Paratiroidi
Diagnostica generale dell'ipofisi

Generalità di diagnostica biochimica e ormonale ipotalamo-ipofisaria
Imaging neuroradiologico ipotalamo-ipofisario
Campimetria ottica e potenziali evocati visivi
Anatomia chirurgica della regione sellare e sovrasellare
Giovanni Lasio, Martina Revay
Neurochirurgia, Istituto Clinico Humanitas, Rozzano (MI)
L'osso sfenoide, situato al centro della base cranica, è in stretto rapporto con le cavità nasali inferiormente e con l'ipofisi superiormente, contenuta nella struttura ossea, detta sella, situata centralmente nel corpo dello sfenoide e che aggetta nel seno sfenoidale. I nervi olfattori, il giro retto, la parte posteriore del lobo frontale, il pavimento del III ventricolo, il ponte, il mesencefalo ed il chiasma ottico sono le strutture nervose a più stretto contatto con lo sfenoide. I nervi cranici dal II al VI sono pure in stretti rapporti con l'osso sfenoide, in quanto i forami da cui escono/entrano nel cranio sono tutti localizzati nell'osso sfenoide stesso: canale ottico (II), fessura orbitaria superiore (III, IV, VI e branca orbitaria del V), forame rotondo (branca mascellare del V), forame ovale (branca mandibolare del V).
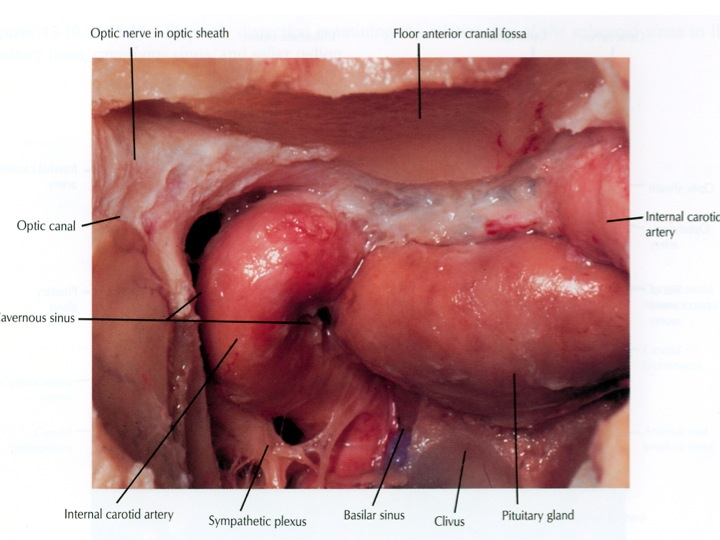
Oltre che importanti rapporti con strutture nervose, l'osso sfenoide ha intimi rapporti con strutture vascolari arteriose e venose. Le arterie carotidi attraversano lo sfenoide prima di entrare nel cranio, l'arteria basilare è addossata alla sua faccia posteriore, mentre il poligono di Willis è situato al di sopra della sua porzione centrale. I seni cavernosi sono appoggiati all'osso sfenoide, che ne forma la parete laterale, mentre la dura sellare ne forma la parete mediale; protrudono all'interno del seno sfenoidale. I seni cavernosi sono uniti dai seni inter-cavernoso superiore ed inferiore, che delineano la parete anteriore della sella e la parete inferiore. Il seno basilare poi connette i seni cavernosi posteriormente al dorsum sellae ed è usualmente il seno di maggiori dimensioni. I seni petrosi inferiori e superiori si uniscono al seno basilare.
La parte successiva è divisa in 3 sezioni: anatomia della regione sellare propriamente detta (vista dal basso) e della regione soprasellare (vista dall'alto), anatomia della ghiandola ipofisaria.
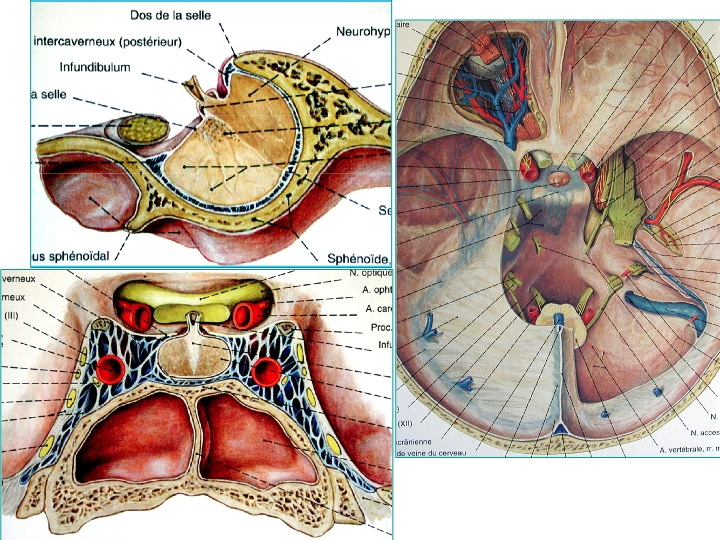
Anatomia della regione sellare in proiezione sagittale (in alto a sinistra), coronale dal basso (in basso a sinistra) e assiale dall' alto (a destra).
