Diagnostica per la midollare surrenalica

Biochimica:
Strumentale:
Metanefrine
Letizia Canu
Dipartimento di Scienze Biomediche Sperimentali e Cliniche “Mario Serio”, Università di Firenze
(aggiornato all’11 maggio 2021)
Nelle 48 ore prima e per tutta la durata della raccolta urinaria delle 24 ore, bisogna evitare l’assunzione di alimenti ricchi di amine: caffè, thè, cioccolata, vaniglia, banane, agrumi, legumi.
| Alterazioni delle concentrazioni di metanefrine | |
| Aumentate da | |
| Cause fisiopatologiche | Stress. Dolore. Esercizio fisico. Freddo. Fumo. Fase luteinica del ciclo mestruale. Ipoglicemia. Interventi chirurgici. Scompenso cardiaco. IMA. Ictus. Cirrosi epatica. Insufficienza renale. Sindrome delle apnee notturne. Ipotiroidismo. Neuroblastoma e (raramente) ganglioneuroma. Feocromocitoma e paragangliomi. |
| Modificazioni farmaco-indotte | Caffeina. Teofillina. Alcool. Cocaina e metamfetamina. Anti-depressivi triciclici: amitriptilina, clomipramina, imipramina, maprotilina, mianserina, noritriptilina, trimipramina. Anti-psicotici: clozapina, quetiapina, amisulpiride, aripiprazolo, olanzapina, risperidone, clorpromazina, flufenazina, periciazina, trifluoperazina. Inibitori della ricaptazione della noradrenalina: bupropione, reboxetina, proclorperazina, reserpina. Αlfa2-bloccanti: fenossibenzamina, fentolamina. β-bloccanti: carvedilolo, labetalolo, acebutolo, bisoprololo, celiprololo, metoprololo, pindololo, propanololo, sotalolo. Calcio-antagonisti (in acuto). Nitroglicerina. Αlfa-metil-DOPA, levo-DOPA e carbi-DOPA. MAO-inibitori: tranilcipromina. ASA e paracetamolo. Furosemide. Eritromicina. Simpatico-mimetici: pseudoefedrina, fenilefrina, dobutamina, isoprenalina, salbutamolo, terbutalina. |
Test con clonidina per catecolamine
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
| Test con clonidina per catecolamine | |
| Indicazioni | Da eseguire nei pazienti che hanno livelli di metanefrine plasmatiche e/o urinarie persistentemente superiori alla norma ma nella zona di incertezza diagnostica, anche dopo l’esclusione di fattori interferenti. |
| Meccanismo d’azione | La clonidina è un agonista selettivo dei recettori alfa2-adrenergici, che inibisce il rilascio di catecolamine nei pazienti senza feo/PGL e con attivazione cronica simpatica, ma non esercita alcun effetto sui livelli aumentati di metanefrine dovuti alla secrezione autonoma di catecolamine dei feo/PGL. |
| Contro-indicazioni |
Anziani.
|
| Materiale necessario per l’esecuzione | Cp di clonidina (da 150 o 300 μg). |
| Relazione con età, sesso, peso corporeo, gravidanza | Dosaggio di 300 μg/70 kg peso corporeo. |
| Esecuzione |
I campioni ematici appena prelevati devono essere conservati in ghiaccio:
|
| Dosaggio | Noradrenalina o normetanefrina plasmatica. |
| Possibili effetti collaterali | Ipotensione, nausea. |
| Parametri da monitorare durante l’esecuzione | PA e frequenza cardiaca. |
| Valutazione risultati |
Sono indicativi di feo/PGL:
|
| Interpretazione | Utilizzando entrambi i criteri sensibilità 97% e specificità 98%. |
| Giudizio complessivo costo beneficio e costo-efficacia | Non rientra fra i testi biochimici di prima scelta ed è da eseguirsi in casi selezionati (per ridurre il numero di falsi positivi del dosaggio basale di catecolamine/metanefrine), dal momento che il suo uso non è stato effettivamente validato in alcuno studio prospettico. |
| Bibliografia |
|
Feocromocitoma e paragangliomi
Classificazione feocromocitomi e paragangliomi
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
(aggiornato al 20 marzo 2020)
I feocromocitomi e i paragangliomi sono tumori che originano dalle cellule endocrine di derivazione dalla cresta neurale o da organi, i gangli, presenti lungo il sistema nervoso simpatico (in sede para-aortica/peri-cavale a livello addominale e mediastinico, o alla biforcazione dell’iliaca – organo dello Zuckerkandl -, più raramente nel tessuto perineale, nella vescica, nell’uretere), e parasimpatico, questi ultimi a distribuzione più ristretta, associati alle branche toraciche e craniali di nervo vago e glosso-faringeo (collo: glomo carotideo e gangli vagali; testa: gangli del bulbo giugulare o giugulo-timpanici).
Nel 2004 l'Organizzazione Mondiale della Sanità ha definito il feocromocitoma (FEO) un “paraganglioma simpatico intra-surrenalico”, classificando invece i tumori che insorgono nei paragangli extra-surrenalici, simpatici e parasimpatici, come “paragangliomi extra-surrenalici” (PGL) (1).
Nel 2017 tutti i paragangliomi sono stati classificati dalla WHO tra i tumori maligni (ICD-O/3) perchè tutti possiedono un potenziale metastatico (2). Quindi oggi questi tumori non si classificano più in benigni e maligni, ma in non metastatici e metastatici.
La distinzione tra feocromocitoma e paraganglioma è importante per le implicazioni cliniche che comporta: rischio di malignità, associazione con altre neoplasie, valutazione genetica.
Feocromocitoma
Costituisce l’80-85% dei casi e ha un comportamento non aggressivo nell’85-90% dei casi; l’incidenza nella popolazione generale è 0.8 casi/100.000/anno, con una prevalenza nei pazienti ipertesi di 0.2-0.4%. Ha maggiore frequenza nella 4°-5° decade per le forme sporadiche, senza differenze significative nei due sessi, mentre nelle forme ereditarie l’insorgenza può essere anticipata. Le forme sporadiche costituiscono circa il 65-70%. La prevalenza del feocromocitoma è riportata nel 1.1–11% dei pazienti con incidentaloma surrenalico, mentre nel 4–14% dei pazienti affetti da feocromocitoma la neoplasia viene rilevata casualmente in corso di indagini strumentali effettuate per altre patologie.
Negli ultimi anni una quota ragguardevole di feocromocitomi/ paragangliomi apparentemente sporadici, variabile nelle diverse casistiche da 7.5 a 32%, si è rivelata essere parte di una sindrome ereditaria (3-6). Le forme familiari di feocromocitoma si presentano nell’ambito di sindromi (tabelle 1 e 2) (vedi capitolo “genetica”).
- MEN-2A e MEN-2B (Neoplasie Endocrine Multiple tipo 2A e 2B): solitamente la prima manifestazione clinica della sindrome è il carcinoma midollare della tiroide, mentre il feocromocitoma è la prima manifestazione in una percentuale che non supera il 20-25% dei casi.
- Sindrome di von Hippel-Lindau, suddivisa nei sottotipi:
- VHL tipo 1, senza feocromocitoma;
- VHL tipo 2 (2A, 2B, 2C), con presenza di feocromocitoma.
- Neurofibromatosi tipo 1.
- Sindromi PGL, legate a mutazioni delle diverse subunità del gene SDHx:
- PGL1: SDHD;
- PGL2: SDHAF2;
- PGL3: SDHC;
- PGL4: SDHB;
- PGL5: SDHA.
- Feocromocitoma familiare – casi caratterizzati da mutazioni di due geni:
- TMEM127 (7-9);
- MAX (10,11)
| Tabella 1 Sindromi associate a Feocromocitoma/Paraganglioma e loro frequenza |
||||
| Sindrome | Gene | Feocromocitoma | Paraganglioma | |
| Simpatico | Parasimpatico | |||
| MEN 2A | RET | ~50% | raro | rarissimo |
| MEN 2B | RET | ~50% | raro | rarissimo |
| Von Hippel-Lindau | VHL | 10-20% | + | raro |
| Neurofibromatosi tipo I | NF1 | 5% | - | - |
| Feocromocitoma familiare TMEM127 | TMEM127 | 100% | raro | rarissimo |
| Feocromocitoma familiare MAX | MAX | 100% | + | rarissimo |
| PGL1 | SDHD | + | + | + |
| PGL2 | SDHAF2 | - | - | + |
| PGL3 | SDHC | - | raro | + |
| PGL4 | SDHB | raro | + | raro |
| PGL5 | SDHA | raro | + | raro |
| + presente; - assente | ||||
Paragangliomi extra-surrenalici
Rappresentano il 15-20% dei casi, e sono suddivisi in due categorie.
- Paragangliomi “simpatici”, per la maggior parte caratterizzati da iperproduzione di catecolamine, con conseguente sintomatologia analoga a quella sostenuta dal feocromocitoma. La maggioranza dei PGL simpatici extra-surrenalici è localizzata nell’addome (circa il 75%) e origina dal tessuto cromaffine para-aortico inferiore (specie organo di Zuckerkandl e gangli para-vescicali) e para-aortico superiore, adiacente a reni e surreni; solo il 10-12% è situato a livello toracico (compresa la rara localizzazione intra-pericardica). L’incidenza dei PGL simpatici non è definita, in quanto storicamente sono stati raggruppati con i feocromocitomi; l’incidenza annua combinata feocromocitoma/ PGL simpatici è di circa 0.8/100.000 persone/anno.
- Paragangliomi “parasimpatici”, in genere non producono catecolamine, anche se nel 5% dei casi è stato descritto rilascio e conseguente sintomatologia tipica (12). Nella maggior parte dei casi la sintomatologia dipende da effetto massa. I PGL parasimpatici, con incidenza di 1/1.000.000/anno, sono prevalentemente localizzati a livello di collo (glomo carotideo, sede più frequente, circa 60% dei casi, e gangli vagali) e testa (gangli del bulbo giugulare o giugulo-timpanici). La metà dei casi circa si sviluppa nell’ambito di una sindrome genetica.
Le forme sporadiche hanno maggiore frequenza di presentazione nella 5° decade, quasi sempre come neoplasie singole e raramente sono metastatiche (circa il 4% dei casi).
Nelle tabelle 1 e 2 sono riportati i caratteri fenotipici delle forme ereditarie di PGL (vedi genetica).
I pazienti con PGL familiare sviluppano la malattia ad un’età più precoce.
Nei bambini il feocromocitoma/paraganglioma è una patologia rara, costituendo meno del 20% di tutti casi (13), ma la prevalenza nei bambini ipertesi cresce a 0.8-1.7%. Nel bambino e nell’adolescente aumenta la probabilità che si tratti di una forma ereditaria. Il rischio di malattia metastatica è superiore nel bambino rispetto all’adulto.
| Tabella 2 Aspetti fenotipici delle forme ereditarie più frequenti di Feocromocitoma/Paraganglioma (14-17) |
|||
| Gene | Età media alla diagnosi (anni) | Bilateralità/multifocalità della neoplasia | Prevalenza forme maligne |
| RET | 37 | + | < 3% |
| VHL | 26 | + | 5% |
| NF1 | 42 | - | 11% |
| TMEM127 | 45 | + | rara |
| MAX | < 30 | + | ~10% |
| SDHD | 32 | + | ~5% |
| SDHAF2 | 35 | + | * |
| SDHC | 40 | - | * |
| SDHB | 28 | + | ~40% |
| SDHA | 43 | - | 10-30% |
| FH | 39 | - | ~40% |
| *ancora non noto; + possibile, - assente | |||
Bibliografia
- DeLellis RA, Lloyd RV, Heitz PU, Eng C, eds. Pathology and genetics of tumours of endocrine organs. Lyon, France: IARC Press; 2004. World Health Organization Classification of Tumours.
- Tischler AS, de Krijger RR. Pheochromocytoma. In: WHO Classification of Tumours of Endocrine Organs, 4th; Lloyd RV, Osamura RY, Kloppel G, Eds; IARC Press: Lyons France, 2017: pp. 183-189.
- Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002, 346: 1459-66.
- Amar L, Bertherat J, Baudin F, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005, 23: 8812-8.
- Korpershock E, Van Nederveen FH, Dannenberg H, et al. Genetic analyses of apparently sporadic pheochromocytoma: the Rotterdam experience. Ann N Y Acad Sci 2006, 1073: 138-48.
- Mannelli M, Castellano M, Schiavi F, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 2009, 94: 1541–7.
- Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nature Genet 2010, 42: 229–33.
- Yao L, Schiavi F, Cascon A, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 2010, 304: 2611-9.
- Abermil N, Guillaud-Bataille M, Burnichon N, et al. TMEM127 screening in a large cohort of patients with pheochromocytoma and/or paraganglioma. J Clin Endocrinol Metab 2012, 97: E805-9.
- Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature Genet 2011, 43: 663–7.
- Burnichon N, Cascón A, Schiavi F, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 2012, 18: 2828-37.
- Barontini M, Levin G, Sanso G. Characteristic of pheochromocytoma in a 4- to 20-year-old population. Ann N Y Acad Sci 2006, 1073: 30-7.
- Bausch B, Schiavi F, Ni Y, et al; European-American-Asian Pheochromocytoma-Paraganglioma Registry Study Group. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol 2017, 3: 1204–12.
- van der Tuin K, Mensenkamp AR, Tops CMJ, et al. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: a nationwide study. J Clin Endocrinol Metab 2018, 103: 438–45.
- Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 2014, 23: 2440-6.
- Clark GR, Sciacovelli M, Gaude E, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 2014, 99: E2046-50.
Overview su feocromocitomi e paragangliomi
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
(aggiornato al 20 marzo 2020)
Il feocromocitoma e i paragangliomi sono tumori rari di origine neuroendocrina. Possono essere di natura:
- simpatergica, secernenti catecolamine, localizzati nei surreni e nei paragangli addominali e toracici;
- parasimpatergica, generalmente non secernenti, localizzati nei paragangli della regione testa/collo.
Il quadro clinico delle forme secernenti è estremamente variabile, legato al rilascio di catecolamine ed all’attivazione dei recettori adrenergici. Possono causare crisi sintomatologiche, con palpitazioni, cefalea, sudorazione ed ansia, crisi ipertensive anche gravi, ma possono anche associarsi a sintomi sfumati, aspecifici e non causare innalzamento pressorio. Molti di questi tumori sono scoperti incidentalmente in corso di indagini radiologiche.
I paragangliomi della regione testa/collo si presentano come tumefazioni latero-cervicali e, in rapporto alle loro dimensioni, possono causare sintomi da compressione sui nervi adiacenti (ipoacusia, disfagia, anisocoria, ipofonia).
Tranne nei casi associati a chiare crisi sintomatologiche ed ipertensione, specialmente se a crisi o resistente, il sospetto diagnostico è molto difficile. In caso di sospetto, la conferma diagnostica va ricercata nella misura delle metanefrine (metanefrina e normetanefrina) nel plasma o nelle urine. Questi dosaggi sono infatti quelli con la più alta sensibilità diagnostica.
La localizzazione del tumore si avvale di tecniche di diagnostica per immagini:
- morfologiche (TC o RMN), con una sensibilità pari quasi al 100%;
- funzionali, da riservare ai casi con sospetto di metastasi: scintigrafia con metaiodobenzilguanidina, capace di concentrarsi nel tumore e in eventuali ripetizioni metastatiche in circa l’80% dei casi; PET con Gallio con sensibilità pari al 93%; PET con F-DOPA con sensibilità dell’80%; FDG-PET con la minor sensibilità (74%).
È ormai provato che l’insorgenza di questi tumori è legata in circa il 40% dei casi a mutazioni in uno dei numerosi geni di suscettibilità finora scoperti. In questo gruppo, accanto ai ben noti geni VHL (responsabile della sindrome di von Hippel-Lindau), RET (responsabile della sindrome MEN-2), e NF1 (responsabile della neurofibromatosi di tipo 1), negli ultimi 10 anni sono stati inclusi anche i geni che codificano per le 4 subunità della succino-deidrogenasi mitocondriale (SDHA, SDHB, SDHC, SDHD), il gene responsabile della flavinazione della subunità SDHA (SDHAF2), il gene TMEM127, il gene MAX ed altri geni di più recente scoperta (EGLN1, EPAS1, FH, KIF1B, MDH2).
Le mutazioni di questi geni predispongono all’insorgenza dei paragangliomi, talvolta nell’ambito di quadri sindromici diversi che, per quanto variabili, devono essere a conoscenza del clinico, sia perché utili ai fini diagnostici, sia perché, individuato il paraganglioma e la sindrome, l’indagine clinica sia completata con la ricerca delle altre lesioni sindromiche potenzialmente associate.
L’esecuzione dell’analisi genetica è divenuta un elemento indispensabile nella gestione clinica del paziente.
La presenza di una mutazione comporta l’obbligo dell’inserimento del paziente in un follow-up a vita per il rischio di recidive e consiglia l’estensione dell’analisi ai familiari, ai fini dell’individuazione dei portatori della mutazione che sono a rischio di sviluppare la malattia.
La terapia dei paragangliomi è chirurgica. Per le forme addominali è generalmente raccomandato l’approccio laparoscopico. Un approccio open è riservato a lesioni con dimensioni > 6 cm o che interessano le strutture circostanti, al fine di assicurare la completa resezione del tumore ed evitare una recidiva locale.
