Scheda irinotecan

Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
Citostatico della classe delle camptotecine, che agisce inibendo l'enzima topo-isomerasi I, bloccando la replicazione del DNA delle cellule tumorali.
Indicazioni approvate
Cancro del colon-retto in mono-terapia o in associazione con 5-FU e acido folinico.
Cancro metastatico del colon-retto che esprime il recettore per EGF-R, senza mutazioni di K-RAS, senza precedente trattamento o dopo fallimento di una terapia citotossica.
Trattamento di prima linea del carcinoma metastatico del colon-retto in associazione con cetuximab, 5-FU, acido folinico e bevacizumab.
Trattamento di prima linea del carcinoma metastatico del colon-retto in combinazione con capecitabina, con o senza bevacizumab.
Controindicazioni
Malattia infiammatoria intestinale cronica e/o ostruzione dell’intestino.
Ipersensibilità al principio attivo o uno qualsiasi degli eccipienti.
Allattamento.
Valori di bilirubina > 3 volte il limite superiore dell’intervallo di normalità.
Grave insufficienza midollare.
Capacità funzionale > 2 secondo l’OMS.
Uso concomitante di iperico (erba di San Giovanni).
Uso di vaccini vivi attenuati.
I pazienti con intolleranza ereditaria al fruttosio non devono essere trattati se non strettamente necessario.
Preparazioni, via di somministrazione, posologia
Irinotecan (1.5 mg/mL, 4.3 mg, 20 mg/mL, 40 mg) per infusione ev della durata di 30-90 minuti.
In mono-terapia (per pazienti precedentemente trattati) la dose raccomandata è 350 mg/m2 ogni 3 settimane.
In associazione con 5-FU e acido folinico (per pazienti non trattati in precedenza) la dose raccomandata è di 180 mg/m2 ogni 2 settimane come infusione ev, seguita da infusione di acido folinico e 5-FU.
In associazione con il cetuximab, l’irinotecan deve essere somministrato alla stessa dose somministrata negli ultimi cicli del precedente trattamento, non prima che sia trascorsa un’ora dalla fine dell’infusione di cetuximab.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Effetti collaterali comuni: sono diarrea, nausea e vomito, fatica e debolezza, neutropenia, anemia e trombocitopenia (aumentato rischio di infezioni e sanguinamenti); può inoltre provocare alopecia, stomatite, perdita di appetito e perdita di peso.
Effetti collaterali meno comuni: sudorazione, lacrimazione e salivazione eccessive, crampi addominali e disturbi visivi durante o poco dopo l'infusione. Altri effetti collaterali sono costipazione, tosse, vertigini, pelle secca e pruriginosa.
In rari casi: insufficienza renale, ipotensione o insufficienza circolatoria in pazienti che hanno presentato episodi di disidratazione associata a diarrea e/o vomito o sepsi.
Le donne in età fertile e gli uomini devono fare uso di adeguati metodi contraccettivi durante il trattamento e, rispettivamente, fino a 1 mese e 3 mesi dopo il trattamento.
Farmaci concomitanti: la somministrazione concomitante con un potente inibitore (per es. ketoconazolo) o un induttore (per es. rifampicina, carbamazepina, fenobarbital, fenitoina, iperico) del CYP3A4 può alterare il metabolismo dell’irinotecan e deve essere evitata. L’uso concomitante con antagonisti della vitamina K può determinare aumentato rischio di emorragia ed eventi trombotici in patologie tumorali. In caso di associazione con agenti immuno-soppressori (ad esempio ciclosporina, tacrolimus) è possibile eccessiva immuno-soppressione con rischio di linfo-proliferazione.
Modalità prescrittive
Prescrivibile in ambiente ospedaliero.
Scheda sali di platino
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
I sali di platino (cisplatino, carboplatino, oxaliplatino) agiscono legandosi covalentemente al DNA, in particolare alle basi azotate delle cellule tumorali. Questi legami crociati distorcono la doppia elica del DNA, bloccando i processi di replicazione e trascrizione, e innescando l'apoptosi.
Indicazioni approvate
Cisplatino:
- carcinoma del testicolo avanzato o metastatico;
- carcinoma ovarico avanzato o metastatico;
- carcinoma della vescica avanzato o metastatico;
- carcinoma a squamocellulare della testa e del collo avanzato o metastatico;
- carcinoma polmonare non a piccole cellule avanzato o metastatico;
- carcinoma polmonare a piccole cellule avanzato o metastatico;
- carcinoma della cervice uterina in associazione con altri chemioterapici o con la radioterapia.
Carboplatino:
- carcinoma dell’ovaio di origine epiteliale in fase avanzata in prima linea o in seconda linea, dopo il fallimento di altri trattamenti;
- carcinoma del polmone a piccole cellule.
Oxaliplatino:
- in associazione con 5-FU e acido folinico per il trattamento adiuvante del cancro al colon di stadio III (C di Duke) dopo resezione completa del tumore primario;
- cancro colo-rettale metastatico.
Controindicazioni
Ipersensibilità al principio attivo (anche come classe) o a uno qualsiasi degli eccipienti, allattamento, mielo-soppressione.
Cisplatino: disfunzione renale pre-esistente; disidratazione; alterazione pre-esistente dell’udito; neuropatia causata dal cisplatino; in associazione con vaccini vivi, incluso il vaccino per la febbre gialla; in associazione con fenitoina in uso profilattico.
Carboplatino: tumori con emorragia; grave compromissione renale pre-esistente (creatinina clearance ≤ 20 mL/minuto).
Oxaliplatino: neuropatia sensoriale periferica con alterazione funzionale antecedente al primo ciclo; funzionalità renale gravemente compromessa (creatinina clearance ≤ 30 mL/minuto).
Preparazioni, via di somministrazione, posologia
Cisplatino (0.5 mg/mL, 1 mg/mL, 50 mg/50 ml): la soluzione diluita deve essere somministrata esclusivamente per infusione. Per la mono-terapia, sono raccomandati due regimi posologici: singola dose da 50 a 120 mg/m² ogni 3-4 settimane; da 15 a 20 mg/m²/die per cinque giorni, ogni 3-4 settimane. Se il cisplatino viene utilizzato in una chemioterapia combinata, la dose di cisplatino deve essere ridotta: la dose abituale è 20 mg/m² o più una volta ogni 3-4 settimane. Per il trattamento del carcinoma della cervice uterina, il cisplatino viene utilizzato in associazione alla radioterapia, alla dose abituale di 40 mg/m2 ogni settimana per 6 settimane.
Carboplatino (10 mg/mL, 50 mg/5 mL, 150 mg/15 mL, 450 mg/45 mL) deve essere somministrato esclusivamente per via ev. La dose raccomandata di carboplatino negli adulti non trattati precedentemente e con funzionalità renale normale (creatinina clearance > 60 mL/min) è 400 mg/m², in un’unica dose somministrata con infusione ev della durata da 15 a 60 minuti.
Oxaliplatino (5 mg/mL): la somministrazione di oxaliplatino deve sempre precedere quella delle fluoropirimidine – per esempio 5-FU. Deve essere somministrato in infusione ev della durata di 2-6 ore in 250-500 mL di soluzione glucosata al 5% (50 mg/mL) per ottenere una concentrazione tra 0.2 e 0.7 mg/mL (che rappresenta la concentrazione più alta riportata nella pratica clinica per una dose di oxaliplatino di 85 mg/m2).
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Effetti collaterali (variabili tra i farmaci, ma comuni al gruppo):
- gastro-intestinali: nausea e vomito (molto comuni, richiedono anti-emetici potenti), anoressia, diarrea;
- ematologici: leucopenia, trombocitopenia, anemia, con aumentato rischio di infezioni e sanguinamenti;
- neuropatia periferica con intorpidimento, formicolio, dolore a mani e piedi (specialmente con cisplatino e oxaliplatino), talvolta irreversibile;
- oto-tossicità con acufeni, perdita uditiva per frequenze alte (specialmente con alte dosi di cisplatino);
- nefro-tossicità: insufficienza renale, gestita con idratazione intensiva;
- cardio-vascolari: aritmie, ipertensione, raramente infarto miocardico (più con oxaliplatino);
- reazioni allergiche: eruzione cutanea, orticaria, prurito.
Cisplatino: forte tossicità renale, uditiva, neurologica.
Carboplatino: meno tossico per i reni ma più mielo-soppressivo.
Oxaliplatino: è caratteristica la neuropatia periferica indotta dal freddo.
Modalità prescrittive
Prescrivibili in ambiente ospedaliero.
Scheda bevacizumab
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
Anticorpo monoclonale che blocca l’angio-genesi legandosi al VEGF (fattore di crescita vascolare endoteliale), privando i tumori di nutrimento e ossigeno.
Indicazioni approvate
In associazione con chemioterapia a base di fluoropirimidine: trattamento di adulti con carcinoma metastatico colo-rettale.
In associazione con paclitaxel: trattamento in prima linea di adulti con carcinoma mammario metastatico.
In associazione con capecitabina: trattamento in prima linea di adulti con carcinoma mammario metastatico, in cui una terapia con altri chemioterapici, inclusi quelli a base di taxani o antracicline, non è considerata appropriata.
In aggiunta a chemioterapia a base di platino: trattamento in prima linea di adulti con carcinoma polmonare non a piccole cellule, non resecabile, avanzato, metastatico o recidivante, con istologia a predominanza non squamo-cellulare.
In associazione con erlotinib: trattamento in prima linea di adulti affetti da carcinoma polmonare non a piccole cellule, non squamo-cellulare, avanzato, non resecabile, metastatico o recidivante, con mutazioni attivanti del recettore del fattore di crescita epidermico (EGF-R).
In associazione con interferone alfa-2a: trattamento in prima linea di adulti con carcinoma renale avanzato e/o metastatico.
In associazione con carboplatino e paclitaxel: trattamento in prima linea di pazienti adulte con carcinoma ovarico epiteliale, carcinoma alle tube di Falloppio o carcinoma peritoneale primario in stadio avanzato (stadio III B, III C e IV).
In associazione con carboplatino e gemcitabina o in combinazione con carboplatino e paclitaxel: trattamento di pazienti adulte con prima recidiva di carcinoma ovarico epiteliale, carcinoma alle tube di Falloppio o carcinoma peritoneale primario platino-sensibili, che non hanno ricevuto una precedente terapia con bevacizumab o altri inibitori del VEGF o altri agenti mirati al recettore VEGF.
In associazione con paclitaxel, topotecan o doxorubicina liposomiale pegilata: trattamento di pazienti adulte con recidiva di carcinoma ovarico epiteliale, carcinoma alle tube di Falloppio o carcinoma peritoneale primario platino-resistenti, che hanno ricevuto non più di due precedenti regimi chemioterapici e che non hanno ricevuto una precedente terapia con bevacizumab o altri inibitori del VEGF o altri agenti mirati al recettore VEGF.
In associazione con paclitaxel e cisplatino o, in alternativa, a paclitaxel e topotecan: in donne che non possono essere sottoposte a terapia a base di platino, per il trattamento di carcinoma della cervice persistente, recidivante o metastatico.
Controindicazioni
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti; ipersensibilità ai prodotti derivati da cellule ovariche di criceto cinese (CHO) o ad altri anticorpi ricombinanti umani o umanizzati.
Gravidanza.
Preparazioni, via di somministrazione, posologia
Bevacizumab (Avastin e altri, 25 mg/mL, 400 mg) mediante infusione endovenosa:
- carcinoma metastatico colo-rettale: la dose raccomandata è di 5 mg/kg o 10 mg/kg ogni 2 settimane oppure 7.5 mg/kg o 15 mg/kg ogni 3 settimane;
- carcinoma mammario metastatico: la dose raccomandata è di 10 mg/kg ogni 2 settimane oppure 15 mg/kg ogni 3 settimane;
- carcinoma polmonare non a piccole cellule (NSCLC) non squamo-cellulare: la dose raccomandata è di 7.5 mg/kg o 15 mg/kg ogni 3 settimane (dopo 6 cicli di trattamento a base di platino);
- NSCLC non squamo-cellulare con mutazioni attivanti dell’EGFR in associazione con erlotinib: la dose raccomandata è 15 mg/kg ogni 3 settimane;
- carcinoma renale avanzato e/o metastatico: la dose raccomandata è 10 mg/kg ogni 2 settimane;
- carcinoma ovarico epiteliale, carcinoma alle tube di Falloppio e carcinoma peritoneale primario in aggiunta a carboplatino e paclitaxel (dopo 6 cicli di trattamento): la dose raccomandata è 15 mg/kg ogni 3 settimane;
- recidiva di malattia platino-sensibile in associazione a carboplatino e gemcitabina (dopo 6 cicli di trattamento fino a un massimo di 10 cicli) oppure in associazione a carboplatino e paclitaxel (dopo 6 cicli di trattamento fino a un massimo di 8 cicli): la dose raccomandata è 15 mg/kg ogni 3 settimane;
- recidiva di malattia platino-resistente in associazione a paclitaxel, topotecan o doxorubicina liposomiale pegilata: la dose raccomandata è 10 mg/kg ogni 2 settimane;
- recidiva di malattia platino-resistente in associazione a topotecan: la dose raccomandata è 15 mg/kg ogni 3 settimane;
- carcinoma della cervice in associazione con paclitaxel e cisplatino o paclitaxel e topotecan: la dose raccomandata è 15 mg/kg ogni 3 settimane.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Il bevacizumab non deve essere somministrato o miscelato con soluzioni di destrosio.
Effetti collaterali comuni: ipertensione, ritardo nella guarigione delle ferite, astenia, diminuzione dell'appetito, trombosi, emorragie.
Bevacizumab può compromettere la guarigione delle ferite: la terapia non deve essere iniziata per almeno 28 giorni dopo un intervento di chirurgia maggiore o finché la ferita chirurgica non sia completamente guarita. In caso di chirurgia elettiva, il trattamento deve essere sospeso circa 4-6 settimane prima.
È stato osservato un aumento dell'incidenza di ipertensione arteriosa: la pressione deve essere adeguatamente controllata prima di iniziare il trattamento e monitorata regolarmente (ogni 2-3 settimane) durante e dopo la terapia.
Il farmaco aumenta il rischio di sanguinamento grave: è necessaria cautela nei pazienti con pregressi episodi emorragici o fattori di rischio.
Sussiste un rischio (raro ma grave) di sviluppare perforazioni del tratto gastro-intestinale o fistole.
Esiste il rischio di sviluppare eventi trombo-embolici arteriosi e venosi, come ictus, infarto miocardico.
Il bevacizumab può alterare la funzione renale, causando proteinuria.
Come per altri anticorpi monoclonali, possono verificarsi reazioni da ipersensibilità durante o dopo l'infusione.
Non sono note interazioni farmacologiche clinicamente significative con la maggior parte degli agenti chemioterapici comunemente usati in associazione (come paclitaxel, carboplatino, fluoropirimidine). Tuttavia, l'uso concomitante di bevacizumab con sunitinib o bisfosfonati per via ev può aumentare il rischio di osteonecrosi della mascella. Si consiglia un esame odontoiatrico preventivo prima del trattamento.
Modalità prescrittive
Prescrivibile in ambiente ospedaliero.
Terapia chirurgica per i NET pancreatici
Stefano Partelli & Massimo Falconi
Clinica Chirurgia del Pancreas, Università Politecnica delle Marche, Ancona
INTRODUZIONE
Le neoplasie pancreatiche neuroendocrine (PNEN) sono malattie rare, clinicamente classificate come funzionanti (F-PNEN) o non funzionanti (NF-PNEN) sulla base della presenza o meno di una sindrome correlata a una inappropriata secrezione ormonale (1). La Società Europea per i Tumori Neuroendocrini (ENETS) ha proposto un sistema di stadiazione basato su dimensione del tumore, coinvolgimento linfonodale e presenza di metastasi (TNM), oltre a un sistema di classificazione dell’aggressività biologica basato sulla valutazione dell’attività proliferativa delle cellule neoplastiche (2). La classificazione delle PNEN proposta dall’Organizzazione Mondiale della Sanità (WHO) ha come primo obiettivo quello di assegnare una categoria diagnostica con significato clinico (3). Il trattamento chirurgico di queste neoplasie può essere molto variabile a causa di presentazione eterogenea e differente comportamento biologico.
VALUTAZIONE PRE-OPERATORIA
Diagnostica strumentale pre-operatoria
La localizzazione del tumore primitivo e la valutazione della sua estensione sono fondamentali in ogni fase del trattamento dei pazienti con PNEN. Sono state utilizzate molte tecniche diagnostiche, tra cui la tomografia computerizzata, la risonanza magnetica nucleare, l’ecografia, l’angiografia, la scintigrafia con recettori per la somatostatina, l’ecoendoscopia ed esami di localizzazione funzionale che misurano il gradiente ormonale, fino al recente uso della tomografia a emissione di positroni (4) (figura 1).
Durante l’atto chirurgico si raccomanda l’utilizzo routinario dell’ecografia intra-operatoria, soprattutto nei casi di insulinoma per i quali è indicata un’enucleazione. Nel caso di piccoli tumori duodenali (specialmente gastrinomi duodenali) si raccomanda l’utilizzo di routine dell’endoscopia con transilluminazione (5).
Stadiazione tumorale
La classificazione attuale dell’Organizzazione Mondiale della Sanità dei tumori neuroendocrini classifica le PNEN sulla base del grado di differenziazione (3). Nel 2006 un gruppo di lavoro dell’ENETS ha pubblicato una proposta di stadiazione TNM per la classificazione delle PNEN (6). In questa classificazione sono considerati pT1 i tumori 4 cm. Nel 2009 l’AJCC/UICC ha proposto una propria classificazione TNM per le PNEN (2). Le due classificazioni proposte dall’AJCC/UICC e dall’ENETS si differenziano nella definizione dello stadio T. In particolare, il sistema della AJCC/UICC distingue le neoplasie pT2 da quelle pT3 sulla base del coinvolgimento del tessuto peri-pancreatico, mentre la classificazione degli ENETS basa questa distinzione sulla dimensione tumorale, stabilendo un cut-off di 4 cm. Entrambe le classificazioni sono uno strumento diagnostico accurato per la stadiazione delle PNEN (7,8).
TRATTAMENTO CHIRURGICO DELLE PNEN SPORADICHE
pNEN localizzate
Il miglioramento e lo sviluppo delle tecniche diagnostiche hanno portato a un significativo incremento nell’identificazione di piccoli NET non funzionanti (1). Un recente studio di Bettini et al (9) ha dimostrato che la prevalenza delle forme maligne nei pazienti con PNEN < 2 cm riscontrata accidentalmente è del 6% (9). Le attuali linee guida dell’ENETS raccomandano una strategia di “wait and see” per questi piccoli tumori accidentali e comunque il rischio di un eventuale intervento deve essere sempre valutato anche in relazione alle comorbilità del paziente e alla sua aspettativa di vita (10). Sebbene non sia stato indicato un protocollo specifico di follow-up, sembra ragionevole eseguire un controllo strumentale annuale con un primo controllo a sei mesi dalla diagnosi.
Al contrario, la chirurgia rappresenta il trattamento di scelta per PNEN > 2 cm e/o per le forme sintomatiche.
La chirurgia radicale per le PNEN prevede sia resezioni tipiche sia resezioni atipiche. Le prime variano secondo il sito di neoplasia: le lesioni della testa del pancreas sono trattate attraverso un intervento di duodeno-cefalo-pancreasectomia (DCP), mentre le lesioni del corpo e della coda sono trattate mediante pancreasectomia distale (PD). Generalmente, se eseguite in centri ad alto volume, il tasso di mortalità si riduce a < 5%, anche se rimane elevata la percentuale di complicanze, che varia dal 40% al 50% (11,12).
Le resezioni pancreatiche tipiche sono associate a un’elevata incidenza d’insufficienza pancreatica: endocrina dal 10% al 24% dopo DCP e dall’8% al 60% dopo PD; esocrina si presenta nel 30-60% dei pazienti dopo DCP e nello 0-40% dei casi dopo PD (13).
Il rischio d’insufficienza pancreatica a lungo termine ha incrementato lo sviluppo di tecniche chirurgiche volte al risparmio di parenchima pancreatico, come le resezioni atipiche quali l’enucleazione e la pancreasectomia intermedia, che consiste nella resezione della parte centrale della ghiandola. Generalmente, queste tecniche sono indicate solo per i piccoli tumori (< 2 cm) associati a sindrome funzionale (14). Le forme non funzionanti dovrebbero essere trattate in maniera conservativa come prima descritto. Il vantaggio principale delle resezioni atipiche è un minor rischio di sviluppo d’insufficienza pancreatica endocrina/esocrina rispetto alle resezioni standard (15,16). D’altra parte, le resezioni limitate sono associate con una più alta percentuale di fistola pancreatica, sebbene si tratti solitamente di fistole con un basso impatto clinico (15).