Anatomia della regione sellare propriamente detta (vista dal basso)
Il corpo dello sfenoide contiene il seno sfenoidale, che è in comunicazione con le cavità nasali tramite due osti, simmetrici. Il seno sfenoidale è soggetto a una considerevole variabilità di dimensione, forma e pneumatizzazione. Nell'adulto esistono tre tipi di seno sfenoidale: concale, presellare e sellare, così definiti in base alla pneumatizzazione, assente nel tipo concale e ben presente nel tipo sellare (75%). Il seno sfenoidale è poi attraversato da setti, assai variabili come numero, forma, direzione, spessore, ecc. I canali ottici protrudono nella porzione supero-laterale del seno sfenoidale, mentre la II branca del trigemino protrude nella sua parte infero-laterale. Il recesso ottico-carotideo è situato lateralmente e superiormente fra canale ottico e protuberanza carotidea.
La struttura ossea definita sella turcica, contenuta nella parte centrale del corpo dell' osso sfenoide, sporge all'interno del seno sfenoidale e ha confini anatomici precisi:
- postero-superiormente il dorso della sella, il cui margine libero termina lateralmente con i processi clinoidei posteriori e si continua inferiormente nel clivus;
- i seni cavernosi, che contengono sangue venoso, sono situati lateralmente alla sella. All'interno di essi decorre la porzione orizzontale della carotide interna ed un segmento del VI nervo cranico, mentre il III, il IV e la I branca del V nervo cranico si trovano nel tetto e nella parete laterale del seno cavernoso. Dal segmento intra-cavernoso della carotide nascono l'arteria meningo-ipofisaria, l'arteria del seno cavernoso inferiore e le arterie capsulari. La parete mediale del seno cavernoso è costituita da dura sottile, ma può presentare dei buchi od essere del tutto assente. La distanza fra arteria carotide e faccia laterale dell'ipofisi in condizioni normali varia fra 1 e 3 mm;
- anteriormente e superiormente il tubercolo della sella e il piano sfeno-etmoidale, separati dal solco chiasmatico;
- inferiormente il pavimento osseo della sella, che si continua con il clivus. Anteriormente la parete anteriore della sella, il clivus e la parete anteriore ossea dei seni cavernosi aggettano nella cavità del seno sfenoidale;
- il tetto della sella è costituito dal diaframma sellare, un setto orizzontale di dura che divide la loggia sellare dalla regione sovra-sellare, con un foro centrale per il passaggio del peduncolo ipofisario, struttura che connette l’ipotalamo all’ipofisi. Il diaframma è più sottile attorno al peduncolo e la sua apertura centrale è di solito più ampia di quanto necessario per il passaggio del peduncolo. In questi casi è presente un'invaginazione dell' aracnoide sovrasellare. La sella è internamente rivestita da periostio e contiene l'ipofisi.
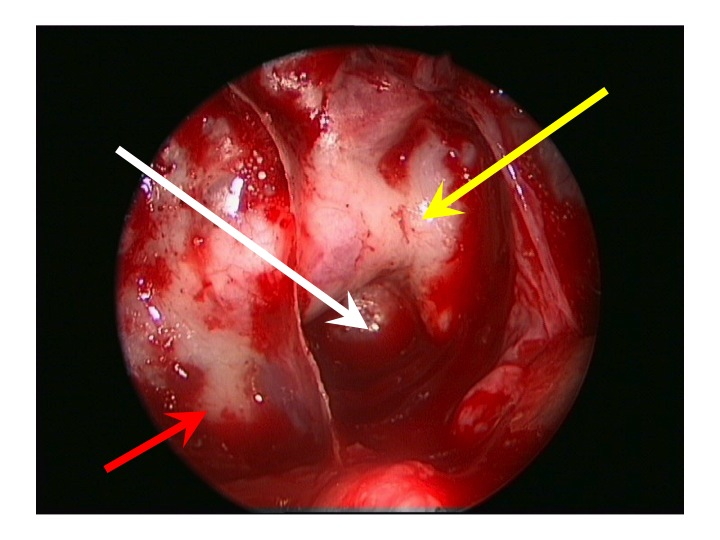
Visione endoscopica della sella, seno sfenoidale e carotidi clivali e cavernose dall’accesso TNS
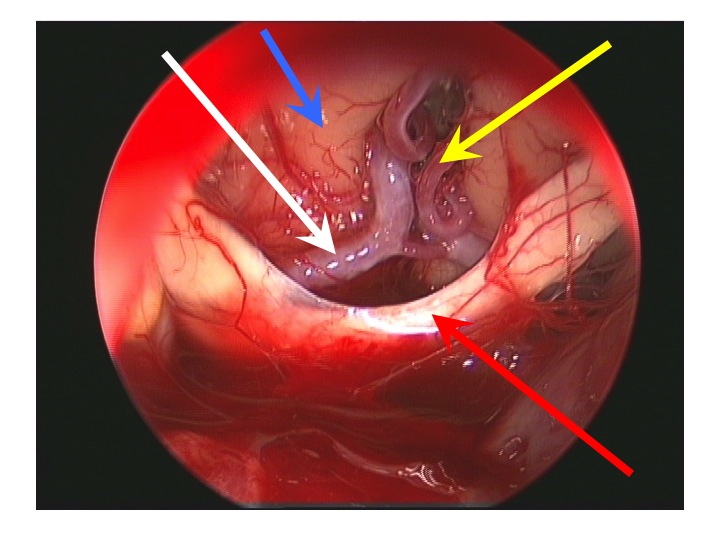
Visione endoscopica della regione sovrasellare (via TNS extended).
