Manifestazioni cardio-vascolari dell'iperparatiroidismo primario

Alessandro Piovesan
Endocrinologia Oncologica, Città della Salute e della Scienza, Torino
(aggiornato al 2 maggio 2017)
L’iperparatiroidismo primitivo (IPT) sintomatico è stato, storicamente, associato ad un’aumentata mortalità rispetto alla popolazione generale. In recenti studi di coorte europei è stata riportata una maggior mortalità, soprattutto per cause cardiovascolari, anche nei pazienti con IPT asintomatico o pauci-sintomatico (1-3), non confermata in studi epidemiologici nordamericani (4).
| Tasso di mortalità standardizzato (SMR), aggiustato per età e sesso, in pazienti con iperparatiroidismo primario lieve tra il 1997 e il 2006 (modif da 3) |
|||||
| Causa di morte | Eventi totali | Morti osservate | Morti attese | SMR | IC 95% |
| Tutte le cause | 46221 | 502 | 191.8 | 2.62 | 2.39-2.86 |
| Cardiovascolari | 22214 | 227 | 84.8 | 2.68 | 2.34-3.05 |
| Cancro | 12390 | 137 | 46.4 | 2.95 | 2.48-3.49 |
Alcune manifestazioni cliniche più frequenti inell’IPT (diabete e/o insulino-resistenza, compromissione renale) potrebbero contribuire a incrementare la morbilità cardiovascolare nei pazienti affetti. Un recente studio epidemiologico su vasta scala ha dimostrato come l'IPT sia un fattore predittivo indipendente del rischio di ipertensione arteriosa (5).
Sia la calcemia che i livelli di PTH sono stati riportati correlare in maniera negativa con la sopravvivenza e la morbilità cardiovascolare nella popolazione generale (6), così come nei pazienti con IPT anche in studi recenti (4,7).
Nell’IPT sintomatico è stato dimostrato l’effetto favorevole della paratiroidectomia (PTX) sulla sopravvivenza e su end-point cardiovascolari importanti (2), mentre i dati sull’IPT asintomatico o pauci-sitomatico sembrano meno univoci (8).
Le complicanze cardiovascolari più frequentemente riportate nell’IPT sono: ipertensione arteriosa, aritmie cardiache, alterazioni metaboliche e alterazioni funzionali e strutturali cardiache (ipertrofia, disfunzione diastolica, calcificazioni valvolari) e dei vasi (elasticità vasale e disfunzione endoteliale).
Alterazioni funzionali e strutturali cardiache e vascolari
Sono state descritte nell’IPT sintomatico. Possono essere presenti le alterazioni elettrocardiografiche (accorciamento del QT, PQ prolungato, onde J) determinate dall’ipercalcemia, così come le calcificazioni valvolari.
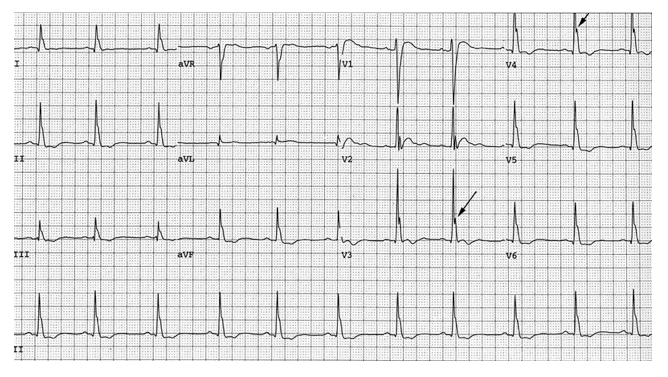
Entrambe sono corrette dalla PTX. Nell’IPT asintomatico o pauci-sitomatico la reale entità del coinvolgimento cardiaco è più discussa; anche i documenti di consenso più recenti non inseriscono le complicanze cardiovascolari tra i criteri di indicazione all’intervento chirurgico (9,10). Nell’IPT paucisintomatico, infatti, le alterazioni cardiovascolari sono state studiate in piccole serie di pazienti, talora con metodiche di non facile riproducibilità e non sono risultate reversibili unicamente dopo risoluzione chirurgica dell’IPT. L’ipertrofia cardiaca è stata descritta in piccole serie di pazienti e non in tutte le serie è stato considerato l’effetto confondente di eventuali fattori di rischio cardiovascolari pre-esistenti (11-13). Sebbene sia stata ipotizzata una relazione tra livelli di PTH e massa cardiaca, tale relazione non è stata confermata in tutti gli studi; più di recente è stato proposto che bassi livelli di vitamina D possano avere un ruolo nel condizionare l’insorgenza dell’ipertrofia (14).
La PTX non ha indotto in maniera univoca un miglioramento dell’ipertrofia cardiaca. Analogamente, anche la disfunzione diastolica, misurata sia con indici ecografici sia con angiocardio-scintigrafia, è stata riportata come più frequente nei pazienti con IPT attivo (15). Anche per questo parametro, tuttavia, i dati non sono conclusivi, vuoi per la difficile standardizzazione nelle misurazioni, vuoi per la difformità di risposta alla risoluzione chirurgica dell’IPT nelle diverse casistiche (11,12,16).
Per quanto riguarda l’effetto dell’IPT sulle alterazioni vasali, è stata riportata una maggior rigidità di parete arteriosa in piccoli gruppi di pazienti con IPT. Anche per questa alterazione non è del tutto confermato il miglioramento dopo PTX.
Ancora più scarni i dati sulla disfunzione endoteliale (16,17).
I meccanismi eziopatogenetici che potrebbero spiegare le complicanze cardiovascolari segnalate nell’IPT non sono ancora del tutto chiariti: elevati livelli ematici di PTH e calcio potrebbero avere effetti inotropi diretti, che a lungo andare potrebbero promuovere ipertrofia cardiaca; i livelli ematici di vitamina D sembrano avere un ruolo nel promuovere tale complicanza.
In generale, si può concludere dicendo che le alterazioni strutturali e funzionali cardiache e vasali descritte nell’IPT non hanno ancora confermato una reale rilevanza clinica e non sembrano modificarsi favorevolmente dopo la risoluzione chirurgica dell’IPT. Non possono pertanto essere considerati, allo stato attuale, elementi clinici da tenere in considerazione nelle indicazioni all’intervento chirurgico nella pratica clinica routinaria.
Ipertensione arteriosa
Da molto tempo è stata riportata un’aumentata prevalenza di ipertensione arteriosa nei pazienti con IPT (18-20). Anche in studi di coorte nella popolazione generale era stata osservata una correlazione diretta tra i livelli di PTH e i valori tensivi (21), con un possibile ruolo rilevante svolto dalla vitamina D3 (22). In considerazione dell’assenza di un chiaro meccanismo fisiopatologico che giustifichi questa associazione, non può essere esclusa la possibilità di un’associazione casuale tra due patologie relativamente frequenti nella stessa fascia di età.
Anche per quanto riguarda l’ipertensione arteriosa, nelle casistiche finora riportate l’effetto della PTX sui livelli tensivi appare favorevole solo in circa il 50% dei pazienti con IPT (19,23,24).
È stato ipotizzato che l’effetto vasodilatatorio del PTH, dimostrato in condizioni fisiologiche attraverso un legame recettoriale diretto alle cellule muscolari lisce, possa essere perso nell’IPT, in particolare quando siano da tempo presenti altri fattori di rischio cardiovascolare e danno endoteliale (25). Si è altresì speculato che la correlazione tra IPT e ipertensione possa avvenire attraverso l’azione su altri sistemi ormonali. Malgrado sia nota la correlazione inversa tra livelli di vitamina D e attivazione del sistema renina-angiotensina-aldosterone, è stata anche riportata una debole azione diretta del PTH sullo stesso sistema (26,27). Inoltre, la risposta vascolare all’azione ipertensiva di epinefrina sembra maggiore nei pazienti con IPT ipertesi rispetto ai normotesi (28).
Alla luce di tali osservazioni, tuttavia, si può concludere che non si sia ancora dimostrata con certezza una relazione causale tra IPT e ipertensione arteriosa: i risultati non univoci sull’efficacia della PTX nel migliorare l’ipertensione nell’IPT fanno concludere in tutti i documenti di consenso che l’ipertensione arteriosa non possa costituire un’indicazione all’intervento chirurgico.
Bibliografia
- Nilsson IL, Yin L, Lundgren E, et al. Clinical presentation of primary hyperparathyroidism in Europe-nationwide cohort analysis on mortality from nonmalignant causes. J Bone Min Res 2002, 17 (suppl 2): N68-74.
- Vestergaard P, Mollerup CL, Frokjaer VG, et al. Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg 2003, 27: 216–22.
- Yu N, Donnan PT, Murphy MJ, Leese GP. Increased mortality and morbidity in mild primary hyperparathyroid patients. The PEARS study. Clin Endocrinol 2010, 73: 30-4.
- Wermers RA, Khosla S, Atkinson EJ, et al. Survival after the diagnosis of hyperparathyroidism: a population-based study. Am J Med 1998, 104: 115–22.
- Kalla A, Krishnamoorthy P, et al. Primary hyperparathyroidism predicts hypertension: Results from the National Inpatient Sample. Int J Card 2017, 227: 335–337.
- Hagstrom et al. Plasma Parathyroid Hormone and the Risk of Cardiovascular Mortality in the Community. Circulation 2009, 119: 2765-71.
- Yu N, Leese GP, Donnan PT. What predicts adverse outcomes in untreated primary hyperparathyroidism? The Parathyroid Epidemiology and Audit Research Study (PEARS). Clin Endocrinol (Oxf) 2013, 79: 27–34.
- Bollerslev J, Rosen T, Mollerup CL, et al. Effect of surgery on cardiovascular risk factors in mild primary hyperparathyroidism. J Clin Endocrinol Metab 2009, 94: 2255-61.
- Bilezikian JP, Khan AA, Potts JT, on behalf of the Third International Workshop on the Management of Asymptomatic Primary Hyperthyroidism. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 2009, 94: 335-9.
- The American Association of Clinical Endocrinologists and the American Association of Endocrine Surgeons position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Pract 2005, 11: 49-54.
- Stefenelli T, Mayr H, Bergler-Klein J, et al. Primary hyperparathyroidism: incidence of cardiac abnormalities and partial reversibility after successful parathyroidectomy. Am J Med 1993, 95: 197-202.
- Dalberg K, Brodin LA, Juhlin-Dannfelt A, et al. Cardiac function in primary hyperparathyroidism before and after operation. An echocardiographic study. Eur J Surg 1996, 162: 171-6.
- Piovesan A, Molineri N, Casasso F, et al. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin Endocrinol (Oxf) 1999, 50: 321–8.
- Walker MD, Fleischer JB, Di Tullio MR, et al. Cardiac structure and diastolic function in mild primary hyperparathyroidism. J Clin Endocrinol Metab 2010, 95: 2172–9.
- Almqvist EG, Bondeson AG, Bondeson L, et al. Cardiac dysfunction in mild primary hyperparathyroidism assessed by radionuclide angiography and echocardiography before and after parathyroidectomy. Surgery 2002, 132: 1126–32; discussion 1132.
- Nuzzo V, Tauchmanovà L, Fonderico F, et al. Increased intima-media thickness of the carotid artery wall, normal blood pressure profile and normal left ventricular mass in subjects with primary hyperparathyroidism. Eur J Endocrinol 2002, 147: 453-9.
- Nilsson IL, Aberg J, Rastad J, et al. Left ventricular sistolic and diastolic function and exercise testing in primary hyperparathyroidism—effects of parathyroidectomy. Surgery 2000, 128: 895–902.
- Rapado A. Arterial hypertension and primary hyperparathyroidism. Incidence and follow-up after parathyroidectomy. Am J Nephrol 1986, 6 (suppl 1): 49-50.
- Feldstein CA, Akopian M, Pietrobelli D, et al. Long-term effects of parathyroidectomy on hypertension prevalence and circadian blood pressure profile in primary hyperparathyroidism. Clin Exp Hypertens 2010, 32: 154-8.
- Lind L, Hvfarner A, Palmer ME. Hypertension in primary hyperparathyroidism in relation to histopathology. Eur J Surg 1991, 157: 457-9.
- Jorde R, Svartberg J, S, Unsfjord J. Serum parathyroid hormone as a predictor of increase in systolic blood pressure in men. J Hypertens 2005, 23: 1639-44.
- Guixiang Z, Ford SL, Chaoyang L, et al. Independent associations of serum concentrations of 25-hydroxyvitamin D and parathyroid hormone with blood pressure among US adults. J Hypertens 2010, 28: 1821-28.
- Broulik PD, Brouliková A, Adámek S, et al. Improvement of hypertension after parathyroidectomy of patients suffering from primary hyperparathyroidism. Int J Endocrinol 2011, 2011: 309068.
- Heyliger A, Tangpricha V, Weber C, et al. Parathyroidectomy decreases systolic and diastolic blood pressure in hypertensive patients with primary hyperparathyroidism. Surgery 2009, 146: 1042–7.
- Nilsson IL, Aberg J, Rastad J, et al. Endothelial vasodilatory dysfunction in primary hyperparathyroidism is reversed after parathyroidectomy. Surgery 1999, 129: 1049–55.
- Kovacs L, Goth MI, Szabolcs I, et al. The effect of surgical treatment on secondary hyperaldosteronism and the relative hyperinsulinemia in primary hyperparathyroidism. Eur J Endocrinol 1998, 138: 543–7.
- Bernini G, Moretti A, Lonzi S, et al. Renin-angiotensin-aldosterone system in primary hyperparathyroidism before and after surgery. Metabolism 1999, 48: 298–300.
- Gennari C, Nami R, Gonnelli S. Hypertension and primary hyperparathyroidism: the role of adrenergic and renin-angiotensin aldosterone systems. Miner Electrolyte Metab 1995, 21: 77–81.
Altre manifestazioni dell'iperparatiroidismo primario
Elena Castellano
Endocrinologia, Ospedale S. Croce & Carle, Cuneo
(aggiornato al 5 giugno 2019)
Le manifestazioni non classiche del PHPT includono alterazioni cardiache, metaboliche, neuro-muscolari, gastro-intestinali, neuro-psichiche e cognitive.
Il PHPT sintomatico con manifestazioni classiche di malattia (osteite fibroso-cistica, nefrolitiasi e sintomi di ipercalcemia) è stato storicamente associato a un’aumentata mortalità rispetto alla popolazione generale (1). In studi di coorte europei (2,3) è stata riportata una maggior mortalità, soprattutto per cause cardio-vascolari (CV), anche nei pazienti con PHPT pauci-asintomatico (aPHPT), non confermata in studi epidemiologici nordamericani (4).
Negli ultimi anni diversi studi hanno suggerito un’associazione tra PHPT e malattie metaboliche, CV, neuro-muscolari, peggiore qualità della vita (QoL) e persino aumentato rischio di alcuni tumori (5). Al momento attuale però, nessuna di queste manifestazioni non classiche di PHPT viene presa in considerazione dalle linee guida (LG) per l’inquadramento clinico della malattia, né per stabilire l’indicazione alla paratiroidectomia (PTX).
Alterazioni funzionali e strutturali cardiache e vascolari
I livelli di calcemia e PTH sono correlati con la sopravvivenza e la morbilità CV nella popolazione generale, così come nei pazienti con PHPT (6). Il PTH, infatti, causa vaso-dilatazione e ha effetto cronotropo e inotropo indiretto. Sebbene sia stata ipotizzata una relazione tra livelli di PTH e massa cardiaca, tale relazione non è stata confermata in tutti gli studi; più recentemente è stato ipotizzato un ruolo dell’ipovitaminosi D nell’insorgenza dell’ipertrofia cardiaca. L’ipercalcemia determina invece alterazioni ECG (accorciamento del QT, PR prolungato, onde J) e calcificazioni valvolari. Nei pazienti con PHPT è stato riportato un certo grado di disfunzione diastolica: anche per questo parametro, tuttavia, i dati non sono al momento conclusivi. Ancora più scarni sono i dati sulla disfunzione endoteliale.
Gli effetti benefici della PTX su endpoint CV riportati da diversi studi osservazionali (6,7) in pazienti con PHPT sintomatico non sono stati ad oggi confermati da studi randomizzati controllati.
L’entità del coinvolgimento cardiaco nell’aPHPT è discussa (5); per questo, anche le più recenti LG non inseriscono le complicanze CV tra i criteri di indicazione all’intervento chirurgico.
Ipertensione arteriosa
È riportata in letteratura un’aumentata prevalenza di ipertensione arteriosa. Studi di coorte sulla popolazione generale hanno inoltre evidenziato una correlazione diretta tra i livelli di PTH e i valori tensivi, con un possibile ruolo della vitamina D3: bassi livelli di 25OH-vitD sembrano attivare il sistema renina-angiotensina-aldosterone ed è stata evidenziata una debole azione diretta del PTH sullo stesso sistema (6).
Alla luce di tali osservazioni, non risulta tuttavia ancora dimostrata una relazione causale tra PHPT e ipertensione arteriosa; peraltro l’effetto della PTX sui livelli tensivi appare favorevole solo in circa la metà dei pazienti con PHPT.
Alterazioni metaboliche
Nel PHPT sintomatico è stata riportata una maggior frequenza di alterazioni metaboliche, quali diabete mellito (DM) e ridotta tolleranza glucidica (IGT); sono inoltre state segnalate dislipidemie e maggior frequenza di obesità. Uno studio (8) ha confermato una riduzione della sensibilità insulinica basale e stimolata e una correlazione significativa tra livelli di calcemia e insulino-resistenza (IR) nei soggetti con PHPT.
Nei pazienti con aPHPT i dati sono meno evidenti (9): alcuni studi hanno riportato un’aumentata prevalenza di DM2 e di IGT, che sembrerebbero dipendere da una riduzione nella sensibilità insulinica. Tuttavia, in osservazioni più recenti non è stata confermata una maggior prevalenza di sindrome metabolica, solitamente espressione clinica di IR. Uno studio di popolazione su donne in post-menopausa ha dimostrato la presenza di una dislipidemia pro-aterosclerotica (riduzione HDL, incremento di trigliceridi totali, trigliceridi-VLDL e colesterolo-VLDL) nell’aPHPT, responsiva alla PTX (10).
Alterazioni neuro-muscolari e articolari
Debolezza muscolare e artralgie sono riportati frequentemente nei pazienti con PHPT, con prevalenza estremamente variabile (13-93%) tra le diverse casistiche in letteratura, talvolta anche valutate tramite la somministrazione di questionari. È stato ipotizzato che tali sintomi siano da correlare primariamente all’ipercalcemia (11): il calcio, infatti, non solo è implicato nella contrazione muscolare, ma sembra esercitare un ruolo anche nella funzione neuronale e nella plasticità sinaptica. In secondo luogo, l’ipovitaminosi D sembra ridurre la performance muscolo scheletrica. Anche i livelli di PTH sono stati associati in studi sperimentali alla perdita muscolare. I livelli di PTH sono stati inoltre correlati, nel PHPT e nella popolazione generale, ad aumento delle concentrazioni di acido urico e alla condro-calcinosi.
Risultati contrastanti sono riportati riguardo al miglioramento delle alterazioni neuro-muscolari ed articolari dopo PTX, che sembra essere particolarmente efficace per il miglioramento della debolezza muscolare.
Alterazioni gastro-intestinali
Nei quadri di PHPT sintomatico sono descritti sintomi gastro-intestinali, quali stipsi, nausea, vomito e algie addominali, ricondotti all’ipercalcemia (12). Gli elevati livelli di calcemia possono favorire la comparsa di ulcere peptiche o di pancreatite, riducendo la contrattilità della muscolatura liscia, stimolando l’ipergastrinemia e la secrezione acida, promuovendo la deposizione di calcoli nei dotti pancreatici.
La risposta clinica all’intervento chirurgico negli studi più datati era netta. In studi più recenti, in cui l’attività di malattia è meno spiccata, la frequenza delle complicanze gastro-intestinali è ridotta e l’effetto della PTX appare contestualmente meno evidente.
Alterazioni neuro-psichiatriche
L’ipercalcemia severa può accompagnarsi a quadri neuro-psichiatrici conclamati (13), che includono confusione mentale, psicosi, letargia, fino al coma. La correzione dell’ipercalcemia solitamente porta alla rapida risoluzione di queste manifestazioni.
Nel PHPT pauci-asintomatico, in cui la calcemia è meno elevata, le manifestazioni neuro-psichiatriche sono meno evidenti e del tutto aspecifiche (9), in particolare nei pazienti più anziani.
Qualità di vita, decadimento cognitivo e depressione
La letteratura è concorde nel rilevare una percezione peggiore della QOL da parte dei pazienti con PHPT, sia sintomatico che asintomatico. Diversi RCT hanno valutato gli effetti della PTX versus l’osservazione sulla QOL (attraverso il questionario QoL SF-36): dopo PTX Rao et al (14) hanno riportato un miglioramento per alcuni aspetti (funzionamento sociale, vissuto emotivo) e un significativo peggioramento di altri (emotività, funzionamento sociale, percezione di salute) nei pazienti non trattati.
Ambrogini et al (15) hanno valutato gli effetti della PTX in soggetti con aPHPT che non soddisfacevano i criteri per la chirurgia proposti dalle LG internazionali (categoria oggi definita “mild” PHPT): ad un anno dalla PTX i punteggi miglioravano in 4 ambiti (dolore, salute generale, vitalità e salute mentale) e rimanevano invariati nei non operati. Al contrario il gruppo di Bollerslev (16), in pazienti con aPHPT non ha evidenziato alcun miglioramento nei parametri del QoL a due anni da PTX rispetto ai non sottoposti a intervento.
È stato recentemente introdotto un questionario specifico per la popolazione PHPT (PHPQoL), validato per la popolazione spagnola (17), che ha mostrato una buona correlazione con SF-36 ed è risultato mediamente peggiore nei soggetti con PHPT sintomatico versus aPHPT.
Nel PHPT è stato inoltre segnalato un decadimento neuro-cognitivo, particolarmente evidente nella malattia sintomatica e nei pazienti più anziani (14-16). I pochi studi condotti sistematicamente su questo aspetto confermano un’aumentata prevalenza di deficit cognitivi e depressione nei pazienti con PHPT.
Diversi autori hanno valutato l’effetto della PTX su questi sintomi: gli studi non controllati di Roman (18) e di Shah-Becker (19) hanno mostrato un significativo miglioramento di ansia e depressione dopo PTX in casistiche miste di pazienti con PHPT sintomatico o asintomatico; Walker (20), che ha riportato peggiori livelli di ansia e depressione nei soggetti con PHPT rispetto ai controlli, ha evidenziato un miglioramento significativo della sola depressione dopo PTX.
Rischio tumorale
Alcuni studi hanno ipotizzato un legame tra PHPT e incrementato rischio oncologico, non organo-specifico (21). Si ipotizza che il PTH possa avere un effetto indiretto di promozione neoplastica tramite lo stimolo di produzione di IGF-1; peraltro, esistono diverse evidenze sperimentali che mostrano l’espressione tumorale di PTHrp e di recettori per il PTH/PTHrp, che medierebbero un effetto mitogenico ed anti-apoptotico (22). Un'altra ipotesi, più recente, contempla un possibile difetto del recettore della vitamina D (23), che promuoverebbe da un lato la predisposizione al PHPT, dall’altro la perdita dell’effetto pro-apoptotico della vitamina D. Sono certamente necessari altri studi per confermare queste ipotesi.
Conclusioni
Nel PHPT moderato-severo è stato riportato un incremento della mortalità CV. L’ipertrofia ventricolare sinistra è l’unica complicanza CV associata al PHPT che sembra migliorare dopo PTX, mentre i dati riguardanti la reversibilità di ipertensione arteriosa e calcificazioni vascolari sono al momento inconsistenti. Allo stato attuale, nel paziente con PHPT non è raccomandata una valutazione CV e la presenza di malattia cardiaca non costituisce un’indicazione alla PTX.
Il sintomo neuro-muscolare più frequentemente riportato nel PHPT è la debolezza neuro-muscolare, che sembra migliorare dopo PTX. Al momento anche questo disturbo non è contemplato dalle LG come criterio per la chirurgia.
Inoltre, nel PHPT sintomatico le disfunzioni neuro-cognitive sono prevalenti rispetto alla popolazione generale, così come la QoL sembra più compromessa: entrambi migliorano dopo PTX, ma il dato è più controverso nel soggetto con PHPT mild.
Infine è verosimile che esista un legame tra PHPT e incrementato rischio oncologico, ma al momento i dati non risultano conclusivi.
Bibliografia
- Nilsson IL, Yin L, Lundgren E, et al. Clinical presentation of primary hyperparathyroidism in Europe-nationwide cohort analysis on mortality from nonmalignant causes. J Bone Min Res 2002, 17 (suppl 2): N68-74.
- Vestergaard P, Mollerup CL, Frokjaer VG, et al. Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg 2003, 27: 216–22.
- Yu N, Donnan PT, Murphy MJ, Leese GP. Increased mortality and morbidity in mild primary hyperparathyroid patients. The PEARS study. Clin Endocrinol 2010, 73: 30-4.
- Wermers RA, Khosla S, Atkinson EJ, et al. Survival after the diagnosis of hyperparathyroidism: a population-based study. Am J Med 1998, 104: 115–22.
- Chiodini I, Cairoli E, Palmieri S, et al. Non classical complications of primary hyperparathyroidism. Best Pract Res Clin Endocrinol Metab 2018, 32: 805-20.
- Pepe J, Cipriani C, Sonato C, et al. Cardiovascular manifestations of primary hyperparathyroidism: a narrative review. Eur J Endocrinol 2017, 177: R297-308.
- Walker MD, Silverberg SJ. Cardiovascular aspects of primary hyperparathyroidism. J Endocrinol Invest 2008, 31: 925-31.
- Tassone F, Procopio M, Gianotti L, et al. Insulin resistance is not coupled with defective insulin secretion in primary hyperparathyroidism. Diab Med 2009, 26, 968-73.
- Silverberg SJ, Clarke BL, Peacock M, et al. Current issues in the presentation of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab 2014, 99: 3580-94.
- Hagström E, Lundgren E, Lithell H, et al. Normalized dyslipidaemia after parathyroidectomy in mild primary hyperparathyroidism: population-based study over five years. Clin Endocrinol 2002, 56: 253-60.
- Pappu R, Jabbour SA, Regianto AM, et al. Musculoskeletal manifestations of primary hyperparathyroidism. Clin Rheumatol 2016, 35: 3081-7.
- Agarwal A, George RK, Gupta SK, et al. Pancreatitis in patients with primary hyperparathyroidism. Indian J Gastroenterol 2003, 22: 224-5.
- Joborn C, Hetta J, Johansson H, et al. Psychiatric morbidity in primary hyperparathyroidism. World J Surg 1988, 12: 476-81.
- Rao DS, Phillips ER, Divine GW, et al. Randomized controlled clinical trial of surgery versus no surgery in patients with mild asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab 2004, 89: 5415-22.
