Rachitismo/osteomalacia: sistematica delle diverse forme

Gregorio Guabello
Ambulatorio di Patologia Osteo-Metabolica, UO Reumatologia, IRCCS Istituto Ortopedico Galeazzi; Ambulatorio di Endocrinologia Oncologica, IRCCS Ospedale San Raffaele; Milano
(aggiornato al 2/9/2020)
1. RACHITISMO/OSTEOMALACIA VITAMINA D-DIPENDENTE: FORMA CALCIOPENICA CON IPERPARATIROIDISMO SECONDARIO
La tabella 1 riporta la classificazione ezio-patogenetica e le caratteristiche biochimiche di questa forma di rachitismo.
| Tabella 1 Rachitismo calciopenico con iperparatiroidismo secondario |
|||||||||||
| Tipo | Patogenesi | Gene | Ca | P | ALP | CaU | TmP/GFR | FGF-23 | PTH | 25OHD3 | 1,25OHD3 |
| Nutrizionale | Deficit di vitamina D | - | N/B | N/B | A | B | B | N | A | B | V |
| Tipo 1A | Difetto 1-alfa-idrossilasi | CYP27B1 | N/B | N/B | A | B | B | N | A | N | B |
| Tipo 1B | Difetto 25-idrossilasi | CYP2R1 | N/B | N/B | A | B | B | N | A | B | B |
| Tipo 2A | Difetto VDR | VDR | N/B | N/B | A | B | B | N | A | N | A |
| Tipo 2B | Difetto signalling VDR | HNRNPC | N/B | N/B | A | B | B | N | A | N | A |
| Tipo 3 | Aumento catabolismo 25OHD3 | CYP3A4 | B | B | A | B | B | N | A | B | B |
| A=alto, B=basso, N=normale, V=variabile | |||||||||||
1.1. Rachitismo nutrizionale (1)
La prevenzione del rachitismo infantile si basa sulla supplementazione con vitamina D a partire dall’età neonatale. Negli ultimi anni si è posta molta attenzione all’alimentazione del lattante con latte materno, carente di vitamina D, e pertanto possibile causa di deficit. È opportuna in tal caso un'adeguata integrazione.
Il fabbisogno quotidiano è insufficiente in condizioni di ridotta esposizione ai raggi solari, di deficit nutrizionale (assoluto o relativo) o nelle sindromi da malassorbimento (celiachia, fibrosi cistica). Gli anziani sono particolarmente a rischio di sviluppare carenza di vitamina D. Con l’avanzare degli anni si ha, infatti, sia ridotto assorbimento intestinale età-dipendente sia riduzione della sintesi cutanea (calcolata riduzione del 75% a 70 anni). Le creme solari ad alto fattore di protezione riducono la penetrazione cutanea delle radiazioni ultraviolette, potendo quindi causare deficit di vitamina D.
Il trattamento di scelta del rachitismo prevede l’uso della vitamina D2 o D3.
Dosaggi nel bambino:
- < 1 mese: 1.000 UI/die;
- da 1 a 12 mesi: da 1.000 a 5.000 UI/die;
- > 1 anno: da 5.000 a 10.000 UI/die.
È necessario inoltre somministrare supplementi di calcio, alla dose di 30-75 mg/kg/die, per evitare la “sindrome dell’osso affamato”.
Proseguire il trattamento fino all’evidenza della risoluzione del quadro radiologico (da verificare dopo 3 mesi), poi ridurre a 400 UI/die la posologia. Sospendere la supplementazione di calcio alla ricomparsa di calcio nelle urine.
Rachitismo causato da insufficiente apporto di calcio, con vitamina D nella norma: è efficacemente corretto dalla somministrazione di 1000 mg/die di calcio per os.
Nell’osteomalacia uno schema comunemente utilizzato per il trattamento della carenza nutrizionale di vitamina D è il seguente: 50.000 UI di vitamina D2 (ergocalciferolo) o D3 (colecalciferolo) per os 1 volta alla settimana per 6-8 settimane; successivamente 800 UI/die di vitamina D3.
1.2. Rachitismo tipo 1A (1)
Epidemiologia
La prevalenza alla nascita è stimata in circa 1/2000.
Clinica
La malattia esordisce nel primo anno di vita con ipotonia, tetania, convulsioni, debolezza muscolare e scarsa crescita. I pazienti sviluppano progressivamente deformità da rachitismo (gambe incurvate, rosario rachitico). Raramente è presente ipoplasia dello smalto dentario.
Eziologia
La malattia, a trasmissione autosomica recessiva, è causata dalle mutazioni inattivanti del gene CYP27B1, che codifica per la 1-alfa-idrossilasi che converte il calcifediolo in calcitriolo. Questo difetto della sintesi della vitamina D provoca ridotto assorbimento intestinale di calcio e fosfati.
Diagnosi
Si basa sui segni biochimici e radiologici.
I segni radiologici classici sono le anomalie scheletriche a livello dei piatti di crescita e delle ossa metafisarie, l'osteomalacia e l'osteoporosi.
I segni biochimici comprendono ipocalcemia grave e ipofosfatemia moderata. Altre caratteristiche biochimiche comprendono la normalità dei livelli sierici di calcifediolo (25-OHD), associati a livelli sierici bassi di calcitriolo (1,25-OHD), aminoaciduria e concentrazioni elevate di PTH. La biopsia ossea può rivelare osteomalacia. La diagnosi è confermata dalle analisi molecolari.
Trattamento
L'obiettivo è migliorare la crescita, ripristinare i normali livelli sierici di calcio, fosforo, fosfatasi alcalina (ALP) e PTH e prevenire le deformità scheletriche. Consiste nella somministrazione giornaliera di forti dosi di vitamina D e di dosi fisiologiche di calcitriolo. Sono state descritte come complicazioni del trattamento nefrocalcinosi, ipercalciuria e ipercalcemia. È perciò necessario programmare un monitoraggio regolare (esame clinico e biochimico, radiografie delle mani, ecografia renale).
Prognosi
In presenza di trattamento, la prognosi è buona.
1.3. Rachitismo tipo 1B (1)
Si tratta di una rara forma di rachitismo dovuta a mutazione inattivante la 25-idrossilasi epatica. I livelli sierici di 25OHD3 sono molto bassi o addirittura non dosabili. La presentazione clinica è simile alla forma da carenza di vitamina D 1A.
Nei pazienti affetti da grave insufficienza epatica, in cui c’è un deficit acquisito della 25OHD3 è indicato l’uso del calcidiolo, che non richiede l’attivazione a livello del fegato. Il dosaggio è pari a 50-200 µg/die. In questi casi si può anche usare il calcitriolo, alla dose di 0.25-1 µg/die.
1.4. Rachitismo tipo 2 (resistenza ereditaria alla 1,25OHD3) (1)
Epidemiologia
La prevalenza è sconosciuta.
Clinica
Durante i primi anni di vita si manifesta progressivo rachitismo, con ritardo di crescita e deformità scheletriche. Nelle forme gravi della malattia è presente alopecia generalizzata. In alcuni casi sono presenti cisti epidermiche. Esiste una stretta correlazione fra fenotipo e genotipo.
Eziologia
La patologia dipende da una mutazione inattivante il recettore della vitamina D (VDR), con conseguente ridotto assorbimento intestinale di calcio e fosforo. La trasmissione è autosomica recessiva.
Diagnosi
La diagnosi si basa sulle caratteristiche cliniche, biochimiche e radiologiche.
Sono presenti severa ipocalcemia, ipofosforemia, iperparatiroidismo secondario, normali valori di 25OHD3, elevati valori di calcitriolo e ALP e aminoaciduria.
I radiogrammi documentano stigmate di rachitismo (deformità scheletriche che interessano le cartilagini di accrescimento a livello metafisario, trabecolizzazione delle ossa lunghe, deformità vertebrali). È comune l’osteite fibroso-cistica.
Terapia
Scopo del trattamento è la promozione della crescita, la correzione dell’ipocalcemia e dell’iperparatroidismo e la mineralizzazione della matrice osteoide. Consiste nella somministrazione quotidiana di alte dosi di calcitriolo e calcio (può rendersi necessaria la supplementazione endovena). La risposta al trattamento dipende in larga misura dalla gravità della patologia. La forma con alopecia in genere non risponde alla terapia orale e necessita di infusione di calcio endovena. È necessario il monitoraggio periodico dei parametri biochimici.
Prognosi
Dipende dalla severità della patologia e dalla possibile correzione chirurgica delle deformità scheletriche.
1.5. Rachitismo tipo 3 (1)
Farmaci, come alcuni anti-convulsivanti (fenitoina, fenobarbital, carbamazepina) e anti-microbici (rifampicina), possono determinare deficit di vitamina D attraverso l'attivazione a livello epatico di alcuni enzimi appartenenti alla famiglia del citocromo P450, che ne provocano il catabolismo. Un meccanismo simile si verifica anche nell’ipertiroidismo.
2. RACHITISMO/OSTEOMALACIA VITAMINA D-RESISTENTE: FORMA FOSFOPENICA
2.1. Rachitismo/osteomalacia da ridotto introito, ridotta biodisponibilità, malassorbimento intestinale
Patogenesi
Allattamento al seno di bambini con basso peso alla nascita.
Uso di formula ipoallergenica o elementare per l’allattamento artificiale.
Nutrizione parenterale.
Uso eccessivo di chelanti del fosforo.
Sindromi da malassorbimento.
La tabella 2 riporta le caratteristiche biochimiche di questa forma di rachitismo (1).
| Tabella 2 Rachitismo fosfopenico |
||||||||
| Ca | P | ALP | CaU | TmP/GFR | FGF-23 | PTH | 25OHD3 | 1,25OHD3 |
| N | B | A | N | A | N | N | N | N |
| A=alto, B=basso, N=normale | ||||||||
2.2. Rachitismo/osteomalacia da perdita renale di fosforo mediata da FGF-23
La tabella 3 riporta le caratteristiche biochimiche delle forme appartenenti a questa tipologia di rachitismo.
| Tabella 3 Rachitismo da perdita renale di fosforo mediata da FGF-23 |
||||||
| Ca | P | CaU | TmP/GFR | PTH | 25OHD3 | 1,25OHD3 |
| N | B | N/B | B | N/A | N | N/B |
| A=alto, B=basso, N=normale | ||||||
2.2.1. Ipofosforemia X-linked (dominante) XLH (2)
Eziologia
Dipende da una mutazione inattivante del gene codificante la endopeptidasi PHEX (PHosphate regulating gene with homology to Endopeptidases on the X chromosome), enzima presente negli osteociti e negli odontoblasti deputato alla fisiologica degradazione di FGF-23, con conseguente aumento dei livelli plasmatici dell’ormone. Sono note ad oggi 170 mutazioni (missenso, non-senso, delezioni, mutazioni del sito di splicing) e sono descritti anche casi sporadici.
Epidemiologia
Si tratta di un disordine raro, ma fra le forme genetiche è il più frequente, con prevalenza stimata di 1:20000.
Clinica
La XLH determina rachitismo nel bambino e osteomalacia nell’adulto. Le manifestazioni cliniche sono numerose e variabili, alcune strettamente legate alle azioni extra-scheletriche di FGF-23. Possono essere così schematizzate a seconda della sede anatomica interessata:
- cranio: cranio-sinostosi (per azione di FGF-23 sulle suture craniali), malformazione di Chiari, ipoacusia neuro-sensoriale eventualmente associata a sindrome vertiginosa e tinnitus (per osteosclerosi ed ispessimento dell’osso petroso con restringimento del meato uditivo interno);
- denti: ascessi spontanei e perdita di denti (per scarsa mineralizzazione della dentina e del cemento e anomalie dello smalto, che favoriscono l’ingresso di micro-organismi nella cavità pulpare, con conseguente formazione di ascessi);
- scheletro: curvatura degli arti inferiori (genu varum, genu valgus), bassa statura, deambulazione scorretta, dolore osseo agli arti inferiori, osteomalacia;
- muscolo: ridotta forza muscolare agli arti inferiori, mialgia;
- articolazioni: artrosi, artralgie arti inferiori (per inibizione da parte di FGF-23 della proliferazione condrocitaria);
- tendini: entesopatia (per azione di FGF-23 sulle entesi, con conseguenti calcificazioni).
Diagnosi
In genere la diagnosi viene fatta entro i primi 2 anni di vita, tuttavia lo spettro clinico ampiamente variabile della patologia rende ragione di casi diagnosticati anche in età adulta.
Laboratorio
Le alterazioni dei parametri biochimici del metabolismo fosfo-calcico, per le note azioni di FGF-23 a livello renale (effetto fosfaturico + inibizione della 1alfa-idrossilasi) sono le seguenti: ipofosfatemia con TmP/GFR basso, 1,25OHD3 normale o inappropriatamente basso, 25OHD3 normale, calcemia normale, calciuria normale/bassa, PTH normale/alto, FGF-23 elevato ma anche solo nel range alto della norma (si tratta di una secrezione svincolata dai meccanismi di feed-back e quindi non necessariamente aumentata), ALP elevata (come nei casi di osteomalacia).
Terapia
In età pediatrica è obbligatorio il trattamento convenzionale (supplementazione con fosforo elementare + calcitriolo) fino al completamento della crescita delle ossa lunghe.
In età adulta il proseguimento della terapia convenzionale (fosforo + calcitriolo) è controverso: non esiste un consenso unanime sulle indicazioni al trattamento, in quanto bisogna attentamente valutare il bilancio fra benefici ed effetti collaterali della terapia (iperparatiroidismo, ipercalciuria/nefrocalcinosi). Nei pazienti adulti asintomatici, in assenza di una chiara evidenza del reale beneficio della terapia convenzionale con fosforo e calcitriolo e in presenza di noti possibili effetti collaterali, è consigliato un atteggiamento “wait and see”. Nei pazienti adulti sintomatici (dolore osseo, osteomalacia, fratture spontanee da insufficienza) è indicato il trattamento con fosforo e calcitriolo; in caso di artrosi e/o entesopatia sono utili gli analgesici e la FKT (la terapia con fosforo e calcitriolo non sembra migliorarne il decorso). Esistono poi condizioni particolari che rappresentano un’indicazione alla terapia con fosforo e calcitriolo: pazienti candidati a chirurgia ortopedica (osteotomia correttiva, protesi di anca e ginocchio) o a implantologia dentale, in cui si consiglia di iniziare la terapia nei 3-6 mesi antecedenti la procedura e di proseguirla nei 6-9 mesi successivi; donne in gravidanza/allattamento e menopausa, in cui è consigliato il trattamento per l’aumentato fabbisogno di calcio e fosforo (non ci sono tuttavia studi ad hoc).
I dosaggi usati negli studi clinici sono molto variabili e possono essere così schematizzati:
- adulto: fosforo elementare 0-2 g/die in 2-4 dosi + 1alfa-calcidiolo 0-1.5 mg/die in 1-2 dosi;
- gravidanza: fosforo elementare 2 g/die in 2-4 dosi + 1alfa-calcidiolo 1-1.5 mg/die in 1-2 dosi;
- menopausa: fosforo elementare 0-2 g/die in 2-4 dosi + 1alfa-calcidiolo 0-1.5 mg/die in 1-2 dosi (la dose di calcitriolo corrisponde a metà dose di 1alfa-calcidiolo).
La supplementazione con fosforo può determinare in alcuni pazienti spiacevoli effetti collaterali di tipo gastro-intestinale (dolore addominale, diarrea), per cui si raccomanda di incrementare lentamente il dosaggio. Inoltre, un eccesso di fosforo può determinare un iperparatiroidismo secondario (che può essere anche pre-esistente all’inizio della terapia) e nel corso degli anni un iperparatiroidismo terziario, per cui si raccomanda di tenere il livello di fosforemia nel range basso della norma. Un eccesso di dosaggio di calcitriolo può invece determinare nel tempo ipercalcemia/ipercalciuria e nefrocalcinosi.
Il trattamento con burosumab (anticorpo monoclonale neutralizzante anti-FGF23) è approvato da FDA, EMA e AIFA per l’età pediatrica, mentre per il soggetto adulto è ad oggi approvato solo da FDA (in Italia è possibile fare una richiesta a scopo compassionevole).
Monitoraggio della terapia
Vanno monitorati i parametri del metabolismo fosfo-calcico (calcemia, fosforemia, creatininemia, ALP ossea, PTH, calciuria 24 ore) ogni 3-4 mesi nel I anno di terapia, quindi ogni 6-12 mesi e comunque a ogni cambio di dosaggio della terapia o di apporto alimentare di calcio. In gravidanza è raccomandato un monitoraggio più stretto ogni 3 mesi.
Ogni 2-5 anni deve essere eseguita ecografia renale per diagnosticare un’eventuale nefrocalcinosi.
