Deficit di StAR e di P450scc

Stefano Tumini
Dipartimento di Pediatria, Università di Chieti.
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
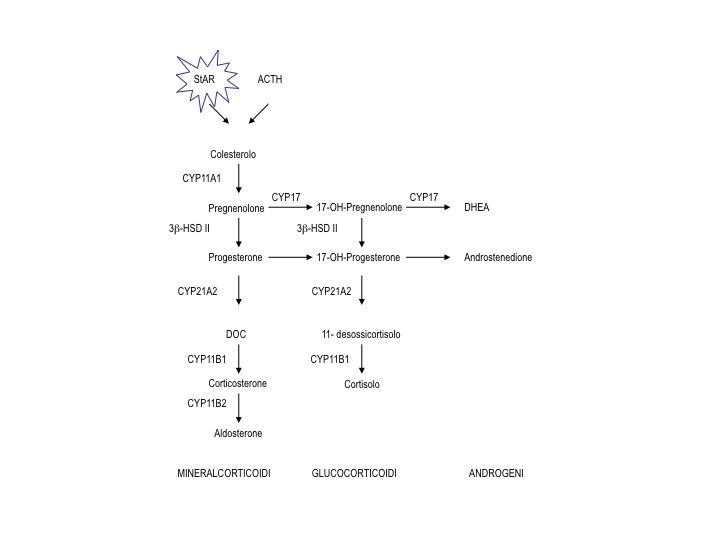
Deficit di StAR (Steroidogenic Acute Regulatory protein)
L’iperplasia congenita lipoidea (Congenital Lipoid Adrenal Hyperplasia – LCAH) (OMIM: 201701) è una malattia autosomica recessiva, molto rara in Europa e Nord America, mentre nei Paesi Asiatici la sua prevalenza è di circa 1:300.000 nati. Determina blocco completo della steroidogenesi. Il difetto fu identificato inizialmente come deficit di conversione del colesterolo in pregnenolone e definito deficit di 20-22 desmolasi. L’osservazione che P450scc è normale nei soggetti LCAH portò all’individuazione nel gene della Steroidogenic Acute Regulatory protein (StAR) di mutazioni responsabili di LCAH (1). StAR regola il passaggio del colesterolo dalla superficie esterna della membrana mitocondriale a quella interna, dove si avvia la steroidogenesi per azione di P450scc. Il corrispondente gene è situato sul locus 8p11.2, è costituito da sette esoni e sei introni ed è espresso nel surrene, nel testicolo, nel cervello ma non nella placenta (2). Sono state descritte oltre 40 mutazioni.
Il modello “two-hits” spiega la sequenza delle manifestazioni cliniche (3); la deposizione di colesterolo nelle cellule steroidogenetiche distrugge (4):
- nel maschio le cellule di Leydig a causa della stimolazione fetale da parte di hCG;
- in entrambi i sessi precocemente la zona fetale del surrene e alla nascita la zona glomerulare e fascicolata;
- nella femmina alla pubertà l’ovaio in seguito alla stimolazione di LH e FSH.
LCAH si manifesta clinicamente nei primi mesi di vita con insufficienza surrenalica severa (vomito, diarrea, disidratazione, iponatremia, ipopotassiemia) (5). Ci sono tuttavia casi in cui sono presenti genitali maschili normali e l’insufficienza surrenalica si manifesta tardivamente (non-classical LCAH) (6).
Nelle forme classiche l’insufficienza surrenalica con perdita di sali presenta manifestazioni cliniche sovrapponibili nei soggetti 46,XX e 46,XY e l’insufficienza testicolare si manifesta nei soggetti 46,XY con “sex reversal” (7). Nei soggetti 46,XX la funzione ovarica può permettere la comparsa di pubertà e menarca con cicli anovulatori e si associa ad ipogonadismo ipergonadotropo (8). Dopo l’inizio della pubertà è stata descritta la comparsa di cisti follicolari con possibilità di torsione ovarica (9) e l’associazione con quadri malformativi del SNC e la sindrome di Chiari tipo 1 (7).
Deficit di P450scc (Cholesterol Side-Chain Cleavage Enzyme)
L’enzima P450scc è codificato dal gene CYP11A1, situato sulla parte interna della membrana mitocondriale e converte il colesterolo in pregnenolone, dando inizio alla steroidogenesi (10). Il deficit di P450scc (OMIM: 118485) compromette la steroidogenesi surrenalica e gonadica. Sono stati riportati solo 19 casi in letteratura (11). Le cellule surrenaliche e gonadiche che esprimono StAR esprimono anche P450scc. Il quadro clinico è indistinguibile dai pazienti con LCAH classica e non classica. Le forme con parziale compromissione di p450scc possono presentarsi tardivamente senza DSD.
Nei pazienti LCAH i surreni appaiono massivamente aumentati di volume, mentre nei pazienti con deficit di P450scc sono di volume diminuito. La diagnosi definitiva viene posta dopo conferma genetica.
Bibliografia
- Lin D, Sugawara T, Strauss J, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 1995, 267: 1828–31.
- Sugawara T, Holt JA, Driscoll D, et al. Human steroidogenic acute regulatory protein: functional activity in COS-1 cells, tissue-specific expression, and mapping of the structural gene to 8p11.2 and a pseudogene to chromosome 13. Proc Natl Acad Sci USA 1995, 92: 4778–82.
- Miller WL. Mitochondrial specificity of the early steps in steroidogenesis. J Steroid Biochem Mol Biol 1995, 55: 607–16.
- Metherell LA, Naville D, Halaby G, et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J Clin Endocrinol Metab 2009, 94: 3865–71.
- King SR, Bhangoo A, Stocco DM. Functional and physiological consequences of StAR deficiency: role in lipoid congenital adrenal hyperplasia. Endocr Dev 2011, 20: 47–53.
- Baker BY, Lin L, Kim CJ, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab 2006, 91: 4781–5.
- Sertedaki A, Dracopoulou M, Voutetakis A, et al. Long-term clinical data and molecular defects in the STAR gene in five Greek patients. Eur J Endocrinol 2013, 168: 351–9.
- Bhangoo A, Buyuk E, Oktay K, Ten S. Phenotypic features of 46, XX females with StAR protein mutations. Pediatr Endocrinol Rev 2007, 5: 633–41.
- Kaku U, Kameyama K, Izawa M, et al. Ovarian histological findings in an adult patient with the steroidogenic acute regulatory protein (StAR) deficiency reveal the impairment of steroidogenesis by lipoid deposition. Endocr J 2008, 55: 1043–9.
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011, 32: 81–151.
- Tee MK, Abramsohn M, Loewenthal N, et al. Varied clinical presentations of seven patients with mutations in CYP11A1 encoding the cholesterol side-chain cleavage enzyme, P450scc. J Clin Endocrinol Metab 2013, 98: 713–20.
- Bose HS, Sugawara T, Strauss III JF, Miller WL. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med 1996, 335: 1870-9.
- Fujieda K, Okuhara K, Abe S, et al. Molecular pathogenesis of lipoid adrenal hyperplasia and adrenal hypoplasia congenita. J Steroid Biochem Mol Biol 2003, 85: 483-9.
Overview sugli iperaldosteronismi primari
Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 28 gennaio 2020)
L’iperaldosteronismo primario è attualmente la causa più frequente di ipertensione arteriosa secondaria. In passato era considerata una patologia rara, benigna, caratterizzata da ipertensione associata ad ipopotassiemia e causata, nella maggior parte dei casi, da un adenoma surrenalico secernente aldosterone (morbo di Conn). Negli ultimi 20 anni, invece, la diffusione di un semplice test di screening (determinazione ARR, Aldosteron-Renin Ratio), ha permesso di identificare tra i pazienti ipertesi un numero crescente di pazienti affetti da IAP, con percentuali che oscillano dal 2% in presenza di ipertensione di primo grado (< 160/100 mmHg) sino al 10-20% in caso di ipertensione resistente (1). L’ipopotassiemia, nelle casistiche più recenti, è presente solo in una minoranza di casi (meno di un terzo) e spesso si associa alle forme più severe (2). La forma più comune di presentazione è quella dell’ipertensione sisto-diastolica con normali livelli di potassio, spesso causata da un’iperplasia surrenalica bilaterale (60-70% dei casi di IAP).
L’entità del problema è rilevante, non solo per la diffusione di questa condizione clinica, ma soprattutto perché l’IAP si associa a una morbilità e mortalità cardio-vascolare superiore a quella dei pazienti con ipertensione essenziale, a parità di sesso, età e livelli pressori. Nei pazienti con IAP, infatti, è stata riscontrata, rispetto ai pazienti con ipertensione essenziale, una frequenza aumentata di ictus (x 4), infarto miocardico (x 6), fibrillazione atriale (x 12) (3). Le complicanze cardio-vascolari dell’IAP sono infatti legate non solo all’ipertensione, ma anche ai danni diretti provocati dall’aldosterone sull’apparato cardio-vascolare (effetti pro-infiammatori e pro-fibrotici dell’aldosterone), che portano a ipertrofia ventricolare sinistra e disfunzione diastolica, disfunzione endoteliale, alterato rimodellamento e fibrosi a livello vascolare (4).
A differenza dell’ipertensione essenziale, l’ipertensione sostenuta da IAP, se diagnosticata precocemente e con accuratezza, è potenzialmente guaribile o trattabile con farmaci “mirati”. Il processo diagnostico prevede tre fasi distinte:
- fase di screening (determinazione ARR);
- fase di conferma mediante un test;
- diagnosi del sotto-tipo di IAP.
Le recenti linee guida dell’Endocrine Society forniscono precise indicazioni a questo riguardo (2), tuttavia ancora molti punti restano da chiarire e sono tuttora oggetto di dibattito tra gli endocrinologi:
- deve ancora essere standardizzato un corretto livello di cut-off dell’ARR; inoltre l’ARR secondo le linee guida dovrebbe essere eseguito in condizioni standard, dopo aver eliminato i fattori interferenti; lavori recenti, tuttavia, mettono in guardia sui potenziali rischi di un wash-out farmacologico troppo rigoroso, specie nei pazienti con ipertensione grave o complicata (5);
- la diagnosi di IAP deve sempre essere confermata mediante un test (test di soppressione al fludrocortisone, test da carico acuto salino endovenoso, test al captopril); resta da definire quale sia il test migliore, anche se il più diffuso è il carico salino ev;
- il cateterismo delle vene surrenaliche (AVS, Adrenal Venous Sampling) è indicato dalle linee guida come il gold standard per la diagnosi di sotto-tipo dell’IAP e dovrebbe essere eseguito in tutti i pazienti con IAP prima di un eventuale intervento di surrenectomia; si tratta comunque di un esame invasivo e costoso e sono pochi i centri d’eccellenza che garantiscono alte percentuali di successo (> 90%); recentemente è stata messa in discussione l’indicazione di eseguire l’AVS in tutti i pazienti con IAP (6,7) e la procedura andrebbe valutata caso per caso, in base all’età del paziente (superiore o inferiore ai 40 anni), al quadro radiologico (presenza o meno di formazione surrenalica tipica per morbo di Conn), al quadro clinico (entità dell’ipertensione e presenza di ipopotassiemia), al desiderio del paziente di essere operato e alla presenza di comorbilità.
Sicuramente un corretto inquadramento diagnostico è fondamentale per poi scegliere il trattamento più efficace (8). L’IAP, infatti, comprende un ampio spettro di condizioni cliniche, che vanno dall’adenoma surrenalico secernente aldosterone all’iperplasia surrenalica bilaterale, con molti fenotipi intermedi. Nel caso di un adenoma secernente aldosterone, l’opzione terapeutica di prima scelta è la surrenectomia, mentre nei casi in cui l’intervento chirurgico è rischioso o viene rifiutato, è consigliata la terapia medica con anti-aldosteronici. Dopo la surrenectomia, circa il 50% dei pazienti (range 30-70%) ottiene la normalizzazione dei livelli tensivi; la persistenza di ipertensione generalmente si associa a età più avanzata, familiarità per ipertensione e lunga durata di malattia. La terapia medica rappresenta invece il trattamento di scelta nel caso di iperplasia surrenalica bilaterale o nel caso di iperaldosteronismo sensibile ai glucocorticoidi (GRA). La surrenectomia monolaterale può anche essere presa in considerazione in casi selezionati di iperplasia surrenalica bilaterale che presentino scarsa risposta al trattamento medico e/o importanti effetti collaterali in corso di terapia con anti-aldosteronici. Prospettive future nel trattamento dell’iperaldosteronismo sono rappresentate da nuovi farmaci, non steroidei, antagonisti dei recettori per i mineralcorticoidi e da farmaci in grado di inibire l’aldosterone-sintetasi (9,10).
Bibliografia
- Mulatero M, Monticone S, Veglio F. Diagnosis and treatment of primary aldosteronism. Rev Endocr Metab Disord 2011, 12: 3-9.
- Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 1889-916.
- Milliez P, Girerd X, Plouin PF, et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005, 45: 1243-8.
- Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab 2009, 94: 3623-30.
- Fischer E, Beuschlein F, Bidlingmaier M, Reincke M. Commentary on the Endocrine Society Practice Guidelines: Consequences of adjustment of antihypertensive medication in screening of primary aldosteronism. Rev Endocr Metab Disord 2011, 12: 43-8.
- Rossi GP, Barisa M, Allolio B, et al. The Adrenal Vein Sampling International Study (AVIS) for identifying the major subtypes of primary aldosteronism. J Clin Endocrinol Metab 2012, 97: 1606-14.
- Cicala MV, Mantero F. Primary aldosteronism: what consensus for the diagnosis. Best Pract Res Clin Endocrinol Metab 2010, 24: 915-21.
- Quinkler M, Stewart PM. Treatment of primary aldosteronism. Best Pract Res Clin Endocrinol Metab 2010, 24: 923-32.
- Kolkhof P, Borden SA. Molecular pharmacology of the mineralocorticoid receptor: prospects for novel therapeutics. Mol Cell Endocrinol 2012, 350: 310-7.
- Colussi G, Catena C, Sechi LA. Spironolactone, eplerenone and the new aldosterone blockers in endocrine and primary hypertension. J Hypertens 2013, 31: 3-15.
Classificazione ed epidemiologia degli iperaldosteronismi primari
Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 28 gennaio 2020)
Definizione ed epidemiologia
L’iperaldosteronismo primario (IAP) attualmente rappresenta la più frequente causa di ipertensione arteriosa secondaria, con un rischio di morbilità e mortalità cardio-vascolare maggiore rispetto all’ipertensione essenziale.
L'IAP è caratterizzato da un’ipersecrezione autonoma di aldosterone, cioè non conseguente a un aumento dei suoi fattori di stimolo, in particolare della renina. Questo conduce a un’aumentata attività mineral-corticoide, con aumento del riassorbimento del sodio e dell’escrezione del potassio a livello del tubulo renale. Ne consegue un’espansione del volume circolante e ipertensione arteriosa con tendenza a ipopotassiemia.
Non molti anni fa si pensava che l’IAP fosse patologia rara (≤ 1% dei casi di ipertensione) e benigna (complicanze simili a quelle degli ipertesi essenziali). Inoltre l’ipopotassiemia era un requisito fondamentale per avviare gli accertamenti diagnostici. Negli ultimi anni si è assistito a una revisione epidemiologica della patologia. L’applicazione su vasta scala negli ipertesi di un semplice test di screening (determinazione del rapporto Aldosterone/Renina) ha infatti dimostrato una prevalenza di IAP nettamente superiore rispetto al passato, con percentuali variabili nelle diverse casistiche dal 3 al 32% (1,2). L’ampio range di prevalenza riportato in letteratura può essere, almeno in parte, spiegato con i diversi criteri di inclusione dei pazienti negli studi e con i differenti criteri diagnostici utilizzati. Uno studio prospettico multicentrico italiano (studio PAPY, Primary Aldosteronism Prevalence in Hypertension), condotto su un’ampia casistica di nuovi ipertesi al fine di accertare la frequenza della patologia in Italia, ha riportato una prevalenza di iperaldosteronismo dell’11.2% (3). Un bias di selezione, in questo studio come in molti altri riportati in letteratura, deriva dal fatto che i test di screening sono stati effettuati su una popolazione di ipertesi afferente a centri di riferimento per l’ipertensione. L’attuale prevalenza dell’IAP tra pazienti ipertesi non selezionati resta pertanto ancora oggi oggetto di dibattito, con una stima approssimativa del 4% (4).
La prevalenza dell’IAP aumenta con la severità dell’ipertensione, dal 2% in pazienti con ipertensione di grado 1 sino al 20% in pazienti con ipertensione resistente (4).
L’ipopotassiemia nelle casistiche più recenti è presente in una minoranza di pazienti affetti da IAP (5,6) e spesso si associa alle forme più severe; la presentazione più comune dell’IAP è pertanto quella dell’ipertensione con livelli normali di potassio (3).
Classificazione
Dalla prima descrizione di iperaldosteronismo causato da un adenoma aldosterone-secernente, fatta da Conn nel 1954, negli anni le acquisizioni fisiopatologiche hanno permesso di identificare altre 6 condizioni cliniche che possono causare la patologia (7).
| Frequenza delle cause di iperaldosteronismo primario | ||
| Sottotipo | Frequenza | |
| Adenoma surrenalico o morbo di Conn | 30-40% | |
| Iperplasia surrenalica bilaterale idiopatica | 60-70% | |
| Cause rare | Iperplasia surrenalica primaria unilaterale | 2% |
| Carcinoma surrenalico aldosterone-secernente | < 1% | |
| Iperaldosteronismo familiare (FH tipo I) o iperaldosteronismo sensibile ai glucocorticoidi (GRA) | < 1% | |
| FH tipo 2, 3, 4 | 5% | |
| Adenoma o carcinoma ectopico aldosterone-secernente (es. neoplasie ovariche o renali) | 0-1% | |
La classificazione si basa su criteri clinici, anatomo-patologici e morfologici. Talvolta la distinzione non è netta e la diagnosi differenziale tra aldosteronoma (APA) e iperplasia idiopatica è complicata dal fatto che esistono forme di APA bilaterali e varianti micro- o macro-nodulari di iperplasia bilaterale.
La diagnosi differenziale tra le diverse forme di IAP non rappresenta un mero esercizio accademico, ma è fondamentale per la scelta della terapia più appropriata.
L’APA rispetto all’iperplasia surrenalica generalmente si manifesta prima (30-50 anni), l’ipertensione può esser più severa e l’ipopotassiemia più frequente.
L’iperplasia surrenalica bilaterale idiopatica attualmente è considerata la forma più frequente di IAP. La variante nodulare può, a sua volta, essere micro- o macro-nodulare.
Recentemente la netta distinzione tra aldosteronoma unilaterale e iperplasia bilaterale è stata messa in discussione dalla dimostrazione della presenza di zone di iperplasia della glomerulosa e di APCCs (aldosterone-producing cell clusters) adiacenti all’aldosteronoma dominante (8).
Il carcinoma surrenalico secernente aldosterone fortunatamente è molto raro, generalmente di dimensioni maggiori rispetto all’adenoma; spesso è in grado di secernere anche altri ormoni surrenalici (soprattutto androgeni), dando origine a quadri clinici più complessi.
