Orbitopatia
Overview sulla gestione clinica dei tumori differenziati della tiroide

Marco Capezzone
UOSD Endocrinologia, Ospedale Misericordia di Grosseto, USL Toscana sud-est Grosseto
(aggiornato al 30/6/2025)
(per la gestione del paziente con tumore a basso rischio, aggiornamento 2026)
INTRODUZIONE
I tumori maligni della tiroide comprendono carcinomi derivati dalle cellule follicolari (carcinoma papillare, carcinoma follicolare e carcinoma anaplastico) e dalle cellule para-folllicolari (carcinoma midollare). Forme più rare sono i linfomi primitivi della tiroide e i sarcomi. La tiroide inoltre può essere sede di metastasi da parte di neoplasie maligne di altri organi (mammella, rene, colon, melanoma).
Questo capitolo è dedicato alla gestione clinica dei tumori differenziati tiroidei (DTC). Per specifici approfondimenti si rimanda ai capitoli su manifestazioni cliniche, chirurgia, terapia radiometabolica, radioterapia esterna, follow-up.
Benché vi siano alcune differenze rilevanti nel comportamento biologico del carcinoma papillare (PTC) rispetto al follicolare (FTC, che compare in media in età più avanzata e si associa più frequentemente a metastasi a distanza), ai fini della pratica clinica la gestione dei DTC può essere trattata in comune (1), perchè la prognosi è simile se valutata per stadio (2,3).
L’incidenza dei DTC ha mostrato negli ultimi trent’anni un incremento superiore a quello di altre neoplasie (4,5). L’aumento è in parte conseguenza delle migliorate capacità diagnostiche per la diffusione dell’ecografia (US) e dell’agoaspirato della tiroide (FNA) (fenomeno della sovra-diagnosi) (6). Tuttavia, l’aumentata incidenza dei DTC riguarda non solo i microPTC, ma anche tumori di maggiori dimensioni e pertanto le cause sono multi-fattoriali (7). A fronte della più elevata frequenza dei DTC, la mortalità tumore-specifica è rimasta invariata nel tempo, confermando l’importanza della diagnosi precoce e della gestione terapeutica integrata.
1. TERAPIA CHIRURGICA
La chirurgia è la principale modalità terapeutica per i DTC. L’intervento chirurgico dovrebbe essere eseguito da chirurghi con esperienza nella chirurgia della tiroide, per minimizzare il rischio di ipoparatiroidismo e di lesioni a carico dei nervi ricorrenti. Sono disponibili opzioni terapeutiche diverse, in rapporto a tipo istologico, età, estensione della malattia e alla preferenza e condizioni generali del paziente (1).
La scelta dell’approccio chirurgico deve essere sempre preceduta dall’attenta stadiazione pre-operatoria, basata principalmente sulla valutazione US dei linfonodi latero-cervicali e del comparto centrale, al fine di pianificare la corretta procedura chirurgica. L’US del collo può arrivare a identificare linfonodi patologici in circa il 20-30% dei pazienti (8). Per confermare il sospetto di malignità, nei linfonodi di diametro > 10 mm dovrebbe essere eseguito FNA eco-guidato con dosaggio della tireoglobulina (Tg) sull’eluato. In pazienti con malattia locale potenzialmente avanzata potrebbero essere richiesti altri metodi di diagnostica per immagini (RM, TC con contrasto, laringoscopia ed endoscopia), per definire in modo accurato l’estensione del coinvolgimento tracheale, esofageo, laringeo o vascolare (9). Nei casi localmente avanzati, la TC del collo-mediastino con MdC permette di valutare in fase pre-operatoria il coinvolgimento delle catene linfonodali e pianificare il trattamento chirurgico ottimale. In questi casi il beneficio di un’accurata pianificazione chirurgica supera il ritardo temporale della somministrazione del radioiodio necessario dopo l’utilizzo del MdC iodato.
In assenza di studi prospettici, le conclusioni riguardo l’approccio chirurgico ottimale sono basate su analisi retrospettive e su opinioni degli esperti. Nei pazienti con PTC o FTC sono indicate queste procedure.