In caso di ipercalcemia/ipercalciuria, si raccomanda di ridurre la dose di calcitriolo (per ipercalciuria molto elevata, è possibile valutare l’idroclorotiazide); in caso invece di iperparatiroidismo, si raccomanda di ridurre la supplementazione con fosforo o aumentare la dose di calcitriolo (per una condizione di iperparatiroidismo secondario/terziario significativo e persistente, è possibile valutare il cinacalcet).
2.2.2. Rachitismo ipofosforemico autosomico dominante ADHR (3)
Eziologia
Mutazione attivante di FGF-23, che diventa resistente alla degradazione proteolitica, cui consegue aumento dei livelli plasmatici di FGF-23.
Clinica
Le manifestazioni cliniche sono molto variabili e simili a quelle della TIO: rachitismo/osteomalacia ipofosforemica, dolore osseo, debolezza muscolare, fratture/pseudo-fratture. La comparsa della ciclicità mestruale alla pubertà può essere un trigger per la comparsa della sintomatologia, in quanto una condizione di sideropenia può determinare una maggiore espressione dell’RNA messaggero di FGF-23.
Laboratorio
Nella forma attiva di malattia, le alterazioni biochimiche sono sovrapponibili a quelle della XLH. Può non essere presente ipofosfatemia, in quanto si possono alternare periodi di ipofosfatemia con periodi di normofosfatemia, a seconda delle differenti e fluttuanti concentrazioni di FGF-23.
Terapia
Come nella XLH (terapia convenzionale).
2.2.3. Rachitismo ipofosforemico autosomico recessivo ARHR (4)
Eziologia
Esistono due varianti della patologia:
- tipo 1, dipendente da una mutazione inattivante di DMP1 (dentin matrix protein 1), che determina un’alterata differenziazione degli osteociti con aumentata produzione di FGF-23;
- tipo 2, dipendente da una mutazione inattivante di ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase 1), con aumentata produzione di FGF-23.
In entrambi i casi il preciso meccanismo che porta all’aumento di FGF-23 non è chiaro.
Clinica
Le manifestazioni cliniche sono simili a quelle di XLH e ADHR (deformità scheletriche, difetti dentali, lesioni osteosclerotiche, entesopatia) ma con valori di densitometria ossea relativamente più conservati.
Laboratorio
Simile alla XLH con FGF-23 normale/alto.
Terapia
Come nella XLH (terapia convenzionale).
2.2.4. Tumor-induced osteomalacia (TIO)
2.2.5. Displasia fibrosa/Mc Cune-Albright (5)
Dipende da una mutazione acquisita somatica (non ereditaria) attivante la subunità alfa della proteina G stimolatoria (con produzione aberrante di cAMP) nelle cellule staminali mesenchimali, con formazione di osteoblasti aberranti (cellule osteogeniche) che:
- depongono matrice osteoide anomala (tessuto osseo fibroso con trabecole anormali in numero, distribuzione e forma);
- in metà dei casi producono FGF-23, con conseguente osteomalacia ipofosforemica;
- producono IL-6;
- up-regolano RANKL, con conseguente osteoclastogenesi inappropriata.
Si parla di Sindrome di McCune-Albright quando alla displasia fibrosa si accompagnano manifestazioni cutanee (macchie cutanee caffè-latte) ed endocrinopatie (pubertà precoce, ipertiroidismo, acromegalia, Cushing).
2.3. Rachitismo/osteomalacia da perdita renale di fosforo non mediata da FGF-23
La tabella 4 riporta le caratteristiche biochimiche delle forme appartenenti a questa tipologia di rachitismo.
| Tabella 4 Rachitismo da perdita renale di fosforo non mediata da FGF-23 |
||||||||
| Patologia | Ca | P | CaU | TmP/GFR | PTH | 25OHD3 | 1,25OHD3 | Altre |
| HHRH | N | B | A | B | N/B | N | A | - |
| Sindrome di Fanconi | N/B | B | A | B | N/A | N | N | glicosuria, aminoaciduria, proteinuria tubulare, acidosi metabolica (bicarbonati venosi < 20 mEq/L), disionie (ipoNa, ipoK, ipoCa) |
| Acidosi tubulare distale | N | B | A | B | N/B | N | N | Acidosi metabolica (bicarbonati venosi < 20 mEq/L), ipoK, pH urinario > 5.5-6 |
| HHRH = Hereditary hypophosphataemic rickets with hypercalciuria; A=alto, B=basso, N=normale | ||||||||
2.3.1. Rachitismo ereditario ipofosforemico con ipercalciuria HHRH (6)
Eziologia
Dipende da una mutazione inattivante del co-trasportatore Na/P NPT IIc (SLC34A3) del tubulo contorto prossimale (perdita renale di fosforo primaria). La trasmissione è autosomica recessiva.
Clinica
Le manifestazioni cliniche comprendono rachitismo/ osteopenia/ osteomalacia e nefrolitiasi (calcoli di fosfato di calcio).
Laboratorio
In questo caso la perdita renale primaria di fosforo è un potente stimolo della sintesi di 1alfa-idrossilasi a livello renale, con conseguente aumento della 1,25OHD3, che a sua volta determina un aumento dell’assorbimento intestinale di fosforo e calcio. Le alterazioni biochimiche sono quindi: 1,25OHD3 elevata con PTH e FGF-23 normali/bassi (FGF-23 appropriatamente down-regolato), calcemia normale, ipercalciuria.
Terapia
In questo caso, a differenza dei precedenti, si deve somministrare al paziente solo ed esclusivamente fosforo elementare (1-2.5 g/die in 4-5 dosi), allo scopo di ridurre i livelli di 1,25OHD3. Il calcitriolo non deve essere somministrato per il rischio di nefrocalcinosi. Utile infine la dieta iposodica.
2.3.2. Sindrome di Fanconi (7)
Eziologia
Si tratta di una disfunzione complessa della capacità di riassorbimento del tubulo contorto prossimale, che oltre al fosforo, può riguardare glucosio, aminoacidi, proteine, bicarbonati (acidosi tubulare prossimale tipo II), acido urico ed elettroliti (calcio, sodio, potassio).
Sono note 2 mutazioni genetiche:
- inattivante del co-trasportatore Na/P NPT IIa (SLC34A1) del tubulo contorto prossimale (sindrome di Fanconi reno-tubulare tipo 2, autosomica recessiva), in cui l’attivazione della 1alfa-idrossilasi può causare ipercalcemia e ipercalciuria;
- inattivante di CLCN5 o OCRL del tubulo contorto prossimale, X-linked recessiva (sindrome di Dent).
Molti farmaci possono determinare disfunzione mitocondriale renale, con riduzione della produzione di ATP, ridotta attività della pompa sodio/potassio ATPasi della membrana baso-laterale e quindi minor disponibilità di energia per i trasportatori della membrana luminale:
- inibitori delle tirosin-chinasi: sorafenib, ibrutinib, imatinib, sunitinib, dasatanib, dabrafenib, ceritinib, nilotinib, regorafenib;
- inibitori di mTOR: everolimus, temsirolimus, sirolimus;
- chemioterapici: ifosfamide, cisplatino, ciclofosfamide, streptozocina, azacitidina, 6-mercaptopurina, suramina;
- anti-epilettici: valproato;
- antibiotici: aminoglicosidi, rifampicina, tetracicline;
- inibitori della trascrittasi inversa: adefovir, tenofovir, cidofovir, abacavir;
- inibitori delle proteasi: parunavir, lopinavir, atazanavir;
- anti-HCV: ribavirina, interferone;
- anti-HBV: lamivudina, entecavir;
- anti-herpes: acyclovir;
- anti-psoriasici: acido fumarico, apremilast.
Possono inoltre essere coinvolti altri meccanismi:
- intossicazione da metalli pesanti;
- errori congeniti del metabolismo: cistinosi, malattia di Wilson, glicogenosi tipo I, galattosemia;
- disordini ematologici: mieloma multiplo, amiloidosi, Sjogren.
Clinica
Le manifestazioni cliniche sono principalmente rappresentate da osteomalacia ipofosforemica, acidosi metabolica e sintomi da deplezione proteica.
Laboratorio
In questo caso all’ipofosfatemia si possono variamente associare ipercalciuria, glicosuria, aminoaciduria, proteinuria tubulare, acidosi metabolica (bicarbonati venosi < 20 mEq/L), disionie (iposodiemia, ipopotassiemia, ipocalcemia).
Terapia
Si basa sulla supplementazione delle sostanze perse a livello renale (fosforo, aminoacidi/proteine, bicarbonato sodio/potassio citrato).
2.3.3. Acidosi tubulare distale (tipo I) (8)
Eziologia
Dipende da un difetto della rigenerazione distale dei bicarbonati, con acidosi metabolica che causa perdita renale di fosforo. Può essere:
- genetica: autosomica dominante o recessiva;
- secondaria: malattie autoimmuni, granulomatosi, farmaci, trapianto di rene, ipertiroidismo, iperparatiroidismo, infezioni croniche delle vie urinarie, epatite cronica attiva, nefrocalcinosi, anemia a cellule falciformi.
Clinica
Le principali manifestazioni cliniche sono rachitismo/osteomalacia e nefrolitiasi/nefrocalcinosi.
Laboratorio
Le principali alterazioni plasmatiche e urinarie sono: bicarbonati venosi < 20 mEq/L, ipofosfatemia, ipopotassiemia, pH urinario > 5.5-6 e ipercalciuria.
Terapia
Si basa sulla correzione dell’acidosi metabolica (bicarbonato sodico o citrato sodico per os) e sulla supplementazione di potassio (potassio citrato che ha anche effetto ipocalciurico).
3. RACHITISMO/OSTEOMALACIA DA INIBIZIONE DIRETTA DELLA MINERALIZZAZIONE
3.1. Ipofosfatasia
BIBLIOGRAFIA
- Carpenter TO, Shaw NJ, Portale AA, et al. Rickets. Nat Rev Dis Primers 2017, 3: 17101.
- Haffner D, Emma F, Eastwood DM, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol 2019, 15: 435-55.
- Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab 1997, 82: 674–81.
- Levy-Litan V, Hershkovitz E, Avizov L, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet 2010, 86: 273–8.
- Boyce AM, Florenzano P, de Castro LF, Collins MT. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. Fibrous Dysplasia/McCune-Albright Syndrome. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. [last revision 2019 Jun 27].
- Lorenz-Depiereux B, Benet-Pages A, Eckstein G, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet 2006, 78: 193–201.
- Karatzas A, Paridis D, Kozyrakis D, et al. Fanconi syndrome in the adulthood. The role of early diagnosis and treatment. J Musculoskelet Neuronal Interact 2017, 17: 303-6.
- Seidowsky A, Moulonguet-Doleris L, Hanslik T, et al. Tubular renal acidosis. Rev Med Interne 2014, 35: 45-55.
Scheda burosumab
Gregorio Guabello
Ambulatorio di Patologia Osteo-Metabolica, UO Reumatologia, IRCCS Istituto Ortopedico Galeazzi; Ambulatorio di Endocrinologia Oncologica, IRCCS Ospedale San Raffaele; Milano
Meccanismo di azione
Anticorpo monoclonale anti-FGF23, che ne contrasta l’effetto fosfaturico determinando un aumento della fosfatemia.
Indicazioni
Trattamento dell’ipofosfatemia X-linked (XLH) con evidenza radiografica di malattia ossea, nei bambini di età > 1 anno e negli adolescenti con sistema scheletrico in crescita.
Controindicazioni
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti.
Somministrazione concomitante con fosfato e analoghi della vitamina D attiva per via orale.
Fosfatemia a digiuno superiore all’intervallo della norma per l’età, a causa del rischio di iperfosfatemia.
Pazienti con compromissione renale severa o malattia renale in fase terminale.
Preparazioni, vie di somministrazione, posologia
Crysvita, flac 10, 20, 30 mg.
La dose iniziale raccomandata è 0.8 mg/kg, somministrati ogni due settimane. Le dosi devono essere arrotondate ai 10 mg più vicini. La dose massima è 90 mg. È necessario un aggiustamento del dosaggio in base ai livelli di fosfatemia in corso di terapia.
Effetti collaterali
Quelli segnalati più comunemente (> 10%) nei pazienti pediatrici trattati per un periodo fino a 64 settimane durante gli studi clinici sono stati: reazioni in sede di iniezione (56%), tosse (56%), cefalea (50%), piressia (43%), dolore alle estremità (40%), vomito (39%), ascesso dentario (35%), diminuzione della vitamina D (32%), diarrea (25%), eruzione cutanea (24%), nausea (15%), stipsi (11%), carie dentaria (11%), mialgia (11%).
Limitazioni prescrittive
Classe H, obbligatoria compilazione scheda di monitoraggio.
Altre malattie metaboliche dell'osso
Malattia di Paget
Chiara Carzaniga1 & Daniela Di Sarra2
1Unità di Endocrinologia, Istituto Auxologico Italiano, Milano
2Divisione di Medicina Interna, AULSS 9 Scaligera-Ospedale di Legnago (VR)
(aggiornato al 28 agosto 2019)
INTRODUZIONE
La malattia di Paget è una patologia che colpisce il tessuto scheletrico, caratterizzata da alterazioni focali del rimodellamento osseo a carico di uno (mono-ostotico) o più (poli-ostotico) segmenti ossei. La normale architettura è sovvertita dalla formazione di tessuto osseo non organizzato, con conseguente tendenza alla comparsa di dolore, deformità e fratture a carico dei segmenti ossei interessati e artrosi secondaria nelle articolazioni adiacenti. I siti maggiormente colpiti sono bacino, rachide, femore, tibia e cranio.
EPIDEMIOLOGIA
Raramente compare prima dei 40 anni, la prevalenza tende a raddoppiare a ogni decade dopo i 50 anni. Studi recenti suggeriscono una prevalenza dell’8% tra i maschi e del 5% tra le femmine nell’ottava decade di vita (il rapporto uomo-donna è di 3:2). La prevalenza della malattia in Italia è stimata tra 0.7% e 1.4%. La malattia è più comune (con prevalenza > 1%) in Europa occidentale, Nord America, Australia e Nuova Zelanda, mentre è più rara nei paesi scandinavi, nel subcontinente indiano e in generale nelle popolazioni asiatiche.
Negli ultimi 30 anni si sono osservati riduzione della prevalenza e dell’incidenza della malattia, aumento dell’età alla diagnosi e riduzione del numero di segmenti scheletrici coinvolti e dei livelli di fosfatasi alcalina (ALP) alla diagnosi (1-2). Le motivazioni alla base di questo rapido cambiamento delle caratteristiche epidemiologiche sono sconosciute, ma sono in parte legate a un cambiamento dell’esposizione a fattori ambientali specificamente correlati alla malattia (2-3).
EZIOLOGIA
Rimane tuttora sconosciuta, ma due ipotesi sono più accreditate: genetica e virale e/o ambientale.
L’ipotesi genetica si basa sull’evidenza di una chiara familiarità in numerosi casi: circa il 12-40% dei pazienti presenta una storia familiare positiva per tale patologia (4). Il rischio di malattia è di circa 7-10 volte maggiore nei parenti di primo grado di soggetti affetti. L’affezione in alcuni casi appare trasmessa come carattere autosomico dominante con penetranza variabile (2). L’alterazione genetica di più frequente riscontro, sia nei casi familiari (40-50%) che sporadici (5-10%), riguarda il gene Sequestosome 1 (SQSTM1), che codifica per p62, una proteina coinvolta nella cascata del segnale del fattore nucleare kappa B (NF-kB). Mutazioni a carico di questo gene provocherebbero un aumento dell’osteoclasto-genesi, meccanismo peculiare della malattia ossea di Paget classica (5-6).
Raramente, possono verificarsi disturbi familiari da Paget (o simili a Paget) in associazione con mutazioni in altri geni (7).
Tuttavia, l’ipotesi genetica non spiega i rapidi cambiamenti epidemiologici osservati negli ultimi anni (3).
Il rilievo di inclusioni nucleari e citoplasmatiche simili ai nucleo-capsidi della famiglia dei paramixovirus (virus del morbillo, virus respiratorio sinciziale) ma anche del virus cimurro canino all’interno dell’osso pagetico e il riscontro di proteine del nucleo-capside e di mRNA nei precursori e negli osteoclasti maturi, hanno permesso di ipotizzare che anche un’infezione virale possa, insieme ad altri fattori ambientali scatenanti (dieta a basso contenuto di calcio e vitamina D durante l’adolescenza e stretto contatto con animali), produrre ulteriori modificazioni qualitative e quantitative delle cellule osteoclastiche (2-7).