Per i paragangliomi della regione testa/collo va attentamente valutato il rapporto rischio/beneficio offerto dalla chirurgia. Nelle forme già voluminose e ritenute non operabili è possibile ricorrere alla radioterapia esterna stereotassica.
La terapia medica è molto importante per la preparazione all’intervento chirurgico dei paragangliomi secernenti. Si basa sull’uso degli alfa-antagonisti. Il farmaco generalmente consigliato è la doxazosina, il cui dosaggio va modulato in base al quadro cardio-circolatorio del paziente, che va portato alla chirurgia in condizione di normotensione.
I paragangliomi metastatici sono molto rari, definiti dalla presenza di localizzazioni a distanza e generalmente legati a mutazioni del gene SDHB; al giorno d’oggi, l’individuazione precoce è possibile grazie a tecniche di medicina nucleare specifiche, che permettono anche di individuare le forme trattabili con terapia radiometabolica.
Genetica di feocromocitomi e paragangliomi
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
(aggiornato al 20 marzo 2020)
I feocromocitomi e i paragangliomi possono insorgere come sporadici o su base familiare. Fino all’inizio degli anni 2000 si riteneva che le forme genetiche fossero circa il 10% e legate alle mutazioni dei geni VHL (1,2), RET (1-3) e NF1 (4), responsabili rispettivamente delle sindromi di von Hippel-Lindau, MEN-2 e neurofibromatosi di tipo 1. Le caratteristiche cliniche di tali sindromi sono riportate nella tabella.
| Quadri sindromici associati a mutazioni dei geni di suscettibilità per feocromocitoma/paraganglioma | |||
| Gene | Sindrome | Tipo | Quadro clinico |
| VHL | von Hippel-Lindau | 1 | Carcinoma renale a cellule chiare, emangioblastomi retinici e cerebellari |
| 2A | Carcinoma renale a cellule chiare, emangioblastomi retinici e cerebellari, feocromocitoma (spesso bilaterale) | ||
| 2B | Emangioblastomi retinici e cerebellari, feocromocitoma (spesso bilaterale) | ||
| 2C | Feocromocitoma (spesso bilaterale), rari HNPGL | ||
| RET | MEN-2 | 2A | Carcinoma midollare tiroideo, feocromocitoma (spesso bilaterale), iperparatiroidismo primario |
| FMTC | Carcinoma midollare tiroideo isolato | ||
| 2B | Carcinoma midollare tiroideo, feocromocitoma (spesso bilaterale), neuromi multipli, habitus marfanoide | ||
| NF1 | Neurofibromatosi 1 | Neuromi multipli di cute e mucose, macchie cutanee caffè-latte, feocromocitomi | |
| SDHD | PGL1 | HNPGL multipli, feocromocitomi, PGLs | |
| SDHAF2 | PGL2 | HNPGL | |
| SDHC | PGL3 | HNPGL, PGLs (rari) | |
| SDHB | PGL4 | PGLs maligni nel 40% dei casi, HNPGL (rari), carcinomi renali (rari) | |
| SDHA | PGL5 | PGLs | |
| FMTC: familial medullary thyroid carcinoma; HNPGL: paragangliomi testa-collo; PGLs: paragangliomi secernenti | |||
Nel 2000 Baysal (5) dimostrò che le mutazioni del gene che codifica per la subunità D della succino-deidrogenasi mitocondriale (SDH) o complesso mitocondriale II sono responsabili dell’insorgenza di una sindrome familiare, denominata PGL1, caratterizzata da paragangliomi della regione testa-collo, spesso multipli e che possono talora essere associati anche a feocromocitomi o PGL extra-surrenalici. Da quel momento non è stato più possibile considerare i tumori cromaffini surrenalici ed extra-surrenalici producenti catecolamine e di origine simpatergica un’entità separata dal gruppo dei PGL della regione testa-collo, di origine parasimpatergica e generalmente non producenti catecolamine. Nello stesso anno fu scoperto che anche mutazioni della subunità SDHB (6) erano responsabili della comparsa di PGL extra-surrenalici e negli anni successivi anche le altre subunità SDHx sono state riconosciute geni di suscettibilità per i feocromocitomi/PGL.
La SDH mitocondriale è formata da quattro subunità: le subunità D e C ancorano alla membrana mitocondriale interna le subunità catalitiche A e B. La SDH partecipa sia alla catena di trasporto elettronico sia alla funzione del ciclo di Krebs, dove trasforma il succinato in fumarato.
Pertanto i geni SDHA (7), SDHB, SDHC (8) e SDHD devono essere annoverati fra i geni di suscettibilità assieme a un altro gene denominato SDHAF2 (9), responsabile della flavinazione della subunità A, necessaria per la sua funzione enzimatica.
Le caratteristiche delle rispettive sindromi legate alle mutazioni di questi geni SDH sono riportate nella tabella.
Tutte le sindromi PGL vengono trasmesse con meccanismo autosomico dominante, ma le sindromi PGL1 e 2, legate rispettivamente a mutazioni del gene SDHD e SDHAF2, presentano il cosiddetto fenomeno dell’imprinting materno, per il quale solo chi riceve il gene mutato dal padre è a rischio di sviluppare la malattia. Tuttavia, va precisato che sono stati segnalati casi con convincente documentazione di presenza di malattia in pazienti con copia del gene mutato ereditata dalla madre (10,11). Si tratta di casi ancora ritenuti estremamente rari, verificatisi per una concomitante perdita di più loci genici.
Un altro elemento di rilevanza clinica è l’alta frequenza di forme metastatiche presenti nella PGL4, sindrome legata a mutazioni del gene SDHB (12). A tutt’oggi non sono noti i meccanismi che condizionano la maggior aggressività dei feocromocitomi/PGL con mutazioni SDHB. Sappiamo inoltre che in alcuni casi questi tumori possono accompagnarsi anche ad altri tumori solidi, tra cui i più frequenti sono il carcinoma renale (RCC), il gastrointestinal stromal tumor (GIST) e l’adenoma ipofisario.
Nel 2010 e nel 2011 sono stati descritti altri 2 geni di suscettibilità.
Il gene TMEM127 è causa di sviluppo di feocromocitomi in circa il 3% dei pazienti affetti (13). Il quadro clinico è caratterizzato dalla presenza di feocromocitomi spesso bilaterali.
Il gene MAX è stato trovato mutato in circa l’1% della popolazione affetta da feocromocitoma ed anche in questo caso i pazienti mutati presentavano sempre una localizzazione surrenalica del tumore, spesso bilaterale e qualche volta associata anche a PGL extra-surrenalici (14).
Nel 2014 è stato scoperto il gene della fumarato idratasi (FH), dimostrato essere responsabile di forme metastatiche (15).
Non si conoscono i meccanismi cellulari attraverso cui le mutazioni geniche portano alla formazione di feocromocitomi/PGL. Tuttavia, studi di biologia molecolare hanno delineato sostanzialmente due diversi profili di espressione genica per questi tumori (16):
- il sottogruppo 1 comprende i tumori causati da mutazioni del gene VHL e dei geni SDH (17). Questi tumori presentano attivazione, indotta dall’ipossia, dei geni dell’angiogenesi, attivazione dei geni codificanti le molecole di adesione, e attivazione della via glicolitica. Hanno un fenotipo biochimico di tipo noradrenergico (secernono cioè noradrenalina) e appaiono come meno differenziati dal punto di vista neuronale di quelli del secondo gruppo. In questo gruppo rientrano anche i geni FH, MDH2 e HIF2A;
- i tumori del sottogruppo 2 comprendono i tumori con mutazioni di RET, NF1, TMEM127 e MAX. Questi si caratterizzano per un fenotipo adrenergico (rilasciano cioè anche adrenalina) e si caratterizzano per un’attivazione delle vie di replicazione cellulare attivate dalle chinasi (18-21).
Recentemente è stato identificato un terzo sottogruppo, nel quale rientrano tumori legati a mutazioni dei geni CSDE1 (Cold Shock Domain-containing E1) e MAML3 (mastermind-like transcriptional coactivator 3), che si caratterizzano per elevato indice proliferativo e comportamento aggressivo.
Importanza della valutazione genetica
Nei pazienti affetti da feocromocitoma/PGL l’indagine genetica è importante sia per il paziente che per i familiari.
Il paziente portatore di mutazione dovrà essere inserito in un percorso di follow-up per tutta la vita, con dosaggio delle metanefrine da effettuarsi annualmente (o ogni 6 mesi nei pazienti a maggior rischio) per la diagnosi precoce di un’eventuale recidiva. Inoltre, nel caso, peraltro raro, che il feocromocitoma sia la prima manifestazione di una forma sindromica, come la VHL o la MEN-2, l’indagine genetica permette la diagnosi e il trattamento precoci delle altre neoplasie associate (vedi tabella).
Nei familiari l’analisi genetica consente di scoprire i portatori della mutazione e permette la diagnosi precoce di feocromocitoma/PGL attraverso un programma di screening (22).
In passato era la clinica a suggerire al genetista quali geni analizzare per primi avendo la maggiore probabilità di essere mutati: ad esempio, in caso di tumore surrenalico bilaterale, i geni di prima analisi erano VHL, RET, TMEM127 e MAX; mentre nel caso di PGL della regione testa-collo, specie se multipli o recidivanti o associati a PGL secernenti, i geni della succinato-deidrogenasi. Nel caso di pazienti affetti da PGL extra-surrenalici metastatici, il primo gene da considerare per l’analisi era SDHB. Queste indicazioni sono presenti nelle ultime linee guida del 2014, ma da allora le cose sono notevolmente cambiate; l’analisi, infatti, non viene più condotta tramite metodica Sanger di sequenziamento diretto, bensì con l’utilizzo della Next generation sequencing (NGS) (23), una metodica di sequenziamento di seconda generazione massiva e ad alta resa, che permette a minor costo l’analisi di pannelli di geni di suscettibilità.
Bibliografia
- Neumann HP, Berger DP, Sigmund G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel- Lindau disease. N Engl J Med 1993, 329: 1531–8.
- Eng C, Smith DP, Mulligan LM, et al. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum Mol Genet 1994, 3: 237-41. Erratum in: Hum Mol Genet 3: 686.
- Eng C. Seminars in medicine of the Beth Israel Hospital, Boston: the RET proto-oncogene in multiple endocrine neoplasia type 2 and Hirschsprung's disease. N Engl J Med 1996, 335: 943–51.
- White R, O'Connell P. Identification and characterization of the gene for neurofibromatosis type 1. Curr Opin Genet Develop 1991, 1: 15–9.
- Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287: 848–51.
- Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 2001, 69: 49–54.
- Burnichon N, Brière JJ, Libé R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Molec Genet 2010, 19: 3011–20.
- Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nature Genet 2000, 26: 268–70.
- Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325: 1139–42.
- Pigny PP, Vincent A, Cardot Bauters C, et al. Paraganglioma after maternal transmission of a succinate dehydrogenase gene mutation. J Clin Endocrinol Metab 2008, 93: 1609–15.
- Yeap PM, Tobias ES, Mavraki E, et al. Molecular analysis of pheochromocytoma after maternal transmission of SDHD mutation elucidates mechanism of parent-of-origin effect. J Clin Endocrinol Metab 2011, 96: E2009-13.
- Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res 2003, 63: 5615–21.
- Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nature Genet 2010, 42: 229–33.
- Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature Genet 2011, 43: 663–7.
- Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 2014, 23: 2440-6.
- Dahia LM, Ross KN, Wright ME, et al. A HIF1α regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 2005, 1: 72–80.
- Favier J, Brière JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 2009, 4: e7094.
- Califano D, Rizzo C, D'Alessio A, et al. Signaling through Ras is essential for ret oncogene-induced cell differentiation in PC12 cells. J Biol Chem 2000, 275: 19297–305.
- Martin GA, Viskochil D, Bollag G, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63: 843–9.
- Johannessen CM, Reczek EE, James MF, et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Nat Acad Sci USA 2005, 102: 8573–8.
- Johannessen CM, Johnson BW, Williams SG, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol 2008, 18: 56–62.
- Mannelli M, Castellano M, Schiavi F, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 2009, 94: 1541–7.
- NGS in PPGL (NGSnPPGL) Study Group, Toledo RA, et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol 2017, 13: 233-47.
Clinica e diagnostica feocromocitomi e paragangliomi
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
(aggiornato al 20 marzo 2020)
FEOCROMOCITOMA
La diagnosi viene di solito presa in considerazione:
- nei pazienti con una sintomatologia accessionale suggestiva per tale patologia;
- in presenza di incidentaloma surrenalico;
- in base all'anamnesi familiare, se il paziente fa parte di una famiglia con una forma sindromica di feocromocitoma.
Il feocromocitoma è caratterizzato da presentazione clinica molto variabile, con segni e sintomi frequentemente aspecifici e rilevabili in altre patologie (1).
La sintomatologia da eccesso di catecolamine comprende:
- ipertensione arteriosa (80-90% dei casi)
- cefalea (60-90%)
- tachicardia (50-70%)
- iperidrosi (55-75%)
- tremori, pallore (40-45%)
- ansia e anche attacchi di panico (20-40%).
Possono presentarsi alterazioni del metabolismo glucidico (40%) (insulino-resistenza, iperglicemia a digiuno, diabete mellito tipo 2), perdita di peso, acidosi lattica; meno frequenti risultano flushing, nausea, vomito, ipostenia. Altre manifestazioni cliniche comprendono febbre, angina, infarto miocardico (tipicamente a coronarie indenni), aritmie ipercinetiche, edema polmonare acuto, shock.
L’ipertensione arteriosa è il sintomo più caratteristico del feocromocitoma e può presentarsi in forma parossistica in circa il 30% dei pazienti, mentre nella maggioranza si rileva ipertensione stabile; a volte parossismi ipertensivi si verificano in pazienti con ipertensione stabile (da ricordare che ciò accade anche in pazienti ipertesi non portatori di feocromocitoma). Episodi di ipotensione ortostatica, inspiegabile in un paziente iperteso, devono fare sospettare la patologia. I pazienti con tumore a prevalente o sola produzione di adrenalina possono presentare episodi di severa ipotensione o shock (2).
La triade sintomatologica classica è costituita da cefalea, cardiopalmo e iperidrosi: se associati a crisi ipertensiva e pallore cutaneo, devono indurre a sospettare il feocromocitoma (specificità > 90%), ma va considerato che la maggioranza dei pazienti con feocromocitoma non presenta la triade sintomatologica.
Le crisi parossistiche, con improvvisi rialzi pressori e aritmie da rilascio di catecolamine, a volte costituiscono una vera emergenza e possono essere fatali per aritmie ipercinetiche, IMA, ictus, edema polmonare acuto. Possono insorgere spontaneamente, o in seguito a diverse condizioni scatenanti:
- esercizio fisico intenso;
- stimolanti: nicotina, caffeina, teofillina;
- assunzione di alimenti ricchi in tiramina (es. vino rosso, cioccolato, formaggio);
- traumi;
- farmaci: ß-bloccanti, anti-depressivi triciclici, metoclopramide, metil-DOPA;
- parto;
- induzione di anestesia;
- chirurgia o altre procedure invasive (endoscopie, cateterismi).
La durata dei parossismi è molto variabile, ma nella maggior parte dei pazienti va da pochi minuti a un’ora.
Nella MEN-2 il feocromocitoma è asintomatico in percentuali variabili dal 30 al 50% (3,4), verosimilmente in quanto la diagnosi è posta in seguito allo screening; anche un terzo dei pazienti con malattia di von Hippel-Lindau è asintomatico, con normali valori pressori (la liberazione di catecolamine è costante e in gran parte vengono trasformate in metanefrine nel tumore).
PARAGANGLIOMI SIMPATICI
Sono pressochè sempre caratterizzati da liberazione di catecolamine e i pazienti presentano metanefrine elevate.
Questi PGL secernenti sono in prevalenza situati a livello addominale e pelvico e, oltre alla sintomatologia tipica da aumentato rilascio di catecolamine, possono presentarsi con algia toracica o addominale, ematuria (in caso di localizzazione vescicale, con episodi parossistici che possono essere indotti da distensione vescicale o da minzione).
I PGL simpatici legati a mutazioni del gene SDHB possono presentarsi in forma subclinica, per basso contenuto di catecolamine, e scarsa differenziazione (5).
I pazienti con PGL dopamino-secernente sono in genere normotesi, a volte ipotesi.
PARAGANGLIOMI PARASIMPATICI
Nella maggior parte dei casi non liberano catecolamine (anche se nel 5% dei casi è stato descritto aumentato rilascio e conseguente sintomatologia tipica, vedi “Classificazione feocromocitoma e paragangliomi") e la diagnosi viene posta per la sintomatologia da effetto massa della lesione, o per riscontro incidentale nel corso di indagini di diagnostica per immagini, effettuate per altre patologie.
I PGL parasimpatici di testa e collo presentano una crescita lenta e possono rimanere clinicamente silenti per anni. La sintomatologia è caratterizzata da deficit dei nervi cranici, in particolare del nervo vago: acufeni, disfonia, senso di ripienezza faringea, paralisi facciale, difficoltà di movimento della lingua. I paragangliomi giugulo-timpanici presentano invece una sintomatologia precoce, rappresentata da acufene pulsatile, in alcuni casi associato a ipoacusia di tipo conduttivo.
