immunostaining of cytologic specimens. Am J Clin Pathol 1996, 105: 139-40.
- Mataraci EA, Ozgüven BY, Kabukçuoglu F. Expression of cytokeratin 19, HBME-1 and galectin-3 in neoplastic and non neoplastic thyroid lesions. Pol J Pathol 2012, 63: 58-64.
- Rimm DL, Camp RL, Charette LA, et al. Tissue microarray: a new technology for amplification of tissue resources. Cancer J 2001, 7: 24-31.
- Ministero del Lavoro, della Salute e delle Politiche Sociali - Dipartimento della Qualità, Direzione Generale della Programmazione Sanitaria, dei Livelli di Assistenza e dei Principi Etici di Sistema - Ufficio III. Manuale per la sicurezza in sala operatoria: raccomandazioni e checklist. Ottobre 2009: 1-62.
- National Institutes of Health. Biospecimen Working Group. Guidelines for human biospecimen storage and tracking within the NIH. Intramural Research Program 2013.
Introduzione alle tecniche di biologia molecolare

Minidizionario di genetica
Alberto Falchetti
Centro Hercolani e Villa Alba (GVM), Bologna
EndOsMet, Villa Donatello, Firenze
(aggiornato al 19 dicembre 2018)
ACCOPPIAMENTO ASSORTATIVO: scelta di un accoppiamento in base a un particolare carattere. L’accoppiamento assortativo è positivo quando le persone tendono a scegliere accoppiamenti con individui di caratteristiche simili ed è il caso più frequente. L’accoppiamento assortativo negativo avviene quando le persone tendono a scegliere accoppiamenti con individui differenti ed è meno comune.
ACRO-CENTRICO: cromosoma il cui centromero è molto vicino alla parte terminale, dando al cromosoma una forma “a V”. Cromosomi acrocentrici sono il 13, 14, 15, 21 e 22. Le braccia corte 'p' sono molto corte e solitamente hanno piccole appendici sferiche sul peduncolo, note come “satelliti”.
AFLP (AMPLIFIED FRAGMENT LENGTH POLYMORPHISM): metodica basata su PCR, utilizzata nella ricerca genetica, nell'analisi del DNA e nella pratica dell'ingegneria genetica. È una metodica altamente sensibile per la rilevazione di polimorfismi nel DNA.
AGGREGAZIONE O CLUSTERING FAMILIARE: prevalenza della malattia tra i membri della famiglia che eccede quella attesa nella popolazione generale.
ALLELI: forme alternative di un gene.
ALLELI NULLI: mutazioni che non si traducono in un prodotto genetico o in assenza di funzione a livello fenotipico.
ANALISI DELL’ETERODUPLEX: metodo di rilevazione delle differenze di sequenza tra il DNA normale e il DNA da testare. Viene utilizzato comunemente come metodo di screening per individuare le mutazioni potenziali in un gene.
ANALISI DI LINKAGE: tecnica di “caccia” genetica, che traccia modelli di malattia nelle famiglie ad alto rischio. Cerca di localizzare un gene che provoca una malattia, identificando marcatori genetici di posizione cromosomica nota, che sono co-ereditati con il caratteristico tratto d’interesse.
ANALISI DI SEGREGAZIONE: in accordo alla genetica classica, i membri di una data coppia di alleli si separano l’uno dall’altro, quando un individuo forma le proprie cellule germinali. Negli organismi diploidi (2n) come l’uomo, la segregazione descrive il processo di separazione dei due geni codificanti per lo stesso carattere presenti su cromosomi omologhi al momento della produzione dei gameti (Prima legge di Mendel). La separazione di geni diversi è invece regolata dalla Seconda legge di Mendel. Aiuta a determinare il modo più probabile di eredità per il tratto o la malattia in studio.
ANTICIPAZIONE DI MALATTIA: al susseguirsi delle generazioni i pazienti mostrano forma più grave della malattia ed esordio più precoce.
ANTICIPAZIONE GENETICA: fenomeno in cui i segni e i sintomi di alcune condizioni genetiche tendono a diventare più gravi e/o appaiono a un'età precedente, poiché il disordine è passato da una generazione all’altra. La malattia di Huntington è un esempio di disordine genetico in cui il meccanismo biologico di questo fenomeno è stato ben documentato. In altri casi, può essere dovuto a fattori quali aumentata sorveglianza o altre cause non genetiche.
APLO-INSUFFICIENZA: situazione che si verifica quando una copia di un gene è inattivata o soppressa e la copia funzionale restante del gene non è adeguata per produrre il prodotto geneticamente necessario per preservare la normale funzione.
APLOTIPO: set di alleli strettamente legati su un cromosoma, normalmente ereditato come un blocco.
AUTOZIGOTE: individuo che riceve due geni identici per discendenza.
BANDEGGIO: serie di strisce scure e chiare attraverso un cromosoma, prodotte dal trattamento dei cromosomi con diverse soluzioni chimiche. Esiste un numero differente di tecniche di colorazione che producono diversi aspetti (es. G-banding, R-banding, ecc).
BANDEGGIO G: pattern di bandeggio sui cromosomi che rende più facile esaminare i cromosomi sotto il microscopio per anomalie nella struttura e nel numero. Le bande G sono un tipo di pattern di bandeggio, indotto per apparire sui cromosomi attraverso la loro colorazione con un reagente chiamato Giemsa.
BIALLELICO: di o pertinente a entrambi gli alleli di un singolo gene (paterni e materni). Per esempio, i portatori biallelici di mutazione hanno una mutazione (non necessariamente la stessa mutazione) in entrambe le copie di un gene specifico (una paterna e una mutazione materna).
CENTROMERO: parte ristretta dei cromosomi che li separa in due braccia. Il braccio corto è chiamato braccio 'p' (per 'piccolo'); il braccio lungo è chiamato 'q' (perché q segue p nell’alfabeto).
CLONE: una copia identica di una sequenza di DNA o di un intero gene; una o più cellule derivate e identiche a una singola cellula antenata o per isolare una sequenza specifica di DNA o un gene.
CO-DOMINANTE: nel caso in cui lo stato eterozigote esprima un fenotipo distinto da quello dei due stati omozigoti.
CODONI: combinazioni di “parola” di 3 lettere chimiche A, G, C e T. Queste parole costituiscono il codice genetico (DNA) che informano la cellula nel produrre un prodotto (proteina) che le cellule possono utilizzare. Noti anche come triplette di nucleotidi.
COEFFICIENTE DI CONSANGUINEITÀ: probabilità che due gameti portino alleli ad un locus autosomico identici per discendenza. Tra estranei è 0.
COEFFICIENTE DI INBREEDING (F): probabilità che un individuo sia autozigote.
COEFFICIENTE DI ININCROCIO (F): probabilità che un individuo omozigote abbia ricevuto ambedue gli alleli di un paio da un antenato (omozigote per discendenza o identità per discendenza). Oppure, proporzione dei loci a cui una persona è omozigote per discendenza.
COEFFICIENTE DI PARENTELA: geni identici condivisi tra due parenti = 2F.
COLLO DI BOTTIGLIA: la popolazione subisce un drammatico calo di dimensioni.
COMPLEMENTAZIONE: incrocio, fra di loro, di individui che presentano mutazioni che danno luogo allo stesso fenotipo. Ad esempio, considerando geni che regolano singoli eventi di una catena metabolica, la presenza di un fenotipo mutato può essere causata da una mutazione su uno qualsiasi dei geni che appartengono alla catena metabolica che risulta essere mutata. Il test della complementazione permette di evidenziare se due individui, o due cellule, che presentano la stessa mutazione fenotipica abbiano subito una mutazione sullo stesso gene o su geni diversi. In maniera più complessa e tecnica, è l’incrocio di due fenotipi recessivi che dà un fenotipo dominante, perché gli individui sono omozigoti recessivi su geni diversi in condizioni di eterogeneità genetica.
CONGENITO: presente alla nascita, non necessariamente ereditato.
CONSANGUINEITÀ O ININCROCIO: accoppiamenti fra individui che sono imparentati per discendenza di un progenitore comune (es. primi cugini). Malattie rare recessive sono più comuni nella progenie di accoppiamenti consanguinei.
COPY NUMBER VARIANT (CNV): si riferisce al tratto genetico che comporta il numero di copie di un particolare gene presente nel genoma di un individuo. Le varianti genetiche, comprese le inserzioni, le delezioni e le duplicazioni di segmenti di DNA, sono anche collettivamente denominate varianti di copie. Le varianti di copie rappresentano una percentuale significativa della variazione genetica tra i singoli.
