EGDS nella diagnostica dei NET

Debora Berretti
Gastroenterologia, Azienda Ospedaliero - Universitaria "S. Maria della Misericordia", Udine
L’EGDS con biopsie gastriche è necessaria per rilevare i NET gastrici.
I NET gastrici di tipo 1 e tipo 2 si presentano generalmente (65-77% dei casi) come piccole rilevatezze della mucosa (< 2 cm).
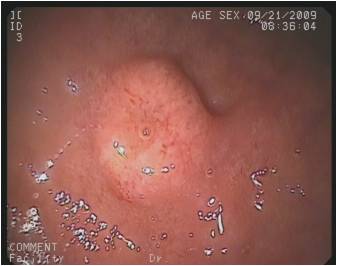
Piccola formazione ombelicata congesta di 5 mm a livello della seconda porzione duodenale: gastrinoma
In caso di piccoli (< 1 cm) NET gastrici (soprattutto se tipo 1 e 2), la EGDS con biopsia è usualmente l’unica indagine diagnostica raccomandata: i prelievi bioptici devono essere effettuati a livello di antro (2 biopsie), al corpo-fondo (4 biopsie) e sui polipi di maggiori dimensioni (1,2). L’eco-endoscopia (EUS) rappresenta un esame complementare (3).
In caso di NET duodenali, la EGDS con biopsia è il metodo diagnostico di scelta (4,5), seguita dalla EUS (3).
Bibliografia
- Vannella L, Lahner E, Annibale B. Risk for gastric neoplasias in patients with chronic atrophic gastritis: a critical reappraisal. World J Gastroenterol 2012, 18: 1279-85.
- Delle Fave G, Kwekkeboom DJ, Van Cutsem E, et al. ENETS consensus guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology 2012, 95: 74-87.
- Merola E, Sbrozzi-Vanni A, Panzuto F, et al. Type I gastric carcinoids: a prospective study on endoscopic management and recurrence rate. Neuroendocrinology 2012, 95: 207-13.
- Ruszniewski P, Amouyal P, Amouyal G, et al. Localization of gastrinomas by endoscopic ultrasonography in patients with Zollinger- Ellison syndrome. Surgery 1995, 117: 629–35.
- Yoshikane H, Suzuki T, Yoshioka N, et al. Duodenal carcinoid tumor: endosonographic imaging and endoscopic resection. Am J Gastroenterol 1995, 90: 642–4.
Diagnostica endoscopica dell'intestino tenue
Silvia Nasoni
SC Gastroenterologia ed Epatologia, Ospedale Regina Apostolorum, Albano Laziale (RM)
L’intestino tenue, ossia il tratto di intestino compreso tra il duodeno ed il colon, è lungo alcuni metri e non è visibile con le più comuni procedure endoscopiche, la gastroscopia e la colonscopia. Per la sua esplorazione sono disponibili due tecniche di recente introduzione: l’enteroscopia con videocapsula e l’enteroscopia a doppio pallone.
L’enteroscopia con videocapsula consente l’esplorazione e la visione del piccolo intestino, ma non permette di effettuare manovre operative.
L'enteroscopia a doppio pallone consente non solo di visualizzare l’intestino tenue e di effettuare biopsie, ma permette anche di eseguire degli interventi, come l’asportazione di eventuali polipi, la dilatazione di restringimenti, il trattamento delle emorragie, la rimozione di corpi estranei, ecc.
Capsula endoscopica nella diagnostica dei NET
Silvia Nasoni
SC Gastroenterologia ed Epatologia, Ospedale Regina Apostolorum, Albano Laziale (RM)
Procedura
La capsula contiene al suo interno una microscopica telecamera, una batteria, una piccola fonte luminosa e una trasmittente che invia al computer le immagini di tutto il percorso seguito nel tratto gastrointestinale, evidenziando possibili anomalie.
La preparazione suggerita dai produttori di videocapsula endoscopica (VCE) consiste solo in una dieta composta da liquidi chiari e 8 ore di digiuno.
La capsula viene ingerita bevendo dell'acqua. La persona potrà bere dopo 2 ore e nutrirsi solo quando sono trascorse 4 ore dall'ingestione della capsula. Al termine del procedimento la capsula rimane all'interno della persona, per essere poi espulsa attraverso le feci, senza possibilità di riutilizzo. La durata totale dell'endoscopia capsulare è di circa 8 ore, durante le quali la persona può esercitare le sue normali attività.
Indicazioni e risultati
L’indicazione più frequente è il sanguinamento gastrointestinale di origine ignota (Obscure Gastrointestinal Bleeding, OGIB), ma la VCE viene utilizzata anche per indagare il potenziale ruolo delle patologie del piccolo intestino nell’anemia da carenza di ferro e nella diagnosi e follow-up del morbo di Crohn. Dopo l’introduzione della VCE nella pratica clinica, è stato dimostrato che la frequenza di tumori del piccolo intestino è più alta di quanto creduto in precedenza: tra il 2.4% e il 9.6% dei pazienti sottoposti a VCE vs il classico 2%.
La resa diagnostica della VCE in pazienti con OGIB è significativamente più alta nei pazienti con sanguinamento in atto rispetto a quelli con sanguinamento occulto intermittente o pregresso. La resa diagnostica è anche maggiore quando l’esame viene eseguito entro le 48 ore di ospedalizzazione dei pazienti per l’episodio emorragico.
Gli studi prospettici e una meta-analisi comparativa tra VCE ed enteroscopia push in pazienti con OGIB hanno mostrato una resa diagnostica significativamente migliore per la VCE rispetto all’enteroscopia (63% vs 23%). Nell’individuazione di una fonte di sanguinamento la VCE ha una resa diagnostica superiore anche rispetto alla TC, all’angio-TC (72% vs 24%) e all’angiografia standard (72% vs 56%). In confronto all’endoscopia intra-operatoria utilizzata come riferimento, la VCE ha sensibilità, specificità e valori predittivi positivi e negativi rispettivamente del 95%, 75%, 95% e 86%. Gli studi finora pubblicati hanno dimostrato che la resa diagnostica della VCE è maggiore anche rispetto a quella dell’enteroscopia a doppio pallone (Double Balloon Enteroscopy, DBE). In uno studio multicentrico americano, la concordanza tra VCE e DBE è risultata circa del 74% per le angio-displasie, 96% per le ulcerazioni, 94% per i polipi e 96% per i tumori di grandi dimensioni.
Giudizio complessivo
La videocapsula presenta alcune limitazioni e alcuni rischi di cui bisogna essere consapevoli: l’esame non deve essere eseguito in pazienti con disturbi della deglutizione, a causa del rischio di aspirazione, o con stenosi accertate del piccolo intestino. La videocapsula non è controindicata nei pazienti con pace-maker cardiaco o defibrillatore impiantabile cardiaco.
La VCE rappresenta un’innovazione che a tutt'oggi non sostituisce comunque né la colonscopia né la gastroscopia diagnostica.
Due fattori compromettono la resa diagnostica della metodica: la presenza di residui o bolle endoluminali e i tempi di transito gastrico (se rallentato) o intestinale (se eccessivamente rapido), poiché impediscono un’adeguata visualizzazione della mucosa nei diversi distretti.
Bibliografia
- Ladas SD, Triantafyllou K, Spada C, et al, and the ESGE Clinical Guidelines Committee. European Society of Gastrointestinal Endoscopy (ESGE): Recommendations (2009) on clinical use of video capsule endoscopy to investigate small-bowel, esophageal and colonic diseases. Endoscopy 2010, 42: 220-7.
- Teshima CW, Kuipers EJ, van Zanten SV, Mensink PB. Double Balloon enteroscopy and capsule endoscopy for oscure gastrointestinal bleeding: an updated meta-analysis. J Gastroenterol Hepatol 2011, 26: 796-801.
- Apostolopoulos P, Liatsos C, Gralnek IM, et al. The role of wireless capsule endoscopy in investigating unexplained iron deficiency anemia after negative endoscopic evaluation of the upper and lower gastrointestinal tract. Endoscopy 2006, 38: 1127-32.
- Muhammad A, Pitchumoni S. Evaluation of iron deficiency anemia in older adult: the role of wireless capsule endoscopy. J Clin Gastroenterol 2009, 43: 627-31.
- Riccioni ME, Urgesi R, Spada C, et al. Unexplained iron deficiency anemia: is it worthwhile to perform capsule endoscopy? Dig Liver Dis 2010, 42: 560-6.
Enteroscopia a doppio pallone nella diagnostica dei NET
Silvia Nasoni
SC Gastroenterologia ed Epatologia, Ospedale Regina Apostolorum, Albano Laziale (RM)
Strumento
L’enteroscopio a doppio pallone è un video-endoscopio ad alta risoluzione, con una lunghezza operativa di 200 cm, un diametro esterno di 8.5 mm e un canale operativo di 2.2 mm. È fornito di un overtube di 140 cm di lunghezza e diametro esterno di 12 mm.L’endoscopio flessibile scivola all’interno dell’overtube. Sulla punta sia dell’endoscopio che dell’overtube si trova un palloncino che viene gonfiato con aria (da qui il nome di enteroscopia a doppio pallone). Facendo avanzare alternativamente l’endoscopio e l’overtube e gonfiando e sgonfiando i due palloncini sarà possibile far procedere l’endoscopio lungo tutto l’intestino. In questo modo, raccogliendo man mano il piccolo intestino sullo strumento, si possono percorrere lunghissimi tratti, a volte anche l'intero tratto intestinale.
Preparazione ed e esecuzione
L’esame viene eseguito con paziente in sedazione cosciente (benzodiazepine e oppiacei) o eventualmente profonda (propofol) fino alla narcosi. Il tipo di sedazione e la necessità di assistenza dell’anestesista viene valutata caso per caso.
Nell’enteroscopia dall’alto il paziente non ha bisogno di nessuna preparazione, mentre per via anale sarà necessaria una preparazione come la colonscopia.
L’introduzione dell’enteroscopio per via anale viene effettuata con uguali modalità. Una volta superata la valvola ileo-cecale con l’enteroscopio (cosa che spesso è più difficile che con il colonscopio), l’overtube viene fatto avanzare sin dentro l’ileo terminale e quindi l’endoscopio più l’overtube, entrambi a pallone gonfio, vengono ritirati.
Il raggiungimento della valvola ileo-cecale per via orale è possibile solo in una minoranza di casi. Se è richiesta l’esplorazione completa del piccolo intestino e questa non sia ottenibile per via orale, si può marcare il punto raggiunto dall’endoscopio con un tatuaggio con china e quindi completare l’esplorazione dell’intestino per via anale sino a raggiungere il punto segnato. Questa tecnica combinata sarebbe in grado di consentire la completa esplorazione del viscere fino all’80% dei casi, anche se questa percentuale così alta è stata messa in discussione da dati più recenti. In realtà solo in una piccola percentuale di casi vi è necessità di esplorare l’intero intestino.
La durata media della metodica per l’esplorazione completa dell’intestino tenue è di circa 120 minuti da dividere tra l’approccio orale (circa 70 minuti) e quello anale (circa 50 minuti).
Risultati e rischi
La metodica è nel complesso ben tollerata e raramente causa complicanze gravi.Rischi potenziali derivano dall’uso di sedativi in pazienti anziani o con patologie respiratorie e cardiache. Le complicanze legate all’atto endoscopico diagnostico sono rare (0.002%-2.4%) e consistono in traumi della mucosa, piccole emorragie, che solitamente si arrestano spontaneamente (ma, se necessario, possono essere fermate con tecnica endoscopica), e perforazione, che rende necessario l’intervento chirurgico.
Ancora non si hanno informazioni definitive sul valore diagnostico della metodica. Nel caso di sanguinamento oscuro, sembra garantire un guadagno diagnostico tra il 50 e il 70%, paragonabile ai risultati ottenuti dalla videocapsula. Tale valore potrebbe essere addirittura maggiore rispetto a quello della capsula per le stenosi e le neoplasie, per le quali l’uso della capsula è addirittura controindicato. Un ulteriore vantaggio dell’enteroscopio è dato dalla possibilità di prelevare biopsie e trattare alcune lesioni del piccolo intestino. L’enteroscopio a doppio pallone attraverso l’uso di accessori idonei consente la terapia iniettiva e termica delle emorragie digestive, la rimozione di polipi e l’effettuazione di mucosectomie, la dilatazione di stenosi, la rimozione di corpi estranei, il posizionamento di endoprotesi.
Bibliografia
- Pohl J, Delvaux M, Ell C, et al. European Society of Gastrointestinal Endoscopy (ESGE) Guidelines: flexible enteroscopy for diagnosis and treatment of small-bowel diseases. Endoscopy 2008, 40: 609-18.
- Hirai F, Beppu T, Nishimura T, et al. Carbon dioxide insufflation compared with air insufflation in double-balloon enteroscopy: a prospective, randomized, double-blind trial. Gastrointest Endosc 2011, 73: 743-9.
Pancolonscopia nella diagnostica dei NET
Silvia Nasoni
SC Gastroenterologia ed Epatologia, Ospedale Regina Apostolorum, Albano Laziale (RM)
La colonscopia è una tecnica introdotta nella pratica clinica sul finire degli anni ´60, allorché furono disponibili i primi colonscopi flessibili a fibre ottiche.
Indicazioni
In generale la colonscopia è indicata:
- se il trattamento del paziente sarà probabilmente influenzato dal risultato della indagine;
- quando si presume l’esistenza di una patologia che può contemplare una procedura di endoscopia terapeutica.
Le indicazioni diagnostiche all’esecuzione dell’esame sono quindi costituite da:
- anomalie rilevate al clisma opaco, all’ecografia, alla TC o alla RMN che richiedano un’ulteriore valutazione;
- positività della ricerca del sangue occulto nelle feci e/o anemia sideropenica di origine non nota;
- emorragia digestiva distale;
- melena (dopo aver escluso l’eziologia a carico del tratto digestivo superiore);
- calo ponderale significativo (dopo aver escluso altre eziologie);
- modificazioni significative e persistenti dell’alvo;
- valutazione pre-operatoria dei pazienti con carcinoma del colon operabile per la ricerca di cancro sincrono o polipi;
- follow-up dei pazienti già sottoposti a polipectomia o intervento chirurgico per carcinoma del colon;
- screening delle neoplasie coliche in pazienti ad alto rischio (per es. pancoliti ulcerose insorte da almeno 7 anni, o coliti sinistre insorte da almeno 10 anni, storia familiare di poliposi o di cancro);
- nelle malattie infiammatorie dell’intestino quando una migliore definizione diagnostica o la valutazione della estensione e dell’attività della malattia influenzerà il trattamento del paziente.
Le indicazioni terapeutiche all’esecuzione dell’esame sono quindi costituite da:
- polipectomia;
- emostasi di lesioni sanguinanti;
- tatuaggio di lesioni da individuare intra-operatoriamente;
- decompressione di megacolon acuto non tossico o riduzione di volvolo;
- dilatazione di stenosi;
- rimozione di corpi estranei.
Controindicazioni
Sono rappresentate da:
- peritonite;
- perforazione intestinale;
- colite tossica o fulminante;
- diverticolite acuta;
- scompenso cardiaco grave e/o insufficienza respiratoria grave;
- diatesi emorragica grave non correggibile.
Preparazione
Una buona preparazione intestinale è un requisito indispensabile allo svolgimento di una colonscopia di qualità. La toilette intestinale, effettuata un tempo con clisteri, mannitolo o sale inglese, è stata da anni sostituita dal lavaggio intestinale per os a base di polietilenglicole (PEG), che per l’elevato volume è scarsamente tollerato dai pazienti ma offre risultati ottimali se ben eseguito, o dalla assunzione di Fosfato di Sodio, più accettabile ma da utilizzare con cautela nei pazienti affetti da insufficienza renale o cardiopatie. Tali preparazioni possono essere integrate e migliorate con l’aggiunta di piccole quantità di altri lassativi.