FORME METASTATICHE
Complessivamente circa il 10% dei tumori secernenti catecolamine è metastatico.
Le metastasi possono presentarsi anche dopo 20 anni dalla rimozione del tumore primitivo. Questo è un argomento ancora dibattuto, in quanto se è vero che l’invasività locale ha uno scarso valore predittivo di metastasi nelle forme sporadiche, o in forme familiari come la MEN-2 (se l’intervento è stato adeguato pur in presenza di invasione locale estesa), in un paziente con mutazione di SDHB anche minimi aspetti invasivi assumono sicuramente un valore predittivo più severo.
Le grandi dimensioni della neoplasia (> 5-6 cm) sono più frequentemente associate a metastasi a distanza.
Nel 2002 Thompson ha proposto il sistema PASS (Pheochromocytoma Adrenal Scaled Score), basato sulla valutazione di vari caratteri istopatologici, che poi si è rivelato di scarsa utilità, per l’ampia variabilità inter- e intra-valutatore (6).
| Sistema PASS | |
| Caratteristiche microscopiche | Punti |
| Invasione capsulare | 1 |
| Invasione vascolare | 1 |
| Invasione del tessuto adiposo peri-surrenalico | 2 |
| Presenza di nidi cellulari o crescita diffusa (in > 10% del volume tumorale) | 2 |
| Necrosi centrale o confluente | 2 |
| Alta cellularità | 2 |
| Aspetto fusato delle cellule tumorali anche se focale | 2 |
| Monotonia cellulare | 2 |
| Aumento del numero delle figure mitotiche | 2 |
| Figure mitotiche atipiche | 2 |
| Pleomorfismo nucleare accentuato | 1 |
| Ipercromasia nucleare | 1 |
| Punteggio massimo totale | 10 |
Ciò che è emerso da molti studi clinici è che i maggiori fattori predittivi di metastatizzazione sono:
- la localizzazione extra-surrenalica dei tumori,
- la presenza di mutazione SDHB,
- le dimensioni della neoplasia.
Inoltre, valori particolarmente elevati di metossitiramina plasmatica rappresentano un marcatore predittivo di malattia aggressiva in relazione al suo basso grado di differenziazione (7).
In presenza degli elementi sopra descritti, è indicato valutare i pazienti con tecniche di immagine morfologiche e funzionali per l’individuazione di lesioni secondarie.
La sopravvivenza a 5 anni delle forme metastatiche è stimata tra 34 e 60%, in base alla localizzazione delle metastasi: l’aspettativa di vita è minore se sono interessati sia scheletro che fegato, rispetto alla sola localizzazione scheletrica (8).
DIAGNOSI BIOCHIMICA
La diagnosi di feocromocitoma/paraganglioma si fonda sull’evidenza di alterata secrezione di catecolamine e/o metanefrine.
Il metabolismo delle catecolamine avviene principalmente a livello intra-tumorale, per metilazione ad opera della catechol-O-metiltransferasi, con formazione di metanefrina da adrenalina, normetanefrina da noradrenalina e metossitiramina da dopamina. Il rilascio di catecolamine può essere modesto, assente o parossistico, mentre il rilascio di metanefrine è costante, essendo continuo il processo di metilazione, e pertanto la determinazione di metanefrine, libere plasmatiche o frazionate urinarie, è più sensibile rispetto a quella di catecolamine.
Nel sospetto di feocromocitoma/paraganglioma la raccomandazione quindi, confermata dalle ultime Linee Guida dell'Endocrine Society (9), è di iniziare la valutazione biochimica con determinazione di metanefrine libere plasmatiche e/o metanefrine frazionate urinarie, in base alla disponibilità del laboratorio di riferimento. Numerosi studi hanno stabilito una sensibilità diagnostica particolarmente elevata delle metanefrine libere plasmatiche rispetto alle urinarie (10). La fase pre-analitica è delicata e vanno osservate alcune precise regole per garantire la correttezza dell'esame: dosaggio in posizione supina, dopo almeno 30 minuti di riposo a letto per limitare l'influenza della postura eretta e dell'attivazione simpatica sui livelli di catecolamine; evitare il prelievo in posizione seduta (11,12). Tuttavia, tale metodo non è universalmente disponibile; in caso di indisponibilità, si può ricorrere alla determinazione di metanefrine frazionate urinarie, gravate da minor sensibilità rispetto alle plasmatiche ma da un'accuratezza diagnostica nettamente superiore rispetto alla determinazione delle catecolamine (8,13). Nonostante la maggiore praticità che comporterebbe il dosaggio delle metanefrine sulle urine spot del mattino, viene raccomandato il dosaggio sulle urine delle 24 ore; il contestuale dosaggio della creatinina urinaria può rivelarsi utile per accertare l’adeguatezza della raccolta. Valori di metanefrine plasmatiche superiori a 2 volte il range di norma sono da considerarsi altamente sospetti per feocromocitoma/paraganglioma, anche in presenza di una bassa probabilità pre-test (14). Valori normali di metanefrine escludono il feocromocitoma, con la sola eccezione delle forme che producono unicamente dopamina: se vengono determinate solo le metanefrine, si può correre il rischio di non diagnosticare queste neoplasie. Pertanto, se il sospetto clinico di feocromocitoma/ paraganglioma è elevato (familiarità per feocromocitoma/ paraganglioma, sindrome genetica che predispone a feocromocitoma/ paraganglioma, incidentaloma surrenalico con caratteri TC o RM suggestivi per feocromocitoma) e le metanefrine sono normali, si deve ricorrere al dosaggio di metossitiramina plasmatica o urinaria (15). È recente il rilievo che oltre il 60% dei pazienti con mutazione SDHB o SDHD presenta anche un aumento di metossitiramina plasmatica (16). Il dosaggio di metossitiramina viene effettuato ancora in pochi laboratori.
Qualora il test basale dia un risultato non sicuramente diagnostico, vanno considerati possibili falsi positivi, e la determinazione va ripetuta evitando fattori interferenti, se possibile. Va ricordato che un rialzo di catecolamine e dei loro metaboliti, può essere indotto da:
- altre condizioni fisiologiche o patologiche: stress, dolore, esercizio fisico, fumo, freddo, fase luteinica del ciclo mestruale, ipoglicemia, insufficienza renale cronica, interventi chirurgici, IMA, ictus, sindrome delle apnee notturne, ipotiroidismo, neuroblastoma e raramente ganglioneuroma;
- farmaci: clozapina, α-bloccanti (in particolare la fenossibenzamina), β-bloccanti, calcio-antagonisti se somministrati in acuto, nitroglicerina, α-metil-DOPA, inibitori MAO, simpatico-mimetici, levo-DOPA, carbi-DOPA, ASA, furosemide, eritromicina;
- caffeina, teofillina, alcool, cocaina.
Nonostante l’alta sensibilità delle metanefrine (sono elevate in quasi tutti i casi di PPGL), non sempre la loro positività implica la presenza di un PPGL (falso positivo, FP). I FP sono comuni: rappresentano il 19-21% sia nel dosaggio delle metanefrine plasmatiche che urinarie. Pertanto, nei pazienti che hanno livelli di metanefrine plasmatiche e/o urinarie persistentemente superiori alla norma ma nella zona di incertezza diagnostica, anche dopo l’esclusione di fattori interferenti, si può ricorrere al test di soppressione con clonidina, descritto per la prima volta nel 1981 (17). Il test consiste nella somministrazione di clonidina per os, 300 μg, con successivi prelievi ematici per determinazione di noradrenalina o normetanefrina. Il suo uso non è stato effettivamente validato in alcuno studio prospettico.
Alcuni autori suggeriscono di effettuare anche il dosaggio plasmatico di cromogranina A, che è co-rilasciata con le catecolamine, al fine di incrementare l’accuratezza diagnostica, nei casi in cui le metanefrine risultano elevate ma non sicuramente diagnostiche (18). Va ricordato che elevati valori di cromogranina A in assenza di feocromocitoma (FP) possono riscontrarsi in pazienti che assumono inibitori di pompa protonica (da sospendere per due settimane prima della determinazione), o con gastrite cronica atrofica o con insufficienza renale.
I test di stimolo non sono più utilizzati non solo in quanto obsoleti, ma anche perchè pericolosi, potendo indurre crisi ipertensive.
Il tipo di secrezione del tumore ci può indirizzare sulla localizzazione della lesione e sul tipo di sindrome ereditaria:
- nella MEN-2, la localizzazione è quasi sempre surrenalica e si rileva incremento di metanefrina e normetanefrina;
- nella malattia di von Hippel-Lindau i feocromocitomi/paragangliomi producono solo noradrenalina;
- le neoplasie correlate a mutazione dei geni SDHB o SDHD sono caratterizzate da rilascio di noradrenalina, con riscontro di valori aumentati di normetanefrina e talora di metossitiramina.
Una volta diagnosticata una forma sindromica, è indicato procedere allo screening delle altre neoplasie endocrine e non endocrine che si possono associare e allo screening genetico dei familiari.
DIAGNOSTICA PER IMMAGINI
Effettuata la diagnosi biochimica di feocromocitoma o paraganglioma, è necessario localizzare la neoplasia e ci si avvale di metodiche di immagine anatomica e funzionale.
Per le prime si ricorre a TC o RMN. La TC è in genere la metodica più utilizzata. Il feocromocitoma presenta elevati valori di HU (Unità Hounsfield: 40-50), quando non sono presenti aree emorrragiche o necrotiche. La vascolarizzazione è intensa, con marcato enhancement durante la fase contrastografica. La TC multistrato è preferibile per individuare piccoli PGL toracici o addominali: la sensibilità complessiva è variabile dall'85 al 94% (19), mentre per il feocromocitoma la sensibilità è del 98%, con specificità del 92%. Recente è la segnalazione della non necessità di preparare i pazienti con alfa-litici per l'effettuazione di TC con gli attuali mezzi di contrasto non ionici a bassa osmolarità (20).
Per i PGL della testa collo si ricorre a RM, ed eventualmente ad angioTC del collo; la RM è anche la metodica da utilizzare per le rare forme cardiache, e l’indagine di prima scelta nei bambini, giovani adulti, donne in gravidanza o durante l'allattamento, per minimizzare l’esposizione a radiazioni ionizzanti. I feocromocitomi/paragangliomi mostrano un elevato segnale nelle sequenze RM T2-pesate: la sensibilità varia da 93 a 100% per i feocromocitomi, è di circa 90% per i PGL, e la specificità è estremamente variabile (50-100%) (18).
Le indagini funzionali possono avere un doppio significato (21): in passato venivano richieste durante l’inquadramento diagnostico al fine di confermare, se positive, la natura cromaffine della massa evidenziata alla TC o RM. In realtà queste indagini sono anche in grado di dare informazioni circa la presenza/assenza di altre lesioni cromaffini o metastatiche e ad oggi l’utilizzo delle tecniche di medicina nucleare durante la fase diagnostica è da riservare principalmente a pazienti con elevato rischio di malattia metastatica, in cui la 123I-MIBG e la PET con 68Ga-DOTANOC permettono di valutare la possibilità di utilizzare la terapia radiometabolica (131I-MIBG e 90Y-DOTATOC o 177Lu-DOTATATE) (22,23). Recenti studi hanno infatti dimostrato che l’utilità in particolare della 123I-MIBG durante la fase diagnostica è limitata, in quanto raramente incrementa l’accuratezza diagnostica della TC o RM, potendo causare una non corretta interpretazione dei risultati (24).
La scelta dei traccianti da utilizzare dipende dalle caratteristiche genetiche del paziente: mentre la PET con Gallio rappresenta la prima scelta nei pazienti con forme sporadiche, multifocali, metastatiche e legate a mutazioni dei geni della succinato deidrogenasi, la F-DOPA è la prima scelta in pazienti con forme familiari, fatta eccezione per i tumori SDHx.
La scintigrafia con 123I-MIBG (da preferire a 131I-MIBG per la maggiore sensibilità e minore esposizione a radiazioni) ha una sensibilità variabile dal 77 al 98% per le neoplasie non metastatiche (25), sensibilità che si riduce al 50-79% nelle lesioni metastatiche (con una sensibilità inferiore per le neoplasie extra-surrenaliche), e specificità prossima al 100%. Falsi negativi alla scintigrafia con 123I-MIBG sono più comunemente associati a feocromocitomi/paragangliomi correlati a mutazione del gene SDHB (26). Nella valutazione dei pazienti va ricordato che la 123I-MIBG evidenzia le ghiandole surrenaliche normali nel 50-80% dei casi.
La 18F-DOPA-PET ha dimostrato un’elevata sensibilità sia per i feocromocitomi sia per i PGL (25,27,28), anche di collo e testa: 81–100% (29,30). L’accuratezza diagnostica di 18F-DOPA-PET è potenziata dall’utilizzo di carbiDOPA, inibitore della DOPA-decarbossilasi. La sensibilità di questa diagnostica si riduce in modo significativo nelle forme metastatiche di feocromocitoma/paraganglioma (45%) e ancor più in quelle sostenute da mutazione del gene SDHB (20%) (25).
La 18F-FDG-PET ha dimostrato una sensibilità dell’88-90% nella localizzazione delle neoplasie non metastatiche, e una maggiore sensibilità (97%) nel rilevare quelle metastatiche, in particolare se correlate a mutazione del gene SDHB (31); la specificità è prossima al 90%.
Nei pazienti con neoplasie che esprimono recettori per la somatostatina, la PET con 68Ga-DOTATOC o DOTANOC (analoghi della somatostatina) può rivelare un’accuratezza diagnostica superiore alla scintigrafia con 123I-MIBG, alla TC o alla RM, fornendo importanti informazioni nella stadiazione pre-terapeutica (32,33) e permettendo di individuare i pazienti con malattia metastatica che potrebbero essere trattati con terapia radiometabolica con 90Y-DOTATOC o 177Lu-DOTATATE.
BIBLIOGRAFIA
- Lenders JWM, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet 2005, 366: 665-75.
- Olson SW, Deal LE, Piesman M. Epinephrin-secreting pheochromocytoma presenting with cardiogenic shock and profound hypocalcemia. Ann Intern Med 2004, 140: 849-51.
- Nguyen L, Niccoli-Sire P, Caron P, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. Eur J Endocrinol 2001, 144: 37-44.
- Brunt M, Lairmore TC, Doherty GM, et al. Adrenalectomy for familial pheochromocytoma in the laparoscopic era. Ann Surg 2002, 235: 713-21.
- Mannelli M, Lenders JW, Pacak K, et al. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab 2012, 26: 507-15.
- Thompson LD. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002, 26: 551-66.
- Eisenhofer G, Lenders JWM, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012, 48: 1739-49.
- Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 2007, 3: 92-102.
- Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014, 99: 1915–42.
- Eisenhofer G, Prejbisz A, Peitzsch M, et al. Biochemical diagnosis of chromaffin cell tumors in patients at high and low risk of disease: Plasma versus urinary free or deconjugated O-methylated catecholamine metabolites. Clin Chem 2018, 64: 1646–56.
- Boyd J, Leung AA, Sadrzadeh H, et al. A high rate of modestly elevated plasma normetanephrine in a population referred for suspected PPGL when measured in a seated position. Eur J Endocrinol 2019, 181: 301-9.
- Darr R, Pamporaki C, Peitzsch M, et al. Biochemical diagnosis of phaeochromocytoma using plasma-free normetanephrine, metanephrine and methoxytyramine: Importance of supine sampling under fasting conditions. Clin Endocrinol 2014, 80: 478–86.
- Grossman A. Pacak K, Sawka A, et al. Biochemical diagnosis and localization of pheochromocytoma: can we reach a consensus? Ann N Y Acad Sci 2006, 1073: 332-47.
- Eisenhofer G, Klink B, Richter S, et al. Metabologenomics of phaeochromocytoma and paraganglioma: An integrated approach for personalised biochemical and genetic testing. Clin Biochem Rev 2017, 38: 69–100.
- Eisenhofer G, Siegert G, Kotzerke J, et al. Current progress and future challenges in the biochemical diagnosis and treatment of pheochromocytomas and paraganglioma. Horm Metab Res 2008, 40: 329-37.
- Eisenhofer G, Lenders JWM, Timmers H, et al. Measurements of plasma metoxytyramine, normetanephrine, and metanephrine as a discriminators of different hereditary forms of pheochromocytoma. Clin Chem 2011, 57: 411-20.
- Bravo EL, Tarazi RC, Fouad FM, et al. Clonidine-suppression test: e useful aid in the diagnosis of pheochromocytoma. N Engl J Med 1981, 305: 623-6.
- Algeciras-Schimnic A, Preissner CM, Young WF, et al. Plasma chromogranin A or urine fractionated metanephrines follow-up testing improves the diagnostic accuracy of plasma fractionated metanephrines for pheochromocytoma. J Clin Endocrinol Metab 2008, 93: 91-5.
- Ilias I, Pacak K. Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab 2004, 89: 479-91.
- Baid SK, Lai EW, Wesley RA, et al. Radiographic contrast infusion and catecholamine release in patients with pheochromocytoma. Ann Intern Med 2009, 150: 27-32.