CORPO DI BARR: è la cromatina altamente conservata del cromosoma X inattivo, presente in ciascuna cellula somatica femminile.
COSEGREGAZIONE: la trasmissione, insieme, di due o più geni sullo stesso cromosoma, a causa della loro vicinanza fisica (cioè “linked”).
COUNSELING GENETICO: serve per:
- valutare la storia familiare e i dati clinici;
- ordinare il test specifico;
- valutare i risultati del test;
- aiutare i pazienti e i loro familiari a comprendere e raggiungere la decisione sul da farsi.
CROMATINA: DNA e proteine istoniche e non, si distingue in:
- eucromatina: meno condensata, corrisponde a zone in cui vi è un'intensa attività di trascrizione per la sintesi proteica;
- eterocromatina: più condensata, non sembra presentare attività di trascrizione. A sua volta si divide in facoltativa o costitutiva.
CROMOSOMA: struttura a bastoncello trovata nel nucleo di tutte le cellule (eccetto i globuli rossi) consistente di DNA e proteine. Ciascun cromosoma può essere pensato come un filo di sfere in cui ciascuna sfera rappresenta un gene.
CROSSING-OVER: quando le coppie di cromosomi si uniscono insieme durante la meiosi (il processo di divisione che produce gli oociti e le cellule spermatiche), i due cromosomi possono scambiarsi materiale genico: parte di un cromosoma 'incrocia sopra' e scambia pezzi con la corrispondente parte sul suo cromosoma partner.
DELEZIONE: la perdita di parte di materiale genetico da un cromosoma. Se la delezione è ampia, può essere osservata nel cariotipo come materiale cromosomico perso. Se piccola, può essere identificata solo con l’analisi del DNA.
DERIVA GENETICA: fluttuazioni casuali irregolari delle frequenze geniche di una popolazione, causate da una diminuzione drastica della popolazione, dovuta a una catastrofe o a un isolamento o a una migrazione. Può causare fissazione di un allele.
DNA: base molecolare dell’eredità, codifica le informazioni genetiche incaricate dello sviluppo e della funzione di un organismo e permette di trasmettere le informazioni genetiche da una generazione all'altra. La molecola di DNA è strutturata come una doppia elica, tenuta insieme da legami deboli di idrogeno tra le coppie di base nucleotidiche di purine - pirimidina: adenina (A) accoppiata con timina (T) e guanina (G) accoppiata con citosina (C).
DNA BARCODING: metodo tassonomico che utilizza una parte di geni specifici, codice a barre, per identificare un organismo rispetto alle specie. I codici a barre sono talvolta utilizzati per identificare specie sconosciute, parti di un organismo o semplicemente per catalogare quanti più “taxa” esistenti.
DNA FINGERPRINT: processo per determinare le caratteristiche del DNA di un individuo, che sono uniche come le impronte digitali.
DNA SATELLITE: ripetizione di sequenze di lunghezza variabile (5-200 coppie di basi, bp). Si estendono per centinaia di Kbp (10% del DNA umano). Si trovano nelle regioni di eterocromatina e nelle zone centromeriche (non sono trascritte).
DOMINANTE: tratto/carattere che si esprime nell’eterozigote, cioè, nel caso di malattia, in cui sia sufficiente una sola copia del gene difettoso per esprimere il fenotipo affetto.
DUPLICAZIONE: una parte del cromosoma è presente in due o più copie. Se la duplicazione è ampia, può essere osservata nel cariotipo come cambiamento in un cromosoma; una piccola duplicazione può essere osservata solo esaminando la struttura del DNA del cromosoma o di un gene.
EFFETTO DEL FONDATORE: uno o più individui fondano una nuova popolazione. Definito come un’alta frequenza di un gene mutante in una popolazione fondata da un piccolo gruppo ancestrale, quando uno o più fondatori sono portatori dell’allele mutante. Se uno dei fondatori è portatore di un allele relativamente raro, quell’allele si potrà fissare nella popolazione in un tempo relativamente breve.
EMIZIGOTE: individuo che ha un solo membro di una coppia di cromosomi o di un segmento di cromosoma piuttosto che i soliti due. Spesso l'emizigosità è usata per descrivere geni X-linked nei maschi che hanno un solo cromosoma X. Questo termine è usato a volte nella genetica della cellula somatica, in cui le linee cellulari sono spesso emizigoti per taluni alleli o regioni cromosomiche.
ENZIMA DI RESTRIZIONE: particolare tipo di enzima in grado di riconoscere specifiche sequenze di basi lungo il filamento di DNA, sequenza di riconoscimento o sito di restrizione, e di "tagliare" esattamente in corrispondenza di queste sequenze. È così possibile scegliere con elevata precisione dove tagliare un campione di DNA. I tratti di DNA che si ottengono sono chiamati frammenti di restrizione, che possono avere lunghezze diverse e, pertanto, separabili gli uni dagli altri.
EPIGENETICA: la produzione di cambiamenti ereditabili nel modo in cui un gene lavora o funziona, senza alterare la sequenza del DNA. La modalità con cui ciò può essere cambiato è quella di cambiamenti strutturali. Tra gli esempi figurano la metilazione del promoter e le modifiche degli istoni.
EPISTASI/IPOSTASI: interazione di 2 geni, non allelici, ovvero non codificanti lo stesso prodotto proteico, in cui un gene altera l’espressione dell’altro che non è ad esso legato fisicamente (determinazione multigenica di un fenotipo). Il gene “mascherato” si dice in ipostasi rispetto al gene epistatico e viceversa. Una relazione epistatica-ipostatica tra 2 loci è simile alla relazione dominante-recessivo tra alleli ad un determinato locus.
EQUILIBRIO DI HARDY-WEINBERG: si realizza se:
- c’è accoppiamento casuale;
- non c’è selezione per un particolare allele;
- non ci sono mutazioni;
- non c’è immigrazione;
- c’è una grande popolazione;
- c’è fitness, cioè capacità di un particolare genotipo di sopravvivere e riprodursi.
EREDITÀ: il passaggio di caratteristiche genetiche da un genitore ai figli.
EREDITABILITÀ: percentuale di variazione in un tratto di popolazione che può essere attribuito a fattori genetici ereditati. Le stime di eredità variano da 0 a 1 e sono spesso espresse in percentuale. Un numero vicino a 1 può essere indicativo di un tratto di caratteri molto ereditabile all'interno di una popolazione. Non deve essere utilizzata per stimare il rischio su base individuale.
EREDITARIETÀ: frazione della varianza fenotipica totale di un tratto quantitativo causato da geni. È quindi una misura della grandezza con cui alleli differenti a vari loci sono responsabili della variabilità in un dato tratto nella popolazione.
EREDITARIETÀ COMPLESSA: geni e fattori ambientali sono ambedue importanti. Non c’è ovvio pattern d’ereditarietà. Molti dei geni determinanti queste patologie non sono stati identificati.
EREDITARIETÀ MENDELIANA: mutazioni in un gene malattia possono tipicamente spiegare perché individui sviluppano la malattia. Il pattern d’ereditarietà è facilmente riconoscibile. La maggior parte dei geni che causano queste malattie sono stati identificati.
EREDITARIO: il trasferimento di un gene da un genitore a un figlio, attraverso il DNA delle cellule germinali.
ESOMA: l’insieme di tutte le porzioni del genoma che “codificano” per proteine. Pur rappresentando una quota vicina all’1% dell’intero genoma, è al suo interno che avviene oltre l’85% delle mutazioni clinicamente rilevanti. Le tecniche di sequenziamento di nuova generazione (NGS) consentono di analizzare le sequenze codificanti degli oltre 20.000 geni di un individuo in un unico test.
ESONE: sequenza di DNA presente in un RNA messaggero maturo, che codifica gli aminoacidi di una proteina. Molti geni hanno più esoni intercalati con introni.
ESPRESSIVITÀ: indicazione della natura e gravità del fenotipo a parità di genotipo, grado con cui una caratteristica ereditata è espressa in una persona.
ESPRESSIVITÀ VARIABILE: esprime la gravità fenotipica di un certo genotipo nell’ambito di una famiglia [es: distrofia miotonica: alcuni pazienti possono avere solo alcuni sintomi clinici (cataratta)].
ETEROGENEITÀ ALLELICA: differenti mutazioni nello stesso gene possono determinare fenotipi diversi (es. gene LRP5 può dare High Bone Mass Syndrome, Osteoporosis pseudoglioma; gene FGFR3 può dare Acondroplasia, Displasia Tanatoforica I, Ipocondroplasia).