Sedazione
La sedazione associata o meno all’analgesia, è una condizione farmacologicamente indotta volta a ridurre il disagio percepito da un paziente durante una procedura invasiva come l’endoscopia. Nella pratica il paziente viene sedato poco prima di iniziare l’esame e viene risvegliato subito dopo il termine. Con i farmaci attualmente disponibili, che hanno una durata d’azione brevissima, il recupero delle condizioni pre-esame avviene in pochi minuti (in genere dai 10 ai 30 min) ed il paziente può immediatamente tornare a casa. Durante l’esame vengono controllati i parametri vitali mediante l’utilizzo di un monitor automatico. Esistono due livelli di sedazione che si distinguono soprattutto in base alla risposta del paziente agli stimoli verbali e agli stimoli dolorosi.
- Sedazione cosciente: viene ottenuta somministrando farmaci ipnotici della classe delle benzodiazepine e farmaci analgesici. Il paziente viene sedato, ma rimane comunque sveglio e collaborante durante tutta la procedura. È possibile che durante questo tipo di sedazione il paziente avverta comunque dolore o fastidio se lo stimolo è rilevante. Si ha un buon effetto ansiolitico.
- Sedazione profonda: viene ottenuta mediante la somministrazione da parte dell’anestesista di un farmaco ipnotico a durata brevissima, il propofol. Il paziente è sedato, non si accorge di nulla durante l’esame, non avverte alcun dolore e respira in modo autonomo come se stesse dormendo normalmente e viene prontamente risvegliato al termine della procedura.
Bibliografia
- ASGE Standards of Practice Committee, et al. The role of endoscopy in the management of obscure GI bleeding. Gastrointest Endosc 2010, 72: 471-9.
- ASGE Standards of Practice Committee, et al. The role of endoscopy in the management of patients with known and suspected colonic obstruction and pseudo-obstruction. Gastrointest Endosc 2010, 71: 669-79.
- ASGE Standards of Practice Committee, et al. The role of endoscopy in the management of patients with diarrhea. Gastrointest Endosc 2010, 71: 887-92.
- Raccomandazioni SIED. La colonscopia, a cura di L. Petruzziello.
- Commissione Linee Guida Federazione Malattie Digestive AIGO-SIED-SIGE. Indicazioni alla colonscopia.
- Rex DK, et al. Quality indicators for colonoscopy. Gastrointest Endosc 2006, 63 (4 Suppl): S16-28.
- American Society for Gastrointestinal Endoscopy. Appropriate use of gastrointestinal endoscopy. 1997.
Criteri per l'impostazione terapeutica dei NET
Miriam Cellini & Franco Grimaldi
SOC Endocrinologia Ospedale Santa Maria della Misericordia di Udine
(aggiornato al dicembre 2025)
PREMESSA
I tumori neuroendocrini (NET) o neoplasie neuroendocrine (NEN) rappresentano un gruppo di neoplasie eterogenee per sede primaria, differenziazione, attività proliferativa, secrezione ormonale e decorso clinico. Nonostante in numerosi casi la patologia si manifesti in maniera indolente, molti pazienti presentano una malattia avanzata al momento della diagnosi, rendendo la scelta terapeutica una sfida complessa, che richiede un approccio multi-disciplinare. Negli ultimi anni il crescente avanzamento diagnostico e la disponibilità dei risultati di studi clinici hanno migliorato la capacità di stratificare i pazienti in termini prognostici e di personalizzare l’approccio terapeutico. I fattori che guidano le scelte terapeutiche nei NET sono l’adeguata stadiazione, la caratterizzazione cito-istologica e funzionale della neoplasia nonché le condizioni cliniche del paziente (1-3).
CRITERI DI IMPOSTAZIONE TERAPEUTICA
L’iter terapeutico dei NET dipende principalmente dai seguenti criteri:
- stadio della neoplasia e sede del tumore primitivo;
- differenziazione morfologica e grading;
- carico neoplastico, estensione e velocità di crescita;
- espressione dei recettori per la somatostatina;
- funzionalità ormonale e sintomatologia;
- stato del paziente;
- obiettivi terapeutici.
Stadio e sede del tumore primitivo
La stadiazione della neoplasia mediante il sistema di classificazione AJCC/TNM, che utilizza il criterio T (dimensione del tumore primario), N (coinvolgimento dei linfonodi regionali) e M (presenza di metastasi a distanza), consente di definire la malattia come locale, loco-regionale o metastatica e condiziona sia la prognosi che la strategia terapeutica. In presenza di una neoplasia potenzialmente resecabile e non metastatica, la chirurgia o la resezione endoscopica con intento curativo costituiscono per molte NEN la prima opzione terapeutica (3).
Anche dimensioni e sede del tumore primitivo condizionano la scelta terapeutica. Ad esempio, per i NET pancreatici non funzionanti deve essere utilizzato un trattamento conservativo se ≤ 2 cm, mentre l’approccio chirurgico rimane di prima scelta se > 2 cm (4). Nelle NEN localmente avanzate e/o metastatiche sono indicati principalmente i trattamenti farmacologici come gli analoghi della somatostatina, la chemioterapia, le terapie a bersaglio molecolare e la terapia radio-recettoriale (PRRT), riservando la chirurgia ad alcuni contesti specifici (es. controllo dei sintomi da effetto massa o da ipersecrezione ormonale, intento di debulking e scopo palliativo) (3).
Differenziazione morfologica e grading
La classificazione WHO del 2022 consente di distinguere i NET ben differenziati dai carcinomi neuroendocrini (NEC) scarsamente differenziati (in base alla differenziazione morfologica) e i NET in G1, G2 e G3 (in base al grading valuto mediante Ki-67 e indice mitotico). Entrambi questi fattori riflettono la velocità di crescita e l’aggressività della neoplasia e hanno un valore prognostico e predittivo che influenza la scelta terapeutica, riservando generalmente le chemioterapie più aggressive ai pazienti con NET G2 ed elevato Ki67%, NET G3 e NEC (5).
Carico neoplastico, estensione e velocità di crescita della neoplasia
Il carico di malattia rappresenta un parametro prognostico cruciale e orienta la scelta di terapie sistemiche o loco-regionali ad alta intensità nei casi con burden elevato, mentre un approccio terapeutico più conservativo o a minore intensità può essere riservato al paziente con basso carico di malattia.
L’estensione della neoplasia, determinata dalla localizzazione del tumore primitivo, dalla presenza di metastasi linfonodali e/o a distanza (in particolare epatiche, ossee o peritoneali), condiziona la fattibilità e l’efficacia di strategie loco-regionali (chirurgia, ablazione, radioterapia) nonché delle terapie sistemiche.
La velocità di crescita della neoplasia, stimata con l’indice proliferativo Ki67 e con le modifiche all’imaging radiologico, fornisce un’informazione predittiva essenziale. Tumori con basso indice proliferativo (G1 o G2 con Ki67 < 10%) o crescita lenta possono essere gestiti con sorveglianza attiva o terapie meno aggressive, mentre quelli con elevata proliferazione (G2 con elevato Ki67 e G3) o rapida progressione impongono interventi terapeutici tempestivi.
L’integrazione di questi parametri permette di stratificare i pazienti in sotto-gruppi prognostici e terapeutici (6,7):
- nei pazienti con elevato carico di malattia e/o in rapida progressione si utilizzeranno trattamenti più aggressivi (es. chemioterapia e terapie loco-regionali per le metastasi epatiche);
- nei pazienti con basso carico e/o lenta progressione di malattia si utilizzeranno approcci terapeutici meno aggressivi (es. analoghi della somatostatina, PRRT, terapie a bersaglio molecolare).
Espressione dei recettori per la somatostatina
L’espressione dei recettori per la somatostatina, valutata mediante imaging funzionale come la PET/TC con 68Gallio, condiziona la scelta terapeutica nei NET. Tale espressione consente di utilizzare in prima linea la terapia con analoghi della somatostatina per i NET ben differenziati (efficace sia per il controllo ormonale sia per l’effetto anti-proliferativo, 8,9) e la PRRT per i GEP-NET ben differenziati in progressione dopo analogo della somatostatina (10,11).
Funzionalità ormonale e sintomatologia
La scelta terapeutica nei pazienti con NET è condizionata dalla natura funzionante della neoplasia. Nei NET funzionanti la terapia medica e/o chirurgica e/o i trattamenti loco-regionali hanno l’obiettivo principale di controllare l’iperproduzione ormonale oltre alla crescita tumorale. In presenza di sintomi da ipersecrezione ormonale, come per esempio nella sindrome carcinoide, la terapia precoce e di prima linea è rappresentata dagli analoghi della somatostatina, che bloccando il rilascio di ormoni consentono di controllare la sintomatologia clinica e le complicanze associate, quali la cardiopatia del cuore destro (12).
Stato del paziente
La pianificazione ottimale della strategia terapeutica nei NET richiede una valutazione multi-dimensionale che integri non soltanto i parametri biologici e radiologici della neoplasia, ma anche le caratteristiche cliniche del paziente, tra cui il performance status, le comorbilità, l’età e le preferenze individuali.
Il performance status rappresenta un indicatore prognostico e un determinante cruciale della tollerabilità terapeutica. Pazienti con performance status conservato presentano maggiore probabilità di beneficiare di approcci intensivi, incluse strategie sequenziali, mentre condizioni funzionali compromesse impongono un orientamento verso trattamenti a minore intensità o a profilo di tossicità più favorevole.
Le comorbilità contribuiscono alla definizione del profilo di eleggibilità ai diversi interventi terapeutici. Patologie cardio-vascolari, epatiche e renali possono influenzare in maniera critica la sicurezza di impiego delle terapie target (in particolare inibitori di mTOR o di tirosin-chinasi), delle procedure loco-regionali e della PRRT, richiedendo spesso una valutazione multi-disciplinare e l’eventuale rimodulazione posologica o la scelta di strategie alternative.
Infine, le preferenze del paziente hanno un ruolo cruciale nella scelta tra strategie con diverso impatto sulla qualità di vita, diverso profilo di effetti collaterali o differente impegno organizzativo e familiare (es. terapie endovenose vs trattamenti orali, frequenza dei follow-up, gestione degli eventi avversi, presenza di un care-giver) (3,13).
Obiettivi terapeutici
Gli obiettivi terapeutici nei pazienti con NET possono essere inclusi nei seguenti gruppi:
- curativo;
- cito-riduzione/debulking;
- controllo della progressione di malattia e dei sintomi;
- palliativo.
La definizione di tali obiettivi condiziona la strategia terapeutica dei pazienti con NEN.
L’obiettivo curativo è riservato ai NET operabili e resecabili, nei quali la terapia chirurgica radicale del primitivo e delle eventuali metastasi rappresenta la prima opzione terapeutica.
La cito-riduzione/debulking mediante approccio chirurgico o terapia sistemica trova indicazione nella malattia metastatica o localmente avanzata, con l'obiettivo di ridurre i sintomi correlati alla neoplasia, per migliorare la risposta ai successivi trattamenti e la sopravvivenza.
Il controllo della progressione di malattia e dei sintomi correlati da ipersecrezione ormonale con l’utilizzo della terapia farmacologica (analoghi della somatostatina, terapia a bersaglio molecolare e chemioterapia), della PRRT e dei trattamenti loco-regionali (es. per le metastasi epatiche) può rappresentare un ulteriore obiettivo terapeutico nella malattia metastatica e/o funzionante.
Infine, l’intento palliativo, che si propone di migliorare o mantenere un’adeguata qualità di vita, è riservato ai casi di malattia metastatica avanzata con elevata aggressività o scarsa tolleranza ai trattamenti (3,14).
Gli obiettivi terapeutici immediati e tardivi andrebbero definiti preferibilmente nell’ambito di un gruppo multi-disciplinare, sebbene in alcuni casi il quadro clinico non lo consenta. Ad esempio, nel paziente fortemente sindromico o sintomatico la cura dei sintomi è sicuramente un obiettivo immediato da considerare, anche prima di discutere il caso collegialmente. Infatti, per il controllo della sindrome carcinoide va avviato immediatamente l’analogo della somatostatina, mentre in un paziente sintomatico con NET metastatico ed elevato carico di malattia la chemioterapia. Infine, la definizione dell’obiettivo tardivo può condizionare l’impostazione terapeutica e la sequenza delle successive terapie. In tal caso sarà necessario considerare la tossicità tardiva e la tossicità dose-cumulativa di alcune terapie (es. chemioterapici e PRRT) per evitare di precludere terapie future (13).
CONCLUSIONI
La gestione terapeutica dei pazienti con NET richiede un approccio personalizzato e multi-dimensionale, che integri le caratteristiche isto-patologiche, molecolari e funzionali della neoplasia con le condizioni cliniche del paziente. La personalizzazione del trattamento deve avvenire all’interno di un percorso multi-disciplinare che coinvolga oncologi, chirurghi, endocrinologi, gastro-enterologi, radiologi, medici-nucleari e radio-terapisti, in modo da garantire decisioni condivise, tempestive e coerenti con gli obiettivi terapeutici a breve e lungo termine, che tengano conto anche delle caratteristiche e delle preferenze del paziente.
BIBLIOGRAFIA
- Yin F, Wu ZH, Lai JP. New insights in diagnosis and treatment of gastroenteropancreatic neuroendocrine neoplasms. World J Gastroenterol 2022, 28: 1751-67.
- Borga C, Businello G, Murgioni S, et al. Treatment personalization in gastrointestinal neuroendocrine tumors. Curr Treat Options Oncol 2021, 22: 29.
- ISS. Linee Guida Neoplasie Neuroendocrine. 2024.
- Kos-Kudła B, Castaño JP, Denecke T, et al. European Neuroendocrine Tumour Society (ENETS) 2023 guidance paper for nonfunctioning pancreatic neuroendocrine tumours. J Neuroendocrinol 2023, 35: e13343.
- Rindi G, Mete O, Uccella S, et al. Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocr Pathol 2022, 33: 115-54.
- Spada F, Gelsomino F, Rinzivillo M, et al. Clinical practice guidelines for the management of non-functioning advanced GEP-NENs: a GRADE approach for evidence evaluation and recommendations by the Italian Association of Medical Oncology (AIOM) in collaboration with the Italian Association for Neuroendocrine Tumors (ITANET). ESMO Open 2025, 10: 105878.
- Cives M, Pellè E, Quaresmini D, et al. The role of cytotoxic chemotherapy in well-differentiated gastroenteropancreatic and lung neuroendocrine tumors. Curr Treat Options Oncol 2019, 20: 72.
- Caplin ME, Pavel M, Cwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014, 371: 224-33.
- Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol 2009, 27: 4656-63.
- Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med 2017, 376: 125-35.
- Panzuto F, Albertelli M, De Rimini ML, et al. Radioligand therapy in the therapeutic strategy for patients with gastro-entero-pancreatic neuroendocrine tumors: a consensus statement from the Italian Association for Neuroendocrine Tumors (Itanet), Italian Association of Nuclear Medicine (AIMN), Italian Society of Endocrinology (SIE), Italian Association of Medical Oncology (AIOM). J Endocrinol Invest 2025, 48: 23-36.
- Spada F, Rossi RE, Modica R, et al. Functioning neuroendocrine tumors (NET): minimum requirements for a NET specialist. Cancer Treat Rev 2025, 135: 102907.
- Riechelmann RP, Taboada RG, de Jesus VHF, et al. Therapy sequencing in patients with advanced neuroendocrine neoplasms. Am Soc Clin Oncol Educ Book 2023, 43: e389278.
- Lamarca A, Bartsch DK, Caplin M, et al. European Neuroendocrine Tumor Society (ENETS) 2024 guidance paper for the management of well-differentiated small intestine neuroendocrine tumours. J Neuroendocrinol 2024, 36: e13423.
Analoghi della somatostatina nella terapia dei NET
Franco Grimaldi
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria Santa Maria della Misericordia di Udine
(aggiornato al 19 marzo 2017)
Nelle neoplasie neuroendocrine (NEN) gli obiettivi terapeutici sono il controllo della sintomatologia secondaria all’iperincrezione ormonale e l’inibizione della crescita neoplastica (1). Più dell’80% delle NEN esprime sulla superficie di membrana cellulare i recettori della somatostatina (SSTR), in particolare SSTR2 nelle NEN di basso grado (1). Per superare il problema della brevissima emivita della somatostatina nativa, sono stati sintetizzati analoghi della somatostatina (SSA) a più lunga durata d’azione.