Il fatto che i fattori ambientali svolgano un ruolo nell’eziopatogenesi della malattia di Paget è dimostrato dal riscontro di una riduzione nella prevalenza e nella gravità osservata negli ultimi 25 anni in molti paesi, soprattutto in quelle regioni che in precedenza avevano un'alta prevalenza (8). In linea con questo, negli ultimi anni è diminuita anche la prevalenza dell'osteosarcoma negli adulti (una complicanza della malattia di Paget).
FISIOPATOLOGIA
Sulla base di dati soprattutto radiologici sono state distinte 3 fasi nell’evoluzione delle lesioni pagetiche:
- prima fase osteolitica o distruttiva (mediata dagli osteoclasti), con eccessivo riassorbimento osseo;
- seconda fase intermedia o mista, in cui a una disordinata ed eccessiva neoformazione ossea (mediata dagli osteoblasti) si associa un contemporaneo riassorbimento dell’osso;
- terza fase (sclerotica), in cui diviene prevalente l’apposizione ossea, sicché le ossa aumentano di dimensioni (di larghezza e non di lunghezza).
Le fibre di collagene non si organizzano nella tipica forma lamellare, ma a mosaico (ciò radiologicamente si traduce in un aspetto “a fiocchi di cotone”). Si ritiene che ciò sia dovuto all’aumento di numero e dimensioni degli osteoclasti.
L’osso pagetico è sede di intensa attività metabolica ed è riccamente vascolarizzato, ha volume superiore alla norma. La mineralizzazione spesso è deficitaria e possiede scarsa efficienza biomeccanica, in quanto l’organizzazione architetturale è impropria e non adeguata al carico meccanico variabile per ogni specifico segmento scheletrico. Questo comporta la tendenza a comprimere le strutture che decorrono all’interno dell’osso, la tendenza a deformarsi, il maggiore rischio di fratture, la frequente associazione con forme di osteo-artrosi secondaria ad anomalo carico articolare conseguente alle suddette deformità (9,10).
QUADRO CLINICO
Circa il 70% dei soggetti affetti è del tutto asintomatico; in questi casi la diagnosi viene posta casualmente tramite il riscontro di elevati livelli plasmatici di ALP ad esami di routine oppure delle tipiche anomalie scheletriche rilevate in esami radiologici eseguiti per altri motivi.
I siti scheletrici più comunemente colpiti sono pelvi (67%), rachide (34%), femore (32%), tibia (25%), cranio (23%), omero (11%); la malattia può interessare uno (forma mono-ostotica) o più segmenti scheletrici (forma poli-ostotica). Negli ultimi anni si è assistito a una progressiva riduzione della gravità della patologia ossea nei pazienti neo-diagnosticati, al punto che in circa il 40% dei casi è coinvolto un unico osso.
La presentazione clinica può essere variabile in funzione dell’estensione della malattia, dei particolari segmenti scheletrici interessati e delle eventuali complicanze associate. Oltre il 30% dei pazienti sviluppa complicanze: deformazioni ossee, fratture, sordità e artrosi secondaria (2-10).
Sintomi e complicanze muscolo-scheletriche
Dolore osseo: è il sintomo più comune (40-45%), che insorge in genere nelle fasi tardive della malattia. Può essere di origine scheletrica, neurologica, muscolare o articolare ed è legato a vari fattori, tra cui l’intensa attività metabolica e l’aumento della vascolarizzazione delle lesioni pagetiche, ma più frequentemente è secondario all’instaurarsi di complicanze che si osservano nei casi di lunga durata (distorsione del periostio, lesioni litiche, micro-fratture, deformità e artrosi). È generalmente sordo e continuo, talvolta associato a sensazione di caldo urente e, diversamente dal dolore tipico dell’artrosi, peggiora con il riposo, nelle ore notturne e con il carico, laddove concomita sovrappeso se la malattia ha colpito lo scheletro portante. Tuttavia, nella maggior parte dei casi il dolore osseo risulta spesso indistinguibile dal dolore articolare con il quale generalmente coesiste (10).
Dolore articolare: compare quando vengono coinvolte le articolazioni localizzate vicino al segmento osseo colpito (in particolare l’articolazione dell’anca che è la più colpita).
Deformazioni ossee: un aspetto tipico della malattia di Paget è l’aumento delle dimensioni e/o l’alterazione della forma tipica dell’osso, originalmente definita “osteitis deformans”. Sono molto frequenti, con prevalenza del 12-36%, localizzate prevalentemente a carico delle ossa lunghe (femore, tibia), del cranio e delle clavicole. Se colpiscono le ossa lunghe degli arti inferiori, possono causare un’alterazione dell’andatura secondaria ad asimmetria di lunghezza o ad alterazione della distribuzione dei carichi (10).
Artrosi: il coinvolgimento delle articolazioni (prevalentemente dell’anca) è dovuto a un’alterata distribuzione del carico meccanico, secondario alle deformazioni dei vari segmenti scheletrici affetti.
Cefalea: dovuta al coinvolgimento delle ossa del cranio.
Fratture patologiche: interessano prevalentemente le ossa lunghe. Sono poco comuni, generalmente trasversali e possono essere complete o semplici fissurazioni della corticale. Queste fratture trasversali vengono denominate fratture “a banana” o “a gessetto” e riflettono la mancanza della qualità della matrice di collagene. Le fratture trasversali parziali o “fissurazioni” possono verificarsi lungo la piega della curva esterna dell’osso (2). La maggior parte delle fratture incomplete si verifica durante la fase osteolitica, mentre quelle complete sono più frequenti nella fase osteoblastica. La guarigione delle fratture nell’osso pagetico non è ritardata, pur essendo possibile un’incompleta saldatura dei segmenti ossei (10).
Cifosi: legata alla deformazione delle vertebre colpite.
Alterazione delle ossa facciali: il loro coinvolgimento determina dismorfismi, caduta dei denti e malocclusione.
Complicanze neurologiche
Ipoacusia: è la complicanza più frequente (prevalenza 2.4-13.5%), dovuta sia all’ingrandimento delle ossa craniche, con conseguente compressione del nervo acustico, sia alla perdita di massa ossea a livello cocleare.
Stenosi del canale spinale: è poco comune, nonostante il frequente coinvolgimento vertebrale, legata alla compressione sul midollo spinale da parte di vertebre deformate, con conseguente paraplegia.
Deficit dei nervi cranici: possono essere compressi il II, V e VII, con alterazioni del visus e paralisi facciali.
Sindrome da furto vascolare: il coinvolgimento della colonna vertebrale può portare a sintomi neurologici che possono interessare anche i nervi periferici e possono derivare dalla lesione diretta o dall’ischemia di alcuni nervi, dovuta a una sindrome da “furto arterioso”, che in alcuni casi porta ad una vera e propria paraplegia.
Complicanze metaboliche
L’ipercalcemia è poco frequente, può verificarsi soprattutto nei pazienti con lesioni poli-ostostiche ed è la conseguenza di immobilizzazione prolungata. Nel 12-18% dei pazienti affetti da malattia di Paget sono presenti aumentati livelli di PTH: si ritiene che questo rifletta la necessità di incrementare la disponibilità di calcio nelle fasi di attiva formazione dell’osso pagetico. L’aumento del riassorbimento osseo può determinare un aumento dell’escrezione urinaria di calcio (ipercalciuria), che non si riflette però in un aumento dell’incidenza di nefrolitiasi (2-10).
Complicanze cardio-vascolari
Nei casi più severi, quando cioè la malattia è diffusa e coinvolge un terzo o più (15-35%) dello scheletro, l’aumento del flusso sanguigno (tipico del Paget) può essere associato a scompenso cardiaco cronico a elevata gittata. Tale complicanza è comunque rara e si sviluppa generalmente in pazienti con sottostante cardiopatia (9).
Altre complicanze descritte più raramente sono diffuse calcificazioni vascolari e più frequentemente stenosi aortica.
Neoplasie ossee
Generalmente si tratta di tumori rari, la cui incidenza non è del tutto definita (~1%), con un alto grado di malignità: osteosarcoma (~86% dei casi), fibrosarcoma (~5%) e condrosarcoma (~2.5%). Occasionalmente si riscontrano inoltre tumori benigni a cellule giganti (~2.5%), clinicamente non sempre distinguibili dai sarcomi, ma istologicamente costituiti da agglomerati di osteoclasti (osteoclastomi), che interessano solitamente il cranio e le ossa facciali, ma possono essere anche extra-scheletrici, e sono spesso sensibili alla terapia con glucocorticoidi (11,12).
DIAGNOSI
Può essere effettuata sulla base della comparsa di sintomi e segni specifici o essere del tutto casuale e viene comunemente fatta attraverso la radiografia del segmento osseo interessato, che mostra le tipiche caratteristiche radiologiche di osteosclerosi alternata ad aree di osteolisi ed espansione ossea, dipendenti dalle fasi della malattia.
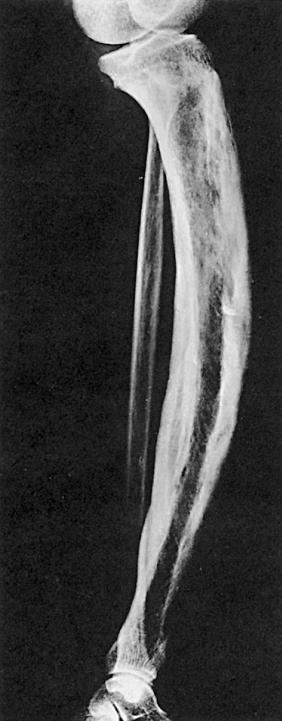
Immagine radiologica della malattia di Paget della tibia (da Williams Textbook of Endocrinology, 12° Edizione)
La scintigrafia ossea è più sensibile della radiografia nell’identificare le lesioni pagetiche, perché è in grado di individuare l’aumentata attività cellulare dell’osso, ma è sicuramente meno specifica. La scintigrafia va quindi raccomandata in seguito al rilievo radiologico di un segmento scheletrico interessato ed è utile per definire l’estensione della malattia. Non è necessario ripetere la scintigrafia nel tempo, ma è bene sapere che, in corso di terapia, la captazione del radionuclide a livello della lesione è ridotta (13).
Biopsia ossea, RMN, TC o PET possono essere riservate a quei rari casi in cui è particolarmente difficile la diagnosi differenziale con le metastasi osteosclerotiche o nei casi in cui si sospetti un’evoluzione verso l’osteosarcoma (14,15). Anche secondo le nuove linee guida non devono essere utilizzate routinariamente (2).
Il morbo di Paget si associa a incremento del turnover osseo e della maggior parte dei suoi marcatori. ALP rappresenta quello con maggior sensibilità e specificità diagnostica, correla direttamente con la gravità e l’estensione del coinvolgimento scheletrico e riflette l’efficacia dell’intervento farmacologico (13). È aumentata nel 95% dei pazienti pagetici, sebbene livelli normali non possano escludere la malattia, dato che può essere normale nelle forme mono-ostotiche o nei pazienti con malattia metabolicamente inattiva. Lo svantaggio della fosfatasi alcalina totale è la sua sovrapposizione con i livelli di fosfatasi alcalina epatica. Quando non fosse possibile dosare la fosfatasi alcalina ossea e qualora la funzione epatica di un paziente affetto fosse alterata, dovrebbero essere presi in considerazione marcatori differenti:
- tra quelli di neoformazione, il propeptide N terminale del collagene di tipo 1 è quello dotato di maggiore sensibilità e specificità; l’osteocalcina è meno utile, poco sensibile per valutare l’attività pagetica;
- tra i marcatori di riassorbimento, le recenti linee guida suggeriscono il dosaggio del βCTX sierico o NTx urinario (rispettivamente prodotto di degradazione ß-CrossLap del propeptide C terminale del collagene di tipo 1, telopeptide C terminale di tipo 1) (2-12,16).
Alcuni recentissimi studi hanno valutato la possibilità di diagnosticare la malattia di Paget integrando l’analisi genetica del gene SQSTM1 con alcuni marcatori genetici e l’utilizzo di marcatori biochimici (17). Sono ovviamente necessari ulteriori studi per permettere l’utilizzo futuro di test molecolari e genetici nella pratica clinica.
TERAPIA
In passato vi era largo consenso sul fatto che il dolore a carico dell’osso pagetico costituisse una chiara e incontrovertibile indicazione alla terapia farmacologica. La terapia veniva quindi consigliata a pazienti affetti da malattia attiva o a rischio di sviluppare complicanze. Recentemente, in considerazione dell’evidenza di una remissione di almeno 6 mesi dopo terapia con bisfosfonati e della conseguente riduzione dei costi del follow-up, unitamente alla dimostrazione di un miglioramento della qualità della vita, la terapia viene consigliata a tutti i pazienti affetti.
Lo scopo della terapia è ottenere la completa remissione, definita da:
- normalizzazione di ALP, che preferibilmente dovrebbe raggiungere un nadir nella metà inferiore del range di riferimento (18);
- regressione dei sintomi;
- miglioramento del quadro radiologico;
- prevenzione delle complicanze (19). È tuttora controverso l’utilizzo della terapia per prevenire complicanze quali artrosi, fratture, sordità o complicanze neurologiche (20).
Per quanto riguarda invece la sintomatologia, e nello specifico il dolore, è importante distinguere il dolore causato dall’attività di malattia a carico dell’osso pagetico, normalmente presente a riposo, dal dolore provocato dalle ossa o articolazioni deformate dalla malattia, che invece compare durante la mobilizzazione. Tale distinzione è fondamentale, in quanto alcuni studi dimostrano che i dolori osteo-articolari rispondono agli analgesici ma non ai bisfosfonati.
La prima terapia utilizzata fu la calcitonina, abbandonata oggi per la risposta parziale, la resistenza acquisita e la breve emivita. Attualmente i bisfosfonati costituiscono la terapia specifica per il Paget, trattandosi di farmaci che riducono il turnover osseo, in particolare il riassorbimento osseo da parte degli osteoclasti. Approvati per tale malattia sono quelli di prima (etidronato e clodronato) e ultima generazione (alendronato, ibandronato, risedronato, neridronato e zoledronato). Tra questi il farmaco che ha permesso di ottenere migliori risultati sul lungo termine è senza dubbio lo zoledronato, per il quale si consiglia una singola dose di 5 mg ev nei pazienti che non presentano controindicazioni (21). A distanza di 6 anni dalla terapia l’87% dei pazienti è ancora in remissione (21). Si ricorda che lo zoledronato è potenzialmente nefrotossico, per cui non dovrebbe essere somministrato a pazienti con filtrato glomerulare < 35 mL/min (19).
Nei pazienti con problematiche a livello gastrico sono da preferire i farmaci ev. Questi possono provocare una sindrome post-infusione, con sintomi simil-influenzali nel 15% dei pazienti trattati durante il primo ciclo. Per questo motivo è consigliata la somministrazione di paracetamolo (es. 1 g x 2/die) durante il giorno dell’infusione e nei primi giorni successivi.
La terapia con bisfosfonati è controindicata nei pazienti anziani asintomatici, in quelli con malattia metabolicamente inattiva e nei pazienti con deficit di vitamina D non corretto da adeguata terapia, dato il rischio di insorgenza di ipocalcemia. Durante l’assunzione di bisfosfonati è pertanto raccomandato un adeguato apporto alimentare di calcio (1000-1500 mg/die) e vitamina D (400-800 U/die).
Non sono ancora disponibili dati riguardanti l’utilizzo di denosumab, inibitore del RANK-L, che blocca la maturazione e l’attivazione degli osteoclasti. Case reports hanno dimostrato una normalizzazione dei livelli di ALP dopo 4-8 mesi di terapia, associati a miglioramenti clinici e scintigrafici (13).
La tabella mostra gli intervalli di dose e di applicazione dei farmaci usati per la terapia del morbo di Paget (13).
| Raccomandazioni riguardanti la dose dei farmaci utilizzati nella malattia di Paget | |
| Zoledronato | 5 mg in unica infusione per 15 minuti Raramente è richiesto un nuovo trattamento entro 5 anni |
| Alendronato | 40 mg/die per 6 mesi Nuovo trattamento in genere richiesto tra 2 e 6 anni |
| Risedronato | 30 mg/die per 2 mesi Nuovo trattamento richiesto in genere tra 1 e 5 anni |
| Ibandronato | 6 o 12 mg ev (pubblicati solo dati riguardanti effetti a breve termine) |
| Denosumab | 60 mg sc ogni 6 mesi (solo case report) Nuovo trattamento richiesto dopo 6 mesi |
Nuovi possibili farmaci in fase di studio sono gli inibitori dell’interleukina-6 e di Dickkopf (22).