Esistono poi le rare forme iperaldosteronismo familiari (FH), generalmente associate con iperplasia surrenalica:
- FH tipo I o iperaldosteronismo sensibile ai glucocorticoidi (GRA - glucocorticoid-remediable aldosteronism), in cui la secrezione di aldosterone è regolata dall'ACTH per un difetto genetico autosomico dominante (gene chimerico CYP11B1/CYP11B2);
- FH tipo II, causato da mutazioni germinali nel gene CLCN2 che codifica per il canale del cloro ClC-2 espresso nella zona glomerulosa del surrene;
- FH tipo III, causato da mutazioni germinali nella subunità KCNJ5 del canale del potassio;
- FH tipo IV, causato da mutazioni germinali nel gene CaCNA1H, che codifica la subunità alfa di un canale del calcio di tipo L, voltaggio-dipendente.
Infine è da ricordare che l’IAP, sostenuto da APA o da iperplasia bilaterale, è stato descritto anche in quadri di MEN tipo 1 (9).
Bibliografia
- Kaplan NM. The current epidemic of primary aldosteronism: causes and consequences. J Hypertens 2004, 22: 863-9.
- Olivieri O, Ciacciarelli A, Signorelli D, et al. Aldosterone to Renin ratio in a primary care setting: the Bussolengo study. J Clin Endocrinol Metab 2004, 89: 4221-6.
- Rossi GP, Bernini G, Caliumi C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. PAPY Study Investigators. J Am Coll Cardiol 2006, 48: 2293-300.
- Mulatero M, Monticone S, Veglio F. Diagnosis and treatment of primary aldosteronism. Rev Endocr Metab Disord 2011, 12: 3-9.
- Mulatero P, Stowasser M, Loh KC, et al. Increased diagnosis of primary aldosteronism, including surgically correctable forms, in centers from five continents. J Clin Endocrinol Metab 2004, 89: 1045-50.
- Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 1889-916.
- Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 2007, 66: 607-18.
- Omata K, Tomlins SA, Rainey WE. Aldosterone-producing cell clusters in normal and pathological states. Horm Metab Res 2017, 49: 951-6.
- Gatta-Cherifi B, Chabre O, Murat A, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol 2012, 166: 269-79.
Clinica e diagnostica generale iperaldosteronismi primari
Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 14 gennaio 2020)
CARATTERISTICHE CLINICHE
Solo una minoranza di pazienti affetti da iperaldosteronismo primario (IAP) si presenta con la classica triade di ipertensione, ipopotassiemia e alcalosi metabolica; spesso, infatti, i pazienti hanno livelli di potassio normali e alcuni possono anche essere ipopotassiemici ma normotesi.
L’ipertensione inizialmente dipende dall’espansione del pool sodico e del volume plasmatico con aumento della portata cardiaca ma, successivamente, entra in gioco un meccanismo di “escape”, che previene un’eccessiva ritenzione sodica e la formazione di edemi. In una prima fase l’ipertensione può essere prevenuta negli animali e trattata nell’uomo con restrizione del sodio nella dieta (1). Nel tempo la persistenza di ipervolemia porta a un aumento delle resistenze periferiche vascolari, che sostengono poi l’ipertensione. L’ipertensione generalmente è sisto-diastolica, di grado medio e talora elevato, spesso resistente (PA > 140/90 mmHg con 3 farmaci a dose piena, incluso un diuretico). Raramente si può associare a forme di ipertensione maligna (2). Spesso i pazienti ipertesi con IAP sono asintomatici o hanno sintomi aspecifici dell’ipertensione (cefalea, vertigini, astenia). I livelli pressori variano ampiamente tra i pazienti affetti, indipendentemente dal fatto che presentino un adenoma secernente aldosterone (APA) o un’iperplasia surrenalica bilaterale, anche se una condizione ipertensiva più severa si associa più frequentemente all’APA. Nelle forme familiari (FH1/GRA) l’ipertensione ha frequentemente un esordio più precoce e può associarsi a ictus emorragico, spesso fatale.
L’ipopotassiemia è stata per molto tempo considerata come una delle principali caratteristiche dell’IAP, ma studi più recenti hanno evidenziato come sia presente solo in una minoranza di pazienti (9-37%) (3,4). L’ipopotassiemia probabilmente si associa ai casi più gravi di ipertensione, ed è stata riportata nel 50% dei pazienti con APA e solo nel 17% dei pazienti con iperplasia bilaterale (5). La presenza di ipopotassiemia pertanto presenta bassa sensibilità e specificità per la diagnosi di iperaldosteronismo primario (4). Può essere peggiorata o slatentizzata dall’uso di diuretici o da un eccessivo/inadeguato introito di sodio con la dieta (6) e può associarci ad alcalosi metabolica. Questa alterazione è dovuta all’escrezione urinaria di ioni idrogeno, mediata sia dall’ipopotassiemia sia dall’azione diretta dell’aldosterone sull’acidificazione distale delle urine. Clinicamente l’ipopotassiemia può manifestarsi con astenia, crampi muscolari, parestesie e cardiopalmo. Nei casi più gravi si possono anche riscontrare anomalie elettrocardiografiche, come onda U prominente e alterazioni del ritmo cardiaco, quali extrasistoli sopra-ventricolari e ventricolari, tachicardie e tachi-aritmie.
Sempre a livello renale l’eccesso di aldosterone può determinare un aumento del filtrato glomerulare (GFR) e della pressione di perfusione renale, indipendentemente dall’ipertensione sistemica. Inoltre è frequente il riscontro di un’aumentata escrezione di albumina nelle urine (micro- e macro-albuminuria). Queste alterazioni possono essere in gran parte reversibili dopo trattamento medico o chirurgico (7, 8). Talvolta il trattamento dell’IAP (surrenectomia o terapia medica con anti-aldosteronici), riducendo l’iperfiltrazione, può determinare un aumento della creatininemia e smascherare una sottostante insufficienza renale (9).
Il rischio cardio-vascolare è aumentato nei pazienti con IAP rispetto ai pazienti affetti da ipertensione di altra natura, compresa l’ipertensione essenziale, l’ipertensione sostenuta da feocromocitoma e la sindrome di Cushing, a parità di età, livelli tensivi e durata di malattia. Recenti studi hanno, infatti, evidenziato come l’eccesso di aldosterone possa determinare un danno cardio-vascolare e renale indipendentemente dai livelli tensivi, attraverso meccanismi di flogosi e successiva fibrosi (10). È stato osservato che i pazienti con IAP rispetto ai pazienti con ipertensione essenziale presentano:
- aumentato spessore dell’intima-media a livello carotideo;
- ridotta funzione endoteliale (misurata mediante vaso-dilatazione flusso-mediata);
- aumento dell’ipertrofia ventricolare sinistra e ridotta funzione diastolica.
In uno studio retrospettivo Milliez et al. (11) hanno riportato in pazienti con IAP una frequenza di ictus, infarto del miocardio e fibrillazione atriale rispettivamente di 4, 6 e 12 volte superiore rispetto ai pazienti con ipertensione essenziale. L’aumentato rischio cardio-vascolare (aumento di ictus, IMA, procedure di rivascolarizzazione e aritmie sostenute), è stato confermato anche da studi prospettici (12).
Il trattamento medico con anti-aldosteronici (o la surrenectomia in caso di APA) è in grado di migliorare/far regredire il danno d’organo e ridurre il rischio cardio-vascolare (10). Anche la dieta iposodica (valutabile con la determinazione della sodiuria 24h) potrebbe essere efficace nel ridurre in modo significativo e indipendente dai livelli tensivi il danno d’organo causato dall’eccesso di aldosterone (13).
Più discussa è la relazione tra iperaldosteronismo primaro e sindrome metabolica, che potrebbe concorrere all’aumentato rischio cardio-vascolare (10). Le alterazioni del metabolismo glicidico e l’insulino-resistenza legata all’ipersecrezione dell’aldosterone sembrano le anomalie metaboliche più rilevanti, suscettibili di miglioramento/risoluzione dopo trattamento, ma per confermare i dati preliminari sono necessari studi prospettici su casistiche numerose (14).
DIAGNOSI
La diagnosi di IAP è un processo che prevede 3 fasi successive:
- screening
- test di conferma
- diagnosi differenziale dei sotto-tipi.
Fase 1: screening
Le linee guida dell’Endocrine Society (4) raccomandano lo screening dell’IAP nei pazienti con ipertensione arteriosa:
- sostenuta (PA > 150/100 mmHg in ciascuna delle tre misurazioni previste, da effettuarsi in giorni differenti);
- resistente (PA > 140/90 mmHg con 3 farmaci a dose piena, incluso un diuretico);
- controllata (PA < 140/90 mmHg) solo con l'impiego di 4 o più farmaci anti-ipertensivi;
- associata a ipopotassiemia (spontanea o indotta da diuretici);
- associata a riscontro di incidentaloma surrenalico;
- associata a sindrome delle apnee notturne (OSAS);
- associata a storia familiare di ipertensione a esordio giovanile e/o ictus in soggetti < 40 anni;
- con un familiare di I grado affetto da IAP.
Un'importante novità delle attuali LG rispetto alle precedenti è lo screening nei pazienti ipertesi affetti da OSAS. Questo, insieme all'abbassamento della soglia dei valori tensivi ritenuti sospetti (attualmente > 150/100 mmHg, in precedenza > 160/100 mmHg), determina un consistente ampliamento della popolazione oggetto di screening. Tale modifica delle indicazioni allo screening è da attribuirsi al costante aumento delle segnalazioni di un’aumentata incidenza di IAP in pazienti ipertesi con sindrome metabolica, diabete e OSAS (15).
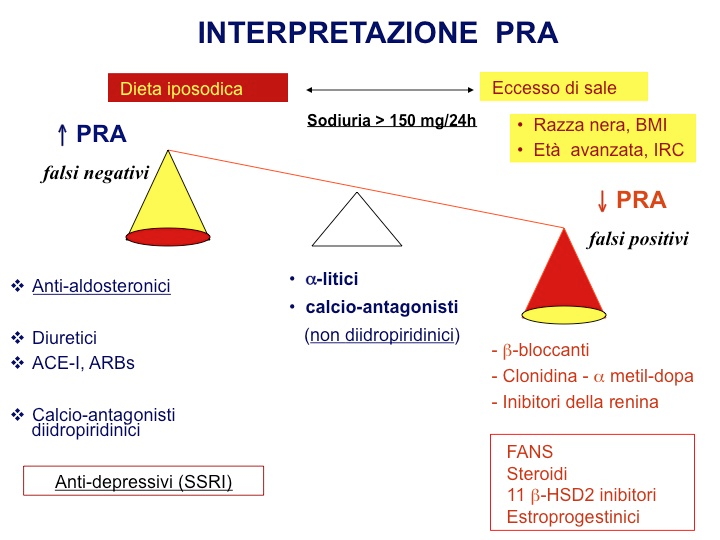
Il test di screening attualmente più utilizzato è costituito dalla determinazione del rapporto tra aldosterone plasmatico e PRA (attività reninica plasmatica), denominato ARR (Aldosteron-Renin Ratio). Un riscontro isolato di PRA soppressa o un singolo rilievo di aldosterone elevato, infatti, non forniscono elementi sufficienti per sospettare l’IAP, ma è necessaria la contemporanea determinazione di entrambi i parametri, misurati al mattino, in ortostatismo. L'ARR viene considerato positivo se superiore a 30-50 (misurando l'aldosterone in ng/dL e la PRA in ng/mL/h). Il cut-off, tuttavia, al momento non è ancora standardizzato e dovrebbe essere stabilito in base all’apporto sodico della dieta della popolazione locale e delle caratteristiche del dosaggio di PRA nel singolo laboratorio, in particolare il minimo valore misurabile di PRA. Il sospetto di IAP, infatti, è tanto maggiore quanto più la PRA è soppressa (< 1 ng/mL/h o indosabile se misurata come renina attiva, DRC) e l'aldosterone plasmatico elevato (> 15-20 ng/dL) (3,4,16).
Numerosi fattori possono interferire con la determinazione del rapporto ARR (17). Le linee guida dell’Endocrine Society (4), a questo proposito per evitare falsi positivi o falsi negativi, raccomandano prima del prelievo:
- dieta con libera assunzione di sodio (la restrizione sodica può dare falsi negativi);
- supplementazione con potassio se necessaria (ipopotassiemia responsabile di falsi negativi);
- almeno 4 settimane prima del prelievo, sospensione di MRA (spironolattone, eplerenone, amiloride, triamterene), diuretici potassio-disperdenti e prodotti derivati dalla liquirizia (rischio di falsi negativi);
- almeno 2 settimane prima del prelievo, sospensione di:
- ACE-inibitori, sartani, inibitori della renina e calcio-antagonisti diidropiridinici (rischio di falsi negativi);
- ß-bloccanti, alfa-agonisti centrali e FANS (rischio di falsi positivi);
- inibitori della renina (abbassano la PRA, ma aumentano la DRC dando, a seconda di cosa si dosa, rispettivamente falsi positivi o falsi negativi).
Nel paziente con difficile controllo pressorio, è possibile utilizzare alcuni farmaci anti-ipertensivi con minori effetti sul sistema renina-angiotensina-aldosterone: verapamil slow-release, idralazina, prazosina, doxazosina, terazosina. Se il wash-out dai farmaci anti-ipertensivi precedentemente utilizzati e il passaggio a questi farmaci consigliati si presentano problematici, anche considerando il rischio di ipopotassiemia, crisi ipertensive e complicanze cardiache, soprattutto nei pazienti con ipertensione severa e/o comorbilità (18), l'ARR deve essere comunque dosato e l'interpretazione del risultato dovrà tenere conto dei fattori interferenti già citati, cui possono aggiungersi altre condizioni che possono determinare falsi positivi (età > 65 anni, fase luteale nelle donne in età fertile, insufficienza renale) o falsi negativi (gravidanza, ipertensione nefro-vascolare o ipertensione maligna).
Fase 2: test di conferma
Data l’alta prevalenza di ipertensione a bassa renina (19), l’aumento dell’ARR non è di per sè diagnostico di IAP, ma deve essere confermato con un test che indichi che la secrezione di aldosterone è inappropriata e non sopprimibile. Sebbene le linee guida dell’Endocrine Society (4) raccomandino l’esecuzione del test di conferma, non è possibile definire con certezza il test migliore: la scelta del test dovrebbe essere basata sulle caratteristiche del paziente, sull’esperienza del centro e sulla semplicità d’esecuzione del test. I test più utilizzati e approvati dalle linee guida sono: il test di soppressione con fludrocortisone (FST), il test da carico salino (endovena o per os) e il test al captopril (4, 20). L'esecuzione di più test, in casi dubbi, non sembra indicata.
Una novità introdotta dalle recenti linee guida (4) rispetto al passato è la possibilità di valutare solo l'ARR, evitando il test di conferma e passando direttamente alla diagnostica strumentale, qualora il paziente presenti ipopotassiemia spontanea, bassi livelli plasmatici di renina e valori plasmatici di aldosterone > 20 ng/dl (550 pmol/L), caratteristiche biochimiche che conferiscono un certo grado di sicurezza. Tutto ciò, in questi casi particolari, ha lo scopo di rendere più rapido l'iter diagnostico. In tutti gli altri casi, rimane la raccomandazione di effettuare uno dei test di conferma.
Il test di soppressione al fludrocortisone (9-α-fluoroidrocortisone) è considerato attualmente il gold standard per la conferma dell’IAP, ma richiede il ricovero del paziente per 4 giorni ed è contro-indicato in pazienti con ipertensione severa, insufficienza cardiaca congestizia, ictus o infarto miocardico. Il test è considerato positivo quando i valori di aldosterone al termine del test siano ≥ 6 ng/dL, con PRA < 1.0 ng/mL/h.
Il test da carico acuto salino endovenoso (test NaCl) è quello più frequentemente utilizzato nella pratica clinica, anche se contro-indicato in pazienti con ipertensione severa o storia di scompenso cardiaco congestizio e grave IRC. Il test viene considerato positivo (diagnostico d'IAP) se i valori di aldosterone alla fine dell’infusione risultano > 10 ng/dL, negativo quando i valori di aldosterone risultano < 5 ng/dL. Livelli plasmatici di aldosterone compresi tra 5 e 10 ng/dL sono “non determinati”, ossia costituiscono una “zona grigia” tra ipertensione essenziale a bassa renina e IAP. La maggior parte degli autori sembra comunque concorde nel considerare questi pazienti come affetti da iperaldosteronismo, più spesso sostenuto da un’iperplasia surrenalica bilaterale che da un adenoma.
Il test da carico salino orale non viene utilizzato in Europa, ma è il test preferito negli USA (3). È considerato positivo qualora l’escrezione urinaria di aldosterone delle 24 ore sia superiore a 10-15 μg/die, con una concomitante escrezione urinaria di sodio > 200 mmol/die. I limiti del test consistono nella scarsa tolleranza del paziente alla dieta ipersodica, nell’attendibilità della raccolta della diuresi e nell’affidabilità del dosaggio dell’aldosteronuria.
Il test con captopril è probabilmente il test di conferma meno standardizzato. Può essere indicato in pazienti che non possono tollerare l’espansione acuta del volume plasmatico. In condizioni normali l'aldosterone è soppresso dopo captopril (> 30%), mentre nei casi di IAP resta elevato con PRA soppressa (4). Il test viene considerato positivo se ARR (determinata dopo 2 ore dalla somministrazione di captopril 25-50 mg) risulta > 30-50 ng/mL (20).
Fase 3: diagnosi dei sotto-tipi di iperaldosteronismo
L’identificazione di un quadro biochimico di IAP con la positività del test di screening e del test di conferma deve dare l’avvio a un iter diagnostico in ambito specialistico, volto a differenziare lesioni surrenaliche aldosterone-secernenti che possano essere asportate chirurgicamente (adenoma, iperplasia surrenalica primitiva unilaterale e carcinoma surrenalico aldosterone-secernente) dall’iperplasia surrenalica bilaterale, suscettibile di terapia medica.
La TC ad alta risoluzione, con mdc e con tagli sottili (2.5-3 mm), mirata al surrene, è la tecnica di imaging più sensibile per identificare i noduli surrenalici, tuttavia non è in grado di diagnosticare i micro-adenomi aldosterone-secernenti (< 1 cm); inoltre non è in grado di distinguere tra un adenoma funzionante e un adenoma non secernente. Nell’iperplasia bilaterale idiopatica il reperto TC può essere del tutto normale o evidenziare surreni ingranditi in toto o con aspetti nodulari. L'esame è fondamentale anche per individuare le rare forme dovute a un carcinoma surrenalico (frequentemente con diametro > 4 cm) e per fornire indicazioni ai radiologi interventisti o ai chirurghi per il management successivo.
La RM surrenalica non è consigliata, perché non offre vantaggi nella diagnosi differenziale tra sotto-tipi, a dispetto di costi più elevati oltre che minore risoluzione spaziale, sensibilità e specificità rispetto alla TC. Pertanto, dovrebbe essere riservata solo ai pazienti che presentano contro-indicazioni all’uso del mezzo di contrasto iodato.
La scintigrafia surrenalica con iodo-colesterolo, peraltro di difficile attuazione, ha una risoluzione spaziale insufficiente per riconoscere la maggior parte degli adenomi aldosterone-secernenti e attualmente non viene più utilizzata nella diagnostica dell'iperaldosteronismo.