- Ambrogini E, Cetani F, Cianferotti L, et al. Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab 2007, 92: 3114-21.
- Bollerslev J, Jansson S, Mollerup CL, et al. Medical observation, compared with parathyroidectomy, for asymptomatic primary hyperparathyroidism: a prospective, randomized trial. J Clin Endocrinol Metab 2007, 92: 1687-92.
- Webb SM, Puig-Domingo M, Villabona C, et al. Validation of PHPQoL, a disease-specific quality-of-life questionnaire for patients with primary hyperparathyroidism; PHPQoL validation group. J Clin Endocrinol Metab 2016, 101: 1571-8.
- Roman SA, Sosa JA, Pietrzak RH, et al. The effects of serum calcium and parathyroid hormone changes on psychological and cognitive function in patients undergoing PTX for primary hyperparathyroidism. Ann Surg 2011, 253: 131-7.
- Shah-Becker S, Derr J, Oberman BS, et al. Early neurocognitive improvements following parathyroidectomy for primary hyperparathyroidism. Laryngoscope 2018, 128: 775-80.
- Walker MD, McMahon DJ, Inabnet WB, et al. Neuropsychological features in primary hyperparathyroidism: a prospective study. J Clin Endocrinol Metab 2009, 94: 1951-8.
- Palmieri S, Roggero L, Cairoli E, et al. Occurrence of malignant neoplasia in patients with primary hyperparathyroidism. Eur J Intern Med 2017, 43: 77-82.
- Carron JA, Fraser WD, Gallagher JA. PTHrP and the PTH/PTHrP receptor are co-expressed in human breast and colon tumours. Br J Cancer 1997, 76: 1095-8.
- Carling T, Rastad J, Szabo E, et al. Reduced parathyroid vitamin D receptor messenger ribonucleic acid levels in primary and secondary hyperparathyroidism. J Clin Endocrinol Metab 2000, 85: 2000-3.
Iperparatiroidismo in gravidanza
Giorgio Borretta
Endocrinologia, AO S Croce e Carle, Cuneo
Il PHPT in gravidanza è di rara osservazione (< 1% dei casi) ma assume una notevole rilevanza clinica per la frequenza e la gravità delle complicanze ad esso correlate (1,2). Le poche casistiche sinora pubblicate riportano infatti fino al 67% di complicanze materne (nefrolitiasi, osteite fibroso-cistica, disturbi gastro-intestinali, ipertensione arteriosa, pre-eclampsia, eclampsia, crisi ipercalcemiche, polidramnios e aborto) e fino all’80% di complicanze fetali e neonatali (morte intra-uterina, parto pretermine, basso peso alla nascita, ipocalcemia severa neonatale con o senza tetania, morte neonatale). E’ quindi indicata la misurazione della calcemia nelle donne con storia anamnestica di aborto che intendono iniziare una gravidanza o, quanto prima, durante la gravidanza (1,2). In queste ultime però è consigliabile il dosaggio della calcemia ionizzata o corretta per le proteine circolanti, in quanto nelle forme lievi di PHPT la calcemia totale potrebbe risultare normale (pseudo-normocalcemia) in conseguenza della fisiologica emodiluizione indotta dalla gravidanza (3).
Il trattamento di elezione del PHPT in gravidanza è chirurgico; la PTX, infatti, si è rivelata più efficace del trattamento conservativo nel ridurre le complicanze materne e soprattutto quelle fetali/neonatali (2). La PTX va eseguita nel secondo trimestre, in quanto il rischio operatorio è minore; poiché l’incidenza di aborto è massima tra la 10° e la 15° settimana, la PTX andrebbe praticata all’inizio del trimestre. La sicurezza della PTX nel terzo trimestre è tuttora dibattuta (4).
Nel primo trimestre, invece, è raccomandato il trattamento conservativo, che prevede l’impiego, in regime di ricovero, di terapia ipocalcemizzante nei casi sintomatici e con calcemia > 12 mg/dL, mediante somministrazione ev di liquidi, furosemide ed eventualmente calcitonina (fig 1 e 2) (1).
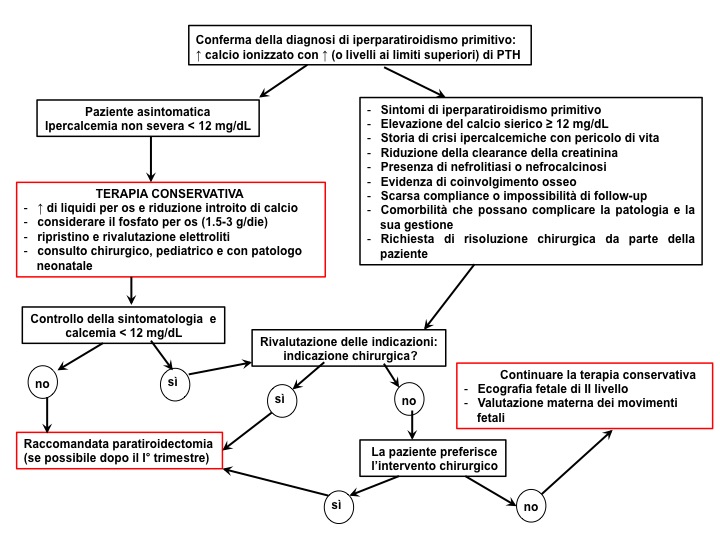
Figura 1. Flow-chart sulla gestione dell'iperparatiroidismo in gravidanza (modificata da 1)
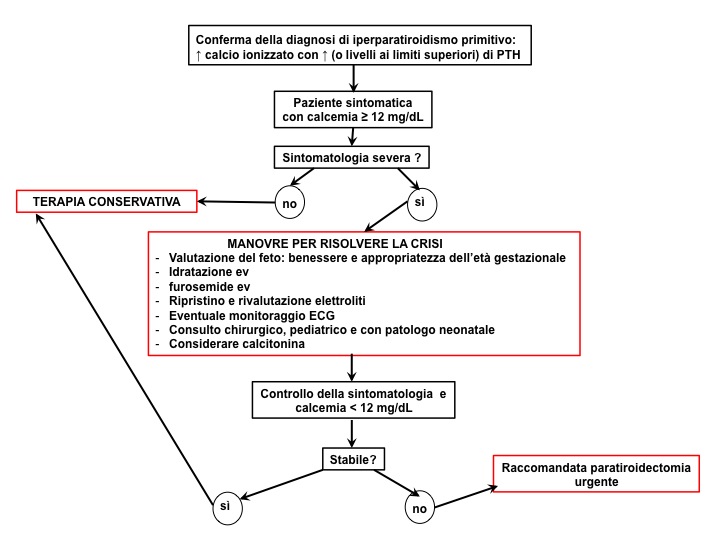
Figura 2. Flow-chart sulla gestione dell'iperparatiroidismo grave in gravidanza (modificata da 1)
L’ecografia è l’unica indagine indicata in gravidanza per la localizzazione pre-operatoria delle paratiroidi patologiche; nei casi dubbi può essere praticata la FNAB della lesione ecograficamente sospetta, con dosaggio di PTH su aspirato (5).
Quando indicata, la MIP risulta la tecnica chirurgica più appropriata in gravidanza, in quanto riduce il tempo chirurgico, il dolore ed il rischio di ipocalcemia post-operatori (6).
Nelle gravide con PHPT mild nella maggioranza dei casi viene considerato appropriato il trattamento conservativo mediante idratazione; il ricorso a PTX, o eventualmente alla terapia con cinacalcet, sono consigliati in caso di peggioramento dell'ipercalcemia (7).
Bibliografia
- Schnatz PF, Curry SL. Primary hyperparathyroidism in pregnancy: evidence-based management. Obstet Gynecol Surv 2002, 57: 365–76.
- Norman J, Politz D, Politz L. Hyperparathyroidism during pregnancy and the effect of rising calcium on pregnancy loss: a call for earlier intervention. Clin Endocrinol 2009, 71: 104-9.
- Kovacs CS, Kronemberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium and lactation. Endocr Rev 1997, 18: 832-72.
- Schnatz PF, Taxton S. Parathyroidectomy in the third trimester of pregnancy. Obstet Gynecol Surv 2005, 60: 672-82.
- Pothiwala P, Levine SN. Parathyroid surgery in pregnancy: review of the literature and localization by aspiration for parathyroid hormone levels. J Perinatology 2009, 29: 779-84.
- Zini M, Attanasio R, Cesareo R, et al. AME position statement: primary hyperparathyroidism in clinical practice. J Endocrinol Invest 2012, 35 (7 suppl): 2-21.
- Marcocci C, Brandi ML, Scillitani A, et al. Italian Society of Endocrinology Consensus Statement: Definition, Evaluation and Management of Patients with Mild Primary Hyperparathyroidism. J Endocrinol Invest 2015, 38: 577-93.
Forme familiari di iperparatiroidismo primario
Alberto Falchetti
Servizio Malattie del Metabolismo Minerale ed Osseo, Ospedale San Giuseppe, Piancavallo (VB) e Diabete & Laboratorio di Ricerca Endocrina e Metabolica, Istituto Auxologico Italiano
(aggiornato al 5 gennaio 2021)
INTRODUZIONE
Esistono diverse forme sindromiche familiari di iperparatiroidismo primario (FPHTP). Mutazioni germinali di specifici geni (tabella 1) spiegano la suscettibilità a sviluppare tumori paratiroidei e anche altri tumori, endocrini e non endocrini. Le sindromi FPHPT a tutt’oggi note sono:
- MEN-1, MEN-2A, MEN-4 e Sindrome Iperparatiroidismo Primitivo-Tumori Mascellari (HPT-JT), in cui il PHPT si associa ad altre manifestazioni cliniche, tumorali e non;
- Iperparatiroidismo Primitivo Isolato Familiare (FIHP), Ipercalcemia Ipocalciurica Familiare (FHH) e Iperparatiroidismo Neonatale severo (NSHPT), in cui il solo PHPT può "semplicemente" costituire l'unica anomalia proliferativa familiare endocrina iperfunzionante.
| Tabella 1 Forme familiari di PHPT, geni e loci, ereditarietà |
|||
| Disordine | Gene | Locus cromosomico | Ereditarietà |
| Sindromiche | |||
| MEN-1 | MEN1 | 11q13.1 | AD |
| MEN-2A | RET | 10q11.21 | AD |
| MEN-4 | CDKN1B | 12p13.1 | AD |
| HPT-JT | CDC73 (HRPT2) | 1q31.2 | AD |
| Non sindromiche | |||
| FIHP | MEN1, CDC73, CaSR (e altri non noti) | 11q13.1, 1q31.2, 3q13.3-q21.1 | AD |
| FHH tipo 1 | CaSR | 3q13.3-q21.1 | AD |
| FHH tipo 2 | GNA11 | 19p13.3 | AD |
| FHH tipo 3 | AP2S1 | 19q13.32 | AD |
| NSHPT | CaSR | 3q13.3-q21.1 | AR |
| AD = autosomica dominante; AR = autosomica recessiva | |||
Ricordare: le forme sindromiche di FPHPT rappresentano il 2-5% (comunque < 10%) dell’Iperparatiroidismo Primitivo (PHPT) complessivo.
CARATTERISTICHE TIPICHE DI FPHPT
La tabella 2 riporta le caratteristiche clinico-patologiche generali, comuni a quasi tutte le forme sindromiche di FPHPT.
| Tabella 2 Caratteristiche clinico-patologiche generali delle forme di FPHTP |
| Coinvolgimento multi-ghiandolare |
| Età di esordio precoce rispetto alla forma sporadica, non sindromica, di PHPT |
Esistono differenze tra le varie forme di FPHPT e la diagnosi clinica precoce è fondamentale per un’adeguata gestione clinica e per prevenirne le complicanze, come nefrolitiasi, nefrocalcinosi, osteoporosi ed osteite fibroso-cistica; il franco PHPT verosimilmente influenza anche disfunzioni neuro-cognitive, morbilità e mortalità cardio-vascolare e, forse, incidenza di tumori.
Ricordare: importante eseguire lo screening clinico per tutte queste complicanze anche nelle forme di FPHPT, pure se colpiscono generalmente soggetti più giovani rispetto al PHPT sporadico.
Aspetti generali
La diagnosi clinica, biochimica e radiologica delle complicazioni non differisce sostanzialmente fra FPHPT e PHPT sporadico. Esistono somiglianze e differenze clinico-patologiche tra le sindromi FPHPT. È sempre presente una franca crescita del tessuto paratiroideo affetto, tranne che in FHH dove può essere scarsa/assente. Tuttavia, l'espressione clinica di FPHPT può presentare un'ampia gamma di gravità e di età di esordio, come riportato nella tabella 3.
| Tabella 3 Diversa espressione di gravità ed esordio nelle forme di FPHPT |
||
| Gravità | Pericolosa per la vita | HPT-JT e NSHPT |
| Generalmente asintomatica | FHH | |
| Esordio | In utero | NSHPT |
| Neonatale | NSHPT e FHH | |
| Intorno/dopo l'infanzia | MEN-1, MEN-2A, HPT-JT | |
| Tarda età adulta | FIHP | |
SISTEMATICA DELLE DIVERSE FORME
PHPT nell’ambito di MEN-1
È caratterizzato da esordio precoce, a 20-30 anni contro i 50-75 anni di età del PHPT sporadico. Circa il 50% dei bambini e adolescenti con MEN-1 ha già sviluppato PHPT al momento dell’osservazione clinica e fino al 75% dei pazienti con MEN-1 di età < 21 anni può avere PHPT. La penetranza di PHPT nell’ambito di MEN-1 è ≌100% (entro i 50 anni) e frequentemente è la prima anomalia biochimico-clinica di presentazione.
In buona parte dei casi di età > 10 anni PHPT può essere asintomatico e rilevato solo in corso di screening biochimico. Tuttavia, il 9-14% presenta urolitiasi e il 22-30% dei pazienti è sottoposto a chirurgia paratiroidea a causa di sintomi o alti livelli di calcemia.
In linea con la trasmissione autosomica dominante, si osserva un'equa distribuzione fra i sessi. La malattia è caratterizzata da coinvolgimento paratiroideo multi-ghiandolare, asimmetrico e asincrono, che può portare a possibile “confusione” con un adenoma singolo quando al momento della chirurgia iniziale viene macroscopicamente identificata solo una ghiandola anormale.
I livelli di calcemia e di PTH sono correlati con l'età, mentre non lo sono quelli di calciuria, i calcoli renali o il coinvolgimento osseo. In quasi il 40% dei casi di età < 50 anni sono presenti livelli di PTH inappropriatamente normali. È stato suggerito che un paziente MEN-1 di età < 50 anni con livelli di PTH nella norma ha una probabilità di sviluppare PHPT 13.5 volte maggiore rispetto a un paziente, non MEN-1, di età > 50 anni di avere livelli elevati di PTH e che, in questa popolazione, l'analisi genetica mutazionale germinale del gene MEN1 potrebbe essere cost-effective.
Nonostante una presentazione biochimica apparentemente più lieve (livelli di calcemia e PTH inferiori rispetto al PHPT sporadico), sono segnalati un coinvolgimento osseo precoce e grave, nonché complicanze più frequenti. Nei pazienti MEN-1 con sindrome di Zollinger-Ellison, l’ipercalcemia, agendo sul recettore sensibile al calcio (CaSR) espresso dalle cellule del gastrinoma, potrebbe aumentare la secrezione di gastrina e quindi i sintomi gastro-intestinali correlati. Pertanto, la concomitante presenza di gastrinoma/i potrebbe rappresentare un criterio aggiuntivo per la paratiroidectomia (PTX).
Considerando l'elevato tasso di recidiva, anche nei pazienti sottoposti con successo a PTX deve essere eseguita annualmente una valutazione biochimica che includa almeno calcemia ionizzata, fosfatemia e calciuria 24 ore.
Ricordare: le linee guida europee suggeriscono che nei pazienti con MEN-1 senza PHPT già diagnosticato si deve valutare la calcemia corretta per l’albumina e il PTH sierico. Tuttavia, considerando la frequente presenza di livelli di PTH inappropriatamente normali, specialmente nei pazienti più giovani e con livelli di calcemia solo leggermente elevati, l'estensione della valutazione a calcio ionizzato, fosfato e calciuria 24 ore può facilitare il riscontro biochimico di forme di MEN-1/PHPT più lievi ma che potrebbero aver già causato complicanze ossee o renali asintomatiche.
PHPT nell’ambito di MEN-2A
Compare in circa il 10-25% dei pazienti con mutazioni RET specifiche. La sua penetranza è inferiore ad altre manifestazioni di MEN-2. È più frequentemente associato a (qualunque) mutazione del codone 634 del gene RET, anche se esiste un'ampia differenza nella prevalenza tra le famiglie, suggerendo un coinvolgimento di fattori ambientali, o modificanti, nella sua espressività clinica.
Come in altre forme di FPHPT, è caratterizzato da esordio precoce rispetto al PHPT sporadico (40 e 50-75 anni, rispettivamente), tipicamente con malattia paratiroidea multi-ghiandolare.
In circa il 50% dei casi non si osserva ipercalcemia. Clinicamente è una forma moderata, spesso asintomatica, soprattutto in età più giovane, con segni clinici di malattia solo nel 15-25% dei casi.
Si consiglia screening per MEN2A-PHPT nei membri delle famiglie affette entro gli 8 anni di età, ripetendo annualmente, qualunque sia il codone mutato, almeno calcemia, albuminemia, PTH, fosfatemia e calciuria delle 24 ore.
Ricordare: anche se spesso lieve e asintomatico, i criteri chirurgici sono gli stessi di quelli applicati al PHPT sporadico, ma, considerando il coinvolgimento multi-ghiandolare, è solitamente obbligatoria un'esplorazione bilaterale dei compartimenti del collo. Il tasso di recidiva, dopo PTX subtotale apparentemente riuscita, è inferiore a quello del PHPT nell’ambito di MEN-1. Attualmente non è consigliabile eseguire PTX sistematica nei pazienti MEN-2A che richiedono chirurgia tiroidea per carcinoma midollare e con livelli normali di calcemia e PTH.
PHPT nell’ambito di MEN-4
Ha predominanza femminile e colpisce approssimativamente l’80% dei casi segnalati fino ad oggi della sindrome. Rispetto al PHPT nell’ambito di MEN-1, quello nell’ambito di MEN-4 si verifica a un'età successiva (media d’insorgenza 56 anni). Sono documentati casi di PHPT, apparentemente non familiari, i cui adenomi paratiroidei ospitano mutazioni di CDKN1B, somatiche e germinali, suggerendo quindi essere MEN-4. Ad oggi, nessuno dei casi di MEN-4 ha mostrato recidiva di PHPT dopo chirurgia, suggerendo che la forma di PHPT nell’ambito di MEN-4 possa rappresentare uno spettro di malattia paratiroidea complessivamente più mite rispetto a quello nell’ambito di MEN-1.
Ricordare: non esiste, ad oggi, alcuna linea guida specifica sulla gestione di PHPT nell’ambito di MEN-4 e le indicazioni per la chirurgia paratiroidea si sovrappongono a quelle della MEN-1.
PHPT nell’ambito di HPT-JT
L'ipercalcemia è comune nel primo decennio di vita. Tra gli adulti, è segnalato PHPT nell'80% dei casi ed è abbastanza frequente il riscontro di calcemia > 12 mg/dL (3 mmol/L). Tuttavia, relativamente all'ipercalcemia (presente in > 90% dei soggetti MEN-1), la diagnosi differenziale tra HPT-JT e MEN1-PHPT è difficile e può dipendere da risultati istologici, così come anche da altre lesioni caratteristiche specificamente associate a ciascuna sindrome.
PHPT è in genere la prima manifestazione, associata o meno a tumori ossificanti della mascella e mandibola (30%), a tumori renali (tumore di Wilms, cisti, amartomi, 15%) e a fibromiomi uterini. Tuttavia, i pazienti HPT-JT hanno spesso anche ipercalciuria. L'allargamento dimensionale della ghiandola paratiroidea in HPT-JT è altamente asimmetrico e non è infrequente la scoperta alla chirurgia di una sola paratiroide anomala.
La maggior parte delle famiglie HPT-JT condivide le caratteristiche con FIHP e la distinzione tra FIHPT e HPT-JT è particolarmente difficile in assenza di tumori alla mascella, anche se tale distinzione è estremamente importante poiché i pazienti HPT-JT hanno un rischio più elevato di sviluppare carcinoma paratiroideo (CP) (vedi sotto). In questi casi, l'identificazione di ulteriori caratteristiche cliniche, quali adeno-carcinomi pancreatici, tumori testicolari a cellule germinali miste (componente seminomatosa prevalente) e adenomi tiroidei a cellule di Hürthle, può essere utile per distinguere i soggetti affetti da HPT-JT da quelli con FIHPT.
Ricordare: raramente la sola ecografia del collo è inizialmente in grado di identificare una neoplasia paratiroidea in un portatore normocalcemico. Il tumore paratiroideo, benigno o maligno, è spesso cistico o microcistico e, in generale, è una lesione monoclonale.
FIHPT
La diagnosi di FIHPT è principalmente di esclusione, poiché può rappresentare casi di espressione incompleta di una forma PHPT sindromica, come MEN-1, HPT-JT o FHH, e/o del verificarsi di due casi di PHPT che non condividono mutazione germinale. In oltre 40 casi di FIHPT sono state identificate mutazioni germinali dei geni MEN1, CDC73 o CaSR. I meccanismi che determinano l'espressione fenotipica “anomala” di tali mutazioni devono essere ancora chiariti e nella maggior parte delle famiglie è ancora sconosciuta l'eziologia genetica del FIHPT, forma apparentemente non sindromica.
Frequentemente, la diagnosi clinica di PHPT, in pazienti geneticamente definiti FIHPT, avviene in età più avanzata, in media a 55 anni, rispetto ad altre forme di FPHPT e condivide la frequenza abituale di caratteristiche "classiche" associate a PHPT, come nefrolitiasi e osteopenia.
PHPT e FHH
In tutte le forme di FHH, l'inattivazione della via correlata al CaSR, a livello di paratiroidi, reni e scheletro, spiega lo squilibrio verso il lato destro della curva del set-point del calcio PTH-dipendente (il valore della concentrazione extra-cellulare di calcio a cui la secrezione di PTH è al 50% della massimale). Nell’FHH la gestione renale del calcio è “anormale” e, anche se è normale l'entità della soppressione del PTH per l’aumento dei livelli di calcemia, generalmente manca la risposta ipercalciurica all'ipercalcemia: il riassorbimento tubulare di calcio è alto e persisterebbe ancora alto dopo eventuale “inopportuna” PTX totale.
Sono geneticamente riconosciute 3 forme distinte di FHH, in rapporto a mutazioni germinali inattivanti di specifici geni (tabella 1): FHH-1 è la forma più frequente che rappresenta oltre il 65% dei casi, mentre FHH-2 e FHH-3 rappresentano < 5% e > 5% dei casi, rispettivamente.
È descritta una penetranza quasi completa per l'espressione fenotipica dell'ipercalcemia, che può essere trovata fin dalla prima settimana di vita, rimanendo stabile per tutta l’età adulta. L'ampiezza dell'ipercalcemia è simile a quella del PHPT tipico e i sintomi generalmente correlati sono solitamente assenti/lievi, ad eccezione della pancreatite occasionale. Membri familiari, o soggetti da diverse famiglie FHH che condividono la stessa mutazione germinale, mostrano un clustering del livello medio di calcemia.
I pazienti FHH sono, in genere, clinicamente asintomatici e presentano inappropriata ipocalciuria nel contesto d'ipercalcemia e valori normali/elevati del PTH sierico.
Il rapporto tra clearance del calcio e della creatinina (CCR) rappresenta un indice utile per differenziare l'FHH rispetto al PHPT “classico”: CCR < 0.01 indica FHH. Tuttavia, il 20% dei soggetti FHH può avere CCR > 0.01, diventando, quindi, indistinguibile dal PHPT sporadico. Inoltre, un livello inferiore di CCR è riportato anche in pazienti afro-americani con PHPT sporadico e carenza di vitamina D o insufficienza renale.
Nell’FHH-1, i livelli di PTH sono normali o leggermente aumentati in circa l'80% dei pazienti, ma nel restante 20% il PTH sierico può essere elevato, rendendo questi soggetti a rischio di ricevere una diagnosi erronea di PHPT e quindi di essere sottoposti a un “inutile” intervento chirurgico al collo. Tuttavia, è importante ricordare che nell’FHH i livelli di fosfatemia sono generalmente normali e può essere presente lieve ipermagnesiemia. Queste alterazioni possono aiutare a distinguere FHH da PHPT in gran parte dei casi dubbi. L’FHH deve essere esclusa nei soggetti con ipercalcemia ed ipocalciuria, naturalmente dopo esclusione di interferenze farmacologiche (es. litio, tiazidici). I livelli di PTH possono essere particolarmente elevati nell’FHH-3 rispetto alle altre forme ed essere associati anche a moderata ipofosfatemia.
A differenza delle altre sindromi FPHPT, la dimensione delle paratiroidi è solitamente normale o solo minimamente ingrandita.
Ricordare: in casi di mancanza di chiaro fenotipo biochimico di FHH, potrebbe essere utile nella diagnosi differenziale l'identificazione di mutazioni germinali inattivanti di uno dei geni noti che rappresentano FHH 1-3.
NSHPT
La presentazione clinica avviene poco dopo la nascita, con severa ipercalcemia, ipocalciuria e franco PHPT, anomalie biochimiche che devono essere rapidamente rilevate e corrette perché pericolose per la vita.
Le ghiandole paratiroidi affette sono ingrandite, con aspetto iperplastico e ipercellularità.
Questa malattia paratiroidea multi-ghiandolare è determinata da mutazioni germinali inattivanti, in omozigosi, del gene del recettore sensibile al calcio (CaSR). Valutare sempre la possibilità di consanguineità nei genitori di un neonato affetto (autozigosi).
Ricordare: differentemente dall’FHH-1, nei pazienti con NSHPT è generalmente richiesta una PTX totale urgente per prevenire un esito fatale.