ETEROGENEITÀ GENETICA: di locus o allelica.
ETEROGENEITÀ GENICA: geni diversi responsabili dello stesso fenotipo. Caratteri che hanno più cause geniche (es. sordità profonda).
ETEROZIGOTE: porta 2 alleli differenti ad un locus.
ETEROZIGOTE DOPPIO: soggetto con presenza di due alleli mutanti mutati in due loci genetici distinti.
FENOCOPIA: un individuo ha la malattia, ma non ha il gene-malattia. Fenotipo che ha cause non geniche, indistinguibile da fenotipi controllati geneticamente (esempio: sordità causata da infezione di rosolia nella madre, presenta lo stesso fenotipo della sordità profonda che è ereditaria).
FENOTIPO: caratteristica osservabile di un individuo, o di una cellula. Risultato osservato d’interazione genotipo/fattori ambientali.
FENOTIPO MOSAICO: fenotipo diverso nelle diverse cellule di un individuo.
FITNESS: capacità di un particolare genotipo di sopravvivere e riprodursi. È contesto specifica e relativa, cioè va misurata in relazione ad altri genotipi.
FLUORESCENCE IN SITU HYBRIDIZATION (FISH): tecnica utilizzata per identificare la presenza di cromosomi specifici o regioni cromosomiche mediante ibridizzazione (attaccamento) di sonde di DNA marcate con fluorescenza con DNA cromosomico denaturato. L'esame attraverso un microscopio sotto illuminazione a fluorescenza rileva la presenza del segnale ibridato colorato (e quindi la presenza del materiale cromosomico) o l’assenza del segnale ibridato (e quindi assenza di materiale cromosomico).
FREQUENZA DI RICOMBINAZIONE (%): probabilità di ricombinazione tra due loci, minore è la frequenza di ricombinazione, più questi sono vicini.
GENE: unità di base di eredità, che occupa una posizione specifica su un cromosoma. Ciascuno è costituito da nucleotidi disposti in modo lineare. La maggior parte dei geni codifica per una proteina o un segmento specifico di proteina, che porta a una particolare caratteristica o funzione.
GENE DI SUSCETTIBILITÀ: alterazione genetica che aumenta la suscettibilità o la predisposizione di un individuo a una determinata malattia o disordine. Quando una variante (o mutazione) è ereditata, lo sviluppo dei sintomi è più probabile, ma non certa. Chiamata mutazione deleteria, mutazione patogena, variante patogena e mutazione predisponente.
GENE ONCO-SOPPRESSORE: tipo di gene che regola la crescita delle cellule. Quando si verifica una mutazione di un gene soppressivo del tumore, si può verificare una crescita cellulare mutata e incontrollata. Ciò può contribuire allo sviluppo del cancro. Noto anche con il nome di "anti-oncogene”.
GENETICA CLASSICA: processi genetici che riguardano i singoli individui, come i geni vengono trasmessi da un individuo all’altro. L’unità di studio è l’individuo.
GENETICA DI POPOLAZIONE: ereditarietà di caratteri determinati da uno o pochi geni in gruppi di individui.
GENETICA MOLECOLARE: natura molecolare dell’eredità. Studia come l’ereditarietà viene codificata nel DNA e come i processi biochimici la traducano in un fenotipo. L’interesse è concentrato sulla cellula.
GENETICA QUANTITATIVA: ereditarietà di caratteri determinati dall’azione simultanea di più geni, all’interno di gruppi di individui.
GENOME-WIDE ASSOCIATION STUDY (GWAS): studio di associazione di genoma, è un modo per identificare le varianti genetiche ereditarie associate a rischi di malattia o di particolari tratti. Questo metodo indica l'intero genoma dei polimorfismi genetici, tipicamente singoli polimorfismi nucleotidici (SNPs), che si verificano più frequentemente nei casi (persone con malattia o tratti valutati) che nei controlli (persone senza malattia o tratti specifici).
GENOTIPIZZAZIONE: processo di laboratorio in cui è analizzato il DNA di un singolo individuo, per determinare se sono presenti determinate varianti. La genotipizzazione differisce dalla sequenza in cui tutti i nucleotidi comprendenti una lunghezza specifica del DNA sono valutati (ad esempio, all'interno di un gene, di un esoma o di un genoma).
GENOTIPO: costituzione genetica di un soggetto, totale o riferita a un gene specifico.
GRUPPO ETNICO: origine degli 8 bisnonni, in pratica si usano i 4 nonni.
HNPCC (Hereditary nonpolyposis colorectal cancer): tumore del colon-retto non poliposico ereditario o sindrome di Lynch. Condizione genetica autosomica dominante, con rischio elevato per cancro del colon e altri tumori tra cui carcinoma endometriale, di ovaio, stomaco, intestino tenue, tratto epato-biliare, tratto urinario superiore, cervello e cute. L'aumento del rischio per questi tumori è dovuto a mutazioni ereditarie che alterano la possibilità di riparare un accoppiamento errato di basi (“mismatch”) del DNA al momento della replicazione cellulare.
IMPRINTING GENOMICO o IMPRINTING GENETICO: modulazione dell’espressione di una parte del materiale genetico. Tale modifica può riguardare l'uno o l'altro dei due corredi parentali. Si tratta di un meccanismo di regolazione genica che riguarda circa un centinaio di geni conosciuti, molti dei quali hanno un ruolo rilevante nel differenziamento e nello sviluppo. Alcune malattie risultano ereditabili solo se la mutazione si verifica sul gene paterno o materno, in casi rispettivamente di imprinting materno e paterno: ad esempio, la sindrome di Prader-Willi, la sindrome di Angelman e la sindrome di Russel-Silver. Più “tecnicamente”, è un processo epigenetico che porta all'inattivazione di un allele, a seconda da quale genitore è stato ereditato. L'imprinting genomico può avere rilevanza clinica, perché può influire sull’espressione di una mutazione genica (cioè il fenotipo) nella progenie di un genitore affetto controllante influenzata, a seconda da quale genitore sta passando la mutazione.
INFORMATIVO: in test genetici, un risultato di prova che rivela definitivamente la presenza o l'assenza dell’alterazione genetica germinale associata al disturbo ereditario da valutare. In analisi di linkage, la capacità di distinguere tra marcatori ereditati dal padre e dalla madre (polimorfismi) all'interno o vicino a un gene di interesse.
INSERZIONE: tipo di cambiamento genetico che comporta l'aggiunta di un segmento di DNA. Può essere piccola come una base unica, ma può variare in misura significativa.
INSORGENZA TARDIVA O VARIABILE: stato in cui si esprime un tratto genetico nella vita o espresso in un tempo non fisso nella storia della vita.
INTERAZIONI GENICHE: vedi epistasi/ipostasi.
INTRONE: sequenza di DNA tra gli esoni inizialmente copiata nell'RNA, è tagliata fuori dalla traduzione definitiva dell’RNA e pertanto non modifica il codice aminoacidico. Alcune sequenze introniche sono note per influenzare l'espressione genica.
INVERSIONE: difetto cromosomico in cui un segmento del cromosoma si rompe e si attacca nella direzione opposta.
LEGGE DI HARDY-WEINBERG: esiste una relazione matematica tra frequenze geniche, genotipiche e fenotipiche. Queste frequenze rimangono inalterate di generazione in generazione se non intervengono: migrazione, mutazione, selezione, deriva genica, consanguineità.
LINKAGE: fenomeno per cui due o più geni situati vicini sullo stesso cromosoma vengono trasmessi sempre insieme, ovvero co-presenza, in uno stesso cromosoma, di due o più geni che vengono ereditati insieme. In pratica, misura la segregazione degli alleli e di un fenotipo entro una famiglia. La segregazione è il processo di separazione dei due alleli per lo stesso carattere al momento della produzione di gameti. In virtù di essa, metà dei gameti di un individuo eterozigote possiederà un allele e metà l'altro allele. Questo è possibile perché i gameti sono aploidi e, a differenza di tutte le cellule somatiche, hanno una sola copia del patrimonio genetico.
LINKAGE DISEQUILIBRIUM: associazione statistica tra specifici alleli relativi a due o più loci, che costituiscono di solito un particolare aplotipo ancestrale, diffuso nella popolazione in cui è rilevato, perché trasmesso lungo la discendenza da un comune progenitore. Il linkage disequilibrium si ha quando c'è un'associazione non casuale.
LOCUS: sito fisico o ubicazione di un gene specifico su un cromosoma.