Assenza di estensione extra-tiroidea, assenza di metastasi linfonodali o a distanza:
- tumori unilaterali ≤ 1 cm: l’approccio preferito è la lobectomia, salvo che non ci siano chiare indicazioni a rimuovere anche il lobo contro-laterale (ad esempio cancro controlaterale clinicamente evidente, storia pregressa di irradiazione nella regione testa-collo, forte familiarità per carcinoma tiroideo). In casi selezionati in alternativa all’intervento chirurgico può essere proposta la sorveglianza attiva (10);
- tumori di 1-4 cm: la procedura chirurgica iniziale può essere sia la tiroidectomia totale che la lobectomia. La tiroidectomia totale dovrebbe essere scelta in base a preferenza del paziente, presenza di anomalie ecografiche nel lobo controlaterale (noduli, tiroiditi, linfoadenopatie aspecifiche, che potrebbero rendere difficoltoso il follow-up) o al potenziale beneficio del radio-iodio sia come terapia adiuvante sia per facilitare il follow-up (11,12);
- tumori > 4 cm: è infrequente il riscontro di un PTC > 4 cm senza coinvolgimento linfonodale o estensione extra-tiroidea. Pertanto, la maggioranza di questi pazienti viene sottoposta a tiroidectomia totale, anche se in pazienti attentamente selezionati a basso rischio potrebbe essere presa in considerazione la lobectomia (1).
In pazienti selezionati in modo appropriato la lobectomia, con o senza istmectomia, è un’alternativa accettabile alla tiroidectomia totale, per l’evidenza di dati che riportano bassa mortalità, bassi tassi di recidiva e minore tasso di complicanze chirurgiche. Nei pazienti con tumore intra-tiroideo studi retrospettivi e meta-analisi hanno mostrato tassi di sopravvivenza simili tra i pazienti sottoposti a tiroidectomia totale e a lobectomia (13,14). Sebbene nei pazienti trattati con lobectomia tiroidea il rischio di recidiva loco-regionale sia leggermente aumentato, queste recidive possono essere trattate chirurgicamente in maniera efficace al momento del riscontro.
Presenza di estensione extra-tiroidea e/o metastasi (linfonodali e/o a distanza) in tumori di ogni diametro: questi pazienti devono essere sottoposti a tiroidectomia totale.
Storia di pregressa irradiazione sul collo e sulla testa in tumori di ogni diametro: dato l’elevato rischio di recidiva tumorale che questi pazienti presentano con interventi chirurgici meno estesi, deve sempre essere eseguita tiroidectomia totale (1).
MicroPTC multi-focale:
- se foci < 5 mm la lobectomia con istmectomia è una procedura appropriata;
- se foci > 5 mm, soprattutto se con diametro di 8-9 mm di, è preferibile eseguire il completamento della tiroidectomia.
Le indicazioni per il completamento della tiroidectomia includono:
- carcinoma tiroideo poco differenziato;
- invasione linfatica o invasione vascolare;
- malattia multi-focale con foci > 10 mm.
Dissezione linfonodale:
- del compartimento centrale del collo (livello VI):
- deve essere eseguita quando vi è evidenza di impegno linfonodale alla stadiazione pre-operatoria o all’esplorazione intra-operatoria;
- è controversa in assenza di metastasi evidenziabili (dissezione profilattica). Consente una stadiazione isto-patologica più completa, definendo il pN e orientando più precisamente verso l’opportunità di un trattamento ablativo con radio-iodio. Tuttavia non si associa a riduzione significativa della mortalità a lungo termine, mentre è seguita da incremento delle complicanze permanenti (ipoparatiroidismo e danno del nervo laringeo ricorrente). Dovrebbe essere presa in considerazione solo nei DTC di ampie dimensioni (> 4 cm) o con estensione extra-capsulare, caratteristiche associate con elevata frequenza a metastasi linfonodali (15). Deve comunque essere eseguita in ambienti chirurgici con specifica competenza e alto volume di interventi di tiroidectomia;
- latero-cervicale: deve essere eseguita in presenza di metastasi linfonodali ecograficamente o clinicamente accertate. La dissezione deve essere funzionale (risparmiando l’integrità di muscoli, fibre nervose e vasi del collo) ed estesa ai compartimenti II, III, IV e V del collo (16,17).
L’impiego dell’ecografia intra-operatoria o della chirurgia radio-guidata potrebbe essere utile in caso di re-intervento per recidiva linfonodale o nel letto tiroideo in pazienti già sottoposti a precedente linfoadenectomia, per ridurre i tempi operatori e minimizzare il rischio di complicanze.