GESTIONE CLINICA DI UN PROBANDO FPHPT E DEI SUOI PARENTI DI PRIMO GRADO
Probando
Pur esistendo diversa penetranza del PHPT nelle sindromi familiari, PHPT rappresenta, generalmente, la prima manifestazione clinica, a un’età d’insorgenza precoce rispetto al PHPT sporadico. Di conseguenza, in un probando sintomatico il riconoscimento biochimico-clinico del PHPT è facilitato (molto frequenti ipercalcemia e aumentati livelli sierici di PTH) (tabella 4).
| Tabella 4 Ricerca/screening di anomalie tumorali/cliniche da eseguire nei casi di FPHPT |
|
| MEN-1 | Tumori neuroendocrini ipofisari, gastro-intestinali e disturbi proliferativi non endocrini (es. lipomi, angio-fibromi facciali e collagenomi) |
| MEN-2A | Carcinoma midollare tiroideo e feocromocitoma |
| MEN-4 | Tumori pancreatici, cervicali uterini (più rari), testicolari, feocromocitoma, paraganglioma e tumori renali |
| HPT-JT | Tumori fibro-ossei mascellari, tumori dell'utero, tumori/cisti/amartomi/adenomi corticali/papillari/carcinomi renali, tumore di Wilms |
| FHH | Ipocalciuria inappropriata (rispetto ai livelli ipercalcemici) |
È necessario ricostruire con precisione, tanto nel probando che nei parenti di primo grado, una storia clinica familiare dettagliata, considerando il possibile spettro variabile di presentazione biochimica-clinica delle patologie/alterazioni associate al PHPT per ciascuna sindrome FPHPT. Nello specifico, le domande descritte nella tabella 5 dovrebbero sempre essere poste e formulate con la massima chiarezza possibile, in base al grado di istruzione di ogni soggetto da intervistare.
| Tabella 5 Esempio di domande utili nella valutazione clinica di (F)PHPT |
| Ha mai sofferto o sono mai stati sospettati calcoli renali? |
| Ha parenti che hanno sofferto di calcoli renali, anche in giovane età? |
| Ci sono stati casi di osteoporosi o fratture ossee da fragilità in soggetti giovani adulti della sua famiglia? |
| Si sono verificati ipercalcemia grave e/o carcinomi paratiroidei in uno o più membri della famiglia? |
| Si sono verificati disturbi funzionali o altri disordini proliferativi endocrini/non endocrini, in uno o più membri della famiglia? (es. alterazioni dell’alvo/diarrea, eruzioni cutanee, ulcere gastro-duodenali, …) |
Ricordare: naturalmente, il probando deve effettuare lo screening biochimico-clinico-radiologico anche per tutti gli altri possibili disturbi proliferativi non funzionanti, che caratterizzano ogni specifica sindrome FPHPT.
Parenti di primo grado del probando con sospetto FPHPT
Non sempre emerge dallo screening una forma biochimica-clinica chiara e distinta di PHPT, talvolta si osserva PHPT normocalcemico. Se il test del DNA nel probando ha rivelato una mutazione germinale causale in uno dei geni responsabili di FPHPT, la sua identificazione nei parenti di primo grado è molto utile nella conferma ed eventualmente nella precocità della diagnosi clinica, guidando sia un'adeguata gestione medica che le tempistiche di follow-up per ciascuna sindrome FPHPT.
Ricordare: accurata e adeguata sotto-fenotipizzazione, con integrazione dei dati genetici e patologici con i dati biochimici e radiologici, per stabilire con il maggior grado di successo un adeguato processo decisionale, anche sul trattamento.
RISCHIO DI CARCINOMA PARATIROIDEO NELLE FORME FAMILIARI
MEN-1: pochissimi casi descritti di CP. Benché estremamente rara in generale, la presenza di CP è sempre possibile e deve essere considerata in pazienti con FPHPT e PHPT sintomatici.
MEN-2A/MEN-4: CP non segnalato.
HPT-JT: in circa il 5% dei casi può essere trovato un grande adenoma paratiroideo atipico, ma nel 20% dei casi può esserci un CP, che si dice essere presente già a 8 anni di età. Naturalmente, l'identificazione di un grande tumore della mascella può essere centrale per la diagnosi, ma a volte il CP può essere isolato, senza alcuna evidenza clinica di tali tumori (un piccolo tumore alla mascella può, comunque, essere facilmente trascurato).
La tabella 6 riassume alcune caratteristiche generali per sospettare CP.
| Tabella 6 Caratteristiche generali per sospetto di CP |
| Livelli di PTH 5-10 volte superiori al limite superiore dell'intervallo di riferimento |
| Livelli di calcemia > 12-12.8 mg/dL (3.0-3.2 mmol/L) |
| Coinvolgimento osseo e renale al momento della diagnosi |
| Massa palpabile del collo, presenza o forte sospetto clinico di tumori della mascella e storia familiare di PHPT o altri disturbi endocrini, correlati/correlabili allo spettro clinico, tipico o sospetto di una specifica sindrome FPHT |
| Grande/i lesione/i paratiroidea/e all'ecografia del collo |
Ricordare: in caso di sospetto CP, non deve essere eseguito agoaspirato.
SPUNTI PRATICI NELLA GESTIONE CLINICA DEI CASI DI FPHPT
Nei pazienti con lieve alterazione biochimica, è della massima importanza la ricerca di complicanze asintomatiche (osteoporosi, fratture vertebrali morfometriche, nefrolitiasi asintomatica) per raggiungere la migliore decisione sulla tempistica dell'intervento chirurgico. Un intervento chirurgico precoce può essere più difficile perché le ghiandole sono solo minimamente ingrandite ed esiste il rischio di predisporre il paziente a recidive e conseguente reintervento anche in tenera età.
Ricordare: un PHPT di lunga data potrebbe predisporre il paziente a patologia scheletrica più grave. In tutti i casi di PHPT, indipendentemente dall'età alla diagnosi, sospettare sempre un possibile FPHPT, in particolare di fronte a malattia paratiroidea multi-ghiandolare e/o inappropriata escrezione urinaria di calcio rispetto ai valori calcemici. In tutte le sindromi FPHPT esiste aumentato rischio di recidiva/persistenza di malattia dovuto al coinvolgimento paratiroideo multi-ghiandolare; sono quindi necessari accurata sorveglianza clinica e monitoraggio biochimico.
Indagini biochimiche
In tutti i pazienti con PHPT, con forma nota o sospetta di FPHPT, valutare e monitorare attentamente il metabolismo calcio-fosfato e il turnover osseo, anche in rapporto alle condizioni generali del soggetto e/o alla storia familiare.
Nella maggior parte dei casi di FHH, la sindrome potrebbe essere accertata/esclusa semplicemente utilizzando il rapporto fra le clearance del calcio e della creatinina (CCR). In soggetti privi di chiaro spettro biochimico FHH, è, comunque, molto utile, se non necessario, il test genetico per i geni CaSR (FHH-1), GNA11 (FHH-2) e AP2S1 (FHH-3).
Quando si sospetta CP sembra molto utile nella pratica clinica la possibilità di determinare il PTH sierico con test di ultima generazione. Infatti, un rapporto > 1 dei test PTH di II e di III generazione, insieme ad altri risultati biochimici, clinici e radiologici correlati, è utile per prevedere/sospettare CP prima dell'intervento chirurgico. La logica si basa sul fatto che la maggior parte dei CP produce una quantità eccessiva di amino-PTH, riconosciuto da saggi di PTH di III generazione, ma non di II. Tuttavia, non è stato raggiunto un consenso universale su questo approccio metodologico, anche perché esistono rari CP "non funzionanti".
Ricordare: quando si sospetta FPHPT, misurare la calcemia anche nei parenti di primo grado, insieme ad altre indagini biochimiche e cliniche in rapporto al possibile spettro fenotipico di presentazione per ciascuna sindrome.
Indagini radiologiche
È opportuno eseguire ecografia del collo, scintigrafia paratiroidea, Rx di cranio/mani/pelvi/femore e colonna e DXA lombare (se possibile applicare anche il TBS)-femorale-terzo distale dell'avambraccio non dominante.
L’imaging paratiroideo consente la localizzazione e la caratterizzazione delle anomalie paratiroidee al tempo pre-operatorio, permettendo l'identificazione e la caratterizzazione di una malattia singola della ghiandola paratiroidea (adenoma o CP), così come il rilevamento di malattia paratiroidea multi-ghiandolare (talvolta indicata all’indagine anatomo-patologica come “iperplasia” diffusa o iperplasia adenomatosa o pseudo-adenomatosa), aiutando un’appropriata strategia dell'approccio chirurgico. Tuttavia, dove esiste una predisposizione a malattia paratiroidea multi-ghiandolare, come si verifica nelle sindromi MEN, l'imaging paratiroideo è probabilmente più utile per localizzare la malattia paratiroidea recidivante/persistente dopo paratiroidectomia sub-totale o totale, quest'ultima seguita da auto-trapianto di frammenti di tessuto paratiroideo (tra i fasci muscolari brachio-radiali dell’avambraccio non dominante).
L’ecografia del collo e la scintigrafia 99Tecnezio SestaMIBI (99mTc-SestaMIBI) sono ancora gli approcci radiologici più comunemente usati in PHPT. È utile considerare che l'assorbimento di 99mTc-SestaMIBI è generalmente aumentato e prolungato nelle paratiroidi neoplastiche o iperplastiche funzionanti e può anche rivelare tessuto paratiroideo localizzato ectopicamente. Quando un PHPT sospetto/noto si verifica insieme a condizioni di nodularità della tiroide, l'uso della tecnica a doppio tracciante con sottrazione di immagine (99mTc-sestaMIBI combinato con iodio) potrebbe essere utile per tentare di “separare” la captazione di sestaMIBI paratiroidea da quella tiroidea, per raggiungere una migliore visualizzazione possibile del tessuto paratiroideo iperfunzionante.
Imaging extra-paratiroideo è necessario per valutare adeguatamente le complicanze PTH-correlate a livello scheletrico e renale, poiché risultati positivi possono giustificare un intervento chirurgico anche in pazienti con PHPT asintomatico. La tabella 7 riassume le indagini più utili.
| Tabella 7 Imaging extra-paratiroideo in corso di (F)PHPTH |
|
| Ecografia reni e vie escretrici renali | Per escludere nefrocalcinosi e nefrolitiasi clinicamente "silenti" (si verificano fino al 20% dei casi, come descritto in particolare in MEN-1). |
| DXA lombare-femorale-avambraccio non dominante | Valuta se presente ridotta densità minerale ossea (BMD), con aumentato rischio per fratture da fragilità. Guida per interventi terapeutici appropriati, per ridurre il rischio della sindrome "dell’osso affamato" post-PTX. PHPT è una condizione specifica in cui è elettivamente suggerita la scansione DXA del terzo distale dell'avambraccio (arricchito nella componente ossea corticale). La valutazione a livello lombare, più ricca di osso trabecolare, di solito rivela una BMD preservata, mentre può mostrare risultati intermedi al femore, mix di osso corticale e trabecolare. Circa il 15% dei pazienti con PHPT può presentare osteopenia vertebrale o osteoporosi. |
| Radiografie "classiche" e TC | Come per PHPT sporadico: fratture patologiche, aspetto "sale e pepe" del cranio, rastremazione del terzo distale delle clavicole, lesioni litiche al bacino e ossa lunghe, localizzazioni alle spalle, erosioni ossee sub-periostali nelle falangi distali (in particolare nei pazienti con FPHPT severo). Metastasi a distanza di CP possono essere sospettate a livello radiografico e identificate anche alla scintigrafia sestaMIBI o alla TC, poi confermate da biopsia. |
Ricordare: le indagini radiologiche, in combinazione con i risultati clinici e biochimici, sono necessarie per guidare il processo decisionale sul trattamento nelle forme di FPHPT.
QUANDO CONSIDERARE IL TEST GENETICO IN SOGGETTI CON PHPT?
La tabella 8 riassume i principali punti pratici.
| Tabella 8 Aspetti pratici per considerare analisi genetica mutazionale in pazienti con PHPT |
|
| Geni da considerare | MEN1, CaSR, AP2S1, GNA11, CDC73, CDKN1A, CDKN1B, CDKN2B, CDKN2C, RET, GCM2, PTH |
| Diagnosi | Malattia paratiroidea multi-ghiandolare |
| Esordio anticipato | 2-3 decenni nelle forme familiari rispetto alla forma sporadica nella popolazione generale (quest'ultima è più frequentemente mono-ghiandolare) |
| Caratteristiche cliniche | Sintomi variabili, dipendono dal contesto della sindrome. La storia familiare è molto importante |
| Caratteristiche biochimiche | Variabili (ipercalcemia da moderata a grave). Se è presente ipocalciuria, considerare FHH |
| Caratteristiche radiologiche | Gli studi di localizzazione sono molto utili nel PHPT recidivante, dopo che la maggior parte del tessuto patologico è stato rimosso |
| Trattamento | Esplorazione di tutte le paratiroidi e PTX totale o sub-totale |
| Prognosi | Rischio di recidiva e/o persistenza dopo intervento chirurgico (specie in casi FHH) |
Ricordare: l'identificazione di una mutazione germinale comporta l'istituzione di un programma clinico, con periodici screening biochimici e radiologici. Si raccomanda una maggiore sorveglianza e monitoraggio per FPHPT, ereditario, dato l'elevato rischio di malattia recidivante o persistente causato dal frequente coinvolgimento multi-ghiandolare. Sono riportate mutazioni de novo in circa il 10% dei pazienti con PHPT, o in presenza di anamnesi familiare sconosciuta o non accertata.
CONSIDERAZIONI GENERALI SUGLI APPROCCI TERAPEUTICI
Le sindromi FPHPT influenzano la strategia chirurgica per questo tipo di PHPT, a parte la FHH in cui la maggior parte dei pazienti non beneficia della PTX.
Paratiroidectomia
La chirurgia paratiroidea è obbligatoria nei pazienti con FPHPT sintomatici e/o con ipercalcemia severa. La tempistica ottimale della chirurgia nei pazienti asintomatici è ancora dibattuta e deve essere valutata individualmente. L'approccio chirurgico per i pazienti FPHPT può essere impegnativo a causa del coinvolgimento multi-ghiandolare, spesso asincrono e con possibile presenza di ghiandole paratiroidi ectopiche e/o soprannumerarie, con alto tasso di persistenza/recidiva. Per queste ragioni, è adeguato anche un approccio farmacologico nei pazienti con diagnosi precoce di PHPT.
Ricordare: una volta accertato (F)PHPT, la terapia elettiva consiste nell'exeresi chirurgica del tessuto paratiroideo patologico. Le linee guida internazionali suggeriscono che un imaging negativo non debba rappresentare un ostacolo nell’inviare il paziente a un chirurgo paratiroideo esperto, generalmente disponibile in centri di endocrinologia di terzo livello.
Approccio farmacologico
Se l'intervento chirurgico non è fattibile, a causa dell'inoperabilità del paziente, o del suo rifiuto o della mancata identificazione della fonte di PHPT persistente, si possono utilizzare approcci farmacologici, riassunti nella tabella 9.
| Tabella 9 Approcci farmacologici utilizzabili in (F)PHPT “inoperabile” |
|
| Cinacalcet (calci-mimetico) | Può ridurre calcemia, ma non calciuria né formazione di calcoli renali o perdita ossea e/o rischio di frattura. L'uso prolungato si associa a perdita ossea continua in alcuni pazienti, probabilmente a causa dei livelli elevati persistenti di PTH. Carenza di dati a lungo termine in pazienti con FPHPT. |
| Diuretici tiazidici | Approccio farmacologico alternativo in pazienti FPHPT con diagnosi precoce, in cui l'ipercalciuria rappresenta la principale alterazione biochimica. Disponibili dati solo in pazienti con PHPT sporadico, in cui riducono calciuria senza però indurre cambiamento sostanziale nella calcemia, indipendentemente dalla dose. Mancano dati sulla formazione di calcoli renali. Non ci sono studi clinici randomizzati che mostrino effetto protettivo sul rischio di frattura. |
| Vitamina D | Non ci sono studi conclusivi sul beneficio a livello di PTH e BMD in pazienti con (F)PHPT e livelli circolanti carenziali. |
| Terapia ormonale sostitutiva/SERM | Suggeriti per ridurre turnover osseo e aumentare BMD. Gli effetti collaterali non scheletrici ne precludono l'uso per questo scopo. |
| Bisfosfonati (BP) | Riducono il turnover osseo e aumentano la BMD. Un recente studio su più di 6000 pazienti con PHPT sporadico, non sindromico, ha però dimostrato che PTX si associa a ridotto rischio di frattura, mentre il trattamento con BP non si è dimostrato, comunque, significativamente superiore a nessuna terapia farmacologica. |
| Combinazione di calci-mimetico e BP | Può essere un'opzione terapeutica (non documentata, che necessita di ulteriore valutazione) in pazienti con PHPT ipercalcemico sintomatico e ridotta BMD, inoperabili. |
| Denosumab | Mancano dati su FPHPT, ma può essere un possibile approccio alternativo in pazienti PHPT più anziani con elevato rischio fratturativo, inoperabili. Dopo 24 mesi, ha ottenuto un aumento significativo di BMD in pazienti con PHPT, maggiore di quello ottenuto in pazienti della stessa età con sola osteoporosi primaria. Nei pazienti con PHPT la prevalenza di risposta inadeguata della BMD (in qualsiasi sito scheletrico) e di fratture da fragilità incidenti erano simili a quelle osservate nei pazienti con osteoporosi primaria. |
BIBLIOGRAFIA DI RIFERIMENTO
- Eller-Vainicher C, Falchetti A. Management of familial hyperparathyroidism syndromes: MEN1, MEN2, MEN4, HPT-Jaw tumour, Familial isolated hyperparathyroidism, FHH, and neonatal severe hyperparathyroidism. Best Prac Res Clin Endocrinol Metab 2018, 32: 861-75.
- Falchetti A. Genetics of parathyroids disorders: overview. Best Prac Res Clin Endocrinol Metab 2018, 32: 781-90.
- Thakker RV. Genetics of parathyroid tumours. J Intern Med 2016, 280: 574-83.
- Bilezikian JP, Cusano NE, Khan AA, et al. Primary hyperparathyroidism. Nat Rev Dis Primers 2016, 2: 16033.
- Bilezikian JP, Marcus R, Levine M, et al. The parathyroids: basic and clinical concepts. 3rd Academic Press 2014.
Incidentaloma paratiroideo
Andrea Frasoldati
Endocrinologia, Arcispedale S. Maria Nuova, Reggio Emilia
Il termine “incidentaloma paratiroideo” indica il riscontro casuale di adenoma (o iperplasia) delle paratiroidi in corso di interventi di tiroidectomia o più in generale di cervicotomia, in totale assenza di sospetto clinico e/o laboratoristico di patologia paratiroidea. L’osservazione risale originariamente agli anni ‘60’ (1), ma l’introduzione del termine incidentaloma è più recente, derivando per analogia dall'analogo termine utilizzato nella patologia surrenalica e ipofisaria (2).
Occorre sottolineare che le casistiche chirurgiche pubblicate in letteratura sono limitate per numero e dimensioni (il numero dei casi complessivamente riportati è inferiore a 150); ciò limita la possibilità di derivarne dati di forte consistenza. Tenendo presente tale limite, la frequenza dell’incidentaloma paratiroideo “chirurgico” risulta compresa tra 1% e 4.5% (2-8); tuttavia, solo un terzo dei pazienti con incidentaloma, una volta specificamente investigato, presenta segni clinici e/o biochimici di iperparatiroidismo, come documenta uno studio francese prospettico su 748 pazienti sottoposti a screening per iperparatiroidismo primitivo prima dell'intervento di tiroidectomia (9). E' stato pertanto ipotizzato che nella maggior parte dei casi, l’incidentaloma chirurgico non corrisponda ad un adenoma funzionalmente attivo, ma ad alterazioni di tipo iperplastico che ne precedono lo sviluppo. In accordo con tale ipotesi, una delle maggiori casistiche pubblicate documenta un'evidenza istologica di iperplasia in 28/36 incidentalomi (77.7%) (3). Inoltre, i 28 pazienti con incidentaloma descritti nella casistica originale di Carnaille (2) presentavano un'età media inferiore e lesioni paratiroidee di dimensioni più ridotte rispetto a quanto osservato in una casistica di oltre 500 pazienti con iperparatiroidismo primitivo trattati chirurgicamente presso la stessa istituzione.
Il deficit di vitamina D, oggi molto diffuso, e l’iperplasia paratiroidea secondaria che ne consegue potrebbero secondo alcuni autori spiegare una quota rilevante degli incidentalomi paratiroidei descritti nelle casistiche chirurgiche (10).
Una frequenza di incidentalomi paratiroidei sorprendentemente elevata (34%) è stata descritta in una casistica (n = 23) di pazienti sottoposti a tiroidectomia per patologia nodulare, tutti caratterizzati dal dato anamnestico di una pregressa irradiazione della regione cervicale (11). Tale dato è in linea con quanto osservato in un ampio studio californiano condotto retrospetticamente su 800 pazienti sottoposti a chirurgia tiroidea in periodo di tempo di circa 14 anni; oltre un terzo dei 36 pazienti con incidentaloma paratiroideo descritti in questa casistica risultavano essere stati sottoposti a trattamento radiante sul collo per varie patologie (3). Nel loro insieme, queste osservazioni suggeriscono che le radiazioni possono avere un ruolo patogenetico sullo sviluppo degli adenomi paratiroidei: a suffragio di tale ipotesi, la segnalazione che l’incidenza di iperparatiroidismo primitivo è risultata significativamente elevata (11%) nel corso del follow-up in un gruppo di pazienti sottosti a trattamento radiante per linfoadenite tubercolare della regione cervicale (12).
A partire dalla fine degli anni ‘90, con l’avvento dell’impiego su larga scala dell’ecografia, il temine di incidentaloma paratiroideo è stato proposto in un ambito più ampio di quello chirurgico. In particolare, venne definito incidentaloma paratiroideo l’occasionale riscontro di una lesione paratiroidea nel corso di un’indagine ecografica eseguita per patologia nodulare o funzionale della tiroide (13). Nel primo studio, pubblicato nel 1999 su una casistica di 1686 pazienti consecutivamente sottoposti ad ecografia per patologia tiroidea, ed in un secondo, più ampio, di alcuni anni successivo, la prevalenza dell’incidentaloma paratiroideo è risultata rispettivamente pari a 0.53% e 0.62%. (13,14). Un recente studio coreano su 6469 pazienti sottoposti ad agoaspirato tiroideo riporta una prevalenza del tutto simile, corrispondente allo 0.4% (15). Sia nell'esperienza del nostro gruppo, sia in quella coreana, la conferma della natura paratiroidea della lesione individuata all’ecografia veniva conseguita mediante agoaspirato completato da dosaggio intralesionale di PTH (13-15). Il valore predittivo positivo del sospetto ecografico di incidentaloma paratiroideo risulta infatti limitato (20-23%) (13-15), poichè noduli e pseunoduli tiroidei, linfonodi ed altre lesioni cervicali possono presentare caratteri ecografici suggestivi di una lesione paratiroidea. La percentuale di pazienti con incidentaloma paratiroideo ecografico caratterizzati da evidenza biochimica e/o clinica di iperparatiroidismo è più alta rispetto a quanto osservato nei pazienti con incidentaloma “chirurgico”, e raggiunge il 55-66%.
E’ possibile che gli incidentalomi paratiroidei non associati a iperparatiroidismo rappresentino uno stadio iniziale della malattia: in accordo con tale ipotesi, alcuni pazienti con incidentaloma ecografico funzionalmente silente sviluppano ipercalcemia nell’arco di alcuni anni di follow-up (16). Su tali basi, e alla luce della limitata morbilità associata all'intervento di paratiroidectomia, la maggior parte degli autori ritiene appropriato procedere all'exeresi chirurgica degli incidentalomi paratiroidei riscontrati in corso di chirurgia tiroidea o di cervicotomia per altra causa (2, 3, 17, 18). Nel caso degli incidentalomi "ecografici", è sempre opportuno procedere ad una accurata caratterizzazione biochimica e clinico-anamnestica del paziente. Nei casi di iperparatiroidismo primitivo lieve e/o asintomatico, l'adozione di un atteggiamento conservativo o viceversa di una scelta di tipo chirurgico potrà avvalersi dei criteri di consueto riferimento (19).
Bibliografia
- Attie JN, Estrin J, Khafif RA, et al. Parathyroid adenomas discovered incidentally during explorations of the thyroid. Am J Surg 1967, 114: 538-42.
- Carnaille BM, Pattou FN, Ouder C, et al. Parathyroid incidentalomas in normocalcemic subjects during thyroid surgery. World J Surg 1996, 20: 830-4.
- Katz AD, Long LB. Incidental preclinical hyperparathyroidism identified during thyroid operations. Am Surg 1992, 58: 747-9.
- Wahl RA, Hentschel F, Vorländer C, et al. Primary hyperparathyroidism - early diagnosis in patients referred for thyroid surgery. Langenbecks Arch Surg 2000, 385: 515-20.
- Marchesi M, Biffoni M, Benedetti RN, et al. Incidental parathyroid adenomas with normocalcemia discovered during thyroid operations: report of three cases. Surg Today 2001, 31: 996-8.
- Shroff, P, McGrath GA, et al. Incidentalomas of the parathyroid gland: multiple presentations, variable function, and review of the literature. Endocr Pract 2005, 11: 363-9.
- Hotouras A, Sinha P. Parathyroid incidentaloma: case report and literature review. Grand Rounds ENT/Head Neck Surg 2007, 7: 45-7.
- Abboud B, Sleilaty G, Braidy C, et al. Enlarged parathyroid glands discovered in normocalcemic patients during thyroid surgery. Am J Surg 2008, 195: 30-3.
- Denizot A, Dadoun F, Meyer-Dutour A, et al. Screening for primary hyperparathyroidism and parathyroid incidentalomas in patients undergoing thyroid surgery. Surgery 2002, 131: 264-9.
- Kirkby-Bott J, El-Khatib Z, Soudan B, et al. 25-hydroxy vitamin D deficiency causes parathyroid incidentalomas. Langenbeck’s Arch Surg 2010, 395: 919-24.
- Prinz RA, Paloyan E, Lawrence AM, et al. Unexpected parathyroid disease discovered at thyroidectomy in irradiated patients. Am J Surg 1981, 142: 355-7.
- Tisell LE, Hansson G, Lindberg S. et al. Hyperparathyroidism in persons treated with x-rays for tubercolous cervical adenitis. Cancer 1977, 40: 846-54.
- Frasoldati A, Pesenti M, Toschi E, et al. Detection of parathyroid incidentaloma during thyroid sonography. J Clin Ultrasound 1999, 27: 492-8.
- Frasoldati A, Valcavi R. Challenges in neck ultrasonography: lymphadenopaty and parathyroid glands. Endocr Pract 2004, 10: 261-8.
- Kwak JY, Kim EK, Moon HG, et al. Parathyroid incidentalomas detected on routine ultrasound-directed fine-needle aspiration biopsy in patients referred for thyroid nodules and the role of parathyroid hormone analysis in the samples. Thyroid 2009, 19: 743-8.
- Pesenti M, Frasoldati A, Azzarito C, et al. Parathyroid incidentaloma discovered during thyroid ultrasound imaging. J Endocrinol Invest 1999, 22: 796-9.
- Helme S, Lulsegged A, Sinha P. Incidental Parathyroid Disease during Thyroid Surgery: Should We Remove Them? ISRN Surgery 2011, 2011: 962186.
- Benabbad I, Chraibi A, Iraqi H, et al. Parathyroid incidentaloma. Literature review about three case reports. Ann Endocrinol 2011, 72: 30-3.