Gli interventi chirurgici ortopedici in questi pazienti sono rappresentati quasi esclusivamente da correzioni di fratture, osteotomie correttive e artro-plastiche (qualora la terapia medica e la fisioterapia non siano state efficaci). Essi sono spesso complicati da un sanguinamento superiore al normale; la terapia medica sembra ridurre questa complicanza, anche se i risultati sono ancora controversi (23).
FOLLOW-UP
Consiste nel dosaggio di ALP ogni 3 mesi per i primi 6 mesi dopo la terapia e successivamente ogni 6-12 mesi, a seconda della terapia utilizzata. Il raggiungimento di livelli di ALP nella metà inferiore del range di norma dopo 6 mesi dalla terapia con zoledronato si associa a un rischio < 10% di perdere l’efficacia terapeutica (2,21).
Sebbene ALP sia il marcatore migliore e meno costoso per monitorare la risposta al trattamento, vi sono alcuni casi in cui è necessario valutare l’efficacia immediata della terapia (es. compressione spinale). In questi casi è meglio utilizzare i marcatori con risposta più rapida rispetto ad ALP: ad esempio βCTx raggiunge un nadir dopo 10 giorni dalla terapia, a differenza di ALP che impiega circa 2-3 mesi.
Dopo 6 mesi dalla terapia va effettuata scintigrafia ossea che, in caso di lesioni osteolitiche, va ripetuta a un anno di distanza. In caso di ricomparsa di sintomatologia dolorosa o incremento dei parametri biochimici, è possibile valutare un nuovo ciclo di terapia. Secondo le nuove linee guida, la modifica dei marcatori ossei è maggiormente indicativa di recidiva di malattia rispetto alla ricomparsa dei sintomi (2): è generalmente accettato iniziare nuovamente la terapia in seguito al riscontro di un incremento di ALP del 25%.
BIBLIOGRAFIA
- Corral-Gudino L, Garcia-Aparicio J, Sanchez-Gonzalez MD, et al. Secular changes in Paget’s disease: contrasting changes in the number of new referrals and in disease severity in two neighboring regions of Spain. Osteoporos Int 2013, 24: 443–50.
- Singer FR, Bone HG, Hosking DJ, et al. Paget’s disease of bone: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014, 99: 4408–22.
- Bolland MJ, Cundy T. Paget’s disease of bone: clinical review and update. J Clin Pathol 2013, 66: 924-7.
- Divisato G, Formicola D, Esposito T, et al. ZNF687 mutations in severe Paget disease of bone associated with giant cell tumor. Am J Hum Genet 2016, 98: 275–86.
- Britton C, Brown S, Ward L, et al. The changing presentation of Paget’s disease of bone in Australia, a high prevalence region. Calcif Tissue Int 2017, 101: 564–9.
- Tuck SP, Layfield R, Walker J, et al. Adult Paget’s disease of bone: a review. Rheumatology (Oxford) 2017, 56: 2050–9.
- Freidrichs WE, Reddy S, Bruder JM, et al. Sequence analysis of measles virus nucleocapsid transcripts in patients with Paget's disease. J Bone Miner Res 2002, 17: 145-51.
- Roodman GD, Windle JJ. Paget disease of bone. J Clin Invest 2005, 115: 200–8.
- Gennari L, Merlotti D, Martini G, et al. Paget’s disease of bone in Italy. J Bone Miner Res 2007, 21: 14–21.
- Falchetti A, Masi L, Brandi ML. Paget’s disease of bone: there’s more than the affected skeletal: a clinical review and suggestions for the clinical practice. Curr Opin Rheumatol 2010, 22: 410–23.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009, 44: 431–6.
- Wermers RA, Tiegs RD, Atkinson EJ, et al. Morbidity and mortality associated with Paget’s disease of bone: a population-based study. J Bone Miner Res 2008, 23: 819–25.
- Muschitz C, Feichtinger X, Haschka J, Kocijan R. Diagnosis and treatment of Paget’s disease of bone, a clinical practice guideline. Wien Med Wochenschr 2017, 167: 18-24.
- Ralston SH, et al. Diagnosis and management of Paget’s disease of bone in adults: a clinical guideline. J Bone Min Res 2019, 34: 579-604.
- Theodorou DJ, Theodorou SJ, Kakitsubata Y. Imaging of Paget disease of bone and its musculoskeletal complications. AJR Am J Roentgenol 2011, 196 (6 suppl): S64-75.
- Alvarez L, Guanabens N, Peris P, et al. Discriminative value of biochemical markers of bone turnover in assessing the activity of Paget’s disease. J Bone Miner Res 1995, 10: 458–65.
- Guay-Belanger S, Simonyan D, Bureau A, et al. Development of a molecular test of Paget’s disease of bone. Bone 2016, 84: 213-21.
- Reid IR, Hosking DJ. Bisphosphonates in Paget’s disease. Bone 2011, 49: 89-94.
- Josse RG, Hanley DA, Kendler D, et al. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007, 30: E210–23.
- Singer FR. Paget disease: When to treat and when not to treat. Nat Rev Rheumatol 2009, 5: 483–9.
- Reid I, Lyles KW, Su G, et al. Long-term efficacy of zoledronic acid compared with risedronate in Paget’s disease. Bone 2010, 47 suppl 1: S38.
- Michou L, Brown JP. Emerging strategies and therapies for treatment of Paget's disease of bone. Drug Des Devel Ther 2011, 5: 225-39.
- Adami S, Bartolozzi P, Brandi ML, et al. Italian guidelines for the diagnosis and treatment of Paget's disease of bone. Reumatismo 2007, 59: 153-68.
Osteogenesi imperfetta
Bruno Madeo
UOC Endocrinologia e Malattie del Metabolismo, Ospedale (NOCSAE) di Modena
L’osteogenesi imperfetta (OI), o malattie delle ossa fragili, è una patologia congenita caratterizzata da un’aumentata fragilità ossea, che comporta una maggiore predisposizione alle fratture e alla deformabilità scheletrica. L’OI è spesso accompagnata da altre manifestazioni extra-scheletriche: sclere blu, dentinogenesi imperfetta, lassità dei legamenti, perdita dell’udito. Caratteristica è l’estrema variabilità clinica, che va dalla forma più lieve, a volte misconosciuta o confusa con un'osteoporosi post-menopausale, a quella più severa con mortalità perinatale.
EPIDEMIOLOGIA
L’incidenza di OI è stimata 1 su 200.000-500.000 (1 su 10.000 nuovi nati): su 60.000 nuovi nati, 4-5 sono affetti da OI, dei quali 2 con la forma lieve (tipo I), 1 con la forma severa (tipo II) ed 1-2 con le restanti forme.
PATOGENESI
La maggior parte dei casi (circa il 90%) di OI presenta anomalie nei geni che codificano per il collagene tipo I (COLIA 1 e COLIA 2). Negli ultimi anni sono stati scoperti nuovi geni che codificano per proteine coinvolte nella formazione del collagene, la cui mutazione causa un quadro di OI come: cartilage-associated protein (CRTAP), propyl-3-hydroxylase (LEPRE1/P3H1), cyclophilin B (PPIB), collagen chaperone-like protein HSP47 (SERPINH1) e la procollagen chaperone protein FKBP65 (FKBP10).
Le anomalie genetiche possono causare o una ridotta sintesi di collagene strutturalmente normale, come nel caso dell’OI tipo I per effetto di una delezione di uno degli alleli, o alterazioni strutturali di vario tipo del collagene. Tuttavia, non sempre vi è un correlazione genotipo-fenotipo, per cui succede che differenti mutazioni rendano ragione di una diversa espressione clinica, ma allo stesso tempo la medesima mutazione si può accompagnare ad una diversa clinica.
Le sedi prevalentemente interessate dalla malattia sono, con tutta evidenza, quelle in cui il collagene tipo I è maggiormente espresso: oltre alle ossa, i legamenti, la cute, le sclere e i denti.
CLASSIFICAZIONE E ASPETTO CLINICO
La classificazione inizialmente suggerita da Sillence (nel 1979) suddivideva l’OI in 4 tipi (I-IV) sulla base della presentazione clinica e del tipo di trasmissione ereditaria. Successivamente (nel 2004), l’estrema eterogeneità clinica e istopatologica e la scoperta di nuove anomalie genetiche hanno condotto a una estensione fino a 7 tipi (I-VII) e recentemente ne sono state proposte una VIII (2007) ed una IX (2010) (tabella).
| Quadro sinottico forme di osteogenesi imperfetta | |||||||||
| Tipo |
I | II | III | IV | V | VI | VII | VIII | IX |
| Gene mutato | COL1A 1/2 |
COL1A 1/2 |
COL1A 1/2 |
COL1A 1/2 |
? | ? | CRTAP | LEPRE1 |
PPIB- |
| Trasmissione | AD | AD | AD | AD | AD | AR | AR | AR | AR |
| Gravità clinica | + | ++++* | +++ | ++ | ++ | ++/ +++ |
+++/ ++++* |
+++/ ++++* |
++/ +++ |
| Deformità | ± | ++++ | +++ | ++ | ++ | ++ | ++ | ||
| Sclere blu | + | ++ | ± | - | - | - | - | - | |
| Perdita udito | ++ | ++ | + | ||||||
| Dentinogenesi imperfetta | ± | + | + | - | - | ||||
| Bassa Statura | ± | +++ | ++ | ++ | ++ | ++ | +++ | ++ | |
| Lassità legamenti | ± | + | + | + | + | ||||
| Rizomelia | - | - | - | - | - | - | + | + | - |
| Coxa vara | - | - | - | - | - | - | + | - | - |
COL1A1/2: gene del collagene tipo 1; CRTAP: gene della proteina cartilagine-associata;
AD: autosomico dominante; AR: autosomico recessivo
± presente/assente; + lieve; ++ moderato; +++ moderato-severo; ++++severo; ? non noto
*mortalità perinatale
Tuttavia, nonostante la variabilità istologica e genetica, clinicamente questi tipi sono indistinguibili, per cui si sta tentando un ritorno alla semplificazione.
La fragilità ossea è spesso associata a sclere blu, dentinogenesi imperfetta e perdita dell’udito. Si possono osservare inoltre: lassità dei legamenti, ipermobilità delle articolazioni, facilità alla formazione di ecchimosi e cicatrici e raramente manifestazioni cardiovascolari (insufficienza aortica, lassità mitrale, insufficienza mitralica e fragiltà dei grossi vasi).
Non sono presenti deficit intellettivi; al contrario la forzata sedentarietà, in condizioni favorevoli, può favorire lo sviluppo di capacità alternative, a volte con straordinari risultati (vd Malati Famosi).
La storia delle fratture rappresenta l’elemento più importante per la classificazione (vd effetti scheletrici), poi esiste una serie di elementi distintivi. Il riconoscimento di questi è:
- molto semplice: l’assenza di sclere blu e di dentinogenesi imperfetta nei tipi V-IX
- relativamente semplice:
- callo ipertrofico nella sede di frattura, indagabile con RM o TC, calcificazioni delle membrane interossee fra le osse dell’avambraccio, e presenza di una banda metafisaria radiopaca in stretta adiacenza della cartilagine di accrescimento, evidente all’Rx, nel tipo V)
- rizomelia (brevità dell’omero o del femore) nel tipo VII e VIII
- coxa vara (Rx) nel tipo VII
- più complicato o invasivo:
- aspetto istologico a squame di pesce delle lamelle ossee e la presenza di eccessivo accumulo di osteoide sulle superfici di neoformazione ossea (che richiede una biopsia) nel tipo VI
- l’aspetto irregolare tipo a rete dell’organizzazione ossea nel tipo V.
Effetti scheletrici
La fragilità ossea causa un aumento del rischio di frattura per traumi minimi la cui entità varia (Tabella): a) da un grado severo letale pre e perinatale con fratture nel periodo fetale tipicamente costali e delle ossa lunghe (tipo II, VII, VIII); b) ad un grado severo ma compatibile con la vita con numerose fratture e importante deformità scheletrica (tipo III, VI-IX); c) ad un grado moderato-severo con frequenti fratture spesso associate a deformità scheletrica (tipo IV, V, IX); d) ad un grado lieve generalmente senza deformità scheletriche con quadri clinici che a volte sono confusi con una osteoporosi post-menopausale (tipo I). Solitamente le fratture sono più frequenti nella fase neonatale, nella puberale e in età avanzata e nelle donne in gravidanza e dopo la menopausa. La guarigione delle fratture non sembra compromessa. Soprattutto le forme severe sono accompagnate da bassa statura da correlare in parte alla scarsa crescita ed in parte alla deformità scheletrica.
Nelle forme severa, infine, è presente una deformità toracica per una scoliosi con o senza fratture vertebrali spesso associata a pectus excavatum o carenatum. La deformità toracica può causare problemi cardiopolmonari con conseguente ridotta spettanza di vita.
Sclere blu
Le sclere possono essere normali o assumere varie tonalità del blu, per l’assottigliamento dello strato di collagene che lascia trasparire la coroide. Sono altamente suggestive ma non patognomoniche di OI.
Perdita dell’udito
La perdita di udito può essere di tipo conduttivo, neurosensoriale o misto. Generalmente si manifesta dalla seconda decade di vita e ne è affetto il 50% dei pazienti ultra-cinquantenni.
Dentinogenesi imperfetta
La formazione di dentina, ricca in collagene, è spesso anormale, per cui i denti possono assumere un colore differente dall’ambra, bruno-giallastro o grigio azzurognolo traslucido. Più spesso sono interessati i denti decidui, che possono essere più piccoli del normale, mentre quelli permanenti sono spesso a forma di campana stretti alla base. In alcuni casi i denti sono fragili e si rompono facilmente. A volte, anche se i denti sembrano normali, risultano anormali all’Rx o all’esame istologico.
DIAGNOSI
La diagnosi si basa innanzitutto sulla clinica e risulta più facilitata se alla fragilità ossea si accompagnano le manifestazioni extra-scheletriche. Viceversa, la sola presenza delle manifestazioni extra-scheletriche non è patognomonica di OI: infatti, le sclere blu possono essere una variante normale o la dentinogenesi imperfetta può essere ereditaria in famiglie senza associata fragilità ossea.
Esami bio-umorali
L’uso dei marcatori di turn-over osseo non è utile nella diagnosi, ma potrebbe essere di aiuto nel monitoraggio del trattamento.
Densitometria ossea
Solitamente la densità minerale ossea è ridotta in tutte le forme. Pertanto, l’esame può essere di supporto per la diagnosi e può essere utile per il follow-up. Bisogna comunque tenere presente le difficoltà interpretative in alcuni casi: per esempio, in presenza di fratture vertebrali il dato densitometrico può risultare sovrastimato. Infine, può essere di supporto in alcuni casi (es sindrome del bambino maltrattato) per una diagnosi differenziale.
Esami radiologici
Nelle forme moderate l’Rx del cranio può documentare un aspetto a chiazze (Osso Wormiano) per la presenza di piccole aree di ossificazione irregolare. L’Rx delle ossa lunghe nelle forme moderate-severe mostra un aspetto a “popcorn” per depositi di minerali. L’Rx, la RMN e la TC sono utili per identificare le caratteristiche distintive del tipo VII (vd sopra) o necessari per la valutazione di fratture.
Biopsia ossea
Può essere utile per la diagnosi, da limitare evidentemente per la sua invasività.
Le biopsie ossee evidenziano riduzione dello spessore corticale e del volume trabecolare osseo, aumentato turn-over e ridotta formazione ossea nei pazienti con OI tipo I. Il disordine in un sottogruppo di pazienti con basso turn-over e calcificazione dei legamenti è stato classificato come OI tipo V.
Analisi molecolare
Lo studio quantitativo e strutturale del collagene tipo I estratto da colture di fibroblasti della cute o la ricerca di mutazioni genetiche non sono necessari per la diagnosi, ma possono essere utili per una conferma o in caso di consulenze genetiche. E’ possibile una diagnosi prenatale, con un prelievo dei villi corionici alla 8°-12° settimana di gravidanza.
DIAGNOSI DIFFERENZIALE
Vanno escluse altre condizioni che causano fratture come: sindrome del bambino maltrattato, deficit nutrizionali, tumori, ipofosfatasia infantile, osteoporosi giovanile idiopatica, ipogonadismo, iperfosfatasia idiopatica autosomica recessiva (o malattia giovanile di Paget).
TERAPIA
I pazienti conducono spesso una vita attiva e di soddisfazione e la loro qualità di vita è strettamente legata a una adeguata gestione della malattia. Negli ultimi anni, i buoni risultati ottenuti con i bisfosfonati hanno finalmente offerto un'opportunità farmacologica, anche se per un adeguato trattamento sono fondamentali i presidi non-farmacologici.