Approccio chirurgico più aggressivo: deve essere impiegato nei tumori avanzati della tiroide che coinvolgono i muscoli e le strutture vitali del collo. L’intervento deve consentire il miglioramento dell’aspettativa e/o della qualità di vita e non deve essere causa di alterazioni anatomiche o funzionali invalidanti. In queste circostanze è necessaria un’accurata stadiazione pre-operatoria da condurre con TC o RM del collo e torace con MdC, studio endoscopico delle vie aeree e digestive superiori, e, ove possibile, PET-TC con 18F-fluorodeossiglucosio (18,19).
2. STADIAZIONE
La stadiazione post-operatoria dei pazienti con DTC riveste un ruolo fondamentale per la gestione nel tempo della malattia. Dopo l’intervento chirurgico, deve essere valutata l’assenza o la presenza di malattia persistente e il rischio di recidiva, per individuare la necessità di trattamento terapeutico aggiuntivo, in particolare la terapia ablativa con radioiodio. Sebbene la stratificazione iniziale del rischio possa essere utilizzata per guidare le decisioni iniziali sulla strategia di follow-up diagnostica e terapeutica, è importante ricordare che le stime iniziali del rischio potrebbero cambiare man mano che vengono accumulati nuovi dati durante il follow-up (stratificazione dinamica del rischio) (1,20). Pertanto, nella pratica clinica è opportuno utilizzare tre sistemi di stadiazione, in grado di guidare la condotta clinica in fasi diverse.
Il rischio di mortalità tumore-specifico può essere definito sulla base dei dati isto-patologici disponibili dopo l’intervento chirurgico (tabella 1). Il sistema di stadiazione più diffuso e accettato è il TNM, adottato dalla UICC e dall’AJCC (21). Si distinguono 4 stadi, caratterizzati da un rischio crescente di mortalità, sulla base di età, dimensioni del tumore, estensione locale di malattia e presenza di metastasi a distanza (tabella 2). La predittività del TNM è soddisfacente nel definire la mortalità ma, essendo basata sui soli dati anatomo-patologici, è meno precisa nel definire il rischio di recidiva o persistenza di malattia.
| Tabella 1 Stadiazione TNM per il tumore della tiroide (papillare, follicolare, scarsamente differenziato, a cellule di Hürthle e anaplastico) (modificato da 21) |
||
| Categoria | Criteri | |
| T (tumore primitivo)* | Tx | Il tumore primitivo non può essere valutato |
| T0 | Non evidenza di tumore primitivo | |
| T1 | Tumore ≤ 2 cm, limitato alla tiroide | |
| T1a | Tumore ≤ 1 cm | |
| T1b | Tumore > 1 cm ma ≤ 2 cm | |
| T2 | Tumore > 2 cm ma ≤ 4 cm, limitato alla tiroide | |
| T3 | Tumore > 4 cm limitato alla tiroide, o con macroscopica estensione extra-tiroidea, con invasione solo dei muscoli anteriori del collo | |
| T3a | Tumore limitato alla tiroide | |
| T3b | Tumore di qualunque dimensione, con estensione extra-tiroidea macroscopica solo nei muscoli anteriori del collo (sterno-ioideo, sterno-tiroideo, omo-ioideo) | |
| T4 | Tumore di qualsiasi dimensione, con invasione macroscopica extra-tiroidea | |
| T4a | L’estensione extra-tiroidea invade tessuto sotto-cutaneo, trachea, laringe, esofago, o nervo ricorrente | |
| T4b | L’estensione extra-tiroidea infiltra la fascia pre-vertebrale o l’arteria carotidea o i vasi mediastinici | |
| N (linfonodi)** | Nx | Linfonodi regionali non valutabili |
| N0 | Assenza di metastasi ai linfonodi regionali | |
| N1 | Presenza di metastasi ai linfonodi regionali | |
| N1a | Uni- o bilaterali ai linfonodi del VI o VII livello | |
| N1b | Ai linfonodi del collo omolaterali, bilaterali o controlaterali (livelli I, II, III, IV o V) o ai linfonodi retro-faringei | |
| M (metastasi) | M0 | Assenti metastasi a distanza |
| M1 | Presenti metastasi a distanza | |
|
*Tutte le categorie possono essere suddivise in:
**I linfonodi regionali sono il compartimento centrale laterale del collo e i linfonodi del mediastino superiore |
||
| Tabella 2 Raggruppamento in stadi (modificato da 21) |
||||||
| Stadio | T | N | M | T | N | M |
| Età < 55 anni |
Età ≥ 55 anni |
|||||
| I | Qualsiasi | Qualsiasi | M0 | T1/T2 | N0/Nx | M0 |
| II | Qualsiasi | Qualsiasi | M1 | T1/T2 | N1 | M0 |
| T3a/b | Qualsiasi | M0 | ||||
| III | T4a | Qualsiasi | M0 | |||
| IVa | T4b | Qualsiasi | M0 | |||
| IVb | Qualsiasi | Qualsiasi | M1 | |||
Il rischio di malattia persistente e/o di recidiva è meglio definito dal sistema di stadiazione clinico-patologica dell’American Thyroid Association (ATA). Questo sistema stratifica i pazienti principalmente sulla base delle caratteristiche clinico-patologiche. Il rischio di recidiva è dinamico, distinto in tre categorie (basso, intermedio e alto) (tabella 3) (1,20).