LOD SCORE: stima statistica di due loci genetici fisicamente vicini fra di loro (o "linked) su un particolare cromosoma, che probabilmente saranno ereditati insieme. Un punteggio di 3 è generalmente inteso a voler dire che due geni sono situati vicino sul cromosoma. In termini di significatività, un punteggio di 3 corrisponde a 1000:1 probabilità che i geni siano in linkage e quindi ereditati insieme.
MALATTIA MENDELIANA: malattia che risulta da mutazione di un singolo gene, che ha un vasto effetto sul fenotipo ed è ereditata con modalità semplici. Le malattie sono:
- autosomiche se alterano geni presenti su cromosomi non sessuali;
- legate al cromosoma X se interessano geni di questo cromosoma.
MAPPATURA GENETICA: determinazione delle posizioni relative di geni su un cromosoma e misura della distanza fra loro.
MAPPATURA GENICA: determinazione delle relative posizioni di geni diversi sui cromosomi.
MARCATORE GENETICO: segmento identificabile di DNA, ad esempio polimorfismo nucleotidico singolo (SNP), Restriction Fragment Length Polymorphism (RFLP), Variable number of Tandem Repeats (VNTR), microsatelliti, con sufficiente variazione tra i singoli individui, in modo che può essere rintracciata la sua eredità e la sua co-eredità con alleli di un gene dato; utilizzati in analisi di linkage.
METABOLOMA: insieme di tutti i metaboliti di un organismo biologico, ovvero di tutte le sostanze in grado di partecipare ai processi di un organismo. Tra di essi figurano sia gli intermedi metabolici (substrati necessari alle reazioni biochimiche e prodotti da esse derivati), sia ormoni e altre molecole di segnale.
MICROARRAY: un “microarray” di DNA (noto anche come “gene chip”) è un insieme di microscopiche sonde (segmenti) di DNA attaccate a una superficie solida come vetro, plastica, o chip di silicio, che formano una matrice o “array”. Gli “arrays” permettono di esaminare simultaneamente la presenza di moltissimi geni all'interno di un campione di DNA (spesso anche tutto il genoma o il trascrittoma di un organismo). Utilizzo tipico è il confronto del profilo di espressione genica di un individuo malato con quello di uno sano, per individuare quali geni sono coinvolti nella malattia.
MICROBIOMA/MICROBIOTA: insieme di micro-organismi (il microbiota) che vivono stabilmente in alcuni dei nostri organi e tessuti - come il tratto gastro-intestinale, la pelle, la bocca, gli occhi, il naso, i polmoni, e i genitali - e di tutto quanto essi producono.
MICRO-RNA: piccola molecola di RNA non codificante (composta da circa 22 nucleotidi) presente in animali, piante e alcuni virus. Funziona nel silenziamento dell'RNA e nella regolazione post-trascrizionale dell'espressione genica.
MICRO-SATELLITE (MSI): segmenti ripetitivi di DNA sparsi in tutto il genoma nelle regioni non codificanti tra geni o entro i geni (introni). Spesso vengono usati come marcatori per l'analisi di linkage, a causa della loro elevata variabilità in termini di ripetizione tra i singoli individui. Queste regioni sono intrinsecamente instabili e suscettibili alle mutazioni. Infatti, si parla d’instabilità dei MSI quando le cellule contengono un difetto nella riparazione di DNA. Ad esempio, la presenza di MSI nel tessuto di tumore del colon può essere utilizzata come marcatore per le mutazioni germinali in uno dei geni di riparazione del DNA associati all'HNPCC. Anche l'MSI può avvenire sporadicamente, e in questi casi è legato all’ipermetilazione del gene.
MOSAICISMO GERMINALE: quando le cellule germinali (spermatozoi o oociti) hanno un differente make-up genetico rispetto alle restanti cellule del corpo.
MULTIPLEX LIGATION-DEPENDENT PROBE AMPLIFICATION (MLPA): metodo di laboratorio comunemente usato per individuare le variazioni di numero insolito (inserzioni o delezioni) delle sequenze genomiche.
MUTAZIONE: cambiamento nella costituzione genetica di una cellula (nelle cellule somatiche = non ereditabile; nelle cellule germinali = ereditabile). Può interessare:
- genoma = mutazioni genomiche;
- cromosoma = mutazioni cromosomiche;
- gene = mutazioni geniche.
MUTAZIONE DELETERIA: alterazione genetica che aumenta la suscettibilità o la predisposizione di un individuo a una certa malattia o disordine. Quando una variante (o mutazione) è ereditata, lo sviluppo dei sintomi è più probabile, ma non certa. Denominata mutazione patogena, variante patogena, mutazione predisponente e gene di suscettibilità.
MUTAZIONE DE NOVO: mutazione che avviene durante la meiosi a livello delle cellule gametiche parentali (mosaicismo germinale), oppure mutazione che avviene nelle prime divisioni dello zigote (post-zigotica).
MUTAZIONI DINAMICHE: mutazioni da espansione di sequenza tri-nucleotidica ripetute nel DNA (es. corea di Huntington, distrofia miotonica, sindrome di Martin-Bell).
MUTAZIONE FONDATRICE O “FOUNDER MUTATION”: modifica genetica osservata con alta frequenza in un gruppo che è o è stato geograficamente o culturalmente isolato, in cui uno o più antenati era un portatore di gene alterato. Questo fenomeno viene spesso definito effetto fondatore. Detta anche variante fondatrice.
MUTAZIONE FRAMESHIFT: inserzioni o delezioni che coinvolgono una serie di coppie di basi che non sono multiple di 3, che pertanto interrompono il quadro di lettura di triplette di una sequenza di DNA. Tali varianti (o mutazioni) generalmente portano alla creazione di un codone prematuro (stop), e si traducono in un prodotto di proteine troncate (più corte del normale). Detta anche variante frameshift.
MUTAZIONE GERMINALE: un cambiamento ereditabile nel DNA, che si è verificato in una cellula germinale (una cellula destinata a divenire oocita o spermatozoo).
MUTAZIONE MISSENSE: modifica genetica in cui una sola sostituzione di base altera il codice genetico, in modo tale da produrre un aminoacido diverso dall'usuale aminoacido in tale posizione. Alcune varianti “missense” (o mutazioni) altereranno la funzione della proteina.
MUTAZIONE NONSENSE: modifica genetica che causa la cessazione prematura di una proteina. Le proteine alterate possono essere parzialmente o completamente inattivate, con conseguente cambiamento o perdita di funzione proteica. Detta anche variante “nonsense”.
MUTAZIONE PREDISPONENTE: alterazione genetica che aumenta la suscettibilità o la predisposizione di un individuo a una determinata malattia o disordine. Quando una variante (o mutazione) è ereditata, lo sviluppo dei sintomi è più probabile, ma non certo. Chiamata mutazione deleteria, mutazione delle malattie, variante patogena e gene di sensibilità.
MUTAZIONE PUNTIFORME: alterazione genetica causata dalla sostituzione di un nucleotide unico per un altro nucleotide. Detta anche variante puntiforme.
MUTAZIONI SITO DI SPLICING: alterazione genetica nella sequenza di DNA che si verifica al confine fra un esone e un introne (sito di splicing). Questo cambiamento può sconvolgere l'RNA, con perdita di esoni o inclusione di introni, e un’alterata codifica di sequenza proteica. Detta anche variante del sito di splicing.
NEXT GENERATION SEQUENCING (NGS): metodo a elevata efficienza per determinare una parte della sequenza nucleotidica del genoma di un individuo. Questa tecnica utilizza le tecnologie sequenziali di DNA, in grado di elaborare sequenze di DNA multiple in parallelo. Denominato anche sequenziamento massiccio.
NORTHERN BLOT: tecnica che permette di visualizzare e identificare RNA purificato da un campione, trasferito su di un supporto solido, come ad esempio una membrana di nylon moderatamente caricata elettricamente o nytran, per studiare l'espressione genica.
NUCLEOTIDE: molecola costituita da una base contenente azoto (adenina, guanina, timina o citosina nel DNA; adenina, guanina, uracile o citosina in RNA), un gruppo fosfato e uno zucchero (deossiribosio nel DNA; ribosio in RNA).
NUMERO APLOIDE: è il numero dei cromosomi nelle cellule sessuali (spermatozoi o oociti), in cui esiste una sola copia di ciascun cromosoma. Nell’uomo, il numero aploide è 23.
NUTRIGENOMICA: studio dell'interazione tra i fattori dietetici e genetici e il loro effetto sul metabolismo, su stato di salute e rischio di malattia.
OMOZIGOTE: portatore di 2 alleli identici ad un locus.