Nell'immagine in alto, la freccia rossa indica la carotide clivale dx, la gialla il seno cavernoso sinistro, la bianca attraversa la sella e indica il clivus.
Nell'immagine in basso, la freccia rossa indica il bordo anteriore del chiasma, la gialla l'arteria di Heubner sinistra, la bianca il tratto A1 della cerebrale anteriore dx, la blu la faccia inferiore del lobo frontale dx.
Anatomia della regione soprasellare (vista dall'alto)
La regione sovrasellare ha limiti più imprecisi. Può essere definita come la regione delimitata:
- anteriormente dal tubercolo sellare;
- inferiormente dal diaframma;
- posteriormente dalla membrana di Lillequist, una membrana aracnoidea che decorre fra il dorso della sella e la faccia anteriore dei corpi mammillari e che separa la cisterna chiasmatica dalla cisterna inter-peduncolare;
- superiormente dai nervi ottici, dal chiasma e dal pavimento del III ventricolo;
- lateralmente dalle carotidi.
Nervi e vasi della regione sono contenuti all'interno di cisterne limitate da aracnoide e contenenti liquor.
La regione sovrasellare è generalmente approcciata attraverso le cisterne che circondano la parte anteriore dell'incisura tentoriale, definita come uno spazio triangolare situato fra i margini liberi del tentorio. Il chiasma ed i nervi ottici attraversano lo spazio incisurale anteriore, i nervi ottici entrano nel cranio dai canali ottici medialmente alle clinoidi anteriori ed alle carotidi e sono diretti medialmente, posteriormente e superiormente verso il chiasma. Il chiasma è normalmente situato al di sopra del diaframma sellare.
Vista dall'alto la parete laterale del seno cavernoso si estende dalla fessura orbitaria superiore anteriormente, fino all'apice della porzione petrosa dell'osso temporale, posteriormente. Il III nervo cranico entra nel seno dal tetto, lateralmente al dorso sellare, il IV entra in posizione più posteriore e laterale, la branca oftalmica del V entra dalla parte inferiore della parete laterale e il VI entra dalla parete posteriore del seno fra carotide medialmente e III lateralmente.
Una volta entrate nel cranio inferiormente e poi lateralmente al nervo ottico, le carotidi si dirigono lateralmente e danno origine nell'ordine all'arteria oftalmica, alla comunicante posteriore, alla corioidea anteriore, per poi biforcarsi in cerebrale media e cerebrale anteriore, connessa alla cerebrale anteriore controlaterale dalla comunicante anteriore. Dalla carotide nasce l'arteria ipofisaria superiore, che raggiunge il tuber cinereum e si connette alla sua omologa controlaterale per formare un anello attorno all'infundibolo, cui è attaccata l'ipofisi tramite il peduncolo. Gli spazi subaracnoidei della regione sellare e parasellare sono poi attraversati da vasi perforanti che irrorano fra l'altro i nervi ottici, il chiasma, i tratti ottici, le pareti del III ventricolo e l'ipotalamo. Le vene della regione sellare e sovrasellare sono di piccolo calibro e la regione sovrasellare è quasi completamente drenata da tributarie delle vene basali.
Il III ventricolo è situato al centro della testa, superiormente alla sella ed è in stretto rapporto con il poligono di Willis e con il sistema venoso profondo del cervello. La manipolazione delle pareti del III ventricolo può causare disturbi ipotalamici (coscienza, termoregolazione, respirazione, secrezione ormonale, memoria...). E' delimitato da un pavimento, da un tetto, da pareti anteriori, posteriori e laterali. Visto dal basso il pavimento si estende dal chiasma all'acquedotto di Silvio, la metà anteriore è formata da strutture diencefaliche, la metà posteriore da strutture mesencefaliche. Dall'avanti verso l'indietro si trovano: il chiasma, l'infundibolo dell' ipotalamo, il tuber cinereum, i corpi mammilllari, la sostanza perforata posteriore e il tegmento del mesencefalo. L'infundibolo è una struttura imbutiforme localizzata fra il chiasma ed il tuber cinereum. L'ipofisi è connessa all'infundibolo e gli assoni infundibolari si estendono fino al lobo posteriore dell'ipofisi. Il tuber cinereum si fonde nell'infundibolo. La parete anteriore del III ventricolo si estende dai forami di Monro al chiasma inferiormente. L'unica parte visibile è la lamina terminale, un fine strato di sostanza grigia e pia, attaccata alla superficie superiore del chiasma e che riempie il gap fra chiasma e rostro del corpo calloso.
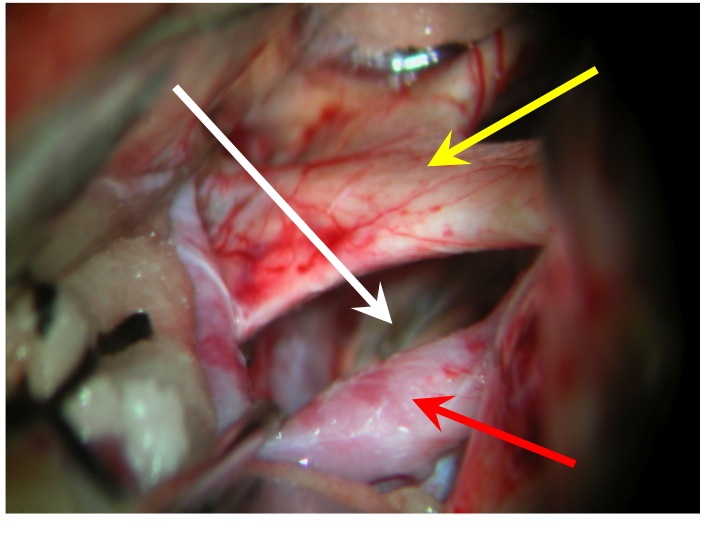
Vista della regione sovrasellare (via pterionale dx).