| Tabella 3 Rischio iniziale di recidiva secondo American Thyroid Association (modificato da 1) |
|
| Basso |
PTC con le seguenti caratteristiche:
FTC intra-tiroideo ben differenziato, con invasione capsulare e invasione vascolare assente o minima (< 4 foci). |
| Intermedio | Tumore con invasione microscopica dei tessuti lassi peri-tiroidei. Tumore con istologia aggressiva (a cellule alte, insulare, a cellule colonnari, a cellule di Hürthle, carcinoma follicolare, variante hobnail). PTC con invasione vascolare. N1 clinico o riscontro istologico N1 con > 5 metastasi (< 3 cm di diametro max). MicroPTC multi-focale con estensione extra-tiroidea (inclusi i casi con mutazione BRAF V600E). Presenza di iodio-captazione nel collo alla scintigrafia post-dose di 131I. |
| Alto | Tumore con invasione macroscopica dei tessuti lassi peri-tiroidei. Resezione tumorale incompleta con residuo macroscopico. Presenza di metastasi a distanza. Tg post-operatoria suggestiva di metastasi a distanza. Riscontro istologico N1 con metastasi > 3 cm di diametro max. FTC con invasione vascolare massiva > 4 foci. |
La stratificazione dinamica del rischio del Memorial-Sloan Kettering Cancer Center consente di modificare nel tempo il rischio di recidiva o decesso del paziente sulla base della risposta alla terapia nel corso del follow-up. La ri-stratificazione, condotta sulla base dei risultati dei primi due anni, permette di descrivere lo stato clinico in qualsiasi momento durante il follow-up. A ogni visita, i pazienti vengono classificati in base a uno dei seguenti risultati clinici (tabella 4):
- risposta eccellente: nessuna evidenza clinica, biochimica o strutturale di malattia;
- risposta incompleta biochimica: Tg aumentata o valori in aumento degli Ab anti-Tg, in assenza di malattia localizzabile;
- risposta strutturale incompleta: persistenza o recidiva loco-regionale o metastasi a distanza;
- risposta indeterminata: reperti non specifici biochimici o strutturali, che non possono con sicurezza essere classificati né benigni né maligni. Questo include pazienti con livelli di Ab anti-Tg stabili o in riduzione, senza sicura evidenza strutturale di malattia.
| Tabella 4 Classificazione dinamica del rischio di recidiva o mortalità |
|||
| Risposta | Intervento iniziale | ||
| Tiroidectomia totale + ablazione con 131I | Tiroidectomia totale senza 131I | Lobectomia | |
| Eccellente | Ab anti-Tg indosabili e imaging negativo. | ||
| Tg non stimolata < 0.2 ng/mL o stimolata < 1 ng/mL. | Tg stabile non stimolata < 30 ng/mL. | ||
| Biochimica incompleta | Livelli di Ab anti-Tg in aumento e imaging negativo. | ||
| Tg non stimolata > 1 ng/mL* o stimolata > 10 ng/mL*. | Tg non-stimolata > 5 ng/mL* o stimolata > 10 ng/mL* o livelli in aumento con livelli simili di TSH. | Tg non-stimolata > 30 ng/mL* o livelli in aumento con livelli simili di TSH. | |
| Strutturale incompleta | Evidenza strutturale o funzionale di malattia, indipendentemente da Tg o Ab anti-Tg. | ||
| Indeterminata | Risultati non specifici negli studi di imaging, in assenza di malattia strutturale o funzionale, o livelli di Ab anti-Tg stabili o in riduzione. | ||
| Debole captazione nel letto tiroideo al WBS. | |||
| Tg non stimolata tra 0.2-1 ng/mL* o stimolata tra 1-10 ng/mL*. | Tg non stimolata tra 0.2-5 ng/mL* o stimolata tra 2-10 ng/mL*. | ||
| *in assenza di Ab anti-Tg interferenti | |||
3. TERAPIA RADIOMETABOLICA
Il trattamento ablativo con 131I provoca, attraverso l’emissione di radiazioni β, la distruzione del tessuto tiroideo residuo e degli eventuali residui microscopici di malattia; consente inoltre di visualizzare la persistenza di malattia e le eventuali metastasi a distanza con la scintigrafia whole-body post-dose, completando la stadiazione della neoplasia, e rende il dosaggio della Tg sierica un marcatore di malattia sensibile e di semplice impiego per il follow-up.