- Timmers HJ, Taieb D, Pacak K. Current and future anatomical and functional imaging approaches to pheochromocytoma and paraganglioma. Horm Metab Res 2012, 44: 367-72.
- Taïeb D, Hicks RJ, Hindié E, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2019, 46: 2112-37.
- Nölting S, Ullrich M, Pietzsch J, et al. Current management of pheochromocytoma/ paraganglioma: a guide for the practicingclinician in the era of precision medicine. Cancers (Basel) 2019, 11: 1505.
- Rao D, van Berkel A, Piscaer I, et al. Impact of 123I-MIBG scintigraphy on clinical decision making in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2019, 104: 3812-20.
- Timmers HJ, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 2009, 94: 4757-67.
- Fonte J, Roblets J, Reynolds J, et al. False-negative 123I-MIBG SPECT is most commonly found in SDHB-related pheochromocytoma or paraganglioma with frequency to develop metastatic disease. Endocr Relat Cancer 2012, 19: 83-93.
- Taieb D, Tessonnier L, Sebag F, et al. The role of 18F-FDOPA and 18F-FDG-PET in the management of malignant and multifocal phaechromocytomas. Clin Endocrinol 2008, 69: 580-6.
- Fiebrich HB, Brouwers AH, Kerstens MN, et al. 6-[F-18]Fluoro-L-dihydroxyphenylalanine positron emission tomography is superior to conventional imaging with 123I-metaiodobenzylguanidine scintigraphy, computed tomography, and magnetic resonance imaging in localizating tumors causing catecholamine excesse. J Clin Endocrinol Metab 2009, 94: 3922-30.
- Hoegerle S, Ghanem N, Altehoefer C, et al. 18F-DOPA positron emission tomography for the detection of glomus tumours. Eur J Nucl Med Mol Imaging 2003, 30: 689-94.
- King KS, Chen CC, Alexopoulos DK, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-Metaiodobenzylguanidine scintigraphy, and 111I-pentetreotide scintigraphy. J Clin Endocrinol Metab 2011, 96: 2779-85.
- Timmers HJ, Kozupa A, Chen CC, et al. Superiority of fluorodeoxyglucose positron emission tomography to other functional imaging techniques in the evaluation of metastatic SDHB-associated pheochromocytoma and paraganglioma. J Clin Oncol 2007, 25: 2262-9.
- Kroiss A, Putzer D, Uprimny C, et al. Functional imaging in phaeochromocytoma and neuroblastoma with 68Ga-DOTA-Tyr3-octreotide positron emission tomography and 123I- metaiodobenzylguanidine. Eur J Nucl Med Mol Imaging 2011, 38: 865-73.
- Nawsa N, Sharma P, Nazar AH, et al. Prospective evaluation of 68Ga-DOTA-NOC PET-CT in phaeochromocytoma and paraganglioma: preliminary results from a single centre study. Eur Radiol 2012, 22: 710-9.
Feocromocitoma e gravidanza
Letizia Canu
Dipartimento di Scienze Biomediche Sperimentali e Cliniche “Mario Serio”, Università di Firenze
(aggiornato all'11 maggio 2021)
Uno studio internazionale, multicentrico, retrospettivo (1) su 232 pazienti e 249 gravidanze ha cercato di identificare i fattori associati all’esito materno e fetale, valutando la morte materna e fetale e lo sviluppo di severe complicanze cardio-vascolari materne dovute all’eccesso di catecolamine durante la gravidanza, al momento del parto o entro 3 giorni dal parto.
La maggior parte delle pazienti presentava una singola lesione surrenalica (62%), < 10% presentava lesioni secondarie e il 66% sono risultate positive all’analisi genetica. Quarantanove pazienti (25%) avevano familiarità positiva per PPGL. Come atteso in relazione alla sede prevalente delle lesioni, la maggior parte erano lesioni funzionanti (95%). Nella metà dei casi i livelli di catecolamine erano più di 10 volte sopra il limite superiore di riferimento (ULN). Quarantadue donne (18%) sono state sottoposte a intervento chirurgico durante la gravidanza (tabella 1).
| Tabella 1 | |
| Diametro della lesione (n = 190): mediana 53 mm (range 13-310) | |
| Localizzazione (n =230) | FEO unilaterale: 142 (62%) FEO bilaterale: 19 (8%) PGL testa-collo: 5 (2%) PGL toracico: 6 (3%) PGL addominale o pelvico: 27 (12%) PPGL multipli: 11 (5%) PPGL metastatico: 20 (9%) |
| Secrezione (n = 232) |
Non funzionanti: 12 (5%)
|
| Storia familiare per PPGL (n = 194) | Positiva: 49 (25%) Negativa: 145 (75%) |
| Eccesso di catecolamine (rispetto a ULN) (n = 178) | 2x: 34 (19%) 5x: 29 (16%) 10x: 26 (15%) > 10x: 89 (50%) |
| Chirurgia (n = 231) | Durante la gravidanza: 42 (18%), (mediana 20° settimana di gestazione, range 10-35) Dopo la gravidanza:161 (70%), (mediana 8° settimana post-partum, range 0-224) Non chirurgia: 28 (12%) |
| Analisi genetica (n = 232) | RET: 28 (12%) SDHB: 27 (12%) VHL: 18 (8%) SDHD: 8 (3%) NF1: 5 (2%) MAX: 2 (1%) SDHC: 2 (1%) SDHA: 1 FH: 1 CDKN2B: 1 Triade di Carney: 2 (1%) Analisi negativa: 49 (21%) Analisi non eseguita: 88 (38%) |
In più della metà dei casi la diagnosi è stata posta durante la gravidanza (54%), mentre nel 31% dopo il parto. Circa la metà delle pazienti con malattia nota prima della gravidanza presentava metastasi (46%) e il 54% di queste non era stato precedentemente sottoposto a intervento chirurgico.
Più dell’80% delle pazienti con malattia funzionante presentava segni e sintomi riferibili agli alti livelli di catecolamine (83%), in particolare ipertensione (93%), palpitazioni (57%), cefalea (50%), sudorazione (42%).
In 11 casi si è verificato un aborto spontaneo (4%), mentre in 8 pazienti è stato necessario un aborto programmato (3%); 168 pazienti (74%) hanno portato a termine la gravidanza, la maggior parte delle quali con parto cesareo. Cinquantotto pazienti hanno avuto un parto pre-termine: cesareo (90%) o naturale (10%). Nelle pazienti con diagnosi pre-gravidanza, la scelta del parto cesareo è stata effettuata prevalentemente per coloro che presentavano sintomi da eccesso di catecolamine (p = 0.014) e livelli > 10 volte ULN (p 0.0023).
La maggior parte delle pazienti (161, 70%) è stata sottoposta a intervento chirurgico dopo il parto, 42 durante la gravidanza (40/42 con forma secernente). Solo 33/40 pazienti sono state preparate all’intervento con l’utilizzo di alfa-bloccanti.
I dati relativi all’esito materno e fetale erano disponibili per 230 pazienti: decesso materno in 3 casi (1%), decesso fetale in 20 casi (9%), complicanze cardio-vascolari severe in 15 (7%) (tabella 2).
| Tabella 2 (n = 249 gravidanze) | |
| Età alla gravidanza | 28 (15-46)* |
| Anno della gravidanza (n = 214) | 2012 (1980-2019)* |
| Segni e sintomi da eccesso di catecolamine | Sì: 206 (83%) No: 43 (17%) |
| Diagnosi di PPGL |
Prima della gravidanza: 37 (15%) |
| Gestione durante la gravidanza (n = 146) | Chirurgia: 42 (17%), 20° settimana di gestazione (10-35)* Terapia medica (alfa-bloccante): 104 (42%) (8 settimane di terapia, 0-40)* |
| Tipo di parto | Parto cesareo: 146 (59%), 36° settimana di gestazione (25-41*) Parto naturale:76 (31%), 38° settimana di gestazione (28-41*) Non noto: 3 (1%), 38° settimana di gestazione (36-39*) Parto naturale di urgenza: 1 (< 1%), 20° settimana di gestazione** Interruzione di gravidanza programmata: 8 (3%), 9° settimana di gestazione (3-22*) Aborto spontaneo: 11 (4%), 18° settimana di gestazione (8-37*) Gravidanza in corso: 2 (1%), 25° e 26° settimana di gestazione** |
| Esito materno (n = 230) | Decesso: 3 (1%) Complicanze cardio-vascolari severe: 15 (7%) Nessuna complicanza: 212 (92%) |
| Esito fetale (n = 230) | Decesso: 20 (9%) Nessuna complicanza: 210 (91%) |
| *mediana (range); **valori per le singole gravidanze | |
Fattori di rischio per un esito peggiore
L’esito sfavorevole ha interessato solo pazienti con forme funzionanti e con diagnosi durante o dopo la gravidanza: il mancato riconoscimento di malattia durante la gravidanza e la localizzazione addominale sono risultati fortemente associati alla comparsa di complicanze materne e fetali (OR 27, IC95% 3.5-3473 e OR 11.3, IC95% 1.5-1440). In particolare, sono risultati fattori prognostici sfavorevoli il livello di catecolamine > 10 volte ULN (OR 4.7, IC95% 1.8-13.8) e la mancanza di terapia con alfa-bloccante (OR 3.6, IC95% 1.1-13.2). La chirurgia durante la gravidanza non è risultata in grado di influenzare positivamente l’esito della gravidanza (OR 0.9, IC95% 0.3-3.9). Neanche l’età della madre, l’anno di gravidanza, le dimensioni del tumore e il tipo di parto sono risultate associate a esito migliore. Il numero di complicanze è stato maggiore nelle pazienti senza forme familiari note (OR 2.7, IC95% 1.1-6.6) e senza malattia metastatica (OR 9.9, IC95% 1.3-1268).
Nel corso degli anni si è assistito a un miglioramento del decorso delle gravidanze in donne con malattia cromaffine. Gli autori hanno considerato diversi fattori come possibili responsabili dell’esito della gravidanza:
- la diagnosi di PPGL prevede il dosaggio dei metaboliti delle catecolamine sul plasma o sulle urine delle 24 ore. Poiché la gravidanza non ne altera la concentrazione, gli intervalli di riferimento durante la gestazione non differiscono da quelli comunemente utilizzati (3);
- visto che nella maggior parte dei casi le lesioni secernenti si localizzano a livello surrenalico, è indicato richiedere un’ecografia dell’addome superiore in quelle donne che durante la gravidanza presentino ipertensione o altra sintomatologia non altrimenti spiegabile (4);
- le dimensioni tumorali (mediana di circa 5 cm) spiegano come nella maggior parte delle pazienti i valori di catecolamine fossero > 10 volte ULN, anche se il diametro di per sé non è risultato associato a peggior esito, diversamente dalla localizzazione addominale della lesione (associato a lesioni di natura simpatergica e quindi funzionanti). Appare meno chiara la mancanza di sintomatologia nonostante valori così elevati di catecolamine;
- l’esito migliore in pazienti con forme sindromiche o metastatiche è verosimilmente legato al fatto che nella maggior parte dei casi si trattava di pazienti con diagnosi di malattia pre-gravidanza, quindi già sottoposte a follow-up e terapia medica quando necessaria;
- in molti casi i pazienti con forme metastatiche presentano un decorso di malattia indolente e le donne affette che decidono di cercare una gravidanza sono solitamente quelle con minor carico di malattia. Infatti, anche se circa la metà delle pazienti con secondarismi non erano state sottoposte a chirurgia, verosimilmente in relazione all’impossibilità di un intervento radicale, si trattava comunque di pazienti in buon controllo medico di malattia;
- meno chiaro è perché solo 33/40 pazienti sottoposte a chirurgia siano state preparate con alfa-bloccanti, visto che l’instabilità legata agli elevati livelli di catecolamine rappresenta la motivazione che più frequentemente induce all’intervento durante la gravidanza. L’utilizzo di alfa-bloccanti si è rivelato in grado di migliorare l’esito sia materno che fetale. Tali esiti sono risultati migliori rispetto a quanto riportato in precedenza (2), con la mortalità materna che è passata dal 48% all’1% e quella fetale dal 54% al 9%, verosimilmente in relazione al miglioramento delle conoscenze in campo ostetrico, chirurgico, anestesiologico e farmacologico;
- nelle donne che presentano un eccesso di catecolamine durante la gravidanza è necessario raggiungere un delicato equilibrio tra la riduzione degli effetti delle catecolamine che possono causare la compromissione del flusso placentare (vaso-costrizione) e l’utilizzo di una dose eccessiva di alfa-bloccante che potrebbe abbassare i livelli pressori a valori tali da compromettere la circolazione utero-placentare (5). I maggiori rischi per il feto derivano dalla vaso-costrizione da eccesso di catecolamine che può causare distacco placentare, ipossia, ritardo di crescita intra-uterina e morte fetale (6);
- non sono stati riportati effetti collaterali nei bambini nati da madri trattate con alfa-litico, in particolare doxazosina (7), nonostante questo farmaco attraversi la barriera placentare e potenzialmente possa indurre ipotensione fetale e depressione respiratoria (8). Il farmaco è classificato dall’FDA come classe C in gravidanza (non è possibile escludere che l’assunzione possa causare effetti collaterali; l’uso è indicato solo qualora i potenziali benefici superino i rischi);
- l’uso della doxazosina è controindicato in allattamento, perché è dimostrato che il farmaco si accumula nel latte di modelli murini (8).
Conclusioni
Non ci sono dati riguardanti ridotta fertilità in donne affette da patologia cromaffine.
Non ci sono dati che controindichino l’utilizzo della contraccezione ormonale in donne affette da feocromocitoma/paraganglioma.
È necessaria un’attenta valutazione clinica, biochimica e radiologica nelle pazienti affette da patologia cromaffine in previsione di una gravidanza.
Non sono disponibili dati per consigliare la scelta di un particolare tipo di alfa-bloccante e il suo dosaggio, che deve avere lo scopo di raggiungere un adeguato controllo pressorio.
L’intervento chirurgico durante la gravidanza non ne migliora l’esito.
Non è possibile fare una raccomandazione specifica sul tipo di parto consigliato, anche se il parto naturale è considerato sicuro in pazienti selezionate (in buon controllo pressorio).
La maggior parte delle gravidanze ha esito positivo.
La somministrazione di doxazosina in allattamento è controindicata a causa dei dati relativi al suo accumulo nel latte di modelli murini. Sono stati riportati livelli indosabili di doxazosina nel latte materno a distanza di 30 ore dall’ultima assunzione del farmaco. Considerando l’emivita della doxazosina, gli autori hanno concluso che i livelli del farmaco fossero bassi durante l’assunzione del farmaco (8).
Bibliografia
- Bancos I, Atkinson E, Eng C, et al; International Pheochromocytoma and Pregnancy Study Group. Maternal and fetal outcomes in phaeochromocytoma and pregnancy: a multicentre retrospective cohort study and systematic review of literature. Lancet Diabetes Endocrinol 2021, 9: 13-21.
- Dong D, Li H. Diagnosis and treatment of pheochromocytoma during pregnancy. J Matern Fetal Neonatal Med 2014, 27: 1930-4.
- Mannelli M, Bemporad D. Diagnosis and management of pheochromocytoma during pregnancy. J Endocrinol Invest 2002, 25: 567-71.
- Schenker JG, Chowers I. Pheochromocytoma and pregnancy. Review of 89 cases. Obstet Gynecol Surv 1971, 26: 739-47.
- Iijima S. Impact of maternal pheochromocytoma on the fetus and neonate. Gynecol Endocrinol 2019, 35: 280-6.
- Lenders JWM. Pheochromocytoma and pregnancy: a deceptive connection. Eur J Endocrinol 2012, 166: 143-50.
- Wing LA, Conaglen JV, Meyer-Rochow GY, Elston MS. Paraganglioma in pregnancy: a case series and review of the literature. J Clin Endocrinol Metab 2015, 100: 3202-9.
- Versmissen J, Koch BC, Roofthooft DW, et al. Doxazosin treatment of phaeochromocytoma during pregnancy: placental transfer and disposition in breast milk. Br J Clin Pharmacol 2016, 82: 568-9.
Terapia feocromocitomi e paragangliomi
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
(aggiornato al 20 marzo 2020)
PREMESSE E OBIETTIVI DEL TRATTAMENTO
La gestione del paziente con feocromocitoma/paraganglioma deve avvalersi della stretta collaborazione fra clinico, anestesista e chirurgo, tutti con esperienza nel trattamento di questa patologia.