Terapia non-farmacologica
Per prevenire o ridurre le fratture e per un miglioramento della qualità della vita sono fondamentali: conoscenza della malattia da parte del paziente e della sua famiglia, ambienti (casa, scuola, lavoro) senza barriere architettoniche o possibili ostacoli rischiosi, gestione multidisciplinare (fisiatrica, ortopedica, internistica, odontostomatologica e otorinolaringoiatrica), presidi di supporto (protesici o chirurgici) per il superamento o il miglioramento di limitazioni funzionali, interventi ortopedici correttivi, esercizi fisioterapici e riabilitativi, esercizio fisico adeguato.
Terapia farmacologica
Il trattamento con rhGH ha dato in alcuni casi un aumento dell’altezza finale.
Numerosi sono stati i tentativi per ridurre le fratture, con risultati spesso deludenti. La terapia anti-riassorbitiva con i bisfosfonati sia in bambini che in soggetti adulti è risultata la sola efficace nel ridurre le fratture, aumentare la densità minerale ossea, ridurre il dolore osseo, aumentare il senso di benessere. Non sono noti gli effeti sulla prevenzione della deformità delle ossa lunghe e sulla scoliosi. Non sembra vi sia un effetto negativo sulla crescita staturale. La molecola più utilizzata in letteratura è il pamidronato ev, ma sono stati condotti studi anche con la terapia orale (alendronato, risedronato, olpadronato). In Italia l’unico farmaco registrato per il trattamento dell’OI è il neridronato somministrato ev a cicli di 3 mesi, ad un dosaggio di 2 mg/kg fino ad un massimo di 100 mg.
Gli effetti collaterali più frequentemente segnalati sono una sintomatologia simil-influenzale immediatamente dopo la somministrazione e il giorno dopo; raramente ipocalcemia. Non sono stati descritti ad oggi casi di osteonecrosi della mandibola.
Per quanto tempo? Gli effetti a lungo termini non sono noti.
Qual è il farmaco migliore? I dati più consistenti in letteratura sono con il pamidronato ev. Non esistono grossi studi di confronto.
Qual è la via di somministrazione migliore? I risultati migliori sono stati ottenuti con la somministrazione ev, ma va detto che non ci sono studi di confronto. Inoltre la somministrazione ev ha una migliore compliance, non richiede il rituale di somministrazione della via orale e può essere assunto anche da pazienti con problemi gastrointestinali.
MALATI FAMOSI
Michel Petrucciani (pianista jazz), Fabiano Lioi (attore e musicista), Samuel L. Jackson nei panni di Elijah Price in Unbreakable.
LETTURE CONSIGLIATE
- Osteogenesis Imperfecta. Harrison's Online. Chapter 363. Part 16.
- Glorieux FH. Osteogenesis imperfecta. Best Pract Res Clin Rheumatol 2008, 22: 85-100.
- Marini J. Ostegenesi Imperfecta. Chapter 16. Endotext.org
Osteopetrosi
Bruno Madeo
UOC Endocrinologia e Malattie del Metabolismo, Ospedale (NOCSAE) di Modena
L’osteopetrosi (OPT), o “malattia delle ossa di marmo”, è un insieme clinicamente eterogeneo di patologie genetiche molto rare, caratterizzate da un notevole incremento della massa ossea.
Epidemiologia
È una condizione molto rara. L’incidenza della forma autosomica recessiva è di circa 1 su 250.000 nuovi nati, mentre quella autosomica dominante è di 5 su 100.000.
Patogenesi
È causata da un’anomalia funzionale o della differenziazione degli osteoclasti, cellule cruciali per il riassorbimento osseo, in presenza di una normale attività osteoblastica. I geni coinvolti nella patogenesi, descritti sinora, sono una decina (tabella), elegantemente semplificati in una figura contenuta in un’esaustiva review sull’argomento di Del Fattore et al (2008).
Classificazione e aspetto clinico
L’OPT comprende un gruppo clinicamente e geneticamente eterogeneo di condizioni classificate in 4 tipi:
- autosomica recessiva severa (ARS)
- autosomica recessiva intermedia (ARI)
- autosomica dominante (malattia di Albers-Schönberg) (AD)
- legata al cromosoma X (LX).
Il grado di severità è molto variabile e va da forme asintomatiche, come in casi di AD, a forme letali precoci, come l’ARS o l’LX. Caratteristico è l’aumento di massa ossea, focale o diffuso a seconda del tipo, che può portare ad avere:
- un osso più duro ma allo stesso tempo più fragile (aumentato rischio fratturativo);
- caratteristiche alterazioni scheletriche: macrocefalia, particolare aspetto cranio-facciale, bassa statura;
- effetti indiretti, generalmente compressivi, su altri distretti come il midollo osseo (pancitopenia, anemia) o il sistema nervoso (compressione, in particolare dei nervi cranici, con cecità, sordità, paralisi facciali, disfagia).
La tabella mostra i vari quadri clinici.
| Caratteristiche dei diversi tipi di osteopetrosi | |||||||||
| Forma ed ereditarietà | ARS | ARI | AD | X-linked | |||||
| Gravità clinica | +++/++++ | ++/+++ | ++/+++ | ++++ | |||||
| Esordio | Neonatale | Infantile | Adolescenziale | Neonatale | |||||
| Aspettativa di vita | Mortalità infantile | Ridotta | Normale | Mortalità infantile | |||||
| Gene mutato | TCIRGI CLCN7 OSTMI RANK-L RANK |
CLCN7 PLEKHMI |
CLCN7 | IKBKG (NEMO) |
|||||
| Manifestazioni scheletriche | Osteosclerosi | diffusa | diffusa | focale | diffusa | ||||
| Fratture patologiche | + | + | + | +/- | |||||
| Osteomielite | + | + | + | +/- | |||||
| Anomalie dentizione | + | + | +/- | +/- | |||||
| Manifestazioni extra-scheletriche | Severa anemia, pancitopenia, emopoiesi extra-midollare, metaplasia mieloide | + | + | - | - | ||||
| Modesta anemia | - | - | + | - | |||||
| Compressione nervi cranici | + | +/- | + | - | |||||
| Immuno-deficienza, infezioni gravi, linfedema | - | - | - | + | |||||
| Epatosplenomegalia, macrocefalia, idrocefalia, ritardo mentale, ipocalcemia | + | - | - | - | |||||
+/- presente/assente; + presente/lieve; ++ moderato; +++moderato-severo; ++++severo
Diagnosi
Si basa essenzialmente sulla clinica e sull’aspetto radiologico dello scheletro.
Esami bioumorali. I livelli di fosfatasi acida tartrato-resistente (TRAP) e dell’isoenzima cerebrale della creatinchinasi (CK-BB) sono spesso aumentati nel tipo AD. A volte nelle forme severe si riscontrano ipocalcemia associata a valori aumentati di PTH e 1,25(OH)2-vitamina D.
Densitometria ossea (DEXA). Non è necessaria per la diagnosi. Documenta naturalmente valori densitometrici aumentati.
Esami radiologici. L’Rx è fondamentale, poiché consente di evidenziare una serie di aspetti radiologici tipici:
- sclerosi diffusa, in particolare in sede vertebrale, nelle ossa lunghe e nel cranio ⇒ ARS
- aspetto di “osso nell’osso” soprattutto nelle falangi e nelle vertebre ⇒ ARS
- sclerosi focale, soprattutto della base del cranio, del bacino e della colonna vertebrale (aspetto “a sandwich” per addensamento del piatto superiore ed inferiore vertebrale, aspetto “a maglia da rugby” del rachide) ⇒ AD
- Alterazioni metafisaria delle ossa lunghe:
- “aspetto ad imbuto” ⇒ AD;
- caratteristiche bande radiolucenti ⇒ ARS
Biopsia ossea. Può essere utile per la diagnosi soprattutto nell’ARS, ovviamente da limitare per la sua invasività.
Analisi molecolare. Lo studio genetico può essere utile per una conferma diagnostica e per eventuali consulenze genetiche. È possibile una diagnosi pre-impianto o prenatale nelle famiglie a rischio.
Diagnosi differenziale
Vanno escluse, clinicamente e con gli appropriati esami, le altre condizioni di osteosclerosi (picnodisostosi, osteopoichilosi, disosteosclerosi, osteopatia striata) e le condizioni di osteosclerosi secondaria (forma sclerotica della malattia di Paget, fluorosi, mielofibrosi, linfoma, metastasi osteoblastiche, avvelenamento da berillio, da piombo o da bismuto).
Terapia
Al momento non esistono terapie efficaci.
L’unico trattamento possibile è il trapianto di cellule staminali emopoietiche, che è tanto più efficace quanto più è precoce (entro i primi 3 mesi di vita), ma è da limitare alle forme severe (ARS) considerata l’elevata mortalità e morbilità (rigetto, ritardo nella ricostituzione emopoietica, malattia veno-occlusiva, ipertensione polmonare, crisi ipercalcemiche).
Sono stati utilizzati anche trattamenti farmacologici, con risultati interessanti nel caso del gamma-interferon 1b, o poco più che deludenti, con prednisone, calcitriolo, ormone paratiroideo.
Pertanto, il trattamento è essenzialmente di supporto e richiede un approccio multidisciplinare con figure specialistiche esperte per la gestione delle varie complicanze.
- Fratture e artrite richiedono un ortopedico esperto soprattutto per la gestione delle complicanze secondarie: mancata saldatura delle fratture, osteomielite.
- Le crisi ipocalcemiche richiedono ovviamente un supplemento di calcio e vitamina D.
- L'insufficienza midollare può richiedere trasfusioni di sangue intero o di piastrine.
- Un ritardo dello sviluppo o crisi convulsive normocalcemiche richiedono un’attenta valutazione neurologica.
- L’atrofia del nervo ottico o la necessità di decomprimere il nervo ottico richiedono un attento follow-up oculistico.
- I problemi dentari richiedono una corretta igiene orale e un’adeguata valutazione odontostomatologica.
Letture consigliate
- Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone 2008, 42: 19-29.
- Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis 2009, 4: 5.
Ipofosfatasia
Flavia Pugliese
Unità Operativa di Endocrinologia, IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG)
(aggiornato al 17/3/2025)
DEFINIZIONE
L’ipofosfatasia (HPP) è una malattia genetica causata da mutazioni del gene ALPL, responsabile di una riduzione dell'attività della fosfatasi alcalina tessuto-non specifica (TNSALP), che determina alterazioni della mineralizzazione scheletrica.
EPIDEMIOLOGIA
È una malattia rara: in Europa si stima che la forma grave della patologia abbia una prevalenza di 1:300.000 nati vivi.
FISIOPATOLOGIA
La fosfatasi alcalina (ALP) è un enzima quasi ubiquitario, in grado di defosforilare una serie di substrati extra-cellulari. Di essa sono noti quattro iso-enzimi, codificati da quattro differenti geni, espressi in diversi tessuti:
- intestinale, codificato dal gene ALPI;
- placentare, codificato dal gene ALPP;
- germinale, codificato dal gene ALPPL2;
- tessuto non-specifico, codificato dal gene ALPL, localizzato sul cromosoma 1, espresso in molti tessuti, tra cui osso, fegato, rene, denti.
La TNSALP rappresenta il 95% dell’attività totale dell'ALP misurata nel siero. È un omodimero, composto da due subunità identiche, con siti di legame per lo zinco (catalizzatore enzimatico), per il magnesio (cofattore) e per il calcio.
Le mutazioni del gene ALPL, che determinano la riduzione dell’attività della TNSALP, provocano un accumulo dei tre substrati:
- il pirofosfato inorganico (PPi) è un potente inibitore della mineralizzazione scheletrica e il suo accumulo da insufficiente attività della TNSALP osteoblastica determina ridotta mineralizzazione di ossa e denti, con sviluppo di rachitismo o osteomalacia di grado variabile. La ridotta mineralizzazione scheletrica può causare anche una serie di alterazioni del metabolismo minerale, in particolare ipercalcemia, con conseguente riduzione del PTH e ipercalciuria;
- il piridossal-5-fosfato (PLP) (metabolita attivo della vitamina B6), la cui mancata defosforilazione a piridossale impedisce a questo metabolita di attraversare la barriera emato-encefalica, dove è cofattore essenziale per la sintesi dei neurotrasmettitori GABA, serotonina e dopamina. La mancanza di vitamina B6 a livello encefalico è associata a crisi epilettiche nei bambini;
- la fosfoetanolamina (PEA), di cui determina la defosforilazione e del cui accumulo non sono note le conseguenze.
La ridotta attività della TNSALP, insieme all’eccesso di PPi e alla riduzione del PTH, possono comportare un incremento del riassorbimento renale del fosfato, causando iperfosfatemia e formazione di cristalli di pirofosfato di calcio di-idrato, che possono portare allo sviluppo di pseudo-gotta e nefrocalcinosi. TNSALP è coinvolta anche nella proliferazione e nella differenziazione delle cellule staminali neurali, nella mielinizzazione e nella crescita assonale, e nella maturazione e mantenimento delle sinapsi.
GENETICA
Sono state descritte più di 400 mutazioni di ALPL causanti ipofosfatasia, ereditabile come malattia autosomica recessiva o dominante:
- le forme gravi sono più spesso sostenute da mutazioni in omozigosi o in eterozigosi composta;
- le forme moderate sono più spesso associate a varianti missenso, con effetto dominante negativo;
- le forme più lievi dipendono in genere da meccanismi di aplo-insufficienza.
Non esiste però una diretta correlazione genotipo-fenotipo e l’espressione clinica è variabile anche in presenza della stessa mutazione. Sono descritte inoltre numerosissime varianti di incerto significato (VUS), con un database in continua espansione e periodiche riclassificazioni per valutare i punteggi fenotipici registrati per ogni variante. In caso di mancata identificazione di una mutazione o VUS del gene ALPL, è opportuno prendere in considerazione diagnosi alternative, sebbene alcune varianti possano non essere identificate con le tecniche di sequenziamento comunemente utilizzate.
CLINICA
La presentazione clinica dell’HPP può essere estremamente variabile.
Le principali manifestazioni cliniche sono correlate al difetto di mineralizzazione scheletrica e alle sue conseguenze, con quadri che possono andare da gravi deformazioni scheletriche, condizionanti insufficienza respiratoria per alterazioni della gabbia toracica, a quadri di rachitismo/osteomalacia con deformità ossee, bassa statura, fratture recidivanti (specie metatarsali), fratture atipiche e pseudo-fratture. Il difetto di mineralizzazione a livello dentario comporta spesso la perdita precoce della dentatura decidua, l’edentulia precoce nell’adulto e parodontopatie ricorrenti.
Sono frequenti il dolore muscolo-scheletrico cronico e le calcificazioni ectopiche, in particolare lo sviluppo di nefro-calcinosi e condro-calcinosi.
Tra le manifestazioni neurologiche nei bambini sono tipiche le crisi epilettiche responsive a vitamina B6; negli adulti si osservano invece manifestazioni neuro-psichiatriche, con astenia/affaticamento, cefalea, disturbi del sonno, disturbi del cammino, vertigini, depressione e ansia.
Sono stati descritti anche casi pauci- o asintomatici, in cui la diagnosi è stata effettuata incidentalmente osservando bassi livelli ALP, associati a incremento dei substrati e a riscontro di mutazioni della ALPL.
L’attuale classificazione clinica dell’ipofosfatasia si base sull’età di insorgenza e sulla gravità della sintomatologia. Si distinguono quindi le forme:
- peri-natali, con manifestazioni cliniche gravi evidenti già durante la gestazione o alla nascita, caratterizzate principalmente da deficit di mineralizzazione e importanti deformità scheletriche, cranio-sinostosi, insufficienza respiratoria, a prognosi sfavorevole;
- neonatale, con esordio entro i primi 6 mesi di vita, con mineralizzazione e deformità scheletriche simili alla forma peri-natale, insufficienza respiratoria, ipercalcemia, crisi epilettiche, con prognosi sfavorevole;
- pre-natale benigna, con evidenza di deficit di mineralizzazione e deformità scheletriche già durante la gestazione o alla nascita, ma senza manifestazioni cliniche gravi, in particolare con minor difetto di mineralizzazione e senza alterazioni della gabbia toracica, con prognosi favorevole;
- infantile grave, a esordio dopo i 6 mesi di vita, con manifestazioni cliniche gravi ma senza prognosi infausta;
- infantile lieve, con manifestazioni spesso aspecifiche, tra cui principalmente dolore muscolo-scheletrico e fratture dopo traumi lievi;
- dell’adulto, con manifestazioni tipiche dell’osteomalacia, tra cui fratture da stress, in particolare metatarsali, pseudo-fratture, fratture femorali atipiche, osteomalacia, condro-calcinosi, artrosi, dolori ossei e muscolari, pseudo-gotta, nefro-calcinosi; possono essere presenti anomalie dentarie;
- odonto-ipofosfatasia, caratterizzata da interessamento esclusivo dei denti, con perdita precoce dei denti decidui (prima dei 4 anni), odontopatie/parodontopatie recidivanti e edentulia precoce.