È impiegata nel DTC come trattamento adiuvante dopo l’intervento di tiroidectomia totale, perché è in grado di ridurre significativamente il rischio di recidiva di malattia a 10 anni e di ridurre, in minor misura, la mortalità tumore-specifica.
Attualmente è riservata solo ai pazienti ad alto rischio e ad alcuni selezionati a rischio intermedio, perchè non sembra migliorare la prognosi nei pazienti con micro-carcinoma o con stadiazione iniziale a basso rischio (1,20,22-24).
Per le indicazioni, modalità di preparazione ed esecuzione, norme protezionistiche, risultati e complicanze vedi i capitoli dedicati. Gravidanza e allattamento costituiscono una controindicazione assoluta alla terapia con 131I.
Nel tentativo di standardizzare la terminologia, un gruppo di lavoro inter-societario, con rappresentanti di ATA, European Thyroid Association, European Association of Nuclear Medicine e Society of Nuclear Medicine and Molecular Imaging ha raggiunto il seguente consenso riguardo agli obiettivi della terapia con 131I nel DTC.
Ablazione del residuo post-chirurgico: l'obiettivo primario è la distruzione del tessuto tiroideo residuo, presumibilmente benigno, dopo la tiroidectomia totale, al fine di facilitare la stadiazione iniziale e il follow-up. Questo, a sua volta, consentirà di:
- migliorare la specificità delle misurazioni della Tg come marcatore tumorale;
- aumentare la specificità della scintigrafia con 131I per l'individuazione di malattia recidivante o metastatica, eliminando l'assorbimento del tessuto sano residuo.
Nei pazienti con basso rischio ATA (T1a/T1b, N0/Nx, M0/Mx), l'ablazione del residuo non è generalmente raccomandata. Tuttavia, la valutazione di caratteristiche specifiche potrebbe portare a prenderla in considerazione in singoli pazienti.
Nei pazienti a rischio intermedio o basso-intermedio (T1/T2, N1a/N1b, M0/Mx), l'ablazione dovrebbe essere presa in considerazione, in particolare in presenza di caratteristiche avverse (età avanzata, tumori di dimensioni maggiori, linfonodi macroscopici o clinicamente evidenti o presenza di estensione extra-tiroidea, o istologia aggressiva o invasione vascolare) (25).
Nei pazienti ad alto rischio o a rischio intermedio-alto (T3(T4, qualsiasi N, qualsiasi M), l'ablazione è raccomandata di routine.
Trattamento adiuvante: l'obiettivo primario è la distruzione dei depositi tumorali subclinici (che possono essere presenti o meno dopo la resezione chirurgica di tutto il tessuto tumorale primario) e dei focolai metastatici. Poiché il trattamento adiuvante viene somministrato in base al rischio di persistenza/recidiva di malattia in assenza di evidenza di malattia biochimica o strutturale, è ipotizzabile che alcuni pazienti selezionati per il trattamento adiuvante possano essere già stati trattati in modo adeguato con l'intervento chirurgico primario. Pertanto, la decisione di raccomandare il trattamento adiuvante richiede un bilanciamento tra il rischio oncologico (rischio di persistenza/recidiva di malattia e mortalità specifica per malattia) e i rischi associati al trattamento adiuvante (rischi a breve e lungo termine di 131I) e il potenziale beneficio del trattamento adiuvante (potenziale di riduzione delle recidive, miglioramento della sopravvivenza libera da progressione e/o miglioramento della mortalità specifica per malattia). Pertanto, in pazienti opportunamente selezionati, i potenziali benefici potrebbero includere:
- distruzione dei focolai microscopici subclinici di malattia residui dopo chirurgia;
- riduzione del rischio di recidiva;
- miglioramento della sopravvivenza specifica per malattia;
- miglioramento della sopravvivenza libera da progressione.