La terapia è chirurgica (1). Nel caso di tumore sporadico non metastatico, che rappresenta l’evenienza largamente più frequente, la completa asportazione del tumore determina quasi sempre la guarigione del paziente. Va comunque ricordato che a tutt’oggi non è possibile differenziare da un punto di vista anatomo-patologico le forme indolenti da quelle aggressive; è possibile, infatti, solo effettuare una stratificazione del rischio di metastasi con l’utilizzo di punteggi anatomo-patologici (il più utilizzato è il PASS score).
| Sistema PASS | |
| Caratteristiche microscopiche | Punti |
| Invasione capsulare | 1 |
| Invasione vascolare | 1 |
| Invasione del tessuto adiposo peri-surrenalico | 2 |
| Presenza di nidi cellulari o crescita diffusa (in > 10% del volume tumorale) | 2 |
| Necrosi centrale o confluente | 2 |
| Alta cellularità | 2 |
| Aspetto fusato delle cellule tumorali anche se focale | 2 |
| Monotonia cellulare | 2 |
| Aumento del numero delle figure mitotiche | 2 |
| Figure mitotiche atipiche | 2 |
| Pleomorfismo nucleare accentuato | 1 |
| Ipercromasia nucleare | 1 |
| Punteggio massimo totale | 10 |
Come detto in precedenza, oggi non è più valida la differenziazione tra forme benigne e maligne di feocromocitoma/paraganglioma. In passato la diagnosi di malignità si basava sulla presenza di metastasi, cioè di tessuto cromaffine in tessuti che normalmente ne sono privi, come i linfonodi, il fegato, il polmone, le ossa. Ad oggi tutti i feocromocitomi/paragangliomi sono classificati come tumori potenzialmente maligni.
TERAPIA CHIRURGICA
Nei casi di feocromocitoma sporadico isolato la tecnica chirurgica di prima scelta è quella laparoscopica. Chirurghi esperti sono capaci di adottare questa tecnica anche per tumori di dimensioni > 6 cm, anche se le linee guida consigliano un approccio open in tumori di tali dimensioni (2,3).
Nel caso di feocromocitoma a localizzazione surrenalica bilaterale o recidivante, come nelle forme familiari, è da consigliare, ove possibile, l’asportazione del tumore con conservazione della corticale del surrene almeno da un lato (adrenal sparing), per evitare l’insorgere di insufficienza surrenalica cronica (4,5).
TERAPIA FARMACOLOGICA
Ruolo
È di enorme importanza per preparare il paziente all’intervento chirurgico, per il trattamento delle complicanze operatorie e per la terapia dei feocromocitomi metastatici o comunque non operabili.
Farmaci
Gli alfa-bloccanti sono il cardine della terapia medica: si distinguono quelli selettivi (alfa-1 antagonisti), come la doxazosina, e quelli non selettivi (alfa-1 e alfa-2 antagonisti), come la fenossibenzamina (6,7).
La doxazosina è un inibitore competitivo e, pur avendo emivita simile a quella della fenossibenzamina (circa 20-24 ore), può essere spiazzata dai recettori da alti livelli circolanti di catecolamine tumorali. Ne consegue che è raccomandabile somministrare il farmaco in più dosi giornaliere.
La fenossibenzamina, non disponibile in Italia, è un inibitore non competitivo somministrabile per os, che comporta un blocco, oltre che degli alfa-recettori post-sinaptici vascolari (alfa-1) anche di quelli pre-sinaptici (alfa-2).
Questi farmaci non interferiscono sul rilascio delle catecolamine tumorali, ma ne limitano gli effetti periferici, inibendo l’azione delle catecolamine sui recettori vascolari alfa-adrenergici che mediano la vaso-costrizione. Gli alfa-antagonisti inducono, infatti, vaso-dilatazione, con conseguente normalizzazione della pressione arteriosa. Essi permettono inoltre l’espansione del volume circolante, che risulta ridotto nei pazienti con feocromocitoma, e annullano la ridotta espressione (down-regulation) dei recettori alfa vascolari determinata dall’eccessiva esposizione agli alti livelli di catecolamine. Tali azioni sono fondamentali per impedire le gravi crisi ipotensive, talora irreversibili, che possono seguire all’asportazione del tumore.
Altro farmaco ipotensivo somministrabile per os è la nifedipina (tabella).
| Gestione peri-operatoria del paziente con feocromocitoma/paraganglioma | ||
| Fase | Farmaci | Posologia e modalità di somministrazione |
| Pre-operatoria | Doxazosina oppure Fenossibenzamina |
Iniziare con 2 mg/die (dose massima 16-20 mg/die per os in 2-3 somministrazioni) |
| Iniziare con 10 mg/die per os (dose massima 1 mg/kg/die, in mono-somministrazione) § | ||
|
Solo dopo aver praticato un adeguato trattamento con alfa-bloccanti Propranololo oppure Atenololo oppure Metoprololo |
|
|
| Iniziare con 10 mg x 4/die per os (dose massima 240 mg/die in 4 somministrazioni) | ||
| Iniziare con 25 mg/die (sino a massimo 100 mg/die per os in unica somministrazione) | ||
| Iniziare con 50 mg x 2/die (dose massima 200-300 mg/die in 2 o 3 somministrazioni) | ||
|
Nifedipina oppure Amlodipina |
30-90 mg/die per os; la formulazione gocce sublinguali (20 gocce = 10 mg) è utile per crisi ipertensive trattabili in ambulatorio | |
| 10–20 mg/die | ||
| Soluzione fisiologica (sodio cloruro 0.9%) | Infusione ev lenta di 1-2 L/die nei 2-4 giorni precedenti l’intervento chirurgico | |
| Intra-operatoria (o crisi da trattare in regime di ricovero) |
Crisi ipertensiva | |
| Fentolamina | Fl 5 mg in 10 mL di soluzione fisiologica, infusione ev lenta di 1 mg/min: se non risposta dopo 3-5 min, infusione di 2.5-5 mg in 2-5 min, eventualmente seguita da infusione di 10-40 mg/h | |
| Urapidil | 25 mg ev in bolo: se non risposta in 2 min, altro bolo di 25 mg ev, oppure infusione ev di 50-100 μg/kg/min | |
| alcuni autori (10) suggeriscono l’utilizzo anche di magnesio solfato | Bolo di 2-4 g ev, seguito da infusione ev 1-2 g/ora | |
| Aritmie | ||
| Esmololo | Dose attacco 500 μg/kg ev in 1 minuto, seguito da 50 μg/kg/min per 4 min: se non risposta dopo 5 min, si ripete l’intero schema della dose di attacco, ma aumentando a 100 μg/kg/min per 4 min, seguito, in base alla risposta, da infusione di 50-200 μg/kg/min | |
| Post-operatoria (raccomandata degenza in UO post-intensiva per 24-48 ore) | Ipotensione: plasma expander | |
| Dopamina | Infusione ev 5-12.5 μg/kg/min | |
| Noradrenalina | Infusione ev 1-12 μg/min | |
| Vasopressina | Infusione ev 0.01-0.04 UI/min | |
| Corticosteroidi | Se necessario: idrocortisone dose massima 200 mg/24 ore ev | |
| Ipoglicemia | ||
| Soluzione Glucosata | Concentrazione e quantità da somministrare vanno stabilite in base alla gravità dell’ipoglicemia | |
Preparazione alla chirurgia
Le dosi di alfa-bloccante vanno progressivamente aumentate fino al raggiungimento di una condizione pressoché continua di normotensione. Il blocco dei recettori alfa-2 causa un maggior rilascio dai terminali nervosi simpatici di noradrenalina, che a livello cardiaco determina tachicardia di tale entità che quasi sempre è necessario aggiungere in terapia un ß-bloccante. È di estrema importanza ricordare che nel paziente con feocromocitoma non vanno mai usati i ß-bloccanti prima di un adeguato alfa-blocco. Infatti, un blocco dei recettori ß vasodilatanti, non accompagnato dal blocco degli alfa, vasocostrittori, può comportare l’insorgere di gravi crisi ipertensive indotte da un improvviso rilascio di catecolamine tumorali.
Durante e dopo la chirurgia
Nel caso di crisi ipertensiva acuta, è necessario ricorrere alla somministrazione, se possibile endovenosa, di alfa-bloccanti, come l’urapidil o la fentolamina (8,9). Quest’ultima va somministrata diluita 1:10, lentamente, monitorando la pressione arteriosa. Una somministrazione rapida in bolo può, infatti, determinare una crisi ipotensiva grave.
Nel caso di ipotensione post-chirurgica, il trattamento si avvale di plasma expanders, di corticosteroidi, di amine per via endovenosa e della somministrazione di vasopressina (tabella).
Dopo l’intervento chirurgico la terapia con alfa-litico può essere sospesa, mentre viene proseguita l’eventuale altra terapia anti-ipertensiva assunta in precedenza.
Dopo circa un mese dall’intervento è indicato il controllo delle metanefrine libere plasmatiche e/o metanefrine frazionate urinarie: dopo un intervento chirurgico radicale devono essere normalizzate.
FEOCROMOCITOMA METASTATICO
Non esiste ad oggi una terapia efficace per il trattamento del feocromocitoma maligno (11). In base alle caratteristiche del singolo paziente abbiamo alcune opzioni terapeutiche tra cui scegliere (12).
La chirurgia del tumore primitivo può avere ancora indicazione se finalizzata alla riduzione della massa del tessuto neoplastico (debulking), con conseguente diminuzione dei livelli di catecolamine tumorali (13). Tale intervento riduttivo può inoltre favorire il risultato della terapia radiometabolica con MIBG, che costituisce l’opzione terapeutica di prima scelta nel caso di metastasi captanti (14-16). Questa terapia si è dimostrata capace di migliorare la condizione clinica del paziente riducendo i livelli di catecolamine, ma non è capace di portare a guarigione il paziente. Insieme alla più recente terapia radio-metabolica con analoghi della somatostatina (90Y-DOTATOC o 177Lu-DOTATATE), rappresenta la terapia di prima scelta nei pazienti con malattia a lenta progressione.
Anche la radioterapia può essere un trattamento indicato in pazienti con forme metastatiche.
La radio-frequenza, la crio-ablazione e la chemio-embolizzazione sono altre manovre finalizzate alla riduzione delle lesioni metastatiche.
Lo schema di chemioterapia più largamente utilizzato è il CVD (ciclofosfamide, vincristina, decarbazina); è stato dimostrato, infatti, che circa il 37% dei pazienti trae beneficio da questo schema terapeutico, che viene solitamente riservato a pazienti con malattia in rapida progressione. Un’altra possibile alternativa è la terapia con temozolomide, che si è dimostrata efficace in particolare in pazienti con mutazioni a carico del gene SDHB (17).
Infine, nei pazienti con malattia metastatica può essere considerato l’utilizzo di farmaci di inibitori delle tirosin-kinasi: il sunitinib è stato il primo farmaco di questa categoria identificato come possibile trattamento in questi pazienti, dando benefici clinici sia in pazienti sporadici che SDHB mutati. Il farmaco è approvato per il trattamento dei GIST, dei tumori renali metastatici e per il trattamento dei tumori neuroendocrini pancreatici. Lo schema di terapia per GIST e carcinomi renali è 50 mg/die per 4 settimane, seguite da 2 settimane di pausa. Per i tumori neuroendocrini del pancreas la dose raccomandata è di 37.5 mg/die senza sospensione.
È indicato un controllo periodico delle metanefrine libere plasmatiche e/o metanefrine frazionate urinarie da effettuare a giudizio clinico ogni 3-6 mesi. Dal punto di vista strumentale, il paziente verrà seguito con TC o RM ed esami funzionali come la FDG-PET, utile nella valutazione della progressione di malattia e della risposta a terapie. La 123I-MIBG e la PET con 68Ga-DOTANOC permettono di valutare la possibilità di utilizzare la terapia radiometabolica.
Nei pazienti con feocromocitoma/paraganglioma metastatico e nei pazienti con feocromocitoma/paraganglioma non metastatico ma non operabile per gravi patologie concomitanti, la terapia medica è finalizzata a limitare gli effetti dannosi delle catecolamine sul sistema cardio-vascolare.
La sopravvivenza dei pazienti con feocromocitoma/paraganglioma metastatico è variabile: alcuni di questi pazienti, generalmente quelli che presentano solo metastasi ossee o linfonodali, sopravvivono spesso anche per molti anni, perché la progressione delle lesioni è lenta e la prognosi è pertanto fortemente condizionata dagli effetti dannosi delle catecolamine. Ne consegue che la terapia medica finalizzata a ridurre gli effetti delle catecolamine riveste un’importanza primaria nella gestione del paziente con feocromocitoma metastatico. I farmaci alfa-bloccanti sono alla base di tale terapia. Altro farmaco di possibile impiego è l’alfa-metil-paratirosina (18), che limita la sintesi delle catecolamine attraverso un’inibizione competitiva con la tirosino-idrossilasi, enzima limitante la sintesi delle catecolamine. I risultati clinici sono buoni, ma a spese di effetti collaterali importanti (per es. l’ipotensione ortostatica), poiché l’inibizione della sintesi non avviene solo a livello del tessuto cromaffine neoplastico, ma anche a livello dei neuroni simpatici.
In caso di persistenza di valori pressori elevati, può essere aggiunto in terapia qualsiasi altro farmaco anti-ipertensivo, tranne i diuretici.
PARAGANGLIOMI DELLA REGIONE TESTA-COLLO
Sono di natura parasimpatergica e generalmente non rilasciano catecolamine. Alcuni autori sostengono che sono capaci di produrre dopamina o il suo precursore DOPA, sostanze che comunque non hanno effetti cardio-vascolari clinicamente rilevanti. La terapia di questi paragangliomi è anch’essa chirurgica quando praticabile e deve avvalersi di chirurghi ORL o vascolari di provata esperienza (19,20). Le lesioni più facilmente aggredibili dal punto di vista chirurgico sono i tumori glomici. La chirurgia dei paragangliomi vagali, giugulari e timpanici, specie se di grandi dimensioni, comporta un alto rischio di provocare quelle lesioni nervose che l’intervento vorrebbe evitare e si presenta purtroppo spesso non praticabile.
Una possibile alternativa terapeutica per questi tumori è il trattamento radioterapico stereotassico (radiochirurgia), che si è mostrato capace di ridurre le dimensioni del tumore (21,22). Un limite della radiochirurgia è legato alla dimensione della lesione da trattare: tumori con diametro > 3 cm sono candidati preferibilmente alla radioterapia convenzionale. Un elemento che però va considerato riguardo all’opzione radioterapica è che, trattandosi frequentemente di pazienti giovani, i potenziali effetti collaterali a lungo termine sulle strutture e gli organi della regione collo/testa non sono attualmente noti (23).
BIBLIOGRAFIA
- Waltz MK, Alesina PF, Wenger FA, et al. Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World J Surg 2006, 30: 899-908.
- Waltz MK, Alesina PF, Wenger FA, et al. Posterior retroperitoneoscopic adrenalectomy – results of 560 procedures in 520 patients. Surgery 2006, 140: 943-50.
- Berber E, Tellioglu G, Harvey A, et al. Comparison of laparoscopic transabdominal lateral versus posterior retroperitoneal adrenalectomy. Surgery 2009, 146: 621-6.
- Dralle H, Scheumann GWF, Kotzerke J, et al. Surgical management of MEN 2. Recent Results Cancer Res 1992, 125: 167-95.
- Brauckhoff M, Gimm O, Thanh PM, et al. Critical size of residual adrenal tissue and recovery from impaired early postoperative adrenocortical function after subtotal bilateral adrenalectomy. Surgery 2003, 134: 1020-8.
- Bravo EL. Pheochromocytoma. Curr Ther Endocrinol Metab 1997, 6: 195-7.
- Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 2007, 92: 4069-79.
- Alijotas-Reig J, Bove-Farre I, de Cabo-Frances F, et al. Effectiveness and safety of prehospital urapidil for hypertensive emergencies. Am J Emerg Med 2001, 10: 130-3.
- Prejbisz A, Lenders JWM, Eisenhofer G, et al. Cardiovascular manifestations of pheochromocytoma. J Hypertension 2011, 29: 2949-60.
- James MF, Cronje L. Pheochromocytoma crisis: the use of magnesium sulfate. Anesth Analg 2004, 99: 680-6.
- Jimenez C. Treatment for patients with malignant pheochromocytomas and paragangliomas: a perspective from the hallmarks of cancer. Front Endocrinol (Lausanne) 2018, 9: 277.
- Corssmit EPM, Snel M, Kapiteijn E. Malignant pheochromocytoma and paraganglioma: management options. Curr Opin Oncol 2020, 32: 20-6.
- Scholz T, Eisenhofer G, Pacak K, et al. Current treatment of malignant pheochromocytoma. J Clin Endocrinol Metab 2007, 92: 1217-25.
- Sisson JC, Shapiro B, Beierwaltes WH. Radiopharmaceutical treatment of malignant pheochromocytoma. J Nucl Med 1984, 25: 197-206.
- Kaltsas GA, Mukherjee JJ, Foley R, et al. Treatment of metastatic pheochromocytoma and paraganglioma with 131I-meta-iodobenzylguanidine (MIBG). Endocrinologist 2003, 13: 321-33.
- Fitzgerald PA, Golsby RE, Huberty JP, et al. Malignant pheochromocytomas and paragangliomas: a phase II study of therapy with high-dose 131I-meta-iodobenzylguanidine (131I – MIBG). Ann N J Acad Sci 2006, 1073: 465-70.
- Hadoux J, Favier J, Scoazec JY, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer 2014, 135: 2711-20.
- Steinsapir J, Carr AA, Prisant LM, et al. Metyrosine and pheochromocytoma. Arch Int Med 1997, 157: 901-6.
- Paris J, Facon F, Thomassin JM. Cervical paragangliomas: neurovascular surgical risk and therapeutic management. Eur Arch Otorhinolaryngol 2006, 263: 860-5.