PCR (POLYMERASE CHAIN REACTION): la reazione a catena della polimerasi è una procedura che produce milioni di copie di un segmento breve di DNA attraverso cicli ripetuti di:
- denaturazione
- annessione
Procedura molto comune in test genetici molecolari, che può essere utilizzata per generare una quantità sufficiente di DNA per effettuare un test (ad esempio amplificazione, amplificazione allele-specifica, quantificazione di ripetizione trinucleotidica).
PENETRANZA: espressione o non espressione di un fenotipo. Può essere:
- completa = il genotipo esprime sempre il fenotipo;
- incompleta = il carattere si manifesta in una proporzione di figli affetti minore di quella attesa dalle proporzioni mendeliane. L’individuo eredita quindi il gene-malattia, ma non sviluppa la patologia.
PERDITA DI ETEROZIGOSITÀ (chiamata anche LOH): se esistono un allele normale e un allele anormale in un particolare locus, come si può osservare in un disturbo di suscettibilità tumorale autosomica ereditaria, allora la perdita del normale allele produce un locus senza funzione normale. Quando la perdita di eterozigosità coinvolge il normale allele, si crea una cellula che ha più probabilità di mostrare crescita maligna se il gene alterato è un gene onco-soppressore.
PLEIOTROPIA: un gene è responsabile di più effetti fenotipici.
POLIMORFISMO BILANCIATO: vantaggio selettivo dell’eterozigote rispetto agli omozigoti. Esistono valori delle frequenze degli alleli “A” e “a” per le quali si raggiunge equilibrio. Esempio: correlazione malaria–anemia falciforme.
POLIMORFISMO DEL DNA: tratto Mendeliano esistente nella popolazione in forma di 2 o più alleli o genotipi con una frequenza minima ≥ 1% (utile marcatore genetico). Viene comunemente interpretato come una presunta mutazione non patogena.
POLIMORFISMO DI SINGOLO NUCLEOTIDE: cfr SNP.
POPOLAZIONE MENDELIANA: trasferimento delle leggi di Mendel dalla famiglia al concetto generale di popolazione. Popolazione con pool di geni in comune, con trasmissione in accordo alle leggi di Mendel. Dovrebbe essere una popolazione molto grande, e in teoria un individuo di un sesso dovrebbe avere la probabilità uguale di accoppiarsi con uno dell’altro (in realtà è falso, una tale popolazione non esiste).
PORTATORE (SANO): eterozigote asintomatico.
PORTATORE DI UN GENE MUTATO: ciascuna cellula contiene due copie (alleli) di ciascun gene. Una copia può essere mutata, mentre l’altra copia è “corretta”. Se l’allele mutato non è espresso nelle cellule (portando a una particolare caratteristica o condizione), l’allele mutato è detto recessivo rispetto all’altra copia corretta del gene. Un individuo che ha una copia genica corretta e una “sbagliata” (recessiva) si definisce 'portatore' per la mutazione che porta a una specifica condizione. I portatori di una mutazione recessiva in un gene sono solitamente non affetti, ma sono a rischio di trasmettere la copia “sbagliata” alla loro progenie.
PORTATORE DI UN RIARRANGIAMENTO CROMOSOMICO: si applica a un individuo che ha un riarrangiamento dei suoi cromosomi, così che la normale informazione genetica è presente (si dice “bilanciata”), ma non nell’usuale pattern di 46 cromosomi.
PORTATORE (FREQUENZA): la percentuale di individui in una popolazione che ha una singola copia di una specifica mutazione genica recessiva, talvolta applicata alla prevalenza di mutazioni in geni di azione dominante, come BRCA1 e BRCA2.
PREDISPOSIZIONE GENETICA: aumento della probabilità o probabilità di sviluppare una particolare malattia dovuta alla presenza di una o più mutazioni geniche e/o di una storia familiare che indichi un aumentato rischio di malattia.
PROTEOMA: insieme di tutte le informazioni necessarie a sintetizzare le proteine di un organismo o di un sistema biologico, ovvero le proteine prodotte dal genoma.
PSEUDO-GENE: sequenza di DNA che assomiglia a un gene, ma è stata mutata in forma inattiva durante l'evoluzione. Spesso manca di introni e altre sequenze di DNA necessarie per funzionare. Sebbene geneticamente simili al gene funzionale originale, gli pseudo-geni non portano alla sintesi di proteine funzionali, sebbene alcuni possano avere effetti regolatori.
RECESSIVO: tratto/carattere che si esprime solo nell’omozigote, cioè, nel caso di malattia, in cui entrambe le copie del gene difettoso devono essere presenti per esprimere un fenotipo affetto.
RESTRICTION FRAGMENT LENGTH POLYMORPHISM (RFLP): tecnica che sfrutta le variazioni nelle sequenze di DNA omologhe provenienti da diverse posizioni dei siti degli enzimi di restrizione. Il campione di DNA viene spezzettato, “digerito”, dagli enzimi di restrizione e i frammenti di restrizione risultanti vengono separati in base alla loro lunghezza mediante elettroforesi su gel. È una tecnica importante nella mappatura del genoma, localizzazione di geni per disordini genetici, determinazione del rischio per la malattia e test di paternità.
RICOMBINAZIONE MEIOTICA: quando il cross-over dei cromatidi scambia frammenti di DNA, i marcatori in linkage rimangono con il cromatide originale.
RISCHIO ATTRIBUIBILE: proporzione di una malattia in soggetti esposti che può essere attribuita a un'esposizione. Nel contesto degli studi genetici, l'esposizione è la frequenza di una specifica variante genetica.
RISCHIO RELATIVO (RR): rischio che il parente di un affetto da malattia ha di sviluppare la stessa malattia rispetto alla popolazione generale. Si definisce anche come il rapporto della prevalenza di una malattia in un parente “r” di una persona affetta e della prevalenza della malattia nella popolazione.
RISCHIO DI RICORRENZA: probabilità che un tratto ereditario o un disordine ereditario presenti in un membro della famiglia si ripetano nuovamente in altri familiari. Questo è distinto dal rischio di ricorrenza/recidiva per il cancro, che è la possibilità che un cancro che è stato trattato si possa ripetere.
RIPETIZIONE DI TRIPLETTE: le cosiddette malattie da espansione di triplette sono circa venti, accomunate dalla stessa causa, ovvero un aumento eccessivo di ripetizioni di triplette nucleotidiche, in genere CGG, in determinate sezioni del DNA, a causa di un errore nella sua sintesi. Le sequenze di 3 nucleotidi sono ripetute in tandem, CGGCGG… n volte, sulla stessa sezione contigua del cromosoma. Una certa variazione di valore normale (polimorfica) in numero di ripetizioni, non è rilevante clinicamente tra gli individui; tuttavia, in alcuni casi, numeri di ripetizione possono portare a effetti negativi sulla funzione del gene, con conseguente malattia genetica.
SCIENZE “OMICHE”: studio del pool di molecole biologiche (ioni, acidi nucleici, proteine, enzimi) in campioni biologici. Esse analizzano, nel loro insieme:
- i geni del DNA (genomica) e le loro funzioni (genomica funzionale);
- i trascritti del DNA, cioè l’RNA (trascrittomica);
- le proteine (proteomica);
- i metaboliti all’interno di un organismo (metabolomica).
Studiano anche le interazioni tra queste molecole (interattomica) e tra queste molecole e i microrganismi della flora intestinale (microbiomica) o dell’ambiente (infettivoma/ infettivomica), i cibi/nutrienti (nutribioma/nutribiomica) e l’ambiente in generale (ambientoma/ambientomica), nonché le modificazioni prodotte da tali interazioni sul DNA (epigenomica).
SCREENING DEL PORTATORE: si testano popolazioni per determinare se singoli individui sono portatori di un gene mutato per una particolare condizione.
SELEZIONE NATURALE: poiché gli organismi competono per le risorse più soddisfacenti, quelli con i geni che si adattano meglio al loro ambiente sopravvivono con maggiore probabilità e trasmettono i loro geni. I geni che danno un fenotipo più vantaggioso aumentano la loro frequenza con il tempo. Effetti della selezione, tenendo presente il grafico della campana di Gauss:
- direzionale: favorisce e stabilizza un fenotipo estremo;
- stabilizzante: favorisce i fenotipi medi;
- differenziale: favorisce i fenotipi estremi.
SINGLE-STRANDED CONFORMATIONAL POLYMORPHISM (SSCP): tipo di scansione delle mutazioni; identificazione di segmenti di DNA a singolo filamento con migrazione anormale su gel elettroforesi. L’analisi mediante SSCP è utilizzata per separare gli acidi nucleici a singolo filamento, basandosi sulle sottili differenze nella sequenza di DNA, spesso una singola coppia, che si traduce in una diversa struttura secondaria e una differenza misurabile di mobilità attraverso un gel.