La freccia rossa indica la carotide dx, la gialla il nervo ottico dx e il chiasma, la bianca il diaframma sellare.
Anatomia dell'ipofisi
L’ipofisi è costituita da un lobo anteriore (adeno-ipofisi), che avvolge la parte più distale del peduncolo ipofisario, costituendo la pars tuberalis, e da un lobo posteriore (neuro-ipofisi), più aderente all'osso della sella di quanto non sia il lobo anteriore. Poichè il lobo anteriore è separato dal lobo posteriore, la pars tuberalis è più frequentemente inserita nel lobo posteriore. Cisti della pars intermedia sono di frequente riscontro.
Nell’adeno-ipofisi sono presenti 6 tipi cellulari diversi:
- le cellule tireotrope, che secernono il TSH;
- le cellule corticotrope, che secernono l’ACTH;
- le cellule lattotrope, che secernono la PRL;
- le cellule somatotrope, che secernono il GH;
- le cellule gonadotrope, che secernono le gonadotropine;
- le cellule follicolo-stellate, che potrebbero rappresentare cellule staminali ipofisarie e la cui funzione sembra importante per la secrezione di fattori di crescita e citochine e per mantenere i corretti rapporti (e quindi l’equilibrio paracrino) fra i diversi tipi cellulari.
Le cellule corticotrope e tireotrope tendono a raggrupparsi insieme nelle zone più centrali della ghiandola, mentre le cellule somatotrope si distribuiscono nelle porzioni più laterali e le cellule gonadotrope, lattotrope e follicolo-stellate sono diffusamente sparse nel parenchima adeno-ipofisario.
La neuro-ipofisi è costituita dalla parte terminale degli assoni delle cellule che secernono vasopressina e ossitocina, i cui corpi cellulari sono contenuti nell’ipotalamo (nei nuclei sopra-ottico e para-ventricolare).
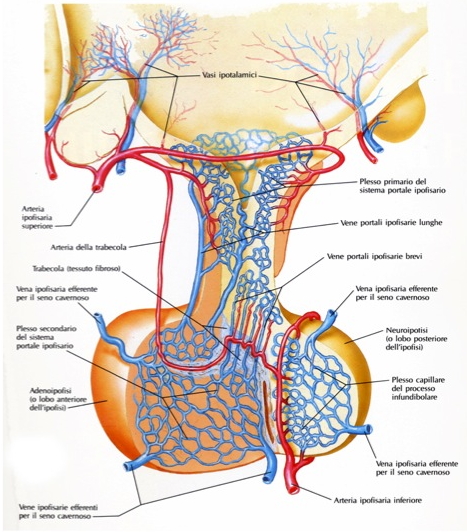
L’ipofisi è vascolarizzata dalle arterie ipofisarie superiori e inferiori e da rami delle arterie comunicanti posteriori, tutte originanti dalla carotide interna o da suoi rami. Le arterie ipofisarie superiori avvolgono la porzione superiore del peduncolo ipofisario, al cui interno comunicano con un plesso primario di capillari sinusoidali spiralizzati. Tali ramuscoli si tramutano in venule e quindi in vene lunghe e sottili che passano sotto il peduncolo ipofisario nella pars distalis dell’adeno-ipofisi, dove si forma un plesso secondario di capillari sinusoidali. Questa disposizione, doppia e parallela, dei plessi capillari prende il nome di sistema portale ipofisario. Il drenaggio venoso fa capo alle vene ipofisarie laterali che si aprono nel seno cavernoso.
Tomografia a coerenza ottica (OCT)
Stefano Amodeo
Facoltà di Medicina e Psicologia UOC di Oftalmologia, Università di Roma Sapienza; Ospedale Sant'Andrea di Roma
(aggiornato al febbraio 2025)
Cosa è
La tomografia a coerenza ottica, comunemente nota come OCT, è una tecnologia di imaging non invasiva, che permette di ottenere immagini ad alta risoluzione delle strutture oculari. Viene utilizzata principalmente in oftalmologia per analizzare la retina, il nervo ottico e la cornea, fornendo informazioni dettagliate sulla loro morfologia e stato di salute.
Come funziona
L'OCT sfrutta la luce per acquisire immagini delle strutture oculari in sezione trasversale, in modo simile a una TC, ma senza l'uso di radiazioni ionizzanti. Questa tecnologia si basa sul principio dell'interferometria a bassa coerenza, che consente di misurare il tempo impiegato dalla luce per riflettersi dai diversi strati dei tessuti oculari. I dati raccolti vengono elaborati da un software dedicato, che genera immagini dettagliate delle strutture osservate.
Il funzionamento dell'OCT si basa sull'uso di un raggio laser a bassa coerenza, solitamente nella banda dell'infrarosso, che viene diviso in due fasci. Uno di questi viene diretto verso il tessuto oculare, mentre l'altro è inviato a uno specchio di riferimento. La luce riflessa dalle diverse strutture dell'occhio viene poi confrontata con quella dello specchio, generando un'interferenza che permette di ricostruire l'immagine dei tessuti. Questa tecnica consente di ottenere immagini con una risoluzione dell’ordine di pochi micron, rendendola uno strumento essenziale per la diagnosi precoce di molte patologie oculari.