Il cateterismo selettivo delle vene surrenaliche (AVS, Adrenal Venous Sampling) rappresenta il gold standard nella diagnosi di sotto-tipo dell’IAP ed è raccomandato dalle linee guida dell’Endocrine Society (4) in tutti i pazienti con diagnosi biochimica di IAP suscettibili d’intervento chirurgico, con l'eccezione di pazienti < 35 anni con ipopotassiemia spontanea e chiara evidenza radiologica di neoplasia unilaterale compatibile con adenoma surrenalico, che pertanto possono subito essere inviati a intervento di surrenectomia monolaterale, senza ulteriori indagini. Si presuppone, infatti, che una massa surrenalica in età giovanile abbia poche probabilità di essere un adenoma non secernente, soprattutto se il sospetto di IAP è sostenuto da chiari dati biochimici e di imaging. Tutti gli altri pazienti devono invece effettuare l’AVS. L’AVS non è ancora standardizzato e la concentrazione ematica di aldosterone va sempre valutata in contemporanea alle concentrazioni di cortisolo.
La stimolazione con ACTH è indicata per i pazienti allergici al mezzo di contrasto, che necessitano della preparazione con steroidi prima di eseguire la procedura (21). In uno studio multicentrico nei pazienti stimolati con ACTH è stato osservato un incremento dell’indice di selettività e una non riduzione significativa dell’indice di lateralizzazione (22). Secondo Monticone et al (21), è preferibile l’infusione continua ai boli per evitare le fluttuazioni dei livelli di aldosterone. Rossi et al (23) sostengono che non ci siano ancora evidenze che la stimolazione con ACTH porti a un esito migliore e che in assenza di infusione di ACTH diventerebbe cruciale l’eseguire un cateterismo simultaneo, sebbene non ci siano dati sufficienti che supportino l’utilizzo del simultaneo piuttosto che del sequenziale.
Il tasso di successo dell’incannulamento delle vene surrenaliche aumenta con l’esperienza del radiologo e può raggiungere il 96%.
L’adeguatezza dell’incannulamento è misurata dal rapporto tra il livello di cortisolo nella vena surrenalica e quello della vena periferica (indice di selettività, SI). Non c’è un consenso relativo al cut-off da utilizzare: per alcuni centri è sufficiente un SI > 1.1, altri usano criteri più restrittivi, quali SI > 2 (preferibilmente > 3) in condizioni basali e > 3 (preferibilmente > 5) durante l’infusione di ACTH (21).
L’indice di lateralizzazione (LI) è il rapporto tra la concentrazione di aldosterone/cortisolo in una vena surrenalica e la concentrazione di aldosterone/cortisolo nella vena surrenalica contro-laterale. Anche in questo caso non vi è omogeneità di interpretazione, dal momento che il rapporto considerato diagnostico varia tra 2 e 5. Monticone et al. suggeriscono di utilizzare un indice di lateralizzazione > 4 come diagnostico di patologia unilaterale e considerare i valori tra 3 e 4 come indeterminati (21).
Alcuni centri al posto dell’indice di lateralizzazione utilizzano un indice ipsi-laterale (rapporto > 2 tra aldosterone/cortisolo di una vena surrenalica e quello di una vena periferica) associato a un rapporto di soppressione contro-laterale (rapporto tra aldosterone/cortisolo della vena surrenalica contro-laterale e aldosterone/cortisolo della vena periferica, CLR) < 1 (21). Per molti autori il CLR può essere utile nei pazienti in cui è stata incannulata una sola vena o che hanno un LI nella zona grigia. A oggi il ruolo del CLR è molto dibattuto, sia nell’indicazione alla surrenectomia che per la sua correlazione agli esiti clinici.
Al fine di ridurre il numero dei pazienti che necessitano di essere sottoposti ad AVS, alcuni studi hanno tentato di identificare una serie di criteri clinici-biochimici che permettano di distinguere tra adenoma e iperplasia bilaterale e quindi di predire le forme di IAP unilaterali. L’APA tipico rispetto all’iperplasia bilaterale è risultato associato più frequentemente a livelli pressori più elevati, a maggiori concentrazioni di aldosterone, ipopotassiemia e a una mancata responsività ai test posturali, ma nessun criterio o punteggio finora proposto è risultato sufficientemente specifico per evitare l'indicazione al cateterismo surrenalico (21,24).
La mancanza di protocolli standardizzati, la disponibilità di pochi centri d’eccellenza dove eseguire l’esame, i costi e la mancata evidenza che l’esame migliori l’outcome, hanno posto recentemente in dubbio le indicazioni delle linee guida di eseguire l’AVS nella maggior parte dei pazienti affetti da IAP (25). Inoltre, recentemente è stato condotto uno studio multi-centrico in cui i pazienti erano randomizzati al trattamento in base ai risultati solo dell'AVS o della TC addominale: solo una delle due metodiche veniva utilizzata per stabilire se il paziente presentasse un adenoma surrenalico responsabile dell'iperaldosteronismo (con conseguente trattamento mediante surrenectomia monolaterale) o se fosse affetto da iperplasia bilaterale (con conseguente trattamento solo con terapia medica, con antagonisti del recettore dei mineralcorticoidi) (26). Lo studio concludeva che, dopo un anno, l'outcome clinico (espresso in termini di controllo pressorio e di qualità della vita) non differiva tra i gruppi di pazienti randomizzati, mettendo quindi in dubbio la necessità di sottoporre i pazienti ad AVS, visto lo scarso vantaggio dal punto di vista pratico. Tale studio è stato tuttavia aspramente criticato perché, nonostante si tratti di uno studio randomizzato e prospettico, è gravato da importanti limitazioni, tra cui bias di selezione, risultati anomali e bassa potenza dello studio per mostrare la differenza negli endpoint più tradizionali (in particolare la cura dell'ipertensione dopo surrenectomia) (27).
Pertanto, ad oggi resta valida l'indicazione delle linee guida dell'Endocrine Society a effettuare, dopo l'esecuzione della TC addome, l'AVS in tutti i pazienti, tranne le eccezioni già citate, raccomandandone l'esecuzione in centri di riferimento e in mani esperte.
Altri esami per la diagnosi di sotto-tipo (il test dell'ortostatismo e il dosaggio del 18-idrossicorticosterone) hanno utilità clinica limitata e non sono a oggi raccomandati.
BIBLIOGRAFIA
- Pedrinelli R, Bruschi G, Graziadei L, et al. Dietary sodium change in primary aldosteronism. Atrial natriuretic factor, hormonal, and vascular responses. Hypertension 1988, 12: 192-8.
- Zarifis J, Lip GY, Leatherdale B, Beevers G. Malignant hypertension in association with primary aldosteronism. Blood Press 1996, 5: 250-4.
- Mulatero P, Monticone S, Veglio F. Diagnosis and treatment of primary aldosteronism. Rev Endocr Metab Disord 2011, 12: 3-9.
- Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 1889-916.
- Rossi GP, Bernini G, Caliumi C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. PAPY Study Investigators. J Am Coll Cardiol 2006, 48: 2293-300.
- Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 2007, 66: 607-18.
- Ribstein J, Du Cailar G, Fesler P, Mimran A. Relative glomerular hyperfiltration in primary aldosteronism. J Am Soc Nephrol 2005, 16: 1320-5.
- Sechi LA, Di Fabio A, Bazzocchi M, et al. Intrarenal hemodynamics in primary aldosteronism before and after treatment. J Clin Endocrinol Metab 2009, 94: 1191-7.
- Reincke M, Rump LC, Quinkler M, et al; Participants of German Conn's Registry. Risk factors associated with a low glomerular filtration rate in primary aldosteronism. J Clin Endocrinol Metab 2009, 94: 869-75.
- Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab 2009, 94: 3623-30.
- Milliez P, Girerd X, Plouin PF, et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol 2005, 45: 1243-8.
- Catena C, Colussi G, Nadalini E, et al. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch Intern Med 2008, 168: 80-5.
- Quinkler M, Stewart PM. Treatment of primary aldosteronism. Best Pract Res Clin Endocrinol Metab 2010, 24: 923-32.
- Fallo F, Pilon C, Urbanet R. Primary aldosteronism and metabolic syndrome. Horm Metab Res 2012, 44: 208-14.
- Monticone S, Viola A, Tizzani D, et al. Primary aldosteronism: who should be screened? Horm Metab Res 2012, 44: 163-9.
- Cicala MV, Mantero F. Primary aldosteronism: what consensus for the diagnosis. Best Pract Res Clin Endocrinol Metab 2010, 24: 915-21.
- Stowasser M, Ahmed AH, Pimenta E, et al. Factors affecting the aldosterone/renin ratio. Horm Metab Res 2012, 44: 170-6.
- Fischer E, Beuschlein F, Bidlingmaier M, Reincke M. Commentary on the Endocrine Society Practice Guidelines: Consequences of adjustment of antihypertensive medication in screening of primary aldosteronism. Rev Endocr Metab Disord 2011, 12: 43-8.
- Olivieri O, Ciacciarelli A, Signorelli D, et al. Aldosterone to Renin ratio in a primary care setting: the Bussolengo study. J Clin Endocrinol Metab 2004, 89: 4221-6.
- Mulatero P, Monticone S, Bertello C, et al. Confirmatory tests in the diagnosis of primary aldosteronism. Horm Metab Res 2010, 42: 406-10.
- Monticone S, Viola A, Rossato D, et al. Adrenal vein sampling in primary aldosteronism: towards a standardised protocol. Lancet Diabetes Endocrinol 2015, 3: 296-303.
- Monticone S, Satoh F, Giacchetti G, et al. Effect of adrenocorticotropic hormone stimulation during adrenal vein sampling in primary aldosteronism. Hypertension 2012, 59: 840-6.
- Rossi GP, Auchus RJ, Brown M, et al. An expert consensus statement on use of adrenal vein sampling for the subtyping of primary aldosteronism. Hypertension 2014, 63: 151-60.
- Mulatero P, Bertello C, Rossato D, et al. Roles of clinical criteria, computed tomography scan and adrenal vein sampling in differential diagnosis of primary aldosteronism subtypes. J Clin Endocrinol Metab 2008, 93: 1366-71
- Stewart PM, Allolio B. Adrenal vein sampling for Primary Aldosteronism: time for a reality check. Clin Endocrinol 2010, 2: 146-8.
- Dekkers T, Prejbisz A, Kool LJS, et al. Adrenal vein sampling versus CT scan to determine treatment in primary aldosteronism: an outcome-based randomised diagnostic trial. Lancet Diabetes Endocrinol 2016, 4: 739–46.
- Beuschlein F, Mulatero P, Asbach E, et al. The SPARTACUS trial: controversies and unresolved issues. Horm Metab Res 2017, 49: 936–42.
Iperplasia surrenalica idiopatica bilaterale
Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 28 gennaio 2020)
Epidemiologia
L’iperplasia surrenalica bilaterale o iperaldosteronismo primitivo (IAP) idiopatico è oggi considerata il più comune sottotipo di IAP (60-70% dei casi). È più frequente nelle donne e si manifesta in età più avanzata rispetto all’adenoma (generalmente dopo i 50 anni) (1).
Patologia
Si caratterizza sul piano morfologico per l’aspetto iperplastico, diffuso o micro-macronodulare, delle zone glomerulari di ambedue i surreni. I nodi iperplastici non sono capsulati e quest’aspetto li differenzia dagli adenomi plurimi. Sono costituiti da cellule chiare, voluminose, ma meno ricche di lipidi rispetto alle cellule dell’adenoma.
Diagnostica
Le alterazioni biochimiche, come l’ipopotassiemia e gli elevati livelli di aldosterone, sono generalmente più attenuate rispetto all’adenoma. Le concentrazioni di aldosterone non presentano ritmo circadiano, possono mantenere la risposta all’ortostatismo e rispondere alla stimolazione da parte dell’angiotensina II. I livelli di DOC e di 18-idrossi-corticosterone risultano per lo più normali (1).
Terapia
Nell'iperplasia surrenalica bilaterale, come per tutte le altre forme di IAP, è fondamentale una rigorosa dieta iposodica (< 2 g/die di NaCl), utile per ridurre al minimo la perdita di potassio e controllare i livelli tensivi (2).
La terapia farmacologica rappresenta il trattamento di prima scelta. I farmaci raccomandati sono rappresentati dagli antagonisti del recettore dei mineralcorticoidi (MR), quali spironolattone, canrenone e il più recente eplerenone (figura) (3). Questi farmaci appaiono efficaci nel controllo dell’ipertensione e nella protezione del danno d’organo, indipendentemente dal controllo pressorio. Infatti, nell'ipertensione da eccesso di mineralcorticoidi, l'obiettivo della terapia medica non è solo quello di controllare i livelli pressori, ma anche quello di inibire gli effetti pro-infiammatori e pro-fibrotici dell'aldosterone, indotti e amplificati dalla presenza del sodio.
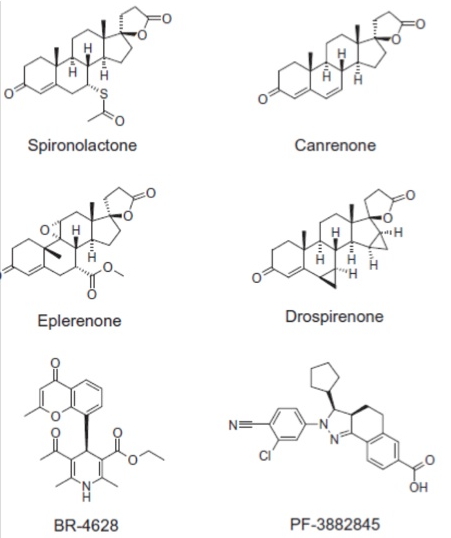
Struttura dei farmaci anti-aldosteronici in uso e sperimentali
Lo spironolattone, anti-aldosteronico di prima generazione, non selettivo, in commercio già dal 1960, è certamente il farmaco più usato nel trattamento dell’IAP. Le sue proprietà terapeutiche sono attribuibili al suo metabolita attivo, il canrenone. Per la sua lunga durata d’azione (emivita 8-12 ore), può essere somministrato in un’unica dose giornaliera. Negli ultimi anni le dosi di spironolattone consigliate nel trattamento dell’IAP sono assai inferiori a quelle utilizzate in passato. Si raccomanda, infatti, di iniziare con dosi di 12.5-25 mg/die, da aumentare gradualmente, circa ogni 2 settimane, sino a normalizzazione dei livelli di potassio; la dose massima consigliata è di 100 mg/die (4).
Al posto dello spironolattone, possono essere utilizzati canrenone o canreonato di potassio, che possono avere minor effetti collaterali rispetto allo spironolattone, ma non ci sono studi di confronto.
L’eplerenone è un anti-aldosteronico di seconda generazione, più selettivo sui MR e pertanto con meno effetti collaterali. Il farmaco tuttavia è più costoso ed è meno efficace dello spironolattone (potenza ridotta al 60%) e deve essere utilizzato in 2 somministrazioni/die. Il minor effetto anti-ipertensivo dell'eplerenone rispetto allo spironolattone era stato dimostrato in uno studio randomizzato, controllato con placebo, condotto su 417 pazienti ipertesi essenziali: 400 mg di eplerenone ottenevano una riduzione dei livelli di pressione sistolica e diastolica sovrapponibile a quella ottenuta con 100 mg di spironolattone (5). La minor efficacia dell'eplerenone nei confronti dello spironolattone è stata recentemente confermata anche in uno studio di confronto condotto su pazienti con IAP (6). Ne consegue che per il trattamento dell’IAP, dovrebbero essere utilizzate dosi di eplerenone più elevate (> 50-100 mg/die) e in duplice somministrazione.
Lo spironolattone o il canrenone potrebbero essere i farmaci da usare all’inizio della terapia, eventualmente sostituibili con l’eplerenone in caso di comparsa di effetti collaterali.
Amiloride e triamterene antagonizzano l’effetto dell’aldosterone a livello renale. L’amiloride, generalmente impiegato alla dose iniziale di 5 mg/die, è ben tollerata e non ha gli effetti collaterali mediati dai recettori steroidei, ma potrebbe avere minor effetto sulla protezione del danno d’organo extra-renale, non riducendo le concentrazioni plasmatiche di aldosterone (7). L’amiloride può essere impiegato in associazione agli anti-aldosteronici, per potenziarne l’effetto ed evitare così gli effetti collaterali dose-correlati, o come alternativa agli stessi.
Se la pressione non si normalizza con l’anti-aldosteronico in mono-terapia, oltre all’amiloride devono essere presi in considerazione anche altri farmaci. L’aggiunta di un diuretico tiazidico a basse dosi (es. 12.5-25 mg/die di idroclorotiazide o clortalidone) può essere di beneficio, poiché l’ipervolemia è spesso la maggior causa di resistenza all’amiloride. Anche i calcio-antagonisti diidropiridinici (riducendo la secrezione dei livelli di aldosterone) possono essere presi in considerazione nelle terapie d’associazione. L’impiego di ACE–inibitori e sartani è controverso, poiché nell’IAP vi è una ridotta produzione di angiotensina I, substrato per l’azione di questi farmaci. Sembrano comunque efficaci nel ridurre i livelli tensivi nelle terapie d’associazione, specie nei pazienti con iperplasia surrenalica bilaterale (7).
Nuovi farmaci in studio sono gli anti-aldosteronici di terza e quarta generazione e gli inibitori dell’aldosterone-sintetasi. Gli anti-aldosteronici di terza generazione sono composti non steroidei, di derivazione dai calcio-antagonisti di-idro-piridinici. Potrebbero evitare gli effetti collaterali di spironolattone e canrenone causati dalla loro interazione con i recettori androgenici e progestinici, mantenendo però le stesse proprietà e la stessa efficacia. Attualmente sono in studio due molecole (BR-4628, PF-3882845), rappresentate in figura.
Sono in fase di studio anche degli anti-aldosteronici di quarta generazione, composti di natura non steroidea, potenti e selettivi, in grado di agire più sugli MR cardiaci e vascolari che sugli MR a livello renale. Potrebbero fornire una buona protezione dal danno d’organo, evitando effetti collaterali quali iperpotassiemia e peggioramento della funzione renale in caso di associazione con ACE-inibitori e sartani. Attualmente è in studio di fase II il composto BAY 94-8862, usato in pazienti con insufficienza renale e insufficienza cardiaca (3).
Gli inibitori dell’aldosterone-sintetasi o CYP11B2, enzima chiave per la sintesi dell’aldosterone, ridurrebbero la produzione di aldosterone a livello della glomerulare del surrene. Sono in studio 2 composti ad azione selettiva sul CYP11B2: FAD 286 e LCI699 (8).
Infine, va segnalato che anche nelle forme di iperaldosteronismo da iperplasia bilaterale potrebbe essere preso in considerazione l’intervento di surrenectomia monolaterale, in casi particolari (es. intolleranza agli anti-aldosteronici e/o inefficacia dei farmaci nel controllo della pressione o presenza di lesioni macro-nodulari di grandi dimensioni) (9). In uno studio retrospettivo condotto su 40 pazienti con iperaldosteronismo idiopatico bilaterale, confermato mediante cateterismo selettivo delle vene surrenaliche, la surrenectomia monolaterale ha guarito dall’ipertensione il 15% dei pazienti dopo 56 mesi dall’intervento e migliorato il controllo dei livelli tensivi nel 20% dei pazienti, già entro un anno dall’operazione chirurgica. La percentuale dei pazienti con ipertensione controllata dopo intervento risultava significativamente superiore a quella prima dell’intervento (65% vs 25%), con miglioramento anche delle complicanze d’organo (10).
Bibliografia
- Young WF. Primary aldosteronism: renaissance of a syndrome. Clin Endocrinol (Oxf) 2007, 66: 607-18.