SINGLE NUCLEOTIDE POLIMORPHISM (SNP): singolo polimorfismo nucleotidico, variazioni di sequenza di DNA che si verificano quando è alterato un nucleotide unico (adenina, timina, citosina o guanina) nella sequenza del genoma; generalmente presente in almeno l'1% della popolazione. È usato per "etichettare” un aplotipo particolare in una regione del genoma. Come sottogruppo di tutti gli SNPs del genoma, gli SNPs marcatori possono essere estremamente utili per testare l'associazione di un marcatore a un locus qualitativo o quantitativo, in quanto non è necessario genotipizzare tutti gli SNPs. Chiamato anche “tag SNP”.
SPLICING: processo con cui gli introni, le regioni non codificanti dei geni, sono tagliati dalla trascrizione principale del RNA messaggero e gli esoni (le regioni codificate) sono unite per generare un RNA messaggero maturo. Quest'ultimo serve come modello per la sintesi di una proteina specifica.
SOUTHERN BLOT: tecnica basata sull'elettroforesi, utilizzata in test genetici per individuare grandi delezioni nel DNA, che possono essere mancate dai metodi di test genetici basati sulla PCR.
SPECIE: gruppo di individui che in teoria possono liberamente accoppiarsi tra di loro originando prole fertile (tutti nella specie umana).
STRATIFICAZIONE DELLA POPOLAZIONE: si definiscono stratificate quelle popolazioni che includono sottogruppi che durante l’evoluzione si sono mantenuti geneticamente distinti. Negli USA esistono numerosi sottogruppi separati geneticamente secondo le origini geografiche degli antenati.
STUDI D’ASSOCIAZIONE CON GENE CANDIDATO: confrontano le frequenze dell’allele-rischio in gruppi etnici diversi e confrontano le frequenze dell’allele-rischio in studi epidemiologici caso-controllo e/o di popolazione. Permettono l’esecuzione di studi di meta-analisi per stimare l’entità dell’effetto. Misurano la segregazione preferenziale di un particolare allele con un fenotipo attraverso le famiglie.
TASSO DI MUTAZIONE DI UN GENE: frequenza di mutazione per locus per generazione oppure in maniera equivalente per locus per gamete.
TELOMERI: parte terminale dei cromosomi. Composti da etero-cromatina, hanno una sequenza comune (nell’uomo TTAGGG). I telomeri hanno un’importante funzione, quale quella di prevenire la fusione termino-terminale dei cromosomi, aiutando il loro accoppiamento alla meiosi ed assicurando una completa replicazione delle estremità.
TEST GENETICO: analisi di geni, dei loro prodotti o funzioni, o analisi del DNA o dei cromosomi per identificare alterazioni del DNA associate a disordini genetici. I test genetici sono classificati in 6 differenti sottotipi:
- diagnostico (es. conferma di sospetto clinico di una malattia monogenica, identificando una mutazione causale in un gene specifico);
- pre-sintomatico (es. quando la mutazione germinale causale, identificata nel probando, è presente nella linea germinale di un soggetto a un’età alla quale quella specifica malattia non è ancora espressa);
- prognostico (es. quando un’alterazione specifica del DNA si associa ad una specifica severità, maggiore o minore, di un fenotipo clinico);
- suscettibilità genetica (es. quando un’alterazione del DNA, una variante o una mutazione, non sono di per sé causali una specifica condizione patologica, ma predispongono al suo sviluppo);
- identificazione dell’eterozigote (è una delle applicazioni pratiche della legge di Hardy-Weinberg, stima la frequenza dell’eterozigote quando esso non può essere identificato con metodi diretti. Importante nelle malattie recessive);
- a scopi forensi (es. test di paternità).
TEST MULTI-GENICO: test genetico che utilizza la “Next Generation Sequencing” per testare simultaneamente geni multipli.
TEST MULTI-GENOMICO: metodo per individuare le alterazioni genetiche multiple (cioè mutazioni geniche o singoli polimorfismi nucleotidici in un singolo gene o attraverso il genoma) simultaneamente.
TEST PRE-SINTOMATICO: analisi genetica di un individuo asintomatico o non affetto che rischia per un disordine genetico specifico.
TRADUZIONE: processo di sintesi di una sequenza di aminoacidi (prodotto proteico) dal codice del RNA messaggero.
TRASCRITTOMA: insieme di tutti i trascritti (mRNA) di un dato organismo o tipo cellulare.
TRASCRIZIONE: processo di sintesi del RNA messaggero (mRNA) dal DNA.
TRASLOCAZIONE: tipo di anormalità cromosomica in cui una frazione cromosomica e una parte di essa raggiungono un altro luogo cromosomico.
TRATTO COMPLESSO O QUANTITATIVO: fenotipo senza classico pattern di ereditarietà Mendeliana (es. “break-down” corrispondenza genotipo/fenotipo). Tratti influenzati da diversi geni e dall’ambiente (colore della pelle e dei capelli, pressione ed altezza).
TRATTO CONTINUO/QUANTITATIVO: tratto misurabile attraverso un range di valori. Questa classe include la vasta maggioranza delle malattie dell’uomo.
TRATTO DISCRETO/QUALITATIVO: tratto che è presente o assente (es. fratture).
VALUTAZIONE DEL RISCHIO: valutazione quantitativa o qualitativa del rischio individuale di trasmissione di una certa mutazione genica o di un particolare disordine, o di avere un bambino con un certo disturbo; a volte fatta ricorrendo a modelli matematici o statistici, che incorporano fattori come storia personale della salute, storia medica familiare e storia etnica.
VANTAGGIO DELL’ETEROZIGOTE: dipende dall’ambiente. Mutazioni sfavorevoli o neutre possono rivelarsi vantaggiose.
VARIABLE NUMBER OF TANDEM REPEATS (VNTR): sono polimorfismi del DNA in cui la differenza tra le diverse varianti non è data da una sostituzione nucleotidica (come per SNP o RFLP), ma da ripetizione in tandem di specifiche sequenze nucleotidiche. Le sequenze ripetute rappresentano circa il 44% del nostro DNA, quelle ripetute in tandem sono caratterizzate dal fatto che le unità di ripetizione si trovano adiacenti l'una all'altra.
VARIANTE DE NOVO: un'alterazione genetica presente per la prima volta in un membro della famiglia a seguito di una variante (o mutazione) in una cellula germinale (oocita o spermatozoo) di uno dei genitori, o una variante che si pone nell’uovo fecondato durante l'embriogenesi precoce. Anche chiamata mutazione de novo, nuova mutazione e nuova variante.
VARIANTE SOMATICA: alterazione del DNA che avviene dopo concepimento e non è presente entro le cellule germinali. Le varianti somatiche possono verificarsi in tutte le cellule del corpo, ad eccezione delle cellule germinali e pertanto non sono trasmesse ai figli. Le varianti somatiche possono (ma non sempre) provocare cancro o altre malattie.
VARIANZA: misura del grado di dispersione dei valori su ambedue i lati della media. Determina la larghezza della curva di distribuzione. La varianza di una quantità misurata nella popolazione è chiamata varianza fenotipica totale.
WESTERN BLOT o IMMUNOBLOT: tecnica biochimica che rileva proteine modificate con acido lipoico usando un anticorpo specifico. Permette l’identificazione di una determinata proteina in una miscela di proteine, mediante il riconoscimento da parte di anticorpi specifici.
WHOLE-EXOME SEQUENCING: processo di laboratorio utilizzato per determinare la sequenza nucleotidica principalmente delle regioni esoniche (o di codifica delle proteine) delle sequenze di un individuo e delle sequenze correlate, che rappresentano circa l'1% della sequenza di DNA completa.
WHOLE-GENOME SEQUENCING: processo di laboratorio utilizzato per determinare quasi tutti i 3 miliardi di nucleotidi di una sequenza completa di DNA di un individuo, inclusa la sequenza non codificante.
X CHROMOSOME INACTIVATION ANALYSIS: si utilizza per determinare il pattern di inattivazione del cromosoma X per femmine portatrici di disturbi legati al cromosoma X e valutare la patogenicità della variante genetica in un gene legato a tale cromosoma. Può trovare ancora applicazione per studiare la clonalità di cellule/tessuti da soggetti di sesso femminile.