Trattamento di malattia nota: l'obiettivo primario in questo caso è la distruzione della malattia macroscopica clinicamente evidente (evidenziata da valori anomali di Tg o da reperti strutturali), non suscettibile di terapia chirurgica (1,20,24). Il trattamento con radioiodio della malattia residua e della malattia metastatica può ridurre il rischio di recidiva e mortalità, soprattutto nelle lesioni di piccole dimensioni avide di radioiodio. La terapia con radioiodio ad alte dosi è un efficace mezzo terapeutico per le metastasi polmonari e, in minor misura, per le altre metastasi a distanza (scheletro, fegato, cervello). Complessivamente, i pazienti trattati con 131I per metastasi a distanza hanno una sopravvivenza a 5 anni che è circa il doppio dei non trattati. La risposta terapeutica è migliore nei pazienti con metastasi polmonari di piccole dimensioni non visualizzabili radiologicamente.
4. TERAPIA CON ORMONE TIROIDEO
Dopo la tiroidectomia iniziale, è necessaria la terapia con levotiroxina (LT4) per prevenire (e curare) l’ipotiroidismo e, nella maggior parte dei pazienti, per ridurre al minimo la potenziale stimolazione del TSH sulla crescita di un eventuale residuo neoplastico. L’ipotesi che valori soppressi di TSH riducano il rischio di mortalità in tutti i pazienti con DTC non è stata dimostrata (26).
È importante sottolineare che la dose di LT4 va adattata all’estensione della malattia e alla probabilità di recidiva. Queste decisioni possono essere basate in parte sulla stadiazione tramite il TNM, in combinazione con il sistema di rischio di recidiva proposto dall’ATA.
La terapia deve essere monitorata con il TSH sierico, misurato annualmente e 6-8 settimane dopo qualsiasi aggiustamento della dose (1).
Pazienti a basso rischio:
- trattati con tiroidectomia e con livelli sierici di Tg dosabili (con o senza ablazione del residuo): mantenere valori di TSH tra 0.1 e 0.5 mU/L;
- trattati con tiroidectomia e con livelli sierici di Tg non rilevabili (con o senza ablazione del residuo): il TSH può essere mantenuto tra 0.5 e 3.0 mU/L;
- trattati con lobectomia: il trattamento con LT4 potrebbe non essere necessario nel paziente il cui TSH si mantiene nell’intervallo desiderato (dal limite inferiore della norma a 3 mU/L) (1).
Pazienti a rischio intermedio: mantenere valori di TSH tra 0.1 e 0.5 mU/L (1).
Pazienti a rischio alto: mantenere valori di TSH < 0.1 mU/L (1).
Adeguamento degli obiettivi di TSH in base alla risposta alla terapia: per il follow-up a lungo termine, bisogna basarsi sulla risposta alla terapia e sulla presenza di comorbilità che aumentano i potenziali rischi di soppressione prolungata del TSH (come menopausa, età avanzata, tachicardia/fibrillazione atriale, osteopenia/osteoporosi) (27).
- Nei pazienti a rischio intermedio che dimostrano eccellente risposta alla terapia durante i primi uno-due anni di follow-up, la dose può essere ridotta per consentire al TSH di tornare nel range di normalità.
- Per i pazienti che inizialmente presentavano malattia ad alto rischio ma che hanno eccellente risposta clinica alla terapia, è accettabile un obiettivo di TSH da 0.1 a 0.5 mU/L fino a cinque anni, dopodiché il grado di soppressione può essere ulteriormente allentato (con sorveglianza continua per il rischio di recidiva).
- Nei pazienti in remissione completa, mantenere i livelli di TSH nel range della norma.
5. RADIOTERAPIA ESTERNA
Ha scarsa indicazione nel trattamento iniziale dei DTC (1,28). Può essere utilizzata come trattamento adiuvante per ridurre/rallentare la recidiva di malattia in pazienti con neoplasie localmente avanzate (pT4). Può essere utile in particolare per i pazienti più anziani con estensione extra-tiroidea macroscopica al momento dell’intervento chirurgico o in pazienti più giovani, selezionati, con malattia estesa e caratteristiche istologiche sfavorevoli (ad esempio, istologia insulare o scarsamente differenziata), la cui malattia viene resecata ma in cui esiste alta probabilità di malattia microscopica residua (29).
La radioterapia esterna è un trattamento palliativo efficace per le metastasi a distanza (prevalentemente cerebrali o scheletriche, soprattutto se iperalgiche), non controllabili dal solo trattamento con radioiodio (30,31).