Nel corso degli anni gli SSA sono divenuti sempre più rilevanti nel trattamento delle NEN. Il presupposto della loro azione è legato alla dimostrazione di SSTR nell’80-90% dei tumori in vivo, attraverso metodiche come la scintigrafia total-body con 111In pentetreotide (Octreoscan), o, se fattibile, con PET 68Ga-DOTANOC/DOTATOC, o in vitro nel preparato istologico mediante la caratterizzazione tissutale dell’espressione dei sottotipi della somatostatina. È stata riscontrata una correlazione intorno all’85% dell’espressione di SSTR su tessuto con quella in vivo a mezzo Octreoscan (2), sebbene attualmente non siano ancora disponibili dati di confronto su ampie casistiche tra l’espressione di SSTR e la risposta terapeutica agli SSA.
Gli SSA attualmente disponibili per questa indicazione sono octreotide e lanreotide. Nuovi SA, con peculiarità dissimili di affinità recettoriale, sono in fase avanzata di sperimentazione, tra i quali pasireotide, caratterizzato da un’elevata affinità per 4 dei 5 SSTR (studi di fase 3 nei NEN)(3).
Gli SSA rappresentano la terapia elettiva della sindrome da carcinoide (2,3) ma vengono suggeriti anche in GEP-NEN funzionanti associate ad altre sindromi, nelle forme non funzionanti evolutive (4) e nelle NEN polmonari: carcinoidi tipici e atipici (5).
Grazie a una concentrazione stabile del farmaco, gli SSA hanno consentito maggior controllo sintomatologico e miglioramento sia della qualità di vita che della condizione fisica. È da segnalare però che circa il 50% delle NEN pancreatiche insulino-secernenti non esprime i recettori SSTR2 e SSTR5; in tale situazione, il trattamento con octreotide e lanreotide può sopprimere gli ormoni che ostacolano l’ipoglicemia (GH e IGF-I, e glucagone), con possibile peggioramento di ipoglicemia severa.
Alle dosi standard, gli SSA inducono una risposta clinica e biochimica nel 66% dei casi e una stabilizzazione della crescita tumorale nel 35-50% dei pazienti trattati. Inoltre, la sopravvivenza media dei pazienti con NEN è notevolmente aumentata dal momento in cui gli SSA sono stati introdotti nella pratica clinica.
Circa il 40% delle sindromi da carcinoide in trattamento con la massima dose di SSA a lento rilascio non è completamente controllata. In questi casi possono essere presi in considerazione un aumento della dose, una riduzione degli intervalli di somministrazione o l’aggiunta di octreotide sottocute (rescue) (6).
Uno dei principali limiti degli SSA nella terapia delle NEN, all’inizio del loro impiego, era il fenomeno della tachifilassi, per il quale un tumore ben controllato per un certo numero di mesi andava successivamente incontro ad “escape”; l’impiego dei nuovi SSA, a lento rilascio, ha consentito di evitare tale inconveniente. Vi sono tuttavia alcuni pazienti che sono poco o per nulla responsivi agli SSA a lunga durata d’azione. I principali meccanismi di resistenza nei pazienti con NEN SSTR-positive sono: la disomogenea distribuzione recettoriale nel tumore, la selezione di cloni SSTR-negativi durante la terapia e l’assenza di SSTR con elevata affinità per l’analogo in uso. In particolare, il profilo recettoriale delle NEN è molto variabile: l’eterogeneità non riguarda solo i diversi istotipi, ma anche le distinte lesioni tumorali nel medesimo paziente. Ciò indica che un paziente può avere un tumore con una scarsa espressione recettoriale SSTR2 e/o SSTR5 e un’elevata espressione recettoriale di differenti SSTR. In tale ipotesi l’efficacia di octreotide e lanreotide sarà molto ridotta, sia in termini anti-proliferativi che anti-secretivi.
Sono stati finora pubblicati due studi prospettici randomizzati vs placebo che hanno valutato l’attività anti-proliferativa degli SSA. Lo studio PROMID (7), in doppio cieco, di fase III, ha confrontato octreotide LAR 30 mg ogni 4 settimane vs placebo in 90 pazienti con NEN del midgut (piccolo intestino + colon prossimale) non pre-trattati. I due bracci di trattamento erano ben bilanciati per età, Ki67 e coinvolgimento epatico, mentre c’era discrepanza per il tempo dalla diagnosi all’inizio del trattamento (7.5 mesi nel braccio di trattamento vs 3.3 nel braccio placebo). Erano includibili pazienti con tumore sia non funzionante che funzionante, purchè il flushing o la diarrea fossero gestibili senza necessità di SSA. Globalmente il 95% delle neoplasie aveva un Ki67 < 2% e il 74% era positivo alla scintigrafia con octreotide marcato. Lo stato di malattia al basale (progressione o stabilità) non è noto. Dei 90 pazienti inclusi, 85 sono stati randomizzati, 42 nel braccio octreotide LAR e 43 nel braccio placebo. Octreotide LAR ha più che raddoppiato il tempo alla progressione (da 6.0 a 14.3 mesi) rispetto al placebo. Il PROMID è stato il primo studio prospettico randomizzato che ha dimostrato superiorità statisticamente significativa per un SSA (nel caso octreotide LAR) rispetto allo standard of care (placebo) in una categoria di GEP-NEN (7).
L’altro studio, denominato CLARINET, di fase III, in doppio cieco, ha incluso non solo NEN del midgut, ma anche NEN intestinali non-midgut e pancreatiche avanzate. Le neoplasie erano tutte non funzionanti e potevano essere pre-trattate (7). Lo studio ha confrontato lanreotide autogel 120 mg ogni 4 settimane con placebo in una popolazione di 204 pazienti (101 dei quali randomizzati a ricevere lanreotide e 103 placebo). Era prevista una stratificazione per progressione verso non progressione al basale e per pre-trattamento verso non pre-trattamento. Il Ki67 (valutato centralmente) doveva essere < 10% e bisognava avere una scintigrafia con octreotide marcato con captazione > 2 (gradi di Krenning). Lo stato del tumore al basale doveva essere valutato con due TC o due RM nei 3-6 mesi antecedenti la randomizzazione per chiarire se il tumore fosse stabile o in progressione RECIST 1.0. Lo studio ha mostrato un vantaggio statisticamente significativo a favore di lanreotide in termini di sopravvivenza libera da progressione (PFS), endpoint primario, che è risultata di 18 mesi nel braccio placebo e non ancora raggiunta nel braccio lanreotide (8). Lo studio CLARINET ha mostrato per la prima volta in prospettico un vantaggio in PFS di un SSA per GEP-NEN avanzate, non funzionanti, con stabilità di malattia al basale, indipendentemente dal carico epatico di malattia.
È controverso l’uso degli SSA dopo trattamento chirurgico, di radiologia interventistica e/o di radioterapia in GEP-NEN metastatici non funzionanti, in pazienti senza evidenza di malattia residua attiva. Non sono stati eseguiti studi per rispondere specificamente a tale quesito. La decisione clinica su singoli casi dovrebbe essere ben ponderata in ambito multidisciplinare, tenendo conto dell’assenza di evidenza specifica, delle caratteristiche della malattia trattata e del rapporto costo/beneficio. L’uso degli SSA come terapia adiuvante nelle GEP-NEN in stadio localizzato o localmente avanzato, radicalmente resecate, non è stato specificamente studiato.
Bibliografia
- Wolin EM. The expanding role of somatostatin analogs in the management of neuroendocrine tumors. Gastrointest Cancer Res 2012, 5: 161–8.
- Oberg K, Kvols L, Caplin M, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol 2004, 15: 966–73.
- Modlin IM, Pavel M, Kidd M, et al. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 2010, 31: 169–88.
- Panzuto F, Di Fonzo M, Iannicelli E, et al. Long-term clinical outcome of somatostatin analogues for treatment of progressive, metastatic, well-differentiated entero-pancreatic endocrine carcinoma. Ann Oncol 2006, 17: 461–6.
- De Dosso S, Bajetta E, Procopio G, et al. Pulmonary carcinoid tumours: indolent but not benign. Oncology 2007, 73: 162-8.
- Ferolla P, Faggiano A, Grimaldi F, et al. Shortened interval of long-acting octreotide administration is effective in patients with well-differentiated neuroendocrine carcinomas in progression on standard doses. J Endocrinol Invest 2012, 35: 326-31.
- Rinke A, Müller HH, Schade-Brittinger C, et al; PROMID Study Group. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009, 27: 4656-63.
- Caplin M, Pavel M, Cwikka JB, et al, for the CLARINET Investigators. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014, 371: 224-33.
Interferone nella terapia dei NET
Franco Grimaldi
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria Santa Maria della Misericordia di Udine
Gli interferoni sono una famiglia eterogenea di citochine, secrete in prevalenza da leucociti, fibroblasti e linfociti. Inizialmente identificati per la loro attività anti-virale, esercitano un ruolo importante in numerosi processi biologici, quali differenziazione e proliferazione cellulare, risposta immunitaria e trasformazione neoplastica.
Noto come agente anti-virale e anti-tumorale, l’interferone alfa ricombinante (IFN-α-2b) è stato introdotto da Öberg nel 1982 nel trattamento dei tumori neuroendocrini (NET) per la sua propensione a controllare la secrezione ormonale, i sintomi clinici e la crescita tumorale, sulla base di dati sperimentali che avevano dimostrato un effetto diretto sulla cellula tumorale e indiretto sulla modulazione del sistema immunitario (1).
I meccanismi d’azione dell’α-IFN sono:
- effetto anti-proliferativo;
- inibizione ormoni e fattori di crescita, tramite blocco cellulare nelle fasi G0 e G1, con inibizione del passaggio alla fase S attraverso il controllo della proteina Rb;
- stimolazione generale del sistema immunitario (induzione di antigeni di classe I sulla superficie cellulare e stimolazione delle cellule natural killer;
- induzione a reazioni fibrotiche intra-tumorali;
- inibizione dell’angiogenesi.
Attualmente un discreto gruppo di pazienti con NET, circa 400, è stato trattato con la forma ricombinante IFN-α-2b, registrata in molti Paesi Europei per il trattamento dei carcinoidi maligni.La terapia con IFN-αumano ricombinante rappresenta un trattamento di terza linea nei NET dopo la target terapia.
La dose standard utilizzata è compresa tra 3 e 9 milioni di unità internazionali, per via sottocutanea, da 3 a 7 giorni la settimana, titolandola sulla base della risposta leucocitaria (la conta dei globuli bianchi non deve scendere sotto 3000/mL), ma va personalizzata per ciascun paziente. In alcuni casi, sono state impiegate anche dosi più elevate.
L’IFN-α è in grado di controllare la sintomatologia associata ai NET nel 15-50% dei casi, ma solo nel 5-15% determina una risposta obiettiva misurabile del tumore.
Tutti gli interferoni di tipo I si legano a uno specifico complesso recettoriale formato dalle catene IFNAR-1 e IFNAR-2.
L’IFN-ß è un’ulteriore molecola, che presenta un’affinità nei confronti delle subunità IFNAR-1 e IFNAR-2 rispettivamente maggiore di 100 e 50 volte dell’IFN-α2. In particolare, l’elevata analogia dell’IFN-ß verso la catena IFNAR-1 pare sia responsabile della maggiore attività anti-proliferativa di questa citochina rispetto all’IFN-α. Di fatto, in diversi modelli cellulari di tumori endocrini (NET del pancreas e carcinoma del surrene), l’IFN-β ha effetti anti-tumorali maggiori dell’IFN-α (2-4).
Terapia combinata: analoghi della somatostatina (SA) e interferone (α-IFN)
L’IFN ha dimostrato una notevole efficacia nei NET, una combinazione α-IFN e SA potrebbe ampliare la risposta terapeutica, consentendo una migliore tollerabilità dell’α-IFN e potrebbe anche essere efficace in caso di progressione della malattia nei pazienti in trattamento con SA. A oggi, la terapia “combinata” non è però ancora validata.
Una review del 2007 (5) ha rivalutato diversi studi, tra cui uno che confrontava 68 pazienti divisi in due gruppi (6), uno trattato con α-interferone e l’altro con α-interferone + octreotide, con differenza statistica a favore dell’associazione nella progressione della malattia e nella sopravvivenza. In un altro studio che includeva 88 pazienti suddivisi in tre gruppi (terapia con solo α-interferone, con solo Lanreotide e con l’associazione) non sono state evidenziate differenze di sopravvivenza (7).
Bibliografia
- Öberg K, Funa K, Alm G. Effects of leukocyte interferon upon clinical symptoms levels in patients with midgut carcinoid tumors and the carcinoid syndrome. N Engl J Med 1983, 309: 129-33.
- Vitale G, de Herder WW, van Koetsveld PM, et al. IFN-beta is a highly potent inhibitor of gastroenteropancreatic neuroendocrine tumor cell growth in vitro. Cancer Res 2006, 66: 554-62.
- Van Koetsveld PM, Vitale G, de Herder WW, et al. Potent inhibitory effects of type I interferons on human adrenocortical carcinoma cell growth. J Clin Endocrinol Metab 2006, 91: 4537-43.
- Vitale G, van Koetsveld PM, de Herder WW, et al. Effects of type I interferons on IGF-mediated autocrine/paracrine growth of human neuroendocrine tumor cells. Am J Physiol Endocrinol Metab 2009, 296: E559-66.
- Fazio N, de Braud F, Delle Fave G, Öberg K. Interferon-alpha and somatostatin analog in patients with gastroenteropancreatic neuroendocrine carcinoma: single agent or combination? Ann Oncol 2007, 18: 13-9.
- Faiss S, Pape UF, Bohming M, et al. Prospective, randomised, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors. The International Lanreotide and Interferon Alfa Study Group. J Clin Oncol 2003, 21: 2689-96.
- Kolby L, Persson G, Franzen S, Ahren B. Randomised clinical trial of the effect of interferon α on survival in patients with disseminated midgut carcinoid tumours. Br J Surg 2003, 90: 687–93.
Scheda interferone α-2b ricombinante
Franco Grimaldi
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria Santa Maria della Misericordia di Udine
Meccanismo d’azione
- Effetto anti-proliferativo: blocco del ciclo cellulare nella fase G0 e G1.
- Inibizione fattori di crescita: induzione di 2’5 A sintetasi.
- Stimolazione del sistema immunitario: induzione di antigeni di classe I e stimolazione delle cellule natural killer.
- Induzione di reazioni fibrotiche
- Induzione dell’angiogenesi.
Farmaci disponibili, via di somministrazione e posologia
IntronA soluzione (Introna). La soluzione è disponibile in diverse formulazioni e dosaggi:
- flaconcino monodose di soluzione iniettabile: 3 milioni UI, 5 milioni UI, 10 milioni UI, 18 milioni UI, 25 milioni UI;
- penna multidose: 18 milioni UI, 30 milioni UI, 60 milioni UI.
Indicazioni
Trattamento di tumori carcinoidi con linfonodi o metastasi epatiche e con "sindrome da carcinoide".
Il trattamento deve essere iniziato da un medico esperto nel trattamento della patologia. La dose usuale è di 5 milioni UI (da 3 a 9 milioni UI) somministrata tre volte la settimana (a giorni alterni) per via sottocutanea. Per gli schemi posologici di mantenimento, a discrezione del medico è consentita la somministrazione attuata direttamente dal paziente.
Pazienti in stadio avanzato possono richiedere dosi giornaliere di 5 milioni UI.
Il trattamento deve essere temporaneamente sospeso durante e dopo intervento chirurgico.
In caso di risposta del paziente, la terapia con α-interferone deve essere protratta sino a progressione.
Controindicazioni
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Grave patologia cardiaca pre-esistente (es. scompenso cardiaco congestizio non controllato, infarto miocardico recente, gravi aritmie).
Grave disfunzione renale o epatica, compresa quella causata da metastasi.
Epilessia e/o funzionalità compromessa del sistema nervoso centrale.
Pre-esistenti gravi malattie psichiatriche o anamnesi positiva di gravi disturbi psichiatrici.
Epatite cronica in presenza di cirrosi epatica scompensata.
Epatite cronica in pazienti contemporaneamente o recentemente trattati con agenti immuno-soppressivi, salvo nel caso di interruzione di una terapia corticosteroidea a breve termine, epatite autoimmune o anamnesi positiva di malattia autoimmune; pazienti trapiantati immuno-depressi.