Non è ancora ben definito il ruolo della linfoadenectomia per i pazienti con PNEN (17). Le metastasi linfonodali si presentano solo nel 30% dei pazienti (17), ma si discute ancora sull’associazione tra presenza di metastasi linfonodali e peggiore sopravvivenza (18). Di conseguenza, è ancora difficile definire il reale vantaggio della linfoadenectomia per queste neoplasie.
La colecistectomia non dovrebbe essere eseguita di routine; infatti, nonostante il trattamento con gli analoghi della somatostatina sia associato con una percentuale elevata di litiasi colecistica, sono rari gli episodi di colecistite.
L’embolizzazione epatica, eseguita in casi molto selezionati, potrebbe molto raramente determinare lo sviluppo di colecistite da reflusso di microsfere.
pNEN localmente avanzata
Un gran numero di pazienti con PNEN presenta alla diagnosi una malattia localmente avanzata. L’asportazione chirurgica radicale della massa tumorale, quando fattibile, si associa a prognosi migliore (19). I criteri di resezione chirurgica per PNEN non escludono la presenza di malattia infiltrante gli organi vicini (stomaco, milza, colon, rene, surrene) o le strutture vascolari. Il trattamento di scelta è sempre una resezione tipica associata a linfoadenectomia, estesa, se necessario, agli organi limitrofi. In caso di tumori neuroendocrini non funzionanti le controindicazioni alla resezione chirurgica sono: l’invasione circonferenziale della vena porta e l’invasione circonferenziale dell’arteria mesenterica superiore. La presenza d’infiltrazione del tronco celiaco non rappresenta una controindicazione assoluta alla pancreasectomia distale (10).
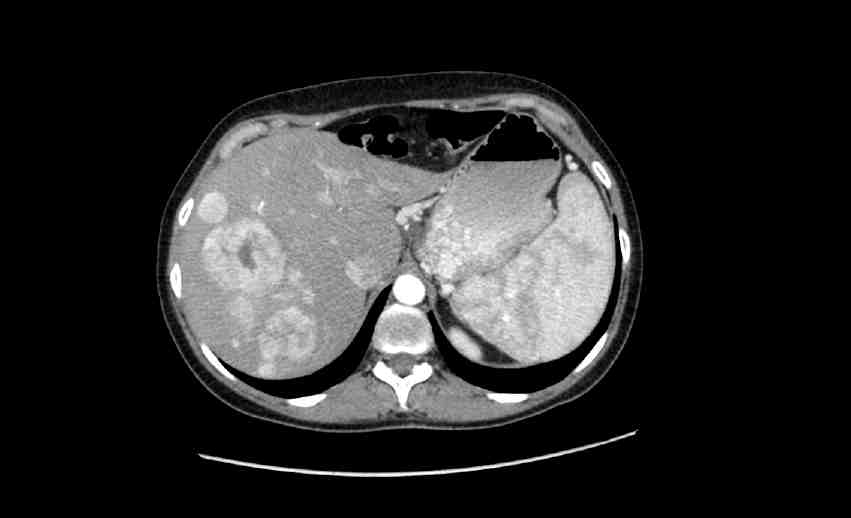
pNEN metastatica
Alla diagnosi, dal 25% al 93% dei pazienti con PNEN hanno metastasi epatiche sincrone al tumore neuroendocrino (20) (figura 2).
In presenza di tumori ben differenziati con metastasi limitate ad un lobo epatico il trattamento di scelta è la resezione radicale del tumore primitivo, associata a una resezione completa delle metastasi epatiche, eventualmente eseguita in più fasi (21,22). L’asportazione radicale del tumore primitivo (resezione R0/R1) e delle lesioni epatiche è efficace nell’alleviare i sintomi in caso di neoplasia funzionante e rappresenta l’unica strategia terapeutica a scopo curativo. Tuttavia, una resezione epatica radicale è possibile in meno del 20% dei pazienti, a causa dell’elevata incidenza di metastasi multifocali e bilobari (23).
Prima di proporre l’intervento chirurgico per le lesioni metastatiche, è fondamentale che siano soddisfatte le seguenti condizioni (24):
- assenza di malattia extra-addominale
- presenza di basso indice proliferativo (Ki-67) valutato sulle cellule prelevate mediante ago-aspirato eseguito pre-operatoriamente
- esistenza di positività per i recettori della somatostatina al fine di somministrare terapie radiorecettoriali, efficaci soprattutto dopo chirurgia citoriduttiva.
Nei pazienti con metastasi bilobari o più del 75% del fegato coinvolto, difficilmente può essere eseguita una chirurgia radicale. In quest’ottica la resezione chirurgica può essere coadiuvata da tecniche di embolizzazione e ablazione (25). Un’opzione alternativa per le metastasi epatiche multifocali consiste nell’approccio integrato di epatectomia parziale e ablazione con radiofrequenza (26).
Il tipo di resezione epatica dipende dal numero delle metastasi, dalla loro localizzazione e dal parenchima epatico residuo. Laddove indicata, la chirurgia può essere una semplice enucleazione, una resezione segmentaria, o un’epatectomia.
Il tasso di sopravvivenza a tre e cinque anni nei pazienti sottoposti a chirurgia epatica radicale per metastasi da tumore neuroendocrino varia dal 79 al 95% (27).
Per quanto riguarda la resezione del tumore primitivo, la chirurgia citoriduttiva è di aiuto per alleviare i sintomi legati alla secrezione ormonale nei carcinomi metastatici funzionanti. Non è stato chiaramente dimostrato un vantaggio in termini di sopravvivenza dopo resezione del tumore primitivo in caso di NF-PNEN (28). In questi casi la resezione chirurgica è giustificata solo per prevenire complicanze ostruttive, sanguinamenti, pancreatite acuta, ittero e ostruzione gastrica (28). D’altra parte, una recente revisione (29) ha dimostrato una migliore sopravvivenza nei pazienti con PNEN metastatico sottoposti a resezione del tumore primitivo rispetto a quelli nei quali il tumore primitivo non viene asportato.
APPROCCIO “OPEN” O LAPAROSCOPICO?
Le procedure laparoscopiche giocano un ruolo importante nel trattamento delle PNENs. È stato dimostrato che la pancreasectomia distale laparoscopica e l’enucleazione sono tecniche sicure e fattibili nei pazienti affetti da queste neoplasie (30). I vantaggi della chirurgia mini-invasiva sono riduzione del dolore post-operatorio, migliori risultati estetici, più breve ospedalizzazione e più rapido ritorno alle normali attività della vita quotidiana, con un tasso di fistola pancreatica paragonabile a quello osservato dopo chirurgia open (30).
Il numero degli insulinomi trattati per via laparoscopica è in progressivo aumento negli ultimi anni. Nell’85% dei pazienti si tratta di tumori singoli, spesso intra-pancreatici, che, se ben localizzati pre-operatoriamente, possono essere trattati nel 70-100% dei casi con questo tipo di approccio (5). Le procedure laparoscopiche dovrebbero essere integrate all’uso dell’ecografia intra-operatoria, per definire correttamente il sito di resezione pancreatica in corso di pancreasectomia distale (31). Il valore della laparoscopia per le lesioni pancreatiche maligne è legato principalmente alle sue capacità diagnostiche e stadiative (32).
CONCLUSIONI
Il trattamento chirurgico delle PNEN rimane ancora un punto cruciale nel trattamento multimodale di questi tumori. Sebbene in passato la chirurgia fosse obbligatoria per tutte le forme di PNEN, attualmente la tendenza è quella di limitarla ai casi di lesioni sintomatiche o in generale > 2 cm. D’altro canto, le resezioni radicali per le forme aggressive rappresentano l’unica strategia di cura per queste patologie, nonostante i recenti progressi delle terapie mediche. Le procedure chirurgiche dovrebbero comunque essere sempre adattate a ogni singolo paziente, sulla base delle comorbilità, dell’estensione dell’eventuale resezione e delle opzioni terapeutiche alternative.
BIBLIOGRAFIA
- Vagefi PA, et al. Evolving patterns in the detection and outcomes of pancreatic neuroendocrine neoplasms. Arch Surg 2007, 142: 347-54.
- Scarpa A, et al. Pancreatic endocrine tumors: improved TNM staging and hystopathological grading permit a clinically efficient prognostic stratification of patients. Mod Pathol 2010, 23: 824-33.
- Bosman. WHO classification of tumor of the digestive system. IARC Press, Lyon 2010.
- Buchmann L, et al. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumors. Eur J Nucl Med Mol Imaging 2007, 34: 1617–26.
- Kulke MH, et al. NANETS treatment guidelines. Well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas 2010, 39: 735-52.
- Rindi G, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch 2006, 449: 395-401.
- Rindi G, et al. TNM staging of neoplasms of the endocrine pancreas: results from a large international cohort study. J Natl Cancer Inst 2012, 104: 1–14.
- Ellison TA. A single institution's 26-year experience with nonfunctional pancreatic neuroendocrine tumors: a validation of current staging systems and a new prognostic nomogram. Ann Surg 2013, doi: 10.1097/SLA.0b013e31828f3174.
- Bettini R, et al. Tumor size correlates with malignancy in non-functioning pancreatic endocrine tumor. Surgery 2011, 150: 75-82.
- Falconi M, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012, 95: 120-34.
- Buchler MW, et al. Changes in morbidity after pancreatic resection: toward the end of completion pancreatectomy. Arch Surg 2003 138: 1310-4.
- Yeo CJ, et al. Six hundred fifty consecutive pancreaticoduodenectomies in the 1990s: pathology, complications, and outcomes. Ann Surg 1997, 226: 248-57.
- Smith JK, et al. Complications after pancreatectomy for neuroendocrine tumors: a national study. J Surg Res 2010, 163: 63-8.
- Falconi M, et al. Parenchyma-preserving resections for small non functioning pancreatic endocrine tumors. Ann Surg Oncol 2010, 17: 1621-7.
- Falconi M, et al. Pancreatic insufficiency after different resections for benign tumours. Br J Surg 2008, 95: 85-91.
- Aranha GV, Shoup M. Nonstandard pancreatic resections for unusual lesions. Am J Surg 2005, 189: 223-8.
- Partelli S, et al. Pattern and clinical predictor of lymph node involvement in nonfunctioning pancreatic neuroendocrine tumors (NF-PanNETs). JAMA Surg 2013, 148: 932-9.
- Bilimoria KY, et al. Prognostic score predicting survival after resection of pancreatic neuroendocrine tumors. Analysis of 3851 patients. Ann Surg 2008, 247: 490–500.
- Fischer L, et al. Clinical outcome and long-term survival in 118 consecutive patients with neuroendocrine tumours of the pancreas. Br J Surg 2008, 95: 627-35.
- Saxena A, et al. Factors predicting response and survival after yttrium-90 radioembolization of unresectable neuroendocrine tumor liver metastases: a critical appraisal of 48 cases. Ann Surg 2010, 251: 910-6.
- Frilling A, et al. Treatment of liver metastases from neuroendocrine tumours in relation to the extent of hepatic disease. Br J Surg 2009, 96: 175-84.
- Fendrich V, et al. An aggressive surgical approach leads to long-term survival in patients with pancreatic endocrine tumors. Ann Surg 2006, 244: 845-51.
- Steinmuller T, et al. Consensus guidelines for the management of patients with liver metastases from digestive (neuro)endocrine tumors: foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2008, 87: 47-62.
- Bloomston M, et al. Cytoreduction results in high perioperative mortality and decreased survival in patients undergoing pancreatectomy for neuroendocrine tumors of the pancreas. J Gastrointest Surg 2006, 10: 1361-70.
- Touzios JG, et al. Neuroendocrine hepatic metastases: does aggressive management improve survival? Ann Surg 2005, 241: 776-83.
- Elias D, et al. Combined liver surgery and RFA for patients with gastroenteropancreatic endocrine tumors presenting with more than 15 metastases to the liver. Eur J Surg Oncol 2009, 35: 1092-7.
- Scigliano S, et al. Clinical and imaging follow-up after exhaustive liver resection of endocrine metastases: a 15-year monocentric experience. Endocr Relat Cancer 2009, 16: 977-90.
- Bettini R, et al. Prognostic factors at diagnosis and value of WHO classification in a mono-institutional series of 180 non-functioning pancreatic endocrine tumours. Ann Oncol 2008, 19: 903-8.
- Capurso G, et al. Role of resection of the primary pancreatic neuroendocrine tumor only in patients with unresectable metastatic liver disease: a systematic review. Neuroendocrinology 2011, 93: 223-9.
- Fernandez-Cruz L, et al. Is laparoscopic resection adequate in patients with neuroendocrine pancreatic tumors? World J Surg 2008, 32: 904-17.
- Iihara M, T Obara. Minimally invasive endocrine surgery: laparoscopic resection of insulinomas. Biomed Pharmacother 2002, 56 Suppl 1: 227s-30.
- Angst E, et al. Laparoscopic surgery for cancer: a systematic review and a way forward. J Am Coll Surg 2010, 211: 412-23.
Terapia chirurgica per i NET intestinali
Emilio Bertani
Divisione di Chirurgia Generale e Laparoscopica, Istituto Europeo di Oncologia, Milano
Introduzione
La terapia chirurgica dei NET intestinali è influenzata dalla modalità della presentazione della malattia.
Per le neoplasie del piccolo intestino, dell’appendice e del colon-retto diagnosticate in stadio I-III la terapia è sempre chirurgica e consiste nella resezione intestinale con ampia linfoadenectomia (1).
Nei NET appendicolari può essere sufficiente l’appendicectomia, mentre l’emicolectomia destra è suggerita in casi particolari (vedi oltre).
In particolare, i NET a primitività digiuno-ileale sono comunemente diagnosticati in stadio IV, con metastasi sincrone a livello epatico (50-75% dei casi), a causa dell’elevato potenziale metastatico di queste neoplasie, indipendente dalla dimensione del tumore primitivo (2, 3). In presenza di metastasi epatiche sincrone, il trattamento chirurgico è ancora fortemente dibattuto. In molti suggeriscono la resezione del tumore primitivo anche in presenza di metastasi epatiche diffuse, per prevenire o curare le complicanze della malnutrizione e l’occlusione intestinale (4-8). Infatti, secondo alcuni con le moderne terapie i pazienti con NET intestinale metastatico possono vivere tanto a lungo da sviluppare un’ischemia mesenteriale o un’occlusione intestinale dovuta alle voluminose metastasi linfonodali all’interno del mesentere (9). Oltre ai casi in cui la resezione del tumore primitivo è indicata per ottenere la radicalità oncologica (assenza di metastasi) o per lo svilupparsi di sintomi, alcuni (10, 11) la ritengono comunque indicata, perchè migliora la sopravvivenza anche nei pazienti asintomatici in cui non è possibile arrivare alla radicalità chirurgica per la presenza di metastasi diffuse (anche se tale risultato non è riproducibile in tutte le casistiche, 12). La resezione del tumore primitivo, quando possibile, è stata suggerita anche dalle linee guida ENETS (1) e nello studio PROMID (13) in pazienti in terapia con octreotide LAR e da diversi altri Autori (10, 14). Recentemente una casistica importante ha riportato risultati diversi (12), dove la resezione del tumore primitivo in un contesto di metastasi epatiche sincrone non ha determinato un miglioramento della prognosi a lungo termine.
Per quanto riguarda la chirurgia delle metastasi epatiche da NET del piccolo intestino, del colon-retto o dell’appendice ciecale, è consigliata la chirurgia cito-riduttiva con intento radicale, anche se l’indicazione dovrebbe essere discussa in ambito multidisciplinare, dato che talora può essere considerata impegnativa per il chirurgo e per il paziente (15, 16). In casi selezionati il trapianto di fegato rappresenta un’opzione percorribile che consente il miglioramento della sopravvivenza (17).
Le resezioni del piccolo intestino
Se l’obiettivo chirurgico è curativo, è sempre indicata la resezione intestinale. La multicentricità della neoplasia, presente in circa il 20% dei casi, non cambia l’indicazione chirurgica.
La resezione curativa del tumore primitivo e delle metastasi linfonodali loco-regionali migliora l’esito a lungo termine, portando a una sopravvivenza del 100% a 5 anni e > 95% a 10 anni per i pazienti in stadio I e II e > 80% per i pazienti con NET digiuno-ileale in stadio III (1).
L’intervento deve sempre comprendere la resezione di un segmento di piccolo intestino con un’ampia linfo-adenectomia (18-25). In caso di adenopatie macroscopiche alla radice dei vasi mesenterici, è raccomandata una dissezione linfonodale alta, ma ciò non sempre è fattibile, a causa di una severa reazione desmoplastica intorno ai vasi (figure 1-2).
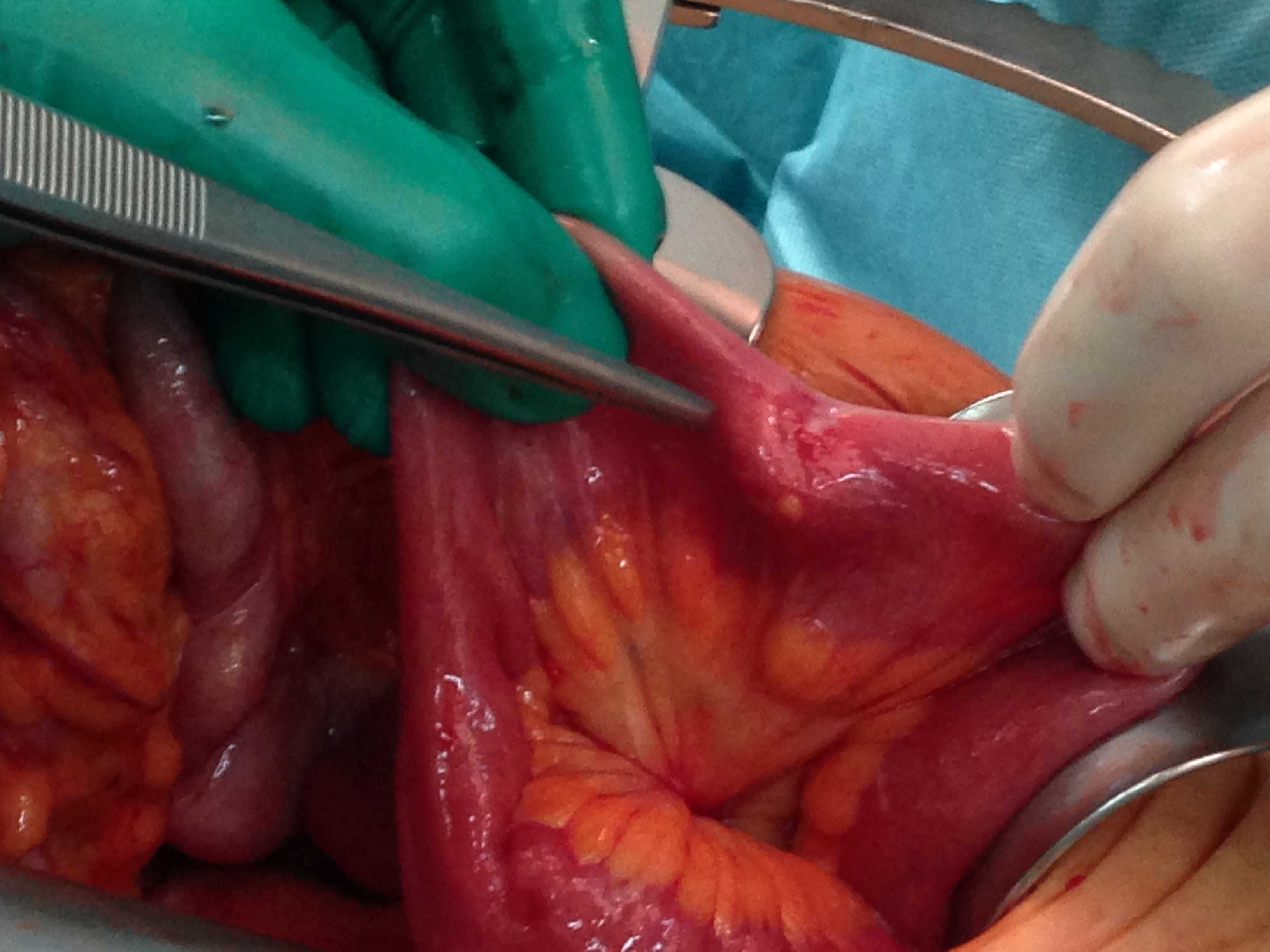
Figura 1: NET primitivo dell’ileo con retrazione mesenteriale
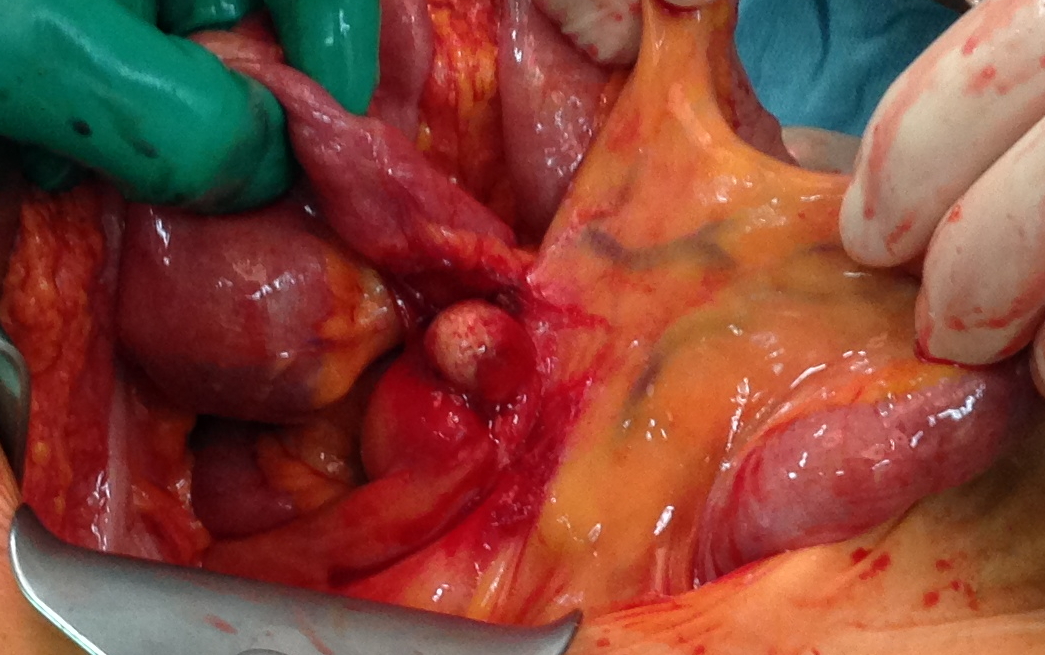
Figura 2: Linfadenopatie mesenteriali da NET ileale
In concomitanza alla resezione intestinale può essere eseguita la colecistectomia, nei pazienti con colelitiasi o nella prevenzione della stessa, dal momento che gli analoghi della somatostatina possono a lungo andare causare una calcolosi della colecisti. Tuttavia, non è mai stato dimostrato il beneficio della colecistectomia associata.