- Quinkler M, Stewart PM. Treatment of primary aldosteronism. Best Pract Res Clin Endocrinol Metab 2010, 24: 923-32.
- Kolkhof P, Borden SA. Molecular pharmacology of the mineralocorticoid receptor: prospects for novel therapeutics. Mol Cell Endocrinol 2012, 350: 310-7.
- Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 1889-916.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002, 15: 709-16.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011, 29: 980-90.
- Moo TA, Zarnegar R, Duh QY. Prediction of successful outcome in patients with primary aldosteronism. Curr Treat Options Oncol 2007, 8: 314-21.
- Colussi G, Catena C, Sechi LA. Spironolactone, eplerenone and the new aldosterone blockers in endocrine and primary hypertension. J Hypertens 2013, 31: 3-15.
- Stowasser M. Update in primary aldosteronism. J Clin Endocrinol Metab 2009, 94: 3623-30.
- Sukor N, Gordon RD, Ku YK, et al. Role of unilateral adrenalectomy in bilateral primary aldosteronism: a 22-year single center experience. J Clin Endocrinol Metab 2009, 94: 2437-45.
Morbo di Conn
Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 28 gennaio 2020)
L’adenoma surrenalico aldosterone-secernente o morbo di Conn è 3 volte più comune nelle donne che negli uomini e si manifesta generalmente tra i 30 e i 50 anni. Solo raramente si presenta nei bambini. L’ipertensione può essere più severa e associarsi a ipopotassiemia con maggior frequenza rispetto all’iperplasia surrenalica bilaterale.
TERAPIA CHIRURGICA
Dovrebbe essere proposta la surrenectomia per via laparoscopica (1).
L’intervento di surrenectomia, eseguito in laparoscopia, generalmente presenta poche complicanze e necessita di pochi giorni di degenza (1). È in discussione la surrenectomia parziale, con rimozione solo dell’adenoma aldosterone-secernente, ma al momento è consigliabile la rimozione totale del surrene, più sicura e più efficace dell’enucleazione dell’adenoma nell’ottenere la guarigione. L’intervento parziale, infatti, potrebbe rimuovere un nodulo surrenalico inattivo e non l’aldosteronoma, che spesso è di piccole dimensioni e non evidenziabile con la TAC pre-intervento (2).
Risultati
Dopo surrenectomia, l’ipertensione e l’ipopotassiemia migliorano nel 100% dei casi, ma solo il 50% circa dei pazienti (range 35-60%) risulta guarito e può sospendere completamente la terapia farmacologica anti-ipertensiva. La pressione generalmente si normalizza o migliora nell’arco di 1-6 mesi dopo l’intervento ma, in alcuni casi, si può osservare una riduzione della pressione anche dopo 12 mesi dall’operazione. La mancata correzione dell’ipertensione può essere attribuita a una diagnosi inaccurata o, più comunemente, alla concomitante presenza di ipertensione essenziale; altri ipotizzano un ruolo del danno vascolare causato dal protratto eccesso di aldosterone.
Fattori predittivi per la guarigione sono (2-4):
- giovane età e breve durata di malattia (< 5 anni);
- familiarità negativa per ipertensione (o al massimo un solo familiare di 1° grado iperteso);
- risposta positiva prima dell'intervento alla terapia con anti-aldosteronici;
- controllo pre-operatorio dell’ipertensione con un numero di farmaci non superiore a 2;
- valori elevati di ARR e di aldosterone urinario pre-intervento;
- funzione renale normale.
Prima di inviare il paziente all’intervento, bisognerebbe quindi considerare anche la probabilità di successo dell’intervento, oltre al grado di ipertensione.
Gestione pre-intervento
Nel pre-intervento, bisogna correggere l’eventuale ipopotassiemia e controllare la pressione con anti-aldosteronici. Si consiglia di trattare il paziente con adenoma aldosterone-secernente per almeno 4 settimane prima dell’intervento, per prevenire l’ipoaldosteronismo post-intervento. Lo spironolattone, per la sua lunga emivita, può essere sospeso 1-3 giorni prima dell’intervento (2).
Gestione post-intervento
In prima giornata post-intervento andrebbero sospesi la supplementazione di potassio e la terapia con anti-aldosteronico, oltre alla riduzione della terapia anti-ipertensiva.
A breve termine, già a partire dal giorno dopo l'intervento, andrebbe effettuato il dosaggio di aldosterone (5) e secondo altri autori anche di PRA (1), per avere un’indicazione precoce di risposta biochimica.
Nelle prime settimane dopo l’intervento si raccomanda una dieta ricca di sodio, per evitare un possibile ipoaldosteronismo secondario alla soppressione cronica del surrene contro-laterale. In alcuni casi può essere necessaria una temporanea terapia con fludrocortisone (0.5-1 mg/die).
TRATTAMENTO FARMACOLOGICO
È consigliato nei pazienti che rifiutano o presentano contro-indicazioni all’intervento chirurgico. I farmaci di scelta sono gli antagonisti del recettore dei mineralcorticoidi (spironolattone, canrenone, eplerenone). Alla terapia farmacologica, come per le altre forme di iperaldosteronismo primitivo, va sempre associata una dieta rigorosamente iposodica (< 2 g/die di sodio), utile a ridurre al minimo la perdita di potassio e controllare i livelli tensivi, oltre a limitare gli effetti tossici dell'aldosterone a livello cardio-vascolare, indipendenti dai livelli pressori (6).
La maggior parte dei pazienti richiede, oltre agli anti-aldosteronici, l’aggiunta di altri farmaci per ottenere un controllo ottimale della pressione (es. amiloride, diuretici tiazidici a basse dosi, calcio-antagonisti diidropiridinici). Infatti, a differenza delle forme di iperaldosteronismo causato da iperplasia bilaterale, l'ipertensione generalmente è più severa.
Secondo alcuni autori l’intervento, laddove indicato, rappresenta un trattamento favorevole in termini di costo/beneficio rispetto alla terapia medica cronica (1,2). Al momento non vi sono ancora sufficienti lavori randomizzati e a lungo termine che dimostrino un miglioramento della qualità di vita, della morbilità e mortalità con il trattamento chirurgico rispetto alla terapia medica con anti-aldosteronici (2,7).
BIBLIOGRAFIA
- Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 1889-916.
- Quinkler M, Stewart PM. Treatment of primary aldosteronism. Best Pract Res Clin Endocrinol Metab 2010, 24: 923-32.
- Moo TA, Zarnegar R, Duh QY. Prediction of successful outcome in patients with primary aldosteronism. Curr Treat Options Oncol 2007, 8: 314-21.
- Mattsson C, Young WF Jr. Primary aldosteronism: diagnostic and treatment strategies. Nat Clin Pract Nephrol 2006, 2: 198-208.
- Mulatero P, Monticone S, Veglio F. Diagnosis and treatment of primary aldosteronism. Rev Endocr Metab Disord 2011, 12: 3-9.
- Pimenta E, Gordon RD, Ahmed AH, et al. Cardiac dimensions are largely determined by dietary salt in patients with primary aldosteronism: results of a case-control study. J Clin Endocrinol Metab 2011, 96: 2813-20.
- Sukor N, KogovseK C, Gordon RD, et al. Improved quality of life, blood pressure, and biochemical status following laparoscopic adrenalectomy for unilateral primary aldosteronism. J Clin Endocrinol Metab 2010, 95: 1360-4.
Glucocorticoid-remediable aldosteronism e novità in ambito genetico
Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 28 gennaio 2020)
L’iperaldosteronismo glucocorticoide-sensibile, noto come GRA (Glucocorticoid Remediable Aldosteronism), è una rara forma di iperaldosteronismo primitivo (IAP), con frequenza del 3% nei bambini ipertesi (1,2). È stato descritto per la prima volta nel 1966 da Sutherland et al (3) in due membri della stessa famiglia che presentavano elevati livelli di aldosterone, iporeninemia, ipertensione e ipopotassiemia. ll GRA è noto anche come iperaldosteronismo primitivo familiare di tipo I (FH-1), in quanto è ereditario ed è trasmesso come carattere autosomico dominante, ad espressione variabile.
Si caratterizza per l’insorgenza in giovane età e per la possibilità di normalizzare la secrezione di aldosterone, e quindi la pressione, con dosi fisiologiche di glucocorticoidi.
Dal punto di vista morfologico, si presenta generalmente con le caratteristiche dell’iperplasia surrenalica bilaterale.
Patogenesi
L’alterazione genetica responsabile della sindrome è stata descritta per la prima volta nel 1992 da Lifton et al. (4) e consiste nella formazione di un gene chimerico o ibrido, risultante da un processo di crossing–over ineguale, durante la meiosi, fra due geni con un’elevata omologia di sequenza (95%): il CYP11B1, che codifica per l’enzima 11ß-idrossilasi, e il CYP11B2, che codifica per l’enzima aldosterone-sintetasi, localizzati entrambi sul braccio lungo del cromosoma 8. Il meccanismo dettagliato di questo processo di ricombinazione genetica è descritto nella fig. 1 (5).
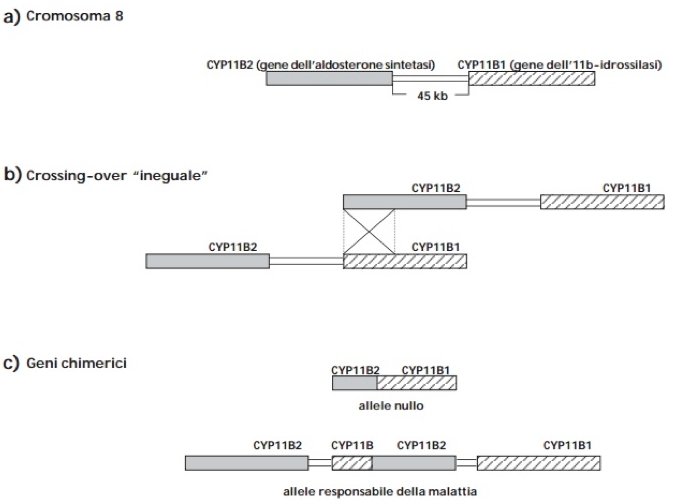
Figura 1. Processo di ricombinazione genetica che porta alla formazione del gene chimerico CYP11B1/B2
In condizioni fisiologiche l’aldosterone viene sintetizzato nella zona glomerulare del surrene e la sua secrezione è regolata dal sistema renina–angiotensina. L’aldosterone-sintetasi è l’enzima chiave nel processo di sintesi dell’aldosterone che avviene in due tappe: l’idrossilazione in C18 del cortico-sterone (sintesi del 18-idrossi-corticosterone) e la successiva ossidazione in C18 dell’idrossile in gruppo aldeidico (sintesi dell’aldosterone). Questo enzima normalmente è presente nella zona glomerulare del surrene, mentre è assente nella zona fascicolata, deputata alla sintesi dei glucocorticoidi (fig 2).
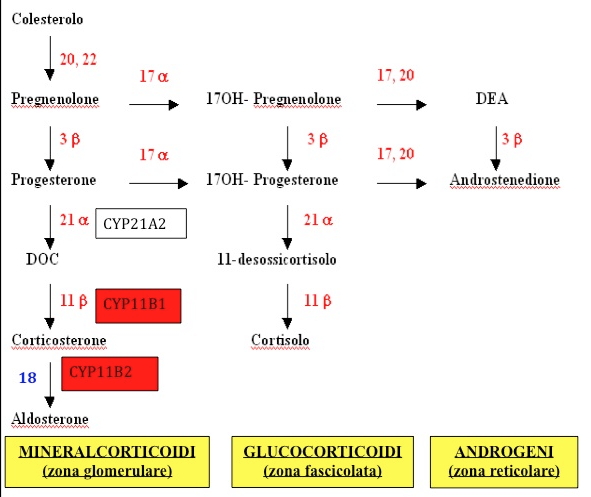
Figura 2
Nell’iperaldosteronismo familiare di tipo 1 invece la formazione del gene chimerico (CYP11B1/B2), derivante dalla fusione della regione regolatrice del gene CYP11B1 (ACTH-sensibile) con la regione del gene CYP11B2 (che codifica per l’aldosterone-sintetasi), si traduce in un’espressione ectopica dell’attività enzimatica dell’aldosterone-sintetasi nella zona fascicolata del surrene, sotto il controllo dell’ACTH. Il risultato finale è la produzione ACTH-dipendente di aldosterone a partire dal corticosterone e di steroidi “ibridi” 18-idrossilati a partire dal cortisolo (18-idrossi-cortisolo e 18-oxo-cortisolo) nella zona fascicolata del surrene (5).
Caratteristiche cliniche
La presenza di questa rara forma di IAP dovrebbe essere sospettata in caso di:
- familiarità per IAP;
- insorgenza precoce dell’ipertensione (< 20 anni);
- ictus emorragico prima dei 40 anni.
Caratteristica clinica peculiare è la pronta e completa risposta al trattamento con desametasone e lo stretto collegamento funzionale con la secrezione di ACTH; per contro, l’iperaldosteronismo è totalmente svincolato dal controllo del sistema renina–angiotensina. L’aldosterone circolante mostra un evidente ritmo circadiano e risponde allo stimolo con ACTH; la caduta dei livelli ormonali dopo somministrazione di desametasone è evidente anche dopo singola dose.
L’esordio e la severità dell’ipertensione possono variare notevolmente, anche tra i soggetti appartenenti allo stesso nucleo familiare. La variabilità di espressione clinica intra-familiare è dovuta al fatto che la penetranza genica può essere incompleta e l’espressività variabile. Questo è dimostrato dal fatto che in una famiglia affetta da GRA, solo uno su tre individui con gene chimerico presenta le caratteristiche cliniche classiche del GRA, mentre gli altri due membri sono normotesi, normokaliemici e con normali livelli di aldosterone e PRA.
L’ipertensione può esordire già nell’infanzia e portare a exitus precoce per complicanze cerebro-vascolari, ma può anche manifestarsi dopo i 50 anni ed essere di grado moderato. In questa sindrome la severità dell’ipertensione si associa al sesso (6): gli uomini infatti presentano un’ipertensione più severa rispetto alle donne. Il cromosoma X si comporterebbe da fattore protettivo, modulando, probabilmente attraverso gli estrogeni, l’espressione del gene ibrido (6).
L’ipopotassiemia non è frequente e i livelli di potassio sono normali nella maggior parte dei casi. Una possibile spiegazione è che la secrezione di aldosterone nel GRA è sotto l’influenza dell’ACTH e pertanto riflette il ritmo circadiano dell’ACTH, con secrezione sovra-normale solo nelle ore diurne. Dopo la somministrazione di un diuretico tiazidico si manifesta frequentemente una marcata ipopotassiemia.
Le complicanze cerebro-vascolari in giovane età, inizialmente derivate da osservazioni aneddotiche, sono poi state confermate su casistiche più ampie, in particolare grazie al registro internazionale per il GRA (7). In questi pazienti il 18% aveva una complicanza cerebro-vascolare, nel 70% dei casi rappresentata da ictus emorragico per rottura di un aneurisma intra-cranico. La causa dell’elevata frequenza di ictus emorragico non è nota, ma è possibile che l’ipertensione congenita durante gli stadi precoci dello sviluppo cerebro-vascolare giochi un ruolo importante. Alcuni autori consigliano di sottoporre tutti i pazienti con GRA a uno screening con angioRM cerebrale alla pubertà e poi ogni 5 anni (7). Il beneficio di tale condotta non è stato dimostrato e va soppesato il rischio di una chirurgia profilattica per piccoli aneurismi che potrebbero anche non rompersi mai.
Diagnosi
La diagnosi va sospettata sulla base delle caratteristiche cliniche sopra riportate. I livelli di aldosterone circolante sono elevati e la PRA soppressa, ma il rapporto ARR tipicamente non è alto come nell’aldosteronoma.
In passato la diagnosi si basava sul test di soppressione con desametasone e sul dosaggio di prodotti del cortisolo ossidati in posizione 18-C (18-idrossi-cortisolo e 18-oxocortisolo). Attualmente l’analisi genetica ha rimpiazzato i test biochimici.
Terapia
Il GRA dovrebbe essere trattato con glucocorticoidi, allo scopo di sopprimere parzialmente la secrezione di ACTH da parte dell’ipofisi. Le linee guida dell’Endocrine Society raccomandano l’uso di glucocorticoidi di sintesi a lunga durata d’azione, come il desametasone o il prednisone, da somministrare alla sera prima di andare a dormire (8). Per evitare una sindrome di Cushing iatrogena, bisognerebbe utilizzare i dosaggi più bassi possibili di glucocorticoidi, sufficienti a normalizzare la pressione e/o il potassio. Negli adulti la dose iniziale di desametasone dovrebbe essere di 0.125-0.250 mg/die, mentre quella del prednisone di 2.5-5 mg/die (9). In alcuni casi, quando la pressione non si normalizza con lo steroide, andrebbero inseriti gli antagonisti dei mineral-corticoidi.
Per valutare l’adeguatezza della terapia e prevenire l’overtreatment, possono essere utili il dosaggio di PRA e aldosterone in corso di terapia (9). Nei bambini può esser utile l’impiego dell’eplerenone, per evitare un ritardo di crescita e/o effetti anti-androgenici secondari a terapia con steroidi e spironolattone.
ALTRE FORME GENETICHE
Le linee guida dell'Endocrine Society (8) suggeriscono l'esecuzione di test genetici nei pazienti con iperaldosteronismo a insorgenza prima dei 20 anni o con storia familiare di IAP o ictus in giovane età (< 40 anni).
Negli ultimi anni la diffusione delle nuove tecniche di indagine genetica (NGS) ha permesso di identificare un maggior numero di forme di IAP familiare (10-12). In particolare, sino a poco tempo fa tutte le forme familiari di IAP non responsive alla somministrazione di glucorticoidi venivano identificate come iperaldosteronismo familiare di tipo II (FH-II), includendo sia pazienti con adenoma che con iperplasia bilaterale. Solo recentemente sono state definite le basi genetiche di FH-II, identificando mutazioni nel gene CLCN2, che codifica per il canale del cloro CIC-2. Le forme di IAP associate a questa mutazione appaiono generalmente bilaterali e responsive alla terapia medica con MR-antagonisti (13).
Una nuova forma di IAP familiare (FH-III) è stata descritta in pazienti molti giovani, con quadri clinici estremamente severi, tali da comportare la surrenectomia bilaterale. La base molecolare della patologia è stata scoperta nel 2011, con l’identificazione di mutazioni germinali nel gene KCNJ5, che codifica per il canale del potassio GIRK4. Il GIRK4 (o KIR 3.4) contribuisce al mantenimento della negatività del potenziale di membrana, pompando il potassio al di fuori della cellula (14). Le mutazioni causano un'alterazione della selettività del filtro del canale, determinando produzione costitutiva di aldosterone e proliferazione cellulare.
Molto rara è la forma di IAP familiare tipo IV (FH-IV), trasmessa come malattia autosomica dominante e causata da una mutazione nel gene CACNA1H, che codifica per canali del calcio voltaggio-dipendenti (subunità alfa1H). Il quadro biochimico è indistinguibile dalla forma sporadica, la TC surreni può presentarsi normale o con minime alterazioni. Dal punto di vista clinico, oltre all’insorgenza di ipertensione in età pediatrica, sono descritti casi con associato lieve ritardo mentale e di sviluppo; buona è la risposta alla terapia con MRA.