Patologia tiroidea pre-esistente, salvo quando controllata con terapia convenzionale.
Effetti collaterali
Nei primi giorni di trattamento: sintomi simil-influenzali (spossatezza, mialgie) ben controllabili con i comuni rimedi sintomatici.
Sintomi di maggiore gravità, legati al prolungamento della terapia, sono connessi alla comparsa di sindrome da affaticamento cronico, anoressia, calo ponderale in circa il 50% dei pazienti; in una percentuale inferiore possono comparire anemia, leucopenia, trombocitopenia, elevazione degli indici di citolisi epatica e manifestazioni allergiche o autoimmuni (tiroiditi con iper o ipotiroidismo).
Precauzioni d’uso
Se durante il trattamento con IntronA si manifestano eventi avversi, per qualsiasi indicazione modificare il dosaggio o sospendere temporaneamente la terapia fino a scomparsa di tali effetti. Nel caso di intolleranza persistente o ricorrente, nonostante l'adeguato aggiustamento posologico, o di progressione della malattia, sospendere il trattamento.
Se durante il trattamento con interferone α2b insorgono gravi eventi avversi, in particolare se i granulociti diminuiscono fino a < 500/mm3 o i livelli di ALT/AST aumentano di almeno 5 volte il limite superiore normale, sospendere temporaneamente il trattamento, fino a risoluzione dell'evento. Il trattamento con interferone α2b deve poi ricominciare al 50% del dosaggio precedente. Se dopo l'aggiustamento della dose, l'intolleranza persiste, o se i granulociti diminuiscono a < 250/mm3 o i livelli di ALT/AST aumentano di almeno 10 volte il limite superiore normale, sospendere la terapia.
Limitazioni prescrittive
Prescrivibile con piano terapeutico.
Inibitori di mTOR nella terapia dei NET
Gabriele Luppi e Fabio Gelsomino
Oncologia Medica, Azienda Ospedaliero-Universitaria di Modena
(aggiornato al 19 marzo 2017)
La via di segnale mTOR
Mammalian target of rapamycin (mTOR) è una serina-treonina chinasi intra-citoplasmatica, che ha un ruolo fondamentale nei processi di regolazione della proliferazione e della crescita cellulare. Fu identificata nel 1994 come bersaglio dell’antibiotico rapamicina, che, legandovisi attraverso un complesso proteico inibitorio (chiamato FKBP12), ne blocca l’attività (1).
mTOR fa parte della via di PI3K/AKT, la cui attivazione si traduce nell’aumentata espressione di proteine fondamentali nel regolare la progressione del ciclo cellulare e la proliferazione (2).
In condizioni fisiologiche mTOR viene attivata, attraverso le chinasi a monte PI3K-AKT, quando le condizioni dell’ambiente extra-cellulare sono favorevoli, ovvero in presenza di ampia disponibilità di glucosio, aminoacidi, ossigeno e fattori di crescita. Quando la disponibilità di tali sostanze è bassa, mTOR viene inibita. L’attivazione avviene prevalentemente in maniera indiretta, attraverso l’inibizione di fattori “upstream” normalmente inibenti mTOR (quindi attraverso il blocco dell’inibizione di mTOR), quali le proteine tuberous sclerosis complex 1 e 2 (TSC1 e TSC2) e PTEN (Phosphatase and Tensin homolog deleted on chromosome ten) (3).
In diverse neoplasie umane questa via di trasduzione del segnale è attivata attraverso vari meccanismi, come mutazioni attivanti in alcune chinasi, la perdita di proteine inibitorie (ad esempio PTEN) o l’overespressione di fattori di crescita come IGF-1 (insulin growth factor 1) o VEGF (vascular endothelial growth factor) (4-5). Questo è particolarmente vero nei tumori neuroendocrini (NET): a riprova di ciò, un’analisi del profilo di espressione genica di una casistica di pNET (NET pancreatici) ha mostrato una ridotta espressione di due geni importanti nella via di mTOR: TSC2 e PTEN (6-9).
Everolimus nei tumori neuroendocrini
Considerato il ruolo chiave di mTOR nel controllo della crescita neoplastica, della proliferazione e dell’angiogenesi, l’inibizione di mTOR ha assunto un importante ruolo nella strategia terapeutica anti-neoplastica. In particolare, la rapamicina (sirolimus) è un antibiotico della famiglia dei macrolidi che lega la proteina citosolica FKBP-12, la quale interagisce con il complesso di mTOR, bloccandone la trasduzione del segnale a valle (10). Gli inibitori di mTOR, tutti derivati della rapamicina, sono everolimus, temsirolimus e deforolimus. Nei NET everolimus è di gran lunga l’agente più diffusamente studiato nei trial clinici.
In studi pre-clinici everolimus ha dimostrato attività immuno-soppressiva (tramite inibizione della proliferazione linfocitaria) e attività anti-tumorale, sia tramite inibizione diretta delle cellule neoplastiche sia attraverso un’azione anti-angiogenetica (11-15).
La sperimentazione clinica di everolimus, oltre ad essere sostenuta dal forte razionale biologico precedentemente descritto, si basa su evidenze pre-cliniche raccolte in modelli cellulari e murini (16), che ne hanno dimostrato l’attività anti-proliferativa e di controllo sulla crescita tumorale.
Una combinazione terapeutica molto interessante e promettente è quella che vede everolimus associato a octreotide. Il razionale di tale combinazione si basa sulla dimostrazione che l’inibizione di mTOR potrebbe essere superata dalla riattivazione della stessa via attraverso Akt (upstream di mTOR), che verrebbe attivata attraverso un aumento dell’espressione del recettore di IGF-1 (17). Sia la somatostatina sia i suoi analoghi, tra cui octreotide, sono in grado di diminuire l’attivazione della via PI3K/Akt/mTOR sia in modo diretto che indiretto. Nel primo caso il cross-talk intracellulare tra le vie attivate a livello della membrana cellulare da parte dei recettori della somatostatina si traduce in segnali che controllano negativamente l’attivazione di mTOR. D’altra parte, l’effetto inibitorio della somatostatina e degli analoghi derivati sul rilascio di fattori di crescita ha un effetto additivo sullo spegnimento delle vie di trasduzione del segnale che convergono su mTOR.
Nell’ambito del principale programma di ricerca su everolimus nei NET, denominato RADIANT (The RAD001 in Advanced Neuroendocrine Tumors), lo sviluppo clinico di questo farmaco è stato portato avanti senza mai escludere la possibilità di combinarlo con gli analoghi della somatostatina ed alcune evidenze cliniche (come quelle degli studi RADIANT-1 e RADIANT-2) suggeriscono che l'efficacia della combinazione dei due agenti possa essere superiore all’uso del singolo agente.
Con questi presupposti è stato condotto il primo studio di fase II, che ha dimostrato l’attività di everolimus in pazienti con NET avanzati, pubblicato da Yao nel 2008 (18). In questo trial pazienti con NET di grado basso o intermedio, metastatici o localmente avanzati, sono stati trattati con octreotide LAR (30 mg intramuscolo ogni 28 giorni) ed everolimus (5 o 10 mg/die). Erano permessi precedenti trattamenti chirurgici, chemioterapici o con octreotide. I pazienti inclusi nel trial erano 60 (30 con carcinoidi e 30 con pNET). Vi sono state 13 risposte parziali (22%) e 42 pazienti con stabilità di malattia (70%). La sopravvivenza mediana libera da progressione è stata di 60 settimane.
Un secondo trial di fase II (denominato RADIANT-1, il primo del progetto di sviluppo RADIANT) con everolimus 10 mg/die è stato condotto in pazienti affetti da pNET avanzati in progressione dopo chemioterapia (19). I pazienti sono stati stratificati sulla base del trattamento o meno con octreotide: nel caso in cui i pazienti fossero in terapia con l’analogo della somatostatina al momento dell’arruolamento nello studio, questo veniva continuato, in associazione ad everolimus. Nei pazienti trattati con solo everolimus è stato osservato un tasso di risposte parziali del 9.6% e di stabilizzazioni di malattia del 67.8%, mentre nei pazienti trattati con everolimus in combinazione con octreotide le risposte parziali e le stabilizzazioni sono state rispettivamente del 4.4% e 80%. La progression-free survival mediana (mPFS) è stata di 9.7 mesi nei pazienti trattati con solo everolimus e 16.7 mesi nel braccio di combinazione, suggerendo un importante effetto di stabilizzazione di malattia ottenuto dall’associazione dei due farmaci, sebbene lo studio non fosse stato disegnato per dimostrare una superiorità della combinazione rispetto alla monoterapia. Come già precedentemente accennato, il razionale alla base di questa combinazione si basa sul fatto che octreotide ha dimostrato di ridurre i livelli di IGF-1 in pazienti con cancro e che la cascata di segnale determinata da IGF-1 è un potenziale meccanismo di resistenza a everolimus. Nello studio in oggetto è stato inoltre osservato che pazienti con precoci risposte biochimiche, valutate mediante normalizzazione o riduzione di almeno il 30% dei livelli di cromogranina A (CgA) e di enolasi neuro-specifica (NSE) dopo 4 settimane di trattamento, avevano una PFS maggiore rispetto a coloro in cui non si verificava tale risposta biochimica precoce.
L’obiettivo del trial di fase III multicentrico RADIANT-2 era quello di valutare l’efficacia di everolimus in aggiunta ad octreotide LAR in pazienti con NET avanzati con sindrome da carcinoide (20). 429 pazienti sono stati randomizzati a ricevere everolimus 10 mg o placebo in combinazione con octreotide LAR 30 mg intramuscolo ogni 28 giorni. I tumori neuroendocrini più rappresentati erano quelli del piccolo intestino (52%), seguiti da quelli polmonari (10%), mentre il 6% dei pazienti in studio aveva un pNET. Tutti i pazienti avevano NET sindromici bene o moderatamente differenziati. La mPFS è stata di 16.4 mesi nel gruppo trattato con everolimus rispetto agli 11.3 mesi nei pazienti trattati con placebo, con una riduzione del rischio di morte del 23% (HR 0.77). Questo risultato, sebbene clinicamente rilevante, non è statisticamente significativo (p=0.026 solo di poco inferiore al limite predefinito di 0.024), anche in conseguenza di significativi sbilanciamenti tra i 2 bracci a sfavore del braccio con everolimus e di incongruenze tra la valutazione radiologica centralizzata rispetto alla valutazione radiologica locale, che hanno portato ad una perdita di potenza dello studio. Analisi di sottogruppo condotte da Anthony e colleghi (21) hanno evidenziato un miglioramento della mPFS sia tra i pazienti che avevano ricevuto analoghi della somatostatina prima dell’arruolamento che tra quelli naive per gli stessi analoghi (in ogni caso nessun sottogruppo ha raggiunto la significatività statistica).
Lo studio di fase III multicentrico randomizzato in doppio cieco RADIANT-3 (22) ha confrontato everolimus 10 mg/die in associazione alla migliore terapia di supporto (best supportive care, BSC) con placebo e BSC in pazienti affetti da pNET avanzato di grado basso o intermedio, in progressione radiologica nei 12 mesi precedenti la randomizzazione. Endpoint primario dello studio era la PFS, mentre obiettivi secondari erano la ORR (overall response rate), la durata della risposta, la sopravvivenza globale e il profilo di sicurezza. Il disegno dello studio prevedeva la possibilità di cross-over per i pazienti randomizzati a placebo nella fase di progressione di malattia. La PFS mediana è stata di 11.4 mesi con everolimus e 5.4 mesi con placebo, con una riduzione del rischio di morte del 66% (HR 0.34). Il beneficio si è mantenuto in tutti i sottogruppi. Anche i risultati in termini di risposta hanno dimostrato l’efficacia di everolimus, con 5% di risposte obiettive (vs 2% con placebo) e 73% di stabilizzazione di malattia (vs 51% con placebo). I risultati di questo trial hanno portato all’approvazione a livello europeo di everolimus nei pNET in progressione. Recentemente è stata pubblicato l’aggiornamento sulla sopravvivenza globale di questo trial, che ha mostrato una sopravvivenza mediana di 44 mesi, la più lunga mai riportata in uno studio di fase III nei pNET avanzati in progressione. Everolimus ha mostrato un vantaggio di sopravvivenza di 6.3 mesi rispetto al placebo, che tuttavia non ha raggiunto la significatività statistica, verosimilmente a causa dell’effetto confondente dell’ampio cross-over (85% dei pazienti inizialmente randomizzati a BSC hanno poi ricevuto everolimus) (23).
Lo studio RADIANT-4 è un trial di fase III in doppio cieco che ha randomizzato pazienti con tumori neuroendocrini bene o moderatamente differenziati di origine polmonare o gastrointestinale (non pancreatici) non funzionanti, avanzati, in progressione, a ricevere everolimus 10 mg versus placebo. In questo trial l’utilizzo concomitante degli analoghi della somatostatina veniva concesso solo in caso di sindrome da carcinoide emergente, non altrimenti controllabile con farmaci sintomatici. Everolimus ha pertanto dimostrato un prolungamento della mPFS (11 mesi vs 3.9 mesi) rispetto al placebo, con una riduzione del rischio di progressione o morte del 52%. I risultati di questo trial potranno portare ad estendere l’indicazione di everolimus al trattamento di tumori neuroendocrini polmonari o di origine gastrointestinale non funzionanti in progressione (24).
Ad oggi non è chiara quale sia la sequenza di trattamento ideale per i pazienti affetti da NET. Tuttavia, in uno studio retrospettivo multicentrico italiano con 169 pazienti trattati con everolimus (85 pNET + 84 non-pNET), il pretrattamento con terapia radiorecettoriale (PRRT) o chemioterapia ha portato ad un incremento di 12 volte del rischio di eventi avversi di grado 3-4 (25). Questo suggerisce pertanto l’importanza di una pianificazione terapeutica strategica in questi pazienti, in una valutazione globale che tenga conto dei trattamenti precedenti e dei rischi di tossicità.
Inoltre al congresso ENETS 2015 sono stati presentati i dati dello studio di fase 2 COOPERATE-2, in cui i pazienti con pNET avanzato in progressione venivano randomizzati a ricevere everolimus vs la combinazione dello stesso con pasireotide, un analogo multi-target della somatostatina. Tale studio non ha raggiunto l’endpoint primario della PFS.
Un altro trial interessante è il CALGB 80701, presentato al congresso ASCO 2015. Si trattava di uno studio randomizzato di fase II in pazienti con pNET localmente avanzato o metastatico, che confrontava everolimus 10 mg/die vs la combinazione di everolimus 10 mg/die e bevacizumab 10 mg/kg ogni 2 settimane. In tale studio il braccio di combinazione ha mostrato un vantaggio non statisticamente significativo in termini di PFS (16.7 vs 14 mesi), con un incremento significativo delle risposte obiettive (31% vs 12%), ma anche un aumento della frequenza degli eventi avversi di grado 3 (diarrea, iposodiemia, ipofosfatemia, proteinuria e ipertensione) (26).
Profilo di tossicità
In base ai dati derivati dagli studi registrativi dei NET (20,22), il profilo di tollerabilità di everolimus appare alquanto accettabile. Gli eventi avversi di grado 3-4, in accordo con la classificazione National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI-CTC AE) versione 4.0, sono poco frequenti (≤ 7%). Per effetti collaterali vedi capitolo relativo.
Prospettive future
Sulla base dei risultati positivi degli studi RADIANT, sono in corso diversi studi che stanno testando il migliore posizionamento di everolimus nella terapia dei NET. Particolarmente interessante è lo studio SEQTOR, che randomizza pazienti con pNET avanzato ben differenziato in progressione a ricevere la sequenza streptotozocina + 5-FU seguita da everolimus alla progressione o la sequenza inversa. Altro aspetto interessante che viene attualmente valutato in uno studio di fase II è il ruolo di everolimus nella terapia di mantenimento dei carcinomi neuroendocrini metastatici di origine polmonare o gastroenteropancreatica (studio MAVERIC).
Bibliografia
- Brown EJ, Albers MW, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369: 756-8.