- Bilezikian JP, Khan AA, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 2009, 94: 335-9.
Carcinoma paratiroideo
Alfredo Scillitani
Endocrinologia, Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG)
Prevalenza
Il carcinoma paratiroideo (CP) è una rara malattia endocrina che verosimilmente rappresenta meno dell’1% di tutte le forme di iperparatiroidismo primitivo (HPT), anche se in alcune casistiche è riportata una prevalenza più elevata (1-7).
Patogenesi
Nella forma familiare più rara di HPT, la sindrome iperparatiroidismo-tumore mascellare (HPT-JT), ben il 15% dei casi è dovuto a CP (6). Il coinvolgimento del gene HRPT2 è dimostrato dall’identificazione di sue mutazioni nel 70% circa dei casi di pazienti con CP metastatico (4). Circa un terzo delle mutazioni identificate non sono somatiche, bensì costituzionali (4). Questo risultato ha un risvolto pratico, perché impone la ricerca della mutazione nei familiari di primo grado dei pazienti con mutazioni costituzionali. Nel caso di positività, questi soggetti dovranno essere seguiti con un follow-up clinico, biochimico (calcemia e PTH) e strumentale (ecografia di collo pelvi e addome superiore, ortopantomografia) allo stesso modo dei “portatori” appartenenti a famiglie HPT-JT.
Quadro clinico
Alcuni autori sostengono che il CP si osservi più spesso in maschi di giovane età, e che sia più frequente il rilievo di valori più alti di calcemia, massa palpabile del collo, coinvolgimento osseo e renale. Il ritorno pratico di questa osservazione è scarso, perchè nessuno di questi dati è altamente predittivo (1). Altri autori hanno osservato che la probabilità di avere un carcinoma è modesta se il valore del PTH (con metodo di II generazione) è meno di 4 volte il limite superiore della norma ed il peso del tumore è < 1.9 g (2). Si deve peraltro osservare che sono stati identificati casi di CP normocalcemico (5,7).
La morbilità del CP è dovuta alla grave ipercalcemia, che può determinare sintomi neuropsichiatrici, aritmie cardiache, insufficienza renale, fratture patologiche (3).
La storia clinica è imprevedibile: più spesso il comportamento è indolente, ma talora è rapido, aggressivo e fatale in breve tempo. Non ci sono indicatori prognostici di aggressività della neoplasia (1).
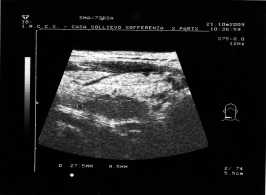
Le dimensioni + i contorni irregolari + la ecostruttura ipoecogena e disomogenea fecero sospettare il carcinoma, poi confermato all'esame istologico
Terapia
Poiché la guarigione dipende dal primo intervento, che potrà essere curativo solo se radicale, nel sospetto clinico di CP (sulla base di calcemia > 14 mg/dL, massa palpabile, paralisi della corda vocale, caratteristiche ecografiche) (2, 3, 6), bisogna pianificare un intervento chirurgico open di emitiroidectomia e paratiroidectomia consensuale “en bloc” (5-7). Il riscontro intra-operatorio di consistenza dura e di invasione macroscopica dei tessuti adiacenti è un indice di malignità (5-7). Poichè bisogna assolutamente evitare la rottura della capsula e la disseminazione delle cellule neoplastiche, il sospetto di CP nel caso si stia operando con tecniche mini-invasive deve fare convertire la procedura chirurgica alla tecnica classica. Per lo stesso motivo, è assolutamente sconsigliabile il FNA nel sospetto di CP e anche nelle forme di HPT persistente o recidivante, in cui è più alto il rischio di avere un carcinoma (3, 6, 7).
Un altro problema pratico è la diagnosi istologica di CP. Oggi si ritiene che:
- solo la presenza di invasione vascolare e/o della capsula a tutto spessore con invasione dei tessuti circostanti o ovviamente la presenza di metastasi a distanza consente la diagnosi di CP
- in presenza di invasione parziale della capsula senza invasione dei tessuti circostanti, in presenza di bande fibrose, di atipie nucleari, di mitosi, di pattern di crescita trabecolare bisogna limitarsi alla diagnosi di adenoma atipico, la cui storia naturale non è ad oggi nota (6-7).
Deve essere sottolineato che in un’ampia casistica la metà dei CP metastatici avevano una diagnosi istologica iniziale di adenoma (1). Secondo alcuni autori, nei casi istologicamente dubbi può essere utile la ricerca sul tessuto neoplastico di mutazioni del gene HRPT2, che è rara in adenomi sporadici e forse anche negli atipici, ma su questo non c’è consenso (7).
Follow-up e terapie adiuvanti
Dopo l’intervento e la normalizzazione dei livelli di calcemia, in caso di diagnosi istologica inaspettata di CP un atteggiamento ragionevole può essere quello di seguire il paziente dal punto di vista biochimico, misurando i livelli di calcemia e PTH ogni 6 mesi (dopo adeguata correzione dell’eventuale ipovitaminosi D). In caso di aumento, bisogna cercare di identificare la lesione causa della recidiva con tutte le tecniche a disposizione (ecografia collo, scintigrafia SestaMIBI, TC ad alta risoluzione e RM). Se possibile questa deve essere asportata (5-7).
La neoplasia recidiva localmente (nei 2/3 dei casi), raramente metastatizza ai linfonodi, mentre più spesso si dissemina a distanza nel polmone, talora nelle ossa. In caso di recidiva, questa si presenterà in media dopo 2-4 anni dall’iniziale intervento (1).
Questi pazienti hanno una sopravvivenza del 76-85% a 5 anni e del 49-77% a 10 anni, con mediana di 6 anni dalla prima diagnosi, ma sono riportate sopravvivenze > 20 anni (1, 3, 5, 6).
La chemioterapia post-intervento non ha dato buoni risultati in termini nè di qualità della vita né di sopravvivenza (5-7), mentre qualche risultato positivo (ma i dati sono scarsi) si è osservato con la radioterapia adiuvante in caso di recidive locali (3, 5-7) .
I pazienti muoiono per l’ipercalcemia incontrollabile, per cui in alcuni centri si suggerisce anche la metastasectomia (3, 6). L’ipercalcemia può anche essere parzialmente controllata farmacologicamente con il cinacalcet e temporaneamente con bisfosfonati ev (5-7).
Bibliografia
- Sandelin K, Tullgren O, Farnebo LO. Clinical course of metastatic parathyroid cancer. World J Surg 1994, 18: 594-8.
- Robert JH, Trombetti A, Garcia A, et al. Primary hyperparathyroidism: can parathyroid carcinoma be anticipated on clinical and biochemical grounds? Report of nine cases and review of the literature. Ann Surg Oncol 2005, 12: 526-32.
- Harari A, Waring A, Fernandez-Ranvier G, et al. Parathyroid carcinoma: a 43-year outcome and survival analysis. J Clin Endocrinol Metab 2011, 96: 3679-86.
- Shattuck TM, Välimäki S, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 2003, 349: 1722-9.
- Owen RP, Silver CE, Pellitteri PK, et al. Parathyroid carcinoma: a review. Head Neck 2011, 33: 429-36.
- Sharretts JM, Kebebew E, Simonds WF. Parathyroid cancer. Semin Oncol 2010, 37: 580-90.
- Marcocci C, Cetani F, Rubin MR, et al. Parathyroid carcinoma. J Bone Miner Res 2008, 23: 1869-80.
Overview terapia iperparatiroidismo primario
Claudio Marcocci
Dipartimento di Endocrinologia e Metabolismo, Università di Pisa, Pisa
L’obiettivo della terapia dell’iperparatiroidismo primario (PHPT) è quello di normalizzare i livelli di calcio e PTH circolanti e di ottenere la regressione delle manifestazioni cliniche conseguenti all’ipercalcemia e all’eccesso di PTH (1).
La paratiroidectomia (PTx) è il solo intervento terapeutico curativo e in mani esperte ha successo in circa il 90-95% dei casi (2). L’intervento chirurgico dovrebbe essere preso in considerazione in tutti i pazienti con diagnosi certa di PHPT e raccomandato nei pazienti con manifestazioni cliniche classiche riferibili alla malattia (nefrolitiasi, osteite fibrosa cistica e sintomi da ipercalcemia). Oggigiorno, nella maggior parte dei casi il PHPT viene diagnosticato in modo fortuito mediante il rilievo di ipercalcemia lieve-moderata in pazienti sostanzialmente asintomatici (PHPT asintomatico). Sono state recentemente pubblicate le nuove linee guida per la terapia del PHPT asintomatico (3). L’intervento di PTx è raccomandato anche nei pazienti con PHPT asintomatico che soddisfano alcuni criteri (tabella). Rispetto alle precedenti linee guida del 2009 (4), rimangono immodificate le indicazioni concernenti l’età, il livello di calcemia, la riduzione della densità minerale ossea e della funzione renale. Nuove indicazioni alla PTx sono rappresentate dal rilievo di ipercalciuria (> 400 mg/24 ore) associata ad un aumentato rischio litogeno, e dall’evidenza strumentale di fratture vertebrali e di nefrolitiasi/nefrocalcinosi. I pazienti con PHPT asintomatico che non presentano alcuna delle suddette indicazioni chirurgiche possono essere classificati come casi di iperparatiroidismo lieve (5).
Nei pazienti che non sono avviati al trattamento chirurgico, è raccomandato un monitoraggio annuale di calcemia, creatininemia, clearance della creatinina/eGFR, ogni 1-2 anni della densità minerale ossea. Nel sospetto di frattura vertebrale (dolore dorso-lombare, riduzione di altezza), si dovrà procedere con uno studio morfometrico vertebrale e nel sospetto di calcolosi/nefrolitiasi ad una valutazione del rischio litogeno e dell'imaging renale.
Nei pazienti non sottoposti a PTx il quadro biochimico e densitometrico rimane stabile per 8-10 anni, anche se in circa un quarto dei casi è possibile documentare una progressione della malattia (6). Va inoltre sottolineato che vi sono evidenze che anche nei pazienti che non soddisfano i criteri chirurgici, la PTx è seguita da un miglioramento della massa ossea e di alcuni parametri della qualità di vita (7,8).
Nel discutere le scelta terapeutica in ogni singolo caso bisogna ricordare che le indicazioni contenute nelle linee guida sono raccomandazioni e non regole assolute, e pertanto in ogni singolo paziente la decisione terapeutica dovrebbe tenere in considerazione la preferenza del paziente, la presenza di concomitanti comorbilità e l’esperienza del chirurgo di riferimento.
Nei pazienti con PHPT seguiti senza intervento chirurgico è opportuno mantenere un adeguato stato vitaminico D (25-OHD > 20 ng/mL, anche se alcuni autori suggeriscono > 30 ng/mL) e un adeguato introito di calcio, poiché sia l’ipovitaminosi D che un ridotto apporto di calcio possono accentuare l’iperproduzione di PTH (3).
In questi pazienti la PTx dovrà essere raccomandata se durante il follow-up si rileva:
- aumento della calcemia > 1mg/dL rispetto al limite superiore della norma;
- riduzione significativa rispetto al basale della BMD, con T score < -2.5 in una delle tre sedi di misura. L’intervento potrà essere suggerito (anche se non raccomandato dalle linee guida) se il valore di T score cade tra -2 e -2.5;
- frattura da fragilità;
- nefrolitiasi;
- clearance della creatinina < 60 mL/min.
| Linee guida per il trattamento dei pazienti con iperparatiroidismo | ||
| Parametro | Criterio per la paratiroidectomia* | Monitoraggio |
| Calcemia | > 1.0 mg/dL al di sopra del limite superiore della norma | Annuale |
| Creatininemia (eGFR) | < 60 mL/min | Annuale |
| Clearance della creatinina | < 60 mL/min | - |
| Densità minerale ossea (DXA) | T-score < -2.5 in qualsiasi sede¶, ° | Ogni 1–2 anni (3 sedi) |
| Morfometria vertebrale (VFA^, TAC, RMN) | Presenza di fratture vertebrali | VFA nel sospetto clinico di frattura vertebrale (ad esempio, dolore dorso-lombare, riduzione di altezza) |
| Urine delle 24 ore | Calciuria > 400 mg/24h ed aumento del rischio litogeno** | sospetto clinico di nefrolitiasi |
| Imaging renale (raggi X, US, CT) | Presenza di nefrolitiasi/nefrocalcinosi | sospetto clinico di nefrolitiasi |
| Età (anni) | < 50 | - |
*La paratiroidectomia dovrebbe essere raccomandata anche nei pazienti nei quali il monitoraggio non è possible
¶Colonna lombare, femore e terzo distale del radio
°Nei soggetti < 50 anni dovrebbe essere usato lo Z-score invece del T-score
^VFA=vertebral fracture assessement con DXA
** calcolato sulla base della misura urinaria di sodio, potassio, calcio, magnesio, ammonio, fosfato, solfato, citrato, ossalato, cloruro e pH (9)
Al momento attuale non esiste una terapia medica definitiva del PHPT. Tuttavia, sono disponibili alcune opzioni che possono essere prese in considerazione in alcuni pazienti che non sono sottoposti a PTx, nei quali esista l’indicazione a ridurre i livelli sierici di calcio, incrementare la massa ossea, o ambedue le indicazioni (10).
Studi controllati hanno dimostrato che gli inibitori del riassorbimento osseo (estrogeni coniugati e alendronato) determinano un aumento della massa ossea a livello lombare e femorale, sovrapponibili a quelli osservati dopo PTx (11-12). Questo trattamento non si accompagna a variazioni significative dei livelli di calcio e PTH.
Un'alternativa medica è rappresentata dai calcio-mimetici. Il cinacalcet, infatti, è efficace nel ridurre i livelli di calcio circolante in varie tipologie di pazienti con PHPT moderato e grave e nei pazienti con carcinoma delle paratiroidi, mentre non sono state osservate significative variazioni della massa ossea (13-15). L’impiego del cinacalcet è stato approvato dall’EMA “nei pazienti con PHPT che dovrebbero essere indirizzati alla paratiroidectomia sulla base dei livelli di calcio (in accordo alle linee guida), ma nei quali l’intervento è controindicato o clinicamente non appropriato”.
Al momento attuale nessuno dei trattamenti medici disponibili è in grado di controllare in modo completo le manifestazioni cliniche di PHPT. Pertanto, la scelta dell’uno e/o dell’altro presidio terapeutico dovrà essere fatta in base al tipo di manifestazione clinica che ci si propone di influenzare (cinacalcet nel caso dell’ipercalcemia; alendronato o estrogeni nel caso della ridotta massa ossea). In particolari casi si potrebbe anche considerare l’associazione dei due trattamenti.
Bibliografia
- Marcocci C, et al. Clinical practice: Primary hyperparathyroidism. N Engl J Med 2011, 365: 2389-97.
- Udelsman R. Six hundred fifty-six consecutive explorations for primary hyperparathyroidism. Ann Surg 2002, 235: 665-72.
- Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyrodism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab 2014, 99: 3561-9.
- Bilezikian JP, Khan AA, Potts JT, on behalf of the Third International Workshop on the Management of Asymptomatic Primary Hyperthyroidism. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 2009, 94: 335-9.
- Marcocci C, Brandi ML, Scillitani A, et al. Italian Society of Endocrinology Consensus Statement: definition, evaluation and management of patients with mild primary hyperparathyroidism. J Endocrinol Invest 2015, 38: 577-93.
- Rubin MR, Bilezikian JP, McMahon DJ, et al. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab 2008, 93: 3462-70.
- Bollerslev J, Jansson S, Mollerup CL, et al. Medical observation, compared with parathyroidectomy, for asymptomatic primary hyperparathyroidism: a prospective, randomized trial. J Clin Endocrinol Metab 2007, 92: 1687–92.
- Ambrogini E, Cetani F, Cianferotti L, et al. Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab 2007, 92: 3114-21.
- Marangella M, Petrarulo M, Daniele PG, Sammartano S. LithoRisk: a software for calculating and visualizing nephrolithiasis risk profiles. G Ital Nefrol 2002, 19: 693-8.
- Marcocci C. Bollerslev J, Khan AA, Shoback DM. Medical management of primary hyperparathyroidism: proceedings of the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab 2014, 99: 3607-18.
- Grey AB et al. Effect of hormone replacement therapy on bone mineral density in postmenopausal women with mild primary hyperparathyroidism. A randomized, controlled trial. Ann Intern Med 1996, 125: 360-8.
- Khan AA, Bilezikian JP, Kung AWC, et al. Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab 2004, 89: 3319–25.
- Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab 2009, 94: 4860–7.
- Marcocci C, Chanson P, Shoback D, et al. Cinacalcet reduces serum calcium concentrations in patients with intractable primary hyperparathyroidism. J Clin Endocrinol Metab 2009, 94: 2766–72.
- Peacock M, Bilezikian JP, Bolognese MA, et al. Cinacalcet HCl reduces hypercalcemia in primary hyperparathyroidism across a wide spectrum of disease severity. J Clin Endocrinol Metab 2011, 96: E9-18.
Terapia chirurgica dell'iperparatiroidismo primitivo
Ignazio Emmolo
Chirurgia della Tiroide e delle Paratiroidi, Casa di Cura “Città di Bra” – Bra (Cuneo)
INTRODUZIONE
L’intervento chirurgico è l’unica terapia in grado di assicurare la guarigione dell'IPT primitivo, ottenuta mediante l’asportazione di tutto il tessuto paratiroideo iperfunzionante. L’endocrinochirurgo esperto guarisce oltre il 95% degli operati, con complicanze inferiori al 3%(1).
Sono di ostacolo alla riuscita dell’intervento:
- la difficoltà di distinguere la malattia unighiandolare (adenoma) dalla multighiandolare (adenomi multipli, iperplasia);
- la possibile ectopia delle paratiroidi;
- l’eventualità di ghiandole soprannumerarie;
- le piccole dimensioni delle paratiroidi;
- i pregressi interventi chirurgici in regione cervicale.
Sono di aiuto al chirurgo:
- la localizzazione pre-operatoria del tessuto paratiroideo patologico (1);
- l’impiego intra-operatorio dell’esame istologico estemporaneo e del dosaggio rapido del PTH.
L’esame istologico estemporaneo, detto anche "al congelatore", è molto affidabile nel confermare la natura paratiroidea della formazione asportata, differenziandola dal tessuto linfonodale, adiposo, tiroideo. Tuttavia, esso presenta importanti limiti nel porre diagnosi differenziale tra adenoma, iperplasia e carcinoma (vedi “Anatomia Patologica”).
Il dosaggio rapido del PTH (PTHio) è reso possibile dalla breve emivita dell’ormone (3-5 minuti). Durante l'intervento, l’entità del crollo del PTH dopo l’asportazione della ghiandola ipersecernente permette di conoscere se il paziente è guarito, cioè se tutto il tessuto iperfunzionante è già stato asportato. Tra i diversi protocolli proposti, il più utilizzato è il “criterio di Miami”: 10 minuti dopo l’asportazione della ghiandola ipersecernente il PTH deve scendere sotto il 50% del più basso tra i due valori basali (prelievo eseguito subito prima dell’incisione cutanea, prelievo eseguito al momento dell’exeresi). Talvolta può rendersi necessario un prelievo anche a 20 minuti. Il PTHio è accreditato del 97% di sensibilità e del 93% di specificità nella malattia unighiandolare, ma risulta meno accurato nella patologia multighiandolare. Esistono purtroppo risposte inappropriate del PTHio, che possono trarre in inganno il chirurgo(2):
- “falsi negativi”, quando il PTH non scende a valori di guarigione, pur essendo già stato asportato tutto il tessuto iperfunzionante;
- i più temibili “falsi positivi”, quando il PTH crolla a valori di guarigione, pur non essendo state asportate tutte le ghiandole patologiche.
LA PREPARAZIONE ALL’INTERVENTO
Il paziente con ipercalcemia moderata (< 11.5-12 mg/dL) non ha bisogno di particolare preparazione all’intervento chirurgico. Valori calcemici più elevati rendono necessaria la terapia ipocalcemizzante.
Bisogna valutare gli effetti dell’ipercalcemia sui vari organi. Riguardo all’apparato cardiovascolare, va posta particolare attenzione alla presenza di turbe del ritmo ed al controllo dell’ipertensione arteriosa.
La funzionalità renale deve garantire una buona diuresi.
L’otorino deve valutare pre-operatoriamente la motilità delle corde vocali.
La copertura antibiotica è indicata solo nel paziente particolarmente esposto alle infezioni (immunodeficienza, diabete non ben controllato, valvulopatia).
In presenza di rischio trombo-embolico va somministrata eparina a basso peso molecolare. Essa sostituisce l’eventuale terapia anti-coagulante o anti-aggregante in corso, che deve essere sospesa almeno una settimana prima dell’intervento.
GLI INTERVENTI CHIRURGICI
Gli interventi di paratiroidectomia vengono distinti in base all'estensione dell’esplorazione, che può essere:
- bilaterale (BNE-Bilateral Neck Exploration), che prevede l’esplorazione delle due logge e, se necessario, dei siti di più frequente ectopia;
- monolaterale (UNE-Unilateral Neck Exploration), che limita l’intervento alla sola loggia dove è stata pre-operatoriamente localizzata la ghiandola patologica, con asportazione della stessa e controllo dell’altra paratiroide omolaterale;
- focale (MIP-Minimally Invasive Parathyroidectomy), che limita l’intervento esclusivamente all’asportazione della ghiandola patologica identificata pre-operatoriamente.
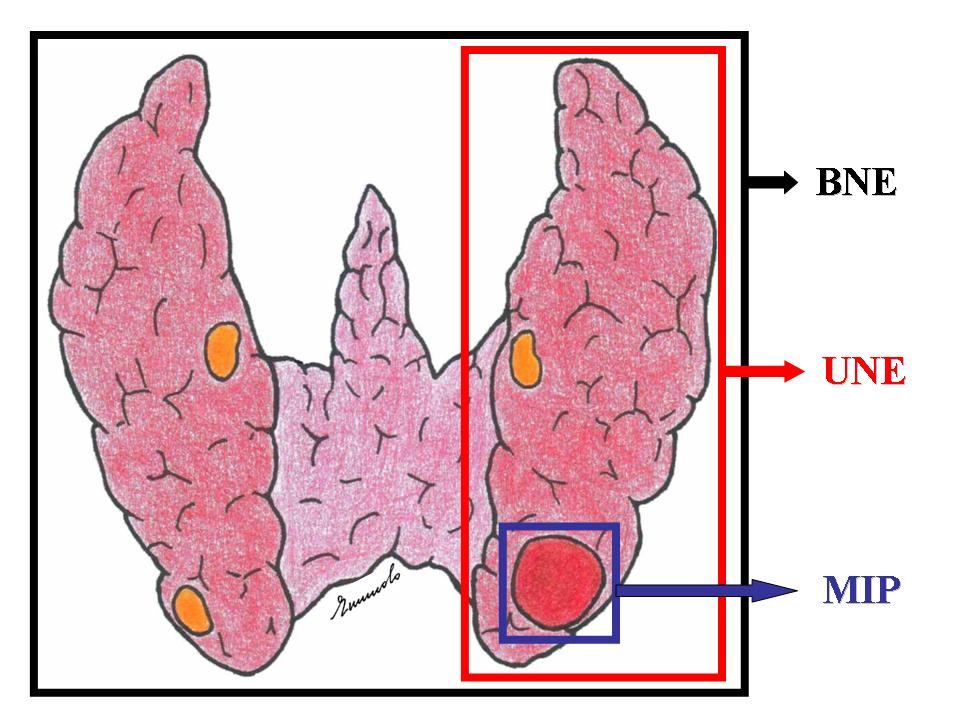
A - Classificazione delle paratiroidectomie.
I diversi interventi di MIP si differenziano tra di loro per
- l’impiego o meno di particolari strumentazioni
- senza l’impiego di strumentazione: MIP-open
- con l’impiego dello strumentario
- della videochirurgia: MIVAP-Minimally Invasive Video Assisted Parathyroidectomy, EP-Endoscopic Parathyroidectomy
- della chirurgia robotica: RP-Robotic Parathyroidectomy
- della sonda per il rilevamento della radioattività: MIRP-Minimally Invasive Radioguided Parathyroidectomy;
- la via di approccio
- anatomica: con incisione cutanea in sede cervicale
- extra-anatomica: con incisione cutanea in sede extra-cervicale (ascellare, mammaria, sotto-claveare, dorsale, trans-orale, ecc.).
La dimensione dell'incisione cutanea è un elemento importante della paratiroidectomia. Essa può essere:
- mini, ovvero ≤ 2.5-3 cm, impiegata nella MIP [3-5]
- tradizionale, più ampia, utilizzata nella BNE.
La chirurgia tradizionale
La BNE (esplorazione cervicale bilaterale) era l’unico intervento praticabile fino ad alcuni anni or sono. Essa comporta la ricerca di tutte le paratiroidi, con l’asportazione di quelle aumentate di volume.
In presenza dell’adenoma si pratica la sua asportazione, seguita dalla biopsia di un’altra ghiandola di aspetto normale necessaria per escludere intra-operatoriamente il coinvolgimento totighiandolare, ovvero l'iperplasia (vedi Anatomia Patologica).
Nell'iperplasia sono possibili due interventi:
- la paratiroidectomia subtotale con asportazione di tre ghiandole e di parte della quarta. Di quest’ultima, generalmente quella meno compromessa, viene lasciata in situ una porzione ben vascolarizzata delle dimensioni approssimative di una ghiandola normale;
- la paratiroidectomia totale con auto-trapianto, con asportazione di tutte le paratiroidi e reimpianto di alcuni piccolissimi frammenti ghiandolari all’interno di apposite tasche ricavate dentro il muscolo dell’avambraccio. L’attecchimento di questi frammenti eviterà l’ipoparatiroidismo.
Entrambi gli interventi sono completati dalla ricerca ed asportazione di eventuali ghiandole sovrannumerarie.
Nel carcinoma è necessaria una chirurgia aggressiva, a causa della sua predisposizione alla recidiva: all’asportazione della neoplasia va associata l’emitiroidectomia con la linfoadenectomia del VI livello omolaterale e l’asportazione di ogni struttura aderente al tumore (muscolo, grasso, nervo, ecc). La linfoadenectomia latero-cervicale è praticata solo in presenza di coinvolgimento di tale comparto.
La chirurgia “meno invasiva”
La UNE (esplorazione cervicale monolaterale) era già stata proposta agli inizi degli anni '80 da alcuni chirurghi che ritenevano eccessiva l’esplorazione bilaterale. L'intervento prevede l’asportazione della ghiandola localizzata preoperatoriamente, l’esecuzione del PTHio ed il controllo ispettivo dell’altra paratiroide omolaterale. Quest’ultimo tempo viene eseguito durante l’attesa della risposta del PTHio e comporta una trascurabile maggiore invasività rispetto all’intervento mini-invasivo (MIP): vedi seguito. Pertanto la UNE, se praticata attraverso una mini-incisione, ha motivo di essere assimilata alla MIP, rispetto alla quale presenta due vantaggi:
- lascia inesplorate solo le due paratiroidi controlaterali, riducendo così il rischio della persistenza della malattia;
- permette di limitare l’eventuale reintervento per IPT persistente alla sola loggia controlaterale vergine.