La chirurgia è essenzialmente eseguita per via laparotomica, in quanto non sempre gli standard oncologici possono essere realisticamente rispettati nella resezione del piccolo intestino, soprattutto in presenza di ampia infiltrazione mesenteriale e multicentricità della neoplasia.
La chirurgia dei NET dell’appendice
La chirurgia delle neoplasie neuroendocrine appendicolari prevede due tipi di intervento: la semplice appendicectomia e l’emicolectomia destra.
Spesso la diagnosi viene posta dal patologo dopo un intervento di appendicectomia laparoscopica o laparotomica per un quadro di appendicite acuta. La strategia successiva, cioè se sottoporre o meno il paziente a emicolectomia destra, viene stabilita da alcuni criteri anatomo-patologici, tuttavia non ancora universalmente riconosciuti e tuttora oggetto di dibattito (1, 26-29).
Per le neoplasie T1 (ENETS) o T1a (UICC/AJCC), cioè 3 mm. In questi casi l’opzione di eseguire un’emicolectomia destra deve essere discussa con il paziente, in quanto non è ancora stato dimostrato il reale beneficio di un allargamento chirurgico, a fronte di un aumento delle complicanze.
Per le neoplasie T2 (ENETS) o T1b (UICC/AJCC), cioè con diametro compreso fra 1 e 2 cm, il rischio di metastasi linfonodali o a distanza sembra essere aumentato, se consideriamo la lunga aspettativa di vita di questi pazienti, spesso giovani alla diagnosi. In questi casi l’emicolectomia destra dovrebbe mettere definitivamente al riparo da recidive di malattia, anche se il rischio di complicanze è sicuramente maggiore rispetto alla semplice appendicectomia. Anche in questo caso, un tumore localizzato alla base dell’appendice resecato in maniera incompleta (resezione R1) o in presenza di un’invasione del mesoappendice > 3 mm devono far propendere per l’intervento chirurgico più ampio, anche se non abbiamo a disposizione dati certi sulla disease-free survival a lungo termine.
Per le neoplasie con diametro > 2 cm con uno stadio ≥ T3 (ENETS) o ≥ T2 (UICC/AJCC) è sempre indicato l’intervento di emicolectomia destra, in considerazione dell’elevato rischio di metastasi linfonodali e di recidiva tumorale con metastasi a distanza.
L’intervento di emicolectomia destra può essere eseguito sia per via laparotomica che laparoscopica, presentando quest’ultima tecnica il vantaggio di un approccio mini-invasivo, con una più rapida ripresa funzionale e meno dolore per il paziente.
La chirurgia dei NET colo-rettali
Per quanto riguarda le indicazioni chirurgiche, vale il discorso fatto per i NET del piccolo intestino: qualora sia fattibile, in assenza di metastasi a distanza, è sempre consigliata la chirurgia con intento radicale (30). In presenza di metastasi epatiche, la chirurgia del tumore colo-rettale primitivo trova indicazione in caso di occlusione intestinale, perforazione o sanguinamento.
I NET del colon con estensione locale vengono trattati analogamente agli adenocarcinomi (31). Le lesioni di diametro < 2 cm possono essere asportate endoscopicamente o tramite mucosectomia. In caso di resezione incompleta o neoplasia G3, è indicata la resezione oncologica standard.
I NET rettali vengono efficacemente curati in stadio iniziale attraverso l’ablazione endoscopica o la resezione chirurgica, mentre negli stadi più avanzati non è chiaro il reale vantaggio della chirurgia. Il fattore prognostico più significativo in queste neoplasie è la dimensione del tumore.
- Le lesioni con diametro < 1 cm dovrebbero essere asportate endoscopicamente o per via trans-anale (32, 33). In questi casi il rischio di metastatizzazione è stato stimato sino a circa il 3%.
- Per le neoplasie di diametro compreso fra 1 e 2 cm, il trattamento è più controverso. Appare tuttavia chiaro come in questi casi sia opportuno procedere a un’accurata stadiazione della neoplasia mediante eco-endoscopia e/o RMN. Il rischio di metastasi a distanza in questa situazione è del 10-15%. Pur non essendoci una forte evidenza, il trattamento di escissione locale sembra essere il più appropriato anche in questi casi, poiché un approccio più aggressivo non sembra vantaggioso in termini di prognosi (34).
- In caso di tumore con diametro > 2 cm, il rischio di metastasi è importante (60-80%) (34-36). In questi casi il paziente è avviato ad una chirurgia rettale con total mesorectal excision, che tuttavia non sempre è in grado di controllare la malattia (32, 37).
Gli interventi di resezione colo-rettale più comunemente eseguiti, a cui deve sempre essere associata una linfoadenectomia standard, sono:
- emicolectomia destra;
- emicolectomia sinistra;
- resezione del colon trasverso;
- sigmoidectomia;
- resezione anteriore del retto con eventuale stomia derivativa;
- amputazione del retto per via addomino-perineale (Miles).
Tutti questi interventi possono essere eseguiti con approccio mini-invasivo laparoscopico (figure 3, 4), qualora indicato, a cui si è affiancato negli ultimi anni quello robotico con il sistema Da Vinci®, che ha dimostrato alcuni vantaggi soprattutto nell’ambito della chirurgia rettale (38, 39).
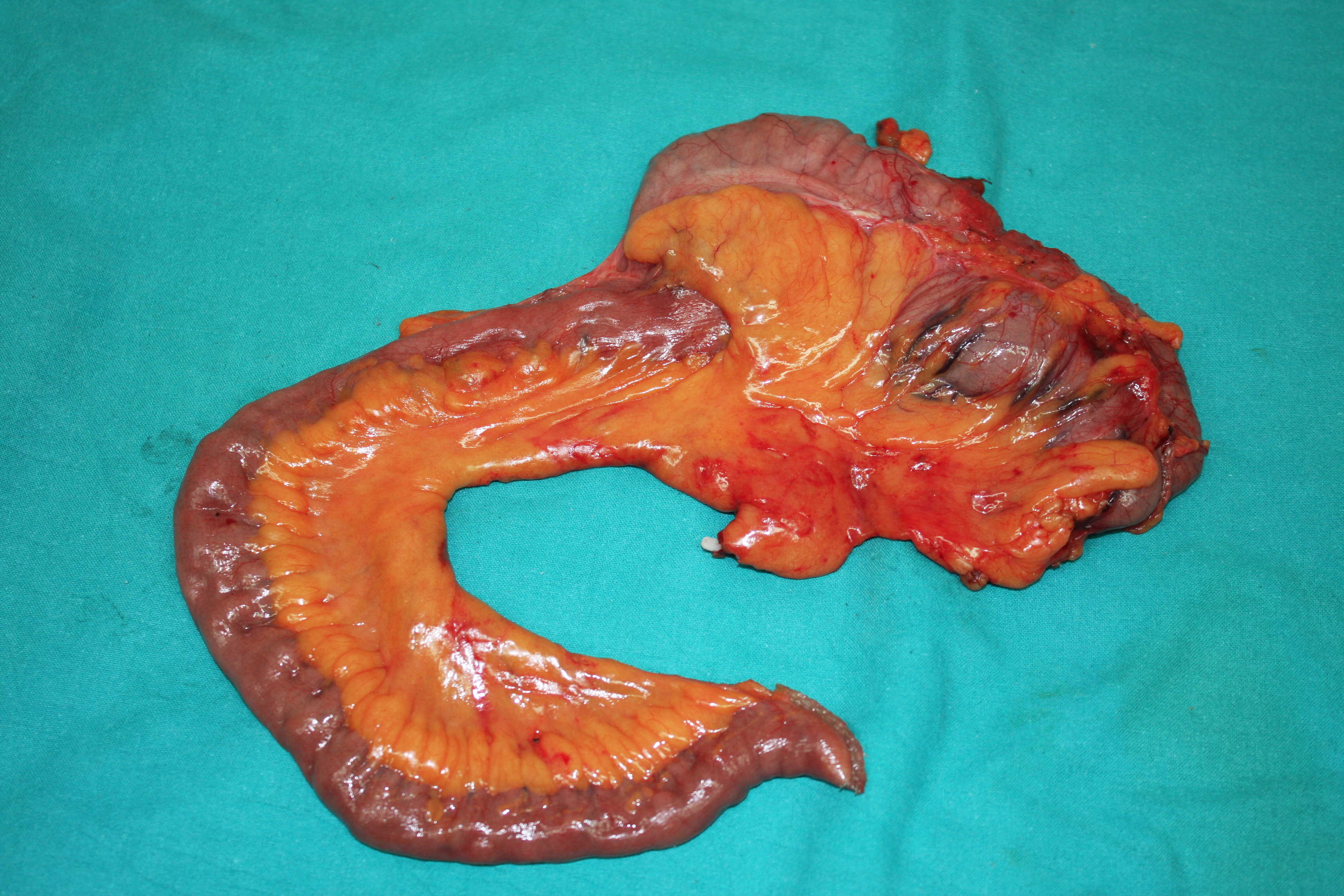
Figura 3: Pezzo asportato dopo emicolectomia destra laparoscopica per NET multipli dell’ultima ansa ileale
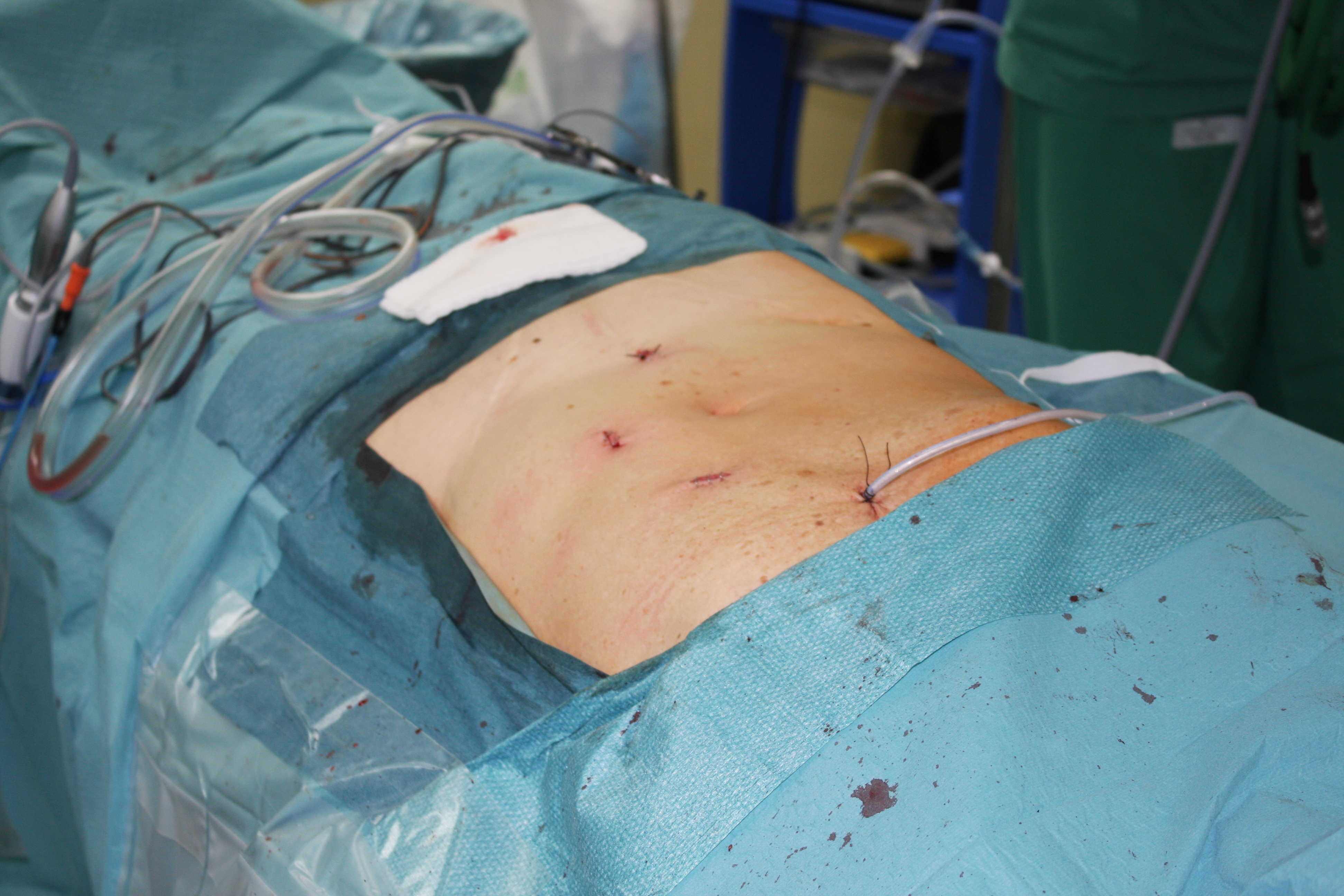
Figura 4: Incisioni chirurgiche al termine di intervento di emicolectomia destra laparoscopica con anastomosi intra-corporea
La chirurgia delle metastasi epatiche da NET intestinale
L’indicazione alla chirurgia epatica resettiva per le metastasi da NET dovrebbe avere sempre un intento di radicalità oncologica, con la rimozione di tutta la malattia a livello epatico, mantenendo una quota sufficiente di parenchima funzionante (40). La possibilità di avere un margine libero dovrebbe essere determinata dal chirurgo e dal radiologo in base alle tecniche di imaging (TC e RMN). Il chirurgo deve stabilire qual è la quota di parenchima residuo accettabile, all’incirca un terzo del volume epatico, o l’equivalente di almeno 2 segmenti di fegato sano. Spesso, nel contesto di una malattia epatica pluri-nodulare, alla chirurgia viene associata la termo-ablazione intra-operatoria con radiofrequenze (TARF) per il trattamento di lesioni profonde < 4 cm e chirurgicamente poco accessibili. Quando fattibile, la chirurgia resettiva con intento radicale è il gold standard per le metastasi epatiche da NET, con sopravvivenza a 5 anni del 60-80%, bassa mortalità post-operatoria (0-5%) e accettabile tasso di complicanze chirurgiche (circa il 30%) (41-44).
I prerequisiti per una chirurgica oncologicamente radicale sono:
- malattia epatica G1-G2;
- assenza di insufficienza cardiaca destra (cardiopatia da carcinoide);
- assenza di metastasi linfonodali ed extra-addominali non resecabili;
- assenza di carcinosi peritoneale diffusa;
- tumore primitivo già resecato o resecabile.
Non è generalmente raccomandata la resezione di metastasi epatiche da NET G3, ma può essere oggetto di trattamento individualizzato per lesioni singole resecabili. In particolari circostanze può essere considerata una valida palliazione la chirurgia epatica di debulking, associata o meno a terapie ablative. In questi casi viene raccomandata l’asportazione di almeno il 90% della malattia a livello epatico (45). È sempre indicata l’asportazione della neoplasia primitiva se ancora in sede, quando possibile, in concomitanza o successivamente all’intervento di resezione epatica.
Bibliografia
- Pape UF, Perren A, Niederle B, et al. ENETS consensus guidelines for the management of patients with neuroendocrine neoplasms from the jejuno-ileum and the appendix including goblet cell carcinomas. Neuroendocrinology 2012, 95: 135–56.
- Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumors. Lancet Oncol 2008, 9: 61-2.
- Dahdaleh FS, Calva-Cerqueira D, Carr JC, et al. Comparison of clinicopathologic factors in 122 patients with resected pancreatic and ileal neuroendocrine tumors from a single institution. Ann Surg Oncol 2012, 19: 966-72.
- Boudreaux JP, Putty B, Frey DJ, et al. Surgical treatment of advanced-stage carcinoid tumors: lessons learned. Ann Surg 2005, 241: 839-45; discussion 845-6.
- Makridis C, Oberg K, Juhlin C, et al. Surgical treatment of mid-gut carcinoid tumors. World J Surg 1990, 14: 377-83; discussion 384-5.
- Makridis C, Rastad J, Oberg K, Akerstrom G. Progression of metastases and symptom improvement from laparotomy in midgut carcinoid tumors. World J Surg 1996, 20: 900-6; discussion 907.
- Partensky C, Landraud R, Velecela E, Souquet JC. Resection of carcinoid tumors of the small intestine is still indicated in the presence of disseminated hepatic metastases. Ann Chir 1990, 44: 34-8.
- Soreide O, Berstad T, Bakka A, et al. Surgical treatment as a principle in patients with advanced abdominal carcinoid tumors. Surgery 1992, 111: 48-54.
- Boudreaux JP, Klimstra DS, Hassan MM, et al. North American Neuroendocrine Tumor Society (NANETS). The NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the jejunum, ileum, appendix, and cecum. Pancreas 2010, 39: 753-66.
- Givi B, Pommier SJ, Thompson AK, et al. Operative resection of primary carcinoid neoplasms in patients with liver metastases yields significantly better survival. Surgery 2006, 140: 891-7; discussion 897-8.
- Ahmed A, Turner G, King B, et al. Midgut neuroendocrine tumours with liver metastases: results of the UKINETS study. Endocr Relat Cancer 2009, 16: 885-94.
- Strosberg J, Gardner N, Kvols L. Survival and prognostic factor analysis of 146 metastatic neuroendocrine tumors of the mid-gut. Neuroendocrinology 2009, 89: 471-6.
- Rinke A, Müller HH, Schade-Brittinger C, et al. PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009, 27: 4656-63.
- Norlén O, Stålberg P, Öberg K, et al. Long-term results of surgery for small intestinal neuroendocrine tumors at a tertiary referral center. World J Surg 2012, 36: 1419-31.
- Norton JA. Surgery for primary pancreatic neuroendocrine tumors. J Gastrointest Surg 2006, 10: 327–31.
- Norton JA. Endocrine tumours of the gastrointestinal tract. Surgical treatment of neuroendocrine metastases. Best Pract Res Clin Gastroenterol 2005, 19: 577–83.
- van Vilsteren FG, Baskin-Bey ES, Nagorney DM, et al. Liver transplantation for gastroenteropancreatic neuroendocrine cancers: defining selection criteria to improve survival. Liver Transpl 2006, 12: 448–56.