Rara è anche la sindrome PASNA (per alcuni autori FH-V), causata da mutazioni germinali nel gene CACNA1D (canali del calcio voltaggio-dipendenti subunità alfa1D), clinicamente caratterizzata da iperaldosteronismo, convulsioni e anomalie neurologiche. Il quadro TC surreni risulta di norma e l’ipertensione non è resistente, ma responsiva a farmaci calcio-antagonisti.
Oltre al riscontro di nuove mutazioni germinali, negli ultimi anni studi multi-centrici hanno identificato mutazioni somatiche nel 10-68% degli APA sottoposti a surrenectomia.
- In soggetti giovani e di sesso femminile, con ipertensione severa e ipopotassiemia, sono state riscontrate frequentemente mutazioni del gene KCNJ5 (8).
- Mutazioni somatiche riguardanti geni codificanti per ATPasi (ATP1A1 e ATP2B3) sono state invece riscontrate più frequentemente in pazienti di sesso maschile, con con APA associato a ipopotassiemia e iperaldosteronemia più marcate rispetto ai non portatori.
- Con frequenze variabili nelle diverse casistiche sono state riportate anche mutazioni somatiche di CACNA1D e CTNNB1 (gene codificante per la ß-catenina)
Nonostante che la ricerca di mutazioni geniche sull'APA asportato chirurgicamente non abbia ancora implicazioni terapeutiche, queste recenti scoperte contribuiscono alla comprensione dei processi fisiopatologici della produzione di aldosterone e aprono possibili scenari per terapie future.
BIBLIOGRAFIA
- Mulatero P, Tizzani D, Viola A, et al. Prevalence and characteristics of familial hyperaldosteronism: the PATOGEN study (Primary Aldosteronism in TOrino-GENetic forms). Hypertension 2011, 58: 797-803.
- Aglony M, Martínez-Aguayo A, Carvajal CA, et al. Frequency of familial hyperaldosteronism type 1 in a hypertensive pediatric population: clinical and biochemical presentation. Hypertension 2011, 57: 1117-21.
- Sutherland DJ, Ruse JL, Laidlaw JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J 1966, 95: 1109-19.
- Lifton RP, Dluhy RG, Powers M, et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat Genet 1992, 2: 66-74.
- Quack I, Vonend O, Rump LC. Familial hyperaldosteronism I-III. Horm Metab Res 2010, 42: 424-8.
- Stowasser M, Bachmann AW, Huggard PR, et al. Severity of hypertension in familial hyperaldosteronism type I: relationship to gender and degree of biochemical disturbance. J Clin Endocrinol Metab 2000, 8: 2160-6.
- Litchfield WR, Anderson BF, Weiss RJ, et al. Intracranial aneurysm and hemorrhagic stroke in glucocorticoid-remediable aldosteronism. Hypertension 1998, 31: 445-50.
- Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016, 101: 1889-916.
- Quinkler M, Stewart PM. Treatment of primary aldosteronism. Best Pract Res Clin Endocrinol Metab 2010, 24: 923-32.
- Seidel E, Schewe J, Scholl UI. Genetic causes of primary aldosteronism. Exp Mol Med 2019, 51: 131.
- Monticone S, Buffolo F, Tetti M, et al. The expanding genetic horizon of primary aldosteronism. Eur J Endocrinol 2018, 178: R101-11.
- Tevosian SG, Fox SC, Ghayee HK. Molecular mechanisms of primary aldosteronism. Endocrinol Metab (Seoul) 2019, 34: 355–66.
- Scholl UI, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 2018, 50: 349-54.
- Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331: 768–72.
Scheda anti-aldosteronici
Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
SPIRONOLATTONE E CANRENONE
Meccanismo d’azione
Lo spironolattone è un diuretico, anti-aldosteronico di prima generazione, non selettivo. Agisce antagonizzando l’azione dell’aldosterone a livello recettoriale, ma può agire anche sui recettori degli androgeni (come antagonista) e del progesterone (come agonista). Il farmaco è assorbito a livello intestinale, viene metabolizzato principalmente nel fegato e le sue proprietà terapeutiche sono attribuibili al suo metabolita attivo, il canrenone. Per la sua lunga durata d’azione (emivita 8/12 ore), può essere somministrato in un’unica dose giornaliera.
Preparazioni, posologia, via di somministrazione
Spironolattone:
- cps 25 mg (Aldactone, Spirolang)
- cps 50 mg (Spirolang)
- cp riv - cps 100 mg (Aldactone, Spirolang, Uractone)
- cp 50 mg (Luvion)
- cp 100 mg (Luvion)
- fl 200 mg/2 mL (Luvion)
Canreonato di potassio:
- cp 25 mg (Kanrenol)
- cp 100 mg (Kanrenol, potassio canrenoato EG, potassio canrenoato Pensa, potassio canrenoato Sandoz)
- cp 200 mg (Kanrenol)
- fiale 200 mg/2 mL (Kanrenol)
Dosi riportate in scheda tecnica nel tratttamento dell'iperaldosteronismo: 100-300 mg, frazionate nelle 24 h. Le recenti linee guida dell'Endocrine Society sull'iperaldosteronismo primario, raccomandano di iniziare con dosi di 12.5-25 mg/die, da aumentare gradualmente, circa ogni 2 settimane, sino a normalizzazione del potassio; dose massima consigliata: 100 mg/die (1).
Indicazioni
Trattamento dell'iperaldosteronismo primario o secondario e dell'ipertensione arteriosa essenziale, laddove altre terapie non sono risultate sufficientemente efficaci o tollerate.
Utilizzato anche nell'edema e ascite da cirrosi epatica o da neoplasia maligne e nella sindrome nefrosica.
Raccomandato inoltre nell'insufficienza cardiaca grave e sintomi di disfunzione ventricolare (2).
Contro-indicazioni
Insufficienza renale cronica di grado avanzato (clearance della creatinina < 30 mL/min), insufficienza renale acuta o anuria.
Condizioni di iperpotassiemia, grave iponatriemia, ipovolemia o disidratazione.
Nelle donne durante la gravidanza e l'allattamento.
Precauzioni d'uso
Il farmaco deve essere inghiottito senza masticare con una sufficiente quantità di liquidi (circa 1/2 bicchiere); consigliata l'assunzione del farmaco con la prima colazione o durante il pranzo, perchè l’assorbimento è aumentato se viene assunto durante il pasto.
Consigliato attento monitoraggio del potassio nei pazienti anziani, specie con ridotta funzionalità renale, e nei pazienti che assumono FANS, ACE-inibitori, sartani (rischio iperpotassiemia).
Effetti collaterali
Sono legati principalmente legati all’azione anti-androgena e all’effetto progestinico:
- nell’uomo ginecomastia, mastodinia, calo della libido e impotenza;
- nella donna irregolarità del ciclo mestruale, tensione mammaria.
La ginecomastia è dose-dipendente, con incidenza dopo 6 mesi di terapia che può andare dal 6.9% con dosi inferiori a 50 mg/die, sino al 52% con dosi superiori a 150 mg/die (1) .
Canrenone e canreonato di potassio, non avendo prodotti intermedi e interferendo meno con gli steroidi sessuali, possono avere minor effetti collaterali rispetto allo spironolattone, ma non ci sono studi di confronto.
Limitazioni prescrittive
No
EPLERENONE
Meccanismo d’azione
Diuretico risparmiatore di potassio ad azione anti-aldosteronica di seconda generazione, più selettivo sui recettori dei mineralcorticoidi. Viene metabolizzato a livello epatico dal CYP3A4. Ha emivita di circa 3 ore e non possiede metaboliti attivi, per cui dovrebbe essere somministrato in 2 somministrazioni/die.
Preparazioni, posologia, via di somministrazione
- cp riv 25: eplerenone Accord, eplerenone DOC, eplerenone KRKA, eplerenone Mylan, eplerenone Tecnigen, Inspra.
- cp riv 50 mg: eplerenone Accord, eplerenone DOC, eplerenone KRKA, eplerenone Mylan, eplerenone Tecnigen, Inspra.
In scheda tecnica consigliata dose iniziale di 25 mg/die, in monosomministrazione, da aumentare a 50 mg/die nell'arco di 4 settimane. Nel caso dell'iperaldosteronismo la dose può essre aumentata sino a 100 mg/die, con duplice soministrazione (1).
Indicazioni
Indicato per la riduzione del rischio di mortalità e morbilità cardiovascolare in pazienti stabili con disfunzione ventricolare sin (LVEF ≤ 40%) ed evidenze cliniche di scompenso cardiaco a seguito di recente infarto del miocardio, in aggiunta alla terapia standard compresi i ß-bloccanti (approvato in USA e altri stati, non in Italia).
Approvato anche nel trattamento dell'ipertensione essenziale (USA e Giappone).
Utilizzato nell'iperaldosteronismo.
Contro-indicazioni
Iperpotassiemia (K+ > 5.0 mmol/L), insufficienza renale moderata-grave (clearance della creatinina < 50 mL/min), insuffiicienza epatica grave (classe C di Child).
Rischio di iperpotassiemia se assunto con: diuretici risparmiatori di potassio, integratori di potassio o potenti inibitori del CYP3A4 (es. itraconazolo, ketoconazolo, ritonavir, claritromicina, nefazodone).
Non vi sono dati sufficienti sull'uso del farmaco in donne in gravidanza.
Precauzioni d'uso
Attento monitoraggio del potassio nei pazienti a rischio di iperpotassiemia, specie nei pazienti (anziani) con compromissione della funzione renale e nei pazienti diabetici.
Effetti collaterali
Nello studio di efficacia e di sopravvivenza sulla scompenso cardiaco conseguente ad infarto (studio Ephesus) condotto su 6632 pazienti, l'incidenza complessiva di eventi avversi con eplerenone è risultata sovrapponibile a quella con placebo.
Eventi avversi comuni: iperpotassiemia, capogiri, ipotensione, diarrea, nausea, alterata funzionalità renale.
Eventi non comuni: ginecomastia, insonnia, prurito, astenia.
Limitazioni prescrittive
No.
AMILORIDE E TRIAMTERENE
Meccanismo d’azione
Diuretici risparmiatori di potassio che agiscono sui canali epiteliali del sodio, a livello del tubulo renale, antagonizzando l’effetto dell’aldosterone a livello renale.
Preparazioni, via di somministrazione, posologia
- Amiloride cp 5 mg (Modamide)*
- Amiloride: 5 mg in associazione a idroclorotiazide 50 mg (Moduretic)
- Triamterene: 25 mg in associazione a furosemide 40 mg (Fluss)
Posologia amiloride: 10 mg/die per os in monosomministrazione o in dose frazionata (5 mg x 2/die); dose massima 20 mg/die.
Indicazioni
Edemi d'origine cardiaca, ipertensione arteriosa, ascite ed edemi in cirrosi epatica.
Contro-indicazioni
Iperpotassiemia, insufficienza renale.
Effetti collaterali
Rash, iponatriemia, confusione, disturbi gastrointestinali, secchezza delle fauci.
Limitazioni prescrittive*
L'amiloride non è in commercio in Italia, ma è in commercio in USA e alcuni paesi europei.
L'amiloride e il triamterene in Italia sono in commercio solo in formulazione d'associazione con altri diuretici.
BIBLIOGRAFIA
- Funder JW, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008, 93: 3266-81.
- McMurray JJ, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012, 33: 1787-847.
Iperaldosteronismi secondari
Classificazione iperaldosteronismi secondari
Sara Cassibba
Endocrinologia, AO S Croce e Carle, Cuneo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
Si definisce iperaldosteronismo secondario una condizione di patologica iperattivazione del sistema renina-angiotensina-aldosterone (RAAS), che caratterizza quadri morbosi differenti per eziologia e clinica. Può associarsi a ipertensione arteriosa o a quadri di normo/ipotensione. Il termine iperaldosteronismo secondario non si applica alle condizioni di attivazione acuta (emorragia, singola dose di diuretico), ma soltanto alle condizioni di attivazione persistente.
IPERALDOSTERONISMO SECONDARIO ASSOCIATO A IPERTENSIONE ARTERIOSA
L'iperaldosteronismo secondario rappresenta il fattore/cofattore eziologico di tre quadri di ipertensione secondaria:
- l'ipertensione nefrovascolare
- il reninoma
- l'ipertensione da estrogeni
L’attivazione intrinseca del RAAS senza reale riduzione del volume circolante effettivo (VCE), né perdita di sali, ha come correlato clinico l’ipertensione e l’ipopotassiemia.
Oltre alle patologie citate, ci sono condizioni in cui l'ipertensione si associa a livelli elevati di renina e aldosterone, ma in cui non è dimostrato che l'attivazione del RAAS abbia un significato patogenetico: 10% delle ipertensioni idiopatiche (ipertensioni ad alta renina) e una quota di casi di ipertensione maligna.
IPERALDOSTERONISMO SECONDARIO SENZA IPERTENSIONE ARTERIOSA
In questo caso l'attivazione del RAAS avviene in risposta a ipovolemia (sindromi edemigene) o in seguito a tubulopatie renali con perdita di sali. I correlati clinici di maggior rilievo in queste condizioni sono l'induzione/aggravamento dell'edema e l'ipopotassiemia che interessano in diversa misura le differenti condizioni morbose.
Le sindromi edemigene comprendono:
- insufficienza cardiaca
- cirrosi epatica
- sindrome nefrosica
- enteropatie proteino-disperdenti
La riduzione del VCE secondario all'edema da stasi e/o all'edema discrasico determina attivazione del RAAS, allo scopo di espandere il volume mediante ritenzione di sodio. Nelle sindromi edemigene l'attivazione del RAAS peggiora l'edema, ma di solito non è causa di ipopotassiemia, in quanto il riassorbimento di sodio a livello della regione prossimale del nefrone è pressoché completo in queste patologie. La somministrazione di diuretici può slatentizzare l'ipopotassiemia da iperaldosteroismo secondario per l'aumento dell'arrivo distale di sodio.
Tra le nefropatie con perdita di sali si distinguono due tubulopatie a trasmissione autosomica recessiva:
- la sindrome di Bartter, da alterazioni di canali ionici localizzati a livello dell’ansa di Henle e bersaglio dei diuretici dell’ansa;
- la sindrome di Gitelman, da alterazioni di canali ionici localizzati a livello del tubulo contorto distale, con effetti simili alla somministrazione cronica di diuretici tiazidici.
Ne segue uno squilibrio idro-elettrolitico differente nelle due patologie.
| Sinossi degli iperaldosteronismi secondari a tubulopatia con perdita di sali | |||||
| Sindrome | Sotto- tipo |
Canale ionico alterato | Effetti | Quadro clinico | |
| Bartter | Ante natale/ neonatale | I |
NKCC2 (cotrasportatore luminale Na+/K+/2Cl-)
|
↓ riassorbimento NaCl nel braccio ascendente spesso dell’ansa di Henle
|
Prenatale: polidramnios I-III trimestre e prematurità Alla nascita:
|
| II |
ROMK (trasportatore luminale di K+)
|
||||
| IV |
ClCKa-ClCKb (ß-subunità del trasportatore basolaterale di Cl)
|
||||
| Classica | III | ClCKb (trasportatore basolaterale di Cl) |
↓ riassorbimento di NaCl nel tubulo contorto distale
|
Età di insorgenza ≥ 2 anni con:
|
|
| Gitelman |
NCCT (cotrasportatore apicale di NaCl) |
↓ riassorbimento di NaCl nel tubulo contorto distale
|
Insorgenza in età variabile ma sempre ≥ 6 anni. Quadri clinici molto diversi: pazienti asintomatici anche da adulti (solo alterazioni di laboratorio);pazienti con sintomi legati prevalentemente a ipomagnesemia e ipocalciuria (raramente all’ipocaliemia)
|
||
Overview sugli iperaldosteronismi secondari
Sara Cassibba
Endocrinologia, AO S Croce e Carle, Cuneo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
L’iperaldosteronismo secondario è una condizione di patologica iperattivazione del sistema renina-angiotensina-aldosterone (RAAS) che caratterizza quadri morbosi, differenti per eziologia e clinica, nei quali può condizionare o meno l’insorgenza di ipertensione arteriosa.
Il RAAS rappresenta uno dei meccanismi di controllo di pressione arteriosa, volemia e bilancio idro-elettrolitico. Si attiva a scopo ipertensivo in caso di reale o avvertita riduzione del volume circolante effettivo (VCE) (attivazione barocettoriale) o per riduzione della concentrazione di NaCl a livello delle cellule della macula densa (attivazione chemocettoriale PGE2-mediata). L’effetto complessivo dell’attivazione del RAAS è l’espansione della volemia mediante la ritenzione di acqua e soluti (NaCl) e l’escrezione di potassio.
IPERALDOSTERONISMO SECONDARIO ASSOCIATO A IPERTENSIONE ARTERIOSA
L’attivazione del RAAS non legata ad una reale riduzione del VCE o a deplezione idro-salina causa ipertensione arteriosa. L’iperaldosteronismo secondario rappresenta il fattore/cofattore eziologico di tre quadri di ipertensione secondaria: l’ipertensione nefrovascolare, l’ipertensione da estrogeni e il reninoma. Oltre a queste, ci sono condizioni in cui l'ipertensione si associa a livelli elevati di renina e aldosterone, ma in cui non è dimostrato che l'attivazione del RAAS abbia un significato patogenetico: 10% delle ipertensioni idiopatiche (ipertensioni ad alta renina) e alcuni casi di ipertensione maligna.
Ipertensione nefro-vascolare
E' legata a stenosi dell’arteria renale o di suoi rami con eziologia diversa (tabella).
Rappresenta l’ipertensione da iperaldosteronismo secondario di maggior rilievo per l’elevata prevalenza (2-5% di tutti i casi di ipertensione arteriosa).
Clinicamente l’ipertensione e l’ipopotassiemia possono essere di grado da moderato a severo.
In ambito diagnostico-terapeutico presenta aspetti peculiari. La diagnosi eziologica di questa condizione può richiedere indagini invasive quali l’arteriografia selettiva, in quanto l’indagine non invasiva (ecodoppler) è strettamente operatore-dipendente.
L’approccio chirurgico di rivascolarizzazione non sempre è risolutivo e nella maggior parte dei casi l’ipertensione nefro-vascolare è ben compensata dalla terapia medica. La difficoltà diagnostica e la frequente buona risposta alla terapia medica implicano che, sebbene l’ipertensione nefro-vascolare sia una condizione relativamente frequente, la valutazione eziologica e l’eventuale terapia chirurgica siano da riservare a casi selezionati: insufficienza renale progressiva, episodi ricorrenti di edema polmonare, ipertensione arteriosa non responsiva ai farmaci. Lo screening di tutti i pazienti ipertesi per questa patologia porterebbe di fatto all’aumento di pratiche terapeutiche invasive e non risolutive, con uno sfavorevole rapporto costo-beneficio1.
Ipertensione da estrogeni
Contrariamente all’ipertensione nefro-vascolare, si caratterizza per un semplice approccio diagnostico-terapeutico. La blanda attivazione del RAAS e l’aumento della produzione di angiotensinogeno a livello epatico legata all’iperestrogenismo non hanno, di solito, rilievo clinico. In donne predisposte la gravidanza o l’assunzione di preparati estro-progestinici possono tuttavia portare a ipertensione. La condizione cessa nel momento in cui ha termine l’esposizione a livelli sovrafisiologici di estrogeni2.