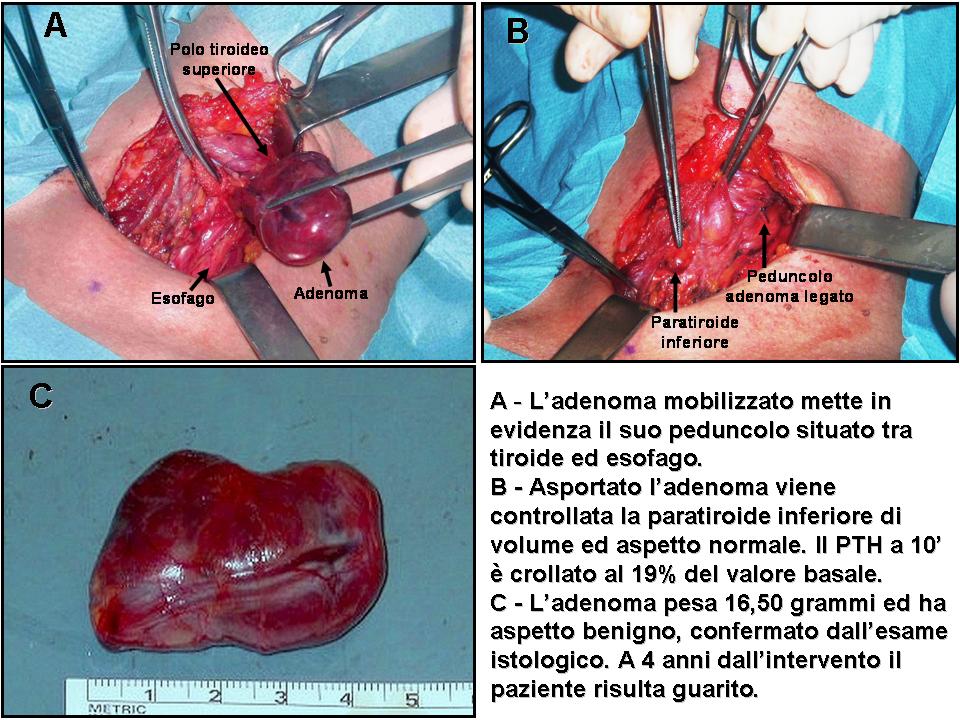
B - Esplorazione cervicale monolaterale (UNE).
Le grosse dimensioni dell'adenoma rendono impossibile la mini-cervicotomia.
La chirurgia mini-invasiva
Le caratteristiche dell’IPT primitivo (malattia unighiandolare nell’80-90% dei casi, lesione di piccole dimensioni, localizzabile pre-operatoriamente, di natura quasi sempre benigna), unitamente alla disponibilità del PTHio, ha fatto sì che negli ultimi tre lustri siano stati sempre più sviluppati interventi di paratiroidectomia mini-invasiva (6). Oltretutto, diversi di questi interventi si prestano meglio della BNE all’anestesia loco-regionale.
La MIP (paratiroidectomia mini-invasiva) ha l'obiettivo di ridurre al minimo il traumatismo. Praticata attraverso una piccola incisione, è un intervento selettivo e mirato esclusivamente all’asportazione della ghiandola patologica già localizzata. L’iperfunzione delle altre paratiroidi viene esclusa con il PTHio. In caso di mancato crollo del PTH, l'intervento è convertito in BNE.
L’indicazione elettiva alla chirurgia mini-invasiva è l’IPT sporadico con adenoma localizzato pre-operatoriamente in un collo mai operato.
Sono controindicazioni alla MIP:
- la malattia multighiandolare
- l’adenoma di grosse dimensioni (> 3 cm)
- la mancata localizzazione pre-operatoria
- il carcinoma accertato o sospetto
- la coesistenza di gozzo di medie/grosse dimensioni
- il sovvertimento aderenziale dell’anatomia del collo (IPT recidivo, pregressi interventi per altre patologie, radioterapia, tiroidite)
- l’indisponibilità del PTHio.
Assolute all’inizio, queste controindicazioni sono divenute meno rigide con il progredire della chirurgia mini-invasiva e, soprattutto, dell’esperienza dei chirurghi. Secondo alcuni, la malattia multighiandolare sporadica, la localizzazione pre-operatoria dubbia e l’indisponibilità del PTHio sono da considerare controindicazioni relative alla MIP.
Gli interventi di MIP (e di UNE) possono essere eseguiti attraverso varie procedure.
La MIP-open (paratiroidectomia mini-invasiva “a cielo aperto”) è l’intervento mini-invasivo più diffuso, praticato attraverso un’incisione cutanea limitata (≤ 2.5-3 cm). Essa prevede l’asportazione mirata della paratiroide già localizzata, seguita dall’esclusione della malattia multighiandolare per mezzo del PTHio. E’ praticamente l’intervento tradizionale ben circoscritto, realizzato senza l’ausilio di particolari attrezzature, se non degli occhiali ingranditori. La sua curva di apprendimento è breve, dal momento che è un intervento già molto familiare all’endocrinochirurgo. E’ ben eseguibile in anestesia loco-regionale. L’incisione cutanea può essere mediana, per rendere possibile l’eventuale esplorazione bilaterale di necessità, oppure mirata sulla sede dell’adenoma per lasciare una cicatrice lievemente più corta.
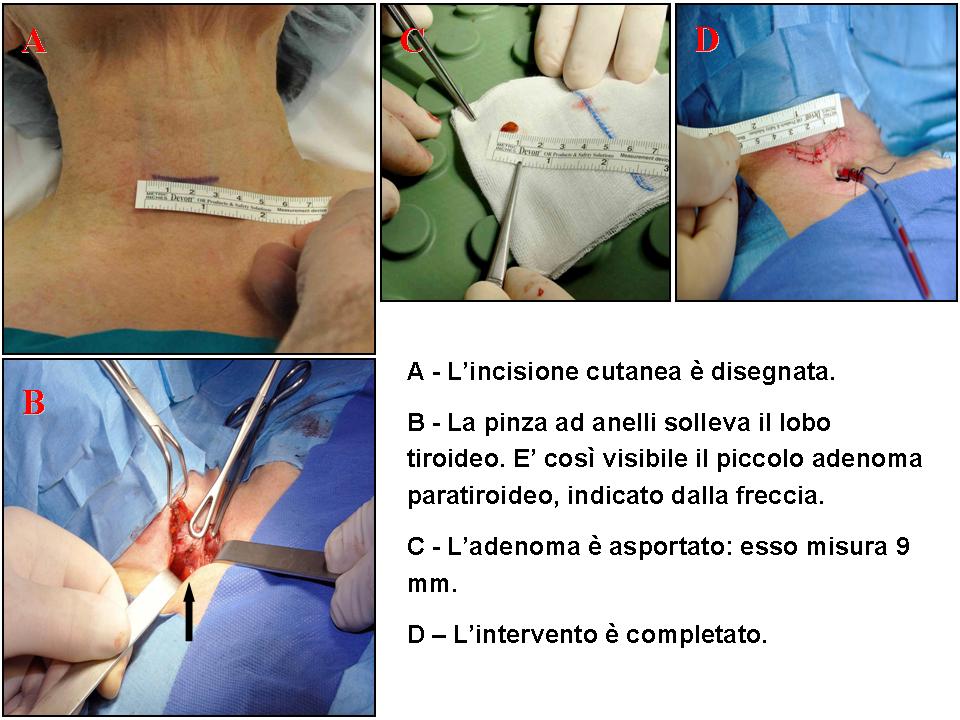
C - Paratiroidectomia mini-invasiva "a cielo aperto" (MIP-open).
Parte I: l'intervento chirurgico.
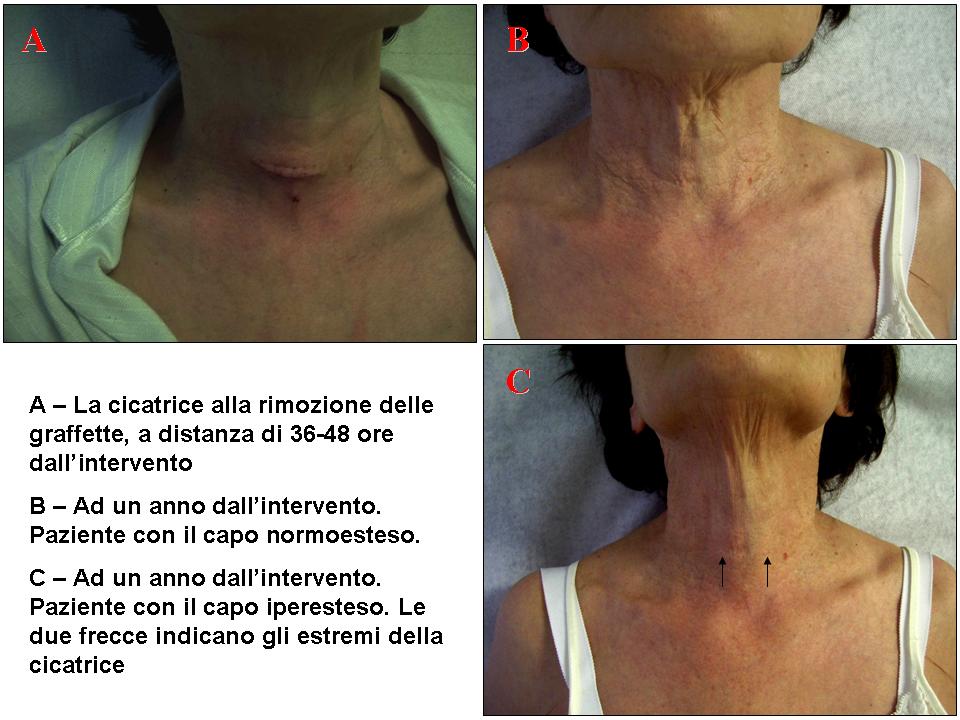
D - Paratiroidectomia mini-invasiva "a cielo aperto" (MIP-open).
Parte II: il risultato estetico.
La MIVAP (paratiroidectomia mini-invasiva video-assistita) è stata messa a punto da Miccoli nel 1997. Essa riproduce i tempi della MIP-open, dalla quale differisce perché, una volta penetrati nella loggia tiroidea, l’intervento prosegue non più sotto visione diretta, ma con visione mediata e ingrandita dall’endoscopio. Attraverso un’incisione cutanea più piccola (≤ 2 cm) vengono introdotti l’endoscopio e gli appositi strumenti che permettono di portare a termine l’intervento. La via di accesso mediana consente di praticare l’esplorazione bilaterale di necessità ed il trattamento della patologia tiroidea associata, purché di dimensioni contenute. Rispetto alla MIP-open si presta meno all’anestesia loco-regionale, necessita di una curva di apprendimento più lunga e di una equipe operatoria formata da almeno tre chirurghi.
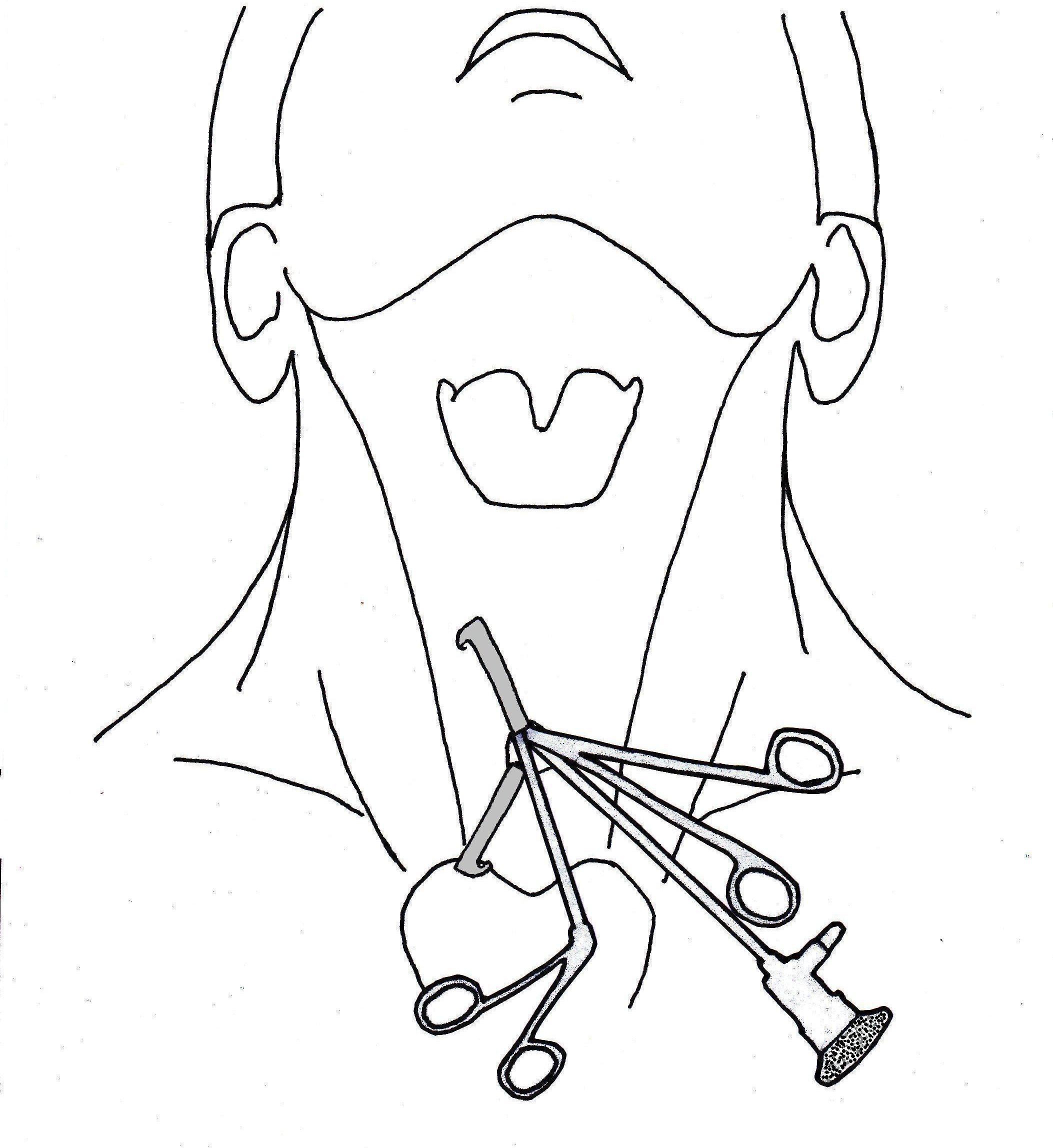
E - Paratiroidectomia mini-invasiva videoassistita (MIVAP).
Attraverso l'unica incisione vengono introdotti gli strumenti operatori e l'ottica, la quale trasmette le immagini al monitor. L'intervento è praticato seguendo le immagini sul monitor.
La EP (paratiroidectomia endoscopica) è stata eseguita per la prima volta da Gagner nel 1996. E’ una tecnica puramente endoscopica, concettualmente paragonabile a quella addominale (laparoscopica). Attraverso tre o quattro mini-incisioni cutanee cervicali (≤ 1 cm) vengono introdotti i trocars, che danno accesso all’endoscopio ed agli strumenti operatori. Le sedi delle incisioni sono generalmente condizionate dalla posizione dell’adenoma: prevalentemente centrali per le ghiandole inferiori, laterali per quelle superiori. L’esecuzione di questo intervento richiede la creazione di una camera operatoria ottenuta con l’insufflazione continua di CO2 (“pneumocollo”) oppure con retrattori meccanici. La EP è poco praticata per la difficile e lunga esecuzione, per la curva di apprendimento prolungata e perché non sempre consente l’ottimale controllo del sanguinamento.
La ricerca esasperata del risultato estetico (nessuna cicatrice cervicale), unitamente alla possibilità di condurre l’intervento completamente per via endoscopica, ha fatto proporre EP con accesso cutaneo al di fuori del collo: sotto-claveare, mammario, ascellare, trans-orale, ecc. Per accedere alla regione cervicale sono però necessarie ampie dissezioni con maggiore invasività, superiore a quella richiesta dall’intervento tradizionale. Per tale motivo, questi interventi non possono essere considerati mini-invasivi. Nonostante la complessità e il maggior rischio di complicanze, questi approcci extra-cervicali hanno trovato applicazione in paesi asiatici dove, per motivazioni sociali e culturali, la cicatrice cervicale non è facilmente accettata dalle donne. La via ascellare sembra fra tutte la più interessante.
F - Paratiroidectomia mini-invasiva endoscopica (EP).
L'intervento è condotto totalmente per via endoscopica. L'accesso ascellare richiede ampi scollamenti, necessari per accedere alla loggia tiroidea.
La RP (paratiroidectomia robotica) è l’intervento endoscopico eseguito con lo strumentario della chirurgia robotica. Ha il vantaggio della visione tridimensionale e della maggiore libertà dei movimenti degli strumenti, con manovre più precise. E’ un intervento praticato da pochissimi e, allo stato attuale, comporta maggiore rischio di complicanze, maggiori costi, maggiore durata dell’intervento e curva di apprendimento decisamente più lunga (7).
La MIRP (paratiroidectomia mini-invasiva radioguidata), sviluppata nel 1996 da Norman, si basa sul principio che l’adenoma trattiene il 99mTc-sestaMIBI in quantità superiore e più a lungo rispetto ai tessuti circostanti. E’ necessaria la scintigrafia pre-operatoria con sestaMIBI, che può essere eseguita poche ore oppure alcuni giorni prima dell'intervento. In quest'ultimo caso è necessario re-iniettare una bassa dose di tracciante all'inizio dell'intervento, minimizzando così l'esposizione dell'equipe operatoria alla radioattività. La ricerca dell’adenoma è effettuata con l’impiego della sonda che rileva la radioattività, misurata dal γ-counter. Il reperimento della paratiroide patologica risulta così più agevole e rapido. Dopo l’asportazione della ghiandola si procede: a) al conteggio della radioattività sul pezzo asportato, per confermare la natura patologica della ghiandola (può sostituire l’esame istologico estemporaneo), b) al conteggio della radioattività residua del campo operatorio per documentare la radicalità (può sostituire il PTHio). Questo intervento può essere eseguito anche in anestesia loco-regionale. Sono controindicazioni alla MIRP: la presenza di nodi tiroidei (relativa), i nodi tiroidei captanti il sestaMIBI (assoluta), la malattia paratiroidea multighiandolare, la mancata localizzazione scintigrafica dell’adenoma (da qualcuno non condivisa) (8). L’impiego routinario della MIRP è comunque poco diffuso (vedi EBM).

G - Paratiroidectomia mini-invasiva radioguidata (MIRP).
LE COMPLICANZE
Le complicanze della paratiroidectomia sono le stesse della tiroidectomia: lesione ricorrenziale, ipoparatiroidismo, sanguinamento. La lesione del n. ricorrente è meno frequente.
Una rara complicanza della chirurgia paratiroidea è la paratireomatosi, che consiste nel reimpianto spontaneo del tessuto paratiroideo in seguito alla rottura della ghiandola durante le manovre di asportazione. Più frequente in corso di intervento mini-invasivo, qualora non siano stati rispettati i criteri di selezione, essa è causa di recidiva dell’IPT.
QUALE INTERVENTO? (Evidence-Based Medicine)
Studi clinici randomizzati hanno messo a confronto la BNE con la MIP (prevalentemente MIP-open e MIVAP) e con la UNE (9): la percentuale di guarigione è uguale, ma MIP e UNE sono associati a minor durata dell’intervento e minor dolore, ipocalcemia e degenza post-operatoria. In presenza delle giuste indicazioni quello mini-invasivo è pertanto divenuto l’intervento di prima scelta.
La MIP è associata a minore dolore post-operatorio se eseguita in anestesia loco-regionale piuttosto che in narcosi (10).
Non esistono studi clinici MIP versus UNE.
Qualche studio randomizzato ha dimostrato la sovrapponibilità della MIP-open alla MIVAP (5). La scelta tra questi due interventi dipende dalle preferenze del singolo chirurgo, tenendo presente che la MIVAP comporta una migliore visione del campo operatorio e una cicatrice un po' più piccola, anche se quest’ultima non necessariamente determina un risultato estetico migliore.
H - Risultato estetico dop MIP e BNE.
L'incisione più ampia della BNE comporta in genere un minor trauma dei suoi bordi; ciò predispone alla cicatrizzazione esteticamente migliore.
I risultati della MIRP sono controversi. Studi retrospettivi sull’impiego routinario di questa metodica riportano risultati eccellenti, non confermati da altri trials. La MIRP può essere preferita in caso di reintervento o di ectopia paratiroidea, sempre che siano rispettate le controindicazioni.
In letteratura sono riportate segnalazioni sulla persistenza dell’IPT dopo MIP/UNE associate a falsi positivi del PTHio, e pubblicazioni che avvalorano la supremazia della BNE sugli interventi mini-invasivi (8). Fra questi uno, condotto su 1158 pazienti, ipotizza un’alta persistenza della malattia in seguito ad interventi limitati: dopo l’asportazione mirata della paratiroide localizzata pre-operatoriamente ed il crollo intra-operatorio del PTH a valori di guarigione, l’intervento è stato proseguito con la BNE. Nel 16% di questi pazienti sono state asportate altre ghiandole ingrossate ed ipercellulari, considerate fonte di potenziale recidiva dell’IPT (11). Alcuni però ritengono che tali caratteristiche macroscopiche ed istologiche delle paratiroidi non esprimano obbligatoriamente significato patologico.
L’esito dello studio di localizzazione pre-operatorio risulta determinante per la scelta del tipo di intervento (12) (tabella).
| Scelta del tipo di intervento in relazione all'esito dello studio di localizzazione pre-operatorio | ||
| Localizzazione | Intervento | |
| Una sola ghiandola | scintigrafia ed ecografia sono concordanti | MIP (o UNE) |
| scintigrafia ed ecografia sono discordanti oppure un solo esame è positivo |
BNE oppure MIP/UNE con PTHio | |
| Più ghiandole oppure nessuna ghiandola | BNE | |
In presenza di IPT primitivo associato a gozzo nodulare la scelta dell’intervento da praticare può essere così riassunta(12):
- MIP/UNE se c’è l’indicazione all’emitiroidectomia, che è omolaterale alla paratiroide patologica localizzata preoperatoriamente;
- BNE in tutte le altre situazioni di IPT primitivo e di gozzo con indicazione chirurgica.
BIBLIOGRAFIA
- Allendorf J, Kim L, Chabot J, et al.The impact of sestamibi scanning on the outcome of parathyroid surgery. J Clin Endocrinol Metab 2003, 88: 3015-8.
- Emmolo I, Dal Corso H, Borretta G, et al. Unexpected results using rapid intraoperative parathyroid hormone monitoring during parathyroidectomy for primary hyperparathyroidism. World J Surg 2005, 29: 785-8.
- Henry JF. Minimally invasive thyroid and parathyroid surgery is not a question of length of the incision. Langenbecks Arch Surg 2008, 393: 621-6.
- Brunaud L, Zarnegar R, Wada N, et al. Incision length for standard thyroidectomy and parathyroidectomy: when is it minimally invasive? Arch Surg 2003, 138: 1140-3.
- Hessman O, Westerdahl J, Al-Suliman N, et al. Randomized clinical trial comparing open with video-assisted minimally invasive parathyroid surgery for primary hyperparathyroidism. Br J Surg 2010, 97: 177-84.
- Bellantone R, Raffaelli M, De Crea C, et al. Minimally-invasive parathyroid surgery. Acta Othorhinolaryngol Ital 2011, 31: 207-15.
- Landry CS, Grubbs EG, Morris GS, et al. Robot assisted transaxillary surgery (RATS) for the removal of thyroid and parathyroid glands. Surgery 2011, 149: 549-55.
- Norman J. Controversies in parathyroid surgery: the quest for a “mini” unilateral parathyroid operation seem to have gone too far. J Surg Oncol 2012, 105: 1-3.
- Russel CF, Dolan SJ, Laird JD. Randomized clinical trial comparing scan-directed unilateral versus bilateral cervical exploration for primary hyperparathyroidism due to solitary adenoma. Br J Surg 2006, 93: 418-21.
- Miccoli P, Barellini L, Monchik M, et al. Randomized clinical trial comparing regional and general anaesthesia in minimally invasive video-assisted parathyroidectomy. Br J Surg 2005, 92: 814-8.
- Siperstein A, Berber E, Barbosa GF, et al. Predicting the success of limited exploration for primary hyperparathyroidism using ultrasound, sestamibi, and intraoperative parathyroid hormone. Analysis of 1158 cases. Ann Surg 2008, 3: 420-8.
- Zini M, Attanasio R, Cesareo R, et al. AME position statement: Primary hyperparathyroidism in clinical practice. J Endocrinol Invest 2012, 35 suppl 7: 2-21.
Terapia farmacologica dell'iperparatiroidismo primario
Laura Gianotti
Endocrinologia, Ospedale S. Croce e Carle, Cuneo
La terapia definitiva/risolutiva del PHPT è chirurgica, sebbene nella forma di PHPT asintomatico possa essere presa in considerazione l'alternativa della sorveglianza clinica con periodico follow-up nel tempo.
La terapia medica trova indicazione nelle seguenti condizioni:
- pazienti non candidabili a chirurgia
- dopo fallimento chirurgico o recidiva
- presenza di comorbilità che controindichino l’intervento
- rifiuto della terapia chirurgica
- crisi ipercalcemica o ipercalcemia severa in attesa della soluzione chirurgica o l'ipercalcemia severa nel carcinoma paratiroideo avanzato.
Obiettivi della terapia sono il controllo della calcemia, la riduzione del riassorbimento osseo e del rischio di frattura, nonché la protezione della funzione renale (1).
Terapia dell’ipercalcemia
Nella gestione terapeutica dell’ipercalcemia occorre in primo luogo sospendere i farmaci che possono indurre ipercalcemia (diuretici tiazidici, sali di calcio, vitamina D o analoghi) e la digitale, che in associazione all’ipercalcemia può indurre aritmie e arresto cardiaco.
In presenza di ipercalcemia sintomatica (solitamente > 12 mg/dL), soprattutto se severa (> 14 mg/dL), la terapia richiede l’espansione volemica con soluzione fisiologica. La dose di carico è pari almeno 200 mL/h nella prima ora e poi si prosegue il trattamento facendo attenzione al volume urinario (che deve essere pari almeno a 100 mL/h). Controverso è l’impiego della furosemide. Questa, comunque, deve essere somministrata a basse dosi (20 mg ev, eventualmente ripetibili) solo dopo aver corretto la volemia e nei pazienti a rischio di scompenso cardiaco e/o con insufficienza renale (2).