- Nourse J, Firpo E, Flanagan WM, et al. Interleukin-2-mediated elimination of the p27 Kip1 cycline-dependent kinase inhibitor prevented by rapamycin. Nature 1994, 372: 570-3.
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004, 18: 1926-45.
- Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6: 91-9.
- Li Y, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast and prostate cancer. Science 1997, 257: 1943-7.
- Von Wichert G, Jehle PM, Hoeflich A, et al. Insulin-like growth factor-1 is an autocrine regulator of chromogranin A secretion and growth in human neuroendocrine tumor cells. Cancer Res 2000, 60: 4573-81.
- Lopez T, Hanahan D. Eleveted levels of IGF-1 receptor convey invasive and metastatic capability in a mouse model of pancreatic islet tumorigenesis. Cancer Cell 2002, 1: 339-53.
- Terris B, Scoazec JY, Rubbia L, et al. Espression of vascular endothelial growth factor in digestive neuroendocrine tumour. Histopathology 1998, 32: 133-8.
- Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: exspression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol 2010, 28: 245-55.
- Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev 2001, 15: 807–26.
- Nashan B. Early clinical experience with a novel rapamycine derivative. Ther Drug Monit 2002, 24: 53-8.
- Eastone JB, Houghton PJ. MTOR and cancer therapy. Oncogene 2006, 25: 6436-46.
- Boulay A, Rudloff J, et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res 2005, 11: 5319-28.
- Haritunians T, Mori A, et al. Antiproliferative activity of RAD001 (everolimus) as a single agent and combined with other agents in mantle cell lymphoma. Leukemia 2007, 21: 333-9.
- Shinoara ET, Cao C, et al. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 2005, 24: 5414-22.
- Capurso G, Fazio N, et al. Molecular target therapy for gastroenteropancreatic endocrine tumours: biological rationale and clinical perspectives. Rev Oncol Hematol 2009, 72: 110-24.
- Pollak MN, Polychronolakos C, et al. Somatostatin analogue SMS 201-995 reduces serum IGF-1 levels in patients with neoplasms potentially dependent on IGF-1. Anticancer Res 1989, 9: 889-91.
- Yao JC, Phan AT, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol 2008, 26: 4311-8.
- Yao JC, Lombard-Bohas C, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J Clin Oncol 2010, 28: 69-76.
- Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomized, placebo-controlled, phase 3 study. Lancet 2011, 378: 2005-12.
- Anthony LB PM, Hainsworth JD, et al. Everolimus plus octreotide LAR versus placebo plus octreotide LAR in patients with advanced neuroendocrine tumors (NET): Effect of prior somatostatine analog therapy on progression-free survival in the RADIANT-2 trial. J Clin Oncol 2011 (Suppl 29): 4078.
- Yao JC, Shah MH, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 514-23.
- Yao JC, Pavel M, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III. RADIANT-3 Study. J Clin Oncol 2016, 34: 3906-13.
- Yao JC, Fazio N, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 2016, 387: 968-77.
- Panzuto F, Rinzivillo M, et al. Real-world study of everolimus in advanced progressive neuroendocrine tumors. Oncologist 2014, 19: 966-74.
- Kulke MH, Niedzwiecki D, et al. Randomized phase II study of everolimus (E) versus everolimus plus bevacizumab (E+B) in patients (Pts) with locally advanced or metastatic pancreatic neuroendocrine tumors (pNET), CALGB 80701 (Alliance). J Clin Oncol 2015, 33 suppl: abstr 4005.
Scheda everolimus
Gabriele Luppi e Fabio Gelsomino
Oncologia Medica, Azienda Ospedaliero-Universitaria di Modena
Meccanismo d’azione
Everolimus è un inibitore orale selettivo di mTOR (mammalian target of rapamycin), una serina-treonina-chinasi intra-citoplasmatica che ha un ruolo importante nella regolazione della proliferazione e della crescita cellulare. Everolimus, come il suo analogo rapamicina, si lega a mTOR, tramite la proteina intra-cellulare FKBP-12, bloccando il complesso mTORC1 e quindi la sintesi proteica mediata dalle proteine S6K1 e 4E-BP1. In studi preclinici everolimus ha dimostrato attività immuno-soppressiva tramite inibizione della proliferazione linfocitaria, e attività anti-tumorale, sia tramite inibizione diretta delle cellule neoplastiche sia attraverso un’azione anti-angiogenetica.
Preparazioni, via di somministrazione, posologia
- Compresse da 0.25 mg (Certican), 0.75 mg (Certican), 2.5 mg (Afinitor), 5 mg (Afinitor, everolimus EthyPharm, everolimus Medac, everolimus Sandoz), e 10 mg (Afinitor, everolimus EthyPharm, everolimus Medac, everolimus Sandoz)
- Compresse dispersibili 0.25 mg (Certican), 2 mg (Votubia), 3 mg (Votubia).
Deve essere somministrato per via orale una volta al giorno alla stessa ora, regolarmente con o senza cibo. Le compresse devono essere inghiottite intere con un bicchiere d’acqua e non devono essere masticate o frantumate.
La dose raccomandata di everolimus è di 10 mg una volta al giorno. Il trattamento deve continuare fino a quando si osserva un beneficio clinico o finché non compaia tossicità inaccettabile.
Indicazioni
Carcinoma renale: trattamento di pazienti con carcinoma renale avanzato, che hanno presentato progressione durante o dopo trattamento con terapia mirata anti-VEGF.
Carcinoma mammario avanzato: trattamento del carcinoma mammario avanzato con stato recettoriale ormonale positivo, HER2/neu negativo, in combinazione con exemestane, in donne in post-menopausa in assenza di malattia viscerale sintomatica dopo recidiva o progressione a seguito di trattamento con un inibitore dell’aromatasi non steroideo.
Tumori neuroendocrini pancreatici: trattamento di tumori neuroendocrini di origine pancreatica, bene o moderatamente differenziati, non operabili o metastatici, in progressione di malattia, negli adulti.
Contro-indicazioni
Ipersensibilità al principio attivo, ad altri derivati della rapamicina o ad uno qualsiasi degli eccipienti.
Precauzioni d'uso
Per i pazienti con moderata compromissione epatica (Child-Pugh class B), la dose deve essere ridotta a 5 mg/die. Everolimus non è stato valutato in pazienti con compromissione epatica grave (Child-Pugh class C) e non è raccomandato per l’uso in questa categoria di pazienti.
Non sono necessarie riduzioni di dose in pazienti con ridotta funzione renale o in pazienti anziani.
Particolare attenzione va posta alle possibili interazioni farmacologiche. Everolimus è un substrato del CYP3A4, e anche un substrato e un moderato inibitore della PgP. Pertanto l’assorbimento e la successiva eliminazione di everolimus possono essere influenzati da sostanze che interferiscono con il CYP3A4 e/o la PgP (vedere le interazioni farmacologiche riportate nella scheda tecnica). Anche alcuni cibi, come il succo di pompelmo, possono aumentare le concentrazioni di everolimus, in quanto inibitori del CYP3A4 e della PgP.
Effetti collaterali
In base ai dati derivati dagli studi RADIANT 2 e RADIANT 3, il profilo di tollerabilità di everolimus appare alquanto accettabile. Gli eventi avversi di grado 3-4, in accordo con la classificazione National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI-CTC AE)- versione 4.0, sono poco frequenti (≤ 7%). Gli effetti collaterali più comuni sono rappresentati da: stomatite, astenia, diarrea, rash cutaneo, prurito, anoressia, nausea, vomito, perdita di peso, alterazione di alcuni parametri di laboratorio (iperglicemia, ipertrigliceridemia, ipercolesterolemia, anemia, piastrinopenia). Un evento avverso potenzialmente serio è la polmonite asettica (12% dei pazienti nel RADIANT-2 e 17% dei pazienti del RADIANT-3, ma raramente di grado severo), caratterizzata dal quadro clinico-radiologico della polmonite interstiziale, associata o meno a segni e sintomi quali versamento pleurico, ipossia, tosse, dispnea e malessere.
Limitazioni prescrittive
La prescrizione è limitata a pazienti adulti. Everolimus non è raccomandato in corso di gravidanza o in donne in età fertile che non usano contraccettivi. Le donne in trattamento con Everolimus non devono allattare al seno.
Ai fini delle prescrizioni a carico del Servizio sanitario nazionale (classe H), i centri utilizzatori dovranno compilare la scheda raccolta dati informatizzata di arruolamento che indica i pazienti eleggibili e la scheda di follow-up e applicare le condizioni negoziali secondo le indicazioni pubblicate sul sito. La risposta deve essere rivalutata a 6 mesi: in assenza di risposta, il farmaco non è più prescrivibile a carico SSN.
Inibitori dell'angiogenesi nella terapia dei NET
Giuseppe Badalamenti
Oncologia medica, Policlinico Universitario P. Giaccone, Palermo
(aggiornato al 18 marzo 2017)
Introduzione
Nell’ambito delle terapie target, i farmaci registrati nelle NEN sono everolimus (EVE, inibitore di mTOR) e sunitinib (SUN, inibitore tirosin-kinasico TKI). Entrambi sono attualmente indicati e registrati nel trattamento delle NEN del pancreas (pNEN) metastatiche o localmente avanzate, avendo determinato un vantaggio statisticamente significativo in termini di Progression Free Survival (PFS, endpoint primario) rispetto al placebo (1,2). SUN, già registrato in altre neoplasie, quali il carcinoma del rene metastatico e i GIST (Gastro-Intestinal-Stromal-Tumors), a differenza di everolimus, è un potente anti-angiogenico.
La neo-angiogenesi tumorale è il processo biologico di formazione e sviluppo di nuovi vasi ematici intra-tumorali ed è essenziale per lo sviluppo e la sopravvivenza del tumore e per il processo di metastatizzazione a distanza. Il processo è mediato da una serie di fattori di crescita, che vengono massivamente prodotti e secreti dalle cellule neoplastiche, dalle cellule infiammatorie reclutate e dalle cellule stromali. I fattori di crescita, una volta rilasciati, attivano la formazione di nuovi vasi ematici, reclutando dai vasi adiacenti i periciti, precursori delle cellule endoteliali, o favorendo la gemmazione dei vasi sanguigni pre-esistenti da bottoni endoteliali (3).
I fattori di crescita maggiormente coinvolti nella neo-angiogenesi sono il bFGF e soprattutto il VEGF, che ha assunto anche una rilevante importanza clinica nella terapia di diverse neoplasie. Il VEGF agisce legandosi al proprio recettore VEGF-R2, non solo localmente sulle cellule endoteliali adiacenti, ma anche a livello del midollo osseo, mobilizzando i precursori degli endoteliociti e favorendo successivamente la proliferazione cellulare e il differenziamento degli stessi. Le metalloproteasi, le angiopoietine, il PDGF (rilasciato dagli stessi endoteliociti attivati per la neo-angiogenesi) e il TGF-β stabilizzano infine i nuovi vasi. La neo-angiogenesi tumorale viene stimolata dal cosiddetto switch angiogenetico, che si verifica quando l’ipossia intra-tumorale, indotta dallo squilibrio tra le dimensioni del tumore e la capacità di nutrizione dello stesso, stimola la produzione di HIF-1, un fattore di trascrizione indotto dall’ipossia, il quale deregola la produzione di VEGF, permettendo alla cellula tumorale di acquisire il cosiddetto “fenotipo neo-angiogenetico” (4).
Sunitinib
SUN inibisce molteplici recettori delle tirosin-chinasi (RTK), che sono coinvolte nella crescita tumorale, nella neo-angiogenesi e nella metastatizzazione. SUN è un inibitore dei recettori del fattore di crescita di derivazione piastrinica (PDGFRα e PDGFRβ), dei recettori del fattore di crescita vascolare endoteliale (VEGFR1, VEGFR2 e VEGFR3), del recettore del fattore della cellula staminale (KIT), del recettore tirosin-chinasico FLT3 (Fms-like tyrosine kinase 3), del recettore CSF-1R (colony stimulating factor receptor CSF-1R) e del recettore del fattore neutrofico di derivazione gliale (RET).
I risultati dello studio di fase III randomizzato hanno portato all’approvazione della terapia con SUN nei pNET non operabili, localmente avanzati o metastatici. Nello studio registrativo a due bracci, controllato in doppio cieco, i pazienti con pNEN avanzata, ben differenziata, in progressione radiologica nei 12 mesi precedenti secondo criteri RECIST, sono stati randomizzati (1:1) a ricevere 37.5 mg di sunitinib una volta al giorno senza un periodo programmato di sospensione (n = 86) o placebo (n = 85). L’obiettivo primario dello studio era la valutazione della PFS, endpoint secondari erano la OS (Overall Survival), la ORR (Overall Response Rate) e la sicurezza.
I 2 gruppi di pazienti erano ben bilanciati. Inoltre, il 49% dei pazienti trattati con SUN e il 52% dei pazienti in placebo avevano tumori non funzionanti e in entrambi i bracci il 92% aveva metastasi epatiche. Nello studio era consentito l’utilizzo di analoghi della somatostatina.
Lo studio è stato interrotto in anticipo, nel 2009, poiché un’analisi non pre-pianificata dell’independent data and safety monitoring board ha trovato una differenza statisticamente significativa in termini di PFS. Ai pazienti del braccio placebo in progressione di malattia veniva permesso di ricevere SUN in uno studio open-label separato, come estensione del protocollo.
Il PFS è risultato di 11.4 mesi nel braccio SUN vs 5.5 mesi nel braccio placebo (HR 0.42, IC95% 0.26–0.66, P < 0.001). In altre parole SUN ha prodotto un prolungamento della PFS nel 68% dei pazienti. Il tasso di RR per SUN è stato di 9.3% (IC95% 3.2–15.4).
I più comuni eventi avversi di grado 3-4 sono stati neutropenia (12%), ipertensione (10%), eritrodisestesia palmo-plantare (6%), diarrea (5%), astenia (5%), dolore addominale (5%), stomatite (4%) e trombocitopenia (4%).
In considerazione della chiusura anticipata dello studio, l’FDA ha commissionato uno studio di fase IV post-marketing, onde acquisire maggiori dati di efficacia e tolleranza, in particolare in prima linea e per meglio studiare le metodiche di valutazione della risposta tumorale. Lo studio, che prevedeva l’assunzione di SUN al dosaggio di 37.5 mg/die per os, è stato completato con 85 pazienti.
Everolimus e Sunitinib: aspetti degli studi registrativi
Entrambi gli studi erano randomizzati 1:1 verso placebo. Il numero totale di pazienti nel braccio di trattamento è stato 410 nello studio con EVE e 171 nello studio con SUN. Il 50% dei pazienti dello studio EVE e il 36% dello studio SUN avevano ricevuto SSA prima dell’inizio dello studio, e 40% e 28% rispettivamente hanno ricevuto SSA durante lo studio. I criteri di inclusione erano pressochè sovrapponibili, con qualche piccola differenza: progressione radiologica entro l’ultimo anno nello studio con EVE e progressione radiologica RECIST nello studio con SUN; tumori ben/moderatamente differenziati nello studio con EVE e ben differenziati nello studio con SUN. I risultati in termini di PFS tra i due studi sono sovrapponibili. In termini di sopravvivenza, nello studio EVE, in relazione al disegno dello studio che prevedeva il cross-over dal braccio placebo al braccio trattamento in caso di progressione, l’endpoint sopravvivenza è risultato non significativo. Per quanto riguarda SUN, i dati riportati nello studio registrativo riguardo al beneficio sulla sopravvivenza nei pazienti trattati rispetto a quelli che ricevevano placebo non sono stati confermati dalla successiva analisi ottenuta con il prolungamento del follow-up.