- Akerstrom G, Makridis C, Johansson H. Abdominal surgery in patients with midgut carcinoid tumors. Acta Oncol 1991, 30: 547–53.
- Rothmund M, Kisker O. Surgical treatment of carcinoid tumors of the small bowel, appendix, colon and rectum. Digestion 1994, 55 (suppl 3): 86–91.
- Ahlman H, Wangberg B, Jansson S, et al. Interventional treatment of gastrointestinal neuroendocrine tumours. Digestion 2000, 62 (suppl 1): 59–68.
- Makridis C, Oberg K, Juhlin C, et al. Surgical treatment of mid-gut carcinoid tumors. World J Surg 1990, 14: 377-83; discussion 384-5.
- Norton JA. Surgical management of carcinoid tumors: role of debulking and surgery for patients with advanced disease. Digestion 1994, 55 (suppl 3): 98–103.
- Goede AC, Winslet MC. Surgery for carcinoid tumours of the lower gastrointestinal tract. Colorectal Dis 2003, 5: 123–8.
- Sutton R, Doran HE, Williams EMI, et al. Surgery for midgut carcinoid. Endocr Relat Cancer 2003, 10: 469–81.
- Han SL, Cheng J, Zhou HZ, et al. Surgically treated primary malignant tumor of small bowel: a clinical analysis. World J Gastroenterol 2010, 16: 1527–32.
- Moertel CG, Weiland LH, Nagorney DM, et al. Carcinoid tumor of the appendix: treatment and prognosis. N Engl J Med 1987, 317: 1699–701.
- Modlin IM, Kidd M, Latich I, et al. Genetic differentiation of appendiceal tumor malignancy: a guide for the perplexed. Ann Surg 2006, 244: 52–60.
- Bamboat ZM, Berger DL. Is right hemicolectomy for 2.0-cm appendiceal carcinoids justified? Arch Surg 2006, 141: 349–52.
- Rossi G, Valli R, Bertolini F, et al. Does mesoappendix infiltration predict a worse prognosis in incidental neuroendocrine tumors of the appendix? A clinicopathologic and immunohistochemical study of 15 cases. Am J Clin Pathol 2003, 120: 706–11.
- Caplin M, Sundin A, Nillson O. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: colorectal neuroendocrine neoplasms. Neuroendocrinology 2012, 95: 88–97.
- Rosenberg JM, Welch JP. Carcinoid tumors of the colon. A study of 72 patients. Am J Surg 1985, 149: 775–9.
- Kwaan MR, Goldberg JE, Bleday R. Rectal carcinoid tumors: review of results after endoscopic and surgical therapy. Arch Surg 2008, 143: 471–5.
- Onozato Y, Kakizaki S, Lizuka H, et al. Endoscopic treatment of rectal carcinoid tumors. Dis Colon Rectum 2010, 53: 169–76.
- Sauven P, Ridge JA, Quan SH, Sigurdson ER. Anorectal carcinoid tumors. Is aggressive surgery warranted? Ann Surg 1990, 211: 67–71.
- Jetmore AB, Ray JE, Gathright JB Jr, et al. Rectal carcinoids: the most frequent NET tumor. Dis Colon Rectum 1992, 35: 717–25.
- Shields CJ, Tiret E, Winter DC. Carcinoid tumors of the rectum: a multi-institutional international collaboration. Ann Surg 2010, 252: 750–5.
- Imada-Shirakata Y, Sakai M, Kajiyama T, et al. Endoscopic resection of rectal carcinoid tumors using aspiration lumpectomy. Endoscopy 1997, 29: 34–8.
- Bertani E, Chiappa A, Biffi R, et al. Assessing appropriateness for elective colorectal cancer surgery: clinical, oncological, and quality-of-life short-term outcomes employing different treatment approaches. Int J Colorectal Dis 2011, 26: 1317-27.
- Baek JH, McKenzie S, Garcia-Aguilar J, Pigazzi A. Oncologic outcomes of robotic-assisted total mesorectal excision for the treatment of rectal cancer. Ann Surg 2010, 251: 882-6.
- Pavel M, Baudin E, Couvelard A. ENETS consensus guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012, 95: 157–76.
- Sarmiento JM, Heywood G, Rubin J, et al Surgical treatment of neuroendocrine metastases to the liver: a plea for resection to increase survival. J Am Coll Surg 2003, 197: 29–37.
- Elias D, Lasser P, Ducreux M, et al. Liver resection (and associated extrahepatic resections) for metastatic well differentiated endocrine tumors: a 15-year single-center prospective study. Surgery 2003, 133: 375–82.
- Chamberlain R, Canes D, Brown K, et al. Hepatic neuroendocrine metastases: does intervention alter outcomes? J Am Coll Surg 2000, 190: 432–45.
- Chen H, Hardacre J, Uzar A, et al. Isolated liver metastases from neuroendocrine tumors: does resection prolong survival? J Am Coll Surg 1998, 187: 88–92.
- Sarmiento J, Que F. Hepatic surgery for metastases from neuroendocrine tumors. Surg Oncol Clin N Am 2003, 12: 231–42.
Terapie loco-regionali per i NET
Sara Massironi1, Antonio Nicolini2, Federica Cavalcoli1,3
1UOC Gastroenterologia ed Endoscopia, Divisioni di 1Gastroenterologia e Epatologia e 2Radiologia, 2Divisione di Radiologia Interventistica, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano
3Dipartimento di Fisiopatologia e dei Trapianti, Università degli Studi di Milano
(aggiornato al 18 marzo 2017)
INTRODUZIONE
Nonostante l’andamento clinico delle neoplasie neuroendocrine (NEN) sia molto variabile a seconda del grado di differenziazione, del comportamento biologico e della sede del tumore primitivo, è esperienza comune che possano presentare un’importante diffusione metastatica già al momento della diagnosi; in corso di malattia metastatica, il fegato rappresenta la principale sede di metastatizzazione (1). Studi recenti riportano la presenza di metastasi a distanza nel 65-90% dei casi di NEN al momento della diagnosi (2,3).
La metastatizzazione a distanza, nel caso delle NEN, correla con il grado istologico, le dimensioni e la sede del tumore primitivo, tuttavia è dimostrato che anche NEN di dimensioni molto ridotte e con basso grado istologico (G1 o G2) possano presentarsi con un importante carico metastatico (4).
La presenza di metastasi a distanza, assieme al grado istologico, rappresenta uno dei più importanti fattori prognostici ed è associata a una riduzione significativa della sopravvivenza globale. La presentazione clinica dei pazienti con NEN e metastasi epatiche può essere caratterizzata da sintomi aspecifici, quali dolore, anoressia, nausea e calo ponderale o da sintomi legati all’effetto massa esercitata dalla neoplasia in sede epatica (ittero/colecistite/colangite); inoltre, nel caso di NEN funzionanti (es. gastrinoma, serotoninoma, VIPoma, ecc), può essere invece secondaria alla secrezione di specifici prodotti ormonali o amine vasoattive in grado di generare sintomi clinicamente rilevanti, dal momento che la presenza di metastasi epatiche riduce la capacità del fegato di metabolizzare tali sostanze (5).
Il trattamento aggressivo dei secondarismi epatici in caso di NEN si è dimostrato efficace nel controllo della sintomatologia e, seppur con minori evidenze cliniche, anche nel determinare un incremento della sopravvivenza globale (2).
Dal punto di vista morfologico, si possono riconoscere tre diverse modalità di infiltrazione epatica (6):
- diffusione metastatica limitata ad un solo lobo epatico o limitata a due segmenti adiacenti (20-25% dei casi);
- pattern metastatico “complesso”, con un lobo primariamente affetto e lesioni focali satelliti controlaterali (10–15% dei casi);
- pattern metastatico diffuso e multilobare (60–70% dei casi).
Il quadro di metastatizzazione è determinante nella scelta del trattamento loco-regionale più appropriato: nel primo caso le localizzazione secondarie possono essere asportate mediante una resezione anatomica standard, nel secondo caso l’approccio chirurgico è possibile solo in casi selezionati, mentre più spesso si possono eseguire trattamenti loco-regionali, mentre nel terzo caso l’approccio chirurgico non è in genere possibile mentre i trattamenti loco-regionali rimangono gli unici applicabili, seppur in un numero selezionato di casi.
TERAPIE LOCO-REGIONALI
Al momento sono disponibili diverse opzioni terapeutiche per i pazienti affetti da NEN con metastasi epatiche, che includono sia approcci sistemici (analoghi della somatostatina –SSA–, terapia radiorecettoriale –PRRT– e le nuove terapie a bersaglio molecolare con Everolimus e Sunitinib) sia approcci loco-regionali (resezione chirurgica, terapie ablative loco-regionali, terapie intra-arteriose e radio-embolizzazione, che combina le terapie intra-arteriose –embolizzazione/chemioembolizzazione– con la radioterapia esercitata da “radioemboli”). Infine, in pazienti selezionati, può essere preso in considerazione anche il trapianto ortotopico di fegato (1).
Quale sia la migliore gestione dei pazienti affetti da metastasi epatiche da NEN risulta ad oggi un punto ancora controverso: ove possibile la resezione chirurgica sembrerebbe essere la migliore scelta terapeutica, anche se risulta fattibile solo in una percentuale limitata di pazienti (4). Generalmente le terapie loco-regionali trovano indicazione in pazienti con NEN avanzate in assenza di diffusione extra-epatica o comunque quando il fegato rappresenta la sede principale di malattia, anche nel caso vi sia malattia extra-epatica (4). Le terapie loco-regionali sono generalmente utilizzate in associazione alla terapia con SSA, tuttavia ad oggi nessuno studio riporta l’utilizzo di terapie loco-regionali in associazione con le terapie sistemiche (SSA, PRTT, Everolimus o Sunitinib) (7). Inoltre, non vi sono studi che forniscano informazioni riguardo il timing ottimale di questi trattamenti rispetto alle altre terapie disponibili.
In generale la scelta delle diverse opzioni terapeutiche in questi pazienti deve essere valutata in modo integrato, considerando diversi fattori quali il grado istologico, la presenza di metastasi extra-epatiche, la presenza di sintomi e il performance status del paziente (8).
Terapie ablative locali
Le terapie ablative includono l’ablazione con radio-frequenza (RFA), l’ablazione mediante micro-onde (MWA), la crio-terapia e l’alcolizzazione.
Le tecniche ablative locali trovano indicazione soprattutto in caso di metastasi non resecabili chirurgicamente, di diametro < 5-6 cm. Attualmente le tecniche più utilizzate sono RFA e MWA, mentre crio-ablazione e alcolizzazione trovano minor applicazione.
La RFA è basata sulla conversione di onde a radiofrequenza in calore, tramite l’utilizzo di correnti alternate ad alta frequenza che causano vibrazioni ioniche. La RFA rappresenta sicuramente la tecnica ablativa più utilizzata, grazie all’ottimo profilo di sicurezza e alla buona tollerabilità (4). Inoltre, la RFA può essere applicata come terapia adiuvante anche in pazienti con malattia epatica diffusa, presenza di metastasi bilobari o localizzazioni anatomiche che risultano difficilmente aggredibili chirurgicamente, consentendo un trattamento citoriduttivo anche a pazienti che non potrebbero beneficiare di un trattamento chirurgico classico.
A seconda della sede delle metastasi, la RFA può essere eseguita con approccio percutaneo o intra-operativamente in corso di interventi laparoscopici o laparotomici (7). La RFA risulta particolarmente efficace in pazienti con lesioni di diametro < 3.5 cm e in numero < 5 o in caso di localizzazioni multiple quando la somma dei diametri risulta comunque < 8 cm (6). In caso di lesioni > 5 cm, localizzazione ilare epatica, o in prossimità di grossi vasi, o dei dotti biliari principali o vicino alla superficie epatica vi è un rischio significativamente aumentato di lesioni termiche e l’utilizzo della RFA è sconsigliato (6).
Dati recenti hanno dimostrato un’elevata efficacia nel controllo locale delle metastasi e dei sintomi che giunge fino al 92% dei casi, con mantenimento della risposta per 14-27 mesi (4). Inoltre, è stata riportata una riduzione significativa dei livelli dei marcatori circolanti. La percentuale di recidiva risulta comunque elevata (63-87% dei casi), con sopravvivenza a 5 anni del 57-80% (9). Nel complesso la RFA è una procedura ben tollerata, con ridotti tassi di morbilità (< 10%) e mortalità (< 1%) e l’efficace controllo dei sintomi la rende un trattamento utile, sia da solo che in combinazione con tecniche resettive (9). In rari casi tuttavia le complicanze possono essere rilevanti e comprendono il sanguinamento intra-epatico e la formazione di ascessi epatici (9,10).
La MWA prevede l’utilizzo di microonde, che applicate al tessuto neoplastico generano calore. Rispetto alla RFA, la MWA presenta un tempo di applicazione più limitato, con una conseguente minore diffusione del calore all’interno del parenchima epatico e rappresenta quindi un’utile opzione terapeutica soprattutto in caso di lesioni localizzate in prossimità dei grossi vasi o dei dotti biliari principali (7).
Terapie intra-arteriose
L’utilizzo di terapie intra-arteriose nei pazienti con NEN e metastasi epatiche è basato sull’osservazione che queste lesioni sono solitamente altamente vascolarizzate, principalmente rifornite da rami dell’arteria epatica, mentre il parenchima epatico, in condizioni fisiologiche, è vascolarizzato dal sistema venoso portale. L’occlusione vascolare arteriosa, volta a indurre ischemia e necrosi nelle lesioni epatiche metastatiche, può essere ottenuta tramite approccio percutaneo, solitamente tramite l’arteria femorale e successiva embolizzazione trans-arteriosa. Esistono diverse tecniche:
- l’embolizzazione trans-arteriosa “semplice” (TAE) utilizza sostanze inerti, come lipiodol, schiume di alcol polivinilico o microsfere;
- la somministrazione intra-arteriosa diretta di chemioterapici citotossici (TACE), consente di raggiungere elevate concentrazioni a livello epatico di sostanze chemioterapiche (doxorubicina, cisplatino, gemcitabina, streptozotocina, o 5-fluorouracile) (7);
- la TACE-DEB prevede l’utilizzo di microsfere che veicolano chemioterapici lentamente, massimizzando l’effetto terapeutico sulle cellule neoplastiche;
- la TARE o radio-embolizzazione trans-arteriosa con Yttrio-90 è una tecnica che è stata sviluppata per colpire multiple aree di malattia a livello epatico, permettendo una sorta di brachiterapia intra-epatica (4,7). Infatti, le microsfere contenenti Yttrio-90 iniettate nell'arteria epatica giungono a livello dei capillari, dove si depositano senza diffondere e continuano a emettere radioattività direttamente nel tumore. La TARE non è quindi limitata dal numero e sede delle metastasi da NEN e trova applicazione anche in caso di malattia bilobare. Sembra presentare alcuni vantaggi rispetto alle altre tecniche intra-arteriose, ad esempio una minor durata di ospedalizzazione (4).
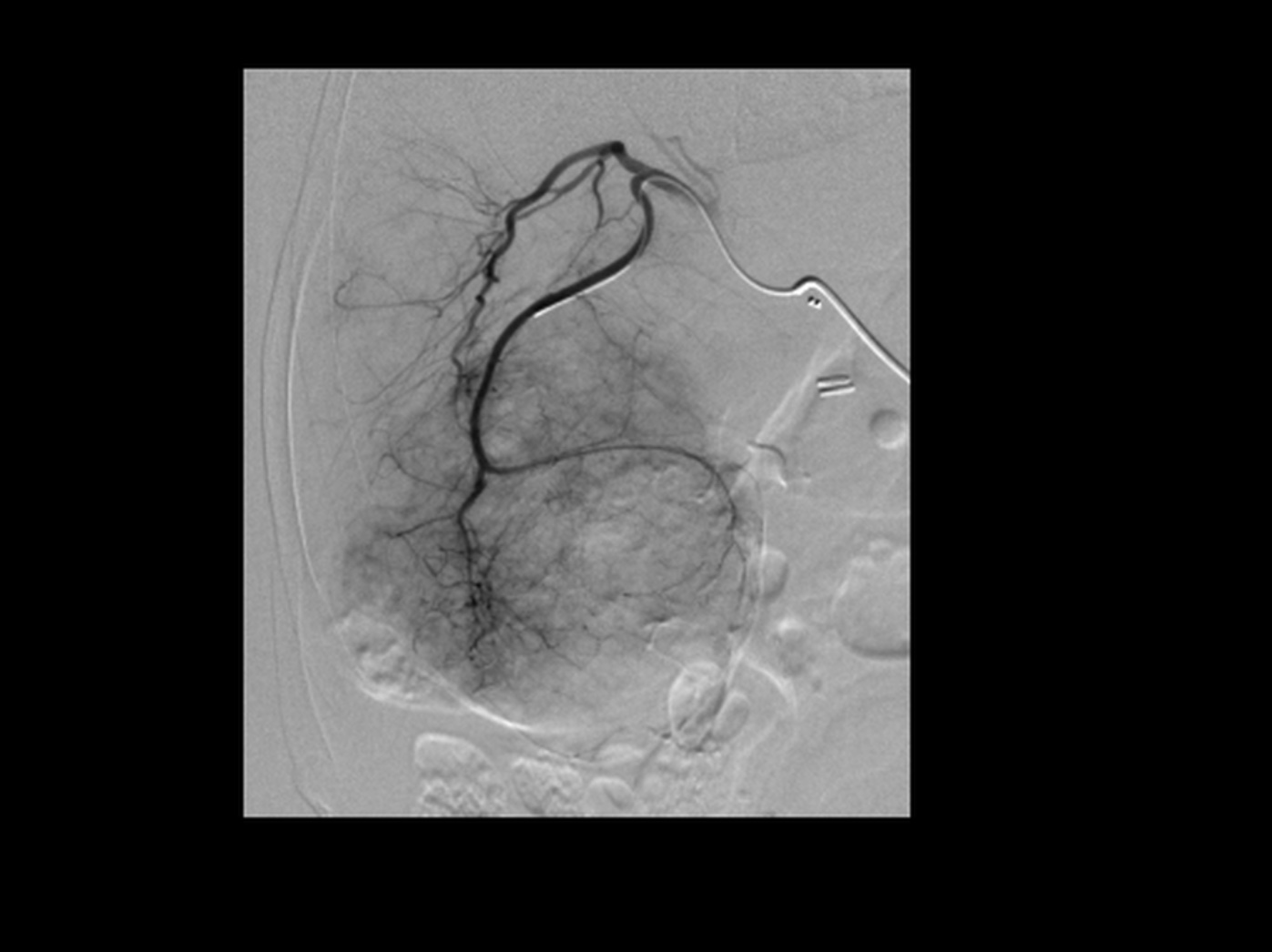
Angiografia arteriosa di voluminose metastasi epatiche ipervascolarizzate ad origine da neoplasia neuroendocrina ileale
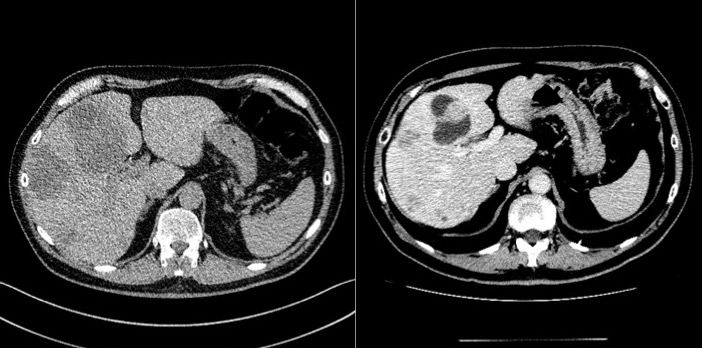
Immagini TC di estese metastasi epatiche da neoplasia neuroendocrina ileale prima (sinistra) e dopo (destra) trattamento mediante chemioembolizzazione epatica
Uno dei principali vantaggi di queste tecniche è che possono essere ripetute fino ad ottenere un’efficace cito-riduzione delle lesioni epatiche. L’utilizzo ripetuto di terapie intra-arteriose si è dimostrato efficace nella palliazione dei sintomi e nel prolungare la sopravvivenza globale in pazienti con metastasi epatiche da NEN. Le principali controindicazioni a queste tecniche sono la presenza di trombosi portale, insufficienza epatica e severe comorbilità (11). Ulteriori controindicazioni sono rappresentate dalla presenza di anastomosi biliari (es. intervento di Whipple), poiché incrementano il rischio di formazione di ascessi epatici legati ad una significativa traslocazione batterica e la presenza di shunt epato-polmonari, in cui si è registrato un incremento di mortalità (4).