Come seconda linea vanno considerati gli steroidi (es. idrocortisone 200 mg ev, ripetibile dopo alcune ore), mentre i bisfosfonati ev (clodronato, ibandronato, pamidronato, zoledronato) vanno presi in considerazione in presenza di ipercalcemia importante o sintomatica e comunque sempre in associazione all’idratazione.
Se l’ipercalcemia si associa a severa acidosi, è opportuno l’impiego di sodio bicarbonato (ovviamente ponendo attenzione alla potassiemia, soprattutto nei pazienti trattati con digitale, per ridurre la tossicità del farmaco che aumenta nel caso di ipopotassiemia e ipercalcemia).
Se l’ipercalcemia è persistente o la diuresi è scarsa si può ricorrere all’emodialisi.
La calcitonina trova uno spazio limitato (2).
Qualora sussistano le condizioni già citate nelle indicazioni (vedi sopra), trova spazio terapeutico il cinacalcet. Appartenente alla famiglia dei calciomimetici – modulatori allosterici del CaSR (situato sulle cellule paratiroidee che attivato inibisce la trascrizione, la sintesi e la secrezione del PTH, nonchè la proliferazione della cellula paratiroidea), il cinacalcet determina una riduzione dei livelli calcemici. È stato approvato nel 2004 per la terapia dell’iperparatiroidismo secondario a IRC e dell’ipercalcemia da carcinoma paratiroideo, con estensione dal 2008 all’impiego nel PHPT (3). In quest’ultima condizione il cinacalcet è stato approvato dall’EMA con l’indicazione: “pazienti con PHPT per cui l’intervento chirurgico sarebbe indicato sulla base dei livelli di calcio, ma in cui la PTX non è clinicamente appropriata o è controindicata”. L’FDA lo ha approvato con l’indicazione: “trattamento dell’ipercalcemia severa in pazienti con PHPT non idonei alla PTX”.
Il cinacalcet è efficace nel ridurre o normalizzare la calcemia in vari gruppi di pazienti con PHPT, dalle forme asintomatiche e lievi a quelle severe del PHPT resistente/recidivo o del raro carcinoma paratiroideo o ancora del PHPT associato alla MEN-1 (4). Il cinacalcet riduce lievemente il PTH, mentre non ha alcun effetto sulla BMD (4,5).
Nello studio di Peacock et al. condotto sino a 5 anni (6), il cinacalcet alla posologia di 30 mg x 2/die ha mostrato, oltre all’effetto ipocalcemizzante, un effetto positivo (di incremento) sulla fosforemia e sui sintomi cognitivi, nonchè sulla qualità di vita. L’efficacia del cinacalcet è stata dimostrata in pazienti con PHPT persistente dopo chirurgia o con controindicazioni alla stessa, caratterizzati da uno stato di ipercalcemia moderata-severa (calcemia > 12.5 mg/dL), impiegando la posologia massima consentita di 90 mg x 4/die (4,5).
Il trattamento con cinacalcet va iniziato alla posologia minima di 30 mg/die, con incrementi posologici graduali nel tempo, in base all’andamento della calcemia e alla tollerabilità individuale.
L’indicazione attuale all’impiego di cinacalcet (7) è in pazienti con PHPT con calcemia > 1 mg/dL oltre il valore superiore di norma e in presenza di almeno una delle seguenti condizioni:
- controindicazione alla chirurgia
- rifiuto dell’intervento chirurgico
- inefficacia della terapia chirurgica
- recidiva di malattia
- in previsione di PTX se prevedibile un periodo di attesa > 6 mesi.
Non è ad oggi definito se la riduzione mediante cinacalcet di ipercalcemie lievi possa determinare effetti positivi sulle potenziali complicanze della malattia (alterazioni neuropsicologiche, cognitive, cardiovascolari e renali), pertanto non è raccomandato l'impiego di cinacalcet in presenza di ipercalcemia lieve (8).
Recentemente è stato descritto l'impiego di denosumab per il controllo dell'ipercalcemia refrattaria nel carcinoma paratiroideo (9).
Bibiliografia
- Khan A, Grey A, Shoback D. Medical management of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 2009, 94: 373-81.
- Ziegler R. Hypercalcemic crisis. J Am Soc Nephrol 2001, 12: S3-9.
- Marcocci C, Chanson P, Shoback D, et al. Cinacalcet reduces serum calcium concentrations in patients with intractable primary hyperparathyroidism. J Clin Endocrinol Metab 2009, 94: 2766–72.
- Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab 2009, 94: 4860–7.
- Marcocci C, Cetani F. Update on the use of cinacalcet in the management of primary hyperparathyroidism. J Endocrinol Invest 2012, 35: 90-5.
- Peacock M, Bilezikian JP, Bolognese MA, et al. Cinacalcet HCl reduces hypercalcemia in primary hyperparathyroidism across a wide spectrum of disease severity. J Clin Endocrinol Metab 2011, 96: E9-18.
- Marcocci C, Bollerslev J, Khan AA, Shoback DM. Medical management of primary hyperparathyroidism: proceedings of the fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab 2014, 99: 3607-18.
- Marcocci C, Brandi ML, Scillitani A, et al. Italian Society of Endocrinology Consensus Statement: definition, evaluation and management of patients with mild primary hyperparathyroidism. J Endocrinol Invest 2015, 38: 577-93.
- Vellanki P, Lange K, Elaraj D, Kopp PA, El Muayed M. Denosumab for management of parathyroid carcinoma-mediated hypercalcemia. J Clin Endocrinol Metab 2014, 99: 387-90.
Terapia di protezione ossea
La terapia di protezione ossea nel PHPT è indirizzata alla riduzione del rischio di fratture e alla riduzione del riassorbimento osseo indotto dall’eccesso di PTH. Va scelta in base al rischio di frattura, combinando il dato della BMD con il profilo di rischio del paziente. L’algoritmo FRAX non è applicabile con certezza al PHPT, anche se il FRAX include il rischio associato all’osteoporosi secondaria (1).
La terapia farmacologica di protezione ossea prevede l’impiego di farmaci anti-riassorbitivi, in prima istanza i bisfosfonati. Tra questi, l’alendronato alla posologia di 10 mg/die per 24 mesi riduce il rimaneggiamento osseo e aumenta significativamente la BMD a livello femorale e lombare nei pazienti con osteopenia secondaria a PHPT (2). La terapia con alendronato non determina variazioni significative di calcemia o PTH (è possibile un transitorio lieve aumento di PTH e una riduzione di calcemia nel primo mese di terapia).
Una recente meta-analisi condotta su circa 40 studi osservazionali e randomizzati-controllati ha confrontato gli effetti del trattamento chirurgico, di quello medico e della semplice sorveglianza clinica sulla variazione di BMD nel PHPT asintomatico. Il risultato dimostra una sostanziale equivalenza del trattamento medico verso quello chirurgico ed entrambe le opzioni risultano preferibili rispetto alla semplice sorveglianza (3).
La terapia medica con alendronato va proposta nei pazienti con PHPT:
- con elevato rischio di frattura prima dell’intervento chirurgico
- dopo l’intervento di PTX qualora il recupero di BMD valutato dopo 12-18 mesi non sia adeguato.
Va detto che non esistono studi RCT sull’efficacia di alendronato in termini di riduzione del rischio di frattura nel PHPT.
La terapia con alendronato va sempre associata al supplemento di vitamina D, sotto forma di vitamina D3 ad una posologia media di 800-1000 U/die. Il deficit di vitamina D è infatti comune nei pazienti con PHPT e si associa alla severità della malattia e ad un aumentato rischio di fratture rispetto ai pazienti repleti (4,5). In talune forme di iperparatiroidismo normocalcemico o ipercalcemico intermittente con ipovitaminosi D, la supplementazione con vitamina D può aiutare nella diagnosi differenziale (6):
- nel caso di una normalizzazione del PTH si viene a delineare un iperparatiroidismo secondario a ipovitaminosi D;
- la persistenza di un eccesso di PTH a fronte di un’adeguata e prolungata supplementazione con vitamina D può condurre verso la diagnosi di PHPT anche in presenza di normocalcemia.
Studi recenti europei (5,6) riportano un’alta prevalenza di ipovitaminosi D nel PHPT (> 80-90%) - sebbene tale alta prevalenza non sia confermata in altri studi (7) - con un’associazione inversa tra bassi livelli di vitamina D e peso dell’adenoma paratiroideo, livelli di PTH e di calcemia, e livelli di BMD femorale e radiale. Si evidenzia una tendenza (non significativa però) all'aumento del rischio di frattura nei pazienti con bassi livelli di 25OH-D (6) e in generale un danno osseo maggiore nei pazienti PHPT con deficit di vitamina D.
Non è del tutto noto il motivo per cui il deficit di vitamina D è così frequente nel PHPT. In parte contribuisce la stimolazione dell’attività enzimatica dell’1-alfa-idrossilasi renale da parte dell’eccesso di PTH e dell’ipofosforemia. L’aumento relativo di 1,25(OH)2D aumenterebbe l’espressione genica della 24-idrossilasi nei tessuti bersaglio della vitamina D, stimolando la degradazione di 25OH-D (6).
Un recente RCT europeo in pazienti con PHPT ha dimostrato che un supplemento con vitamina D sotto forma di D3 (colecalciferolo) alla posologia di 2800 U/die vs placebo per 26 settimane prima dell'intervento chirurgico ha indotto un incremento della vitamina D circolante dell'88%, ha ridotto i livelli di PTH del 17%, e migliorato la BMD a livello lombare, senza modificazioni dei livelli di calcio sierici e urinari nel gruppo trattato con D3 rispetto al gruppo placebo.
La correzione del deficit di vitamina D pre-operatorio è importante, in quanto la carenza risulta associata a un rischio aumentato di ipocalcemia post-PTX (8). La recente consensus SIE sulla gestione del PHPT lieve asintomatico (9), in linea con l'ultimo workshop sul PHPT asintomatico (10), raccomanda di correggere il deficit di vitamina D in tutti i pazienti con PHPT. La supplementazione consigliata è di 800–1000 IU/die di vitamina D (o l'equivalente settimanale o mensile). L'obiettivo di vitamina D circolante da raggiungere si colloca sopra i 20 ng/mL.
Bibliografia
- Kanis JA, Johnell O, Oden A, et al. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008, 19: 385-97.
- Khan AA, Bilezikian JP, Kung AWC, et al. Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab 2004, 89: 3319-25.
- Sankaran S, Gamble G, Bolland M, et al. Skeletal effects of interventions in mild primary hyperparathyroidism: a meta-analysis. J Clin Endocrinol Metab 2010, 95: 1653-62.
- Silverberg SJ, Shane E, Dempster DW, et al. The effects of vitamin D insufficiency in patients with primary hyperparathyroidism. Am J Med 1999, 107: 561–7.
- Nordenstrom E, Westerdahl J, Lindergard B, et al. Multifactorial risk profile for bone fractures in primary hyperparathyroidism. World J Surg 2002, 26: 1463–7.
- Bollerslev J, Marcocci C, Sosa M, et al. Current evidence for recommendation of surgery, medical treatment and vitamin D repletion in mild PHPT. Eur J Endocrinol 2011, 165: 851-64.
- Tassone F, Gianotti L, Baffoni C et al. Vitamin D status in primary hyperparathyroidism: a Southern European perspective. Clin Endocrinol (Oxf) 2013, 79: 784-90.
- Rolighed L, Rejnmark L, Sikjaer T, et al. Vitamin D treatment in primary hyperparathyroidism: a randomized placebo controlled trial. J Clin Endocrinol Metab 2014, 99: 1072–80.
- Marcocci C, Brandi ML, Scillitani A, et al. Italian Society of Endocrinology Consensus Statement: definition, evaluation and management of patients with mild primary hyperparathyroidism. J Endocrinol Invest 2015, 38: 577-93.
- Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab 2014, 99: 3561-9.
Terapia di protezione renale
Non esistono linee guida o RCT indirizzati al trattamento medico dell’IRC associata al PHPT e pertanto si propone di seguire le indicazioni fornite dalla Società Italiana di Nefrologia per il management dell’IRC (1).
Per quanto concerne la terapia della calcolosi renale, è noto che la PTX riduce il rischio di recidiva ma non lo annulla e il rischio permane elevato per oltre 10 anni dopo la PTX (2). Anche ipercalciuria e ipofosforemia non vengono rapidamente corrette dalla PTX (3).
In assenza di evidenze specifiche, ad oggi si consiglia di trattate la nefrolitiasi in corso di PHPT o dopo PTX in modo simile alla calcolosi di altra natura o primitiva/idiopatica.
In generale, in presenza di litiasi renale silente asintomatica non vi è indicazione a terapia specifica, salvo la terapia idropinica (almeno 2 litri/die) e una dieta iposodica (per ridurre la calciuria). L’apporto di calcio nella dieta non va ridotto, perché una dieta ipocalcica può indurre un aumento dell’escrezione di ossalati e di fosfati. Si raccomanda un apporto medio di calcio di 1 g/die anche nei pazienti prima della PTX (4). In uno studio di coorte in pazienti con PHPT, è stato dimostrato che i pazienti in supplemento calcico hanno una prevalenza inferiore di nefrolitiasi rispetto a quelli non supplementati (5). È consigliabile inoltre una dieta povera di ossalati.
In caso di litiasi sintomatica è consigliabile sempre una valutazione urologica. In caso di colica renale da microlitasi o calcoli di diametro < 5 mm, il trattamento prevede idratazione, analgesici FANS o derivati morfinici, insieme a farmaci alfa-litici o calcio-antagonisti (4). In caso di calcoli di diametro superiore, è prevedibile l’impiego di tecniche di litotripsia di competenza urologica (4).
In caso di nefrolitiasi ricorrente, è consigliabile uno screening metabolico per escludere cause sistemiche; utile inoltre un trattamento profilattico con diuretici tiazidici e citrato di potassio.
Bibliografia
- Cianciaruso B e Società Italiana di Nefrologia. Linee guida per la terapia conservativa dell’insufficienza renale cronica. G It Nefrol 2003, 20 (S-24): S48-S60.
- Mollerup CL, Vestergaard P, Frøkjaer VG, et al. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ 2002, 325: 807.
- Cooperberg MR, Duh QI, Stackhouse GB, et al. Oral calcium supplementation associated with decreased likelihood of nephrolithiasis prior to surgery for hyperparathyroidism. Int J Urology 2007, 14: 1113-5.
- Suh JM, Cronan JJ, Monchik JM. Primary hyperparathyroidism: is there an increased prevalence of renal stone disease? AJR Am J Roentgenol 2008, 191: 908-11.
- Rejnmark L, Vestergaard P, Mosekilde L. Nephrolithiasis and Renal Calcifications in Primary Hyperparathyroidism. J Clin Endocrinol Metab 2011, 96: 2377-85.
Scheda cinacalcet
Laura Gianotti
Endocrinologia, Ospedale S.Croce e Carle, Cuneo
Meccanismo d’azione
Il recettore sensibile al calcio (CaSR) sulla superficie delle cellule principali della paratiroide è il principale regolatore della secrezione di paratormone (PTH). Cinacalcet è un calciomimetico che, aumentando la sensibilità del CaSR nei confronti del calcio extra-cellulare, riduce direttamente i livelli di PTH. La riduzione del PTH è associata a un concomitante calo dei livelli sierici di calcio. La diminuzione dei livelli di PTH è correlata alla concentrazione di cinacalcet. Dopo che è stato raggiunto lo steady state, le concentrazioni sieriche di calcio si mantengono costanti nell'intervallo fra le somministrazioni.
Indicazioni terapeutiche
Trattamento dell'iperparatiroidismo secondario in pazienti affetti da insufficienza renale in stadio terminale (end-stage renal disease ESRD) in terapia dialitica di mantenimento. Il cinacalcet può essere usato come parte di un regime terapeutico che includa, secondo necessità, chelanti del fosfato e/o vitamina D.
Riduzione dell'ipercalcemia in pazienti con:
- carcinoma paratiroideo
- iperparatiroidismo primario, nei quali la paratiroidectomia sarebbe indicata sulla base dei valori sierici di calcio (in accordo con le relative linee guida di trattamento), ma nei quali l’intervento chirurgico non è clinicamente appropriato o è controindicato.
Contro-indicazioni
ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Preparazioni, via di somministrazione, posologia
Compresse per uso orale da 30 mg, 60 mg, 90 mg (cinacalcet Aristo, cinacalcet Aurobindo, cinacalcet DOC, cinacalcet Dr.Reddy's, cinacalcet EG, ccinacalcet Mylan, cinacalcet Tillomed, Mimpara)
La biodisponibilità del cinacalcet è maggiore se assunto insieme agli alimenti. Le compresse devono essere assunte intere.
La dose iniziale raccomandata nell'adulto è pari a 30 mg una o due volte al giorno. La dose del medicinale deve essere aumentata gradualmente ogni 2-4 settimane con la seguente sequenza posologica:
- 30 mg una volta al giorno
- 30 mg due volte al giorno
- 60 mg due volte al giorno
- 90 mg due volte al giorno
- 90 mg tre o quattro volte al giorno
come necessario per ridurre la concentrazione di calcio sierica fino al limite superiore della norma o al di sotto di tale valore. La dose massima usata negli studi clinici è stata di 90 mg quattro volte al giorno.
Dopo l'inizio del trattamento con il farmaco o dopo una modifica della dose occorre determinare il calcio sierico entro una settimana.
Dopo aver stabilito la dose di mantenimento, la calcemia dovrà essere misurata ogni 2-3 mesi.
Nel caso in cui non fosse possibile mantenere una riduzione clinicamente rilevante dei livelli sierici di calcio, si dovrà considerare un'interruzione della terapia.
Bambini e adolescenti: non è indicato a causa della mancanza di dati su sicurezza ed efficacia.
Insufficienza epatica: non è necessario modificare la dose iniziale, ma deve essere usato con cautela e il trattamento deve essere controllato attentamente durante l’aumento graduale della dose e nel corso della terapia, perchè nei pazienti con insufficienza epatica moderata o grave (classificazione di Child-Pugh) si possono raggiungere livelli plasmatici di cinacalcet 2-4 volte superiori.
Gravidanza: non vi sono dati clinici. Mimpara deve essere usato durante la gravidanza solo qualora i benefici potenziali giustifichino i potenziali rischi per il feto.
Allattamento: non è noto se cinacalcet venga escreto nel latte materno. Dopo un'attenta valutazione del rapporto rischi/benefici, si dovrà prendere la decisione di interrompere l'allattamento al seno oppure il trattamento con Mimpara.
Avvertenze speciali e precauzioni di impiego
Convulsioni: riduzioni significative dei livelli sierici di calcio determinano un abbassamento della soglia di insorgenza
Ipotensione e/o peggioramento dell’insufficienza cardiaca: sono stati riportati casi isolati, idiosincratici di ipotensione e/o di peggioramento di insufficienza cardiaca in pazienti con ridotta funzionalità cardiaca.
Lattosio: i pazienti con rari problemi ereditari di intolleranza al galattosio, deficit di Lapp lattasi o malassorbimento di glucosio-galattosio, non devono assumere questo farmaco.
Interazioni
Il cinacalcet viene metabolizzato in parte dall'enzima CYP3A4 e in parte dal CYP1A2. Potrebbe essere necessario un aggiustamento della dose nel caso in cui un paziente trattato con Mimpara inizi o interrompa una terapia con un potente inibitore (per es. ketoconazolo, itraconazolo, telitromicina, voriconazolo o ritonavir) o induttore (per es. rifampicina) di CYP3A4, nel caso in cui il paziente inizi o smetta di fumare, oppure in caso di inizio o interruzione di un trattamento concomitante con potenti inibitori del CYP1A2 (per es. fluvoxamina, ciprofloxacina).
La somministrazione concomitante di carbonato di calcio (una dose singola da 1500 mg), sevelamer (2.400 mg x 3/d), pantoprazolo (80 mg/d) non ha alterato la farmacocinetica di cinacalcet.
Cinacalcet è un potente inibitore di CYP2D6. Potrebbero essere necessari aggiustamenti della dose dei medicinali assunti in concomitanza, metabolizzati prevalentemente dal CYP2D6, titolati individualmente e con un ristretto indice terapeutico (per es. flecainide, propafenone, metoprololo, desipramina, nortriptilina, clomipramina).
Cinacalcet non è un induttore di CYP3A4, CYP3A5, CYP1A2 e CYP2C9. Pertanto non sono necessaari aggiustamenti delle dosi di warfarin, midazolam, ciclosporina e tacrolimus.
Effetti collaterali
Alle dosi basse (30-60 mg/die) è ben tollerato, mentre all’aumentare della posologia (oltre 60-90 mg/die) gli effetti collaterali sono frequenti: nausea (36%), artralgie (30%), diarrea (22%), mialgie (22%) e parestesie (22%). Non comunemente sono state riportate convulsioni e casi isolati, idiosincratici di ipotensione e/o di peggioramento dell’insufficienza cardiaca in pazienti con ridotta funzionalità cardiaca. Reazioni allergiche, inclusi angioedema ed orticaria.
Gli effetti collaterali tendono a ridursi dopo 1 mese dall’inizio del trattamento.
Sovradosaggio
In pazienti sottoposti a dialisi sono state somministrate dosi fino a 300 mg una volta al giorno senza problemi di sicurezza.
Il sovradosaggio di Mimpara può portare a ipocalcemia. In caso di sovradosaggio si devono monitorare i pazienti al fine di rilevare eventuali segni e sintomi di ipocalcemia e si deve instaurare un trattamento sintomatico e di supporto. Poiché cinacalcet si lega in larga parte alle proteine, l'emodialisi non rappresenta un trattamento efficace in caso di sovradosaggio.
Limitazioni prescrittive
Prescrivibile in classe A con piano terapeutico
Sorveglianza e follow-up nell'iperparatiroidismo primario
Laura Gianotti
Endocrinologia, Ospedale S. Croce e Carle, Cuneo
LA SORVEGLIANZA CLINICA
Già dal 1990, anno della Prima Consensus sul Management del PHPT asintomatico, venne introdotto il concetto di sorveglianza clinica nel PHPT ad espressione pauci-sintomatica. I primi studi osservazionali sull’andamento clinico del PHPT asintomatico non operato indicavano una sostanziale stabilità della malattia ed una scarsa progressione delle complicanze, anche se è pur vero che circa 1/3 dei pazienti nelle varie casistiche mostrava una progressione del danno osseo, mentre i pazienti operati mostravano un recupero della densità minerale ossea (1-3).
Il più lungo studio prospettico di follow-up sul PHPT asintomatico, durato sino a 15 anni (3), evidenzia un progressivo declino della massa ossea, in particolare a livello corticale, anche a distanza di oltre 10 anni dalla diagnosi, ponendo l’attenzione sull’importanza della sorveglianza densitometrica anche a lungo termine.
Anche i dati sull’aumento della mortalità cardiovascolare del PHPT non operato non sono incoraggianti, così come l’evidenza che alcuni sintomi aspecifici, quali depressione, astenia, apatia e disturbi del sonno, persistano nel tempo e regrediscano solo dopo la terapia chirurgica.
Gli sforzi delle società scientifiche si sono pertanto concentrati nel definire i vantaggi e individuare i criteri della sorveglianza clinica nel PHPT asintomatico (4). Non tutte le società scientifiche sono in linea con questa condotta: le linee guida proposte dall’AACE-AAES (5) propongono ad esempio la paratiroidectomia come unica terapia curativa e risolutiva del PHPT anche nella forma asintomatica, con un rapporto favorevole sia in termini di costo-efficacia che di sicurezza. Laddove si presentino casi di PHPT inoperabile o nei pazienti che rifiutino l’intervento, è evidente però la necessità della sorveglianza clinica giovandosi della terapia medica. Il più recente Workshop del 2014 sul Management del PHPT asintomatico (6) ha sostanzialmente confermato le ultime indicazioni del 2008 (7) e cioè che la sorveglianza clinica e la terapia medica possono costituire una valida opzione in taluni pazienti con PHPT asintomatico, in particolare nei soggetti che non presentino ipercalcemia significativa (< 1 mg/dL oltre il limite massimo di norma), che non abbiano una riduzione della clearance creatininica < 60 mL/min, che abbiano più di 50 anni di età ed infine che non presentino un deficit densitometrico significativo e/o non abbiano una frattura da fragilità.
La sorveglianza clinica va attuata annualmente mediante la misurazione della calcemia e della creatininemia, mentre la densitometria ossea è consigliata ogni 24 mesi, mirata a tutti i siti (colonna lombare, collo femorale e radio ultra-distale). Lo studio Rx del rachide o la morfometria vertebrale tramite DEXA sono consigliati se si sospetta una frattura vertebrale (dorso-lombalgia o calo staturale). Dalle nuove indicazioni del IV Workshop (6) viene reintrodotta la misurazione della calciuria (> 400 mg/24 h) come criterio per la scelta terapeutica, mentre per il follow-up è consigliata l'esecuzione di un profilo urinario delle 24 ore includente la calciuria solo nel sospetto di nefrolitiasi (episodi di colica renale, renella, infezioni ricorrenti delle vie urinarie). Il monitoraggio ecografico renale è proponibile solo nel sospetto di evoluzione litiasica e non routinariamente per lo screening della litiasi renale silente, salvo alla diagnosi della malattia (6). Anche la recente consensus della SIE sulla gestione del PHPT lieve asintomatico (8) propone la misurazione della calcemia e della creatinina annualmente e della BMD ogni 2 anni. L'esecuzione dell'ecografia renale e del profilo di rischio calcolotico sulle urine è consigliato solo in caso di sospetto clinico, mentre non è raccomandato nel follow-up l'impiego routinario dell'ecografia renale per evidenziare la litiasi renale asintomatica.
Bibiliografia
- Rao DS, Phillips ER, Divine GW, et al. Randomized controlled clinical trial of surgery versus no surgery in patients with mild asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab 2004, 89: 5415-22.
- Ambrogini E, Cetani F, Cianferotti L, et al. Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab 2007, 92: 3114-21.
- Rubin MR, Bilezikian JP, McMahon DJ, et al. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab 2008, 93: 3462-70.
- Bilezikian JP, Potts Jr JT, El-Hajj Fuleihan G, et al. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Clin Endocrinol Metab 2002, 87: 5353-61.
- AACE/AAES Task Force on Primary Hyperparathyroidism. The American Association of Clinical Endocrinologists and the American Association of Endocrine Surgeons position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Pract 2005, 11: 49-54.
- Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab 2014, 99: 3561-9.
- Bilezikian JP, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 2009, 94: 335-9.
- Marcocci C, Brandi ML, Scillitani A, et al. Italian Society of Endocrinology Consensus Statement: definition, evaluation and management of patients with mild primary hyperparathyroidism. J Endocrinol Invest 2015, 38: 577-93.