6. FOLLOW-UP
Anche se la maggioranza delle recidive dei DTC ha luogo entro 5 anni dal trattamento iniziale (32), il follow-up deve essere esteso per tutta la vita, perché si possono verificare recidive anche alcune decadi dopo la diagnosi della neoplasia.
Elementi essenziali del follow-up sono la determinazione della Tg e l’ecografia del collo (33,34). La scintigrafia whole-body è importante nella stadiazione post-dose ablativa, ma ha un ruolo limitato nel follow-up a lungo termine: deve essere impiegata nei soli casi ad alto rischio o con sospetta recidiva di malattia (Tg sierica in incremento), in assenza di lesioni cervicali dimostrabili all’esame ecografico (1).
Gli accertamenti da eseguire nel corso del follow-up dipendono dalla risposta clinica iniziale al trattamento e dalla classe di rischio.
Follow-up a breve termine dopo trattamento iniziale (chirurgia e radioiodio)
In tutti i pazienti, nel primo anno dopo il trattamento iniziale (che sia lobectomia o tiroidectomia, con o senza terapia ablativa con 131I) misurare dopo 3 mesi TSH, FT4, Tg e Ab anti-Tg. L’obiettivo di TSH in questa fase andrà modulato sulla base della classe di rischio del paziente (cfr sopra). Successivamente la frequenza del monitoraggio della Tg dipenderà dalla risposta alla terapia e il dosaggio dovrebbe essere eseguito usando sempre lo stesso metodo (preferibile un metodo con sensibilità funzionale di 0.05-0.1 ng/mL). Nei pazienti con presenza di Ab anti-Tg, le concentrazioni sieriche di Tg da sole non possono essere utilizzate come marcatore per rilevare la persistenza o la recidiva di malattia. Con i nuovi e più sensibili test ultrasensibili di Tg (sensibilità funzionale < 0.1 ng/mL), le concentrazioni sieriche di Tg (misurate durante la terapia TSH-soppressiva con LT4) sono correlate alle concentrazioni di Tg stimolate da TSH ricombinante (rhTSH) e, pertanto, possono rendere superflua la necessità di misurazioni stimolate da rhTSH (35). L'interpretazione dei livelli sierici di Tg dipende dalla terapia iniziale e dalla presenza di Ab anti-Tg.
- Tiroidectomia + ablazione con 131I: in questi pazienti la risposta viene considerata ottimale se Tg non stimolata < 0.2 ng/mL (o stimolata con rhTSH < 1 ng/mL).
- Tiroidectomia senza ablazione con 131I: l'interpretazione della Tg dipende dalle dimensioni del residuo tiroideo. Molti pazienti hanno livelli basali di Tg non rilevabili (< 0.2 ng/mL), mentre valori in aumento nel tempo sono sospetti per la crescita del tessuto tiroideo o per il cancro.
- Lobectomia: sebbene non siano stati definiti criteri specifici per distinguere il tessuto tiroideo residuo normale dal cancro tiroideo persistente o recidivante, la maggior parte dei pazienti con risposta eccellente dovrebbe avere un livello di Tg sierica < 30 ng/mL e molti pazienti hanno livelli di Tg compresi tra 2 e 10 ng/mL.
- Presenza di anticorpi anti-Tg elevati: i valori di Tg sierica non sono affidabili in presenza di aumento del titolo di Ab anti-Tg, che può riscontrarsi nel 25% dei pazienti affetti da DTC (1). È opportuno procrastinare di alcuni mesi la ristadiazione prevista a 6-12 mesi, in attesa di una loro possibile normalizzazione. Le variazioni della concentrazione degli Ab anti-Tg possono essere usate come marcatore surrogato di malattia, posto che gli Ab siano determinati costantemente con lo stesso metodo. Generalmente, nei pazienti in remissione di malattia la negativizzazione degli Ab anti-Tg si verifica dopo una mediana di circa tre anni. La ricomparsa degli Ab anti-Tg dopo precedente negativizzazione o l’aumento del titolo anticorpale può essere indicativo di recidiva e/o persistenza di malattia.
- Imaging: nel primo anno dopo il trattamento del DTC (lobectomia, tiroidectomia con o senza ablazione con iodio radioattivo) si deve eseguire ecografia del collo, in genere a intervalli di 6-12 mesi a seconda della valutazione del rischio. Successivamente, la frequenza dell'US dipende dalla risposta alla terapia. La recidiva tumorale nel collo e la presenza di metastasi linfonodali può essere rilevata dall'esame clinico o dall'aumento delle concentrazioni sieriche di Tg, ma l'US è la tecnica più sensibile per la localizzazione (1,36). L’impiego della scintigrafia diagnostica con 131I non è indicata di routine e trova possibile applicazione solo in casi selezionati (1,20,24).