Considerazioni
L’avvento delle terapie target nel trattamento delle pNEN ha sicuramente contribuito a migliorare notevolmente l’aspettativa di vita dei nostri pazienti. Tuttavia, ancora tanto dobbiamo fare nella valutazione corretta dell’attività e dell’efficacia di questi farmaci, nonché nello studio delle sequenze di trattamento. L’attività di SUN nei pNET, valutata in termini di risposte obiettive, è < 10%. Generalmente in oncologia un farmaco si definisce attivo quando determina risposte obiettive in almeno il 25-30% dei casi. Questo significa che SUN non è un farmaco attivo in termini di risposte dimensionali di tipo RECIST, per cui non avrebbe senso il suo utilizzo in un setting neoadiuvante o pre-operatorio, dove l'obiettivo è la riduzione del diametro tumorale. Tuttavia, le cose non stanno così, perché è ormai noto che i classici criteri dimensionali RECIST non valutano correttamente la risposta e quindi l’attività dei farmaci targeted e questo è soprattutto vero nel caso di famaci anti-angiogenici. Infatti, se volessimo usare nella valutazione della risposta criteri più moderni di tipo funzionale (vedi i criteri CHOI nei GIST), la percentuale di risposte positive al trattamento aumenterebbe almeno del 10-20%, rendendo pertanto il farmaco attivo. Per tale motivo, nella valutazione della risposta al trattamento è utile affiancare alla classica TC con mezzo di contrasto e ai comuni criteri RECIST, l’utilizzo di imaging più funzionali, quale la TC in perfusione, la RM in diffusione e la CEUS (ecografia con mezzo di contrasto) (5). Queste indagini valutano precocemente una risposta positiva al trattamento in termini di riduzione della vascolarizzazione e predicono spesso una risposta radiologica di tipo RECIST. Questo è molto importante poiché la valutazione precoce della risposta potrebbe consentire di ridurre gli effetti collaterali e i costi di un trattamento non efficace.
Sicuramente una questione ancora aperta è la valutazione dell’efficacia del trattamento, in termini di OS o del suo surrogato PFS. Sia SUN che EVE hanno fallito nel dimostrare un aumento della sopravvivenza nei pazienti affetti da pNEN. Il disegno degli studi può esser il maggior responsabile di questo dato, ma personalmente ritengo che la OS non sia un parametro migliore della PFS per valutare l’efficacia in neoplasie di questo tipo, poco aggressive e suscettibili di diversi trattamenti in sequenza.
Dopodichè rimane il problema ancora più aperto delle sequenze terapeutiche nelle pNEN localmente avanzate o metastatiche, per le quali esistono diverse opzioni terapeutiche. In questo momento non sappiamo, per esempio, cosa utilizzare tra EVE e SUN e soprattutto quando utilizzare l’uno o l’altro. L'esperienza conferma che SUN può essere efficace in pazienti in progressione ad EVE, ma non è noto se valga il contrario. L’identificazione di biomarcatori predittivi di risposta all’uno o all’atro farmaco potrebbe rappresentare la chiave di volta (6). Nei due studi registrativi solo una parte dei pazienti è stata trattata in associazione con SSA e potenzialmente l’associazione dell’analogo, che agisce anche sulla neo-angiogenesi, con un farmaco anti-angiogenico come SUN potrebbe essere molto vantaggiosa rispetto al SUN in monoterapia.
Consideriamo infine che le pNEN, anche ben differenziate, rispondono molto bene a un trattamento chemioterapico, che sicuramente determina un maggior numero di riposte obiettive rispetto alle terapie target. Questo dato è da considerare attentamente nel caso in cui si programmi un trattamento neoadiuvante o nel caso di malattia fortemente sintomatica.
SUN al dosaggio di 37.5 mg/die continuativamente è generalmente ben tollerato. È molto importante conoscere bene e saper gestire gli effetti collaterali, motivando molto il paziente nell'assumere correttamente il farmaco. In Italia il farmaco è rimborsato solo da poco tempo, per cui l’esperienza nella real life in questo tipo di pazienti è ancora limitata. L’eritrodisestesia palmo-plantare, la diarrea e l’astenia sono effetti collaterali meno importanti per l’oncologo, ma sicuramente pesanti per il paziente, considerato anche la somministrazione continuativa del farmaco. L’eritrodisestesia palmo-plantare può essere in parte prevenuta con manicure e pedicure accurata in profilassi e con l’utilizzo di pomate specifiche. La diarrea può essere trattata con i farmaci convenzionali. La mielotossicità e l’ipertensione sono sicuramente gli effetti collaterali da trattare con più attenzione. È opportuno monitorare l’emocromo almeno ogni 15 giorni per i primi 2 mesi di trattamento e quindi prima di ogni ciclo. La neutropenia in genere si manifesta nel primo periodo di trattamento e diventa meno frequente nel tempo. Prima dell’inizio del trattamento deve essere effettuata una valutazione cardiologica con ECG ed ecocardiogramma, da ripetere in relazione alla clinica. È anche utile invitare il paziente a monitorare quotidianamente i valori pressori (7).
Nel caso in cui gli effetti collaterali non siano gestibili, è indicata la modificazione della posologia. In assenza di suggerimenti condivisi, la riduzione del dosaggio giornaliero o l’introduzione di giorni di pausa sono a discrezione del medico.
Prospettive future
Bevacizumab, un anticorpo anti-VEGF già registrato nel trattamento di diverse neoplasie, è stato utilizzato con risultati interessanti, da solo o in associazione. Uno studio di fase II ha arruolato pazienti con NEN ben differenziate trattate con SSA a ricevere bevacizumab o interferon peghilato α2b: bevacizumab ha mostrato superiorità di ORR e PFS (8,9).
Bevacizumab in associazione con temozolomide in pazienti con pNEN metastatico ha dimostrato un maggior numero di risposte obiettive, con miglioramento di PFS e OS (10).
In un recente studio di fase II in pNEN avanzate, everolimus e bevacizumab hanno mostrato un tasso di risposte obiettive pari al 26% e buona tollerabilità (11).
Pazopanib è un inibitore tirosin-chinasico orale (recettore VEGF 1, 2, 3). Due studi di fase II hanno mostrato una certa attività del farmaco non solo nei pNET, ma anche nei NET gastrointestinali (12,13).
Diversi altri agenti anti-angiogenici sono tuttora oggetto di studio, quali Famitinib (inibitore di c-kit, PDGFR, VEGFR2, VEGFR3, Flt1 e Flt3) (14), Regorafenib (inibitore di c-Raf, BRAF, VEGFR-1,2,3; PDGFRα, FGFR-1 c-kit, RET, Flt-3) (15) e Nintedanib (inibitore di VEGFR, FGFR, PDGFR) (16).
Bibliografia
- Yao JC, Shah MH, Ito T, et al; RAD001 in advanced neuroendocrine tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 514-23.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 501-13.
- Mittala K, Ebos J, Rinia Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin Oncol 2014, 41: 235–51.
- Cella CA, Minucci S, Spada F, et al. Dual inhibition of mTOR pathway and VEGF signalling in neuroendocrine neoplasms: from bench to bedside. Cancer Treat Rev 2015, 41: 754–60.
- Yao J, Phan A, Hess K, et al. Perfusion computed tomography as functional biomarker in randomized run-in study of bevacizumab and everolimus in well-differentiated neuroendocrine tumors. Pancreas 2015, 44: 190–7.
- De Dosso S, Grande E, Barriuso J, et al. The targeted therapy revolution in neuroendocrine tumors: in search of biomarkers for patient selection and response evaluation. Cancer Metastasis Rev 2013, 32: 465–77.
- Vallea JW, Faivreb S, Hubnerc RA, et al. Practical management of sunitinib toxicities in the treatment of pancreatic neuroendocrine tumors. Cancer Treat Rev 2014, 40: 1230–8.
- Yao JC, Phan AT, Hoff PM, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumour: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol 2008, 26: 1316-23.
- Faivre S, Sablin MP, Dreyer C, Raymond E. Novel anticancer agents in clinical trials for well-differentiated neuroendocrine tumours. Endocrinol Metab Clin North Am 2010, 39: 811-26.
- Chan JA, Stuart K, Earle CC, et al. Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J Clin Oncol 2012, 30: 2963-8.
- Yao JC, Phan AT, Fogleman D, et al. Randomized run-in study of bevacizumab (B) and everolimus (E) in low- to intermediate-grade neuroendocrine tumors (LGNETs) using perfusion CT as functional biomarker. J Clin Oncol 2010, 28 suppl 15: abstr 4002.
- Ahn HK, Choi JY, Kim KM, et al. Phase II study of pazopanib monotherapy in metastatic gastroenteropancreatic neuroendocrine tumours. Br J Cancer 2013, 109: 1414-9.
- Phan AT, Yao JC, Fogelman DR, et al. A prospective, multi-institutional phase II study of GW786034 (pazopanib) and depot octreotide (sandostatin LAR) in advanced low-grade neuroendocrine carcinoma (LGNEC). J Clin Oncol 2010, 28 suppl 15: abstr 4001.
- Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumour activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumour progression and angiogenesis. Cancer Res 2004, 64: 7099-109.
- Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumour activity. Int J Cancer 2011, 129: 245-55.
- Bill R, Fagiani E, Zumsteg A, et al. Nintedanib is a highly effective therapeutic for neuroendocrine carcinoma of the pancreas (PNET) in the Rip1Tag2 transgenic mouse model. Clin Cancer Res 2015, 21: 4856-67.
Scheda sunitinib
Giuseppe Badalamenti
Oncologia medica, Policlinico Universitario P. Giaccone, Palermo
Meccanismo d’azione
Sunitinib inibisce molteplici recettori delle tirosin-chinasi (RTK), coinvolte nella crescita tumorale, nella neo-angiogenesi e nella metastatizzazione: PDGFRα e PDGFRβ, VEGFR1, VEGFR2 e VEGFR3, KIT, FLT3, CSF-1R, RET.
Indicazioni
Trattamento delle NEN del pancreas (pNEN) metastatiche o localmente avanzate
Tumore stromale del tratto gastrointestinale (GIST)
Carcinoma renale metastatico
Contro-indicazioni
Ipersensibilità al principio attivo
Preparazioni, via di somministrazione, posologia
Cp 12.5 mg (sunitinib Accord, sunitinib Dr.Reddy's, sunitinib EG, sunitinib Mylan, sunitinib TEVA, sunitinib Zentiva, Sutent), 25 mg (sunitinib Accord, sunitinib Dr.Reddy's, sunitinib EG, sunitinib Mylan, sunitinib Sandoz, sunitinib TEVA, sunitinib Zentiva, Sutent), 37.5 mg (sunitinib Accord), 50 mg (sunitinib Accord, sunitinib Dr.Reddy's, sunitinib EG, sunitinib Mylan, sunitinib Sandoz, sunitinib TEVA, sunitinib Zentiva, Sutent)
Per k renale e GIST 50 mg/die, per pNEN 37.5 mg/die
Effetti collaterali
Mielotossicità
Ipertensione
Eritrodisestesia palmo-plantare, diarrea, astenia
Distiroidismo
Precauzioni d'uso
Monitorare l’emocromo almeno ogni 15 giorni per i primi 2 mesi di trattamento e quindi prima di ogni ciclo.
Prima dell’inizio del trattamento, valutazione cardiologica con ECG ed ecocardiogramma, da ripetere in relazione alla clinica.
Monitorare quotidianamente i valori pressori.
Nel caso in cui gli effetti collaterali non siano gestibili, modificare la posologia a discrezione del medico.
Attenzione alla cosomministrazione di ketoconazolo e rifampicina
Limitazioni prescrittive
Ricetta non ripetibile limitativa, distribuzione ospedaliera
Chemioterapia per i NET
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
(aggiornato al 15/10/2025)
Generalità
Il primo trattamento raccomandato per i NET localizzati, secondo le linee guida (LG) europee e americane, è la resezione chirurgica, indipendentemente dal grado tumorale. La chemioterapia può essere presa in considerazione nei NET G3 quando è necessaria una riduzione del tumore prima dell'intervento chirurgico, perchè potrebbe indurre la riduzione della massa tumorale, consentendo un intervento chirurgico successivo.
La terapia con analoghi della somatostatina resta il cardine per il trattamento iniziale dei NET G1-G2. Tuttavia, sono disponibili ulteriori opzioni terapeutiche per questo gruppo di NET, come everolimus (inibitore di mTOR), sunitinib (inibitore multi-chinasico con attività anti-angiogenica) e la terapia con radionuclidi recettoriali peptidici (PRRT).
Sebbene la chemioterapia venga utilizzata in casi selezionati di NET G1-G2, rappresenta attualmente il trattamento di prima scelta per i NET G3 a causa del loro comportamento aggressivo e dell'elevato tasso di proliferazione tumorale. Infatti, secondo le LG della European Neuroendocrine Tumor Society (ENETS), è raccomandato un trattamento cito-tossico sistemico nei NET avanzati in progressione o voluminosi localizzati nel pancreas o in altre sedi, quando è presente un Ki-67 elevato, una malattia rapidamente progressiva e dopo il fallimento di altre terapie se l'imaging SSTR è negativo.
I chemioterapici disponibili sono:
- gli alchilanti, come streptozocina (STZ), temozolomide (TMZ) e dacarbazina (DTIC);
- gli anti-metaboliti, come 5-fluoro-uracile (5-FU) e capecitabina (CAP);
- gli inibitori della topo-isomerasi, come etoposide e irinotecan;
- i derivati del platino, come oxaliplatino, carboplatino e cisplatino.
Solitamente gli schemi di chemioterapia prevedono l’associazione di due o più farmaci, al fine di potenziarne la risposta tumorale (1-5).
Regimi a base di streptozocina
La STZ, un antibiotico derivato dal batterio Gram-positivo Streptomyces, è stata studiata per la prima volta negli anni ‘80 in uno studio di fase II per il trattamento dei NET pancreatici da sola e in associazione a fluorouracile (5-FU), dimostrando come la terapia di associazione desse un tasso di risposta obiettiva maggiore rispetto alla monoterapia (ORR 63% vs 36%). Ulteriori studi sono disponibili sull’associazione di STZ e doxorubicina (DOXO) versus sia STZ da sola che combinazione STZ-5FU. Sebbene questa combinazione abbia dimostrato tassi di risposta superiori all’associazione STZ-5FU (ORR 69% vs 45%; sopravvivenza mediana 2.2 anni vs 1.4 anni), l’utilizzo è limitato per la grave tossicità gastro-intestinale.
Lo studio FAS ha indagato la combinazione di STZ-DOXO-5FU, riportando una sopravvivenza mediana libera da progressione (mPFS) di 20 mesi con sopravvivenza globale mediana (mOS) di 63 mesi.
L’utilizzo della STZ è stato studiato anche in tumori extra-pancreatici e in associazione con agenti anti-angiogenici, come il sunitinib o il bevacizumab, anticorpo monoclonale umanizzato contro il fattore di crescita endoteliale vascolare (VEGF). Nello studio di fase II BETTER, in 34 pazienti con NET del pancreas la combinazione di STZ-5-FU e bevacizumab ha ottenuto PFS mediana di 23.7 mesi, ORR del 56% (taso di controllo della malattia - DCR - 100%) e OS dell'88% a 2 anni (6-8).
Sono attualmente in corso diversi studi che valutano l’efficacia di STZ in associazione ad altri chemioterapici:
- lo studio SEQTOR (NCT 02246127) randomizzato di fase III, valuta l’efficacia della terapia sequenziale STZ–5FU seguita da everolimus o viceversa nei pazienti con NET pancreatici in progressione;
- lo studio BETTER-2 (NCT 03351296) randomizzato di fase II, confronta STZ+5FU ± bevacizumab vs CAP+TMZ ± bevacizumab in pazienti con NET pancreatici.
Regimi a base di temozolomide
La TMZ è un derivato alchilante della DTIC somministrato per via orale, che viene utilizzato nel trattamento dei NET da solo o in associazione ad altri farmaci come la CAP (CAPTEM). Anche se diversi agenti mirati come talidomide, bevacizumab o everolimus sono stati associati a TMZ, la combinazione di CAP e TMZ è forse quella con il maggior razionale pre-clinico. Si ritiene infatti che la CAP sia in grado di ridurre la produzione di MGMT, l'enzima responsabile della riparazione del danno al DNA causato dalla TMZ.
La combinazione CAPTEM ha riportato in vari studi, sia retrospettivi che prospettici, ORR variabile dal 33 al 70%, mPFS di 18-20 mesi e mOS di 25.3-75.2 mesi.
L’utilizzo di TMZ e CAPTEM è stato studiato anche in associazione ad altre terapie, come gli inibitori delle tirosin-chinasi o gli inibitori del check-point immunitario come nivolumab. Lo studio di fase II CONTROL NET ne ha valutato l’associazione con la PRRT in pazienti affetti da NET pancreatico: la PFS a 27 mesi era maggiore nei pazienti trattati con CAPTEM+PRRT rispetto ai pazienti trattati con solo CAPTEM (61.1% vs 33.3%; ORR 72.2% vs 33.3%).