- Papaspyrou K, Mann WJ, Amedee RG. Management of head and neck paragangliomas: review of 120 patients. Head Neck 2009, 31: 381-7.
- Henzel M, Hamm K, Gross MW, et al. Fractionated stereotactic radiotherapy of glomus jugulare tumors. Local control, toxicity, symptomatology, and quality of life. Strahlenther Onkol 2007, 183: 557-62.
- Wagner RE, Rodriguez KD, Heron DE, et al. Linac-based stereotactic body radiation therapy for treatment of glomus jugulare tumors. Radiother Oncol 2010, 97: 395-8.
- Hu K, Persky MS. Treatment of head and neck paragangliomas. Cancer Control 2016, 23: 228-41.
Terapia radiometabolica per feocromocitomi-paragangliomi
Nadia Cremonini1, Benedetta Zampetti2, Letizia Canu3 & Massimo Mannelli3
1Ambulatorio di Endocrinologia, Clinica Privata Villalba, GVM, Bologna
2SC di Endocrinologia, Ospedale Niguarda, Milano
3Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
(aggiornato al 20 marzo 2020)
La terapia radiometabolica rappresenta un’interessante opzione terapeutica nei pazienti con feocromocitoma/paraganglioma metastatico, effettuata con l’utilizzo di traccianti marcati con metaiodobenzilguanidina (MIBG) o più recentemente con analoghi della somatostatina (90Y-DOTATOC o 177Lu-DOTATATE).
La MIBG presenta un’elevata affinità di struttura con la noradrenalina, che le permette di accumularsi all’interno dei tessuti cromaffini grazie al trasportatore specifico (human norepinephrine transporter, hNET) (1). Il trattamento con 131I-MIBG deve essere preso in considerazione in pazienti con significativa captazione del tracciante alla scintigrafia diagnostica con 131I-MIBG o 123I-MIBG.
Sono stati proposti diversi schemi di trattamento con una dose singola o dosi frazionate (200-1400 mCi) (2,3). Alcuni dati sembrano indicare che la dose iniziale di 131I-MIBG sia un importante determinante della risposta alla terapia e della sopravvivenza: pazienti che ricevono un’alta dose iniziale (> 500 mCi) presentano una maggior sopravvivenza rispetto a quelli trattati con dosi iniziali minori (4). Nonostante questo, un trattamento frazionato (dose cumulativa 1800 mCi) appare la miglior scelta terapeutica, perché riesce ad essere efficace minimizzando gli effetti collaterali (5). Circa il 60% dei siti di metastasi capta il tracciante (6); solitamente si ottengono risposte migliori per localizzazioni a carico dei tessuti molli rispetto a metastasi ossee (7).
Solitamente questa terapia viene ben tollerata e gli unici effetti collaterali sono rappresentati da leucopenia e trombocitopenia dose-dipendenti. Raramente sono stati descritti mielo-soppressione severa, infezioni e insufficienza epatica (8).
Il trattamento con alte dosi di 131I-MIBG determina riduzione delle dimensioni delle lesioni nel 30% dei pazienti, stabilizzazione di malattia nel 57% e progressione nel restante 13% (7). In molti pazienti trattati si ottengono buoni risultati in termini di riduzione della secrezione ormonale e controllo dei sintomi (8,9).
È attualmente in corso uno studio di fase II (ClinicalTrials.gov Identifier NCT00107289), che prevede l’utilizzo della terapia radiometabolica con 131I-MIBG in pazienti affetti da neuroblastoma o feocromocitoma maligno in rapida progressione e resistenti ad altre terapie. Lo scopo dello studio è valutare sicurezza ed efficacia del trattamento.
Nelle preparazioni convenzionali, oltre il 99% delle molecole di MIBG non sono marcate con iodio radioattivo (131I), presentando quindi un’attività specifica molto bassa. Questo comporta la somministrazione di grandi dosi di MIBG non marcata, che competono con il meccanismo di ricaptazione della noradrenalina. Il conseguente aumento delle catecolamine circolanti può causare effetti collaterali come crisi ipertensive acute. Pertanto, una formulazione che contenga una quantità significativamente inferiore di MIBG non marcata potrebbe ridurre questo effetto collaterale. Si tratta della 131I-MIBG ad alta attività specifica (HSA), chiamata anche Iobenguane, che si compone quasi interamente di 131I-MIBG marcata (10,11). In uno studio clinico di fase II (clinicaltrial.gov NCT02961491), oltre il 90% dei pazienti hanno ottenuto una risposta parziale (30%) e stabilizzazione della malattia (68%) con il miglioramento dell'ipertensione. Inoltre, molti pazienti hanno mostrato un effetto anti-neoplastico persistente a lungo termine. Attualmente, la HSA è l'unica terapia approvata negli Stati Uniti per i pazienti con feocromocitoma/PGL metastatico.
L’espressione da parte delle cellule cromaffini di recettori per la somatostatina (SSTR), in particolar modo tipo 2, tipo 3 e tipo 5, rappresenta il razionale sul quale si basa l’utilizzo di traccianti marcati con analoghi della somatostatina, octreotide e lanreotide. Vengono utilizzati traccianti marcati con Ittrio (90Y-DOTA-octreotide, 90Y-DOTA-lanreotide, 90YDOTA-octreotate), Lutezio (177Lu-DOTA-octreotide, 177Lu-DOTA-octreotate) e Indio (111In-pentetreotide, 111In-DOTA-octreotide) (3,6,12,13)). Sono stati applicati diversi protocolli terapeutici che prevedono l’utilizzo di analoghi marcati con Ittrio (12,13). Nei pazienti trattati sono stati ottenuti risultati analoghi a quelli dei pazienti sottoposti a terapia radiometabolica con 131I-MIBG (3,12-14).
Anche in questo caso i principali effetti collaterali sono rappresentati da leucopenia e trombocitopenia. I traccianti marcati con 177Lu possono indurre tossicità renale (15).
Una mancata risposta alla terapia potrebbe essere legata al tipo di SSTR presenti sulle cellule cromaffini; infatti, solo il tipo 2 lega ad alta affinità gli analoghi comunemente utilizzati. In uno studio condotto su 52 feocromocitomi/PGL (16) è stata dimostrata un’espressione del recettore di tipo 3 nel 90% dei tessuti. Per questo motivo alcuni pazienti potrebbero beneficiare della terapia con pasireotide, un analogo con elevata affinità per tutti i sottotipi recettoriali ad eccezione del 4.
È stato condotto uno studio di fase II al fine di valutare meglio l’efficacia della terapia con 90Y- e 177Lu-DOTA-octreotide nella terapia dei tumori neuroendocrini avanzati. In alcuni pazienti si trova un quadro eterogeneo di captazione: alcune lesioni risultano captanti alla scintigrafia con 123I-MIBG, mentre altre non lo sono e risultano invece positive alla scintigrafia con 111In-pentetreotide. In questi casi è possibile che possa risultare efficace una terapia in successione con diversi traccianti marcati (15).
In linea generale i FEO/PGL che liberano catecolamine (addominali e toracici, simpatergici) risultano più spesso positivi alla scintigrafia con MIBG, mentre i PGL della regione testa collo, non attivi e parasimpatergici, mostrano più spesso positività alla scintigrafia con analoghi della somatostatina.
Bibliografia
- Shulkin BL, Ilias I, Sisson JC, et al. Current trends in functional imaging of pheochromocytomas and paragangliomas. Ann N Y Acad Sci 2006, 1073: 374–82.
- Kaltsas GA, Mukherjee JJ, Foley R, et al. Treatment of metastatic pheochromocytoma and paraganglioma with 131I-meta-iodobenzylguanidine(MIBG). Endocrinologist 2003, 13: 321–33.
- Kaltsas GA, Papadogias D, Makras P, et al. Treatment of advanced neuroendocrine tumours with radiolabelled somatostatin analogues. Endocr Relat Cancer 2005, 12: 683–99.
- Safford SD, Coleman RE, Gockerman JP, et al. Iodine-131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery 2003, 134: 956-62.
- Lam MG, Lips CJ, Jager PL, et al. Repeated [131I]metaiodobenzylguanidine therapy in two patients with malignant pheochromocytoma. J Clin Endocrinol Metab 2005, 90: 5888-95.
- Fitzgerald PA, Goldsby RE, Huberty JP, et al. Malignant pheochromocytomas and paragangliomas: a phase II study of therapy with high-dose 131Imetaiodobenzylguanidine (131IMIBG). Ann N Y Acad Sci 2006, 1073: 465–70.
- Loh KC, Fitzgerald PA, Matthay KK, et al. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest 1997, 20: 648–58.
- Mukherjee JJ, Kaltsas GA, Islam N, et al. Treatment of metastatic carcinoid tumours, phaeochromocytoma, paraganglioma and medullary carcinoma of the thyroid with 131I-meta-iodobenzylguanidine (131I-mIBG). Clin Endocrinol (Oxf) 2001, 55: 47-60.
- Troncone L, Rufini V, Daidone MS, et al. [131I] metaiodobenzylguanidine treatment of malignant pheochromocytoma: experience of the Rome group. J Nucl Biol Med 1991, 35: 295-9.
- Pryma DA, Chin BB, Noto RB, et al. Efficacy and safety of high-specific activity 131I-MIBG therapy in patients with advanced pheochromocytoma or paraganglioma. J Nucl Med 2019, 60: 623-30.
- Jimenez C, Pryma DA, Chin BB, et al. AZEDRA® (iobenguane I 131) in patients with metastatic and/or recurrent and/or unresectable pheochromocytoma or paraganglioma: results of a multicenter, open-label, pivotal phase 2b study. NANETS: Philadelphia, PA, 2017: C28.
- Kwekkeboom DJ, Mueller-Brand J, Paganelli G, et al. Overview of results of peptide receptor radionuclide therapy with 3 radiolabeled somatostatin analogs. J Nucl Med 2005, 46 suppl 1: 62S-6S.
- Shapiro B, Gross MD, Shulkin B. Radioisotope diagnosis and therapy of malignant pheochromocytoma. Trends Endocrinol Metab 2001, 12: 469-75.
- Waldherr C, Pless M, Maecke HR, et al. Tumor response and clinical benefit in neuroendocrine tumors after 7.4 GBq 90Y-DOTATOC. J Nucl Med 2002, 43: 610-6.
- Ahlman H. Malignant pheochromocytoma. State of the field with future projections. Ann N Y Acad Sci 2006, 1073: 449-64.
- Mundschenk J, Unger N, Schulz S, et al. Somatostatin receptor subtypes in human pheochromocytoma: subcellular expression pattern and functional relevance for octreotide scintigraphy. J Clin Endocrinol Metab 2003, 88: 5150-7.
Chirurgia surrenalica
Cristian Fiori, Francesco Porpiglia
SCDU Urologia - AOU San Luigi Gonzaga, Orbassano (Torino), Università degli Studi di Torino
INTRODUZIONE
Da quando alla fine dell'800 fu descritta la prima surrenectomia, la tecnica chirurgica ha subito numerose modifiche, tanto che oggi sono possibili numerosi approcci chirurgici alla ghiandola. Tuttavia è l'introduzione della laparoscopia (1992) ad aver letteralmente rivoluzionato il trattamento chirurgico del surrene.
In generale vi sono due indicazioni assolute alla chirurgia del surrene: le lesioni funzionanti (secernenti cortisolo, aldosterone, catecolamine o più raramente androgeni) e il carcinoma del surrene. Esiste poi una terza indicazione, di più complesso inquadramento, che comprende masse non funzionanti in crescita nel tempo o di cui non può essere esclusa la natura neoplastica maligna (vedi incidentalomi).
SURRENECTOMIA LAPAROSCOPICA
La surrenectomia laparoscopica (SL) fu descritta per la prima volta da Gagner nel 1992 [1] e il suo utilizzo in caso di lesioni benigne è cresciuto in modo esponenziale, tanto che oggi la laparoscopia rappresenta l'approccio di scelta per il trattamento della patologia surrenalica benigna [2]. Analogamente a quanto accaduto per la colecistectomia, non sono disponibili studi prospettici randomizzati che paragonino l'approccio open con quello laparoscopico, tuttavia numerosi lavori retrospettivi sottolineano come la SL presenti i vantaggi della chirurgia mini-invasiva [3, 4, 5] senza inficiare sicurezza ed efficacia dell'intervento.
Tecniche chirurgiche
Approccio trans-peritoneale. E' la prima tecnica laparoscopica descritta ed è ancora oggi la più utilizzata. Con il paziente in decubito laterale a 45°, si induce lo pneumo-peritoneo mediante ago di Veress. Una volta raggiunta un'adeguata distensione addominale, si procede a posizionamento di una porta laparoscopica (trocar) del diametro pari a 12 mm a livello ombelicale o para-ombelicale.
A destra si posiziona una porta del diametro di 5 mm caudalmente al processo xifoideo, attraverso la quale si introduce un retrattore per il sollevamento della faccia inferiore del fegato. Successivamente, si posiziona una porta di 12 mm a livello dell'emiclaveare, due dita distalmente all'arco costale ed una porta di 5 mm a livello dell'ascellare anteriore a circa 4 dita dalla porta ottica (fig. 1).
Figura 1. Surrenectomia trans-peritoneale destra. Si notino le posizioni delle porte laparoscopiche. A sinistra il posizionamento delle porte è in genere speculare, anche se non è necessaria la porta sotto-xifoidea.
Brevemente, i successivi step sono: incisione del peritoneo sotto-epatico e latero-cavale, dissezione del margine laterale della vena cava, identificazione della vena surrenalica, generalmente breve e tozza, legatura della stessa mediante clip metalliche o in materiale plastico non riassorbibile e sua sezione (fig. 2).
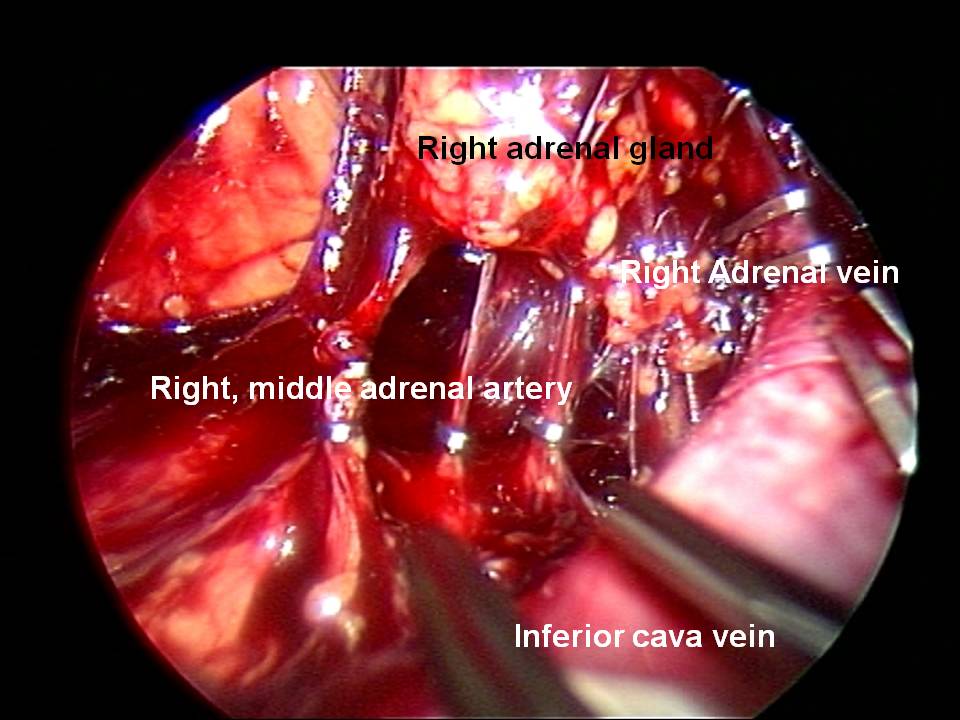
Figura 2. Surrenectomia trans-peritoneale destra. Sono stati legati mediante clip metalliche multipli peduncoli arteriosi e la vena surrenalica, ben evidente fino al suo sbocco nella vena cava. La faccia posteriore della ghiandola è stata completamente dissecata.
La dissezione prosegue sviluppando il piano superiore, posteriore laterale e inferiore (nefrosurrenalico). In tali fasi vengono legati (o coagulati) e sezionati multipli peduncoli arteriosi.
A sinistra l'intervento è in genere condotto mediante posizionamento di tre porte (ombelicale, emiclaveare, ascellare anteriore). I principali step sono: incisione del peritoneo a livello della flessura splenica, mobilizzazione del colon discendente e della milza, identificazione della vena renale e della vena surrenalica principale, che viene legata e sezionata (fig. 3).