I pazienti che vanno incontro a queste procedure possono presentare frequentemente una sindrome post-embolizzazione, caratterizzata da nausea e vomito (50–70%), dolore in ipocondrio destro/epigastrio (50–60%), febbre (30–60%), incremento delle transaminasi (100%). Gli effetti collaterali più rilevanti comprendono: necrosi della colecisti, sindrome epato-renale, pancreatite, formazione di ascessi epatici o aneurismi. Poiché queste procedure possono determinare una significativa morbilità, dovrebbero essere eseguite solamente in centri specializzati. La mortalità risulta ridotta in centri dotati di buona esperienza (0-3.3%) (11).
È riportata una risposta parziale o completa, rispettivamente nel 33-50% come risposta oggettiva radiologica, nel 73-100% come controllo della sintomatologia e 57-91% come riduzione dei marcatori circolanti. La durata media della risposta in termini di controllo dei sintomi risulta essere di 14-22 mesi (11). La sopravvivenza a 5 anni in diversi studi varia tra il 50-83% per la TACE e il 40-67% per la TAE (4). Un recente studio nei pazienti con metastasi epatiche da NEN trattate con terapie intra-arteriose (TAE, TACE, TACE-DEB, TARE) ha dimostrato una sopravvivenza mediana di 34 mesi e una sopravvivenza a 5 anni del 30%, vs 123 mesi e 74% nei pazienti che andavano incontro ad un approccio chirurgico (12). Nello stesso lavoro gli autori non osservavano differenze significative nella sopravvivenza nei pazienti trattati con tecniche intra-arteriose e approccio chirurgico in pazienti sintomatici e con interessamento epatico > 25% (12). Le tecniche intra-arteriose potrebbero essere pertanto particolarmente indicate in pazienti con importante carico metastatico, mentre l’approccio chirurgico dovrebbe essere riservato a pazienti con malattia epatica < 25% o pazienti con sintomi correlati all’effetto massa.
La TAE è stata storicamente confrontata soprattutto con la TACE, ma nessuna delle due tecniche ha dimostrato un significativo miglior beneficio rispetto all’altra (7). Alcuni studi sembrano dimostrare una maggiore efficacia della TACE-DEB rispetto alla TACE tradizionale, tuttavia sono state riportate complicanze severe, come lo sviluppo di biliomi e ascessi intra-epatici, che hanno comportato la precoce interruzione del principale studio di fase II sulla TACE-DEB (13).
Al momento non vi sono studi disponibili che confrontino direttamente queste tecniche, anche se alcuni dati preliminari sembrerebbero suggerire un profilo di tossicità migliore con una minor durate dell’ospedalizzazione per la TARE rispetto a TACE e TAE. Tuttavia, appaiono necessari ulteriori studi per determinare il profilo di sicurezza e di tossicità ed i possibili benefici di una tecnica rispetto all’altra. Inoltre sono necessari studi volti ad identificare il corretto timing dell’utilizzo di tale trattamento rispetto alle altre terapie sistemiche disponibili.
BIBLIOGRAFIA
- Pavel M, O'Toole D, Costa F, et al. ENETS Consensus Guidelines Update for the management of distant metastatic disease of intestinal, pancreatic, bronchial neuroendocrine neoplasms (NEN) and NEN of unknown primary site. Neuroendocrinology 2016, 103: 172-85.
- Saxena A, Chua TC, Sarkar A, et al. Progression and survival results after radical hepatic metastasectomy of indolent advanced neuroendocrine neoplasms (NENs) supports an aggressive surgical approach. Surgery 2011, 149: 209-20.
- Pape UF, Jann H, Müller-Nordhorn J, et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer 2008, 113: 256-65.
- Pavel M, Baudin E, Couvelard A, et al. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012, 95: 157-76.
- Rossi RE, Massironi S, Spampatti MP, et al. Treatment of liver metastases in patients with digestive neuroendocrine tumors. J Gastrointest Surg 2012, 16: 1981-92.
- Steinmüller T, Kianmanesh R, Falconi M, et al. Consensus guidelines for the management of patients with liver metastases from digestive (neuro)endocrine tumors: foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2008, 87: 47-62.
- Kennedy A, Bester L, Salem R, et al. Role of hepatic intra-arterial therapies in metastatic neuroendocrine tumours (NET): guidelines from the NET-Liver-Metastases Consensus Conference. HPB (Oxford) 2015, 17: 29-37.
- Frilling A, Clift AK. Therapeutic strategies for neuroendocrine liver metastases. Cancer 2015, 121: 1172-86.
- Mohan H, Nicholson P, Winter DC, et al. Radiofrequency ablation for neuroendocrine liver metastases: a systematic review. J Vasc Interv Radiol 2015, 26: 935-42.
- Akyildiz HY, Mitchell J, Milas M, et al. Laparoscopic radiofrequency thermal ablation of neuroendocrine hepatic metastases: long-term follow-up. Surgery 2010, 148: 1288-93.
- Roche A, Girish BV, de Baère T, et al. Trans-catheter arterial chemoembolization as first-line treatment for hepatic metastases from endocrine tumors. Eur Radiol 2003, 13: 136-40.
- Mayo SC, de Jong MC, Bloomston M, et al. Surgery versus intra-arterial therapy for neuroendocrine liver metastasis: a multicenter international analysis. Ann Surg Oncol 2011, 18: 3657-65.
- Bhagat N, Reyes DK, Lin M, et al. Phase II study of chemoembolization with drug-eluting beads in patients with hepatic neuroendocrine metastases: high incidence of biliary injury. Cardiovasc Intervent Radiol 2013, 36: 449-59.
Terapia radio-recettoriale nei tumori neuroendocrini gastro-entero-pancreatici (GEP-NET)
Angelina Filice & Annibale Versari
Struttura Complessa di Medicina Nucleare, Azienda USL-IRCCS di Reggio Emilia
(aggiornato al 14 giugno 2021) questo capitolo è in attesa di aggiornamento
Introduzione
La terapia radiorecettoriale (Peptide Receptor Radionuclide Therapy - PRRT) trova il proprio razionale nell’espressione, da parte delle neoplasie neuroendocrine (NEN), dei recettori per la somatostatina (SSTR). Le NEN sono, infatti, caratterizzate da un’elevata espressione dei SSTR sulla membrana delle cellule tumorali. I sottotipi SSTR2 e SSTR 5 sono quelli più frequentemente rappresentati e possono essere usati per l’imaging e per la PRRT (1).
La presenza di tali recettori sulla superficie cellulare può essere documentata tramite indagini di immuno-istochimica o in vivo, mediante l’imaging medico-nucleare che utilizza analoghi della somatostatina radio-marcati. Il primo radio-farmaco a essere impiegato in quest’ambito è stato l’Octreoscan marcato con (111In), un isotopo radioattivo che emette radiazioni gamma e per il quale si utilizza come sistema di rilevazione la gamma-camera. In tempi più recenti c’è stato lo sviluppo di nuovi analoghi della somatostatina, i DOTA-peptidi, marcati con l’isotopo 68Ga, che emette positroni rilevabili in PET/TC. Per quanto nei centri di medicina nucleare vengano ancora entrambi utilizzati con indicazioni sovrapponibili, questi ultimi si sono dimostrati superiori ai precedenti in termini di accuratezza diagnostica (2). La positività dell’imaging medico-nucleare, che documenti un’adeguata captazione del radio-peptide, rappresenta un pre-requisito fondamentale per la selezione del paziente per la PRRT (3).
Radio-farmaci
I radio-farmaci prevalentemente impiegati in PRRT sono attualmente 90Y/177Lu-DOTATOC/DOTATATE, con caratteristiche fisiche diverse. Il vettore è il peptide analogo della somatostatina (DOTATOC o DOTATATE), che spesso è lo stesso impiegato in diagnostica PET/TC marcato con 68Ga. Ovviamente, nel caso della PRRT, l’isotopo radioattivo legato al vettore non è un emettitore di positroni come in diagnostica (68Ga), ma un ß-emettitore, che permette di irradiare la sede di accumulo e cioè la lesione neoplastica:
- 90Yttrio (90Y) è un radio-nuclide con un’emivita di 67 giorni, che emette particelle ß di 2.27 MeV, con una penetrazione nei tessuti di circa 12 mm;
- 177Lutezio (177Lu) ha un’emivita di 6.64 giorni, che emette particelle ß di energia inferiore (0.5 MeV), con capacità di penetrazione nei tessuti di circa 2 mm.
Queste caratteristiche fisiche si traducono, quindi, in una maggior capacità di penetrazione dei radio-farmaci marcati con 90Y, che quindi risultano più efficaci in caso di lesioni di maggiori dimensioni (> 2 cm), ma che al tempo stesso hanno un maggior impatto dal punto di vista dosimetrico sugli organi a rischio (reni e midollo osseo), mentre i radio-farmaci marcati con 177Lu sono più appropriati per le lesioni di piccole dimensioni (< 2 cm), con minor impatto dosimetrico sugli organi a rischio e consentono la somministrazione di dosi più elevate (4). Le conoscenze sul diverso potere di penetrazione degli isotopi radioattivi utilizzati in PRRT hanno portato all’impiego in molti studi dei due radio-farmaci utilizzati in tandem, al fine di sfruttarne le diverse caratteristiche (5,6).
PRRT: studi di efficacia e di tossicità
Nel corso di più di venti anni di esperienza sulla PRRT sono stati pubblicati prevalentemente studi di fase I-II, molto eterogenei per vari aspetti (popolazione di pazienti e tipi di neoplasia neuroendocrina, radio-farmaci e isotopi radioattivi, dosi e schemi terapeutici, ecc) e pertanto difficilmente confrontabili. Nonostante tale eterogeneità, tuttavia, gli studi di efficacia hanno documentato un controllo della malattia in termini di risposta (parziale e completa) e di stazionarietà tra il 66% e il 92% (7-14).
Gli autori di una metanalisi pubblicata nel 2015 (15), che ha incluso 6 studi per un totale di 473 pazienti con NET inoperabili o metastatici sottoposti a PRRT, ne hanno confermato l’efficacia, evidenziando una risposta obiettiva globale del 29% con criteri RECIST e del 23% con criteri SWOG. La percentuale media di controllo di malattia è 81% nel gruppo RECIST e 82% nel gruppo SWOG.
Un lavoro del 2016 in 6 centri in Germania ha analizzato l'efficacia terapeutica di 90Y e 177Lu in 450 pazienti affetti da NEN del pancreas (38%), dell'intestino tenue (30%), a primitività incerta (19%) e NET polmonari (4%). La sopravvivenza globale mediana di tutti i pazienti è stata di 59 mesi. I pazienti affetti da neoplasie di grado II e III hanno mostrato sopravvivenza globale più bassa rispetto a quelli affetti da neoplasie di grado I. La sopravvivenza dei pazienti affetti da tumori di basso grado dell'intestino tenue è significativamente maggiore, rispetto a quella degli affetti da tumori di altri distretti corporei. Una remissione completa del tumore si è avuta nel 5.6% dei casi, mentre il 22.4% dei pazienti ha presentato una risposta parziale, il 47.3% è risultato stabile, il 4% è andato in progressione (16).
Nella gestione dei GEP-NET è attualmente ben noto che ottenere una stabilizzazione di malattia è da considerarsi un buon risultato in termini di controllo della malattia, in quanto, dal punto di vista prognostico, stabilizzazione e risposta al trattamento mostrano un’analoga probabilità di sopravvivenza (13).
A fronte dei risultati ottenuti in termini di efficacia, inoltre, la PRRT negli studi pubblicati nel corso degli anni, si è dimostrata essere ben tollerata in termini di tossicità. Gli effetti collaterali acuti sono generalmente lievi e auto-limitanti. Tra la tossicità acuta e a breve termine, sono più frequenti nausea, vomito e affaticamento, mentre i pazienti riferiscono meno frequentemente dolore addominale, diarrea e si riscontra lieve tossicità ematologica reversibile. L'alopecia e la sindrome carcinoide sono rare. I reni e il midollo osseo sono considerati organi a rischio per la tossicità a lungo termine nella PRRT. I possibili effetti collaterali gravi a lungo termine sono, infatti, l'insufficienza renale, la sindrome mielo-displastica (MDS) o la leucemia acuta (LA). Quindi, prima di iniziare la PRRT devono essere valutate la riserva ematologica e la funzionalità renale.
Come documentato in molti studi, è ormai noto che la somministrazione di aminoacidi, come forma di protezione renale, riduce l’irradiazione dei reni e di conseguenza la probabilità di comparsa di tossicità renale. In una revisione pubblicata pochi anni fa gli autori riportano una tossicità renale severa (grado 3/4) in < 3% dei pazienti qualora si usi un’adeguata protezione renale, che arriva al 15% negli studi in cui si usino protocolli senza somministrazione di aminoacidi (17).
Molti studi hanno riportato effetti a lungo termine sul midollo osseo. LA o MDS sono state riportate in < 3% dei pazienti che hanno ricevuto PRRT. In uno degli studi con una casistica più ampia (807 pazienti) è stata osservata tossicità ematologica lieve/assente nella grande maggioranza dei pazienti (82.2%) e severa nel 9.5%, in particolare 2.35% MDS e 1.1% LA (18). In uno studio comprendente 142 pazienti è stata osservata tossicità ematologica transitoria di grado 3-4 nel 12.8% dei pazienti, mentre LA e MDS sono state osservate rispettivamente nello 0.1% e 0.1% dei casi (19).
Studio di fase 3 NETTER-1 e approvazione del 177Lu-DOTATATE
Nonostante i numerosi studi sopra-citati, solo nel 2017 c’è stata la pubblicazione del NETTER-1, il primo studio multi-centrico, randomizzato di fase 3 sulla PRRT in 229 pazienti con tumori del piccolo intestino in progressione, inoperabili e positivi al recettore della somatostatina (20): sono stati confrontati 177Lu-DOTATATE (4 dosi da 7400 MBq ogni 8 settimane) più 30 mg di octreotide ogni 4 settimane per il controllo dei sintomi, verso una dose elevata di octreotide (60 mg ogni 4 settimane). L'end-point primario dello studio era la sopravvivenza libera da progressione (PFS), che è risultata significativamente diversa (p < 0.0001) tra i gruppi: al momento dell'analisi, la mediana non era stata ancora raggiunta per 177Lu-DOTATATE, mentre era di 8.5 mesi per octreotide LAR. Gli autori hanno riportato una riduzione del 79% del rischio di progressione o morte nei pazienti trattati nel braccio PRRT rispetto al braccio di controllo. Oltre a migliorare la PFS, 177Lu-DOTATATE fornisce un significativo beneficio in termini di qualità di vita (21).
Lo studio Erasmus (22) a supporto del NETTER-1 ha valutato sicurezza ed efficacia di 177Lu-DOTATATE in 1200 pazienti con NET (midgut, foregut, hindgut e a primitività sconosciuta). Efficacia e sopravvivenza sono state analizzate in un sottogruppo di 443 pazienti: il tasso di risposta oggettiva è stato del 39%, la stabilità di malattia è stata raggiunta nel 43% dei pazienti. La PFS e la sopravvivenza globale (OS) per tutti i pazienti erano, rispettivamente, di 29 mesi e 63 mesi. I pazienti con NET pancreatico avevano OS più prolungata (71 mesi). Sono state rilevate LA in quattro pazienti (0.7%) e MDS in nove (1.5%).
In seguito alla pubblicazione dei dati di tali studi, il 177Lu-DOTATATE è stato approvato dalle autorità regolatorie, EMA e successivamente AIFA, con la seguente indicazione: trattamento di tumori neuroendocrini gastro-entero-pancreatici (GEP-NET) non operabili o metastatici, in progressione, ben differenziati (G1 e G2) e positivi ai recettori della somatostatina (fig 1-2). Il radio-farmaco 177Lu-DOTATATE (Lutathera) è stato incluso nell’elenco AIFA dei farmaci innovativi per un periodo di tre anni (30/3/2019 – 29/3/2022).
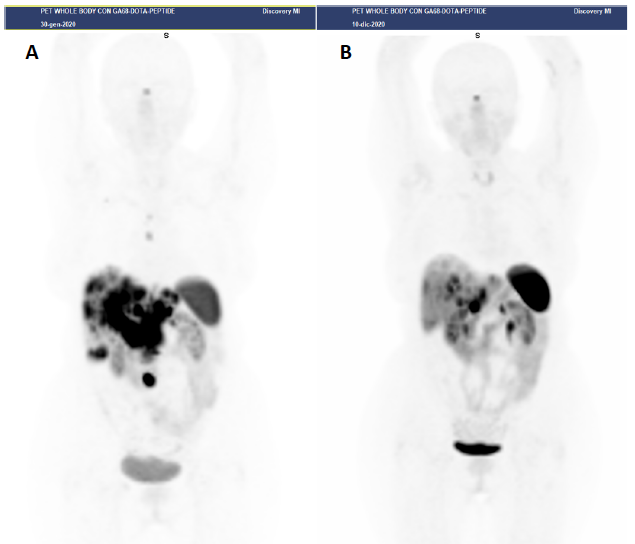
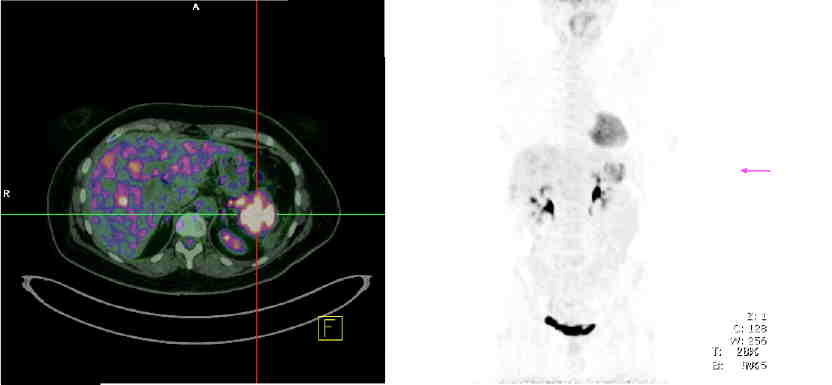
Figura 1
Paziente con NET G2 duodenale, Ki67 = 7%, in progressione dopo terapia con analoghi della somatostatina.
A: 68Ga-DOTATOC PET/TC basale eseguita in previsione di PRRT. Aree di aumentata captazione, compatibili con lesioni neoplastiche ad elevata espressione recettoriale a livello del parenchima epatico, di linfonodi mediastinici e addominali.
B: 68Ga-DOTATOC PET/TC controllo dopo tre mesi dalla fine del trattamento radio-recettoriale con 177Lu-Oxodotreotide. Risposta parziale con riduzione di numero, estensione e intensità di captazione delle lesioni presenti alla PET/TC basale.
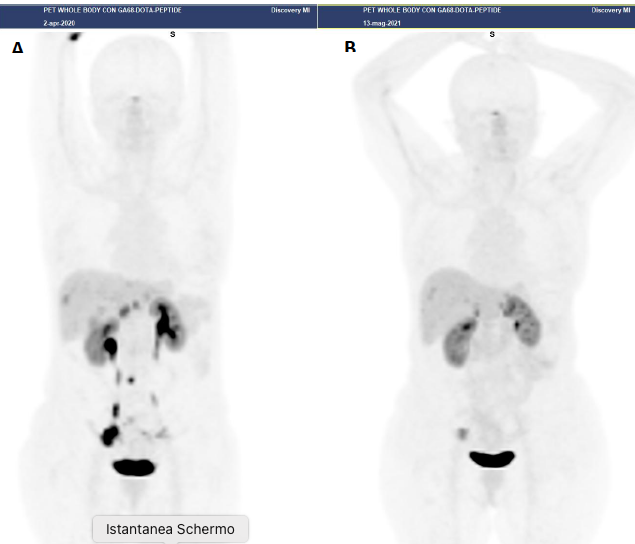
Figura 2
Paziente con insulinoma pancreatico G2 (Ki67 7%), in progressione dopo terapia con analoghi della somatostatina.
A: 68Ga-DOTATOC PET/TC basale in previsione di PRRT. Aree di aumentata captazione, compatibili con lesioni neoplastiche ad elevata espressione recettoriale a livello di pancreas, parenchima epatico, linfonodi addominali e scavo pelvico di destra.
B: 68Ga-DOTATOC PET/TC controllo dopo tre mesi dalla fine del trattamento radio-recettoriale con 177Lu-Oxodotreotide. Risposta parziale alla PRRT, con importante riduzione del numero delle lesioni presenti alla PET/TC basale.