X-LINKED DOMINANTE: eredità dominante del cromosoma X, si riferisce alle condizioni genetiche associate a mutazioni nei geni del cromosoma X. Una sola copia della mutazione è sufficiente a provocare la malattia sia nei maschi (che hanno un solo cromosoma X) che nelle femmine che hanno due cromosomi X. In alcune condizioni, l'assenza di un gene funzionale comporta la morte dei maschi colpiti.
X-LINKED RECESSIVO: eredità recessiva collegata al cromosoma X, si riferisce a condizioni genetiche associate a mutazioni nei geni del cromosoma X. Un uomo che trasporta una tale mutazione ne sarà colpito, perché ha solo un cromosoma X. Una donna che trasporta una mutazione in un gene, con un gene normale sull'altro cromosoma X, è generalmente non affetta.
Z-SCORE: punteggio che indica quante deviazioni standard un risultato si colloca sopra o sotto la media.
Sistematica sulla diagnostica endocrinologica
Dosaggi relativi alle paratiroidi
Dosaggi relativi al metabolismo osseo
Dosaggi relativi a diabete e metabolismo
Metodo di determinazione della PRL
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento agosto 2021)
La prolattinemia viene oggi misurata pressocché universalmente su strumentazione automatizzata, impiegando metodi immunometrici “sandwich”, nei quali un anticorpo, di solito immobilizzato su fase solida, cattura la molecola che viene poi riconosciuta da un secondo anticorpo marcato con enzimi, fluorocromi o (più spesso) sostanze chemiluminescenti. Dopo rimozione dei composti che non hanno reagito, si misura il segnale generato, che è direttamente proporzionale alla concentrazione dell’ormone presente. Nonostante l’adozione universale di uno standard WHO, esiste ancora una notevole variabilità tra metodo e metodo, che è alla base di significative differenze tra gli intervalli di riferimento dei singoli laboratori.
La PRL circola nel sangue periferico in diverse isoforme: la monomerica, la dimerica e la polimerica, tutte dotate di immuno-reattività che, peraltro, è rilevata in modo differente dai diversi metodi. Al di là della possibile interferenza da anticorpi eterofili (da tenere sempre in considerazione nell’interpretazione di tutti i dosaggi immunometrici), il vero problema analitico nella misura della PRL resta la possibile presenza di macroprolattina, cioè di masse molecolari di PRL con PM > 150 kDa. In questi casi, la forma predominante di PRL in circolo è costituita da complessi di PRL monomerica con auto-anticorpi anti-PRL. Ogni laboratorio dovrebbe avere una procedura per la rilevazione di macroprolattinemia. Non è necessario ricorrere alla gel-cromatografia per rivelare la presenza di macroprolattina, ma è sufficiente trattare il campione con poli-etilen-glicole (PEG), in grado di far precipitare le proteine con PM > 100 kDa. Se il recupero di PRL dopo trattamento è < 40%, nel campione c’è una presenza prevalente di macroprolattina, se è > 60% c’è in prevalenza PRL monomerica.
Un’altra possibile inaccuratezza nei metodi di dosaggio può verificarsi nel caso di concentrazioni estremamente elevate di PRL. Il cosiddetto “hook effect” (effetto gancio) consiste nel sotto-stimare la reale elevata concentrazione di ormone in presenza di quantità molto alte di antigene, in grado di saturare tutti i siti di legame anticorpale. Si tratta di eventi eccezionali, perché gli attuali metodi sono molto meno esposti a questo effetto che in passato. L’unico rimedio a tale ormai raro problema è la tempestiva segnalazione da parte del clinico del sospetto e l’immediata attuazione da parte del laboratorista di diluizioni adeguate del campione, che consentano di confermare o escludere la possibilità.
Bibliografia
- Saleem M, Martin H, Coates P. Prolactin biology and laboratory measurement. An update on physiology and current analytical issues. Clin Biochem Rev 2018, 39: 3-16.
- Ferguson J, Douglas T, Rigsby P, Burns C. WHO International Collaborative Study of the Proposed 4th International Standard for Prolactin, Human. 2016.
- Smith TP, Suliman AM, Fahie-Wilson MN, et al. Gross variability in the detection of prolactin in sera containing big big prolactin (macroprolactin) by commercial immunoassays. J Clin Endocrinol Metab 2002, 87: 5410-5.
- Beltran L, Fahie-Wilson MN, McKenna TJ, et al. Serum total prolactin and monomeric prolactin reference intervals determined by precipitation with polyethylene glycol: evaluation and validation on common immunoassay platforms. Clin Chem 2008, 54: 1673-81
Metodo di determinazione del GH
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento agosto 2021)
Il dosaggio del GH rappresenta ancora oggi una sfida impegnativa sulla strada della standardizzazione e dell’armonizzazione dei metodi per la misura degli ormoni peptidici. Con il passaggio ormai consolidato ai metodi immuno-radiometrici, il problema della confrontabilità di risultati ottenuti con metodi diversi non è del tutto risolto.
La secrezione ipofisaria del GH produce una miscela di diverse isoforme molecolari e non una sostanza omogenea. La forma più rappresentata è quella da 22 kDa. Con l’impiego di anticorpi monoclonali, l’impatto sul riconoscimento delle singole isoforme è molto maggiore che utilizzando anticorpi policlonali, in quanto la specificità anticorpale è nettamente cresciuta. In base alla differente specificità degli anticorpi utilizzati, ogni immuno-dosaggio riconosce soltanto determinate molecole. Non tutti i metodi in commercio dichiarano la specificità dei propri anticorpi, anche se si può dare per scontato che tutti riconoscano la 22 kDa; sull’utilità clinica di riconoscere le altre isoforme mancano ancora evidenze convincenti.
Un altro importante fattore in grado di rendere difficile il confronto tra risultati di GH ottenuti con metodi differenti è la presenza delle proteine di legame (Binding Proteins, BP). Oltre il 50% del GH circola nel sangue periferico legato a proteine ad alta affinità, la cui concentrazione dipende da fattori individuali (stato nutrizionale, compenso metabolico, ecc). Dal punto di vista analitico la loro presenza diventa determinante quando il legame con il GH avvenga in prossimità dei siti di legame di uno degli anticorpi utilizzati per la sua rilevazione. L’ingombro sterico potrebbe impedire il legame e portare a una sotto-stima della reale concentrazione in situazioni nelle quali le BP sono elevate. Questa interferenza, molto importante quando si utilizzavano anticorpi policlonali, sembra ridimensionata con i nuovi immuno-dosaggi, ma non ancora del tutto scomparsa.
Altri effetti legati alla matrice sono poco studiati, anche se generalmente si consiglia di utilizzare il siero rispetto al plasma. Infine, sono state segnalate interazioni significative in pazienti in trattamento con pegvisomant.
Nonostante l’adozione di un calibratore comune (IRP 98/574), la concordanza dei risultati tra metodi diversi lascia ancora a desiderare e rappresenta un ostacolo non secondario nella gestione clinica. È necessario che anche le aziende del diagnostico diano maggiori informazioni sulla sensibilità alle diverse isoforme dei propri reagenti e sull’interazione con le BP incomplete.
In conclusione, ancora oggi l’interpretazione dei risultati del dosaggio ematico del GH pone consistenti problemi. La fisiologica variabilità della secrezione, la non perfetta correlazione con altri indicatori metabolici (IGF-1) e gli accennati problemi di standardizzazione ed armonizzazione tra i differenti metodi complicano l’applicazione di linee guida e raccomandazioni internazionali. È importante che il clinico comprenda le difficoltà e i problemi connessi con questo esame e ne discuta con il laboratorista, per ottimizzare la gestione complessiva di queste patologie.
Bibliografia
- Molitch ME, Clemmons DR, Malozowski S, et al. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2006, 96: 1621–34.
- Iwahara K, Tanabe C, Maekawa M. Comparison of access ultrasensitive human growth hormone assay to monoclonal antibody-based immunoradiometric assay. Clin Chim Acta 2007, 376: 248–9.
- Bidlingmaier M, Freda PU. Measurement of human growth hormone by immunoassays: current status, unsolved problems and clinical consequences. Growth Horm IGF Res 2010, 20: 19–25.
- Wieringa GE, Sturgeon CM, Trainer P. The harmonisation of growth hormone measurements: taking the next steps. Clin Chim Acta 2014, 432: 68-71.
- Giustina A, Arnaldi G, Bogazzi F. Pegvisomant in acromegaly: an update. J Endocrinol Invest 2017, 40: 577–89.
- Ribeiro de Oliveira J, Ribeiro Oliveira A, Bidlingmaier M. Growth hormone: isoforms, clinical aspects and assays interference. Clin Diabetes Endocrinol 2018, 4 https://doi.org/10.1186/s40842-018-0068-1.