Il sospetto clinico dovrebbe sorgere in caso di ipofosfatasemia persistente o in presenza di manifestazioni cliniche tipiche della patologia, in particolare, edentulia precoce o perdita precoce della dentatura decidua (prima dei 4 anni), dolore muscolare o osseo cronico, debolezza muscolare, ritardo motorio, fratture da stress, pseudo-fratture, fratture femorali atipiche, fratture da fragilità recidivanti, nefro-calcinosi, pseudo-gotta, calcificazioni ectopiche.
DIAGNOSI
La diagnosi è complessa a causa dell’eterogeneità clinica e il ritardo diagnostico medio è di 5.7 anni. Si basa su una serie di parametri biochimici, radiologici e clinici.
Per la diagnosi è necessaria la dimostrazione di livelli di ALP persistentemente bassi in rapporto a età e sesso (tabella 1).
| Tabella 1 Limite inferiore di normalità dell'ALP |
|
| Età | U/L (M/F) |
| < 1 mese | 60 |
| 1-11 mesi | 70 |
| 1-3 anni | 125 |
| 4-11 anni | 150 |
| 12-13 anni | 160/110 |
| 14-15 anni | 130/55 |
| 16-19 anni | 60/40 |
| > 20 anni | 40 |
Gli altri parametri biochimici, radiologici e clinici sono presenti invece in una percentuale variabile. Recentemente l’HPP International Working Group ha identificato una serie di criteri diagnostici, distinti in maggiori (se descritti in più del 50% dei casi clinici pubblicati o raccomandati da più del 50% degli studi) e minori (tabella 2).
| Tabella 2 Criteri diagnostici |
||
| Adulti | Criteri maggiori |
|
| Criteri minori |
|
|
| Bambini | Criteri maggiori |
|
| Criteri minori |
|
|
In particolare, tra i parametri biochimici tipici della patologia vi sono l’incremento dei substrati, tra cui la vitamina B6 plasmatica (da dosare previa sospensione di eventuali supplementazioni per almeno una settimana), la PEA e il PPi urinari. L’ipercalcemia con PTH soppresso, l’ipercalciuria e l’iperfosfatemia possono essere osservate ma non rappresentano un criterio diagnostico di HPP. L’identificazione di una mutazione del gene ALPL può confermare la diagnosi.
Gli esami radiologici possono mostrare più tipicamente segni di rachitismo, fratture femorali atipiche, pseudo-fratture, fratture da stress (in particolare metatarsali), scarso consolidamento delle fratture, condro-calcinosi, nefro-calcinosi. Non sono invece tipiche dell’HPP bassa massa ossea o fratture vertebrali.
- Forma in utero: può essere individuata ecograficamente durante il secondo trimestre di gestazione; alla radiografia è presente una quasi totale mancanza di mineralizzazione ossea e/o alterazioni tipiche del rachitismo.
- Ipofosfatasia del lattante: l’esame radiologico mostra demineralizzazione scheletrica generalizzata e progressiva, e i segni tipici del rachitismo.
- Pazienti pediatrici: le radiografie delle ossa lunghe possono mostrare difetti nella cartilagine a livello metafisario, denominate lingue; inoltre, in presenza di cranio-sinostosi, il cranio può avere un aspetto a “rame battuto”.
- Forma dell’adulto: la radiologia può mostrare pseudo-fratture indicative di osteomalacia, osteopenia generalizzata e condro-calcinosi. La densitometria (DEXA) può evidenziare la presenza di bassa massa ossea nel femore e nella colonna vertebrale.
- Odonto-ipofosfatasia: la radiografia dentale mostra ridotto osso alveolare, camere pulpari e canali radicolari allargati.
Dal punto di vista clinico, i sintomi più frequentemente descritti, che possono essere utilizzati come criterio diagnostico, sono l’edentulia precoce o la perdita precoce dei denti decidui, il dolore muscolo-scheletrico cronico (per quanto aspecifico), le convulsioni responsive a vitamina B6, il ritardo di crescita e il ritardo motorio.
L’HPP International Working Group propone quindi che, in soggetti con valori persistentemente bassi di ALP, la diagnosi di HPP possa essere posta in presenza di 2 criteri maggiori oppure di 1 criterio maggiore + 2 criteri minori (tabella 1).
Analisi molecolare
Con il sequenziamento, sono state identificate circa il 95% delle mutazioni responsabili delle forme gravi (perinatale e neonatale) della malattia. La consulenza genetica è complicata da variabilità della trasmissione (autosomica dominante o autosomica recessiva), dall'esistenza della forma prenatale benigna e dalla penetranza incompleta del gene-malattia. L'analisi mutazionale del DNA sui villi coriali rende possibile la diagnosi prenatale dell'ipofosfatasia grave.
DIAGNOSI DIFFERENZIALE
Dal punto di vista clinico/radiologico, la diagnosi differenziale dipende molto dalle manifestazioni del soggetto. In base all’età di insorgenza e all’entità del danno osseo e dei sintomi associati, la diagnosi differenziale si pone in genere con altre forme di rachitismo (da deficit di vitamina D, da ipofosfatemia), con l’osteogenesi imperfetta, con l’osteoporosi, tutte forme che in genere, a differenza dell’HPP, presentano livelli di ALP normali o incrementati.
Dal punto di vista biochimico, è necessario escludere altre condizioni associate a bassi livelli di ALP (ipofosfatasemia), le cui principali cause sono elencate nella tabella 3.
| Tabella 3 Cause di ipofosfatasemia |
|
| Farmaci | Anti-riassorbitivi Chemioterapici Vitamina D (intossicazione) |
| Malattie endocrinologiche | Ipoparatiroidismo Ipotiroidismo Ipercortisolismo Ritardo di crescita |
| Malattie ematologiche | Anemia perniciosa Trasfusioni massive Malattie mielo-proliferative Mieloma multiplo |
| Deficit nutrizionali | Magnesio Zinco Vitamina C Vitamina B6 Vitamina B12 Folati (?) Malattia celiaca |
| Alterazioni laboratoristiche | EDTA/citrato/ossalato nella provetta (perché legano il Mg) Emolisi |
| Altro | Osteodistrofia renale con osso adinamico Malattie acute gravi Traumi o chirurgia maggiore Malattia di Wilson Acondroplasia |
Una ipofosfatasemia transitoria è comune in caso di condizioni acute, tra cui sepsi, interventi chirurgici maggiori, traumi. Ipofosfatasemia persistente si osserva in alcune patologie endocrinologiche misconosciute e quindi non in trattamento (ipoparatiroidismo, ipotiroidismo, ipercortisolismo), patologie associate a malassorbimento, deficit di zinco o di magnesio, patologie ematologiche, malattia di Wilson; bassi livelli di ALP possono essere causati da terapia con anti-riassorbitivi, chemioterapici e da intossicazione da vitamina D.
| Tabella 4 Diagnosi differenziale biochimica |
|||||
| Ipofosfatasia |
Rachitismo nutrizionale |
Rachitismo ipofosfatemico X-linked | Osteogenesi imperfetta | Osteoporosi primaria | |
| ALP | diminuita | aumentata | aumentata | = | normale |
| Calcemia | aumentata/= | ridotta | normale | = | normale |
| Fosfatemia | aumentata/= | ridotta | ridotta | = | normale |
| PTH | diminuito/= | aumentato | aumentato/= | = | normale |
| Vitamina D | = | ridotta | ridotta/= | = | normale |
TRATTAMENTO
L’obiettivo è il controllo della sintomatologia e la prevenzione o il trattamento delle complicanze.
Il trattamento deve essere individualizzato in base al quadro clinico: il paziente deve quindi essere seguito in un centro specializzato in malattie osteo-metaboliche, che possa garantire un approccio multi-disciplinare, con il coinvolgimento di diversi specialisti (endocrinologo, ortopedico, odontoiatra, fisiatra e, nelle forme gravi infantili, anche pneumologo, neurologo, neurochirurgo).
I trattamenti di supporto più frequentemente utilizzati sono rappresentati da fisioterapia, riabilitazione, terapia occupazionale, farmaci anti-infiammatori/anti-dolorifici. I livelli di vitamina D devono essere mantenuti nel range di normalità (> 30 ng/mL) per prevenire lo sviluppo di iperparatiroidismo secondario; deve però essere evitato l’eccesso di supplementazione per ridurre il rischio di ipercalcemia e ipercalciuria. Nei soggetti ipercalcemici a volte può essere necessaria una dieta ipocalcica. Nei soggetti con manifestazioni epilettiche può essere utilizzata la piridossina. In base alla evoluzione del quadro clinico possono essere necessari supporto respiratorio, trattamenti ortopedici, trattamenti chirurgici per la cranio-sinostosi.
Nel 2015 è stata approvata per il trattamento dell’ipofosfatasia una terapia enzimatica sostitutiva. Asfotase alfa è una forma di TNSALP umana ricombinante, a somministrazione sotto-cutanea. Il farmaco si è dimostrato efficace nelle forme gravi, determinando miglioramento di sopravvivenza, funzionalità respiratoria, funzionalità motoria, crescita, qualità di vita, dolore, mineralizzazione scheletrica, oltre a miglioramento dei parametri radiologici. I benefici della terapia enzimatica sostitutiva sono stati dimostrati sia per forme a esordio infantile che per le forme gravi dell’adulto. Ancora discusso è invece l’utilizzo di Asfotase alfa nelle forme più lievi, in cui deve essere sempre valutato il rapporto costo/rischio/beneficio, in considerazione anche dei potenziali effetti collaterali della terapia, i più frequenti dei quali sono le calcificazioni ectopiche, le reazioni in sede di iniezione (in particolare lipodistrofia), l’ipocalcemia.
I pazienti in trattamento devono essere monitorati a 3 e 12 mesi, quindi annualmente. Il monitoraggio comprende:
- dosaggio di ALP, che risulta elevata durante il trattamento, come indice di aderenza alla terapia, ma non deve essere usata per la titolazione del trattamento;
- dosaggio di vitamina B6, la cui riduzione dimostra l’efficacia del trattamento;
- dosaggio di calcemia e PTH, per monitorare l’eventuale sviluppo di ipocalcemia da sindrome dell’osso affamato;
- dosaggio di vitamina D per mantenere livelli adeguati e ridurre il rischio di sviluppare ipocalcemia;
- dosaggio fosfatemia per monitorare l’eventuale sviluppo di iperfosfatemia (descritto durante terapia con Asfotase alfa) e ridurre il rischio di calcificazioni ectopiche;
- valutazione di funzione renale ed epatica, emocromo, elettroliti;
- valutazioni cliniche del dolore (con questionari specifici), della funzione motoria (ad esempio test del cammino), della forza muscolare;
- monitoraggio radiologico dell’età ossea e dei segni di rachitismo;
- nei bambini monitoraggio della crescita e delle tappe di sviluppo motorio;
- monitoraggio della funzionalità respiratoria;
- imaging renale e valutazioni oculistiche per valutare lo sviluppo di calcificazioni ectopiche;
- monitoraggio della salute dentale.
La risposta al trattamento è valutata osservando il miglioramento della mineralizzazione scheletrica e dei sintomi clinici, con riduzione dei valori di vitamina B6 e tendenziale incremento del PTH. Rappresentano segni di undertreatment valori persistentemente elevati di vitamina B6, calcemia, calciuria e PTH soppresso, oltre che la scarsa risposta radiologica nei bambini; segni di overtreatment, più difficili da evidenziare, possono essere rappresentati dallo sviluppo di calcificazioni ectopiche.
Negli adulti con ipofosfatasia sono stati utilizzati off-label anche il teriparatide e il romosozumab. Il teriparatide ha mostrato in alcuni casi un incremento degli indici di turn-over scheletrico e della densità minerale ossea e più rapida guarigione delle fratture. Anche il romosozumab ha determinato incremento dei marcatori di osteosintesi e della densità minerale ossea. Tali benefici restano tuttavia limitati al periodo di somministrazione del farmaco e non è noto l’effetto di un utilizzo prolungato e della loro sospensione.
Nei soggetti con ipofosfatasia sono assolutamente controindicati i farmaci anti-riassorbitivi (bisfosfonati, denosumab), che determinano peggioramento della sintomatologia, della mineralizzazione scheletrica e incremento del rischio di fratture atipiche femorali.
BIBLIOGRAFIA
- Montero-Lopez R, et al. Challenges in hypophosphatasia: suspicion, diagnosis, genetics, managementi and follow up. Horm Res Paediatr 2024, DOI: 10.1159/000540692.
- Salles Reis F, Lazaretti-Castro M. Hypophosphatasia: from birth to adulthood. Arch Endocrinol Metab 2023, 67: e000626.
Osteodistrofia renale
Bruno Madeo1 & Fabio Vescini2
1UOC Endocrinologia e Malattie del Metabolismo, Ospedale (NOCSAE) di Modena
2SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria S. Maria della Misericordia, Udine
DEFINIZIONE
L’osteodistrofia renale (ODR) è un’alterazione della morfologia ossea tipica dei pazienti con insufficienza renale cronica (IRC) e rappresenta uno dei componenti di un disordine sistemico definito come alterazioni del metabolismo minerale e osseo secondarie a malattia renale cronica (Chronic kidney disease-mineral and bone disorder - CKD-MBD) (vedi linee guida Kidney Disease: Improving Global Outcomes - KDIGO).
Il CKD-MBD è caratterizzato da una o più dei seguenti aspetti:
- alterazione del metabolismo del calcio, del fosforo, del PTH o della vitamina D;
- alterazione del turnover, della mineralizzazione e del volume osseo, della crescita lineare o della forza muscolare;
- calcificazioni vascolari o di altri tessuti molli.
PATOGENESI
La capacità del rene di eliminare adeguatamente il fosforo inizia a diminuire già da un’IRC allo stadio 3 (tabella). Ciò si accompagna a iperfosforemia, che porta ad aumento del PTH, ridotta conversione di 25(OH)D in 1,25(OH)2D e rialzo dei livelli di FGF-23. La riduzione dell’1,25(OH)2D porta a una riduzione dell’assorbimento intestinale di calcio ed insieme causano un aumento del PTH. Tuttavia, a livello renale, né il rialzo di PTH si accompagna al normale aumento di escrezione di fosforo e riassorbimento di calcio, né quello di FGF-23 all’escrezione di fosforo. In aggiunta, vi sono evidenze di una down-regulation del recettore della vitamina D a livello tissutale periferico e di una resistenza all’azione del PTH.
| Stadi dell’insufficienza renale cronica secondo la Classificazione della KDIGO proposta nel 2002 ed aggiornata nel 2004 |
||
| Stadio | Descrizione | Filtrato glomerulare (GFR mL/min/1.73 m2) |
| 1 | Segni di danno renale con GFR normale o aumentato | ≥ 90 |
| 2 | Segni di danno renale con lieve riduzione del GFR | 60-89 |
| 3 | Riduzione moderata del GFR | 30-59 |
| 4 | Riduzione grave del GFR | 15-29 |
| 5 | Fallimento renale | < 15 |
| 5D | dialisi | |
Si aggiunge T ad ogni stadio (1-5) in caso di trapianto renale
CLASSIFICAZIONE
L’ODR è caratterizzata da differenti alterazioni ossee, suddivise inizialmente in base al turnover in basso (osso adinamico, osteomalacia), moderato (osteopatia lieve) ed elevato (osteopatia mista, osteite fibrosa). Recentemente è stata introdotta, invece, una nuova classificazione (sistema TMV), che tiene conto oltre che del turnover, del volume osseo (basso, medio, alto) e della mineralizzazione (normale e anormale).
ASPETTO CLINICO
Nei bambini l’ODR causa deformità scheletriche tipo il rachitismo e ritardo della crescita. Deformità scheletriche (cifosi/scoliosi, dissoluzione delle porzioni distali delle dita, tumori bruni spesso sede di fratture) sono presenti anche negli adulti in casi di severa osteite fibrosa. Tuttavia, il sintomo più frequente è il dolore, che aumenta con l’attività fisica e il carico articolare, può interessare sedi differenti (rachide inferiore, spalle, mani, anche, ginocchia e gambe) e può variare nello stesso soggetto per presentazione e localizzazione. È presente inoltre astenia, in genere prossimale, caratterizzata da difficoltà ad assumere la posizione eretta, facile affaticabilità e diminuita resistenza allo sforzo. Frequenti sono anche le fratture del femore, che rappresentano la complicanza più comune della ODR.