Uno dei principali vantaggi dell'OCT è la capacità di fornire immagini in tempo reale senza necessità di contatto diretto con l'occhio, rendendola una tecnica sicura e ben tollerata. Inoltre, la rapidità di esecuzione permette di integrare facilmente l'esame nella pratica clinica quotidiana. Tuttavia, come tutte le tecnologie diagnostiche, presenta alcune limitazioni, tra cui la riduzione della qualità dell'immagine in presenza di opacità dei mezzi diottrici, come cataratta avanzata o edema corneale.
Per cosa si usa
L'OCT viene utilizzata principalmente per la diagnosi e il monitoraggio di malattie della retina, come la degenerazione maculare legata all’età, la retinopatia diabetica e il pucker maculare. Inoltre, è fondamentale nello studio del nervo ottico, permettendo di valutare i danni legati al glaucoma, permette di misurare lo spessore delle fibre nervose, evidenziando gli assottigliamenti tipici del glaucoma e permettendo di seguire nel tempo l’evoluzione della patologia nonché di valutare l’idoneità del trattamento terapeutico locale messo in atto.
L’OCT trova applicazione anche nelle patologie della cornea: utile nella valutazione di cheratocono o nel follow-up post-chirurgico di interventi refrattivi.
Prospettive
Negli ultimi anni, l’evoluzione della tecnologia ha portato allo sviluppo dell’OCT ad alta velocità e dell’Angio-OCT, che permette di visualizzare la micro-vascolarizzazione retinica senza l’uso di mezzi di contrasto. Questo ha reso l’OCT uno strumento sempre più potente per la diagnosi precoce e il monitoraggio di numerose malattie oculari, migliorando significativamente la gestione clinica dei pazienti.
In conclusione, l’OCT rappresenta uno dei più importanti, utili e necessari strumenti diagnostici in oftalmologia. La sua capacità di fornire immagini ad alta risoluzione delle strutture oculari interne ha rivoluzionato la diagnosi e il trattamento di molte patologie. Grazie ai continui progressi tecnologici, l’OCT continuerà a giocare un ruolo chiave nella pratica clinica, consentendo diagnosi sempre più precoci e precise. Le nuove tecniche di imaging permetteranno di studiare porzioni di retina sempre più vaste, fino ad arrivare ad ottenere fotografie ad alta risoluzione di tutta la retina; l’applicazione nel segmento anteriore dell’occhio permetterà di comprendere in modo ancor più specifico le alterazioni a carico degli strati profondi della cornea, consentendo diagnosi sempre più precoci e dettagliate.