IL FOLLOW-UP NEI PAZIENTI OPERATI
Biochimico
La calcemia scende rapidamente nelle prime ore e va controllata ogni 6–12 ore dall’intervento. L’ipocalcemia, se presente, è solitamente transitoria; è meno frequente in caso di chirurgia mini-invasiva, ma non è rara dopo la PTX bilaterale e/o in pazienti con PHPT di lunga durata e con ipercalcemia marcata (1-4).
È utile una determinazione di calcemia a 6 mesi dall’intervento nel PHPT sporadico: se è nella norma, non sono necessari ulteriori controlli mirati nel tempo (5), sebbene non sia escludibile una forma multi-ghiandolare o familiare.
I livelli di PTH possono permanere elevati anche a distanza di mesi dalla PTX (9-62%), ma non vanno considerati come una persistenza di malattia. L’incremento del PTH è ritenuto secondario al recupero della mineralizzazione ossea (la “sindrome dell’osso affamato”) e tende a normalizzarsi con la supplementazione di calcio e vitamina D (6).
Clinico
Osso: dopo la PTX è atteso un recupero di BMD già a 12 mesi e quindi a 24 mesi, quando è riportato il recupero del 4% circa a livello femorale e dell’8% a livello della colonna (7-9). Pertanto è consigliabile l’esecuzione di una DEXA a 12 mesi e quindi a 24 mesi.
Rene: la sorveglianza della funzione renale o il controllo ecografico periodico renale dopo la PTX non vanno proposti routinariamente, ma sulla base delle caratteristiche del paziente e delle alterazioni morfo-funzionali pre-esistenti (9-11).
Apparato cardio-vascolare: sebbene la PTX si associ a una riduzione del rischio cardiovascolare (12-17), non è richiesto un monitoraggio o una rivalutazione cardiovascolare routinaria dopo la PTX.
IL FOLLOW-UP NEI PAZIENTI NON OPERATI
PHPT sintomatico:
- monitoraggio semestrale di calcemia e creatininemia;
- monitoraggio di BMD sui 3 siti (colonna lombare, polso, femore) ogni 24 mesi;
- utile la sorveglianza annuale cardio-vascolare e delle funzioni neuropsicologiche.
PHPT asintomatico
Sebbene una larga parte dei pazienti con PHPT asintomatico non mostri un’evoluzione del quadro clinico, una percentuale variabile dal 13.5 al 37.5% può presentare una progressione biochimica/clinica di malattia nell’arco di 10-15 anni (9,11).
Nel breve termine (1-2 anni) la maggior parte degli studi osservazionali o prospettici non evidenzia un declino significativo di BMD nei pazienti con PHPT non operati (7,8), sebbene nell’arco dei 15 anni in oltre il 50% dei pazienti sia riportata una riduzione di BMD del 10% circa a livello del femore e del radio distale (i.e. siti ricchi in osso corticale)(9).
In generale il follow-up dei pazienti non operati prevede:
- indagini biochimiche annuali e misurazione della BMD ogni 2 anni;
- adozione del trattamento farmacologico per il controllo della calcemia e la riduzione del rischio di fratture.
Secondo le ultime indicazioni del IV workshop (18), condivise dalla Consensus della SIE (19) durante il follow-up del paziente con PHPT asintomatico occorre rivalutare i criteri (follow-up vs terapia chirurgica) e proporre la terapia chirurgica, nelle seguenti condizioni:
- aumento della calcemia oltre 1 mg/dL rispetto al range di normalità;
- riduzione della clearance creatininica < 60 mL/min;
- comparsa di calcoli renali o nefrocalcinosi;
- riduzione della BMD (T-score a qualunque sito < -2.5);
- comparsa di una frattura da fragilità, clinica o rilevata con Rx o morfometria.
Bibliografia
- Mittendorf EA, Merlino JI, McHenry CR. Post-parathyroidectomy hypocalcemia: incidence, risk factors, and management. Am Surg 2004, 70: 114-20.
- Kald BA, Heath DI, Lausen I, et al. Risk assessment for severe postoperative hypocalcemia after neck exploration for primary hyperparathyroidism. Scand J Surg 2005, 94: 216-20.
- Norman JG, Politz D. Safety of immediate discharge after parathyroidectomy: a prospective study of 3,000 consecutive patients. Endocr Pract 2007, 13: 105-13.
- Vasher MN, Goodman A, Politz D, et al. Postoperative calcium requirements in 6,000 patients undergoing outpatient parathyroidectomy: easily avoiding symptomatic hypocalcemia. J Am Coll Surg 2010, 211: 49-54.
- Witteveen JE, Kievit J, Morreau H, et al. No recurrence of sporadic primary hyperparathyroidism when cure is established 6 months after parathyroidectomy. Eur J Endocrinol 2010, 162: 399-406.
- Biskobing DN. Significance of elevated parathyroid hormone after parathyroidectomy. Endocr Pract 2010, 16: 112-7.
- Ambrogini E, Cetani F, Cianferotti L, et al. Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab 2007, 92: 3114-21.
- Bollerslev J, Jansson S, Mollerup CL, et al. Medical observation, compared with parathyroidectomy, for asymptomatic primary hyperparathyroidism: a prospective, randomized trial. J Clin Endocrinol Metab 2007, 92: 1687–92.
- Rubin MR, Bilezikian JP, McMahon DJ, et al. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab 2008, 93: 3462-70.
- Mollerup CL, Vestergaard P, Frøkjaer VG, et al. Risk of renal stone events in primary hyperparathyroidism before and after parathyroid surgery: controlled retrospective follow up study. BMJ 2002, 325: 807.
- Yu N, Leese GP, Smith D, et al. The natural history of treated and untreated primary hyperparathyroidism: the parathyroid epidemiology and audit research study. QJM 2011, 104: 513-21.
- Hedbäck G, Odén A, Tisell LE. The influence of surgery on the risk of death in patients with primary hyperparathyroidism. World J Surg 1991, 15: 399–405; discussion 406–407.
- Piovesan A, Molineri N, Casasso F, et al. Left ventricular hypertrophy in primary hyperparathyroidism. Effects of successful parathyroidectomy. Clin Endocrinol (Oxf) 1999, 50: 321–8.
- Vestergaard P, Mollerup CL, Frokjaer VG, et al. Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg 2003, 27: 216–22.
- Nilsson IL, Aberg J, Rastad J, Lind L. Maintained normalization of cardiovascular dysfunction 5 years after parathyroidectomy in primary hyperparathyroidism. Surgery 2005, 137: 632–8.
- Bollerslev J, Rosen T, Mollerup CL, et al. Effect of surgery on cardiovascular risk factors in mild primary hyperparathyroidism. J Clin Endocrinol Metab 2009, 94: 2255-61.
- Farahnak P, Ring M, Caidahl K, et al. Cardiac function in mild primary hyperparathyroidism and the outcome after parathyroidectomy. Eur J Endocrinol 2010, 163: 461-7.
- Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, Potts JT Jr Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab 2014, 99: 3561-9.
- Marcocci C, Brandi ML, Scillitani A, et al. Italian Society of Endocrinology Consensus Statement: definition, evaluation and management of patients with mild primary hyperparathyroidism. J Endocrinol Invest 2015, 38: 577-93.
Aggiornamenti iperparatiroidismo primario
In attesa degli aggiornamenti di Endowiki, si possono trovare articoli aggiornati ai seguenti link:
- Iperparatiroidismo primario mild: operare o non operare? AME News n 27/2016
- Come distinguere iperparatiroidismo e ipercalcemia ipocalciurica? AME News n 36/2016
Iperparatiroidismi secondari
Classificazione e inquadramento generale
Classificazione e inquadramento generale degli iperparatiroidismi secondari
Costanza Santini
UOS Endocrinologia e Diabetologia, Ospedale Bufalini, Cesena
È una condizione endocrino-metabolica in cui la causa dell’eccessiva e continua secrezione di PTH è situata al di fuori delle paratiroidi.
| Cause di iperparatiroidismo secondario (s-PTH) | ||
| Renali | Osteodistrofia renale – ad alto turn-over | |
| Non renali | Deficit di vitamina D |
ridotto apporto alimentare |
| Resistenza alla vitamina D |
rachitismo vitamina D-dipendente di tipo 2 (resistenza al calcitriolo) |
|
| Resistenza al PTH | pseudo-ipoparatiroidismi | |
| Richiesta eccessiva di PTH | pancreatite acuta iperfosforemia acuta grave con perdita rilevante di calcio dal liquido extra-cellulare: da insufficienza renale, ipotermia, insufficienza epatica massiva, neoplasie ematologiche maligne |
|
| Farmaci | ||
| Inquadramento biochimico dell'iperparatiroidismo secondario | |||||
| Calcemia | Fosforemia | PTH | 25-OH-D | Fosfatasi alcalina | |
| Nefropatie croniche | N/↓ | ↑↑ | ↑↑ | N/↓ | ↑↑ |
| Deficit vitamina D | ↓ | ↓ | ↑↑ | ↓↓ | ↑ |
| Malassorbimenti | ↓ | ↓ | ↑ | ↓ | N/↑ |
| Epatopatie | N/↓ | N/↓ | N/↑ | ↓ | ↑ |
| Pseudo-ipoparatiroidismo | ↓ | ↑ | ↑↑ | N/↓ | N/↑ |
Bibliografia
- Martin KJ, Gonzales E. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol 2007, 8: 875-85.
- Lips P. Vitamin D physiology. Progr Biophys Mol Biol 2006, 92: 4-8.
- Adami S, Romagnoli E, et al. Linee guida su prevenzione e trattamento dell'ipovitaminosi con colecalciferolo. Reumatismo 2011, 63: 129-47.
- Holick MF, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 1911-30.
Forme di iperparatiroidismo secondarie a nefropatia
Andrea Guarnieri
SC Nefrologia e Dialisi - AO S. Croce e Carle - Cuneo
L'iperparatiroidismo secondario (sPTH) è una delle complicanze più frequenti in corso di insufficienza renale cronica (IRC) e si manifesta con una classica patologia del metabolismo minerale, associata alla formazione di calcificazioni vascolari, con conseguente aumento della morbilità e della mortalità cardiovascolare.
Per questo motivo la Kidney Disease Improving Global Outcomes (K-DIGO) ha raccomandato di integrare le anomalie del metabolismo osseo e minerale e le calcificazioni extra-scheletriche in un'unica entità clinica denominata Chronic Kidney Disease - Mineral and Bone Disorder (CKD-MBD) (1).
La CKD-MBD è definita come una patologia sistemica del metabolismo minerale e osseo causata dall'IRC, che si può presentare con uno o più dei seguenti segni o sintomi:
- anomalie biochimiche (alterato metabolismo di Ca, P, PTH o vitamina D);
- anomalie del turnover, mineralizzazione, crescita lineare o forza ossea;
- calcificazioni vascolari o extra scheletriche.
Patogenesi
L'sPTH, caratterizzato dall'aumento della sintesi e secrezione del PTH e dall'iperplasia delle paratiroidi, inizia precocemente nel corso della malattia renale cronica e la sua prevalenza aumenta con il declino della funzione renale già da valori di filtrato glomerulare (GFR) < 60 mL/min (IRC Stadio IIIa secondo la classificazione K-DOQI).
Numerose sono le anomalie che contribuiscono alla patogenesi dell' sPTH.
Iperfosforemia. Il declino della funzione renale si accompagna a un’incapacità del rene a mantenere in equilibrio il bilancio del fosforo: l'iperfosforemia causa una riduzione della concentrazione di calcio ionizzato e interferisce con la produzione di 1,25(OH)2 vitamina D, con conseguente stimolo alla secrezione di PTH. Nell'IRC avanzata l'iperfosforemia può anche avere un effetto diretto sulla sintesi e la secrezione del PTH, indipendentemente dalle concentrazioni di calcio e vitamina D (2). I livelli di fosforo condizionano inoltre la sintesi del Fibroblast Growth Factor 23 (FGF-23), un ormone fosfaturico che contribuisce alla regolazione del funzionamento delle paratiroidi nell'IRC avanzata (3) (vedi sotto).
Ridotti livelli di calcitriolo. La concentrazione plasmatica di calcitriolo scende in genere al di sotto dei limiti di norma per valori di GFR < 60 mL/min. La causa iniziale è probabilmente l'aumentata concentrazione di FGF-23 piuttosto che la riduzione della massa renale con conseguente minore attività dell'1-alfa-idrossilasi; la sintesi del calcitriolo è inoltre inibita dall'iperfosforemia. Il deficit di calcitriolo determina una riduzione dell'assorbimento di calcio dall'intestino e del rilascio di calcio dall'osso, con conseguente sviluppo di ipocalcemia (stimolo alla secrezione di PTH), riduce l'effetto di regolazione della sintesi di PTH mediato dal legame con i recettori paratiroidei per la vitamina D (VDR) (4) e, al contempo, ne riduce il numero promuovendo l'iperplasia delle paratiroidi (5).
Ipocalcemia. Le cellule paratiroidee esprimono un recettore per il calcio (calcium-sensing receptor, CaSR), che svolge un ruolo fondamentale nel regolare la trascrizione e le secrezione del PTH e lo sviluppo di iperplasia delle paratiroidi (6). L'ipocalcemia della malattia renale cronica è un potente stimolo alla secrezione di PTH; tale azione può inoltre essere potenziata dalla riduzione del numero dei CaSR presenti nelle paratiroidi ipertrofiche.
Fibroblast Growth Factor 23. La secrezione di FGF-23 da parte degli osteociti e degli osteoblasti è regolata da livelli di calcitriolo, aumento del carico dietetico di fosforo, PTH e calcio. La funzione primaria dell'ormone è il mantenimento della normale concentrazione sierica del fosforo attraverso due meccanismi:
- minore riassorbimento tubulare, effetto mediato dal legame a livello del tubulo prossimale con il recettore specifico (FGFR) e con il co-recettore Klotho;
- minore assorbimento intestinale per ridotta sintesi renale di calcitriolo mediata dalla ipo-espressione dell' enzima 1-alfa-idrossilasi.
Un'altra azione del FGF-23 è la soppressione della sintesi di PTH. I pazienti con IRC hanno elevati livelli di FGF-23 (a causa dell'iperfosforemia e della ridotta clearance renale), che non sono tuttavia in grado di ridurre la sintesi di PTH, probabilmente per la ridotta espressione di FGFR e Klotho.
Resistenza scheletrica al PTH. La resistenza dell'osso all'azione del PTH può contribuire alla patogenesi dell'sPTH. Tale effetto è principalmente dovuto alla riduzione della concentrazione dei recettori per il PTH indotta dagli elevati livelli dell'ormone. Un possibile ruolo è svolto inoltre dal deficit di calcitriolo e dall'iperfosforemia.
Terapia
La terapia dell'sPTH si è sviluppata sulla scorta delle conoscenze dei meccanismi che ne sono alla base. A causa dell’interdipendenza dei vari fattori coinvolti (Ca, P, vitamina D), nessun singolo intervento farmacologico è generalmente sufficiente a ripristinare l'omeostasi del metabolismo calcio-fosforo; il trattamento si fonda pertanto sulla combinazione di più farmaci, in assenza di studi clinici controllati in grado di suggerire un percorso terapeutico applicabile alla maggior parte dei casi.
Dieta ipofosforica. La riduzione dell'apporto alimentare di fosforo a 800-1000 mg/die può ridurre i livelli di PTH (7); è difficile applicare tale regime dietetico senza una dieta a basso tenore proteico.
Chelanti del fosforo. In presenza di una significativa riduzione del filtrato glomerulare, la sola dieta ipofosforica non è in grado di prevenire l'iperfosforemia; pertanto, la maggior parte dei pazienti dovrà assumere farmaci in grado di chelare il fosforo alimentare, impedendone l'assorbimento.
- Sali di alluminio: molto utilizzati in passato per la loro potenza chelante, la tollerabilità e il basso costo; il loro uso, gravato dal rischio di tossicità a carico del sistema nervoso centrale, di osteopatia adinamica e anemia, è attualmente limitato a brevi periodi in caso di iperfosforemia severa (8).
- Chelanti a base di calcio (carbonato e acetato): sono efficaci, generalmente ben tollerati e poco costosi. L'assorbimento di calcio, specialmente se utilizzati a dosi elevate e con contemporanea assunzione di vitamina D, può determinare un bilancio calcico positivo e conseguente ipercalcemia, con rischio di eccessiva soppressione del PTH, osteopatia adinamica e calcificazioni vascolari (9).
- Magnesio carbonato: utilizzato in associazione con il calcio acetato, ha un'efficacia chelante sovrapponibile al sevelamer.
- Sevelamer: è un polimero cationico che lega il fosforo con un meccanismo di scambio ionico ed efficacia sovrapponibile a quella del calcio carbonato.
- Carbonato di lantanio: è un catione metallico facente parte degli elementi rari; ha un'efficacia paragonabile a quella dell'alluminio e superiore a quella del sevelamer e del calcio carbonato.
- Chelanti a base di ferro (ossi-idrossido sucroferrico e citrato ferrico): non ancora in commercio in Italia. Si sono dimostrati efficaci nel controllo dell'iperfosforemia dei pazienti emodializzati. Studi di fase 3 hanno inoltre evidenziato come il citrato ferrico migliori l'assetto marziale, consentendo una riduzione dell'uso di agenti stimolanti l'eritropoiesi.
Al momento nessuno dei chelanti in commercio soddisfa i requisiti di farmaco ideale e i dati a disposizione non mostrano una chiara superiorità di un composto rispetto ad altri; pertanto, la scelta della terapia più appropriata deve tener conto, nel singolo paziente, di una serie di fattori, quali l'età, il bilancio calcico, la presenza o meno di calcificazioni vascolari, la cui progressione può essere accelerata da un eccessivo apporto di calcio.
Vitamina D e analoghi. Il calcitriolo, metabolita attivo naturale della vitamina D, è stato il principale farmaco utilizzato per il controllo dell'sPTH, grazie alla sua efficacia nell'inibire direttamente la sintesi del PTH. Il suo impiego si accompagna peraltro al rischio di ipercalcemia e iperfosforemia, in relazione all'aumentato assorbimento intestinale di calcio e fosforo, con possibile sviluppo di calcificazioni vascolari (10). Sono stati pertanto sviluppati degli attivatori selettivi dei VDR, che, pur mantenendo l'attività inibitoria sulle paratiroidi, hanno minori effetti sul trasporto intestinale di calcio e fosforo. I composti attualmente disponibili per l'uso clinico sono il paricalcitolo (somministrabile per via parenterale o per os), il doxercalciferolo e l'oxacalcitriolo (non disponibili in Europa).
L'alfacalcidolo, 1(OH) vitamina D 25-idrossilata a livello epatico, ha dimostrato una certa efficacia rispetto al placebo nel controllo dell'sPHT (11).
Calciomimetici. Rappresentano un gruppo di farmaci che agiscono come modulatori del CaSR: il legame dei calciomimetici con il recettore ne aumenta la sensibilità al calcio extra-cellulare, con conseguente inibizione della secrezione di PTH. Il cinacalcet è l'unico calciomimetico attualmente in commercio: per il suo meccanismo d'azione è in grado di ridurre i valori di PTH senza aumentare l'assorbimento intestinale di calcio e fosforo.
Paratiroidectomia. L'intervento chirurgico di paratiroidectomia (totale con autotrapianto o subtotale) trova indicazione qualora l'sPTH risulti refrattario alla terapia medica, in genere dopo un tentativo terapeutico che comprenda calcitriolo, analoghi della vitamina D e calciomimetici. I farmaci disponibili hanno reso l'intervento estremamente infrequente.
Prevenzione e terapia dell'iperparatiroidismo secondario nel paziente con IRC non in dialisi
Alterazioni dei parametri di laboratorio (Ca, P, PTH) possono manifestarsi già nello stadio III dell'IRC (GFR compreso tra 30 e 60 mL/min), ma la presenza di valori anomali e la severità delle alterazioni sono estremamente variabili.
Anche modesti incrementi dei valori di PTH sono associati a una significativa perdita ossea nel pazienti con IRC di grado lieve-moderato; è consigliabile pertanto mantenere il PTH all'interno del range di normalità.
Nella tabella sono riportati i parametri suggeriti dalle Linee Guida K-DIGO (8) per i pazienti con IRC non in dialisi.
| Parametri suggeriti dalle Linee Guida K-DIGO (8) per i pazienti con IRC non in dialisi | |
| PTH intatto | Non conosciuto il livello di normalità Prima di ridurre i livelli di PTH, trattare l'iperfosforemia, l'ipocalcemia e/o il deficit di vitamina D Utilizzare gli analoghi della vitamina D se i livelli di PTH rimangono a livelli superiori ai valori di normalità |
| Ca sierico | Mantenere la calcemia nel range di normalità |
| P sierico | Mantenere la fosforemia nel range di normalità |
| Controllo fosforemia | Chelante del fosforo senza calcio se presente ipercalcemia, calcificazioni vascolari, malattia adinamica dell'osso o PTH costantemente ridotto |
Nella pratica clinica:
- il primo passo nel trattamento dell'sPTH è il controllo dell'iperfosforemia con la restrizione dietetica;
- è opportuno correggere con ergocalciferolo o colecalciferolo un eventuale deficit di vitamina D (calcidiolo < 30 ng/mL);
- se dopo due-tre mesi i valori di fosforo rimangono elevati, si introducono i chelanti;
- qualora i valori di PTH si mantengano superiori ai limiti della norma dopo quattro-sei mesi (con valori di calcidiolo > 30 ng/mL), si aggiungono allo schema terapeutico gli analoghi della vitamina D;
- i pazienti che mantengono valori elevati di PTH malgrado la terapia suddetta potrebbero teoricamente essere trattati con il cinacalcet. Attualmente il suo utilizzo è consentito solo nei pazienti in trattamento dialitico, poiché sono pochi i dati presenti in letteratura su efficacia e sicurezza nei pazienti non in dialisi.
Terapia dell'iperparatiroidismo secondario nel paziente in dialisi
L'sPTH rappresenta una complicanza frequente del paziente in dialisi. Circa l'80% dei soggetti con IRC di grado avanzato ha aumentati valori di PTH al momento dell'inizio della terapia sostitutiva, con conseguenze cliniche a carico non solo dello scheletro ma anche dell'apparato cardiovascolare, del sistema nervoso, immunitario, emopoietico ed endocrino(12).
Gli obiettivi terapeutici sono quindi rivolti al mantenimento di valori ottimali di PTH, calcio e fosforo e alla riduzione della morbilità e mortalità cardiovascolare. È necessario evitare in questi pazienti valori eccessivamente alti o bassi di PTH, per prevenire lo sviluppo rispettivamente di osteodistrofia uremica e di osteopatia adinamica. I valori che possono essere suggeriti sono (7): PTH compreso tra 150 e 300 pg/mL, calcemia 8.4-9.5 mg/dL, fosforemia 3.5-5.5 mg/dL.
La gestione ottimale dell'sPTH nei pazienti in dialisi necessita di un trattamento polifarmacologico, con un complesso bilancio tra chelanti del fosforo con o senza calcio, analoghi della vitamina D e calciomimetici. I livelli sierici di PTH, Ca e P devono essere misurati frequentemente: almeno una volta al mese Ca e P, ogni tre mesi PTH; più frequentemente dopo modifiche dello schema terapeutico.
Bibliografia
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes. Kidney Intern 2006, 69: 1945-53.
- Almaden Y, Hernandez A, Torregrosa V, et al. High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid tissue in vitro. J Am Soc Nephrol 1998, 9: 1845-52.
- Saito H, Maede A, Ohtorno S, et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 2005, 280: 2543-9.
- Brumbaugh PF, Hughes MR, Haussler MR. Cytoplasmic and nuclear binding components for 1alpha-dihydroxyvitamin D3 in chick parathyroid glands. Proc Natl Sci USA 1975, 72: 4871-5.
- Denda M, Finch J, Brown AL, et al. 1,25-dihydroxyvitamin D3 and 22-oxacalcitriol prevent the decrease in vitamin D receptor content in the parathyroid glands of uremic rats. Kidney Int 1996, 50: 34-39.
- Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 1998, 139: 4391-6.
- Llach F, Massry SG. On the mechanism of secondary hyperparathyroidism in moderate renal insufficiency. J Clin Endocrinol Metab 1985, 61: 601-6.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention and treatment of Chronic Kidney Disease - Mineral Bone Disorder (CKD-MBD). Kidney Int 2009, 76 (Suppl 113): S1-130.
- Moe SM, Chertow GM. The case against calcium-based phosphate binders. Clin J Am Soc Nephrol 2006, 1: 697-703.
- Cozzolino M, Brancaccio D, Gallieni M, et al. Pathogenesis of vascular calcification in chronic kidney disease. Kidney Int 2005, 68: 429-36.
- Ritzerfeld M, Klasser M, Mann H. Alfacalcidol in the therapy of renal bone disease. Int J Clin Pharmacol Ther 2001, 39: 546-51.
- Bro S, Olgaard K. Effects of excess PTH on nonclassical target organs. Am J Kidney Dis 1997, 30: 606-20.
Scheda paracalcitolo
Andrea Guarnieri
SC Nefrologia e Dialisi - AO S. Croce e Carle - Cuneo
Meccanismo d’azione
Il paracalcitolo è un analogo sintetico, biologicamente attivo, del calcitriolo con modificazioni della catena laterale (D2) e dell’anello A (19-nor).
Il paracalcitolo stimola selettivamente il recettore della vitamina D (VDR) a livello delle ghiandole paratiroidee, non è attivo a livello intestinale e risulta meno attivo sul riassorbimento osseo.
Il paracalcitolo, inoltre, stimola il recettore sensibile al calcio a livello delle ghiandole paratiroidee. Di conseguenza, riduce i livelli di PTH, inibendo la proliferazione delle paratiroidi e diminuendo la sintesi e la secrezione di PTH, con un impatto minimo sui livelli di calcio e fosforo.
Inoltre, il paracalcitolo può agire direttamente sugli osteoblasti per preservare il volume osseo e migliorare le superfici di mineralizzazione.
Il ripristino dei livelli alterati di PTH, unitamente alla normalizzazione dell’omeostasi del calcio e del fosforo, può prevenire o curare la malattia ossea metabolica associata all’insufficienza renale cronica.
Preparazioni, via di somministrazione
- capsule molli 1 µg (Paracalcitolo Teva, Zemplar)
- capsule molli 2 µg (Paracalcitolo Teva, Zemplar)
- fiale da 1 mL contenenti 5 µg/mL (Paracalcitolo Mylan, Paracalcitolo Sandoz, Zemplar)
Le capsule possono essere assunte sia durante che lontano dai pasti.
Indicazioni
Prevenzione e trattamento dell’iperparatiroidismo secondario in pazienti con insufficienza renale cronica (negli stadi 3 e 4) e in pazienti con insufficienza renale cronica allo stadio terminale (stadio 5) sottoposti ad emodialisi o a dialisi peritoneale.