Reninoma
E' una patologia molto rara (descritti 50 casi in letteratura), ma caratterizzata da ipertensione e ipopotassiemia severa, con esigenza di diagnosi e terapia tempestiva e complessa.
Nonostante la differente eziologia, il quadro patologico che ne deriva può essere simile alle forme severe di stenosi dell'arteria renale e la diagnosi differenziale si impone mediante l'esecuzione di arteriografia selettiva. La localizzazione del tumore con metodiche di diagnostica per immagini è di solito non conclusiva per le piccole dimensioni della massa. Spesso è pertanto necessario valutare la lateralizzazione della lesione mediante dosaggio della renina su sangue refluo dalla vena renale e procedere all'esplorazione chirurgica del rene interessato. In caso di mancato reperimento della neoplasia all'esplorazione chirurgica, si pratica la nefrectomia1.
| Sinossi degli iperaldosteronismi secondari associati a ipertensione arteriosa | ||
| Patologia | Attivazione del RAAS | Effetti |
| Ipertensione nefrovascolare (2-5% ipertensioni) Ateromi: soggetti > 55 anni, fattori di rischio per ateromasia (fumo, dislipidemia, diabete mellito), ateromasia poli-distrettuale Fibrodisplasia: soggetti < 30 anni senza fattori di rischio per ipertensione Altro (raro): arteriti, aneurisma, masse a compressione estrinseca |
I fase: ipoperfusione renale -> attivazione di RAAS -> espansione della volemia II fase: compenso da espansione della volemia -> normalizzazione dei livelli di renina-angiotensina-aldosterone |
Ipertensione e ipopotassiemia da moderate a gravi, a insorgenza/ peggioramento improvviso, spesso associata a soffio vascolare |
| Ipertensione da estrogeni: gravidanza o terapia estro-progestinica | Fisiologica modesta attivazione di RAAS e aumento della produzione di angiotensinogeno epatico | In soggetti predisposti: ipertensione di grado lieve moderato |
| Reninoma: neoplasia renino-secernente | Iperattivazione intrinseca | Ipertensione e ipopotassiemia severa |
IPERALDOSTERONISMO SECONDARIO SENZA IPERTENSIONE ARTERIOSA
Nell'iperaldosteronismo secondario senza ipertensione arteriosa l'attivazione del RAAS avviene in risposta a una riduzione del VCE (sindromi edemigene) o in seguito a deplezioni idro-elettrolitiche legate a tubulopatie renali. I correlati clinici di maggior rilievo sono l'aggravamento dell'edema e l'ipopotassiemia, che interessano in diversa misura le differenti condizioni morbose.
Sindromi edemigene
Comprendono l'insufficienza cardiaca, la cirrosi epatica, la sindrome nefrosica e le sindromi proteino-disperdenti intestinali. Nel contesto di queste patologie l’iperattivazione del RAAS da deplezione di VCE contribuisce ad instaurare e aggravare l’edema, ma non comporta ipertensione né, in genere, ipopotassiemia clinica. L’ipopotassiemia può evidenziarsi per l’uso di diuretici che aumentano la delivery distale di sodio.
Nell'insufficienza cardiaca congestizia la riduzione della forza contrattile e/o l'alterazione della fase diastolica ventricolare determinano stasi a livello del compartimento venoso e riduzione del VCE. L’attivazione del RAAS produce ritenzione idro-salina. Se la cardiopatia è grave, il meccanismo di compenso si traduce in un peggioramento della stasi del circolo venoso senza aumento della gittata cardiaca con edema periferico (compromissione prevalente del ventricolo destro) o edema polmonare (insufficienza ventricolare sinistra).
La sindrome nefrosica, l’enteropatia protido-disperdente e l’edema da malnutrizione comportano la diminuzione della pressione colloido-osmotica ematica di origine discrasica: perdita di proteine (sindrome nefrosica, enteropatia protido-disperdente) o ipoproduzione proteica (cirrosi epatica, stati di malnutrizione) generano il passaggio di soluti e liquidi nel terzo spazio, con conseguente riduzione del VCE. Ne segue l'attivazione del RAAS con ritenzione idrosalina e peggioramento dell'edema.
Nel caso della cirrosi epatica l'edema ha componente mista: discrasica da ipoproduzione epatica di albumina e da stasi del circolo a livello epatico. Consegue una notevole riduzione del VCE con attivazione del RAAS. Nella cirrosi l'iperaldosteronismo è accentuato dalla ridotta clearance epatica dell'aldosterone. L'edema, inizialmente localizzato a livello addominale si estende, nelle fasi più avanzate, anche a livello periferico3.
Tubulopatie con perdita di sali
Le modalità di attivazione del RAAS e i suoi effetti sono differenti. La sindrome di Bartter e la sindrome di Gitelman sono patologie rare a trasmissione autosomica recessiva, causate da mutazioni di geni codificanti canali ionici per il trasporto di elettroliti a livello del nefrone distale. L’alterazione di questi trasportatori determina effetti simili all'azione farmacologica della furosemide (sindrome di Bartter) o dei diuretici tiazidici (sindrome di Gitelman). I disordini elettrolitici e i quadri clinici sono differenti nelle due patologie, ma in entrambe la perdita renale di NaCl e la deplezione di volume determinano l’attivazione del RAAS al fine di contrastare l'ipovolemia.
L’attivazione del RAAS è maggiore nella sindrome di Bartter a causa della più spiccata disidratazione – attivazione barocettoriale e tubulare (PGE2-mediata) - più modesta e solo barocettoriale nel Gitelman.
Clinicamente si distinguono la sindrome di Bartter neonatale, la sindrome di Bartter classica e la sindrome di Gitelman.
La sindrome di Bartter neonatale si presenta precocemente, con poli-idramnios tra la 24° e la 30° settimana di gestazione e con grave poliuria, polidipsia e disidratazione dopo la nascita. Nella sindrome di Bartter classica la clinica e le alterazioni biochimiche sono più lievi e compaiono all'età di circa due anni o più tardivamente. Per il tipo d’alterazione tubulare e per l’importante attivazione del RAAS si ha ipopotassiemia. Sono presenti inoltre ipercalciuria (con possibile nefrocalcinosi) ed elevati livelli di PGE2 urinari.
La sindrome di Gitelman presenta quadri clinici che vanno dalle forme asintomatiche, a varianti con sola astenia, a quadri con sintomatologia evidente (crampi tetanici muscolari, nicturia, polidipsia, poliuria, sete, ipotensione, vertigine). Si manifesta in genere dopo i sei anni di età. L’attivazione del RAAS è meno importante che nella sindrome di Bartter e prevalgono i segni e sintomi legati all’ipomagnesemia e all’ipocalciuria (condrocalcinosi) sull’ipokaliemia e l’alcalosi metabolica.
La diagnosi delle sindromi di Bartter e Gitelman si basa su sintomi e alterazioni biochimiche e trova conferma nell'analisi genetica4-5.
BIBLIOGRAFIA
- Mancia G, De Backer G, Dominiczak A, et al. ESH-ESC Task Force on the management of arterial hypertension. 2007 Guidelines for the management of arterial hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007, 25: 1105-87.
- Monaco F. Linee guida per la diagnosi, la terapia e il controllo delle malattie endrocrine e metaboliche. Collana “Progressi in Endocrinologia e Malattie Metaboliche”, See-Firenze. 2000: pg 49.
- Loscalzo J. Harrison, Elementi di medicina interna. XVI ed. Mc Graw Hill. 2005 Vol. I: 248-53.
- Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis 2008, 3: 22.
- Unwin RJ, Capasso G. Bartter's and Gitelman's syndromes: their relationship to the actions of loop and thiazide diuretics. Curr Opin Pharmacol 2006, 6: 208-13.
Fisiopatologia generale iperaldosteronismi secondari
Sara Cassibba
Endocrinologia, AO S Croce e Carle, Cuneo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
Il rene filtra quotidianamente 180 litri di acqua e soluti; i sali, per la loro dimensione, passano nell’ultra-filtrato e devono essere riassorbiti lungo il nefrone, in modo da eliminare solo la minima quota di acqua e soluti che controbilanci gli introiti (< 1% dell’ultra-filtrato). Gli elettroliti vengono per il 65% riassorbiti a livello del tubulo contorto prossimale (TCP), per il 25% dall’ansa di Henle e per il 10% tra tubulo contorto distale (TCD) e dotto collettore corticale (DCC).
Le porzioni del nefrone distale alterate nelle sindromi di Gitelman e di Bartter sono il tratto ascendente spesso dell’ansa di Henle (TAL) e il tubulo contorto distale.
Tratto ascendente spesso dell’ansa di Henle (TAL)
Fisiologicamente TAL è deputata al riassorbimento di circa il 25% dei soluti, in particolare di Na+ e Cl- per via trans-cellulare e di Ca++ e Mg++ per via para-cellulare. Il gradiente per il trasporto passivo di ioni è fornito dall’attività della Na/K ATPasi e mantenuto dai canali apicali elettroneutri NKCC2 (bersaglio della furosemide) e ROMK e dai canali baso-laterali ClCKa e ClCKb. TAL è impermeabile all’acqua e mediante il riassorbimento di soli elettroliti contribuisce alla creazione e al mantenimento di un interstizio iperosmotico indispensabile per gli scambi controcorrente e la concentrazione dell’urina1,2.
Tratto ascendente spesso dell’ansa di Henle
Tubulo contorto distale (TCD)
Il TCD riassorbe soluti (Na+, Cl-, Ca++, Mg++) utilizzando, analogamente a TAL, il gradiente elettrochimico creato dalla Na/K ATPasi e dalla pompa attiva del Ca++, entrambe baso-laterali. Ciò permette il flusso elettroneutro di ioni attraverso i canali apicali NCCT (bersaglio dei diuretici tiazidici), TRPV5, TRPV6 e quelli baso-laterali di cloro (ClCKb) e di scambio Ca++/Na+ e Mg++/Na+.
Il riassorbimento di soluti è quantitativamente inferiore rispetto a TAL, ma a questo livello, insieme con il dotto collettore corticale, si ha la fine regolazione dell’escrezione di acqua e soluti.
Nella porzione più distale di TCD c’è una sovrapposizione tra l’espressione dei canali apicali NCCT ed ENaC (bersaglio dell’amiloride), espressi poi a livello delle cellule principali del DCC. I canali ENaC e la pompa Na/K ATPasi consentono il riassorbimento di Na+ indipendentemente dal Cl- e sono sotto l’influenza dell’aldosterone che ne aumenta l’espressione, incrementando il riassorbimento di sodio a spese del potassio. Il potassio lungo i restanti tratti del nefrone viene prevalentemente assorbito1,2.
Tubulo contorto distale
Cellule principali del dotto collettore corticale
Le tubulopatie distali con perdita di sali
Le sindromi di Gitelman (SG) e di Bartter (SB) sono tubulopatie legate ad alterazioni genetiche di canali tubulari del nefrone distale. Vengono classicamente trattate come distinte per ragioni storiche. Il progresso della patologia neonatale dopo gli anni ’80 ha permesso la sopravvivenza dei neonati prematuri affetti da sindrome di Bartter antenatale (aBS). La valutazione di questi pazienti e l’applicazione delle indagini genetiche ha portato a una conoscenza più approfondita dei meccanismi patogenetici e delle diverse varianti delle due patologie, alla luce delle quali sembra appropriata una trattazione congiunta delle stesse con una suddivisione in base al tratto tubulare funzionalmente alterato e alle manifestazioni cliniche. Si distinguono quindi i disordini di TAL puri o misti che causano una sindrome prenatale di gravità maggiore (SB tipo I, II, IV), i disordini di TCD con presentazione più tardiva e meno severa (SB tipo III e SG), come illustrato in tabella 1.
| Classificazione delle sindromi di Bartter e di Gitelman in base alla porzione di nefrone funzionalmente alterata | ||||
| Alterazione tubulare | Sindrome | Alterazione canale (gene) | Azione diuretico-simile | Clinica |
| Forme prenatali | ||||
| Tratto ascendente spesso dell’ansa di Henle |
Poliuria ipo/isostenuria:
Squilibri elettrolitici: iposodiemia, ipocloremia, ipomagnesemia.
|
|||
| Bartter tipo I | NKCC2 (SLC12A1) | Furosemide | Ipercalciuria e nefrocalcinosi | |
| Bartter tipo II | ROMK (KCNJ1) | Furosemide-tiazide |
Ipercalciuria e nefrocalcinosi |
|
| Tratto ascendente spesso dell’ansa di Henle + tubulo contorto distale | Bartter tipo IV | ClCKa-ClCkb (battrina) | Furosemide-tiazide |
Maggior gravità |
| Forme ad insorgenza tardiva | ||||
| Tubulo contorto distale |
Insorgenza ed entità dei sintomi variabile |
|||
| Gitelman | NTCC | Tiazidici | Ipocalciuria, condrocalcinosi | |
| Tubulo contorto distale (+ tratto ascendente spesso dell’ansa di Henle) | Bartter tipo III | ClCKb (CLCKB) | Tiazidici-furosemide | Generalmente non ipercalciuria, nè nefrocalcinosi |
A questa classificazione si aggiungono le varietà delle due sindromi, a presentazione atipica, di recente descritte e in parte ricondotte a difetti dei canali del tratto distale del nefrone, come la SB tipo V, la sindrome EAST e le forme fruste delle patologie legate a parziale inattivazione dei suddetti canali4.
Bibliografia
- Viganò SM, et al. Trasporto renale tubulare e forme genetiche di ipotensione. G Ital Nefrol 2006, 23: 396-405.
- Graziani G, et al. Gitelman syndrome: pathophysiological and clinical aspects. QJM 2010, 103: 741-8.
- Friis UG, et al. Regulation of renin secretion by renal juxtaglomerular cells. Pflugers Arch Eur J Physiol 26 Jun 2012.
- Fremont OT, Chan JC. Understanding Bartter syndrome and Gitelman syndrome. World J Pediatr 2012, 8: 25-30.
Sindrome di Bartter
Sara Cassibba
Endocrinologia, AO S Croce e Carle, Cuneo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
La sindrome di Bartter (SB) è un insieme eterogeneo di tubulopatie con perdita di sali, patologie autosomiche recessive (eccetto la SB tipo V, autosomica dominante) dovute a mutazione di geni codificanti per canali ionici del nefrone distale.
La tabella 1 e la successiva trattazione forniscono una classificazione delle diverse varianti della patologia con riferimento alla clinica, distinguendo in particolare tra quelle ad insorgenza prenatale e quelle ad insorgenza tardiva1.
| Classificazione delle sindromi di Bartter | ||||
| Tipo |
Alterazione canale(gene) |
Alterazione tubulare | Azione diuretico-simile | Clinica |
| insorgenza prenatale | ||||
| I | NKCC2 (SLC12A1) | TAL | Furosemide | Ipercalciuria e nefrocalcinosi |
| II | ROMK (KCNJ1) | TAL | Furosemide-tiazide | Ipercalciuria e nefrocalcinosi Transitoria iperpotassiemia con successiva ipopotassiemia più lieve che nelle altre s. Bartter prenatali |
| IV | ClCKa-ClCkb (battrina) | TAL + TCD | Furosemide-tiazide | Maggior gravità Normale calciuria, non nefrocalcinosi ma IRC |
| insorgenza tardiva | ||||
| III | ClCKb (CLCKB) | (TAL) + TCD | Tiazidici-furosemide | Generalmente non ipercalciuria, nè nefrocalcinosi |
| V | CaRS | TAL | Furosemide | Ipocalcemia e ipercalciuria |
SINDROME DI BARTTER AD ESORDIO PRENATALE
La Sindrome di Bartter ad esordio prenatale comprende tre tubulopatie differenti per patogenesi ma con analogie cliniche: le sindromi di Bartter tipo I, tipo II e tipo IV.
SB tipo I e tipo II
Si caratterizzano per un difetto esclusivo del tratto ascendente spesso dell’ansa di Henle (TAL):
- nella SB tipo I è alterato il canale apicale NKCC2
- nella SB tipo II il canale apicale ROMK.
In entrambi i casi il mancato funzionamento dei canali determina il dissiparsi del fisiologico gradiente creato dalla NA/K ATPasi baso-laterale. Ne consegue un deficit di riassorbimento di soluti (Na+ e Cl- per via trans-cellulare; Mg++e Ca++ per via para-cellulare) e la mancata formazione di un interstizio iperosmotico indispensabile agli scambi contro-corrente.
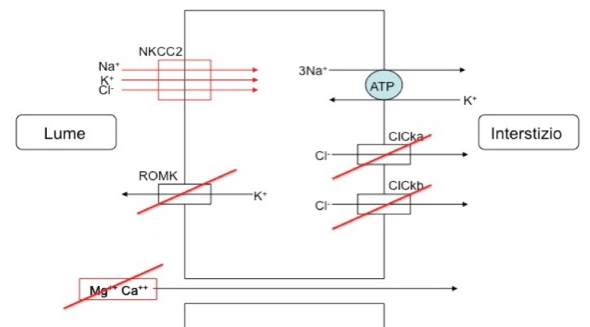
Sindrome di Bartter tipo I
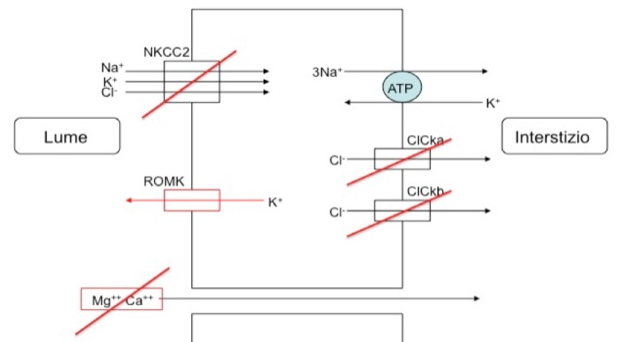
Sindrome di Bartter tipo II
La manifestazione clinica patognomonica delle due sindromi, e delle SB prenatali in genere, è la poliuria, che si manifesta già nel corso della vita intra-uterina e che comporta poli-idramnios alla fine del II trimestre di gravidanza e parto pre-termine all’inizio del III trimestre.
Alla nascita il bambino presenta poliuria, polidipsia e tendenza alla disidratazione. Può sviluppare una nefrocalcinosi. Gli esami di laboratorio evidenziano iso/ipostenuria, ipercalciuria, ipomagnesiemia, iposodiemia, ipocloremia. Sono presenti gli effetti legati al tentativo di compenso della perdita di liquidi e soluti: iperaldosteronismo secondario con ipopotassiemia e alcalosi metabolica e iperprostaglandinuria da aumentata produzione renale di PGE2.
La SB tipo II rispetto alla SB tipo I si differenzia per una transitoria iperpotassiemia nelle prime settimane di vita, cui segue una ipopotassiemia meno pronunciata che nelle altre SB prenatali: l’alterazione di ROMK comporta un parziale risparmio di K+, su cui prevale poi la perdita legata all’iperaldosteronismo secondario1,2,3.
SB tipo IV
E' dovuta all’alterazione dei canali baso-laterali del cloro ClCKa e CLCKb. L’alterazione genetica che la causa può coinvolgere la battrina, una proteina regolatoria indispensabile per il posizionamento dei canali del cloro a livello della membrana, o un difetto digenico di ClCKA e ClCKB, che codificano per i rispettivi canali ionici ClCKa e ClCKb.
I due canali del cloro sono presenti a livello della membrana baso-laterale sia di TAL che del tubulo contorto distale (TCD) e la contemporanea alterazione di entrambi porta alla compromissione della funzione delle due porzioni del nefrone.