Utilizzo in endocrinologia
L’OCT è particolarmente utile nello studio della retina nei pazienti diabetici, permettendo di individuare precocemente segni di retinopatia diabetica, edema maculare e alterazioni micro-vascolari. Ciò consente un monitoraggio più accurato delle complicanze oculari legate al diabete mellito, migliorando la gestione complessiva del paziente.
Inoltre, l’OCT può essere impiegata nella valutazione dei nervi ottici in pazienti con orbitopatia di Graves, per monitorare lo stato delle fibre nervose e l’eventuale compressione orbitale.
Alcuni studi sperimentali stanno anche esplorando il ruolo dell’OCT nel controllo delle complicanze neuro-oftalmologiche delle malattie ipofisarie, fornendo un supporto aggiuntivo alla diagnostica tradizionale.
Grazie alla sua rapidità, sicurezza e precisione, la tomografia a coerenza ottica rappresenta un importante strumento complementare nella pratica endocrinologica moderna, favorendo una diagnosi precoce e un follow-up più mirato delle complicanze sistemiche.
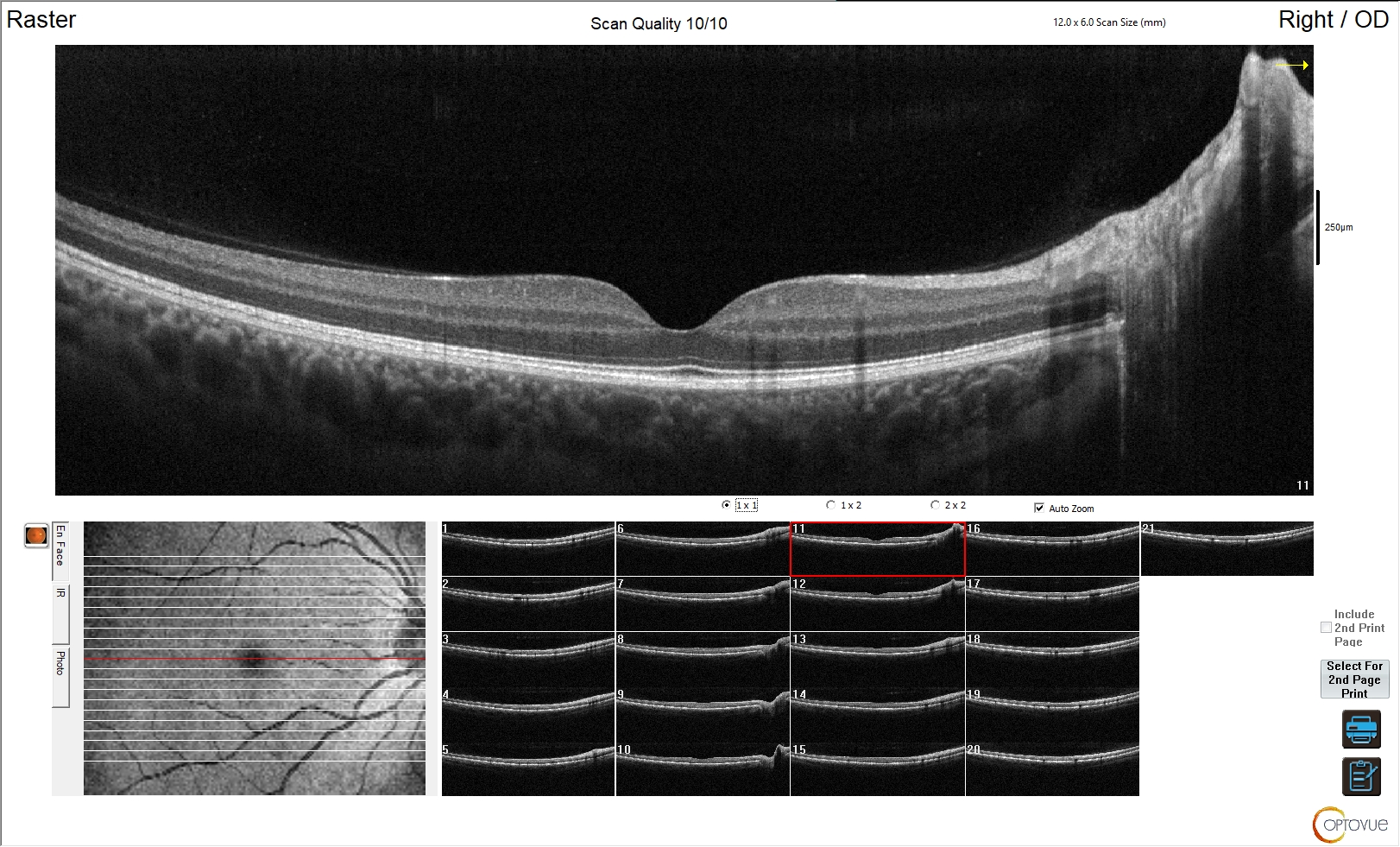
Figura 1. Esame normale
L'immagine mostra un esame OCT perfettamente nella norma, in cui sono riconoscibili i singoli strati retinici ed è mantenuta la corretta morfologia dei singoli strati e della retina in toto; è ben identificabile la fisiologica depressione foveale.
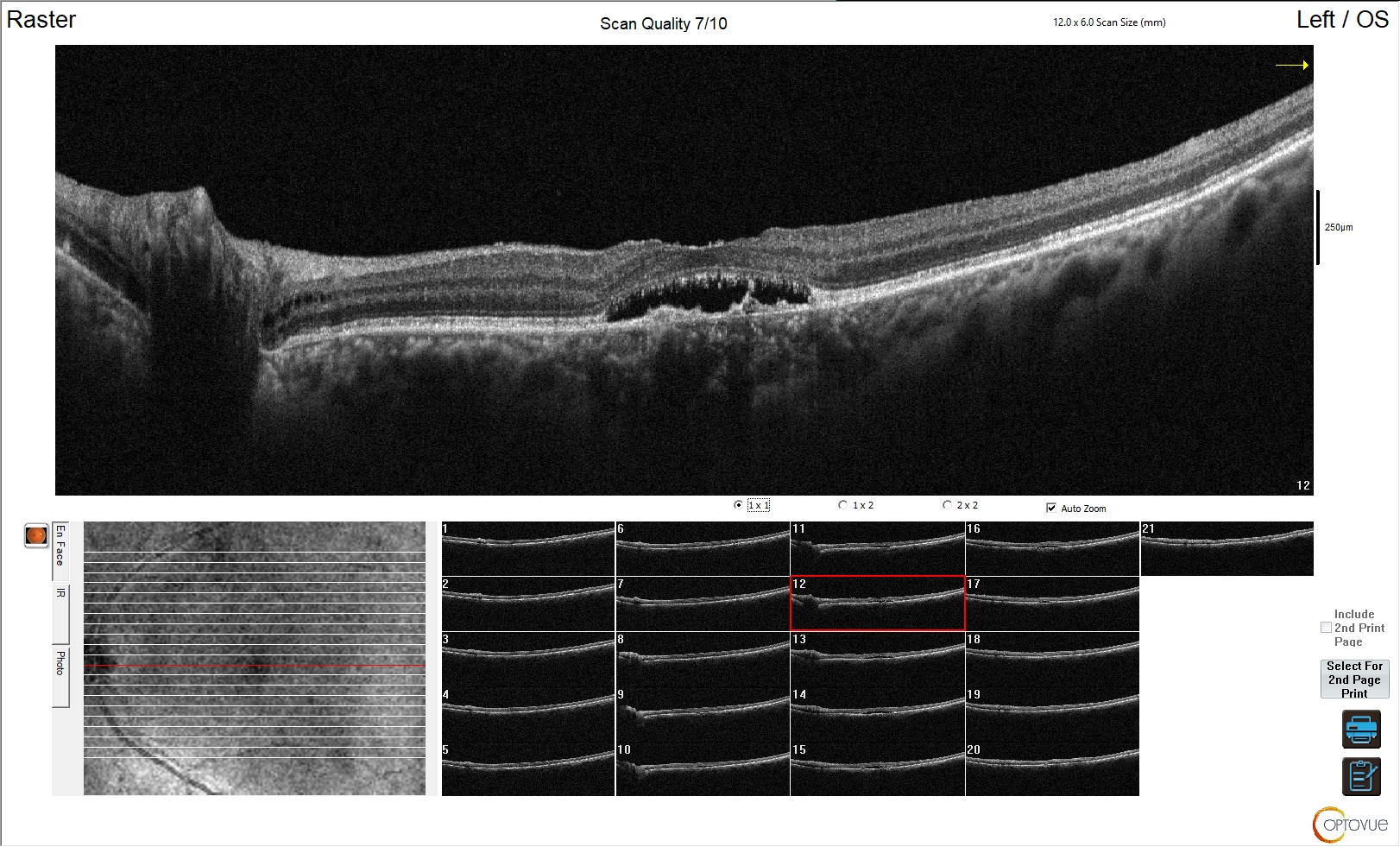
Figura 2. Maculopatia neo-vascolare nel soggetto anziano
L'esame OCT in questa immagine mostra un’alterazione contenuta della morfologia retinica. È scomparsa la fisiologica depressione foveale ed è presente negli strati interni della retina a livello dell'epitelio pigmentato un'area iporiflettente, con irregolarità di morfologia a livello del complesso epicoriocapillare. Tale quadro è riconducibile a una maculopatia senile neo-vascolare.