Schemi alternativi di TMZ, come quello metronomico (somministrazione continuativa di chemioterapici a dosi significativamente al di sotto della dose massima tollerata, senza interruzioni tra i vari cicli), sono stati valutati in una serie di NET G2 e G3 di origine pancreatica e polmonare. Questo schema si è rivelato essere efficace e allo stesso tempo ben tollerato dai pazienti, soprattutto quelli con ridotto performance status (9-11).
Regimi a base di dacarbazina
L’utilizzo della DTIC è stato valutato in uno studio di fase II su pazienti con NET del pancreas, ottenendo ORR del 33% e OS mediana di 19.3 mesi. Nonostante i dati di efficacia, il profilo di tossicità sfavorevole e la mancanza di dati sulla differenziazione e sul grado dei tumori nei pazienti arruolati nello studio ne impediscono l'uso routinario nel trattamento dei NET.
Anche in uno studio di fase II condotto su pazienti affetti da carcinoidi, pur con risposta tumorale efficace, è stato riscontrato un alto tasso di eventi avversi, soprattutto gastro-intestinali come nausea e vomito, che ne limitano pertanto l’utilizzo (12).
Regimi a base di anti-metaboliti
Le combinazioni a base di oxaliplatino con 5-FU (FOLFOX), gemcitabina (GEMOX) o CAP (XELOX/CAPOX) hanno mostrato vari gradi di attività nei NET, soprattutto del tratto gastro-entero-pancreatico (GEP).
Uno studio retrospettivo su 78 pazienti con NET G2 sottoposti a chemioterapia a base di oxaliplatino ha osservata ORR del 26%, mPFS di 8 mesi e mOS di 32 mesi. Uno studio di fase II con XELOX condotto su 40 pazienti con NET (tra cui 11 con NET del pancreas ben differenziato) ha riportato ORR del 27%.
Ulteriori studi di associazione di FOLFOX o CAPOX con bevacizumab hanno evidenziato ORR del 41% e mPFS di 21 mesi nei pazienti con NET pancreatici.
L’associazione di 5-FU e irinotecan (FOLFIRI) ha ottenuto mPFS di 5.0-9.1 mesi e mOS di 15 mesi, ma con maggiore tossicità gastro-intestinale di grado severo. Per tale motivo, lo schema FOLFIRI è utilizzato spesso come alternativa solo dopo fallimento di altre opzioni terapeutiche (13-15).
Chemioterapia nei NEC
Rappresenta il principale approccio terapeutico per i NEC, che, pur risultando chemio-sensibili, presentano prognosi infausta con sopravvivenza mediana generalmente di 7.5 mesi circa. Le prove a supporto per l’utilizzo nei GEP-NEC G3 sono scarse e derivano da limitate casistiche retrospettive e da pochissimi piccoli studi clinici non controllati. Sulla base del loro ruolo consolidato nel microcitoma polmonare metastatico, l’associazione di cisplatino ed etoposide (EP) resta uno dei regimi più utilizzati nei pazienti affetti da NEC, con tassi di risposta che arrivano fino a circa il 30% e sopravvivenza mediana di circa un anno. Uno studio recente ha osservato una significativa associazione del Ki-67 con la risposta alla chemioterapia: i pazienti con Ki-67 > 55% hanno avuto tasso di risposta tumorale maggiore rispetto a quelli con valori di Ki-67 < 55% (42 vs 15%). Sulla base di questi dati, soprattutto in pazienti con Ki-67 > 55%, la prima linea di terapia è solitamente a base di cisplatino/carboplatino + EP. Alternativamente, in pazienti con Ki-67 < 55% vengono utilizzati maggiormente regimi a base di TMZ (da solo o in associazione con CAPE e bevacizumab). Dato il crescente utilizzo del trattamento a base di TMZ e la sua nota associazione con lo stato di MGMT in altri gruppi tumorali come il glioblastoma, studi clinici hanno indagato la relazione tra lo stato di MGMT e la terapia con alchilanti nei pazienti con NET. Lo stato di metilazione del promotore del gene MGMT è un importante fattore che indica la probabilità di migliore risposta al trattamento in conseguenza di una ridotta espressione dell’enzima.
La scelta della terapia più appropriata per i pazienti affetti da NEC, sulla base di studi che hanno messo in evidenza pari efficacia dei regimi cisplatino/EP vs carboplatino/irinotecan, deve tenere conto anche delle possibili tossicità: maggiore incidenza di tossicità renale e gastro-intestinale con l’associazione cisplatino/EP e maggior rischio di tossicità ematologica con l’utilizzo di carboplatino/irinotecan. Ulteriori schemi terapeutici utilizzati sono a base di oxaliplatino (XELOX, FOLFOX) o a base di irinotecan (FOLFIRI, IP), con tassi di risposta del 23-40%. Non esiste ancora, però, un’indicazione univoca di seconda linea nei pazienti affetti da NEC avanzato e in progressione dopo una prima linea con cisplatino/carboplatino + EP. Sulla base dei pochi dati disponibili, in questa tipologia di pazienti può essere presa in considerazione, in caso di progressione di malattia dopo una prima linea di trattamento con cisplatino/carboplatino + EP, una terapia a base di TMZ, irinotecan, oxaliplatino o fluoropirimidine (16,17).
Conclusioni
La chemioterapia rappresenta la principale scelta terapeutica per i pazienti affetti da NEC. A differenza dei NET G1-G2 sia pancreatici che extra-pancreatici, per i quali la chemioterapia è riservata a casi selezionati, anche nei NET G3 la chemioterapia può giocare un ruolo importante, data la maggiore chemio-sensibilità di questo gruppo di tumori. Il ruolo della chemioterapia nei NET in generale richiede ulteriori studi possibilmente prospettici, per poter definire eventuali marcatori di efficacia e possibili schemi terapeutici sequenziali che possano includere non solo la chemioterapia ma anche altre opzioni terapeutiche come, ad esempio, la PRRT o altre terapie mirate, al fine di incrementare il numero di opzioni terapeutiche disponibili per questa tipologia di patologie.
Bibliografia
- Das S, Al-Toubah T, Strosberg J. Chemotherapy in neuroendocrine tumors. Cancers (Basel) 2021, 13: 4872.
- Das S, Dasari A. Epidemiology, incidence, and prevalence of neuroendocrine neoplasms: are there global differences? Curr Oncol Rep 2021, 23: 43.
- Sorbye H, Welin S, Langer SW., et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol 2013, 24: 152–60.
- Mitry E, Baudin E, Ducreux M, et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br J Cancer 1999, 81: 1351–5.
- Thomas KEH, Voros BA, Boudreaux JP, et al. Current treatment options in gastroenteropancreatic neuroendocrine carcinoma. Oncologist 2019, 24: 1076–88.
- Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med 1980, 303: 1189–94.
- Moertel CG, Lefkopoulo M, Lipsitz S, et al. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med 1992, 326: 519–23.
- Rogers JE, Lam M, Halperin DM, et al. Fluorouracil, doxorubicin with streptozocin and subsequent therapies in pancreatic neuroendocrine tumors. Neuroendocrinology 2022, 112: 34–42.
- Al-Toubah T, Pelle E, Valone T, et al. Efficacy and toxicity analysis of capecitabine and temozolomide in neuroendocrine neoplasms. J Natl Compr Canc Netw 2021, 20: 29–36.
- Kunz PL, Graham N, Catalano PJ, et al. A randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors: final analysis of efficacy and evaluation of MGMT (ECOG-ACRIN E2211). J Clin Oncol 2022, 40 suppl 16: 4004.
- Pavlakis N, Ransom DT, Wyld D, et al. Australasian Gastrointestinal Trials Group (AGITG) CONTROL NET Study: 177Lu-DOTATATE peptide receptor radionuclide therapy (PRRT) and capecitabine plus temozolomide (CAPTEM) for pancreas and midgut neuroendocrine tumours (pNETS, mNETS) — Final results. J Clin Oncol 2022, 40 suppl 16: 4122.
- Ramanathan RK, Cnaan A, Hahn RG, et al. Phase II trial of dacarbazine (DTIC) in advanced pancreatic islet cell carcinoma. Study of the Eastern Cooperative Oncology Group-E6282. Ann Oncol 2001, 12: 1139–43.
- Bajetta E, Catena L, Procopio G, et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother Pharmacol 2007, 59: 637–42.
- Spada F, Antonuzzo L, Marconcini R, et al. Oxaliplatin-based chemotherapy in advanced neuroendocrine tumors: clinical outcomes and preliminary correlation with biological factors. Neuroendocrinology 2016, 103: 806–14.
- Brixi-Benmansour H, Jouve J-L, Mitry E, et al. Phase II study of first-line FOLFIRI for progressive metastatic well-differentiated pancreatic endocrine carcinoma. Dig Liver Dis 2011, 43: 912–6.
- Zappi A, Persano I, Galvani L, et al. Chemotherapy in well differentiated neuroendocrine tumors (NET) G1, G2, and G3: a narrative review. J Clin Med 2023, 12: 717.
- Garcia-Carbonero R, Sorbye H, Baudin E; all other Vienna Consensus Conference participants. ENETS consensus guidelines for high-grade gastroenteropancreatic neuroendocrine tumors and neuroendocrine carcinomas. Neuroendocrinology 2016, 103: 186–94.
Scheda streptozocina
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
La molecola subisce una decomposizione spontanea per produrre ioni di metil-carbonio reattivo, che alchilano il DNA e provocano legami crociati inter-filamento. I gravi danni al DNA determinano la morte cellulare per apoptosi o necrosi.
Indicazioni approvate
Terapia sistemica degli adulti con NEN G1 o G2 ben differenziate di origine pancreatica inoperabili, in fase avanzata o metastatica, progressive e/o sintomatiche, in associazione a 5-FU.
Controindicazioni
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Insufficienza renale (GFR < 30 mL/min).
Gravidanza e allattamento.
Preparazioni, via di somministrazione, posologia
Streptozocina Keocyt 1 g di polvere da diluire.
Va somministrata per via endovenosa mediante infusione che deve durare fra 30 minuti e 4 ore. Il medicinale è vescicante per natura e deve essere somministrato con cautela attraverso una linea a flusso libero. In caso di stravaso, la somministrazione va interrotta immediatamente. La dose viene calcolata in base all’area di superficie corporea (m2). È possibile utilizzare due schemi posologici diversi:
- somministrazione ogni sei settimane: 500 mg/m2/die, per 5 giorni consecutivi ogni 6 settimane fino al raggiungimento del massimo beneficio o finché non si osserva tossicità limitante il trattamento;
- somministrazione ogni tre settimane: 500 mg/m2/die, per 5 giorni consecutivi durante il primo ciclo, seguiti da 1000 mg/m2 ogni 3 settimane durante i cicli successivi.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Si raccomanda una pre-medicazione anti-emetica per elevato potenziale emetico (nausea e vomito).
Prima, durante e dopo il trattamento devono essere monitorate attentamente glicemia, funzionalità renale, epatica ed ematologica.
A seconda del grado di tossicità osservato, può essere indicato l’aggiustamento della dose o l’interruzione del farmaco.
La dose deve essere adeguata sulla base della funzionalità renale:
- GFR < 60 mL/min: ridurre la dose del 50%;
- GFR 30-45 mL/min: valutare il rapporto rischio/beneficio;
- GFR < 30 mL/min: controindicata.
In caso di compromissione epatica, considerare una riduzione di dose.
Streptozocina non è stata studiata in pazienti < 18 e > 65 anni.
Può indurre riduzione di ematocrito, formula leucocitaria e conta piastrinica, intolleranza glucidica, confusione, letargia, depressione, nausea, vomito, diarrea, aumento di AST e LDH, epato-tossicità, ipo-albuminemia, proteinuria, insufficienza renale, febbre, reazioni nel sito di iniezione.
Non somministrare insieme a vaccini vivi o attenuati.
Modalità prescrittive
Prescrivibile in ambiente ospedaliero
Scheda dacarbazina
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
Nel fegato viene trasformata nel suo metabolita attivo, il gruppo metilico, che si lega al DNA (e altre macro-molecole), causando danni che impediscono alle cellule tumorali di replicarsi e crescere.
Indicazioni approvate
Trattamento di pazienti con melanoma maligno metastatizzato.
Altre indicazioni nell’ambito di un regime chemioterapico di combinazione sono: morbo di Hodgkin in stadio avanzato e sarcoma dei tessuti molli in stadio avanzato negli adulti (ad eccezione del mesotelioma e del sarcoma di Kaposi).
Controindicazioni
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Gravidanza o allattamento.
Leucopenia e/o trombocitopenia.
Gravi malattie epatiche o renali.
Preparazioni, via di somministrazione, posologia
Dacarbazina 100, 200, 500, 1000 mg di polvere da diluire e somministrare per via ev.
L’uso deve essere limitato a specialisti in oncologia o ematologia.
Morbo di Hodgkin. La dacarbazina viene somministrata, ogni 15 giorni, alla dose giornaliera di 375 mg/m² in combinazione con doxorubicina, bleomicina e vinblastina (regime ABVD). Si raccomandano generalmente 6 cicli di terapia di combinazione ABVD.
Melanoma maligno. Può essere somministrata in monoterapia a dosi di 200–250 mg/m²/die per 5 giorni a intervalli di 3 settimane. In alternativa all’iniezione in bolo, è possibile somministrarla come infusione nell’arco di 15–30 minuti. È anche possibile somministrare 850 mg/m² al giorno 1 e quindi a intervalli di 3 settimane.
Sarcoma dei tessuti molli negli adulti. La dacarbazina viene somministrata a dosi giornaliere di 250 mg/m² (giorni 1–5) in combinazione con doxorubicina a intervalli di 3 settimane (regime ADIC).
Nel melanoma maligno metastatizzato e nel sarcoma dei tessuti molli in stadio avanzato la durata del trattamento dipende dall’efficacia e dalla tollerabilità nel singolo paziente.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Poiché sono frequenti gravi reazioni intestinali, si consiglia l’adozione di misure anti-emetiche e di supporto.
La dacarbazina è sensibile all’esposizione alla luce: tutte le soluzioni ricostituite devono essere tenute al riparo dalla luce anche durante la somministrazione, utilizzando mezzi appropriati (set per infusione resistenti alla luce).
Le preparazioni di dacarbazina 100 mg e 200 mg sono iposmolari (circa 100 mOsmol/kg) e devono pertanto essere somministrate mediante iniezione ev lenta, nell’arco di 1 minuto, anziché mediante bolo ev rapido in alcuni secondi. Le dosi > 200 mg/m² devono essere somministrate mediante infusione ev nell’arco di 15–30 minuti.
Durante l’iniezione si deve procedere con cautela per evitare lo stravaso nei tessuti, che provoca dolore locale e danno tissutale. In caso di stravaso, si deve sospendere immediatamente l’iniezione e si deve introdurre l’eventuale dose restante in un’altra vena.
Poiché possono verificarsi gravi disturbi gastro-intestinali ed ematologici, si raccomanda una valutazione estremamente accurata dei rischi e dei benefici prima di ogni ciclo di terapia.
Effetti collaterali comuni: nausea, vomito, anoressia, fatica, anemia, diarrea, mal di testa, confusione.
Durante il trattamento effettuare un monitoraggio frequente della crasi ematica e della funzione epatica e renale.
Nei casi di sola insufficienza renale o epatica da lieve a moderata, non è generalmente necessaria una riduzione della dose. Nei pazienti con compromissione sia renale che epatica l’eliminazione della dacarbazina richiede più tempo.
Poiché l’esperienza nei pazienti anziani è limitata, non è possibile fornire istruzioni particolari per l’uso in questa popolazione. La sicurezza e l’efficacia della dacarbazina non sono state ancora stabilite nei bambini di età < 15 anni.
Qualora si manifestino sintomi di disfunzione epatica o renale o di una reazione di ipersensibilità, occorre interrompere immediatamente la terapia. In caso di malattia veno-occlusiva del fegato, è controindicato proseguire la terapia ed è risultata efficace una terapia precoce a base di corticosteroidi a dosi elevate (p.e. idrocortisone 300 mg/die), con o senza agenti fibrinolitici come l’eparina o l’attivatore del plasminogeno tissutale.