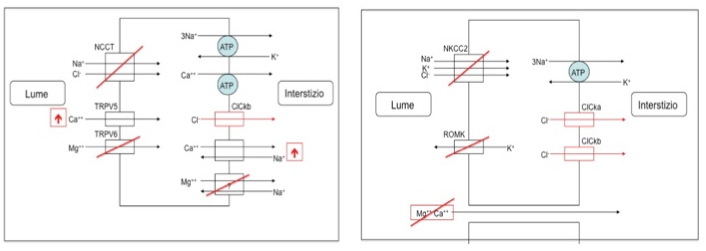
Sindrome di Bartter tipo IV
Le manifestazioni cliniche sono simili a quelle della SB tipo I e II ma più gravi. Le alterazioni elettrolitiche sono in parte analoghe a quelle di SB tipo I e II (iposodiemia, ipocloremia, iso/ipostenuria, ipopotassiemia e alcalosi metabolica da iperaldosteronismo secondario, iperprostaglandinuria) e in parte differenti (manca l’ipercalciuria).
Non si ha sviluppo di nefrocalcinosi, ma è comunque frequente l’evoluzione verso l’insufficienza renale cronica.
La SB tipo IV presenta come elemento clinico distintivo una sordità neurosensoriale, dovuta alla presenza di ClCKa anche a livello della coclea, dove è indispensabile per la corretta composizione dell’endolinfa1,2,3.
SINDROME DI BARTTER A PRESENTAZIONE TARDIVA
SB tipo III o SB classica
La SB tipo III si distingue dalle forme prenatali per la presentazione clinica tardiva e la gravità variabile.
L’alterazione genetica interessa il canale baso-laterale del Cl- ClCKb. Come nel caso della SB tipo IV, il difetto coinvolge sia TAL che TCD, tuttavia in TAL la presenza di ClCKa compensa il ClCKb rendendo la SB tipo III una patologia del TCD.
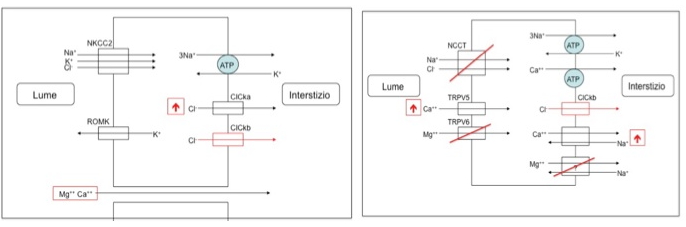
Sindrome di Bartter tipo III
Per la localizzazione del difetto e la clinica risulta più simile alla sindrome di Gitelman che alle altre varianti di SB.
La presentazione è molto eterogenea e la genetica non è in grado di spiegare in modo esauriente lo spettro delle diverse variabili fenotipiche della SB tipo III, che potrebbe riflettere o una variabilità inter-individuale nella distribuzione di ClCKa e ClCKb nel nefrone distale o la diversa capacità di sviluppare e utilizzare vie alternative di escrezione baso-laterale del cloro.
Nella presentazione più tradizionale della SB tipo III si ha un periodo neonatale asintomatico, cui segue un ritardo di crescita nell’infanzia. Agli esami ematochimici si rivela un importante squilibrio elettrolitico: ipocloremia, iponatriemia, iperaldosteronismo secondario con ipopotassiemia e alcalosi metabolica. Generalmente non risulta alterata la capacità di concentrare le urine e non sono presenti poliuria, iso e ipostenuria né nefrocalcinosi, tipici dei disordini di TAL (descritti solo casi molto rari). In anamnesi patologica remota circa il 30% dei pazienti ha un modesto poli-idramnios nel corso del III trimestre, che solitamente non ha richiesto amniocentesi né è stato causa di prematurità1,2.
SB tipo V
La SB tipo V è una rara variante, legata all’alterazione del recettore sensibile al calcio (CaRS) baso-laterale delle cellule di TAL. Le alterazioni elettrolitiche sono: iposodiemia, ipocloremia, ipocalcemia e ipomagnesemia. L’attivazione compensatoria di RAAS causa alcalosi metabolica con ipopotassiemia.
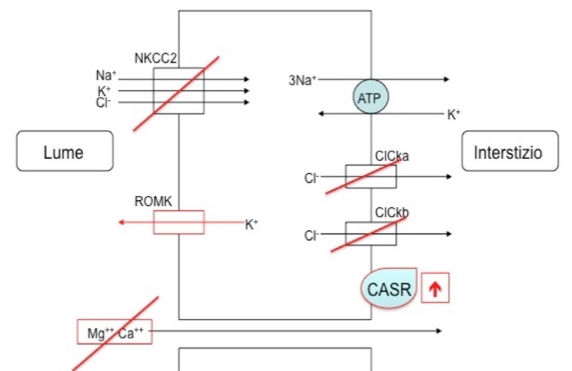
Sindrome di Bartter tipo V
Le manifestazioni cliniche sono simili per tipologia a quelle dei disordini di TAL, ma l’età e la severità della presentazione è molto variabile e dipende dall’entità di attivazione di CaRS4,5.
BIBLIOGRAFIA
- Deschênes G, Fila M. Primary molecular disorders and secondary biological adaptations in Bartter syndrome. Int J Nephrol 2011, 2011: 396209.
- Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol 2011, 26: 1789–802.
- Bhat YR, et al. Antenatal Bartter syndrome: a review. Int J Pediatr 2012, 2012: 857136.
- Watanabe S, et al. Association between activating mutations of calcium-sensing receptor and Bartter's syndrome. Lancet 2002, 360: 692-4.
- Egbuna OI, Brown EM. Hypercalcaemic and hypocalcaemic conditions due to calcium-sensing receptor mutations. Best Pract Res Clin Rheumatol 2008, 22: 129-48.
Sindrome di Gitelman
Sara Cassibba
Endocrinologia, AO S Croce e Carle, Cuneo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
La sindrome di Gitelman (SG) è una tubulopatia con perdita di sali, un disordine del tubulo contorto distale (TCD) con effetti clinici simili a quelli prodotti dall’abuso di diuretici tiazidici1.
Patogenesi
La patologia è autosomica recessiva, dovuta a mutazioni inattivanti il gene SLC12A3, con conseguente alterazione del funzionamento del canale apicale NCCT delle cellule del TCD.
Il TCD, insieme al dotto collettore corticale (DCC), è fisiologicamente deputato al riassorbimento del 10% circa dei soluti dell’ultra-filtrato renale. Ciò è consentito dal gradiente elettro-chimico creato dalla Na/K ATPasi e mantenuto dai canali apicali e baso-laterali che provvedono al riassorbimento di elettroliti.
Tubulo contorto distale
Cellule principali del dotto collettore corticale
La compromissione di NCCT, che provvede al riassorbimento apicale di NaCl, dissipa il gradiente creato dalla Na/K ATPasi baso-laterale del TCD, con mancato riassorbimento trans-cellulare di Na+, Cl+ e Mg++. Le ridotte concentrazioni intra-cellulari di Na+ e Ca++, dovute all’azione della Na/K ATPasi e della pompa del Ca++ baso-laterali, determinano un aumento dell’attività di scambio Na/Ca baso-laterale e dell’ingresso intra-cellulare di Ca++ tramite il TRPV5 baso-laterale. Si ha una down-regulation del TRPV6, con ridotto riassorbimento di Mg++.
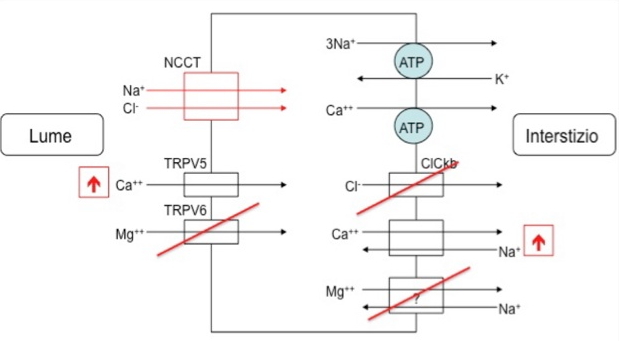
Le suddette modificazioni determinano ipocloremia, iposodiemia, ipomagnesemia, ipocalciuria.
La perdita di acqua e soluti determina l’attivazione del sistema renina-angiotensina-aldosterone (RAAS), con conseguente alcalosi metabolica e ipopotassiemia1,2,3.
Clinica
La storia naturale della SG è molto eterogenea in termini di età della diagnosi, natura e gravità delle alterazioni biochimiche e delle manifestazioni cliniche. Lo stesso paziente può presentare variazioni degli squilibri elettrolitici e della clinica nelle differenti fasi della vita, per l’attuarsi di meccanismi di compenso del nefrone.
Quando fu descritta nel 1966, la SG fu considerata una variante benigna di tubulopatia con perdita di sali, con esordio clinico più tardivo rispetto alla sindrome di Bartter -solitamente nell’infanzia, talvolta nell’adolescenza o nell’età adulta- e con conservata capacità di concentrare le urine. La maggior parte dei pazienti presenta una sintomatologia lieve, con debolezza muscolare e affaticabilità, desiderio di assumere sale, segni di aumentata eccitabilità neuromuscolare come crampi e tetania. Spesso la diagnosi è accidentale durante l’iter diagnostico avviato per ritardo di crescita, costipazione, enuresi, o per familiarità.
In un sottogruppo di pazienti, specialmente giovani maschi, la patologia presenta maggiore gravità, con compromissione severa della qualità della vita, con manifestazioni quali rabdomiolisi ipocaliemica, aritmie con sincope e, in rari casi, morte improvvisa, condrocalcinosi di ginocchia, gomiti e spalle come effetto dell’ipomagnesemia cronica.
La sintomatologia può emergere o aggravarsi nelle condizioni che portano a disidratazione, come febbre, vomito e diarrea1,2,3.
SINDROME EAST
Recentemente sono riportati in letteratura rari casi di un complesso sindromico caratterizzato da epilessia, ritardo mentale, sordità neuro-sensoriale, e perdita renale di sali con squilibri idro-elettrolitici simili a quelli che caratterizzano la sindrome di Gitelman e, quindi, tipiche dei disordini di TCD: iposodiemia, ipocloremia, ipomagnesemia, ipocalciuria, ipocaliemia con alcalosi metabolica da iperattivazione del RAAS e conservata capacità di concentrare le urine.
La sindrome EAST è una patologia autosomica recessiva dovuta alla mutazione del gene KCNJ10, che codifica per il canale Kir4.1, membro della famiglia dei canali del potassio. Nel rene Kir4.1 è espresso nella membrana baso-laterale di TCD e del dotto collettore, dove permette, insieme alla Na/K ATPasi il mantenimeno del gradiente per il riassorbimento di Na+.
Kir4.1 è espresso anche a livello delle cellule gliali di molte parti del SNC e della stria vascolare dell’orecchio interno e ciò spiega il complesso delle restanti manifestazioni4.
BIBLIOGRAFIA
- Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol 2011, 26: 1789–802.
- Fremont OT, Chan JC. Understanding Bartter syndrome and Gitelman syndrome. World J Pediatr 2012, 8: 25-30.
- Graziani G, et al. Gitelman syndrome: pathophysiological and clinical aspects. QJM 2010, 103: 741-8.
- Bandulik S, et al. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch - Eur J Physiol 2011, 461: 423–35.
Terapia dell'iperaldosteronismo secondario
Sara Cassibba
Endocrinologia, AO S Croce e Carle, Cuneo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
L’iperaldosteronismo secondario è parte di diverse patologie, di cui è fattore patogenetico o meccanismo di compenso che si attua in risposta alla riduzione del volume plasmatico o del volume circolante effettivo (VCE).
IPERALDOSTERONISMO SECONDARIO CON IPERTENSIONE ARTERIOSA
Ipertensione nefro-vascolare
Le stenosi dell’arteria renale determinano una riduzione del VCE, che innesca un iperaldosteronismo secondario. La mancanza di edema è legata a meccanismi di escape dall’eccesso di sodio, ma l’iperaldosteronismo causa ipertensione arteriosa.
Il trattamento dell’ipertensione nefro-vascolare è controverso, poiché non sempre le procedure di rivascolarizzazione risultano risolutive. L’approccio chirurgico, ormai quasi esclusivamente rappresentato dall’angioplastica, andrebbe considerato solo in presenza di un’ipertensione resistente (valori pressori elevati nonostante l’associazione con almeno tre farmaci, tra cui un diuretico a dosi adeguate) o di un progressivo deterioramento della funzione renale.
L’angioplastica ha risultati migliori in caso di iperplasia fibro-muscolare e per patologie di durata inferiore a 10 anni, mentre la percentuale di successo si riduce in presenza di malattia vascolare aterosclerotica (elevata incidenza di ristenosi che può essere ridotta grazie all’impiego di uno stent) e se l’ipertensione è presente da più di 10 anni.
Nei pazienti con ipertensione nefrovascolare su base aterosclerotica con lunga durata di malattia è da preferire un trattamento farmacologico anti-ipertensivo, soprattutto se consente un buon controllo dei valori pressori e se la funzionalità renale è normale. Può essere indicato l’impiego di un diuretico tiazidico a dosi adeguate e di un calcio-antagonista. L’associazione con un bloccante del RAAS è da utilizzare solo se sia stata esclusa una stenosi bilaterale dell’arteria renale.
Reninoma
Nel reninoma l’iperaldosteronismo secondario è legato alla produzione afinalistica di renina da parte di una neoplasia renale.
L’ipertensione arteriosa da reninoma non risponde alla terapia farmacologica e richiede sempre la risoluzione chirurgica.
Solitamente le dimensioni di queste neoplasie sono al di sotto del potere di risoluzione delle tecniche di diagnostica per immagini, per cui è necessario provvedere alla lateralizzazione della lesione mediante dosaggio della renina su sangue refluo dalla vena renale, effettuare l’esplorazione chirurgica del rene interessato e praticare, se l’indagine manuale non è conclusiva, una nefrectomia2.
IPERALDOSTERONISMO SECONDARIO SENZA IPERTENSIONE ARTERIOSA
Sindromi edemigene
Nelle patologie edemigene, quali cirrosi epatica, scompenso cardiaco e sindrome nefrosica, l’iperaldosteronismo secondario rappresenta un tentativo di compenso inefficace alla riduzione del VCE per passaggio di liquidi e soluti nel III spazio da riduzione della pressione oncotica/aumento della pressione idrostatica.
Cirrosi epatica con ascite
Le linee guida per il trattamento della cirrosi epatica con ascite inseriscono il trattamento dell’iperattivazione di RAAS tra le principali terapie: “i diuretici di prima scelta nella cirrosi con ascite sono gli antagonisti dell’aldosterone, cui i diuretici dell’ansa possono essere associati. Lo spironolattone può essere iniziato alla dose di 50-100 mg/die e titolato fino a un dosaggio massimo di 400 mg/die. Per aumentare l’effetto diuretico e mantenere la potassiemia nei range di norma può essere aggiunta la furosemide a un dosaggio di 20-40 mg/die3”.
Scompenso cardiaco
Considerando anche gli effetti di renina e angiotensina a livello sistemico e cardiaco, oltre all’effetto di ritenzione di sodio e liquidi, le linee guida per il trattamento dello scompenso cardiaco pongono i farmaci che agiscono sul RAAS tra i farmaci di prima scelta per il controllo dei sintomi e per il miglioramento della prognosi: “gli ACE inibitori [o i sartani se gli ACE inibitori non sono tollerati] insieme ai beta-bloccanti e ai bloccanti il recettore dell’aldosterone sono di importanza fondamentale […] e andrebbero considerati per ogni paziente4”.
Sindrome nefrosica
I farmaci con effetto sul RAAS (ACE-inibitori, sartani e antagonisti recettoriali dell’aldosterone) sono utilizzati per il controllo dell’edema e della progressione della patologia (controllo di proteinuria e ipertensione) anche nella sindrome nefrosica5.
Sindrome di Bartter (SB) e sindrome di Gitelman (SG)
In queste sindromi l’iperaldosteronismo secondario costituisce un meccanismo di compenso alla deplezione di volume da perdita di acqua e soluti legata alle tubulopatie renali. Solitamente l’uso di farmaci attivi sul sistema RAAS non è necessario e può talvolta risultare dannoso; si riserva un tentativo terapeutico con questi farmaci solo a casi limite.
Le tubulopatie con perdita di sali (SLTs) che interessano il tratto ascendente spesso dell’ansa di Henle (TAL) sono patologie i cui segni e sintomi sono particolarmente gravi nel periodo pre- e peri-natale e tendono ad attenuarsi con la crescita.
Le SB tipo I e II solitamente rispondono in modo soddisfacente alla terapia con indometacina, che riduce la risposta compensatoria caratterizzata dalla iperproduzione a livello locale di PgE2. Si ha così un miglioramento degli squilibri idro-elettrolitici, del ritardo di crescita e attenuazione del rischio di progressione verso l’insufficienza renale cronica.
La SB tipo IV, rispetto a SB tipo I e II, non ha una risposta ottimale alla somministrazione di indometacina, per ragioni non chiare. L’insufficienza renale cronica è pertanto più frequente in questa forma.
L’uso prolungato di inibitori della sintesi delle prostaglandine può causare intolleranza gastro-intestinale. Come alternativa è stato valutato l’uso di inibitori selettivi della COX-2, ma questi sembrano aumentare il rischio cardio-vascolare, pertanto l’indometacina rimane il farmaco di prima scelta, eventualmente da abbinare a inibitori di pompa protonica.
Al trattamento con indometacina si associa inizialmente la terapia di supporto che prevede reidratazione e ripristino degli squilibri elettrolitici. Possono essere necessari bloccanti di RAAS. Una volta titolata l’indometacina e raggiunta la minima dose efficace (spesso < 1 mg/kg/die), possono non essere necessarie supplementazioni degli elettroliti né bloccanti del RAAS.
Se c’è persistenza di ipokaliemia nonostante la terapia con indometacina e la supplementazione di sali, si possono considerare trattamenti off label cronici con farmaci bloccanti di RAAS. Il tentativo di trattamento con diuretici risparmiatori di potassio e di calcio (questi ultimi sperimentati solo in SB tipo I e II) può essere dannoso, perché può attenuare o abolire i meccanismi compensatori che si instaurano, peggiorare la deplezione di volume e, nel caso dei diuretici risparmiatori di potassio, portare all’iperkaliemia, specialmente se si ha compromissione della funzione renale e disidratazione. In questi pazienti è indispensabile un attento monitoraggio della funzione renale e della pressione arteriosa e il follow-up va intensificato con il concomitante uso di farmaci. Con la crescita il soggetto può andare incontro a un miglioramento della patologia, per cui può rendersi necessario un aggiustamento posologico dei farmaci.
Per la sordità neurosensoriale di origine cocleare i pazienti possono trarre beneficio dal posizionamento di impianto cocleare, che by-passa il sito dell’alterazione e permette di recuperare acuità uditiva.
Le SLTs del tubulo contorto distale si caratterizzano per un’insorgenza più tardiva, ma per una tendenza al peggioramento della sintomatologia con la crescita del soggetto: asintomatiche nel periodo peri-natale, diventano clinicamente evidenti o si aggravano nell’infanzia e nell’età adulta. Per la SB tipo III e la SG non sempre è necessaria una terapia, ma è sempre consigliata una dieta ricca di potassio e magnesio e supplementi degli stessi elettroliti.