Le calcificazioni dei tessuti molli, dovute all’iperfosforemia e all’aumento del prodotto calcio-fosforo, si osservano più spesso in sede peri-articolare (peri-artrite calcifica), poi a livello parenchimale (cardiaco, polmonare, oculare, renale), ma sono particolarmente pericolose a livello vascolare, poiché, soprattutto in casi di grave iperparatiroidismo secondario, possono portare a ischemia e gangrena.
Il prurito, a volte lamentato dai pazienti, è stato attribuito all’aumentata quantità di calcio cutaneo in casi con ipercalcemia o severa iperfosfatemia.
DIAGNOSI
La diagnosi si basa sullo studio istopatologico, anche se nella pratica clinica sono comunque utili i dati di laboratorio e le indagini radiologiche.
Esami bioumorali
Benchè gli esami bioumorali non siano in grado di identificare tutti i tipi di ODR, il dosaggio dei livelli sierici di calcio, fosforo, paratormone intatto (PTHi) e fosfatasi alcalina è indispensabile per identificare le modificazioni del metabolismo minerale nei vari stadi di IRC e per il monitoraggio della terapia. Le linee guida della KDIGO consigliano di iniziare il monitoraggio dallo stadio 2 nei bambini e 3 negli adulti, da proseguire con una frequenza che varia dai 6-12 mesi iniziali agli 1-3 mesi negli stadi avanzati o in caso di trattamento. Utile anche il dosaggio della 25(OH)D, soprattutto negli stadi avanzati, per documentare e quindi correggere uno stato di insufficienza. Il PTHi e la fosfatasi alcalina ossea sono in grado di fornire informazioni sul turnover osseo, rispettivamente alto o basso, solo in caso valori marcatamente elevati o ridotti. Tutti gli altri marcatori di turnover osseo hanno una scarsa utilità clinica, perché dipendono, per la loro escrezione, esclusivamente dalla filtrazione glomerulare renale. I livelli sierici di alluminio non sono diagnostici nell’ODR da intossicazione di alluminio.
Densitometria ossea (DEXA)
L’esame densitometrico osseo nel paziente con ODR non è in grado di predire il rischio di frattura, né consente l’identificazione dei sottotipi di ODR. Tuttavia può essere utilizzato nella valutazione complessiva del paziente con ODR. Mantiene invece la sua utilità clinica negli stadi iniziali (1-2) di IRC e nei pazienti trapiantati.
Esami radiologici
Lo studio radiologico non consente la diagnosi dei vari sottotipi di ODR, ma può mostrare la presenza di alcune anomalie tipiche:
- riassorbimento sub-periosteo: metafisi prossimale e distale delle falangi, parte distale della clavicola, pelvi e margine inferiore delle coste;
- cranio "sale e pepe”: fenomeni di riassorbimento portano a un aspetto granulare e lacunoso, associato ad aree di osteosclerosi;
- aspetto del rachide a maglie di giocatori di rugby (“rugger-jersey spine”): aspetto sclerotico delle superfici inferiori e superiori delle vertebre;
- manifestazioni osteomalaciche: nel bambino assumono i quadri tipici del rachitismo, mentre nell’adulto l’unico segno possono essere le “strie di Looser” (o pseudofratture).
Biopsia ossea
La diagnosi di ODR si basa sulla biopsia ossea a livello della cresta iliaca. La somministrazione di tetraciclina 3 settimane e 3-5 giorni prima dell’esame consente la valutazione del turnover osseo, sfruttando la capacità del farmaco di depositarsi nell’osso neoformato. I parametri valutati sono il turnover, la mineralizzazione e il volume, utili per classificazione TMV. Essendo un esame invasivo, la biopsia va limitata ai pazienti con stadi avanzati di IRC (> 3) e con particolari condizioni fra le quali: fratture, ipercalcemia o ipofosfatemia non spiegabili, dolore osseo persistente, possibile intossicazione da alluminio, prima di un trattamento con bisfosfonati in pazienti con CKD-MBD.
TERAPIA
La decisione terapeutica va basata sull’andamento più che sui singoli esami bioumorali. Il trattamento può avere degli ottimi risultati se correttamente focalizzato sul meccanismo patogenetico specifico.
L’obiettivo è di mantenere livelli normali di calcio e fosforo e minimizzare l’esposizione all’alluminio.
La restrizione di fosfato dovrebbe iniziare relativamente presto, tuttavia diete povere in fosforo sono difficili da attuare. Pertanto solitamente, già dallo stadio 3, è necessario somministrare sali leganti il fosforo, come il calcio carbonato, acetato, o citrato.
Negli stadi iniziali è sufficiente una modesta supplementazione di vitamina D, per mantenere i livelli di 1,25(OH)2D, ma in caso di progressivo rialzo dei valori di PTH, nonostante le precedenti correzioni, bisogna somministrare calcitriolo a basse dosi (da 0.25 a 0.5 µg/die).
La terapia con calcio e vitamina D va ridotta in caso di ipercalcemia persistente o ricorrente, in presenza di calcificazioni arteriose e/o osso adinamico. In caso invece di progressivo rialzo dei valori di PTH, è possibile aggiungere un calciomimetico (cinacalcet), che può invece causare ipocalcemia.
I livelli ottimali di PTHi non sono noti: nei pazienti dializzati si suggeriscono livelli di PTH fra 2 e 9 volte al di sopra dei limiti di norma, mentre in caso di riduzione dei valori di PTHi due volte inferiori ai limiti superiori è consigliabile ridurre la terapia.
La paratiroidectomia va presa in considerazione in casi di iperparatiroidismo severo che non rispondono alla terapia farmacologica, ipercalcemia persistente in pazienti con fallimento renale, prurito intrattabile, calcificazioni extra-cellulari e lesioni scheletriche severe. Mentre, l’intervento dovrebbe essere evitato in pazienti con osteopatia indotta da alluminio, perché i sintomi possono peggiorare.
I pazienti con osteoporosi e/o alto rischio di frattura secondo la definizione dell’OMS vanno trattati come la popolazione generale nei casi iniziali di IRC (stadio1-2) o allo stadio 3 ma con valori di PTH normali. Negli stadi più avanzati bisogna tenere in considerazione l’aspetto istologico.
Il trapianto renale corregge molte alterazioni biochimiche che portano all’osteodistrofia renale, ma la malattia ossea può progredire. Una delle principali preoccupazioni, in particolare nei pazienti anziani, è l’osteoporosi progressiva peggiorata dall’utilizzo di glucocorticoidi e immunosoppressori.
I bambini con deficit di crescita possono essere trattati con GH ricombinante.
LETTURE CONSIGLIATE
- Block GA, Martin KJ, de Francisco AL, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med 2004, 350: 1516-25.
- Kandula P, Dobre M, Schold JD, et al. Vitamin D supplementation in chronic kidney disease: a systematic review and meta-analysis of observational studies and randomized controlled trials. Clin J Am Soc Nephrol 2011, 6: 50-62.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease–mineral and bone disorder (CKD–MBD). Kidney Int 2009, 76 (Suppl 113): S1–130.
- Malluche HH, Mawad HW, Monier-Faugere MC. Renal osteodystrophy in the first decade of the new millennium analysis of 630 bone biopsies in black and white patients. J Bone Miner Res 2011, 26: 1368-76.
- Moorthi RN, Moe SM. CKD-mineral and bone disorder: core curriculum 2011. Am J Kidney Dis 2011, 58: 1022-36.
- Steddon S, Sharples E. Renal Association Clinical Practice Guideline in mineral and bone disorders in CKD. Nephron Clin Pract 2011, 118 Suppl 1: c145-52.
Metastasi ossee
Stefania Bonadonna e Sonia Baruffi
UOS Dipartimentale di Riabilitazione Neuromotoria a indirizzo Oncologico, Pio Albergo Trivulzio, Milano
Il problema clinico delle metastasi ossee
Le metastasi ossee sono una complicanza frequente della malattia tumorale: si presentano in una percentuale che raggiunge l’80% nella neoplasia mammaria e prostatica e il 40% nei pazienti con tumore del polmone, della vescica, del rene e nei melanomi; inoltre l’espansione dei cloni cellulari nel mieloma multiplo avviene all’interno dell’osso (1).
| Prevalenza di metastasi ossee nei diversi tipi di neoplasia | |
| Neoplasia | Prevalenza |
| Mammella | 65-80% |
| Prostata | 65-80% |
| Mieloma multiplo | 90-100% |
| Polmone | 30-40% |
| Vescica | 40% |
| Rene | 20-25% |
| Melanoma | 14-45% |
Una volta che il tumore ha invaso l’osso, la malattia diviene inguaribile ed il paziente può andare incontro a diversi eventi scheletrici, quali dolore osseo severo, ipercalcemia, sintomi da compressione nervosa e fratture patologiche. Tutto ciò incrementa in modo significativo la morbilità e riduce la qualità di vita del paziente. Pertanto, per sviluppare nuove terapie è fondamentale comprendere le diverse tappe della carcinogenesi e il meccanismo d’invasione e metastasi.
Fisiopatologia
Le fasi iniziali della carcinogenesi sono caratterizzate da crescita iperplastica del clone cellulare tumorale e neo-vascolarizzazione. Le cellule tumorali assumono quindi caratteristiche fenotipiche di cellule mesenchimali in grado di migrare. Lo stadio iniziale della cascata metastatica prevede l’ingresso nel vaso, la circolazione nel torrente sanguigno, l’adesione all’endotelio di organi remoti, la fuoriuscita dal vaso e la colonizzazione a distanza.
Nell’osso numerosi dati hanno suggerito che la metastatizzazione sia un processo che coinvolge diverse fasi coordinate. Infatti, una sottopopolazione specifica di cellule tumorali, una volta acquisita una capacità migratoria, può migrare verso siti in grado di promuoverne la sopravvivenza e la proliferazione. L’osso gioca in questo un ruolo fondamentale, agendo non solo come abitazione ottimale della cellula tumorale (processo promosso da osteoblasti ed osteoclasti), ma anche come fonte di precursori cellulari del midollo, che facilitano lo sviluppo metastatico dapprima nell’osso e poi anche in siti extra-scheletrici. Inoltre, il microambiente osseo ha la capacità assolutamente unica di mantenere le cellule staminali in uno stato di quiescenza, agendo come riserva, capace di far crescere metastasi anche anni dopo la resezione del tumore primitivo. Tutti questi meccanismi sono fortemente regolati da segnali che includono PTH, vitamina D e calcio e una serie di fattori prodotti all’interno della singola unità ossea.
I determinanti della crescita metastatica nell’osso sono poco noti, ma vi è una sostanziale evidenza relativa al ruolo che giocano le cellule tumorali e il tessuto ospite. In particolare, si è definita sia la presenza di cellule tumorali staminali (una sottopopolazione di cellule all’interno della massa tumorale in grado di navigare il torrente sanguigno e colonizzare nuovi siti metastatici) che quella di “nicchie pre-metastatiche”. Queste, definite come microambienti che si formano nell’organo bersaglio di metastasi prima dell’arrivo di cellule tumorali metastatiche, sono costituite da specifiche proteine e da cellule di origine midollare. Tutto ciò garantisce la produzione di VEGF e fibronectina e quindi la neo-vascolarizzazione di nuovi aggregati di cellule tumorali. Le nicchie pre-metastatiche vengono intese, quindi, come regioni fertili di tessuto che facilitano l’invasione, la sopravvivenza e la proliferazione delle cellule tumorali metastatiche, fornendo un nuovo meccanismo per la crescita metastatica. Inoltre, si ritiene che alcuni tessuti siano più recettivi di altri nei confronti di alcune specie tumorali; si pensa che sia presente un cross-talking tra i microambienti da colonizzare e la cellula tumorale staminale che circola nel torrente sanguigno. In particolare, si stanno studiando delle vie di trasduzione del segnale presenti nell’osso (come WNT, SHH, NOTCH e PTEN). Inoltre la presenza nell’osso di chemochine (piccole citochine che si legano a specifiche proteine-G di membrana delle cellule bersaglio) può servire a condurre la cellula staminale tumorale all’osso. Anche il sistema RANK/RANK-L ha un ruolo nella tumorigenesi, essendo coinvolto non solo nel reclutamento del precursore osteoclastico e nella sua maturazione, ma direttamente nell’osteoclastogenesi e nell’osteolisi indotta dalla cellula tumorale (2).
La diagnosi di metastasi ossee
La diagnosi precoce di metastasi ossee ha un ruolo importante nello sviluppo di una terapia ottimale, nella terapia della malattia e nella qualità di vita del paziente.
Molti pazienti con metastasi ossee sono asintomatici e le metastasi vengono riscontrate accidentalmente durante uno screening di routine o perché si osserva un incremento del marcatore tumorale.
Le metastasi ossee possono essere di tipo osteolitico, osteoblastico o miste.
L’imaging è parte essenziale del trattamento delle metastasi ossee. Varie sono le tecniche, sia morfologiche sia funzionali, utilizzate per evidenziare le metastasi ossee. La radiografia convenzionale e la scintigrafia ossea sono le più comunemente usate e le meno costose.
Alla radiografia convenzionale le lesioni osteolitiche appaiono come aree radio-trasparenti, mentre le lesioni addensanti sono aree radio-opache. Il dato significativo è comunque che oltre il 50% delle metastasi scheletriche sono già presenti prima di essere visualizzabili con la radiografia convenzionale. La radiografia diviene tecnica di prima scelta alla comparsa di dolore severo nel sospetto di una frattura ossea.
Lesioni osteolitiche: a sinistra lesioni dell’omero destro che arrivano ad interrompere la corticale in paziente di 59 anni affetta da tumore della mammella; a destra lesioni del femore destro in paziente di 77 anni con neoplasia avanzata della prostata (le frecce evidenziano solo le lesioni maggiori).
Quasi completo sovvertimento del rachide in paziente di 67 anni affetta da neoplasia mammaria.
Lesione osteo-addensante del femore sinistro in paziente di 72 anni affetto da neoplasia della prostata.
La scintigrafia ossea con tecnezio è da sempre la tecnica di prima scelta, perché permette di documentare la presenza di metastasi ossee più precocemente rispetto alla radiografia convenzionale. Le metastasi ossee si presentano all’esame scintigrafico come “aree calde” di ipercaptazione, che devono però entrare in diagnosi differenziale con malattie degenerative ossee, traumi o infezioni.
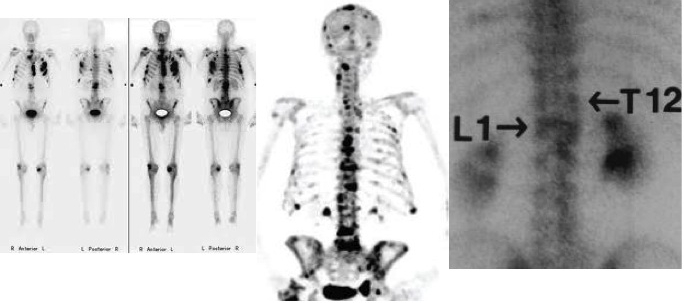
Metastasi ossee. Quadri scintigrafici di metastasi ossee da carcinoma prostatico
La PET con Fluoro-Desossiglucosio (18F-FDG PET) ha maggior sensibilità e specificità della scintigrafia ossea: identifica lesioni di dimensioni inferiori ed è sempre più comunemente usata nella stadiazione tumorale. Se associata alla tomografia computerizzata (TC) con immagini di fusione (PET-TC) diviene poi ancora più precisa nel determinare lesioni anche in fase iniziale.
La risonanza magnetica nucleare (RM) è sicuramente la migliore metodica nello studio di lesioni metastatiche alla colonna vertebrale, anche se i costi fanno sì che rimanga un esame di seconda linea.
In ogni caso, 18F-FDG PET, TC e RM vengono utilizzate con sempre maggior consuetudine. Deng e coll. (3) hanno condotto una meta-analisi di confronto tra le varie tecniche su 145 studi, per un totale di 15.221 pazienti tra i 10 ed i 90 anni. Sia in base alla valutazione per-paziente sia sulla valutazione per-lesione, PET e RM appaiono sostanzialmente sovrapponibili e decisamente più accurate di TC e scintigrafia. La ricerca è attivamente impegnata nel trovare il modo di sovrapporre alle tecniche che forniscono i dettagli di strutture anatomiche (TC e RM) i metodi altamente sensibili quali PET e scintigrafia, che permettono di monitorare l’andamento clinico delle metastasi.
Non esiste consenso sull’utilizzo dei marcatori di rimodellamento osseo nella diagnosi delle lesioni metastatiche. Il riscontro di ipercalcemia e di incremento della fosfatasi alcalina rimane l’indicazione alla ricerca di lesioni secondarie ossee. Diversi marcatori ossei stanno invece assumendo un ruolo sempre più determinante nel monitoraggio del trattamento del paziente con neoplasia metastatica (4-5).