Metodica e modalità di somministrazione del radiofarmaco
La somministrazione di 177Lu-DOTATATE deve essere effettuata esclusivamente da personale autorizzato a manipolare radio-farmaci.
Prima di iniziare e durante il trattamento è necessario esaminare la funzionalità epatica e renale e la riserva midollare. Inoltre, l'imaging recettoriale, ovvero la scintigrafia con 111In-Octreoscan o, meglio, la PET/TC con 68Ga-DOTA-peptide, deve confermare la presenza di SSTR sulle cellule tumorali, con captazione tumorale pari almeno alla normale captazione epatica (captazione tumorale ≥ 2 secondo il punteggio di Krenning).
Lo schema terapeutico raccomandato consiste in 4 infusioni da 7400 MBq ciascuna, a distanza consigliata di 8 settimane, estensibile fino a 16 settimane in caso di tossicità. Lo schema della somministrazione prevede prima una pre-medicazione con anti-emetici, seguita dopo 30 minuti da una soluzione di aminoacidi per via ev per una durata complessiva di 4 ore, seguita a sua volta dall’infusione del 177Lu-DOTATATE.
Valutazione della risposta alla PRRT
È senz’altro uno dei punti di maggiore discussione e criticità. Per quanto, infatti, le linee guida EANM suggeriscano di utilizzare i criteri RECIST per valutare la risposta alla PRRT (23), sono noti i limiti dell’impiego di criteri meramente dimensionali, in particolare in questi tumori per lo più a lenta crescita. Con l’avvento dei farmaci biologici e della stessa PRRT, è sempre più evidente che la sola valutazione dimensionale delle lesioni non è sufficiente per valutare la risposta al trattamento.
Dati recenti della fase 4 dello studio sunitinib nei NET pancreatici hanno indicato una migliore stima della risposta utilizzando i criteri CHOI, basati sulla variazione del 10% delle dimensioni o sulla variazione di densità del tumore alla TC rispetto ai criteri RECIST (24).
Considerando i limiti sovra-esposti sulla valutazione della risposta al trattamento, non è ancora stato raggiunto un consenso tra gli esperti sui metodi e sulle tempistiche. Probabilmente dovranno essere impiegati insieme l’imaging morfologico e quello funzionale, sfruttando anche l’avvento di metodi innovativi di analisi dell’imaging (25). Saranno necessari ulteriori studi per valutare e affermare l’uso nella pratica clinica di approcci quali la biopsia liquida, in particolare del NETest (26).
Conclusioni
La PRRT è attualmente riconosciuta come trattamento efficace e ben tollerato per i GEP-NET ben differenziati, in progressione, non operabili, SSTR-positivi, in progressione dopo i trattamenti di prima linea. Sebbene attualmente tale terapia sia impiegata su larga scala in molti paesi, diverse problematiche sono ancora al centro del dibattito scientifico. Innanzitutto, è di fondamentale importanza definire i criteri appropriati di selezione del paziente, come stabilire la corretta sequenza terapeutica e il corretto posizionamento della PRRT nella sequenza, definire l’opportunità di personalizzare dose e schemi terapeutici utilizzando la dosimetria e inoltre, definire criteri più appropriati per la valutazione della risposta al trattamento. È infine, auspicabile in futuro la possibilità di utilizzare la PRRT anche nella pratica clinica (e non solo in ambito sperimentale), in una fase più precoce della malattia e/o in associazione con altri trattamenti.
Bibliografia
- Oberg KE, Reubi JC, Kwekkeboom DJ, Krenning EP. Role of somatostatins in gastroenteropancreatic neuroendocrine tumor development and therapy. Gastroenterology 2010, 139: 742-53.
- Graham MM, Gu X, Ginader T, et al. 68Ga-DOTATOC imaging of neuroendocrine tumors: a systematic review and metaanalysis. J Nucl Med 2017, 58: 1452-8.
- Virgolini IJ, Gabriel M, von Guggenberg E, et al. Role of radiopharmaceuticals in the diagnosis and treatment of neuroendocrine tumours. Eur J Cancer 2009, 45 suppl 1: 274-91.
- Kam BL, Teunissen JJ, Krenning EP, et al. Lutetium-labelled peptides for therapy of neuroendocrine tumours. Eur J Nucl Med Mol Imaging 2012, 39 suppl 1: S103-12.
- Dannoon SF, Alenezi SA, Elgazzar AH. The efficacy of the available peptide receptor radionuclide therapy for neuroendocrine tumors: a meta-analysis. Nucl Med Commun 2017, 38: 1085-93.
- Kunikowska J, et Tandem peptide receptor radionuclide therapy using 90Y/177Lu-DOTATATE for neuroendocrine tumors efficacy and side-effects - Polish multicenter experience. Eur J Nucl Med Mol Imaging 2020, 47: 922-33.
- Sabet A, Haslerud T, Pape UF, et al. Outcome and toxicity of salvage therapy with 177Lu-octreotate in patients with metastatic gastroenteropancreatic neuroendocrine tumours. Eur J Nucl Med Mol Imaging 2014, 41: 205-10.
- Vinjamuri S, Gilbert TM, Banks M, et al. Peptide receptor radionuclide therapy with 90Y-DOTATATE/90Y-DOTATOC in patients with progressive metastatic neuroendocrine tumours: assessment of response, survival and toxicity. Br J Cancer 2013, 108: 1440-8.
- Waldherr C, Pless M, Maecke HR, et al. Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq 90Y-DOTATOC. J Nucl Med 2002, 43: 610-6.
- Valkema R, Pauwels S, Kvols LK, et al. Survival and response after peptide receptor radionuclide therapy with [90Y-DOTA0,Tyr3]octreotide in patients with advanced gastroenteropancreatic neuroendocrine tumors. Semin Nucl Med 2006, 36: 147-56.
- Filice A, Fraternali A, Frasoldati A, et al. Radiolabeled somatostatin analogues therapy in advanced neuroendocrine tumors: a single experience. J Oncol 2012, 2012: 320198.
- Mariniello A, Bodei L, Tinelli C, et al. Long-term results of PRRT in advanced bronchopulmonary carcinoid. Eur J Nucl Med Mol Imaging 2016, 43: 441-52.
- Bodei L, Cremonesi M, Grana CM, et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE: the IEO phase I-II study. Eur J Nucl Med Mol Imaging 2011, 38: 2125–35.
- Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol 2008, 26: 2124–30.
- Kim SJ, Pak K, Koo PJ, et al. The efficacy of 177Lu-labelled peptide receptor radionuclide therapy in patients with neuroendocrine tumours: a meta-analysis. Eur J Nucl Med Mol Imaging 2015, 42: 1964-70.
- Hörsch D, Ezziddin S Haug A, et al. Effectiveness and side-effects of peptide receptor radionuclide therapy for neuroendocrine neoplasms in Germany: a multi-institutional registry study with prospective follow-up. Eur J Cancer 2016, 58: 41-51.
- van der Zwan WA, Bodei L, Brand JM, et al. GEP-NETs update: Radionuclide therapy in neuroendocrine tumors. Eur J Endocrinol 2015, 172: R1-8.
- Bodei L, Cremonesi M, Ferrari M, et al. Long-term evaluation of renal toxicity after peptide receptor radionuclide therapy with 90Y-DOTATOC and 177Lu-DOTATATE: the role of associated risk. Eur J Nucl Med Mol Imaging 2008, 35: 1847-56.
- Imhof A, Brunner P, Marincek N, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol 2011, 29: 2416–23.
- Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med 2017, 376: 125-35.
- Strosberg J, Wolin E, Chasen B, et al. Health-Related Quality of Life in Patients With Progressive Midgut Neuroendocrine Tumors Treated With 177Lu-Dotatate in the Phase III NETTER-1. J Clin Oncol 2018, 36: 2578-84.
- Brabander T, van der Zwan WA, Teunissen JJM, et al. Long-term efficacy, survival, and safety of [177Lu-DOTA0,Tyr3]octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine Clin Cancer Res 2017, 23: 4617-24.
- Bodei L, Mueller-Brand J, Baum RP, et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging 2013, 40: 800-16.
- Raymond E, Kulke MH, Quin S, et al. Efficacy and safety of sunitinib in patients with well-differentiated pancreatic neuroendocrine tumours. Neuroendocrinology 2018, 107: 237-45.
- Liberini V, Huellner MW, Grimaldi S, et al. The challenge of evaluating response to peptide receptor radionuclide therapy in gastroenteropancreatic neuroendocrine tumors: the present and the future. Diagnostics (Basel) 2020, 10: 1083.
- Bodei L, Kidd MS, Singh A, et al. PRRT genomic signature in blood for prediction of 177Lu-octreotate efficacy. Eur J Nucl Med Mol Imaging 2018, 45: 1155-69.
Overview sui NET
Sara Bianchetti1 & Silvia Nasoni2
1SC Oncologia e 2SC Gastroenterologia ed Epatologia,, Ospedale Regina Apostolorum, Albano Laziale (RM)
I tumori neuroendocrini (NET) sono patologie spesso misconosciute. Il ritardo diagnostico è dovuto a molteplici ragioni tra cui, non ultima, una scarsa familiarità dei clinici con questo tipo di patologia. Inoltre, i NET possono manifestarsi con una sintomatologia sfumata, sub-continua e aspecifica, soprattutto nei tumori non funzionanti.
Particolare attenzione si deve porre nei pazienti con anamnesi positiva per sindromi genetiche, come la MEN-1 e la VHL, che si possono associare ai NET.
La diagnosi dei NET parte, quindi, prima che da un corteo di esami specifici di tipo laboratoristico e strumentale, da un’accurata anamnesi che permetta di identificare la neoplasia in una fase precoce; un ritardo nell’inizio di una terapia adeguata, infatti, potrebbe comportare un peggioramento della prognosi nell’ambito di una classe di tumori che complessivamente hanno una prognosi migliore rispetto ai carcinomi non endocrini: la mediana di sopravvivenza di tutti i NET, infatti, considerati tutti gli stadi e grading, è di 75 mesi (1,2).
Essendo i NET un gruppo di neoplasie molto eterogeneo, si sono susseguiti molti tentativi per identificare fattori prognostici adeguati; tra tutti, il grading e lo stadio di malattia sono tra i fattori di maggiore utilità. Infatti, mentre la mediana di sopravvivenza dei NET, considerati tutti gli stadi e grading, è di 75 mesi, nei G2 e nei G3 tale valore scende rispettivamente a 64 e 10 mesi e sale a 124 mesi nei G1. Per quando riguarda lo stadio, i pazienti affetti da NET G1 o G2 con malattia localizzata e localmente avanzata hanno una sopravvivenza media rispettivamente di 223 mesi e 111 mesi. Questo dato si modifica drasticamente nei pazienti che, a parità di grading, sono in fase metastatica, arrivando fino a 33 mesi. In aggiunta, nei NET G3 localizzati la sopravvivenza mediana è pari a 34 mesi, riducendosi rispettivamente a 14 e a 5 mesi nei casi di malattia localmente avanzata e metastatica (1,2). Utilizzando la classificazione TNM, si ottengono risultati sovrapponibili: i pazienti con malattia allo stadio IV avevano sopravvivenze a 5 anni pari a 57%, un valore intermedio tra quelli con NET G2 (73.4%) e G3 (27.7%)(3,4).
Poiché una diagnosi tardiva di NET potrebbe avere importanti ripercussioni sulla prognosi, è essenziale prendere in considerazione tali neoplasie nella diagnosi differenziale fin dal momento dell’anamnesi.
Bibliografia
- Maggard MA, O’Connel JB, Ko CY. Updated population-based review of carcinoid tumours. Ann Surg 2004, 240: 117-22.
- Yao JC, Hassan M, Phan A, et al. One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008, 26: 3063-72.
- Strosberg JR, Cheema A, Weber J, et al. Prognostic validity of a novel American joint committee on cancer staging classification for pancreatic neuroendocrine tumors. J Clin Oncol 2011, 29: 3044-9.
- Pape UF, Jann H, Muller-Nordhorn J, et al. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer 2008, 113: 256-65.
Approccio diagnostico ragionato al paziente con sospetto NET
Il sospetto di NET può sorgere in 4 scenari clinici differenti, ognuno dei quali richiede un approccio ragionato diverso:
- reperto incidentale in un paziente totalmente asintomatico (con il caso particolare dell'alterazione di laboratorio)
- paziente sintomatico per effetti locali
- paziente con sindrome iperfunzionante
- paziente con metastasi a primitività ignota.
I primi 2 scenari sono tipici dei NET non funzionanti.
Iter diagnostico ragionato nel paziente con reperto incidentale sospetto per NET
Nicola Fazio
Unità Tumori Gastrointestinali e Neuroendocrini, Istituto Europeo di Oncologia, Milano
Introduzione
Il sospetto di NET può nascere a seguito di un riscontro di alterazione radiologica (ecografia, TC o RM) o endoscopica in un paziente senza alcun sintomo o segno clinico di NET (1-3).
Il paziente dovrebbe essere interrogato relativamente a disturbi gastroenterici (diarrea, stipsi, acidità, reflusso) e dovrebbero essere ricercate masse palpabili, alterazioni cutanee e metaboliche suggestive per una sindrome iperfunzionante. È opportuno inoltre raccogliere un’accurata anamnesi familiare per la possibilità di una sindrome ereditaria (3).
Sospetto nato per un'alterazione endoscopica
La diagnosi incidentale di NET spesso segue l’esame istologico di una lesione polipoide riscontrata durante una procedura endoscopica eseguita in un paziente asintomatico. Altra possibilità è che un NET gastroduodenale o colorettale venga sospettato sulla base di una protrusione mucosa polipoide singola o multifocale (4-6). C’è comunque da sottolineare come nessun rilievo endoscopico sia patognomonico di NET.
La prima tappa obbligatoria è la biopsia endoscopica della lesione sospetta. Nel caso questa non sia possibile o non porti a una diagnosi, la tappa successiva sarà uno studio morfologicodi diagnostica per immagini. All’interno del team multidisciplinare dovrà poi essere discussa la possibilità di una biopsia eco/TC-guidata o laparoscopica. Infine, si potrà eseguire una diagnostica per immagini funzionale, come mezzo complementare per la stadiazione e la previsione prognostica.
Non ci sono test di laboratorio indicati per il work-up diagnostico: il rilievo di ipergastrinemia, acloridria, anemia macrocitica, deficit di vitamina B12 e/ anticorpi anti-fattore intrinseco può aiutare nell’inquadramento di un NET gastrico.
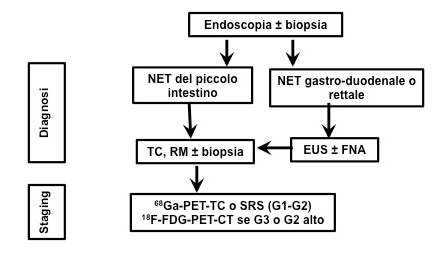
Flow-chart diagnostica per GEP-NET sospettato all’endoscopia
Sospetto nato per un'alterazione radiologica (eco/TC/RM)
Questo rilievo è di solito legato a un tumore pancreatico primitivo o a metastasi epatiche.
Il rilevo all’ecografia (anche con contrasto) di lesione ipoecogena ipervascolarizzata e/o di lesioni ben definite, e alla TC o RM di lesioni ipervascolarizzate che prendono il contrasto può far sospettare un NET pancreatico. Nelle lesioni di grandi dimensioni si possono evidenziare alterazioni cistiche, necrosi o calcificazioni (7).
Specialmente se il sospetto è nato all’ecografia, è importante che il team multidisciplinare escluda i falsi positivi: emangiomi, carcinomi pancreatici, tumori mucinosi pancreatici intra-duttali, carcinomi epatocellulari, adenomi e lesioni metastatiche (8-14).
Bisognerebbe per prima cosa prelevare un campione citologico o istologico (15,16).
Confermata la diagnosi anatomo-patologica di GEP-NET, bisogna passare alla stadiazionemorfologica e funzionale. Se la biopsia non è fattibile o non porta alla diagnosi, bisogna eseguire un altro esame di diagnostica per immagini (ecografia con contrasto, eco-endoscopia, RM con sequenze specifiche per il fegato, ecc) in relazione alla disponibilità ed esperienza locali (3).
Le lesioni metastatiche a primitività occulte richiedono un percorso specifico.
Anche se non esistono test di laboratorio raccomandati nell’iter diagnostico, livelli urinari elevati di 5-HIAAsono altamente specifici di metastasi da GEP-NET e potrebbero orientare la diagnosi in caso di biopsia non conclusiva. Nei pazienti con NET pancreatico, bisogna sempre ricercare con attenzione la presenza di vaghi sintomi e segni funzionali subclinici che possano orientare verso una sindrome funzionale; in tal caso è opportuno eseguire esami ormonali selettivi.
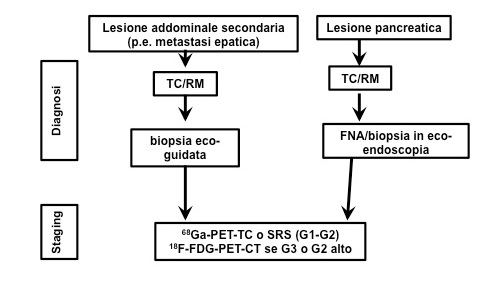
Flow-chart diagnostica per GEP-NET sospettato a un esame radiologico
Bibliografia
- Modlin IR, Lye KD, Kidd M. A 5-decade analysis of 13.715 carcinoid tumors. Cancer 2003, 97: 934-59.
- Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008, 26: 3063–72.
- Öberg K, Knigge U, Kwekkeboom D, Perren A; ESMO Guidelines Working Group. Neuroendocrine gastro-entero-pancreatic tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012, 23 Suppl 7: 124-30.
- Vannella L, Lahner E, Annibale B. Risk for gastric neoplasias in patients with chronic atrophic gastritis: a critical reappraisal. World J Gastroenterol 2012, 18: 1279-85.
- Ruszniewski P, Amouyal P, Amouyal G, et al. Localization of gastrinomas by endoscopic ultrasonography in patients with Zollinger- Ellison syndrome. Surgery 1995, 117: 629–35.
- Scherubl H. Rectal carcinoids are on the rise: early detection by screening endoscopy. Endoscopy 2009, 41: 162-5.
- Haynes AB, Deshpande V, Ingkakul T, et al. Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors. Short-term and long-term patient outcomes. Arch Surg 2011, 146: 534-8.
- Sahani DV, Bonaffini PA, Fernandez-Del Castillo C, Blake MA. Gastroenteropancreatic neuroendocrine tumors: role of imaging in diagnosis and management. Radiology 2013, 266: 38-61.
- Sundin A, Vullierme MP, Kaltsas G, Plöckinger U; Mallorca Consensus Conference participants; European Neuroendocrine Tumor Society. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: radiological examinations. Neuroendocrinology 2009, 90: 167–83.
- Malagò R, D’Onofrio M, Zamboni GA, et al. Contrast-enhanced sonography of non-functioning pancreatic neuroendocrine tumors. AJR Am J Roentgenol 2009, 192: 424–30.
- Quaia E, Stacul F, Gaiani S, et al. Comparison of diagnostic performance of unenhanced vs. SonoVue-enhanced ultrasonography in focal liver lesions characterization. The experience of three Italian centers. Radiol Med 2004, 108: 71–81.
- Mork H, Ignee A, Schuessler G, et al. Analysis of neuroendocrine tumour metastases in the liver using contrast-enhanced ultrasonography. Scand J Gastroenterol 2007, 42: 652–62.
- Hoeffel C, Job L, Ladam-Marcus V, et al. Detection of hepatic metastases from carcinoid tumor: prospective evaluation of contrast-enhanced ultrasonography. Dig Dis Sci 2009, 54: 2040-6.
- Wu W, Chen MH, Yin SS, et al. The role of contrast enhanced sonography of focal liver lesions before percutaneous biopsy. AJR Am J Roentgenol 2006, 187: 752–61.
- Falconi M, Bartsch DK, Eriksson B, et al; Barcelona Consensus Conference participants. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012, 95: 120–34.
- Cheslyn-Curtis S, Sitaram V, Williamson RC. Management of non-functioning neuro-endocrine tumours of the pancreas. Br J Surg 1993, 80: 625–7.
Iter diagnostico ragionato nel paziente che si presenta con alterazione sospetta dei dati di laboratorio
Antongiulio Faggiano
Dipartimento di Medicina e Chirurgia Clinica, Università Federico II, Napoli
Nella pratica clinica può capitare di dover valutare un paziente arrivato per alterazioni di esami di laboratorio richiesti in modo inappropriato o non sufficientemente giustificato. Qui ci concentriamo sul caso della cromogranina A, che è la situazione che può richiedere maggiore riflessione.