In sottogruppi di pazienti con clinica più severa può essere necessario istituire una terapia che, oltre ai supplementi di potassio e magnesio, anche endovenosi, può richiedere l’uso di anti-aldosteronici e risparmiatori di potassio.
Tutti i soggetti con SG dovrebbero effettuare un ECG per la valutazione dell’allungamento del tratto QT da ipokaliemia e ipomagnesemia e in ogni caso dovrebbero essere evitati farmaci che prolunghino il tratto QT, quali macrolidi, anti-istaminici, anti-tussigeni, anti-micotici, psicotropici, beta-2-agonisti6.
BIBLIOGRAFIA
- Mancia G, et al. Practice guidelines 2007 for the treatment of arterial hypertension. G Ital Cardiol (Rome) 2007, 8: 389-479.
- Wong L, et al. Reninoma: case report and literature review. J Hypertens 2008, 26: 368-73.
- Suk KT, et al. Revision and update on clinical practice guideline for liver cirrhosis. Korean J Hepatol 2012, 18: 1-21.
- McMurray JJV, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur Heart J 2012, 14: 803-69.
- Seigneux S, Martin PY. Management of patients with nephrotic syndrome. Swiss Med Wkly 2009, 139: 416-22.
- Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol 2011, 26: 1789–802.
Classificazione e patogenesi pseudoiperaldosteronismi
Nicola Argese
Ospedale Sant’Andrea - Facoltà di Medicina e Psicologia, “Sapienza”, Università di Roma
Il termine pseudoiperaldosteronismo indica una sindrome caratterizzata da ipertensione arteriosa associata a ipokaliemia e bassi livelli di aldosterone e renina.
CLASSIFICAZIONE FISIOPATOLOGICA
Effetto mineralcorticoide diretto
- Terapie con farmaci ad azione mineral-corticoide (9α-fluoroprednisolone, fludrocortisone)
- Sindrome e malattia di Cushing
- Condizioni endogene che determinano un aumento dei livelli plasmatici di DOC e/o altri ormoni mineralcorticoidi endogeni (deficit di 11β-idrossilasi e 17α-idrossilasi, adenomi e carcinomi producenti DOC e/o mineralcorticoidi)
- Uso di contraccettivi contenenti estrogeni e/o progestinici
- Assunzione di carbenoxolone
Blocco o saturazione della 11β-idrossi-steroido-deidrogenasi di tipo 2
- Ingestione di grandi quantità di derivati della liquirizia (acido glicirretico, glicirretinico, carbenoxolone) e flavonoidi
- Sindrome e malattia di Cushing
- Sindrome da apparente eccesso di mineralcorticoidi di tipo 1 (AME1)
- Ridotta sensibilità ai glucocorticoidi per effetto di alterazioni del recettore
- Ipertensione con bassi livelli di renina
Ridotta attività della 11β-idrossi-steroido-deidrogenasi di tipo 2
- Sindrome da apparente eccesso di mineralcorticoidi di tipo 2 (AME2)
Iperespressione dei recettori dell’aldosterone dovuti a mutazioni attivanti
- Alcuni casi di pre-eclampsia
Mutazioni di geni che codificano per canali del sodio a livello del rene
- Sindrome di Liddle
- Pseudoiperaldosteronismo di tipo II (sindrome di Gordon)
- Alcuni casi di ipertensione essenziale a bassa renina
PATOGENESI
Attualmente le cause di pseudoiperaldosteronismo sono per la maggior parte dei casi attribuibili all’assunzione di sostanze esogene mediante un’azione diretta a livello dell’enzima 11β-idrossisteroido-deidrogenasi di tipo 2 (11β-HSD2), altamente espresso nei tessuti in cui agisce selettivamente l’aldosterone (reni, colon, ghiandole salivari e placenta), causando l’inibizione e di conseguenza l’arresto della conversione del cortisolo in cortisone, come avviene con i derivati della liquirizia (l’acido glicirretico e glicirretinico), ma anche con il carbenoxolone, il gossipolo e altre sostanze vegetali contenenti flavonoidi, come ad esempio il pompelmo (1-3).
I corticosteroidi di sintesi solitamente hanno una scarsa capacitá di legare il recettore dei mineralcorticoidi (MR) e rappresentano quindi una causa non frequente di pseudoiperaldosteronismo; fanno tuttavia eccezione il 9α-fluoroprednisolone, uno steroide contenuto in numerose preparazioni ad uso topico (pomate, gocce oculari, spray nasali), e il fludrocortisone, ampiamente utilizzato nella terapia sostitutiva della malattia di Addison con deficit mineralcorticoide e prescritto in alcuni casi di ipotensione ortostatica. Questi due steroidi di sintesi hanno proprietá del tutto simili all’aldosterone e sono quindi capaci di legare il MR con uguale affinitá (1).
I contraccettivi orali contenenti estro/progestinici in alcuni casi attivano il sistema renina-angiotensina, detrminando un incremento dei valori di pressione arteriosa. In altri casi possono avere un effetto diretto sia sul MR che sull’inibizione dell’11β-HSD2, stimolando un aumentato riassorbimento di sodio, ritenzione di liquidi, e quindi ipertensione arteriosa con bassi livelli di renina e aldosterone (1).
Lo pseudoiperaldosteronismo di natura endogena, tra le varie cause, riconosce il deficit congenito di 11β-HSD2, denominata anche sindrome da apparente eccesso di mineralcorticoidi (AME), suddivisa in due forme, AME di tipo 1 e di tipo 2, entrambe trasmesse per via autosomica recessiva. La prima è caratterizzata da un esordio molto precoce con ipertensione arteriosa associata a bassi livelli di renina e aldosterone, poliuria e polidipsia, ritardo di crescita, ipokaliemia grave, alcalosi metabolica e molto spesso nefrocalcinosi. L’AME di tipo 2, pur essendo sempre dovuta ad una mutazione del gene dell’ 11β-HSD2, è associata ad una sintomatologia più lieve e ad un esordio più tardivo (1,4).
Lo pseudoiperaldosteronismo si può instaurare anche nel caso di adenomi e carcinomi secernenti desossicorticosterone (DOC) e nelle sindromi adrenogenitali da deficit di 11β-idrossilasi e di 17α-idrossilasi che, inducendo un aumento della produzione di DOC, provocano una saturazione del MR (1).
Altre cause che possono condurre a tale condizione sono legate a una ridotta sensibilità dei recettori dei glucocorticoidi (GR) e all’azione del cortisolo endogeno come nella sindrome di Cushing, in cui l’eccessiva produzione di cortisolo comporta una completa saturazione dell’ 11β-HSD2. Rarissimi sono i pazienti in cui è stata dimostrata una mutazione attivante del MR, che in alcuni casi puó essere responsabile di pre-eclampsia per via dell'aumentata sensibilitá del recettore, oltre che all'aldosterone, anche al progesterone (1).
In circa il 25 % dei casi di ipertensione definita “essenziale”, si possono riscontrare bassi livelli di renina e normali o bassi livelli di aldosterone. Tali reperti sono più frequenti nella popolazione anziana e tra i soggetti di etnia nera ed hanno alcune caratteristiche cliniche comuni, come una migliore risposta dei valori pressori alle modifiche dello stile di vita, in particolare alla riduzione dell’apporto di sale, riuscendo spesso ad avere un buon controllo della pressione arteriosa con l’assunzione di una sola classe di farmaci (ACE-inibitori o diuretici). Il rischio cardiovascolare è ridotto rispetto ai pazienti con valori di renina normali o elevati. La patogenesi delle ipertensioni definite “a bassa renina” riconosce varie cause, come ad esempio l'aumento della perfusione delle cellule juxta-glomerulari con la conseguente riduzione del rilascio di renina, oppure alterazioni del canale del sodio a livello del tubulo renale, mentre in altri casi è legata a forme lievi di alterata funzionalitá della 11β-HSD2 non imputabili tuttavia alla mutazione di un singolo gene (5).
Una causa di pseudoiperaldosteronismo attribuibile alla mutazione di un singolo gene è invece rappresentata dalla sindrome di Liddle, forma di ipertensione arteriosa ad esordio precoce, trasmessa per via autosomica dominante, il cui quadro clinico spesso è associato a ipokaliemia e alcalosi metabolica. Tale sindrome è provocata dalla mutazione di uno dei geni che codificano per le tre subunità proteiche (alfa, beta e gamma) del canale del sodio aldosterone-dipendente, che è normalmente espresso a livello dell'epitelio del tubulo renale distale. Come conseguenza di tale mutazione vi è una modificazione del canale del sodio, con un eccessivo riassorbimento di liquidi e conseguente espansione plasmatica (6).
Ancora più rara è la sindrome di Gordon o pseudoipoaldosteronismo di tipo II. Si tratta di una patologia trasmessa per via autosomica dominante, caratterizzata da una mutazione del canale che regola il riassorbimento del sodio e potassio a livello del tubulo renale distale e che si sviluppa generalmente nella 2°-3° decade di vita, con ipertensione arteriosa associata ad iperpotassiemia, ipercloremia ed acidosi metabolica (4,7).
BIBLIOGRAFIA
- Armanini D, Calò L, Semplicini A. Pseudohyperaldosteronism: pathogenetic mechanisms. Crit Rev Clin Lab Sci 2003, 40: 295-335.
- Sontia B, Mooney J, Gaudet L, Touyz RM. Pseudohyperaldosteronism, liquorice, and hypertension. J Clin Hypertens (Greenwich) 2008, 10: 153-7.
- Wynn GJ, Davis GK, Maher B. Trick or treat? Pseudohyperaldosteronism due to episodic licorice consumption. J Clin Hypertens (Greenwich) 2011, 13: E3-4.
- Monnens L, Levtchenko E. Distinction between Liddle syndrome and apparent mineralocorticoid excess. Pediatr Nephrol 2004, 19: 118-9.
- Sahay M, Sahay RK. Low renin hypertension. Indian J Endocrinol Metab 2012, 16: 728-39.
- Gao L, Wang L, Liu Y, et al. A family with Liddle syndrome caused by a novel missense mutation in the PY motif of the beta-subunit of the epithelial sodium channel. J Pediatr 2013, 162: 166-70.
- Bogdanović R, Kuburović V, Stajić N, et al. Liddle syndrome in a Serbian family and literature review of underlying mutations. Eur J Pediatr 2012, 171: 471-8.
Overview sugli pseudoiperaldosteronismi
Nicola Argese
Ospedale Sant’Andrea - Facoltà di Medicina e Psicologia, “Sapienza”, Università di Roma
Il termine pseudoiperaldosteronismo indica una sindrome caratterizzata da ipertensione arteriosa associata ad ipokaliemia e bassi livelli di aldosterone e renina.
Da sostanze esogene
Attualmente costituisce la maggior parte dei casi di pseudoiperaldosteronismo:
- derivati della liquirizia, carbenoxolone, gossipolo e altre sostanze vegetali contenenti flavonoidi, come il pompelmo: azione diretta a livello dell’enzima 11β-idrossisteroido-deidrogenasi di tipo 2 (11β-HSD2), causando inibizione della conversione del cortisolo in cortisone (1-3);
- corticosteroidi di sintesi: hanno scarsa capacitá di legare il recettore dei mineralcorticoidi (MR) tranne il 9α-fluoroprednisolone, contenuto in numerose preparazioni ad uso topico, e il fludrocortisone, ampiamente utilizzato nella terapia sostitutiva della malattia di Addison (1);
- contraccettivi orali contenenti estro/progestinici: in alcuni casi attivano il sistema renina-angiotensina, producendo un incremento dei valori della pressione arteriosa; in altri casi possono avere un effetto diretto sia sul MR che sull’inibizione dell’11β-HSD2, stimolando aumentato riassorbimento di sodio, ritenzione di liquidi, e quindi ipertensione arteriosa con bassi livelli di renina e aldosterone (1).
Di natura endogena
- Deficit congenito di 11β-HSD2, forma rara denominata anche sindrome da apparente eccesso di mineralcorticoidi (AME), suddivisa in due forme, trasmesse per via autosomica recessiva:
- AME di tipo 1: esordio molto precoce con ipertensione arteriosa associata a bassi livelli di renina e aldosterone, poliuria e polidipsia, ritardo di crescita, ipokaliemia grave, alcalosi metabolica e molto spesso nefrocalcinosi;
- AME di tipo 2: sintomatologia più lieve ed esordio più tardivo (1,4).
- Adenomi e carcinomi secernenti desossicorticosterone (DOC) e sindromi adrenogenitali da deficit di 11β-idrossilasi e di 17α-idrossilasi: l’aumento della produzione di DOC provoca una saturazione del MR (1).
- Eccessiva produzione di cortisolo (sindrome di Cushing o ridotta sensibilità dei recettori dei glucocorticoidi, GR): comportano una completa saturazione dell’11β-HSD2.
- Mutazione attivante del MR: rarissimi casi (1).
- Ipertensione “essenziale” a bassa renina: più frequente nella popolazione anziana e di etnia nera, con rischio cardiovascolare ridotto rispetto ai pazienti con valori di renina normali o elevati, migliore risposta dei valori pressori alle modifiche dello stile di vita, e buon controllo della pressione arteriosa con l’assunzione di una sola classe di farmaci (5).
- Sindrome di Liddle: provocata dalla mutazione di uno dei geni che codificano per le tre subunità proteiche (alfa, beta e gamma) del canale del sodio aldosterone-dipendente, normalmente espresso a livello dell'epitelio del tubulo renale distale. La modificazione del canale del sodio provoca eccessivo riassorbimento di liquidi e conseguente espansione plasmatica (6). Provoca una forma di ipertensione arteriosa ad esordio precoce, trasmessa per via autosomica dominante, il cui quadro clinico spesso è associato a ipokaliemia e alcalosi metabolica.
- Sindrome di Gordon o pseudoipoaldosteronismo di tipo II: ancora più rara, trasmessa per via autosomica dominante, caratterizzata da una mutazione del canale che regola il riassorbimento del sodio e potassio a livello del tubulo renale distale e che si sviluppa generalmente nella 2°-3° decade di vita, con ipertensione arteriosa associata ad iperpotassiemia, ipercloremia ed acidosi metabolica (4,7).
Bibliografia
- Armanini D, Calò L, Semplicini A. Pseudohyperaldosteronism: pathogenetic mechanisms. Crit Rev Clin Lab Sci 2003, 40: 295-335.
- Sontia B, Mooney J, Gaudet L, Touyz RM. Pseudohyperaldosteronism, liquorice, and hypertension. J Clin Hypertens (Greenwich) 2008, 10: 153-7.
- Wynn GJ, Davis GK, Maher B. Trick or treat? Pseudohyperaldosteronism due to episodic licorice consumption. J Clin Hypertens (Greenwich) 2011, 13: E3-4.
- Monnens L, Levtchenko E. Distinction between Liddle syndrome and apparent mineralocorticoid excess. Pediatr Nephrol 2004, 19: 118-9.
- Sahay M, Sahay RK. Low renin hypertension. Indian J Endocrinol Metab 2012, 16: 728-39.
- Gao L, Wang L, Liu Y, et al. A family with Liddle syndrome caused by a novel missense mutation in the PY motif of the beta-subunit of the epithelial sodium channel. J Pediatr 2013, 162: 166-70.
- Bogdanović R, Kuburović V, Stajić N, et al. Liddle syndrome in a Serbian family and literature review of underlying mutations. Eur J Pediatr 2012, 171: 471-8.
Clinica degli pseudoiperaldosteronismi
Nicola Argese
Ospedale Sant’Andrea - Facoltà di Medicina e Psicologia, “Sapienza”, Università di Roma
Dal punto di vista clinico lo pseudoiperaldosteronismo, analogamente a quanto accade nell’iperaldosteronismo primitivo, si presenta quasi sempre con un quadro rappresentato da ipertensione arteriosa dovuta all’espansione del volume plasmatico e all'aumento delle resistenze periferiche, che tuttavia, a differenza dell'iperaldosteronismo primitivo, è spesso caratterizzato da valori pressori di grado lieve-moderato, responsivi più di frequente ad una sola classe di farmaci.
Spesso non sono presenti edemi declivi, nonostante la ritenzione idrica dovuta al riassorbimento del sodio a livello renale, per l’innescarsi di un meccanismo fisiologico di autoregolazione, per cui, a fronte di un aumentato riassorbimento di sodio, vi è un incremento della diuresi che impedisce un eccessivo accumulo di liquidi extra-cellulari (1-3).
Segno caratteristico è l'affaticamento muscolare che si può presentare quando le concentrazioni plasmatiche di potassio scendono al di sotto di 2.5 mEq/L. L’ipokaliemia è una conseguenza del massivo riassorbimento di sodio a livello del tubulo renale, che provoca un aumento dell'escrezione di potassio e di idrogenioni, con conseguente potassiuria ed alcalosi metabolica; ciò può non verificarsi quando, per via della deplezione di volume, il flusso a livello del tubulo renale distale è ridotto, consentendo così il mantenimento di normali valori di potassio anche a fronte di elevati valori di aldosterone (1).
Per quanto riguarda invece il rischio cardiovascolare e di ictus, alcuni studi mostrano come i pazienti affetti da pseudoiperaldosteronismo abbiano, rispetto alla popolazione sana, un aumento del rischio, che tuttavia risulta notevolmente più basso rispetto ai pazienti con valori normali o elevati di renina e soprattutto rispetto alle condizioni di iperaldosteronismo (4).
Numerosi altri sintomi e segni possono essere associati alle singole patologie di base che provocano lo pseudoiperaldosteronismo, come nel caso della sindrome di Cushing e di tumori producenti steroidi o nel caso di deficit enzimatici specifici come quello della 11β-idrossilasi e della 17α-idrossilasi.
Le forme geneticamente determinate di eccesso apparente di mineralcorticoidi (AME) possono esordire con un ritardo di crescita intra-uterino e, sin dai primi anni di vita, possono presentare nefrocalcinosi, diabete insipido nefrogenico e rabdomiolisi (1,2). La fatigue cronica può essere presente nei casi in cui vi è una congenita alterazione del recettore dei glucocorticoidi, con conseguente resistenza all'azione dei glucocorticoidi e quindi, pur in presenza di un incremento delle concentrazioni di cortisolo ematico, per feed-back positivo, il suo effetto risulta scarsamente efficace a livello tissutale, specialmente a livello di organi come cervello e muscoli scheletrici.
L’ipertensione arteriosa è dovuta alla saturazione dell'11β-HSD2, senza avere alcun aspetto tipico legato alla condizione di ipercortisolismo.
Infine bisogna ricordare come molte di queste condizioni sono caratterizzate da un’elevata familiarità per ipertensione arteriosa, che frequentemente insorge in giovane età (2).
Bibliografia
- Sahay M, Sahay RK. Low renin hypertension. Indian J Endocrinol Metab 2012, 16: 728-39.
- Gupta V. Mineralocorticoid hypertension. Indian J Endocrinol Metab 2011, 15 Suppl 4: S298-312.
- Melcescu E, Phillips J, Moll G, et al. 11Beta-hydroxylase deficiency and other syndromes of mineralocorticoid excess as a rare cause of endocrine hypertension. Horm Metab Res 2012, 44: 867-78.
- Smith JH, Lindor NM, Rabinstein AA. Cerebrovascular consequences of pseudohyperaldosteronism. J Clin Hypertens (Greenwich) 2012, 14: 547-52.