Una terapia a lungo termine può causare tossicità cumulativa del midollo osseo. La tossicità emopoietica può giustificare una sospensione temporanea o la cessazione della terapia.
Interazioni. L’uso concomitante con fenitoina deve essere evitato, perché il ridotto assorbimento di fenitoina da parte del tratto gastro-intestinale può predisporre il paziente a convulsioni. La dacarbazina è un immuno-soppressore moderato. La somministrazione di vaccini vivi a pazienti immuno-compromessi in conseguenza del trattamento con chemioterapici, come la dacarbazina, può causare infezioni gravi e potenzialmente fatali. L’immunizzazione con vaccini vivi deve pertanto essere evitata durante la terapia con dacarbazina. La vaccinazione con vaccini vivi deve essere praticata non prima di 3 mesi dal completamento della chemioterapia. I vaccini inattivati possono essere usati, se disponibili. Dacarbazina non deve essere usate in concomitanza con fotemustina, perché l’uso concomitante può causare tossicità polmonare acuta (sindrome da distress respiratorio dell’adulto), che può avere esito fatale. Durante la chemioterapia evitare l’uso di medicinali epato-tossici e il consumo di alcool.
Modalità prescrittive
Prescrivibile in ambiente ospedaliero.
Scheda 5-Fluoro-uracile
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
Anti-metabolita che agisce come analogo dell'uracile, per interferire con la sintesi di DNA e RNA e causare la morte delle cellule tumorali.
Indicazioni approvate
Trattamento palliativo del carcinoma della mammella, del colon, del retto, dello stomaco e del pancreas in pazienti selezionati, considerati intrattabili chirurgicamente o con altri mezzi.
Controindicazioni
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Grave debilitazione (ad es. in stato di denutrizione).
Diminuita funzionalità midollare, ad es. dopo radioterapia o trattamento con altri agenti anti-neoplastici.
Infezioni gravi (ad es. Herpes Zoster, varicella).
Assenza totale nota di attività della diidropirimidina-deidrogenasi (DPD); mutazioni omozigotiche per la DPD; trattamento recente o concomitante con brivudina, sorivudina ed analoghi (potenti inibitori dell’enzima DPD che metabolizza il 5-FU).
Trattamento di patologia non maligna.
Grave insufficienza epatica.
Allattamento.
Preparazioni, via di somministrazione, posologia
5-FU (50 mg/mL, 1 g/20 mL, 5 g/100 mL) può essere diluito con sodio cloruro 0.9% o con destrosio 5%. La soluzione ottenuta è stabile per 48 ore a temperatura ambiente.
Viene somministrato principalmente per via ev, ma è possibile la somministrazione anche per via endo-arteriosa.
I regimi di trattamento variano con la combinazione di 5-FU con altri agenti cito-tossici o con la dose di acido folinico usato in concomitanza. Il numero di cicli usati dovrebbe essere deciso in base ai protocolli di trattamento locali e alle linee guida, prendendo in considerazione il successo del trattamento e la tollerabilità nei singoli pazienti. Il dosaggio deve essere personalizzato e calcolato sul peso corporeo effettivo del paziente, usando l’indice di massa magra corporea (peso secco) se il paziente è obeso o se il peso risulta artificiosamente aumentato a causa di edema, ascite o altre condizioni di ritenzione idrica anormale.
La dose iniziale è di 12 mg/kg una volta al giorno per 4 giorni successivi. La dose giornaliera non dovrebbe superare gli 800 mg. Se non si nota tossicità, si possono somministrare 6 mg/kg in 6°, 8°,10°, 12° giornata (nessuna somministrazione in 5°, 7°, 9°, 11° giornata). La terapia deve essere sospesa alla fine del 12°giorno, anche se non si manifestano segni di tossicità. La dose totale giornaliera non dovrebbe superare i 400 mg.
Terapia di mantenimento: nei casi in cui la tossicità non rappresenta un problema, si prosegue la terapia ripetendo la somministrazione con il medesimo dosaggio della precedente ogni 30 giorni dall’ultimo trattamento. In caso di tossicità in corso del primo ciclo e quando i segni di tale tossicità sono diminuiti, somministrare una dose di mantenimento di 10-15 mg/kg/settimana in un’unica somministrazione.
Infusione: 15 mg/kg/die (ma non > 1 g/infusione), da diluire in 500 mL di destrosio 5% o sodio cloruro 0.9% e somministrare alla velocità di 40 gocce/minuto in 4 ore. In alternativa, la dose giornaliera può essere infusa per 30-60 minuti, oppure con infusione continua durante le 24 ore. Questa dose giornaliera va somministrata in giorni successivi fino a che non si riscontrano segni di tossicità oppure fino a che non si è somministrata una dose cumulativa di 12-15 g.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
È consigliabile riduzione della dose in pazienti con cachessia, intervento chirurgico maggiore nei 30 giorni precedenti, ridotta funzionalità midollare, epatica o renale.
Prima di ogni dose monitorare per la tossicità ematologica (piastrine e leucociti), gastro-intestinale (stomatite, diarrea, sanguinamento) e neurologica, e, se necessario, ridurre la dose o sospendere la somministrazione.
Effetti collaterali comuni:
- gastro-intestinali: stomatite, nausea, vomito, diarrea;
- ematologici: anemia, leucopenia, trombocitopenia;
- dermatologici: eritro-disestesia palmo-plantare, secchezza cutanea, perdita di capelli, foto-sensibilità;
- neurologici: cefalea, disorientamento (raro).
Modalità prescrittive
Prescrivibile in ambiente ospedaliero.
Scheda capecitabina
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
Anti-metabolita orale, che agisce bloccando la crescita delle cellule tumorali convertendosi in 5-fluoro-uracile (5-FU).
Indicazioni approvate
Terapia adiuvante nei pazienti sottoposti a chirurgia per carcinoma del colon di stadio III (Dukes C).
Carcinoma del colon-retto metastatico.
Trattamento di prima linea del carcinoma gastrico avanzato in associazione con un regime a base di platino.
In associazione a docetaxel nel trattamento di pazienti con carcinoma della mammella localmente avanzato o metastatico dopo fallimento della chemioterapia cito-tossica.
In mono-terapia per il trattamento del carcinoma della mammella localmente avanzato o metastatico dopo fallimento di un regime chemioterapico contenente taxani e un’antraciclina o per le quali non è indicata un’ulteriore terapia con antracicline.
Controindicazioni
Anamnesi di reazioni gravi o inattese alla terapia con una fluoro-pirimidina.
Ipersensibilità alla capecitabina o a uno qualsiasi degli eccipienti.
Assenza totale nota di diidropirimidina-deidrogenasi (DPD).
Gravidanza e allattamento.
Forme gravi di leucopenia, neutropenia o trombocitopenia.
Grave compromissione della funzione epatica.
Grave compromissione della funzione renale (creatinina clearance < 30 mL/min).
Trattamento recente o concomitante con brivudina.
Preparazioni, via di somministrazione, posologia
Capecitabina (Xeloda e altri) cp 150 e 500 mg. Le compresse rivestite con film devono essere ingerite intere con acqua entro 30 minuti dopo un pasto e non devono essere frantumate né tagliate.
Mono-terapia per carcinoma del colon, del colon-retto e della mammella: la dose iniziale consigliata è 1250 mg/m2 per due volte al giorno per 14 giorni, seguita da un intervallo di 7 giorni. La terapia adiuvante nei pazienti con carcinoma del colon in stadio III è raccomandata per 6 mesi.
Terapia di associazione:
- carcinoma del colon, del colon-retto e gastrico: la dose iniziale raccomandata deve essere ridotta a 800-1000 mg/m2, se somministrata due volte al giorno per 14 giorni seguiti da un intervallo di 7 giorni, o a 625 mg/m2 per due volte al giorno se somministrata continuativamente. In associazione a irinotecan (200 mg/m2 il giorno 1), la dose iniziale consigliata è 800 mg/m2 per due volte al giorno per 14 giorni, seguiti da un periodo di 7 giorni di intervallo. La durata raccomandata del trattamento adiuvante nei pazienti affetti da tumore del colon in stadio III è di 6 mesi;
- carcinoma della mammella: la dose iniziale consigliata è 1250 mg/m2 per due volte al giorno per 14 giorni, seguiti da un intervallo di 7 giorni, in associazione a docetaxel 75 mg/m2 in infusione ev di 1 ora ogni 3 settimane.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Effetti collaterali più comuni: diarrea (spesso il sintomo che limita il dosaggio), nausea, vomito, dolori addominali, affaticamento, stomatite, eritro-disestesia palmo-plantare (reazione cutanea caratterizzata da dolore, gonfiore, arrossamento e desquamazione della pelle su mani e piedi, la cui gestione può richiedere riduzione della dose o interruzione del trattamento). È fondamentale un attento monitoraggio e un'adeguata gestione dell'idratazione per prevenire la disidratazione, che può portare a insufficienza renale acuta, potenzialmente fatale. Il medico deve essere contattato immediatamente se il numero di evacuazioni aumenta significativamente.
Associazione con tossicità gravi, talvolta fatali, specialmente in pazienti con specifiche condizioni pre-esistenti:
- pazienti con deficit noto (completo o parziale) dell'enzima DPD sono a rischio significativamente maggiore di tossicità grave o fatale (es. neutropenia, tossicità gastro-intestinale e neurologica). Prima di iniziare il trattamento, può essere presa in considerazione la caratterizzazione genotipica o fenotipica per identificare i pazienti con deficit di DPD;
- cardio-tossicità, simile a quella osservata con altre fluoropirimidine (come 5-FU). I pazienti con storia pre-esistente di coronaropatia devono essere trattati con cautela. Le manifestazioni possono includere angina, infarto miocardico, aritmie e insufficienza cardiaca.
Interazioni farmacologiche:
- brivudina: la somministrazione concomitante è controindicata a causa di un'interazione potenzialmente fatale che aumenta la tossicità della capecitabina. È necessario attendere almeno 4 settimane tra la fine del trattamento con brivudina e l'inizio della capecitabina;
- anti-coagulanti cumarinici (es. warfarin): l'uso concomitante richiede un monitoraggio frequente dei parametri della coagulazione, poiché la capecitabina può aumentarne significativamente l'effetto, con rischio di sanguinamento;
- fenitoina: durante l'uso combinato è stato segnalato un aumento delle concentrazioni plasmatiche, con sintomi di intossicazione, per cui è richiesto un monitoraggio regolare.
Modalità prescrittive
Prescrivibile in ambiente ospedaliero.
Scheda etoposide
Michela Del Prete
ASST Lariana, SC Endocrinologia, Diabetologia, Nutrizione Clinica, Ospedale Sant’Anna, San Fermo della Battaglia (CO)
Meccanismo d’azione
Blocca la topo-isomerasi II, un enzima cruciale per la riparazione del DNA, causando rotture nel DNA cellulare e prevenendo la replicazione delle cellule tumorali, portando alla morte cellulare.
Indicazioni approvate
Tumori del testicolo resistenti non seminomatosi, in associazione con altri agenti chemioterapici.
Carcinoma polmonare a piccole cellule, in associazione con altri agenti chemioterapici.
Leucemia monoblastica acuta e leucemia mielomonocitica acuta, in associazione con altri agenti chemioterapici quando la terapia di induzione standard si sia rivelata inefficace.
Controindicazioni
Ipersensibilità a etoposide o a uno qualsiasi degli eccipienti, podofillotossina o derivati.
Grave compromissione della funzionalità epatica.
Grave mielo-soppressione.
Allattamento.
L’uso concomitante del vaccino per la febbre gialla o di altri vaccini vivi è controindicato.
Preparazioni, via di somministrazione, posologia
Etoposide 20 mg/mL. Il farmaco deve essere diluito in una soluzione di glucosio al 5% o in una soluzione di sodio cloruro allo 0.9%, per ottenere una concentrazione finale di 0.2-0.4 mg/mL (cioè 1 mL o 2 mL di concentrato in 100 mL di diluente, per raggiungere una concentrazione, rispettivamente, di 0.2 mg/mL e 0.4 mg/mL). Questa soluzione viene somministrata come soluzione ev per un periodo non inferiore a 30 minuti e non superiore a 2 ore.
La dose raccomandata negli adulti è 60-120 mg/m2 al giorno, per 5 giorni consecutivi. Gli schemi posologici più frequentemente usati sono 100 mg/m² per 5 giorni o 120 mg/m² a giorni alterni o nei giorni 1, 3 e 5.
Poiché causa mielo-soppressione, il ciclo non deve essere ripetuto con frequenza < 10-20 giorni (o < 21 giorni per le indicazioni non–ematologiche) o comunque prima che il quadro ematico non sia stato controllato per eventuali segnali di mielo-soppressione e sia ritenuto soddisfacente.
Avvertenze speciali, precauzioni di impiego, interazioni, effetti collaterali e tossicità
Effetti collaterali più comuni: modifiche ematologiche (neutropenia, trombocitopenia e anemia), problemi gastro-intestinali (nausea, vomito, perdita dell'appetito, stipsi o diarrea), astenia e fatigue, alopecia e reazioni cutanee (eruzioni cutanee pruriginose, simili all'acne, o rossore localizzato), mucosite (possibile da 5 a 10 giorni dopo la terapia), ipotensione.
Meno comuni: reazioni allergiche sistemiche, con brividi, febbre o rossore al volto, ipertensione, polmonite interstiziale o fibrosi polmonare.
Il farmaco può determinare grave mielo-soppressione (con conseguente infezione o sanguinamento), anche fatale. I pazienti in trattamento con etoposide devono essere strettamente e frequentemente monitorati per la mielo-soppressione, sia durante che dopo la terapia. Se prima dell’inizio del trattamento con etoposide è stata somministrata radioterapia o chemioterapia, deve intercorrere un intervallo di tempo adeguato per consentire il recupero della funzionalità midollare. Dopo la dose iniziale, le dosi successive devono essere adattate se:
- la conta dei neutrofili è < 500 cellule/mm³ per più di 5 giorni o è associata a febbre o infezione;
- la conta piastrinica è < 25.000 cellule/mm³;
- si sviluppa qualsiasi altro effetto tossico di grado 3 o 4;
- la clearance della creatinina è < 50 mL/min.
In pazienti trattati con etoposide in associazione con altri anti-neoplastici sono stati segnalati rari casi di insorgenza di leucemia acuta, che può manifestarsi con o senza una fase pre–leucemica.
Nei pazienti con bassi livelli sierici di albumina il rischio di tossicità associata a etoposide può essere elevato. Se il paziente soffre di disfunzione epatica o renale, la funzionalità epatica e renale deve essere regolarmente monitorata, a causa del rischio di accumulo.
Inoltre, cicli di terapia con etoposide devono essere effettuati solo se non ci sono patologie del sistema nervoso periferico.
L’etoposide è mutageno e cancerogeno, cosa di cui occorre tener conto quando si eseguono trattamenti di lunga durata. Considerato il potenziale mutageno di etoposide, è richiesta una contraccezione efficace da parte dei pazienti di entrambi i sessi durante il trattamento e nei 6 mesi successivi. Si raccomanda una consulenza genetica se il paziente desidera procreare dopo la fine del trattamento. Poiché etoposide può diminuire la fertilità maschile, si può prendere in considerazione la conservazione del seme ai fini di una successiva paternità.
Terapie concomitanti: attenzione a ciclosporina, cisplatino, fenitoina o fenobarbitale. In caso di precedente o concomitante uso di altri farmaci ad azione mielo-soppressiva, si possono prevedere effetti additivi o sinergici. Con l’uso del vaccino contro la febbre gialla, esiste un aumento del rischio che si manifesti la malattia vaccinica sistemica fatale. L’uso di vaccini vivi è controindicato nei pazienti immuno-depressi. Fenilbutazone, sodio salicilato e acido acetilsalicilico possono spostare il legame proteico di etoposide. La terapia concomitante con warfarin può causare un aumento dell’INR, che deve essere strettamente monitorato.
Modalità prescrittive
Prescrivibile in ambiente ospedaliero.