Il dosaggio della CgA non deve mai essere considerato un test diagnostico di prima linea. Cionostante, il sospetto di NET può nascere dal rilievo di livelli elevati di questo marcatore, richiesti sulla base di dati clinici aspecifici.
Prima di avviare un iter con esami radiologici ed endoscopici, è opportuno escludere accuratamente tutti i fattori che possono influenza il dosaggio di CgA. Prima di tutto è sempre opportuno ripetere una seconda volta il dosaggio, anche se eseguito in un laboratorio affidabile; inoltre bisogna ripetere il dosaggio dopo sospensione (se possibile) di tutti i farmaci che interferiscono nei risultati, in particolare gli inibitori di pompa, per almeno 2 settimane.
Se i livelli di CgA si confermano elevati in assenza di fattori interferenti/confondenti, è opportuno eseguire un’ecografia trans-addominale. L’eventuale iter a seguire deve essere discusso in un team multidisciplinare, oppure il paziente può essere inviato a un centro di riferimento.
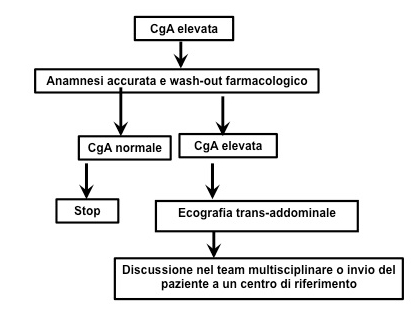
Flow-chart diagnostica per il sospetto di NET insorto dopo il rilievo di elevati valori di CgA
Iter diagnostico ragionato nel paziente sintomatico per effetti locali sospetti per NET
Nicola Fazio
Unità Tumori Gastrointestinali e Neuroendocrini, Istituto Europeo di Oncologia, Milano
Un NET non funzionante può diventare sintomatico quando comprime o invade le strutture adiacenti o quando metastatizza. Il sospetto diagnostico può derivare da alterazioni radiologiche o endoscopiche suggestive e/o dalla progressione apparentemente lenta di una malattia neoplastica (1). Alterazioni di laboratorio (p.e. livelli francamente patologici di CgA in assenza di fattori confondenti) possono corroborare il sospetto, ma la diagnosi può essere fatta solo dal patologo (su materiale istologico o citologico)(2).
Il primo passo è l’anamnesi dettagliata con un esame obiettivo completo.
Si possono distinguere 4 differenti modalità di presentazione clinica: dolore addominale isolato, quadro subocclusivo, ittero ed emorragia gastro-enterica.
Il dolore addominale è la presentazione clinica più comune dei GEP-NET non funzionanti, in relazione al tumore primitivo o alle metastasi (3,4). Bisogna indagare accuratamente caratteristiche e localizzazione del dolore.
Dolore addominale isolato
Un dolore oppressivo e persistente ai quadranti superiori può indicare una massa pancreatica o retro-peritoneale (5,6), mentre un dolore intermittente crampiforme solitamente indica un’origine intestinale (1,7). Nel primo caso è opportuno partire con un esame radiologico, seguito da endoscopia ed eco-endoscopia per indagare lesioni pancreatiche e duodenali. Nel secondo caso è opportuno partire con un esame endoscopico (1,7). Bisogna cercare di prelevare un campione citologico o istologico della lesione sospetta ogni volta che sia possibile (fig 1)(5,6).
Un dolore addominale diffuso e mal definito può essere correlato a metastasi epatiche o linfonodali. In quel caso è opportuno eseguire un’ecografia addominale, seguita da TC total body e biopsia eco-guidata.
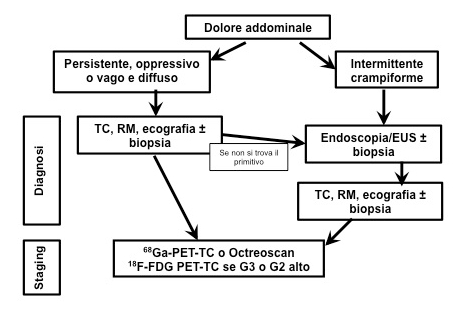
Figura 1
Flow-chart diagnostica per GEP-NET sospettato per dolore addominale
Quadro subocclusivo
Può essere provocato da un voluminoso NET ileale, spesso metastatico, e/o da carcinomatosi peritoneale.
In relazione alla gravità del quadro clinico, bisogna eseguire un Rx addome a vuoto e/o un’endoscopia (1,7).
Se si sospetta un’ostruzione estrinseca, è opportuno eseguire TC addominale; nel sospetto di carcinomatosi peritoneale, può essere utile la valutazione del transito con radiografia con contrasto idrosolubile (fig 2.).
Se possibile, è opportuno prelevare un campione bioptico per via endoscopica, altrimenti discutere nel team multidisciplinare l’esecuzione di una biopsia eco/TC-guidata di una lesione epatica o di un altro sito anatomico coinvolto o una laparoscopia esplorativa.
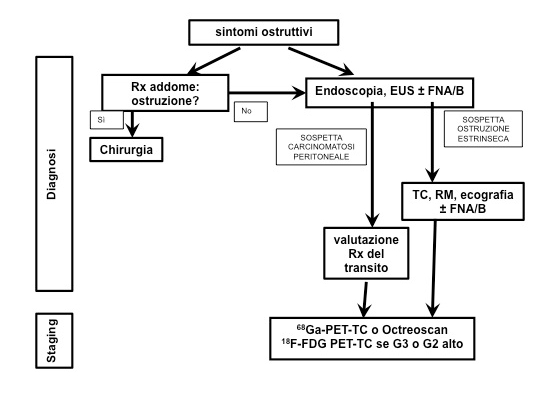
Figura 2
Flow-chart diagnostica nel sospetto di GEP-NET per quadro clinico subocclusivo
Ittero
Questa presentazione clinica fa sospettare il coinvolgimento di fegato, vie biliari o pancreas. È opportuno eseguire una valutazione di funzione e struttura del fegato attraverso esami biochimici ed ecografia, per escludere un’ostruzione biliare. Di solito la dilatazione delle vie biliari extra-epatiche suggerisce effetto compressivo da linfadenopatie o massa pancreatica, quella delle vie intra-epatiche la presenza di metastasi epatiche (5,6). In caso di ittero ostruttivo, può essere indicata l’esecuzione di colangio-RM o ERCP (che permette anche la raccolta di campioni bioptici oppure citologici per spazzolamento). La TC total body e l’endoscopia possono aiutare a definire la sede del tumore primitivo e a stadiare la malattia (fig 3).
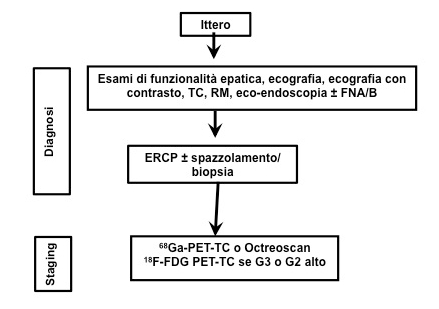
Figura 3
Flow-chart diagnostica nel sospetto di GEP-NET per ittero
Sanguinamento gastro-intestinale
Può dipendere dagli effetti compressivi e infiltrativi di una massa tumorale. Il sanguinamento può essere massivo (ematemesi, melena, proctorragia) oppure occulto o a gemizio. Bisogna eseguire esami per valutare la crasi ematica e le riserve di ferro e un’endoscopia. Il paziente con sanguinamento massivo deve essere sempre ricoverato e può essere necessario eseguire un’angiografia (1,7). Nel caso di lesioni nello stomaco, nel duodeno, nel colon-retto o nel tratto terminale dell’ileo, si può arrivare alla diagnosi istologica attraverso una biopsia endoscopica. Se l’endoscopia è negativa, bisogna discutere nel team multidisciplinare le tappe successive, che potranno essere enteroscopia, enteroTC/RM, o capsula endoscopica, a seconda della disponibilità e dell’esperienza locale (fig 4). Per una lesione localizzata al piccolo intestino, si deve prendere in considerazione un approccio chirurgico a scopo diagnostico e terapeutico.
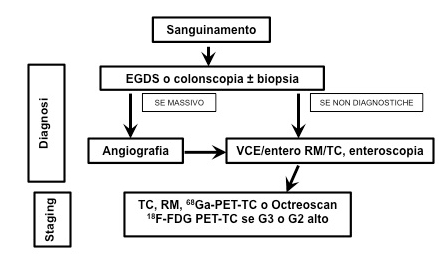
Figura 4
Flow-chart diagnostica nel sospetto di GEP-NET per sanguinamento gastro-intestinale
Bibliografia
- Shah T, Srirajaskanthan R, Bhogal M, et al. Alpha-fetoprotein and human chorionic gonadotrophin-beta as prognostic markers in neuroendocrine tumour patients. Br J Cancer 2008, 99: 72-7.
- Öberg K, Knigge U, Kwekkeboom D, Perren A; ESMO Guidelines Working Group. Neuroendocrine gastro-entero-pancreatic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012, 23 Suppl 7: 124-30.
- Modlin IR, Lye KD, Kidd M. A 5-Decade Analysis of 13.715 Carcinoid Tumors. Cancer 2003, 97: 934-59.
- Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008, 26: 3063–72.
- Falconi M, Bartsch DK, Eriksson B, et al; Barcelona Consensus Conference participants. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012, 95: 120–34.
- Cheslyn-Curtis S, Sitaram V, Williamson RC. Management of non-functioning neuro- endocrine tumours of the pancreas. Br J Surg 1993, 80: 625–7.
- Caplin M, Sundin A, Nillson O, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: colorectal neuroendocrine neoplasms. Neuroendocrinology 2012, 95: 88-97.
Iter diagnostico ragionato nel paziente che si presenta con sindrome iperfunzionante
Dario Giuffrida e Ivana Puliafito
Unità operativa di Oncologia Medica - Istituto Oncologico del Mediterraneo - Viagrande (CT)
Il paziente che giunge alla nostra osservazione lamentando una sintomatologia caratterizzata da diarrea cronica secretoria, non responsiva alla terapia medica, e ripetuti episodi di flushing cutaneo con localizzazione al volto e diffusione al collo ed al tronco superiore, pone il sospetto di una sindrome da carcinoide (1). Nella forma tipica, gli episodi di flushing sono di solito di breve durata, possono verificarsi più volte nell’arco della giornata, spontaneamente o dopo sforzi, emozioni intense, pasti, assunzione di alcolici e si associano a sensazione di calore. Comuni anche le manifestazioni cardiovascolari, che consistono in insufficienza tricuspidale e stenosi polmonare, e le manifestazioni respiratorie, con episodi di broncocostrizione e tosse secca. Nella forma atipica, che caratterizza i carcinoidi secernenti istamina, il flushing è purpureo, di durata prolungata e generalizzato. Sono possibili anche ipotensione, sudorazione, scialorrea, lacrimazione (2). L’aspecificità dei sintomi richiede tuttavia un approfondimento diagnostico, come il dosaggio di alcuni analiti nel plasma e nelle urine. Marcatori generici, utili nella diagnosi e nel follow-up di malattia, includono la cromogranina A (CgA) e l’enolasi neurono-specifica (NSE)(3). Un altro test utile nel sospetto di sindrome da carcinoide è il dosaggio nelle urine delle 24 h dell’acido 5-idrossi-indolacetico (5-HIAA)(4,5).
In presenza di sintomi quali diarrea e algie addominali e di negatività del 5-HIAA, la diagnosi differenziale si pone con ulteriori due sindromi, associate solitamente a NET di origine pancreatica (pNET), la sindrome di Zollinger-Ellison e la sindrome di Verner-Morrison (6,7).
La sindrome di Zollinger-Ellison è tipica dei gastrinomi, che possono originare dalle cellule G del duodeno e del pancreas e si caratterizzano per l’ipergastrinemia, responsabile dell’ipercloridria gastrica. Clinicamente, ai sopracitati sintomi si associa la comparsa di ulcere peptiche, che tipicamente si riscontrano a livello duodenale. In questo caso le indagini di laboratorio prevedono il dosaggio della gastrina e la valutazione della secrezione acida gastrica: valori rispettivamente > 1000 pg/mL e >15 mEq/ora sono tipici del gastrinoma. Nei casi dubbi (gastrinemia < 1000 pg/mL), si ricorre al test dinamico di tipo provocativo con secretina. Il test è positivo in presenza di aumenti del valore di gastrina di almeno 120 pg/mL rispetto al basale.
La sindrome di Verner-Morrison è tipica dei VIPomi, tumori di origine pancreatica caratterizzati da iperproduzione di polipeptide intestinale vasoattivo (VIP). Clinicamente la diarrea acquosa si può complicare con disordini idro-elettrolitici e alterazioni della conduzione elettrica cardiaca, inoltre poichè il VIP inibisce la secrezione acida gastrica un incremento dei suoi livelli circolanti può facilmente associarsi ad ipocloridria o nel 50% dei casi ad acloridria. Il VIPoma si caratterizza per elevati livelli plasmatici di VIP (un incremento del 20-50% rispetto al range normale è già significativo) e per alterazioni elettrolitiche quali ipokaliemia legata alla perdita intestinale ed ipercalcemia, che dipende dalla capacità del VIP di influenzare il metabolismo osseo (8,9). Qualora non fosse possibile dosare il VIP, non è disponibile un test evocativo, ma la combinazione di diarrea acquosa profusa, ipokaliemia, acloridria, ipercalcemia può guidare la diagnosi.
Due ulteriori sindromi iperfunzionanti che si possono associare ai pNET comprendono la sindrome da Insulinoma e la sindrome di Becker. La sindrome da insulinoma è provocata da neoplasie generalmente benigne, che originano dalle ß-cellule delle insule pancreatiche, produttrici di insulina. I sintomi includono crisi ipoglicemiche a digiuno e di conseguenza tremori, sudorazione, palpitazioni, astenia che regrediscono con l’assunzione di cibo. Ipoglicemia e iperinsulinemia caratterizzano l’insulinoma, ma poiché una franca ipoglicemia potrebbe essere assente, in alcuni casi si ricorre al test del digiuno in regime ospedaliero. Il test è positivo se glicemia < 55 mg/dL (3 mmol/L), insulina ≥ 3 μU/mL (18 pmol/L), C-peptide ≥ 0.6 ng/mL (0.2 nmol/L).
La sindrome di Becker caratterizza i glucagonomi, tumori maligni poco frequenti, che derivano dalle alfa-cellule delle insule pancreatiche. L’ipersecrezione di glucagone è causa di iperglicemia e manifestazioni cutanee tipiche. Concentrazioni plasmatiche di glucagone > 500 pg/mL in associazione ad iperglicemia sono patognomoniche di glucagonoma (10, 11). Qualora non fosse possibile dosare il glucagone, non esiste un test dinamico, pertanto il riscontro combinato di diabete, eritema necrolitico migrante e comunemente di metastasi epatiche (poichè clinicamente la sindrome si manifesta tardivamente) orienta nella diagnosi differenziale.
In caso di positività dei test di laboratorio, bisognerà procedere all’individuazione del tumore primitivo e di eventuali metastasi a distanza attraverso le indagini strumentali. La TC con mdc e la RM sono in grado di individuare anche tumori di piccole dimensioni, grazie alla loro ricca vascolarizzazione, e di definire il rapporto del tumore con gli organi e le strutture vascolari vicine, utile in caso di approccio terapeutico chirurgico (12, 13). L’ecografia addominale può essere utile per la ricerca di metastasi epatiche, ma la sua validità è strettamente operatore-dipendente e l’esame può essere inficiato da condizioni come il meteorismo intestinale (14). Gli esami endoscopici consentono l’individuazione diretta della neoplasia in caso di localizzazione a livello gastro-duodenale o colo-rettale, mentre per lo studio dell’intestino tenue si ricorre al clisma del tenue (che potrebbe essere negativo negli stadi iniziali ove la lesione è ancora di piccole dimensioni, limitata alla sottomucosa), all’enteroscopia e alla videocapsula endoscopica, soprattutto quando si è di fronte ad una malattia metastatica in assenza del tumore primitivo. Tuttavia questi esami sono costosi, poco diffusi e necessitano di adeguata pulizia intestinale (15). L’eco-endoscopia è una metodica invasiva, operatore-dipendente, non eseguibile in tutti i centri, che consente di individuare lesioni pancreatiche anche millimetriche singole o multiple, o carcinoidi gastrointestinali localizzati nella parete gastrica, duodenale e colo-rettale. Dovrebbe essere usata dopo le consuete tecniche di imaging, ma i sopracitati limiti non ne consentono un uso routinario (7). Infine i NET sono caratterizzati dall’espressione di recettori per la somatostatina. La diagnostica medico-nucleare è utile nei casi di malattia metastatica da tumore primitivo sconosciuto, per definire la diffusione di malattia e per individuare pazienti candidabili alla terapia radiometabolica. La scintigrafia con Octreoscan è la metodica più diffusa. L’accumulo del radiofarmaco in organi che comunemente non esprimono i recettori per la somatostatina può essere indicativo della presenza di una neoplasia (16-18). La PET con fluorodesossiglucosio (F-FDG) trova scarsa indicazione nei NET ben differenziati per il loro lento metabolismo, mentre la PET con i peptidi Ga–DOTA-TOC, Ga-DOTA-NOC, Ga-DOTA-TATE si sta affermando con un’efficacia superiore a quella dell’Octreoscan, soprattutto nella valutazione di lesioni piccole a livello linfonodale, epatico, scheletrico. Tuttavia l’esame è ancora eseguibile solo in un numero limitato di centri (19).
Bibliografia
- Khan MS, et al. Long-term results of treatment of malignant carcinoid syndrome with prolonged release Lanreotide (Somatuline Autogel). Alim Pharmacol Ther 2011, 34: 235-42.
- Modlin IM, et al. Current status of gastrointestinal carcinoids. Gastroenterology 2005, 128: 1717-51.
- Prestifilippo A, Blanco G, Vitale MP, Giuffrida D. Chromogranin A and Neuroendocrine Tumors. In Neuroendocrine Tumor, cap. 2: pg 11-8.
- Maroun J, et al. Guidelines for the diagnosis and management of carcinoid tumours. Part 1: the gastrointestinal tract. A statement from a Canadian national carcinoid expert group. Curr Oncol 2006, 13: 67-76.
- Feldman JM. Role of neuropeptides and serotonin in the diagnosis of carcinoid tumors. Am J Med 1986, 81: 41-8.
- Oberg KE. Gastrointestinal neuroendocrine tumors. Ann Oncol 2010, 21 (suppl 7): vii72-80.
- Massironi S, et al. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol 2008, 14: 5377-84.
- O’Toole D, et al. ENETS Consensus Guidelines for the standards of care in neuroendocrine tumors: biochemical markers. Neuroendocrinology 2009, 90: 194-202.
- Ardill JE, O'Dorisio TM. Circulating biomarkers in neuroendocrine tumors of the enteropancreatic tract: application to diagnosis, monitoring disease, and as prognostic indicators. Endocrinol Metab Clin North Am 2010, 39: 777-90.
- Anthony LB, et al. North American Neuroendocrine Tumor Society (NANETS). The NANETS consensus guidelines for the diagnosis and management of gastrointestinal neuroendocrine tumors (NETS): well-differentiated NETS of the distal colon and rectum. Pancreas 2010, 39: 767-74.
- Jensen RT, et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 2012, 95: 98-119.
- Sugimoto E, et al. Midgut carcinoid tumours. CT appearance. Acta Radiol 1995, 36: 367-71.
- Owen NJ, et al. MRI of pancreatic neuroendocrine tumours. Br J Radiol 2001, 74: 968-73.
- Doerffel Y, et al. Neuroendocrine tumors: characterization with contrast-enhanced ultrasonography. Ultraschall Med 2008, 29: 506-14.
- Rondonotti E, et al. Small-bowel neoplasms in patients undergoing videocapsule endoscopy: a multicenter European study. Endoscopy 2008, 40: 488-95.
- Ramage JK, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours (NETs). Gut 2012, 61: 6-32.
- Falconi M, et al. Well differentiated pancreatic non functioning tumors/carcinoma. Neuroendocrinology 2006, 84: 196-211.
- Ricke J, et al. Standardisation of imaging in neuroendocrine tumours: results of a European Delphi process. Eur J Radiol 2001, 37: 8-17.
- Gabriel M, et al. 68Ga–DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nucl Med 2007, 48: 508-18.
Iter diagnostico ragionato nel paziente con metastasi a primitività ignota
Franco Grimaldi
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria Santa Maria della Misericordia di Udine
Il tumore metastatico a primitività ignota è una condizione in cui si ha la conferma istologica o citologica di NET su una metastasi, senza evidenza del tumore primitivo, dopo un primo iter diagnostico che comprende TC toraco-addominale, Octreoscan, EGDS e colonscopia.