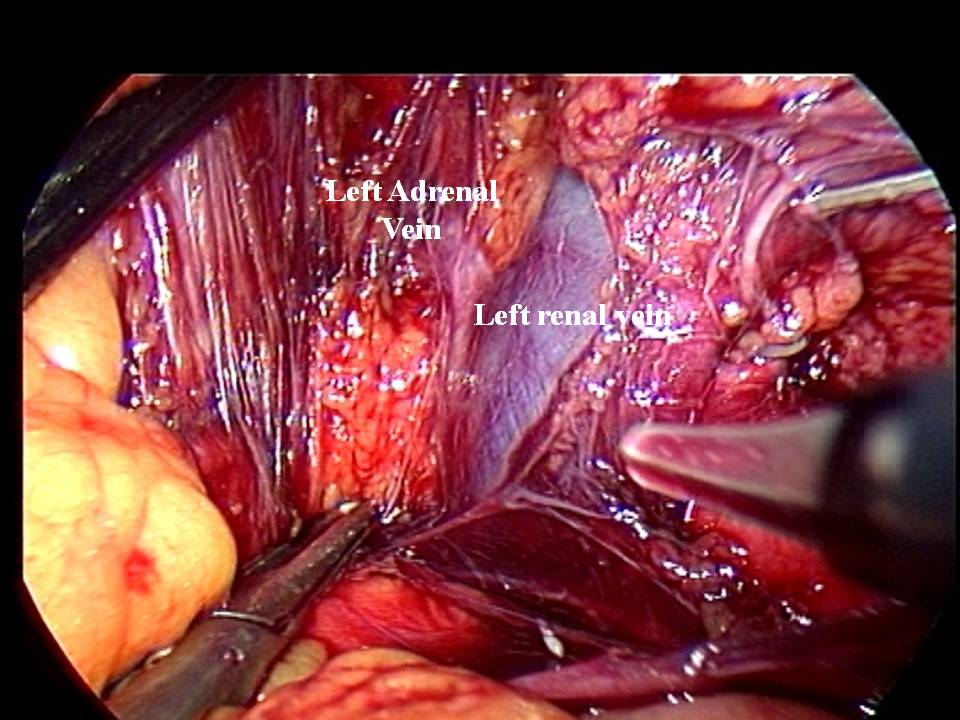
Figura 3. Surrenectomia laparoscopica trans-peritoneale sinistra. Dopo accesso allo spazio retro-peritoneale, si identifica la vena renale. Sono bene evidenti i rapporti fra la vena renale e la vena surrenalica sinistra, che verrà successivamente legata e sezionata.
Approccio retro-peritoneale. Con il paziente in decubito laterale a 90° si accede al retro-peritoneo mediante incisione a livello del triangolo di Petit di circa 15 mm. Si introduce dunque il pallone dilatatore di Gauer, attraverso il quale si espande lo spazio retro-peritoneale (che è solo virtuale). Si posizionano quindi per via digito-guidata una porta di 5 mm anteriormente all'apice della 12° costa e altre due porte a livello dell'ascellare media ed anteriore. Le quattro porte assumono configurazione "a diamante". Infine si induce lo pneumo-retroperitoneo. La dissezione della ghiandola inizia a livello posteriore, poi craniale e anteriore, liberando la ghiandola dal foglietto peritoneale posteriore. Si sviluppa successivamente il piano nefrosurrenalico. Multipli peduncoli vascolari vengono legati e sezionati (fig. 4).
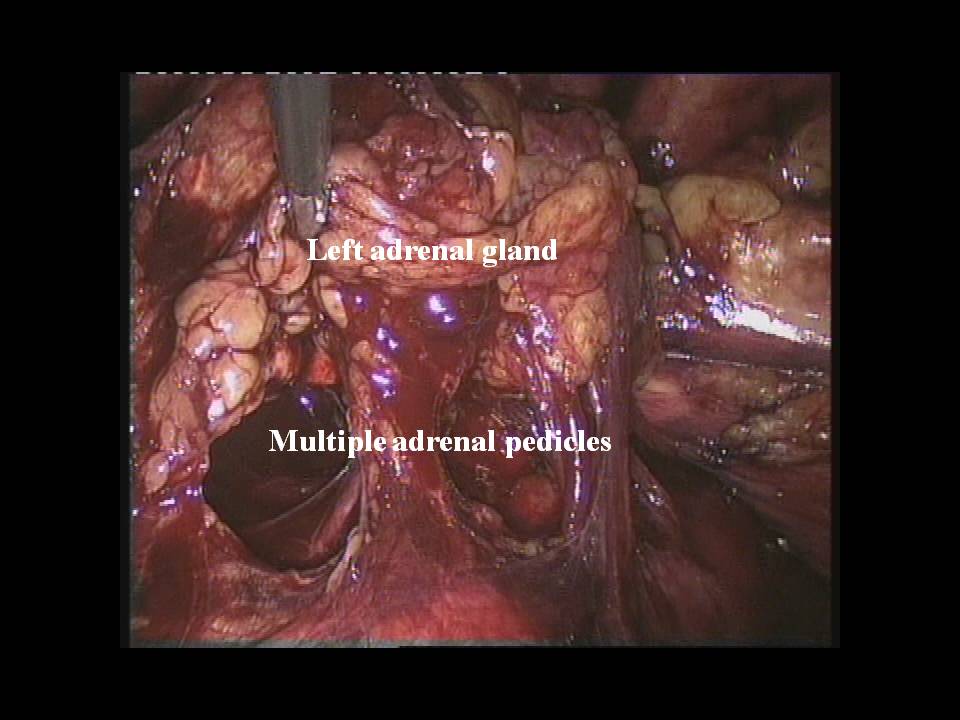
Figura 4. Surrenectomia retro-peritoneale sinistra. La ghiandola è stata completamente dissecata e sono stati isolati multipli peduncoli arteriosi, che verranno legati e sezionati.
Dopo aver identificato il margine laterale della vena cava (a destra) e la vena renale (a sinistra), si reperta la vena surrenalica principale (fig. 5), che viene legata e sezionata.
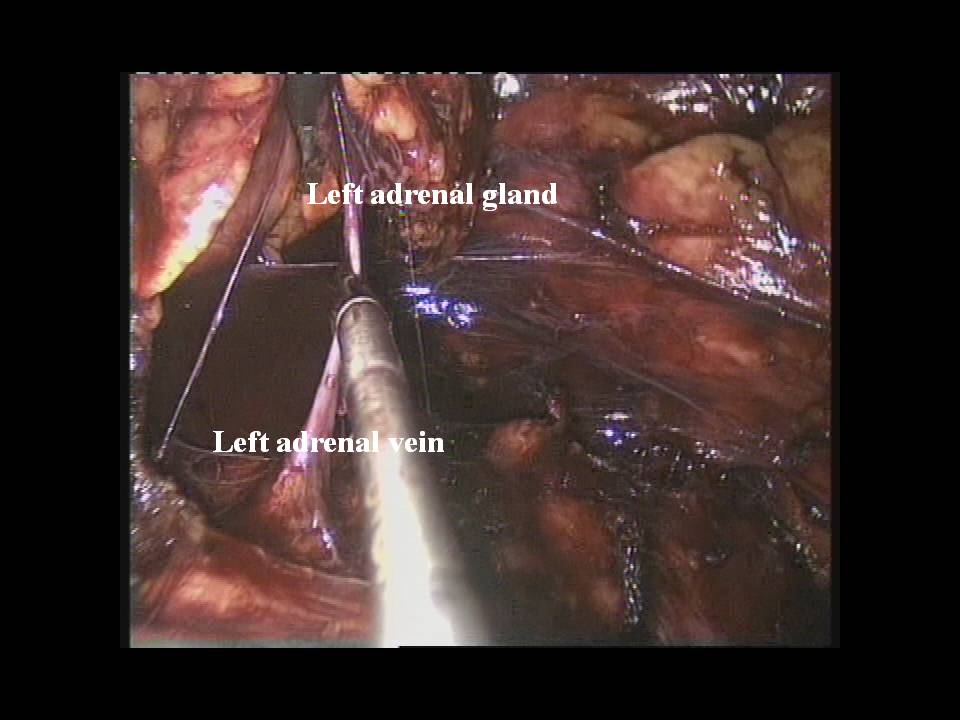
Figura 5. Surrenectomia sinistra retro-peritoneale. Dopo legatura e sezione dei peduncoli arteriosi, si identifica e disseca la vena renale, che viene successivamente legata e sezionata.
Al termine della dissezione (in entrambi gli approcci), la ghiandola viene posta in un endobag (sacchetto laparoscopico) (fig. 6) ed estratta attraverso una piccola incisione, generalmente il prolungamento di una delle incisioni eseguite per il posizionamento delle porte.
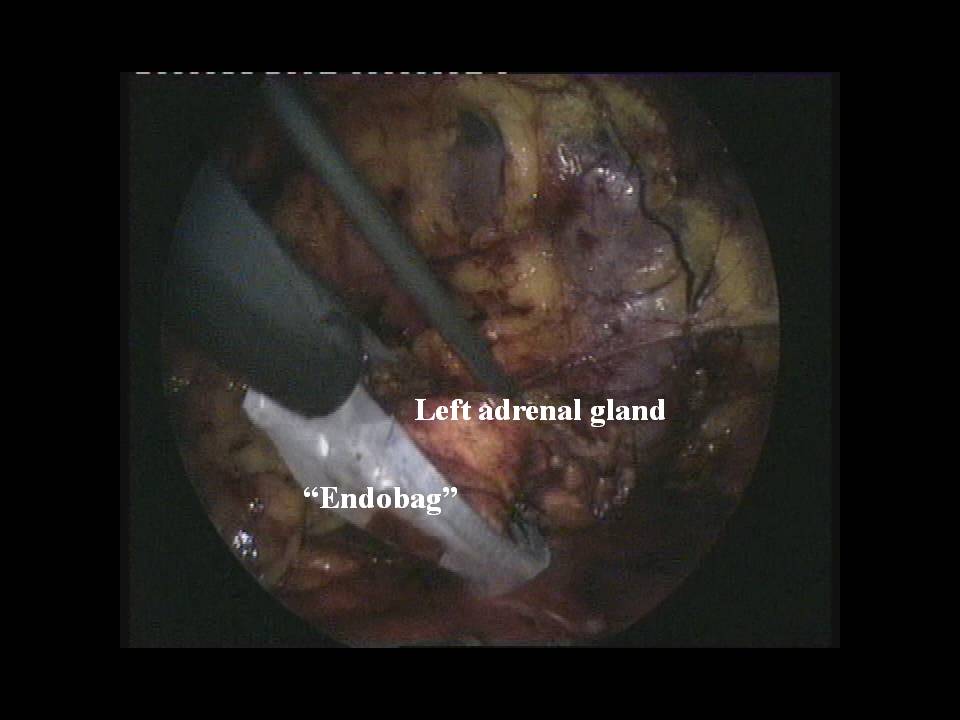
Figura 6. Surrenectomia sinistra. Al termine degli interventi laparoscopici (retro- o trans-peritoneali), il pezzo operatorio viene intrappolato in endobag (sacchetto laparoscopico) ed estratto attraverso la parete addominale.
Pro & contro dei diversi approcci laparoscopici
Una recente metanalisi [6] evidenzia come non vi siano significative differenze in termini di variabili peri-operatorie tra i due approcci (trans vs retro-peritoneale). Vantaggi di quest'ultima tecnica sono: l'accesso diretto alla ghiandola senza manipolazione di organi endo-addominali, la possibilità di trattare pazienti con precedenti chirurgici addominali significativi e i grandi obesi. Per contro, lo svantaggio principale è lo "spazio di lavoro" meno ampio rispetto a quello che si ottiene durante la procedura trans-peritoneale, pertanto quest'ultimo approccio va preferito in caso di lesione voluminosa.
Decorso post-operatorio
Nella nostra esperienza i pazienti rimangono a letto con infusioni per via endovenosa per 24 ore. Successivamente, è consentita la deambulazione e l'alimentazione per os. Il drenaggio chirurgico è rimosso dopo 48 ore e la dimissione, salvo complicanze avviene dopo 72 ore. Terapie specifiche (es. cortisoniche) vengono concordate con i colleghi endocrinologi.
Risultati
Indipendentemente dall'approccio utilizzato, la SL si è dimostrata sicura ed efficace nel trattamento delle lesioni surrenaliche benigne. Come detto, essa presenta tutti i vantaggi della chirurgia mini-invasiva, come ridotto dolore post-operatorio, ridotta degenza post-operatoria, rapido recupero delle normali attività e migliore risultato estetico rispetto alla chirurgia tradizionale [2]. Per tali motivi questo tipo di chirurgia è da considerare il gold standard nel trattamento delle lesioni surrenaliche benigne [3].
Complicanze
Intra-operatoriamente ricordiamo lesioni di organi viciniori (milza, pancreas, fegato, rene e colon) che possono richiedere riparazioni chirurgiche o conversione open dell'intervento (< 1% nella nostra esperienza).
Nel post-operatorio può comparire sanguinamento (tale da richiedere emotrasfusione o, in rari casi re-intervento) e infezione delle ferite chirurgiche. In un recente lavoro retrospettivo su 1980 procedure laparoscopiche (2005-2009), sono state registrate complicanze di alto grado sec. Clavien [7] (grado IV a rischio di vita o grado V con decesso) nell'1.8% dei casi di SL (contro il 7.6% della surrenectomia open) [8].
Controversie
Due argomenti animano il dibattito sulla SL: il trattamento delle lesioni benigne voluminose e il trattamento del carcinoma corticosurrenalico (ACC).
Non vi è accordo su quale sia il diametro delle lesioni surrenaliche oltre il quale è consigliato l'approccio chirurgico tradizionale in luogo di quello laparoscopico. Tale limite, con il miglioramento delle tecniche chirurgiche, della strumentazione e dell'esperienza degli operatori è stato progressivamente innalzato fino a 12-15 cm [9]. In una nostra esperienza abbiamo dimostrato come il diametro della lesione aumenti i tempi operatori, ma non infici sicurezza ed efficacia della tecnica [10]. Va tuttavia sottolineato che masse voluminose sono di difficile handling laparoscopico e vanno trattate in centri di riferimento e da laparoscopisti esperti [10] (fig. 7). Più complesso è il discorso relativo alla sicurezza e all'efficacia della SL nel trattamento delle lesioni maligne, per cui si rimanda al capitolo dedicato all'ACC.
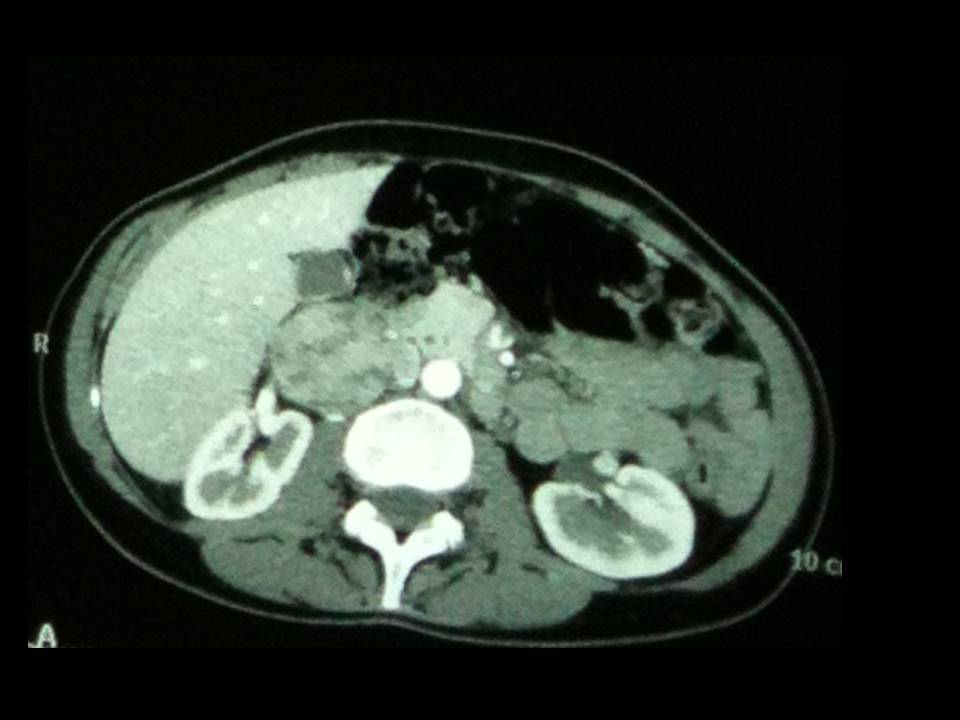
Figura 7. TC addominale che evidenzia voluminosa lesione surrenalica destra asportata fruttuosamente mediante tecnica laparoscopica. Nel caso specifico non sono state registrate complicanze intra- o post-operatorie e l'istologia ha escluso la presenza di ACC.
SURRENECTOMIA CHIRURGICA OPEN
L'enorme diffusione delle tecniche laparoscopiche ha progressivamente eroso le indicazioni alla chirurgia surrenalica "open", limitando queste al trattamento di masse particolarmente voluminose (>12-15 cm, ancorchè benigne), al trattamento dell'ACC e della recidiva di ACC.
Approcci chirurgici
Sono possibili diversi approcci chirurgici open alla ghiandola surrenalica.
Quasi scomparso per la difficoltà tecnica e un campo chirurgico estremamente limitato è l'accesso posteriore realizzato attraverso una incisione sotto-costale e il sacrificio della 11° o 12° costa.
L'accesso laterale (al fianco) è il più familiare per gli urologi, che sono soliti impiegarlo nell'ambito della chirurgia renale. In questo caso la ghiandola è aggredita per via extra-peritoneale.