Contro-indicazioni
Comprovata tossicità alla vitamina D, ipercalcemia, o ipersensibilità al paracalcitolo o a qualsiasi degli eccipienti.
Posologia
Insufficienza renale cronica (CKD) stadi 3 e 4: deve essere somministrato o una volta al giorno, oppure tre volte a settimana, a giorni alterni. La dose iniziale deve essere calcolata tenendo conto dei livelli basali di paratormone intatto (iPTH).
| Tabella 1 Dosaggio iniziale (µg) |
||
| Livello basale di PTH intatto | Giornaliero | Da assumere tre volte a settimana (a giorni alterni, senza superare tale frequenza) |
| ≤ 500 pg/mL (56 pmol/L) | 1 | 2 |
| > 500 pg/mL (56 pmol/L) | 2 | 4 |
La dose deve essere poi personalizzata, ossia determinata individualmente in base ai livelli sierici o plasmatici di iPTH, monitorando calcemia e fosforemia sieriche. La tabella 2 propone un esempio di approccio consigliato per la titolazione della dose.
| Tabella 2 Titolazione della dose (aggiustamento a intervalli di 2–4 settimane) |
||
| Livello di PTH intatto | Dose giornaliera | Dose da assumere tre volte a settimana (a giorni alterni, senza superare tale frequenza) |
| Uguale o in aumento o diminuito di < 30% | Aumentare 1 µg | Aumentare 2 µg |
| Diminuito tra 30% e 60% | Lasciare invariato | Lasciare invariato |
| Diminuito di > 60% o iPTH < 60 pg/mL (7 pmol/L) | Diminuire 1 µg* | Diminuire 2 µg* |
*Se è necessaria una riduzione della dose e il paziente sta già assumendo la dose più bassa di farmaco in regime giornaliero o di tre volte a settimana, può essere diminuita la frequenza della dose
Dopo aver iniziato la terapia e nel corso dei periodi di titolazione della dose, si deve procedere a monitorare attentamente i livelli di calcio sierico: se si osserva ipercalcemia o un prodotto calcio-fosforo persistentemente elevato (> 55 mg²/dL² o 4.4 mmol²/L²), qualora il paziente sia sottoposto a terapia con chelanti del fosforo a base di calcio, si deve:
- ridurre la dose o sospendere la somministrazione del chelante
- in alternativa, ridurre o interrompere temporaneamente la somministrazione di paracalcitolo.
Se la terapia viene sospesa, la somministrazione del farmaco deve essere nuovamente iniziata ad una dose più bassa, quando la calcemia ed il prodotto calcio-fosforo si saranno normalizzati.
CKD stadio 5: deve essere somministrato tre volte a settimana, a giorni alterni.
La dose iniziale deve essere calcolata in base ai livelli basali di paratormone intatto (fino ad una dose massima iniziale di 32 µg):
- µg paracalcitolo = iPTH (pg/mL)/60 = iPTH (pmol/L)/7
La dose dovrebbe essere poi personalizzata, ossia determinata individualmente, sulla base dei livelli sierici di iPTH, calcio e fosforo. Una titolazione della dose di paracalcitolo in capsule consigliata si basa sulla formula seguente:
- µg paracalcitolo = livello più recente di iPTH (pg/mL)/60 = livello più recente di iPTH (pmol/L)/7
Dopo aver iniziato la terapia, durante il periodo di titolazione della dose ed in concomitanza con la somministrazione di potenti inibitori del P450-3A, si deve procedere ad un attento monitoraggio dei livelli di calcio e fosforo. Se si nota la presenza di ipercalcemia o di un elevato prodotto calcio–fosforo e se il paziente è sottoposto a terapia con chelanti del fosforo a base di calcio, si deve procedere ad una riduzione della loro dose o se ne deve sospendere la somministrazione. In alternativa, il paziente può passare ad un chelante del fosforo non a base di calcio.
Se la calcemia risulta > 11.0 mg/dL (2.8 mmol/L) o il prodotto Ca x P > 70 mg²/dL² (5.6 mmol²/L²), o iPTH ≤ 150 pg/mL, la dose deve essere diminuita di 2-4 µg rispetto a quella calcolata in base al livello più recente di iPTH/60 (pg/mL) [iPTH/7 (pmol/L)]. Nel caso in cui si renda necessario un ulteriore aggiustamento della dose, la somministrazione del paracalcitolo in capsule deve essere ridotta o interrotta fino a quando tali parametri non si saranno normalizzati.
A mano a mano che il livello di iPTH si avvicina all’intervallo dei valori di riferimento (150-300 pg/mL), possono essere necessari piccoli aggiustamenti individualizzati della dose per raggiungere un livello di iPTH stabile. Se si presentano situazioni in cui il monitoraggio dei livelli di iPTH, calcio o fosforo può essere effettuato meno frequentemente di una volta a settimana, può essere utilizzato un rapporto più modesto dose iniziale/titolazione della dose.
Effetti collaterali
Non sono state riscontrate differenze statisticamente significative tra i pazienti trattati con il paracalcitolo ed i pazienti trattati con placebo in termini di incidenza di ipercalcemia o di prodotto calcio-fosforo elevato.
La reazione avversa più comune è rappresentata dall’eruzione cutanea, che si è verificata nel 2% dei pazienti.
Tutti gli eventi avversi almeno possibilmente correlati al paracalcitolo sono riportati nelle tabelle 3 e 4 per tipo e frequenza. Sono state utilizzate le seguenti categorie di frequenza:
- molto comune: ≥ 1/10;
- comune: ≥1/100 a <1/10;
- non comune: ≥1/1000 a <1/100;
- raro: ≥ 1/10.000 a <1/1000;
- molto raro: <1/10.000);
- non noto: non può essere calcolata in base ai dati disponibili.
| Tabella 3 Reazioni avverse riportate negli studi clinici CKD stadi 3 e 4 |
|
| Reazione | Frequenza |
| Enzimi epatici anormali | Non comune |
| Capogiri | Non comune |
| Disgeusia | Non comune |
| Imbarazzo intestinale | Comune |
| Costipazione | Non comune |
| Bocca secca | Non comune |
| Eruzione cutanea | Comune |
| Prurito | Non comune |
| Orticaria | Non comune |
| Spasmi muscolari | Non comune |
| Ipersensibilità | Non comune |
| Tabella 4 Reazioni avverse segnalate in uno studio pilota di fase III CKD stadio 5 |
|
| Reazione | Frequenza |
| Capogiri | Comune |
| Diarrea | Comune |
| Disturbi da reflusso gastro-esofageo | Comune |
| Acne | Comune |
| Ipercalcemia | Comune |
| Ipocalcemia | Comune |
| Diminuzione dell’appetito | Comune |
| Sensibilità mammaria | Comune |
Limitazioni prescrittive
Prescrivibile con piano terapeutico
Scheda chelanti del fosforo
Andrea Guarnieri
SC Nefrologia e Dialisi - AO S. Croce e Carle - Cuneo
SEVELAMER CARBONATO
Meccanismo d’azione
Chelante del fosforo alimentare.
Preparazioni, via di somministrazione, posologia
- cp 800 mg (Renvela, Sevelamer Aurobindo, Sevelamer DOC, Sevelamer EG, Sevelamer Mylan, Sevelamer Sandoz)
- bustine 0.8 g (Renvela)
- bustine 2.4 g (Renvela, Sevelamer Aurobindo, Sevelamer DOC, Sevelamer EG, Sevelamer Sandoz)
Assumere tre volte al giorno, con i pasti.
La polvere deve essere dispersa in 60 mL di acqua per ogni bustina, prima della somministrazione; la sospensione deve essere ingerita entro 30 minuti dalla preparazione.
La dose iniziale raccomandata per sevelamer carbonato è 2.4 g o 4.8 g al giorno, sulla base delle esigenze cliniche e dei livelli di fosforo. Titolazione e mantenimento: si devono monitorare i livelli di fosforemia titolando la dose di sevelamer carbonato ogni 2-4 settimane fino a raggiungere un livello accettabile di fosforo sierico, con successivo regolare monitoraggio.
Nella pratica clinica, il trattamento sarà continuo, in base alle esigenze di controllare i livelli di fosforemia; la dose prevista sarà in media circa 6 g al giorno. Non è raccomandato nei < 18 anni.
Indicazioni
Controllo dell'iperfosforemia in pazienti adulti:
- sottoposti ad emodialisi o a dialisi peritoneale
- con patologia renale cronica non sottoposti a dialisi con fosforo sierico ≥ 5.5 mg/dL (1.78 mmol/L).
Utilizzare nel contesto di un approccio politerapeutico, che potrebbe includere integratori di calcio, calcitriolo o uno dei suoi analoghi per controllare lo sviluppo della malattia ossea renale.
Contro-indicazioni
Ipersensibilità al sevelamer o ad uno qualsiasi degli eccipienti.
Ipofosforemia.
Occlusione intestinale.
Effetti collaterali
Molto comuni: nausea, vomito.
Comuni: diarrea, dispepsia, flatulenza, dolore all'addome superiore, stitichezza.
Durante l'uso del medicinale successivamente all'approvazione sono stati riportati casi di prurito, rash, dolore addominale.
Durante il trattamento con sevelamer cloridrato, che contiene la stessa frazione attiva di sevelamer carbonato, in casi molto rari sono stati osservati occlusione intestinale e ileo/subileo, diverticolite e perforazione intestinale. La stipsi può rappresentare un prodromo. I pazienti che soffrono di stipsi devono essere monitorati attentamente. Il trattamento deve essere rivalutato in caso di pazienti che sviluppano grave stipsi o altri sintomi gastrointestinali gravi.
I pazienti affetti da IRC possono manifestare una carenza di vitamine liposolubili A, D, E e K, a seconda della dieta e della gravità della patologia; non è possibile escludere che il medicinale possa legarsi alle vitamine liposolubili contenute negli alimenti ingeriti. Attualmente vi sono dati insufficienti per escludere la possibilità di carenza di folato durante il trattamento a lungo termine.
I pazienti con IRC possono sviluppare ipocalcemia o ipercalcemia. Il medicinale non contiene calcio: monitorare a intervalli regolari i livelli di calcemia e somministrare un integratore di calcio elementare, se necessario.
I pazienti con patologia renale cronica sono predisposti all'acidosi metabolica: nell'ambito della buona pratica clinica, si raccomanda pertanto il monitoraggio dei livelli di bicarbonato sierico.
La peritonite è una complicanza nota nei pazienti sottoposti a dialisi peritoneale e in uno studio clinico su sevelamer cloridrato è stato riferito un maggior numero di casi di peritonite nel gruppo di sevelamer rispetto al gruppo di controllo.
Da uno studio clinico della durata di un anno non è emersa evidenza di accumulo di sevelamer, tuttavia non è possibile escludere del tutto il potenziale assorbimento e accumulo di sevelamer nel trattamento cronico a lungo termine.
Limitazioni prescrittive
Prescrivibile con piano terapeutico.
CARBONATO DI LANTANIO
Meccanismo d’azione
Chelante del fosforo alimentare.
Preparazioni, via di somministrazione, posologia
Compresse masticabili contenti carbonato idrato di lantanio equivalente a 500 mg, 750 mg e 1000 mg di lantanio o bustine da 750 mg e 1000 mg (Foznol).
Deve essere assunto con i pasti o immediatamente dopo; le compresse devoso essere masticate e non ingerite intere.
È necessario controllare i livelli sierici di fosforo e aggiustare la dose ogni 2-3 settimane fino ad ottenere livelli di fosforo accettabili, effettuando successivamente un monitoraggio regolare.
Dosi iniziali di 750 mg/die riescono generalmente a controllare i livelli sierici di fosforo. La dose massima studiata nelle sperimentazioni cliniche, in un numero limitato di pazienti, è di 3.750 mg. I pazienti che rispondono alla terapia normalmente raggiungono livelli sierici di fosforo accettabili con 1500–3000 mg/die.
Indicazioni
Controllo dell'iperfosforemia in pazienti adulti:
- affetti da insufficienza renale cronica emodializzati o in dialisi peritoneale ambulatoriale continua
- con nefropatia cronica non in dialisi, con livelli di fosforemia ≥ 5.5 mg/dL (1.78 mmol/L) nei quali una dieta a basso contenuto di fosfati non sia sufficiente a tenere sotto controllo i livelli sierici del fosforo.
Contro-indicazioni
Ipersensibilità al carbonato idrato di lantanio o ad uno qualsiasi degli eccipienti.
Ipofosfatemia.
Effetti collaterali
Le reazioni avverse più comunemente riportate, ad eccezione dell'ipocalcemia, sono di natura gastrointestinale, minimizzate dall'assunzione del prodotto con il cibo e che generalmente scompaiono col tempo in seguito a somministrazione continua.
Comuni: dolore addominale, stipsi, diarrea, dispepsia, flatulenza, nausea, vomito.
Non comuni: ipocalcemia, ipercalcemia, iperglicemia, iperfosforemia, ipofosforemia, anoressia, aumento dell'appetito, capogiri, cefalea, alterazione del gusto, vertigini, eruttazione, indigestione, sindrome dell'intestino irritabile, secchezza delle fauci, esofagite, stomatite, feci molli, disturbi odontoiatrici, alopecia, pizzicore, prurito, eruzioni eritematose, aumento della sudorazione, artralgia, mialgia, osteoporosi, astenia, dolore toracico, sensazione di stanchezza, senso di malessere, edema periferico, dolore, sete, perdita di peso; aumento dei livelli ematici di alluminio, gamma-GT, transaminasi, fosfatasi alcalina.
Sono state osservate modificazioni transitorie del QT, non associate pero' ad un aumento degli eventi avversi cardiaci.
Limitazioni prescrittive
Prescrivibile con piano terapeutico.
Forme di iperparatiroidismo secondarie a ipovitaminosi D
Giusi Beretta Anguissola1 & Costanza Santini2
1Area di Endocrinologia e Diabetologia, Università Campus BioMedico, Roma
2UOS Endocrinologia e Diabetologia, Ospedale Bufalini, Cesena
L’insufficienza di vitamina D e la conseguente riduzione dell’assorbimento di calcio a livello intestinale determinano un aumento dei livelli sierici di PTH. Tale condizione, definita iperparatiroidismo secondario a ipovitaminosi D, rappresenta una condizione molto frequente ed assume rilevanza clinica per il suo impatto sul sistema muscolo-scheletrico.
Definizione di ipovitaminosi D
Anche se non vi è un consenso uniforme riguardo il livello plasmatico ottimale di 25-OH vitamina D, il deficit di vitamina D è generalmente definito come un valore inferiore a 20 ng/mL (50 nmol/L). Valori compresi tra 21-29 ng/mL sono considerati indice di insufficienza e livelli pari o superiori a 30 ng/mL come normali. Questi cut-off provengono dall'osservazione che l’assorbimento intestinale di calcio aumenta in modo significativo quando si ha un incremento di vitamina D da 20 a 32 ng/mL (80 nmol/L) e che i valori di 25-OH vitamina D sono inversamente correlati al valore di PTH. Il PTH comincia ad aumentare quando la vitamina D risulta inferiore a 30-40 ng/mL (75-100 nmol/L) mentre rimane stabile per valori di vitamina D superiori a tali valori.
Epidemiologia
La carenza di vitamina D è molto frequente in Italia, in cui interessa il 50% dei giovani e la quasi totalità degli anziani durante il periodo invernale (86% delle donne di età > 70 anni). La prevalenza è maggiore in Europa rispetto ad Asia, Australia ed USA; in Europa correla con la latitudine (maggiore nel Sud–Italia, in Spagna e Grecia), in conseguenza delle caratteristiche della dieta a minore contenuto di grassi animali, al non uso di integratori multivitaminici o di alimenti addizionati, alla minor esposizione alla luce solare (anche per uso di creme ad alto fattore di protezione) e alla pelle più scura. Negli immigrati (africani, asiatici), fattori specifici di rischio sono l'abbigliamento e la pigmentazione cutanea. Altre condizioni di rischio sono l'obesità, per sequestro della vitamina liposolubile nel tessuto adiposo, e le condizioni di malassorbimento. Lo screening dell'ipovitaminosi D (mediante il dosaggio della 25OH-vitamina D sierica) è raccomandato nei soggetti a rischio per condizioni cliniche (tabella). Nei soggetti sani, lo screening è giustificato nella fascia di età 60-70 anni, non è necessario per età > 70 anni in cui la carenza vitaminica è vicina al 100%, non è ragionevole per età < 60 anni qualora lo stile di vita preveda una normale esposizione solare.
| Condizioni cliniche che interferiscono con il metabolismo della vitamina D (modificata da 7, 8) |
||
| Diminuita biodisponibilità | Malassorbimento dei grassi |
fibrosi cistica |
| Ridotta biodisponibilità | obesità | |
| Aumentato catabolismo/consumo | farmaci gravidanza allattamento |
|
| Diminuita sintesi di 25(OH)D | insufficienza epatica grave | |
| Perdite urinarie di 25(OH)D | sindrome nefrosica | |
| Diminuita sintesi di 1,25(OH)2-D | insufficienza renale cronica iperfosforemia deficit congeniti 1α-idrossilasi sarcoidosi TBC infezioni fungine croniche linfomi |
|
Patogenesi
Il deficit di vitamina D, evidenziato a livello ematico da una ridotta concentrazione di 25(OH)vitamina D, determina una riduzione della 1,25(OH)2 vitamina D, ossia della sua forma attiva, con conseguente riduzione dell’assorbimento di calcio. Infatti, in assenza di vitamina D solo il 10-15% del calcio introdotto con la dieta viene assorbito. L'interazione della 1,25(OH)2 vitamina D con il suo recettore intestinale determina un aumento dell’assorbimento intestinale di calcio del 30-40%. Quando i livelli di 25(OH)-vitamina D scendono al disotto dei 30 ng/mL l’assorbimento intestinale di calcio si riduce.
Il basso livello di calcemia stimola la secrezione di PTH, che aumenta il riassorbimento tubulare di calcio e stimola il rene a produrre 1,25(OH)2 vitamina D. Attraverso questo meccanismo la 1,25(OH)2 vitamina D viene mantenuta a un livello quasi normale, a spese di un aumento di PTH, e, se il deficit di vitamina D persiste, si determina il quadro dell’iperparatiroidismo secondario. Questo implica che, rispetto alla concentrazione di calcio, il valore di PTH risulti relativamente alto.
La regolazione della sintesi di PTH avviene a livello di trascrizione genica. All’interno del promoter del gene del PTH è presente una regione che lega il recettore della vitamina D (VDR), per cui la 1,25(OH)2 vitamina D ha un'azione regolatoria in senso inibitorio del promoter del PTH. La capacità della 1,25(OH)2 vitamina D di regolare l’espressione del gene del PTH è modulata dalla calcemia. Inoltre, il calcio stesso è un potente inibitore della produzione e secrezione del PTH, agendo presumibilmente attraverso il recettore del calcio sito sulla membrana plasmatica della cellula paratiroidea. La capacità della 1,25(OH)2 vitamina D di inibire la sintesi e secrezione di PTH è la base dell’utilizzo in clinica della vitamina D per il trattamento dell’iperparatiroidismo secondario associato ad insufficienza renale.
Il valore di PTH può risultare normale per valori di vitamina D inferiori a 30 ng/mL a causa della concomitante ipomagnesemia, che bilancia la risposta di ipersecrezione di PTH. Il PTH inoltre accelera il metabolismo della 25OH-vitamina D a 1,25(OH)2 vitamina D e questo contribuisce a peggiorare il deficit di vitamina D. Il PTH stimola la fosfaturia con conseguenti livelli di fosforo normali o bassi.
Come conseguenza della variazione stagionale della 25(OH)vitamina D, legata essenzialmente all’esposizione solare, il deficit è più evidente alla fine dei mesi invernali, mentre il PTH sierico mostra una variabilità stagionale inversa.
Quadro clinico
Il quadro clinico di questa forma di iperparatiroidismo secondario si sovrappone a quello proprio del deficit di vitamina D. La carenza di vitamina D nel bambino determina un rallentamento nella crescita e il quadro clinico del rachitismo, nell’adulto favorisce osteopenia e osteoporosi aumentando il rischio di frattura. Da un punto di vista istopatologico, si osserva una alterata mineralizzazione dell’osteoide che determina, nelle situazioni di ipovitaminosi più marcate e prolungate nel tempo, il quadro dell'osteomalacia. Poiché il recettore della vitamina D è presente anche a livello del muscolo scheletrico, il deficit di vitamina D è associato a debolezza dei muscoli prossimali, dolorabilità muscolare e aumentato rischio di caduta.
È importante sottolineare come la presenza di elevati livelli di PTH abbia un ruolo chiave nelle manifestazioni muscolo-scheletriche proprie di tale condizione.
Il PTH determina un'attivazione dei pre-osteoclasti in osteoclasti maturi, che distruggono la matrice mineralizzata dell’osso, causando osteopenia e osteoporosi e aumentando il rischio di frattura.
L’aumento di PTH sierico è responsabile di un aumento del turn-over osseo, che è solitamente associato con perdita di tessuto osseo soprattutto a livello corticale. Come è noto dalle osservazioni sull’iperparatiroidismo primitivo, l’osso trabecolare è relativamente conservato, così che la densità minerale ossea è generalmente normale a livello della colonna lombare, mentre è ridotta a livello del radio ultra-distale e del femore prossimale, dove la corticale è maggiormente rappresentata.
L’iperparatirodismo secondario a deficit di vitamina D è molto comune negli anziani ed è spesso associato alle fratture dell’anca dell’anziano.
Accanto all’aumento dei livelli sierici di PTH, nei pazienti con deficit di vitamina D si osserva un aumento degli altri marcatori biochimici del turn-over osseo, quali fosfatasi alcalina, osteocalcina e idrossiprolina urinaria, ulteriori indici di aumento del turn-over osseo e riduzione della massa ossea.
Trattamento
Il trattamento dell’iperparatiroidismo secondario ad ipovitaminosi D consiste nella somministrazione di vitamina D, fino ad ottenerne livelli plasmatici normali, con conseguente normalizzazione dei valori di PTH.
Bibliografia
- Holick MF. Vitamin D deficiency. N Engl J Med 2007, 357: 266-81.
- Holick MF, Thai Chen C. Vitamin D deficiency: a worldwide problem with health consequences. Amer J Clin Nutr 2008, 87: 1080S-6S.
- Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implication. Endocr Rev 2001, 22: 477-501.
- Adams JS, Hewlson M. Update in vitamin D. J Clin Endocrinol Metab 2010, 95: 471-8.
- Holick MF, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 1911-30.
- Heaney RP, Holick MF. Why the IOM recommendations for vitamin D are deficient. J Bone Miner Res 2011, 26: 455-7.
- Adami S, Romagnoli E, et al. Linee guida su prevenzione e trattamento dell'ipovitaminosi con colecalciferolo. Reumatismo 2011, 63: 129-47.
Forme di iperparatiroidismo secondario iatrogene
Costanza Santini
UOS Endocrinologia e Diabetologia, Ospedale Bufalini, Cesena
| Classificazione patogenetica delle cause iatrogene di iperparatiroidismo secondario (s-HPT) | ||
| Riduzione dei livelli di vitamina D | Interferenza con l'assorbimento | lassativi a base oleosa e lipidica orlistat colestiramina corticosteroidi |
| Interferenza con il metabolismo | anti-epilettici (fenilidantoina, carbamazepina, barbiturici) rifamipicina cimetidina diuretici tiazidici litio anti-retrovirali |
|
| Riduzione della calcemia | aminoglicosidi anti-convulsivanti (bambino, anziano) inibitori della pompa protonica bisfosfonati chelanti del calcio (citrati) cisplatino chetoconazolo gallio |
|
| Aumento dell'escrezione urinaria di calcio |
litio |
|
| Riduzione della fosfatemia | antiacidi alchilanti il fosforo chemioterapici (cisplatino, bevacizumab, irinotecano, everolimus, imatinib) corticosteroidi diuretici dell'ansa bicarbonati plasma expanders |
|
| Aumento dell'escrezione urinaria di fosfati | corticosteroidi preparati contenenti fosfati |
|
| Altre cause | Malassorbimento | chirurgia bariatrica (in particolare by-pass digiuno-ileale) estese resezioni intestinali e pancreatiche |
| Deficit nutrizionali | alimentazione parenterale digiuno prolungato |
|
| Altre condizioni | trasfusioni ripetute (uso chelanti del calcio) trapianto renale |
|
Si riportano gli effetti dei farmaci di più frequente utilizzo.
Litio: prevalenza elevata 4.3-6.3% (1-4/1000 nella popolazione generale), F/M = 4/1. E' inoltre riportata una aumentata incidenza di iperparatiroidismo primario (p-HPT) nei pazienti in trattamento cronico con sali di litio. Gli adenomi multipli o l'iperplasia sono più frequenti dell'adenoma singolo rispetto al p-HPT sporadico (18-83% vs 5-10%). Non è chiaro se il litio determini la patologia o la slatentizzi: il farmaco sembrerebbe innalzare il set-point inibitorio del calcio sulla secrezione del PTH, agendo sul recettore CaSR. L'aumento di calcemia e PTH si associa ad ipocalciuria, più raramente a ipercalciuria, complicata da nefrolitiasi e osteopenia (in tal caso il p-HPT è verosimilmente pre-esistente). Le alterazioni possono comparire in tempi brevi, entro 1-2 mesi dall'inizio dell'esposizione; è consigliabile controllare calcemia e PTH prima e durante il trattamento con litio. Sia l'incidenza sia la persistenza del p-HPT dopo l'interruzione del litio correlano con la durata di terapia.
Inibitori della pompa protonica: la ionizzazione del calcio preliminare all'assorbimento viene ridotta dall'uso di PPI e sembra associarsi sia ad aumento adattativo del PTH sia ad aumento del rischio fratturativo. Non sono tuttavia disponibili studi a lungo termine.
Diuretici: i diuretici dell'ansa determinano aumento rilevante della calciuria e dei livelli di PTH, mentre i tiazidici hanno effetti modesti; è stato descritto un aumento del rischio di perdita ossea e di frattura con i primi ed un effetto protettivo con i secondi.
Relativamente al trapianto renale, i disordini del metabolismo minerale osseo possono persistere a causa di vari fattori: s-HPT, immobilizzazione prolungata, funzionalità del rene trapiantato, farmaci immuno-soppressori. L'ipercalcemia tende a migliorare fino a normalizzarsi entro un anno; nella forme autonomizzate, l’involuzione dell’iperplasia paratiroidea può richiedere tempi prolungati.
Bibliografia
- Szalat A, Mazeh H, Freund HR. Lithium-associated hyperparathyroidism: report of four cases and review of the literature. Eur J Endocrinol 2009, 160: 317-23.
- Laria A, Zoli A, et al. Proton pump inihibitors in rheumatic disease. Reumatismo 2011, 63: 5-10.
- Mc Knight R, et al. Lithium toxicity profile: a systematic review and metanalysis. Lancet 2012, 379: 721-8.