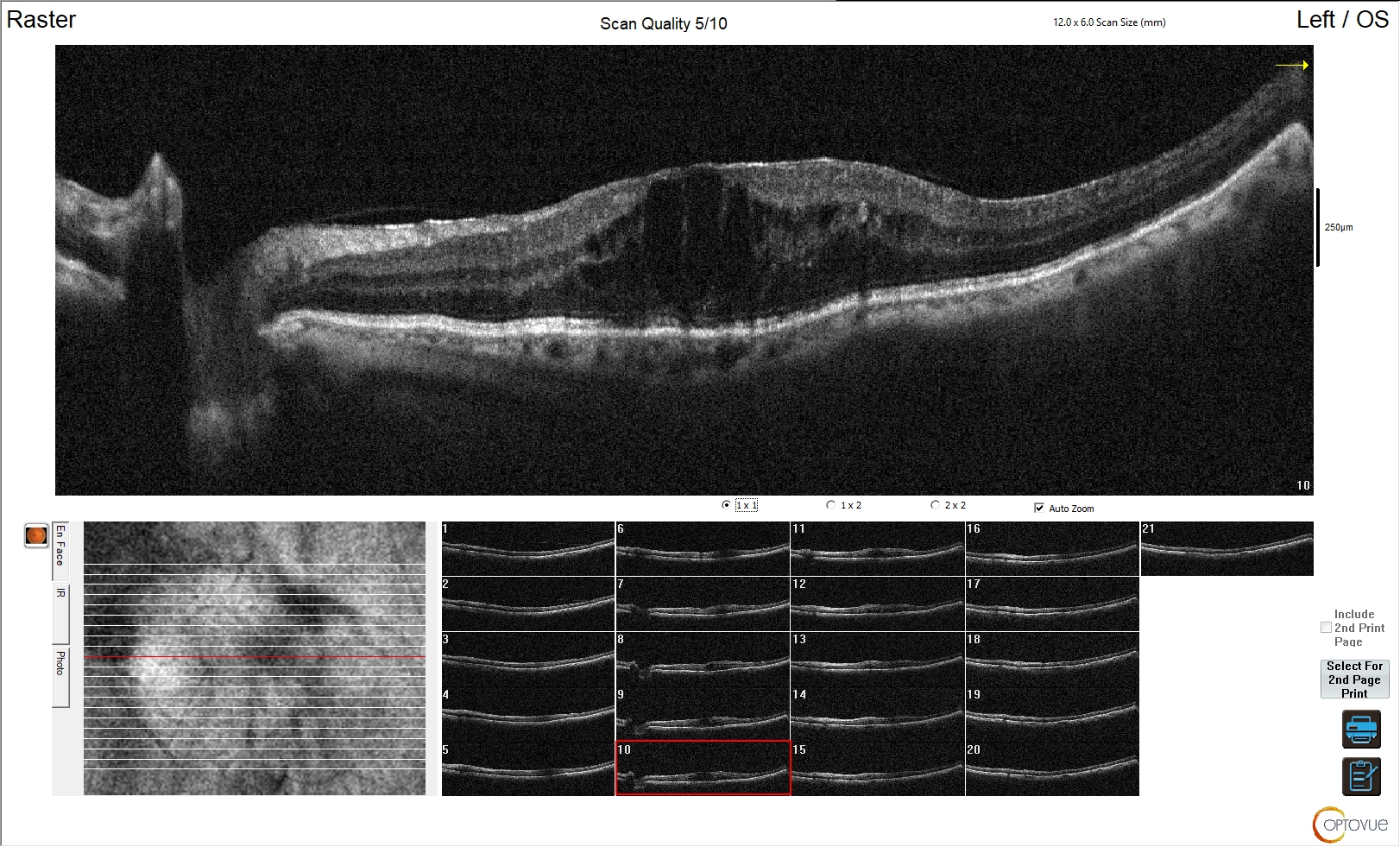
Figura 3. Edema maculare cistoide da retinopatia diabetica
L'esame OCT della regione maculare mostra marcato aumento di spessore della retina. Risulta sovvertita la normale architettura e morfologia della retina, con scomparsa della fisiologica depressione foveale. È evidente nella porzione centrale dell'esame un edema cistico da retinopatia diabetica con interessamento maculare.