Nei tumori più voluminosi, per i quali l'accesso laterale offre un'esposizione inadeguata, l'accesso trans-addominale o trans-toracico possono costituire un'alternativa preferenziale, poichè consentono un'ampia esposizione. L'accesso trans-addominale può essere effettuato con incisione sotto-costale anteriore, trasversa, mediana o chevron (fig. 8).
L'uso di questi accessi è dettato dalle dimensioni e dalle caratteristiche della lesione, così come dalle abitudini del chirurgo [16]. Questo tipo di approccio consente un ampio dominio del surrene, del rene, dei grossi vasi retro-peritoneali e degli organi viciniori (pancreas, milza, fegato in primis).
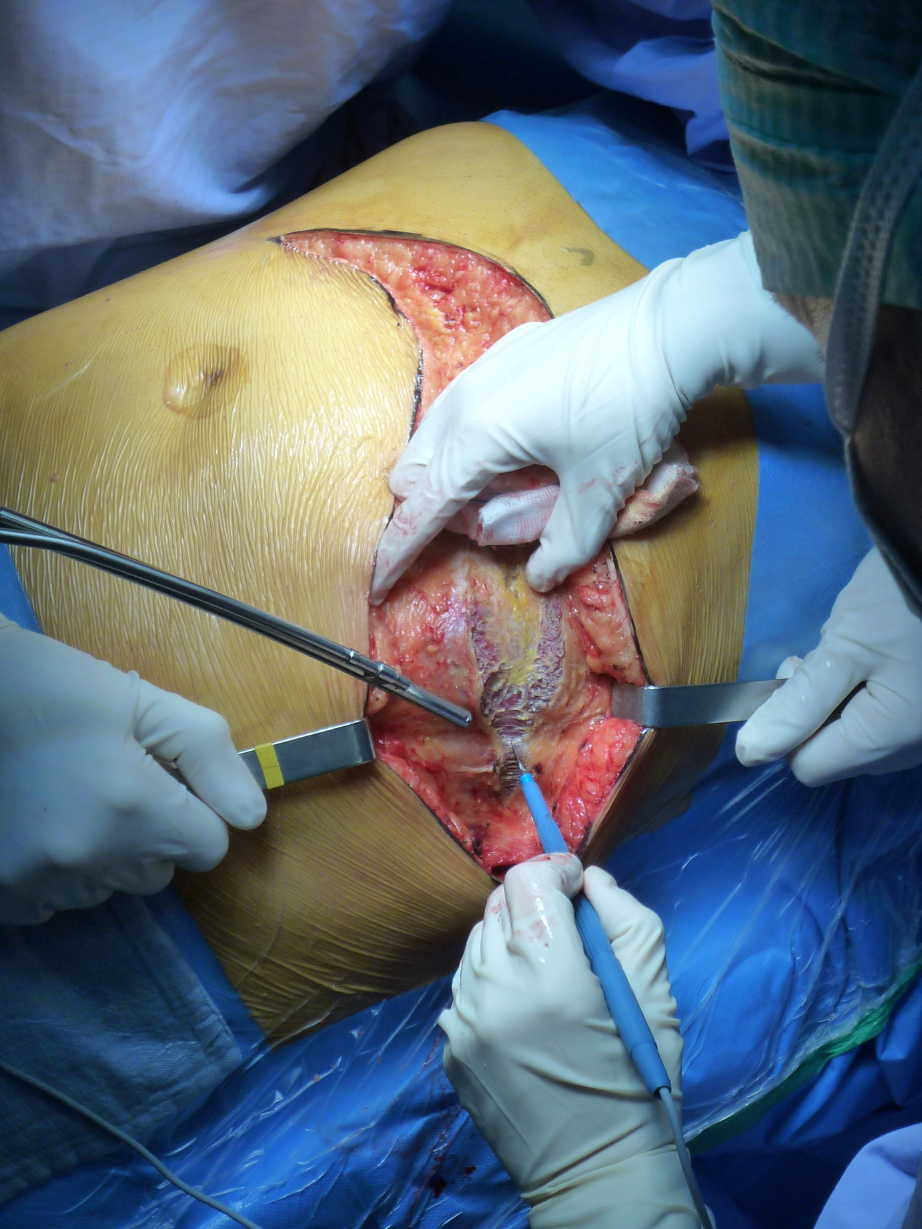
Figura 8. Ampia incisione sotto-costale anteriore sinistra. Questo accesso consente di ottenere un ampio campo operatorio con dominio di rene, surrene, organi addominali e retro-peritoneali, compresi i grossi vasi.
Chirurgia dell'ACC
Un capitolo a parte merita il trattamento chirurgico dell'ACC, per cui si rimanda alla sezione a esso dedicata per l'inquadramento nosologico. Come noto l'ACC è una neoplasia rara (con una prevalenza pari a 0.5-2 per milione di abitanti) e una prognosi sfavorevole. La diagnosi è in genere tardiva, tanto che solo il 30% degli ACC è intra-capsulare, mentre il rimanente si presenta al III o IV stadio già al momento della diagnosi, con una sopravvivenza a 5 anni del 15-50% [17]. Nonostante i progressi della terapia medica, la resezione chirurgica rappresenta in pratica l''unico approccio curativo, per cui la chirurgia deve essere il più possibile radicale. Tradizionalmente la chirurgia open è considerata l'approccio di scelta per l'ACC. Tuttavia, in uno studio retrospettivo su 14 casi abbiamo dimostrato la fattibilità e la sicurezza della laparoscopia nel trattamento di questo tipo di lesione [12]. Più recentemente abbiamo confrontato i risultati oncologici di due gruppi di pazienti affetti da ACC in stadio I/II e trattati mediante chirurgia open (n=25) e laparoscopica (n=18). Dopo un follow-up mediano di 30 mesi, non sono state registrate significative differenze in termini di sopravvivenza libera da malattia e globale [13]. Tuttavia, numerosi altri Autori hanno ottenuto risultati di segno opposto, ovvero hanno osservato risultati oncologici più sfavorevoli in pazienti trattati mediante laparoscopia [14, 15, 16]. A nostro avviso la laparoscopia rappresenta una tecnica sicura ed efficace nel trattamento dell'ACC in stadio I e II (almeno quanto la tecnica open), a patto che siano rispettati i principi della chirurgia oncologica (asportazione en bloc della lesione, integrità della capsula tumorale). Al contrario, in caso di ACC in stadio II particolarmente voluminoso o in stadio > II consideriamo la chirurgia open come l'approccio di scelta.
Nella nostra pratica clinica siamo soliti utilizzare un approccio anteriore trans-addominale, con ampia incisione a lembo sottostale in caso di lesioni maligne voluminose. In questi casi l'intervento deve essere il più radicale possibile e, al fine di garantire tale radicalità, può essere necessario il sacrificio di organi viciniori con contestuali nefrectomia, pancreasectomia parziale, splenectomia o epatectomia parziale. E' inoltre necessaria la linfoadenectomia loco-regionale [18]. Pur in presenza di chirurgia radicale, una percentuale che varia dal 75 all'85% dei pazienti presenta una recidiva di malattia, locale (in loggia surrenalica) fino al 65% dei casi [18]. Anche in questi casi la chirurgia open "reiterata" ovvero il trattamento della recidiva, anche ripetuto, rappresenta un gesto fondamentale per aumentare la sopravvivenza dei pazienti [18, 19](fig. 9).
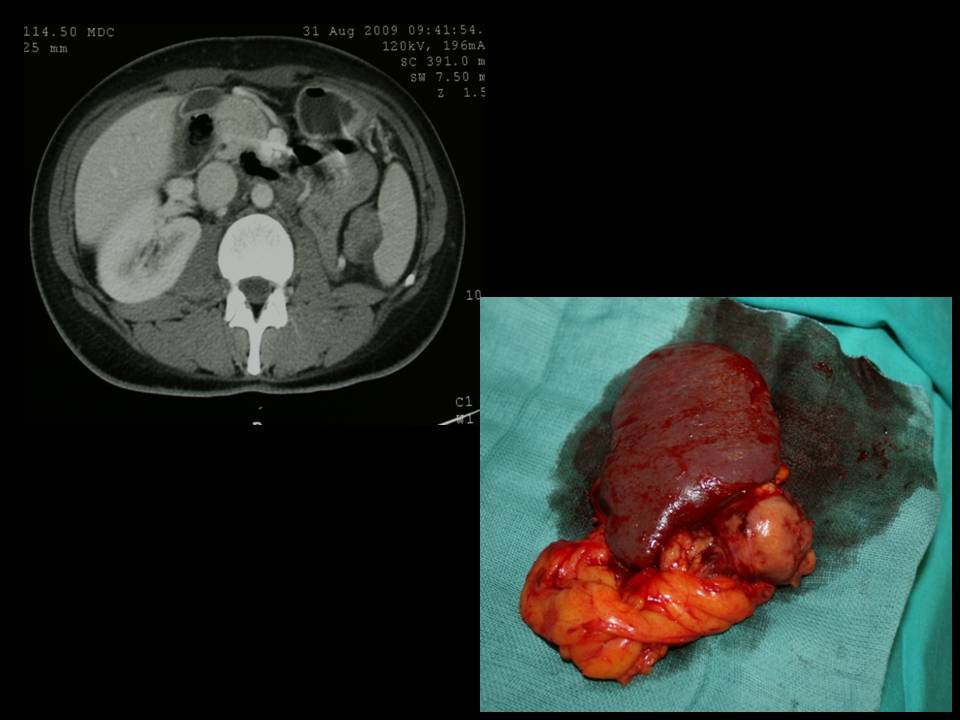
Figura 9. a. Scansione TC addominale che evidenzia voluminosa recidiva di ACC in loggia surrenalica sinistra; b. La paziente è stata sottoposta a exeresi della recidiva e contestuale splenectomia: pezzo operatorio.
NUOVE TECNICHE CHIRURGICHE
Mini-laparoscopia
La miniaturizzazione delle porte e degli strumenti laparoscopici ha consentito lo sviluppo delle tecniche mini-laparoscopiche, ovvero un intervento laparoscopico (uguale in tutto e per tutto agli interventi descritti nella sezione laparoscopia) ma condotto attraverso strumenti di 3 mm (fig. 10) e mini-incisioni cutanee (4 mm circa) che non richiedono sutura cutanea e consentono risultati estetici notevoli. Presso il nostro Centro abbiamo sviluppato tale tecnica e l'abbiamo utilizzata con successo sia in corso di procedure trans- che retro-peritoneali [20], senza aumento di complicanze nè tempi operatori (video 1).
Figura 10. Surrenectomia mini-retro-peritoneoscopica destra. Si noti la disposizione a diamante (romboidale) e il diametro limitato (3.9 mm) delle porte laparoscopiche. Le incisioni cutanee sono così piccole che non richiedono sutura, è in genere sufficiente la medicazione con "steri- strips".
Â
Video 1. Breve estratto da un intervento di surrenectomia mini-retro-peritoneoscopica sinistra. Le tappe dell'intervento sono quelle standard, si noti come i vasi vengano coagulati e sezionati (tecnica clipless) e al termine dell'intervento non sia necessario il drenaggio chirurgico (tecnica tubeless). Non sono state registrate complicanze intra- e post-operatorie e le perdite ematiche sono risultate trascurabili.
Laparoendoscopic single site surgery (LESS)
Durante la surrenectomia LESS l'intervento laparoscopico viene eseguito attraverso una singola incisione cutanea (usualmente 3 cm) in luogo delle tre o quattro piccole incisioni necessarie per la procedura laparoscopica standard. La procedura è fattibile, sicura ed efficace, ma le esperienze riportate in letteratura sono poche [21] e soprattutto non sono emersi vantaggi rispetto alla laparoscopia tradizionale.
Chirurgia robotica
La chirurgia robotica ha avuto ampia diffusione nell'ultima decade, soprattutto in campo urologico. In un recente lavoro Brunaud et al hanno analizzato i dati di 100 pazienti trattati mediante surrenectomia robotica, registrando una percentuale di complicanze pari al 10% e un tasso di conversione (a laparoscopico tradizionale o chirurgia open) pari al 5%. Gli Autori concludono che l'approccio robotico non è nè più sicuro nè più economico rispetto all'approccio laparoscopico standard, ma che consente significativi vantaggi al chirurgo in termini di ergonomicità e visione 3D. L'estrema precisione e i gradi di libertà degli strumenti robotici insieme al miglioramento della visione (3D) facilitano un intervento già descritto mediante tecnica laparoscopica standard [23] ma di una certa complessità, come la surrenectomia parziale in caso di feocromocitoma associato a sindromi genetiche (MEN IIa) [24].
BIBLIOGRAFIA
- Gagner M, Lacroix A, Bolte E. Laparoscopic adrenalectomy in Cushing's syndrome and pheochromocytoma. N Engl J Med 1992, 327: 1033.
- Haveran LA, Novitsky YW, Czerniach DR, et al. Benefits of laparoscopic adrenalectomy: a 10-year single institution experience. Surg Laparosc Endosc Percutan Tech 2006, 16: 217-21.
- Murphy MM, Witkowski ER, Ng SC, et al. Trends in adrenalectomy: a recent national review. Surg Endosc 2010, 24: 2518-26.
- Prager G, Heinz-Peer G, Passler C, et al. Applicability of laparoscopic adrenalectomy in a prospective study in 150 consecutive patients. Arch Surg 2004, 139: 46-9.
- Thompson GB, Grant CS, van Heerden JA, et al. Laparoscopic versus open posterior adrenalectomy: a case-control study of 100 patients. Surgery 1997, 122: 1132-6.
- Constantinides VA, Christakis I, Touska P, et al. Systematic review and meta-analysis of retroperitoneoscopic versus laparoscopic adrenalectomy. Br J Surg 2012, 99: 1639-48.
- Dindo D, Demartines N, Clavien PA. Classification of surgical complications: a new proposal with evaluation in a cohort of 6336 patients and results of a survey. Ann Surg 2004, 240: 205-13.
- Eichhorn-Wharry LI, Talpos GB, Rubinfeld I. Laparoscopic versus open adrenalectomy: another look at outcome using the Clavien classification system. Surgery 2012, 152: 1090-5.
- Dalvi AN, Thapar PM, Thapar VB, et al. Laparoscopic adrenalectomy for large tumours: single team experience. J Minim Access Surg 2012, 8: 125-8.
- Porpiglia F, Destefanis P, Fiori C, et al. Does adrenal mass size really affect safety and effectiveness of laparoscopic adrenalectomy? Urology 2002, 60: 801-5.
- Graham SD, Keane TE. Glenn's Urologic Surgery 2010. Edizioni Lippincott Williams & Wilkins.
- Porpiglia F, Fiori C, Tarabuzzi R, et al. Is laparoscopic adrenalectomy feasible for adrenocortical carcinoma or metastasis? B J U Int 2004, 94: 1026-9.
- Porpiglia F, Fiori C, Daffara F, et al. Retrospective evaluation of the outcome of open versus laparoscopic adrenalectomy for stage I and II adrenocortical cancer. Eur Urol 2010, 57: 873-8.
- Jurowich C, Fassnacht M, Kroiss M, et al. Is there a role for laparoscopic adrenalectomy in patients with suspected adrenocortical carcinoma? A critical appraisal of the literature. Horm Metab Res 2013, 45: 130-6.
- Mir MC, Klink JC, Guillotreau J, et al. Comparative outcomes of laparoscopic and open adrenalectomy for adrenocortical carcinoma: single, high-volume center experience. Ann Surg Oncol 2012 Nov 26.
- Miller BS, Gauger PG, Hammer GD, et al. Resection of adrenocortical carcinoma is less complete and local recurrence occurs sooner and more often after laparoscopic adrenalectomy than after open adrenalectomy. Surgery 2012, 152: 1150-7.
- Lafemina J, Brennan M. Adrenocortical carcinoma: past, present, and future. J Surg Oncol 2012, 106: 586-94.
- Gaujoux S, Brennan M. Recommendation for standardized surgical management of primary adrenocortical carcinoma. Surgery 2012, 152: 123-32.
- Bellantone R, Ferrante A, Boscherini M, et al. Role of reoperation in recurrence of adrenal cortical carcinoma: results from 188 cases collected in the Italian National Registry for Adrenal Cortical Carcinoma. Surgery 1997, 122: 1212-8.
- Porpiglia F, Fiori C. Mini laparoscopy in urology: current indications and future perspectives. Storz Silver book 2010: 6-17.
- Han DH, Lim MS, Seo JW, et al. Laparoendoscopic single site adrenal surgery. Arch Esp Urol 2012, 65: 336-41.
- Brunaud L, Ayav A, Zarnegar R, et al. Prospective evaluation of 100 robotic-assisted unilateral adrenalectomies. Surgery 2008, 144: 995-1001.
- Porpiglia F, Destefanis P, Bovio S, et al. Cortical-sparing laparoscopic adrenalectomy in a patient with multiple endocrine neoplasia type IIA. Horm Res 2002, 57: 197-9.
- Asher KP, Gupta GN, Boris RS, et al. Robot-assisted laparoscopic partial adrenalectomy for pheochromocytoma: the National Cancer Institute Technique. Eur Urol 2011, 60: 118-24.