Metodo di determinazione dell'IGF-I
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento agosto 2021)
L’utilizzo in clinica della concentrazione ematica dell’Insulin-like Growth Factor ha dovuto superare notevoli problemi legati alla difficoltà di dosare la molecola in modo preciso ed accurato, e all'impossibilità di confrontare dati provenienti da laboratori diversi che utilizzavano metodi e strumentazioni diverse. Il principale ostacolo analitico è dovuto al fatto che l’ormone circola nel sangue periferico legato ad almeno 6 differenti proteine di legame (IGF-BP), tutte ad elevata affinità. Più del 75% dell’IGF-I circola come complesso ternario con IGFBP-3 e una subunità ricca di leucina, acido-labile (ALS). Una percentuale minore è legata ad altre proteine leganti e meno dell’1% è libera. Poiché le IGF-BP mascherano gli epitopi dell’IGF-I o competono con questi, i dosaggi immunometrici devono essere preceduti dalla rimozione delle IGF-BP.
Oggi i metodi più utilizzati sono quelli immuno-radiometrici in automazione. È stata proposta anche la doppia cromatografia seguita dalla spettrometria di massa (Tandem-MS), che offre alcuni vantaggi in termini di specificità ed accuratezza, ma resta ancora oggi una soluzione dispendiosa in termini di tempo e risorse e pertanto non trova utilizzi molto estesi.
Nonostante gli sforzi per standardizzare ed armonizzare il dosaggio di IGF-I e la disponibilità di uno standard internazionale ricombinante (IS 02/154), è ancora difficile ottenere risultati comparabili tra laboratorio e laboratorio. L’ostacolo principale è garantire le minori interferenze possibili da parte delle IGF-BP. Per il dosaggio si preferisce utilizzare il siero, e non esistono particolari problematiche analitiche in quanto l’ormone, analogamente al GH, è abbastanza stabile a temperatura ambiente. L’altro aspetto essenziale è la definizione di intervalli di riferimento appropriati per la popolazione afferente al laboratorio, che prenda in considerazione un numero adeguato di soggetti distinti per genere ed età.
Bibliografia
- Clemmons DR. Consensus statement on the standardization and evaluation of Growth Hormone and Insulin-like Growth Factor assays. Clin Chem 2011, 57: 555–9.
- Chanson P, Arnoux A, Mavromati M, et al. Reference values for IGF-I serum concentrations: comparison of six immunoassays. J Clin Endocrinol Metab 2016, 101: 3450–8.
- Katznelson L, Lawson ER, Melmed S et al. Acromegaly: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014, 99: 3933-51.
- Hines J, Milosevic D, Ketha H, et al. Detection of IGF-1 protein variants by use of LC-MS with high-resolution accurate mass in routine clinical analysis. Clin Chem 2015, 61: 990-1.
- Pokrajac A, Wark G, Ellis AR, et al. Variation in GH and IGF-I assays limits the applicability of international consensus criteria to local practice. Clin Endocrinol (Oxf) 2007, 67: 65–70.
Metodo di determinazione del TSH
Marco Caputo
Synlab Med srl, Calenzano (FI)
(aggiornamento agosto 2021)
Quella della misura della concentrazione ematica del TSH è una “storia di successo” per la Medicina di Laboratorio. Tuttavia, il percorso per arrivare all’accuratezza degli attuali dosaggi “di ultima generazione” è stato molto lungo ed accidentato. La sensibilità dei metodi negli ultimi anni è decisamente aumentata: i primi dosaggi con metodi competitivi e anticorpi policlonali non riuscivano a distinguere i pazienti ipertiroidei dai soggetti sani. La sensibilità analitica è la stima della più bassa concentrazione discriminabile dallo zero; quella funzionale è la stima della precisione del metodo a basse concentrazioni (in genere è indicata dalla concentrazione più bassa in cui il coefficiente di variazione è < 20%). Questa percentuale, arbitraria, comprende le variazioni analitica e biologica ed è sempre più alta di quella analitica. Termini come metodo “sensibile” e “ultra-sensibile” vanno abbandonati, mentre sono ormai entrate nell’uso comune espressioni come “di prima, seconda, terza, quarta generazione”, intendendo un aumento di sensibilità funzionale di 10 volte ad ogni passaggio di generazione. Per il TSH si definisce di “terza generazione” un metodo che ha una sensibilità funzionale < 0.02 mU/L. I metodi attuali hanno una riproducibilità garantita fino a concentrazioni di 0.01 mUI/L. Naturalmente è importante che ogni laboratorio monitori regolarmente le prestazioni analitiche in questo intervallo di concentrazioni, cosa non sempre agevole in quanto richiede la disponibilità di un pool di sieri da utilizzare costantemente. Va ricordato che la struttura del TSH circolante nel sangue non è identica a quella contenuta nell’ipofisi o negli estratti ipofisari usati per la messa a punto del metodo, e non si può escludere che gli anticorpi monoclonali impiegati per identificare il TSH possano avere specificità diversa per gli epitopi delle isoforme di TSH nel siero rispetto a quelle ipofisarie. Il TSH è una glicoproteina con struttura simile a quella di altri ormoni dell’ipofisi anteriore. È stato dimostrato, ad esempio, che nel siero di pazienti ipotiroidei circola TSH con oligosaccaridi differenti (acido sialico e residui di galattosio) dalla forma presente nell’ipofisi di soggetti sani. È importante che le aziende produttrici specifichino la sensibilità del proprio metodo alle diverse possibili glicoforme della molecola.
Come per tutti gli immuno-dosaggi, anche per il TSH va sempre tenuta in considerazione la possibile interferenza da anticorpi eterofili. Il laboratorio è in grado di valutarne la presenza mediante la loro determinazione diretta o cimentando il campione con anticorpi in grado di intercettarli.
Un problema emerso recentemente riguarda l’interferenza da biotina. La biotina (vitamina B7) viene oggi spesso assunta in quantità elevate, ma viene anche utilizzata in molti immuno-dosaggi per la sua capacità di formare legami covalenti con molte molecole di varie dimensioni senza alterarne le proprietà antigeniche e biologiche e, complessata alla streptavidina, è in grado di resistere a variazioni di pH e di temperatura anche estreme. Nel caso del TSH, l’anticorpo viene incubato con il siero del paziente e con un secondo anticorpo specifico legato ad un sistema di rilevazine come il rutenio. Una volta avvenuta la reazione, i complessi precipitano su una fase solida rivestita di streptavidina, e il segnale che ne deriva è direttamente proporzionale alla concentrazione dell’ormone cercato. In presenza di grandi eccessi di biotina solubile, si verifica una competizione con la streptavidina e pertanto la reazione non avviene o avviene in modo incompleto. Nel caso di immuno-dosaggi competitivi, l’interferenza esita in una riduzione del segnale e quindi nella generazione di un risultato falsamente elevato. In letteratura sono segnalati casi di Graves misdiagnosticati per colpa della biotina, ed interferenze analoghe sono state riportate per PTH, tireoglobulina, DHEAS, E2 e ferritina. Per questo problema non esistono al momento soluzioni analitiche. Il clinico deve essere pertanto allertato e verificare questa possibilità in anamnesi.
Bibliografia
- Thienpont LM, Van Uytfanghe K, Beastall G, et al. Report of the IFCC working group for standardization of thyroid function tests, part 1: Thyroid-stimulating hormone. Clin Chem 2010, 56: 902–11.
- Spencer CA, Hollowell JG, Kazarosyan M, et al. National Health and Nutrition Examination Survey III thyroid-stimulating hormone (TSH)-thyroperoxidase antibody relationships demonstrate that TSH upper limit reference limits may be skewed by occult thyroid dysfunction. J Clin Endocrinol Metab 2007, 92: 4236–40.
- Beckett G, MacKenzie F. Thyroid guidelines - are thyroid-stimulating hormone assays fit for purpose? Ann Clin Biochem 2007, 44: 203-8.
- Taher J, Brinc D, Gilmour JA, Beriault DR. Validating thyroid-stimulating hormone (TSH) reflexive testing cutpoints in a tertiary care institution. Clin Chem Lab Med 2019, 58: e11-3.
- Kellogg MD, Law TC, Huang S, et al A girl with goiter and inappropriate Thyroid-Stimulating Hormone Secretion. Clin Chem 2008, 54: 1241–4.
- Caputo M. Biotina: repetita juvant. AME Breaking News 9/2019.
- FDA warns that biotin may affect some lab test results. Lab Test Online 2/1/2018.