Il problema della malattia ossea legata alla terapia anti-tumorale
La salute dell’osso viene spesso inficiata nei pazienti in terapia anti-tumorale. L’integrità scheletrica è compromessa dai tumori primitivi che risiedono direttamente nell’osso e dai tumori solidi che metastatizzano all’osso. Inoltre, è in costante incremento la consapevolezza che i pazienti sottoposti a trattamenti anti-tumorali vanno incontro a maggior rischio di perdita ossea, in particolare quelli in terapia con inibitori dell’aromatasi per il tumore della mammella e con deprivazione androgenica per il tumore della prostata. L’ipogonadismo indotto da tali trattamenti ha come risultato la perdita di massa ossea e l’aumentato rischio di osteoporosi e di fratture.
Negli ultimi anni sono stati condotti numerosi studi allo scopo di valutare il meccanismo attraverso il quale le terapie anti-tumorali inducono perdita di massa ossea (6). Senza dubbio, al primo posto si posiziona l’ipogonadismo indotto da terapie specifiche, ma va anche preso in considerazione l’utilizzo di agenti tossici diretti sull’osso (es. corticosteroidi o ciclosporine) o l’utilizzo di radioterapia.
| Terapie anti-tumorali associate con perdita di massa ossea | ||
| Terapia | Neoplasia | |
| Orchiectomia bilaterale | Tumore della prostata | |
| Ovariectomia | Tumore della mammella | |
| GnRH agonisti | Tumore della prostata Tumore della mammella |
|
| GnRH antagonisti | Tumore della prostata | |
| Anti-androgeni | Tumore della prostata | |
| Chemioterapia | Diverse neoplasie | |
| Ciclofosfamide | Tumore della mammella | |
| Methotrexate/ifosfamide | Osteosarcoma | |
| Agenti alchilanti | Linfoma Hodgkin e non Hodgkin | |
| SERM | Tumore della mammella | |
| Inibitori dell’aromatasi | Tumore della mammella | |
| Glucocorticoidi/ciclosporina | Trapianto di midollo per diverse neoplasie | |
| Radioterapia | Diverse neoplasie | |
Conclusioni
L’osso è l’organo più frequentemente sede di processo di metastatizzazione. I tumori della mammella e della prostata sono quelli che maggiormente metastatizzano all’osso. I diversi eventi scheletrici, quali dolore osseo severo, ipercalcemia, sintomi da compressione nervosa e fratture patologiche legati al processo di metastatizzazione, si accompagnano a peggioramento della qualità di vita del paziente e a perdita dell’autonomia motoria. L’attuale trattamento delle metastasi ossee mira a ridurre la morbilità legata agli eventi scheletrici, così da preservare e migliorare la qualità di vita del paziente.
Bibliografia
- Lipton A, Uzzo R, Amato RJ, et al. The science and practice of bone health in oncology: managing bone loss and metastasis in patients with solid tumors. J Natl Compr Canc Netw 2009, 7 Suppl 7: S1-29.
- Coleman RE, Lipton A, Roodman GD, et al. Metastasis and bone loss: advancing treatment and prevention. Cancer Treat Rev 2010, 36: 615-20.
- Wu LM, Gu HY, Zheng J, et al. Diagnostic value of whole-body magnetic resonance imaging for bone metastases: a systematic review and meta-analysis. J Magn Reson Imaging 2011, 34: 128-35.
- Carlson RW, Allred DC, Anderson BO, et al; NCCN Breast Cancer Clinical Practice Guidelines Panel. Breast cancer. Clinical practice guidelines in oncology. J Natl Compr Canc Netw 2009, 7: 122-92.
- Coleman R, Costa L, Saad F, et al. Consensus on the utility of bone markers in the malignant bone disease setting. Crit Rev Oncol Hematol 2011, 80: 411-32.
- Brown SA, Guise TA. Cancer treatment-related bone disease. Crit Rev Eukaryot Gene Expr 2009, 19: 47-60.
Approccio al paziente con BMD aumentata
Gregorio Guabello
Ambulatorio di Patologia Osteo-Metabolica, UO Reumatologia, IRCCS Istituto Ortopedico Galeazzi; Ambulatorio di Endocrinologia Oncologica, IRCCS Ospedale San Raffaele; Milano
(aggiornato al 19 febbraio 2019)
INTRODUZIONE
Non è infrequente nella pratica clinica il riscontro incidentale di elevata BMD alla densitometria ossea: si configura in questo caso un quadro di sclerosi ossea che pone il metabolista dell’osso di fronte a una sfida diagnostica importante. In un’interessante revisione sull’argomento (1), su 335.115 DEXA eseguite in 15 centri nel Regno Unito, gli autori hanno rilevato una prevalenza di sclerosi ossea (utilizzando come cut-off Z-score ≥ +4 alla colonna o al femore) di 5/1000, con il 50% dei casi riconducibili a malattie degenerative del rachide. Non esiste attualmente un consenso unanime sulla definizione densitometrica di aumentata massa ossea: in attesa di una validazione, gli autori consigliano di utilizzare un cut-off di Z-score/T-score ≥ +2 presente sia alla colonna sia al femore.
SISTEMATICA
Artefatti o patologie responsabili di un aumento focale della BMD possono determinarne una sovrastima (tabella 1).
| Tabella 1 Artefatti e cause di aumento focale della BMD |
|
| Artefatti | Malattie degenerative della colonna con/senza scoliosi (50% dei casi) Spondilite anchilosante con sindesmofiti DISH (malattia di Forestier) Calcificazioni vascolari dell’aorta addominale Esiti di vertebroplastica Protesi vascolari |
| Aumento focale della BMD |
Segno della vertebra d’avorio a uno o più livelli:
Metastasi osteo-addensanti (k mammella e prostata) |
La DISH (iperostosi scheletrica idiopatica diffusa o malattia di Forestier) è una rara forma di spondilosi idiopatica, nel 50% dei casi presente in soggetti diabetici/obesi e/o con iperuricemia e/o dislipidemia, associata a calcificazioni tendinee.
La sindrome SAPHO (acronimo che sta per sinovite, acne, pustolosi, iperostosi e osteite) è una malattia auto-infiammatoria, caratterizzata dall'associazione di interessamento dei neutrofili della cute e osteomielite cronica.
Le cause di aumento generalizzato della BMD possono essere genetiche o acquisite (tabella 2).
| Tabella 2 Cause di aumento generalizzato della BMD |
||
| Genetiche | Difetto di riassorbimento osseo |
Basso rimodellamento, quindi ridotta elasticità dell’osso con fragilità scheletrica e aumentato rischio fratturativo:
|
| Aumento della neoformazione ossea |
Aumento dello spessore delle trabecole, quindi senza fragilità scheletrica, con ridotto rischio fratturativo:
|
|
| Acquisite | Fluorosi scheletrica | |
| Osteodistrofia renale | ||
| Endocrinopatie | Acromegalia Ipoparatiroidismo cronico Pseudoipoparatiroidismo |
|
| Malattie ematologiche | Mastocitosi sistemica Sindromi mielo-proliferative (mielofibrosi idiopatica) Leucemie Linfomi Mieloma multiplo sclerotizzante |
|
| Metastasi osteo-addensanti diffuse | ||
| Epatite C | ||
| Osteomalacia assiale | ||
| Obesità | ||
Picnodisostosi
Si tratta di una patologia lisosomiale autosomica recessiva, che dipende da una mutazione del gene della catepsina K (enzima osteoclastico necessario per la degradazione della componente organica della matrice osteoide). Dal punto di vista clinico i pazienti presentano osteosclerosi diffusa, bassa statura e fratture. Sono stati descritti circa 100 casi (3).
Sclerosteosi e malattia di van Buchem
La sclerosteosi dipende da una mutazione con perdita di funzione del gene SOST (sclerostina, fisiologico inibitore della via di segnale Wnt-ßcatenina). Sono noti meno di 100 casi. Colpisce lo scheletro in toto, con un caratteristico aumento volumetrico alla pubertà delle ossa mascellari/mandibolari e cranio-facciali (malocclusione). La sclerosi ossea può determinare compressione di strutture nervose, con sintomatologia neurologica (paralisi del nervo facciale, sordità, atrofia ottica, ipertensione endocranica); sono inoltre presenti alta statura (gigantismo) e sindattilia. Alla densitometria si rilevano alti valori di Z-score (da +5.4 a +14.4 a livello lombare e da +5.2 a +12.1 al collo femorale), con aumento della massa e della resistenza ossea e riduzione del rischio di frattura.
La malattia di van Buchem è causata da una delezione downstream di 52 Kb del gene SOST. Sono noti 30 casi. Le manifestazioni cliniche sono più lievi rispetto a quelle della sclerosteosi, senza alta statura e sindattilia (4).
Sindrome di Worth (iperostosi endostale)
Dipende da una mutazione attivante del gene LRP5 (low-density lipoprotein receptor-related protein 5), che è il co-recettore della via di segnale Wnt-ßcatenina, e si trasmette con modalità autosomica dominante (sono note meno di 10 famiglie). Dal punto di vista clinico si caratterizza per la comparsa in adolescenza di modificazioni cranio-facciali, fra cui la prominenza della fronte, l’ingrossamento della mascella/mandibola, l’allargamento della radice del naso e l’esostosi del palato. L’esecuzione della DEXA ogni 2 anni permette di valutare nel tempo la progressione della malattia (5).
Displasia diafisaria progressiva (malattia di Camurati-Engelmann)
Dipende da una mutazione del gene TGF-ß1, che ne causa l’attivazione prematura. Sono noti più di 300 casi. Dal punto di vista clinico è caratterizzata da iperostosi diffusa (ossa lunghe, cranio, colonna, pelvi), con apposizione di nuovo osso sulla superficie sia endostale sia periostale, disabilità motoria che mima la distrofia muscolare, paralisi dei nervi cranici ed estrema variabilità nella progressione clinica. Può essere efficace la terapia steroidea (6).
Meloreosteosi e osteopoichilosi
La meloreosteosi è una forma sporadica, con meccanismo molecolare non noto. Dal punto di vista clinico, si caratterizza per la comparsa di iperostosi periostale e del canale midollare adiacente, con lesioni singole o multiple, spesso dolorose e ipercaptanti alla scintigrafia ossea (7).
L’osteopoichilosi dipende da una mutazione inattivante del gene LEMD3, codificante una proteina che fisiologicamente antagonizza il segnale del TGF- ß. Dal punto di vista clinico, si caratterizza per la comparsa di piccole aree di osteosclerosi ovalari ben delimitate (spotted bones), di riscontro radiologico incidentale, che possono mimare lesioni metastatiche ma non sono captanti alla scintigrafia ossea (8).
Fluorosi scheletrica
Dipende da un eccessivo apporto di fluoro tramite alimenti (pesce, frutti di mare, latte, carne, formaggio e alimenti di origine vegetale, il cui contenuto di fluoro varia a seconda delle caratteristiche del terreno su cui sono stati coltivati e dell'eventuale uso di fertilizzanti), tè, acqua potabile e fluorizzata, dentifricio e farmaci. È caratterizzata da un indurimento anomalo delle ossa, associato a dolori e rigidità delle articolazioni, debolezza, danni al sistema nervoso e paralisi.
Malattie ematologiche
Nella mastocitosi sistemica si può avere una riduzione grave e veloce della BMD, che colpisce le ossa lunghe e la colonna, in relazione all’effetto osteoclastogenetico di numerose citochine (TNF-alfa, IL-1, IL-6, IL-17) liberate in eccesso dai mastociti, con conseguente osteopenia/osteoporosi e lesioni osteolitiche; valori molto elevati di triptasi possono paradossalmente stimolare l’attività osteoblastica ed essere responsabili di una forma di osteosclerosi che interessa lo scheletro assiale.
Le sindromi mielo-proliferative, in particolare la mielofibrosi idiopatica, possono dare forme di osteosclerosi. Il mieloma multiplo sclerotizzante è molto raro.
Epatite C
L’HCV ha un’azione diretta sull’osso, determinando un’aumentata attività osteoclastica attraverso l’aumentata produzione di citochine pro-infiammatorie: il quadro che ne deriva è rappresentato da osteopenia e osteoporosi. Sono tuttavia noti casi molto rari di osteosclerosi associata ad HCV, a patogenesi non completamente chiara, forse mediata da anomalie della secrezione di IGF-I e II (9).
Osteomalacia assiale
Si tratta di una forma molto rara (descritti 20 casi in letteratura), caratterizzata da un interessamento dello scheletro assiale, con normali valori densitometrici al sito femorale; l’istomorfometria documenta un aumentato spessore delle trabecole, con ridotta mineralizzazione e normale contenuto di fluoro; i parametri biochimici del metabolismo fosfo-calcico sono nella norma (10).
Obesità
L’obesità severa (BMI > 40) può determinare una sovrastima della BMD, per cui in questi pazienti può essere utile la DEXA del terzo distale del radio.
APPROCCIO DIAGNOSTICO AL PAZIENTE CON AUMENTATA MASSA OSSEA
Di fronte al dato densitometrico di aumentata massa ossea, la prima cosa da fare è escludere l’evenienza più frequente nella pratica clinica, cioè la presenza di un artefatto o di una patologia responsabile di un aumento focale della BMD, che possono determinarne una sovrastima.
Dopo tale esclusione e in presenza di aumentata BMD sia a livello lombare sia a livello femorale, è necessario indagare le cause di aumento generalizzato della BMD, evenienza meno frequente nella pratica clinica ma di più difficile valutazione in considerazione delle numerose patologie coinvolte.
L’approccio al paziente con sclerosi ossea diffusa prevede:
- anamnesi accurata per valutare la familiarità per malattie genetiche e/o fratture da fragilità, pregresse fratture ossee, la presenza di dolore osseo, una storia di epatite C, un apporto eccessivo di fluoro;
- attento esame obiettivo per valutare esiti di paralisi di nervi cranici (paralisi facciale), ipoacusia, ipovisus, splenomegalia, decadimento delle condizioni generali, lesioni cutanee, dismorfismo mascellare/facciale e anomalie dentali;
- esami biochimici relativi allo stato di salute generale del paziente (emocromo, elettroforesi proteica, funzione renale ed epatica, indici di flogosi), al metabolismo fosfo-calcico (calcio, fosforo, 25OH-D, fosfatasi alcalina, CTX sierico, PTH) e allo specifico sospetto diagnostico (Ab anti-HCV, fluoro sierico, triptasi);
- radiogrammi per evidenziare la sclerosi ossea diffusa (Rx rachide lombare e dorsale, Rx bacino, Rx cranio) e in casi selezionati TC/RM di segmenti ossei e scintigrafia ossea;
- consulenza genetica nel caso di mutazione genetica identificata.
Qualora tutti gli accertamenti risultino negativi, è necessario ricordare che nella determinazione del quadro clinico del paziente possono essere coinvolte molte malattie mono o poligeniche al momento non conosciute.
La figura riporta una flow-chart per la diagnosi differenziale dell’aumentata massa ossea.
Diagnosi differenziale dell’aumentata massa ossea
BIBLIOGRAFIA
- Paccou J, Michou L, Kolta S, et al. High bone mass in adults. Joint Bone Spine 2018, 85: 693-9.
- Yaga U, Panta P. Osteopetrosis. N Engl J Med 2017, 376: e34.
- Gelb BD, Shi GP, Chapman HA, et al. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996, 273: 1236–8.
- Van Lierop AH, Hamdy NA, van Egmond ME, et al. Van Buchem disease: clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res 2013, 28: 848–54.
- Worth HM, Wollin DG. Hyperostosis corticalis generalista congenita. J Canad Assoc Radiol 1966, 17: 67–74.
- Bhadada SK, Sridhar S, Steenackers E, et al. Camurati-Engelmann disease (progressive diaphyseal dysplasia): reports of an Indian kindred. Calcif Tissue Int 2014, 94: 240–7.
- Freyschmidt J. Melorheostosis: a review of 23 cases. Eur Radiol 2001, 11: 474–9.
- Hellemans J, Preobrazhenska O, Willaert A, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet 2004, 36: 1213–8.
- Khosla S, Hassoun AA, Baker BK, et al. Insulin-like growth factor system abnormalities in hepatitis C-associated osteosclerosis. Potential insights into increasing bone mass in adults. J Clin Invest 1998, 101: 2165–73.
- Demiaux-Domenech B, Bonjour JP, Rizzoli R. Axial osteomalacia: report of a new case with selective increase in axial bone mineral density. Bone 1996, 18: 633–7.