La frequenza di tumori ben differenziati di questo tipo varia fra il 9 e il 19% (1,2). In tutti i tipi di NET la presenza di metastasi epatiche influenza in modo importante la prognosi, che dipende dal sito di origine, dall’estensione tumorale (stadio T) e dalla differenziazione istologica (NET vs carcinoma neuroendocrino, NEC). Il tasso di sopravvivenza a 5 anni nei piccoli NET intestinali e pancreatici G1-G2 è del 54% e 27%, rispettivamente, nel database SEER (3). Inoltre, pur in presenza di metastasi epatiche, la sopravvivenza è peggiore nei pazienti in cui non è stata identificata la sede primitiva (4). Nei pazienti con metastasi epatiche la sopravvivenza è influenzata dalla presenza di sintomi ostruttivi e di quelli correlati all’ipersecrezione ormonale.
La valutazione di questi pazienti deve comprendere un’accurata anamnesi, compresa la familiare per identificare i parenti affetti e il rischio di tumore poliendocrino (MEN-1 o MEN-2) e studi biochimici e strumentali (5). I preparati istologici devono essere rivisti con colorazioni immunoistochimiche per guidare la ricerca del tumore primitivo (6,7):
- TTF-1 per il polmone e il tumore midollare della tiroide;
- CDX-2 per l’intestino;
- PAX-8, istidina-decarbossilasi, polipeptide pancreatico e glucagone per il pancreas;
- xenina per il duodeno;
- gastrina per il gastrinoma occulto.
La valutazione biochimica deve comprendere i dosaggi di gastrina, 5-HIAA e altri marcatori secondo la disponibilità locale (8).
Di recente è stato riportato che la maggior parte dei tumori metastatici a primitività ignota deriva dal pancreas e dal piccolo intestino (9). Di conseguenza, il team multiscipinare deve valutare l’esecuzione di studi di localizzazione da selezionare fra RM addome, eco-endoscopia, enteroTC/RM, PET con Ga-68, capsula endoscopica, enteroscopia a doppio pallone, in relazione al quadro clinico, alla disponibilità e all’esperienza locale (10-12). Nei NEC può essere utile la 18F-FDG-PET.
Flow-chart diagnostica nel paziente con malattia metastatica a primitività ignota
Bibliografia
- Bellizzi AM. Assigning site of origin in metastatic neuroendocrine neoplasms: a clinically significant application of diagnostic immunohistochemistry. Adv Anath Pathol 2013, 20: 285-314.
- Kirshbom PM, Kherani AR, Onaitis MW, et al. Carcinoids of unknown origin: comparative analysis with foregut, midgut, and hindgut carcinoids. Surgery 1998, 124: 1063-70.
- Pavel M, Baudin E, Couvelard A, et al, all other Barcelona Consensus Conference participants. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012, 95: 157-76.
- Catena L, Bichisao E, Milione M, et al. Neuroendocrine tumors of unknown primary site: gold dust or misdiagnosed neoplasms? Tumori 2011, 97: 564-7.
- NCCN Clinical Practice Guidelines in Oncology. Neuroendocrine Tumors. Version 2.2013.
- Anlauf M, Garbrecht N, Bauersfeld J, Schmitt A, Henopp T, Komminoth P, Heitz PU, Perren A, Klöppel G. Hereditary neuroendocrine tumors of the gastroenteropancreatic system. Virchows Arch 2007, 451 Suppl 1: S29-38
- Srivastava A, Hornick JL. Immunohistochemical staining for CDX-2, PDX-1, NESP-55, and TTF-1 can help distinguish gastrointestinal carcinoid tumors from pancreatic endocrine and pulmonary carcinoid tumors. Am J Surg Pathol 2009, 33: 626-32.
- Joseph S, Wang YZ, Boudreaux JP, et al. Neuroendocrine tumors: current recommendations for diagnosis and surgical management. Endocrinol Metab Clin N Am 2011, 40: 205–31.
- Wang SC, Parekh JR, Zuraek MB, et al. Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg 2010, 145: 276-80.
- Putzer D, Gabriel M, Henninger B, et al. Bone metastases in patients with neuroendocrine tumor: 68Ga-DOTA-Tyr3-octreotide PET in comparison to CT and bone scintigraphy. J Nucl Med 2009, 50 (8): 1214-1221.
- Gabriel M, Decristoforo C, Kendler D, et al. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nucl Med 2007, 48: 508-18.
- Wang SC, Fidelman N, Nakakura EK. Management of well-differentiated gastrointestinal neuroendocrine tumors to the liver. Semin Oncol 2013, 40: 69-74.
Approccio razionale alla stadiazione dei NET
Nicola Fazio
Unità Tumori Gastrointestinali e Neuroendocrini, Istituto Europeo di Oncologia, Milano
La valutazione dell’estensione di malattia ha un ruolo chiave nella pianificazione terapeutica.
La stadiazione pre-trattamento dovrebbe comprendere metodiche morfologiche e funzionali di diagnostica per immagini. Le metodiche morfologiche sono necessarie in tutti i NET, indipendentemente dal grado. Per quanto riguarda le metodiche funzionali, nei NET di grado basso-intermedio (G1-G2 secondo la classificazione WHO 2010) bisogna usare quelle basate sulla visualizzazione dei recettori per la somatostatina (Octreoscan o PET con Ga-68 coniugato a DOTA-peptidi), mentre la 18F-FDG-PET andrebbe riservata ai G3 e ad alcuni G2.
Per la stadiazione morfologica, bisogna usare una TC multidetettore toraco-addomino-pelvica oppure una TC toracica senza mdc associata a una RM addomino-pelvica (1). Per la stadiazione funzionale, l’Octreoscan viene attualmente considerato il gold standard. Però, dovrebbe essere usata preferenzialmente la PET/TC con Ga-68 coniugato a DOTA-peptidi se disponibile. In effetti, la PET manca dei dettagli anatomici richiesti perla stratificazione terapeutica (pianificazione chirurgica o calcolo della dose per la radio-embolizzazione con microsfere radio-marcate). Recentemente è stato riportato che la RM con contrasto specifico per il fegato combinata con la PET con Ga-68 coniugato a DOTA-peptidi sarebbe più accurata della PET/TC nella diagnosi di metastasi epatiche da GEP-NET (2). Promettenti ma ancora sperimentali sono le PET/TC con 18F-DOPA e con 11C-5HTP: il loro uso è da ipotizzare in caso di negatività di Octreoscan e PET con Ga-68 coniugato a DOTA-peptidi (3).
NET gastrici: l’EGDS è solitamente l’unica metodica diagnostica raccomandata nei piccoli ( 1 cm. L’EUS è utile anche nella valutazione del coinvolgimento linfonodale regionali e permette la conferma istologica con FNA. TC/RM e metodiche medico-nucleari sono necessarie nella stadiazione dei NET gastrici di tipo 2 e 3, mentre sono superflui nel tipo 1.
NET duodenali: l’EUS è utile prima della resezione delle lesioni polipoidi. Per valutare l’estensione locale e a distanza dovrebbero essere eseguiti TC multislice o RM. Nei pazienti con malattia localmente avanzata e/o metastasi epatiche, è opportuno eseguire scintigrafia ossea e RM del rachide e pelvi (4).
NET digiuno-ileali: la ricerca di metastasi a distanza deve esseere eseguita con TC multidetettore toraco-addomino-pelvica oppure con TC toracica senza mdc associata a RM addomino-pelvica, Octreoscan o PET con Ga-68 coniugato a DOTA-peptidi. La TC del fegsato dovrebbe essere eseguita con tecnica multislice e multifase. È opportuna l’esecuzione di una colonscopia per escludere un carcinoma colorettale sincrono (4).
NET colo-rettali: bisogna eseguire una TC multidetettore toraco-addomino-pelvica. È molto utile l’esecuzione pre-operatoria di un’ecografia endo-anale/endo-rettale per valutare la profondità dell’invasione di parete e il coinvolgimento dei linfonodi loco-regionali (5).
NET pancreatici non funzionanti: per la stadiazione morfologica bisogna eseguire una TC multislice/multifase o una RM con sequenze T1 con saturazione del grasso e a contrasto ritardato e poi una EUS con biopsia (6). Poi Octreoscan o PET con Ga-68 coniugato a DOTA-peptidi.
Bibliografia
- Berna MJ, Hoffmann KM, Serrano J, et al. Serum gastrin in Zollinger-Ellison syndrome. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2,229 cases from the literature. Medicine (Baltimore) 2006, 85: 295–330.
- Giesel FL, Kratochwil C, Mehndiratta A, et al. Comparison of neuroendocrine tumor detection and characterization using DOTATOC-PET in correlation with contrast enhanced CT and delayed contrast enhanced MRI. Eur J Radiol 2012, 81: 2820-5.
- Lindholm DP, Oberg K. Biomarkers and molecular imaging in gastroenteropancreatic neuroendocrine tumors. Horm Metab Res 2011, 43: 832–7.
- Delle Fave G, Kwekkeboom DJ, Van Cutsem E, et al. ENETS consensus guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology 2012, 95: 74-87.
- Caplin M, Sundin A, Nillson O, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: colorectal neuroendocrine neoplasms. Neuroendocrinology 2012, 95: 88-97.
- Ichikawa T, Peterson MS, Federle MP, et al. Islet cell tumor of the pancreas: biphasic CT versus MR imaging in tumor detection. Radiology 2000, 216: 163-71.
Sindrome carcinoide
Franco Grimaldi
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria Santa Maria della Misericordia di Udine
(aggiornato al 19 marzo 2017)
Definizione ed epidemiologia
La sindrome da carcinoide (SC) è un cluster di sintomi riscontrati a volte in pazienti con neoplasie neuroendocrine (NEN), tumori rari, spesso a lenta crescita. La maggior parte delle NEN è localizzata nel tratto gastrointestinale. La sindrome carcinoide si verifica in < 10% dei pazienti con NEN, di solito dopo che il tumore si è diffuso al fegato. I tumori in questi pazienti rilasciano quantità eccessive di serotonina, causando diarrea. Le complicazioni della diarrea incontrollata includono perdita di peso, malnutrizione, disidratazione e squilibrio elettrolitico.
Classificazione
La SC è classificata come:
- tipica, caratterizzata da diarrea, dolori addominali e flushing, nel 95% dei casi;
- atipica nel restante 5%, in cui il quadro clinico è variabile, a causa del tipo di sostanze bioattive secrete (serotonina, tachichinine, callicreine e prostaglandine).
Clinica
La diarrea della SC è probabilmente causata dalla serotonina, che stimola il piccolo intestino e il colon. È fondamentale eseguire un’anamnesi delle caratteristiche della diarrea, valutando l’eventuale associazione con ulteriori manifestazioni della SC. Nella SC la diarrea è cronica, prevalentemente secretoria, non migliora con il digiuno ed è associata a squilibri elettrolitici. Le feci sono solitamente acquose, in relazione a ipermotilità e ipersecrezione. La diarrea notturna e l’incompleta risposta al trattamento specifico avvalorano il sospetto di SC (1,2).
Circa metà dei pazienti con SC ha dolore addominale: è intermittente e crampiforme con coliche, oppure sordo, e non si attenua con la defecazione.
Il flushing è il sintomo più frequente, che si può accentuare con l’assunzione di cibo e alcol, con l’esercizio fisico e gli stati emotivi. Le caratteristiche del flushing sono particolari: viso, collo e parte superiore del tronco diventano di colore rosso con pelle tipicamente asciutta. Il flushing può essere associato a ipotensione transitoria e broncocostrizione. Altre cause di flushing da porre in diagnosi differenziale includono:
- feocromocitoma, menopausa (associata a sudorazione), sindrome di Zollinger-Ellison e carcinoma midollare della tiroide, solitamente associati a flushing intermittente;
- alcolismo, policitemia, stenosi mitralica, associati a flushing costante;
- possono essere implicati anche farmaci (vasodilatatori quali calcio-antagonisti, morfina e altri oppiacei) e mastocitosi (il flushing dura di più e può essere accompagnato da cefalea, dispnea, cardiopalmo)(3).
La SC atipica è associata all’iperproduzione di istamina ed è caratterizzata da rossore prolungato, broncocostrizione, cefalea, lacrimazione, dispnea e ipotensione. La dispnea può suggerire la presenza di asma, ma questa è normalmente associata a tosse, difficoltà respiratoria, senso di costrizione toracica, con sintomi che si verificano o peggiorano durante la notte e sono accentuati dall’aria fredda, dall’esercizio fisico, o dall'esposizione agli allergeni. Per riconoscere l'asma si utilizzano i test di funzionalità respiratoria (4).
La cardiopatia da carcinoide è uno degli aspetti più diffusi e critici della SC, presente nel 10-20% dei pazienti al momento della diagnosi. La SC provoca un ispessimento delle valvole cardiache, alterandone la funzione, con fibrosi cardiaca e conseguente insufficienza cardiaca destra. In 1/5 dei pazienti sono presenti affaticamento, dispnea da sforzo ed edema periferico. Fino al 50% dei decessi della SC sono dovuti all’insufficienza cardiaca (5,6).
La crisi da carcinoide è la manifestazione estrema della SC, pericolosa per la vita, indotta dal massiccio rilascio in circolo di amine dopo anestesia, procedure interventistiche o assunzione di farmaci. Le caratteristiche principali sono: ipotensione (raramente ipertensione), tachicardia, dispnea bronchiale e disfunzione del sistema nervoso centrale.
Diagnosi
La diagnosi è spesso complessa, perchè la sintomatologia percepita dal paziente potrebbe essere ascritta ad altre condizioni, in particolare a disturbi gastrointestinali. Nella diagnosi differenziale devono essere considerate ed escluse malattia infiammatoria intestinale (Crohn e rettocolite ulcerosa), colite microscopica, intolleranza alimentare e allergia, celiachia, pancreatite cronica, altre neoplasie (es. carcinoma del colon, linfoma) e altre situazioni (asma, ansia, alcolismo).
Prima di procedere nel percorso diagnostico di una NEN con SC, si consiglia di escludere attentamente le altre cause che possono determinare flushing e diarrea. La chiave di volta per una gestione ottimale è rappresentata dall’accuratezza dell’anamnesi e dell’esame obiettivo. Per una migliore valutazione delle caratteristiche della sintomatologia, potrebbe essere utile suggerire al paziente di tenere un diario per 2-4 settimane, per registrare gli aspetti qualitativi e quantitativi di flushing, sudorazione e diarrea.
Il test biochimico per confermare la presenza della SC è la determinazione urinaria dell’acido 5-OH-indolacetico (5-HIAA), metabolita della serotonina. È il test più utile nei pazienti con la SC tipica, prevalentemente nelle NEN a sede digiuno-ileale. Questo dosaggio è altamente sensibile (fino al 90%) e specifico (85-90%) per la diagnosi di SC. Nei pazienti con SC, i livelli di 5-HIAA sono di solito almeno il doppio rispetto al limite superiore della norma. Deve essere posta attenzione ai fattori che possono causare livelli falsamente positivi o negativi (tabella).
| Interferenze con dosaggio di 5HIAA | |
| Falsi positivi | Alimenti: banane, avocado, kiwi, ananas, frutta secca, arachidi, susine, pomodori, melanzane, caffè, tè, cacao, cioccolata, vaniglia, dolci Farmaci: paracetamolo, tranquillanti (es. diazepam), anti-tussigeni, fenobarbital, reserpina |
| Falsi negativi | Acido acetilsalicilico, MAO-inibitori, alcol, eparina |
Nella SC atipica dei carcinoidi bronchiali vengono prodotti 5-idrossitriptofano e istamina anziché serotonina, poiché questi sono privi di DOPA-decarbossilasi, l’enzima che converte il 5-idrossitriptofano in serotonina. Purtroppo non sono disponibili in commercio test urinari per il 5-idrossitriptofano, mentre la determinazione dell’istamina è circoscritta a pochissimi centri.
La determinazione nel siero della serotonina non è raccomandata, in quanto può variare notevolmente a seconda dei livelli di attività e dello stress; da rilevare infine che la cromogranina è scarsamente specifica (7).
Nel momento in cui è confermata la diagnosi biochimica di SC, deve essere localizzata la sede della neoplasia. La causa più frequente è la presenza di una NEN nel piccolo intestino associata a metastasi epatiche. Le procedure di imaging dovrebbero includere entero-TC, endoscopia, Octreoscan o PET con Ga-68 ed ecocardiografia (chiedendo specificamente la valutazione delle cavità cardiache di destra) (8).
Terapia
La terapia della SC si avvale degli analoghi della somatostatina (SSA) long-acting (octreotide LAR o lanreotide autogel), sia a dosi standard che ad alte dosi (octreotide LAR 30 mg/28 gg o 40-60 mg/28 gg e lanreotide autogel 120 mg/28 gg o 180 mg/28 gg), dell’alfa-interferone alla posologia di 3-5 MUI x 3/settimana.
Il 2 marzo 2017 FDA ha approvato telotristat etiprate, inibitore dell'enzima triptofano-idrossilasi, in combinazione con SSA per il trattamento di adulti con diarrea da SC non adeguatamente controllata con SSA. La sicurezza e l’efficacia di telotristat sono state stabilite in uno studio di 12 settimane, in doppio cieco controllato con placebo, su 90 adulti con NEN metastatiche ben differenziate e diarrea da SC (4-12 scariche/die nonostante l’uso di SSA a dose stabile per almeno tre mesi). I pazienti che ricevevano la terapia con telotristat in aggiunta a SSA hanno evidenziato una riduzione maggiore nella frequenza media delle scariche (-2/die nel 33% dei trattati vs 4% del gruppo placebo) (9).
Bibliografia
- Modlin IM, Kidd M, Latich I, et al. Current status of gastrointestinal carcinoids. Gastroenterology 2005, 128: 1717-51.
- Van der Lely AJ, de Herder WW. Carcinoid syndrome: diagnosis and medical management. Arq Bras Endocrinol Metab 2005, 49: 850-60.
- Vinik AI, McLeod MK, Fig LM, et al. Clinical features, diagnosis, and localization of carcinoid tumors and their management. Gastroenterol Clin North Am 1989, 18: 865–96.
- Kulke MH, Mayer RJ. Carcinoid tumors. N Engl J Med 1999, 340: 859-68.
- Janson ET, Holmberg L, Stridsberg M, et al. Carcinoid tumors: analysis of prognostic factors and survival in 301 patients from a referral center. Ann Oncol 1997, 8: 685-90.
- Soga J, Yakuwa Y, Osaka M. Carcinoid syndrome: a statistical evaluation of 748 reported cases. J Exp Clin Cancer Res 1999, 18: 133–41.
- Tomassetti P, Migliori M, Lalli S, et al. Epidemiology, clinical features and diagnosis of gastroenteropancreatic endocrine tumours. Ann Oncol 2001, 12 (Suppl 2): S95–9.
- Kaltsas G, Besser GM, Grossman AB. The diagnosis and medical management of advance neuroendocrine tumors. Endocr Rev 2004, 25: 458-511.
- Kulke MH, Hörsch D, Caplin ME, et al. Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome. J Clin Oncol 2017, 35: 14-23.
Scheda telotristat
Franco Grimaldi
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria Santa Maria della Misericordia di Udine
Meccanismo d’azione
Inibitore dell'enzima triptofano-idrossilasi, inibisce la produzione di serotonina da parte dei tumori carcinoidi, riducendo la frequenza della diarrea causata dalla sindrome carcinoide.
Indicazioni
Da utilizzare in combinazione con la terapia con analogo della somatostatina (SSA) nel trattamento di adulti con diarrea da sindrome carcinoide non adeguatamente controllata con la sola terapia con SSA.
Contro-indicazioni
Nessuna.
Preparazioni, via di somministrazione, posologia
Compresse da 250 mg (Xermelo): da assumere oralmente tre volte al giorno durante i pasti.
Effetti collaterali
Nausea, cefalea, stipsi, livelli aumentati di gamma-GT, depressione, edema periferico, flatulenza, diminuzione dell’appetito, febbre.
Precauzioni d'uso
Epatopatia, depressione.
Il rischio di sviluppare stipsi può essere maggiore nei pazienti con frequenza delle scariche < 4/die. I pazienti trattati con un dosaggio più alto di quello raccomandato hanno sviluppato stipsi grave negli studi clinici. I pazienti devono essere monitorati: se si verifica stipsi grave o dolore addominale grave, persistente o che peggiora, sospendere il farmaco e contattare il proprio medico.
Limitazioni prescrittive
Non è ancora in commercio in Italia