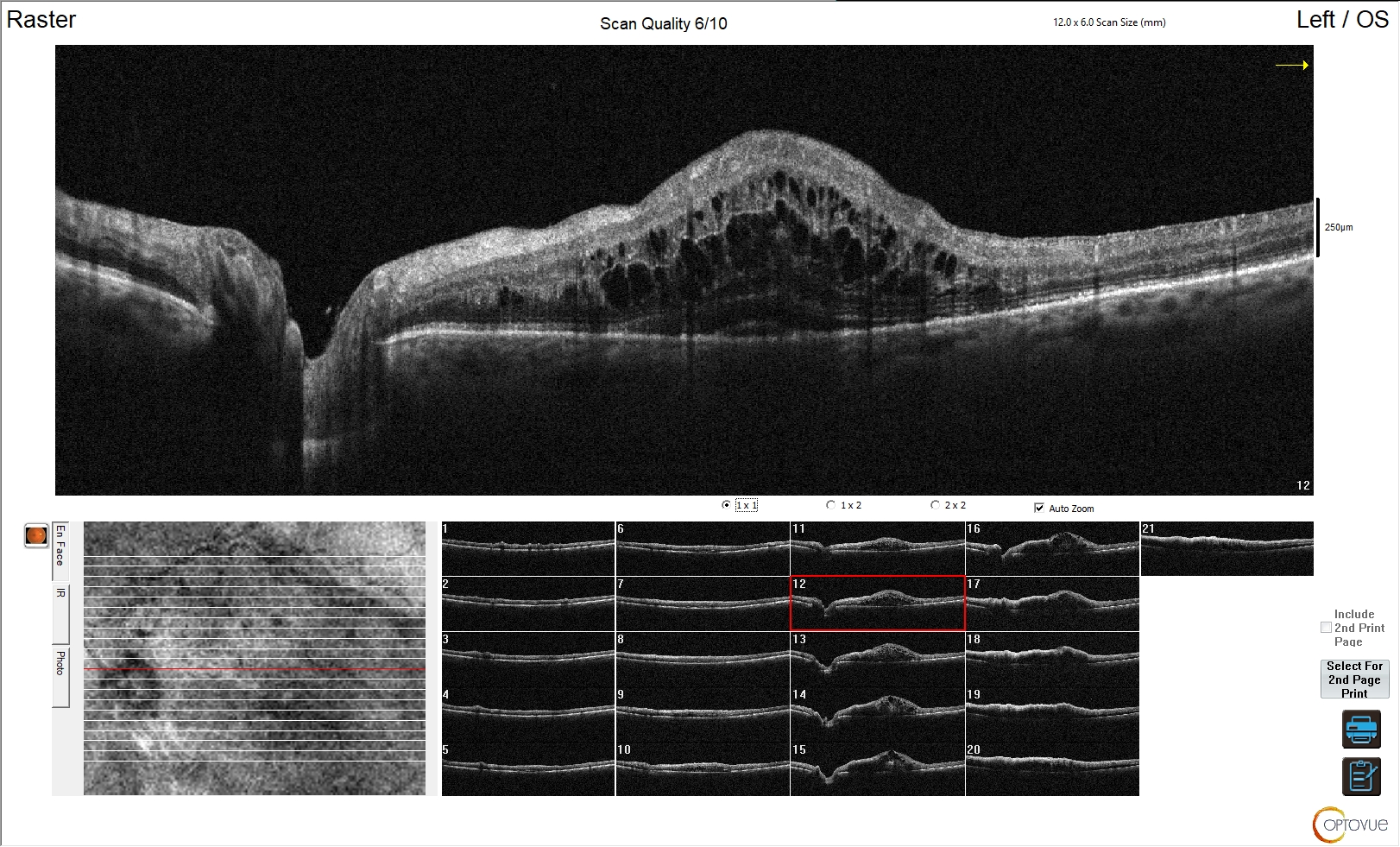
Figura 4. Edema maculare indotto da trombosi vascolare venosa
L'esame OCT in questa immagine evidenzia un’alterazione della fisiologica morfologia sia degli strati esterni che degli strati interni della retina. Non è riconoscibile la fisiologica depressione foveale ed è evidente la presenza di edema maculare ed extra-maculare, con alterazione degli strati interni della retina. Tale quadro è riconducibile a trombosi venosa retinica, con tipico edema intra-retinico diffuso nell'area ischemica.
Bibliografia
- Crincoli E, Sacconi R, Querques L, Querques G. OCT angiography 2023 update: focus on diabetic retinopathy. Acta Diabetol 2024, 61: 533-41.
- Nesper PL, Soetikno BT, Zhang HF, Fawzi AA. OCT angiography and visible-light OCT in diabetic retinopathy. Vision Res 2017, 139: 191-203.
- Reznicek L, Kolb JP, Klein T, et al. Wide-field Megahertz OCT imaging of patients with diabetic retinopathy. J Diabetes Res 2015, 2015: 305084.
Fisiologia ipotalamo-ipofisaria
Asse TRH-TSH-tiroide
Asse CRH-ACTH-surrene
Asse GHRH-GH-IGF
Asse GnRH-gonadotropine-gonade femminile