Follow-up a lungo termine
I controlli successivi dovranno essere previsti basandosi sulla risposta al trattamento iniziale e sui risultati dei controlli successivi, ridefinendo in maniera dinamica lo stato di malattia.
- Pazienti a basso rischio ATA con risposta eccellente alla terapia (Tg on L-T4 indosabile, Tg dopo stimolo < 1 ng/mL e US cervicale negativa per persistenza di malattia nel controllo 6-12 mesi dopo la terapia iniziale): controllo clinico ogni 12-18 mesi, con determinazione della Tg on L-T4 e degli Ab anti-Tg e con l’US del collo.
- I pazienti con risposta indeterminata hanno una minima possibilità di evoluzione verso una persistenza di malattia, per cui non necessitano di controlli frequenti e il trattamento con LT4 dovrà essere di tipo semi-soppressivo.
- I pazienti con risposta incompleta biochimica (in genere 10-20% in tutte le categorie di rischio) dovrebbero eseguire periodiche misurazioni di Tg e Ab anti-Tg on L-T4 e US del collo ogni 6-12 mesi. Il progressivo incremento dei valori di Tg con tempo di raddoppio < 12 mesi indica progressione rapida della malattia e richiede esami morfologici per valutare la presenza di malattia strutturale. Il test di stimolo della Tg con rhTSH può essere utile solo per ri-stadiare i pazienti, dopo terapie aggiuntive. In questi pazienti viene raccomandata la soppressione del TSH moderata (0.1-0.5 mIU/mL) o completa (< 0.1 mIU/mL), da individualizzare in accordo ai livelli di Tg e Ab anti-Tg e al loro trend di incremento.
- I pazienti con risposta strutturale incompleta necessitano di stretto follow-up, con esecuzione periodica di esami morfologici per valutare lo stato di malattia e il bisogno di un appropriato trattamento terapeutico. Il programma individuale di follow-up dovrebbe essere pianificato in base al diametro e al sito delle metastasi, alla captazione con 131I e in base all’intensità di captazione alla FDG-PET. Il TSH dovrebbe essere mantenuto indosabile (< 0.1 mU/L) (1).
Per la gestione del paziente con tumore a basso rischio
BIBLIOGRAFIA
- Haugen BR, Alexander EK, Bible KC, et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: the American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26: 1-133.
- Boucai L, Zafereo M, Cabanillas ME. Thyroid cancer: a review. JAMA 2024, 331: 425-3
- Yamazaki H, Sugino K, Katoh R, et al. Management of follicular thyroid carcinoma. Eur Thyroid J 2024, 13: e240146.
- Li M, Dal Maso L, Pizzato M, Vaccarella S. Evolving epidemiological patterns of thyroid cancer and estimates of overdiagnosis in 2013-17 in 63 countries worldwide: a population-based study. Lancet Diabetes Endocrinol 2024, 12: 824-36.
- Seib CD, Sosa JA. Evolving understanding of the epidemiology of thyroid cancer. Endocrinol Metab Clin North Am 2019, 48: 23-35.
- Vaccarella S, Franceschi S, Bray F, et al. Worldwide thyroid-cancer epidemic? The increasing impact of overdiagnosis. N Engl J Med 2016, 375: 614-7.
- Kitahara CM, Schneider AB. Epidemiology of thyroid cancer. Cancer Epidemiol Biomarkers Prev 2022, 31: 1284-97.
- Zhao H, Li H. Meta-analysis of ultrasound for cervical lymph nodes in papillary thyroid cancer: diagnosis of central and lateral compartment nodal metastases. Eur J Radiol 2019, 112: 14-21.
- Yeh MW, Bauer AJ, Bernet VA, et al. American Thyroid Association statement on preoperative imaging for thyroid cancer surgery. Thyroid 2015, 25: 3-14.
- Cho SJ, Suh CH, Baek JH, et al. Active surveillance for small papillary thyroid cancer: a systematic review and meta-analysis. Thyroid 2019, 29: 1399-1408.
- Barney BM, Hitchcock YJ, Sharma P, et al. Overall and cause-specific survival for patients undergoing lobectomy, near-total, or total thyroidectomy for differentiated thyroid cancer. Head Neck 2011, 33: 645-9.
- Abelleira E, Jerkovich F. Dynamic risk assessment in patients with differentiated thyroid cancer. Rev Endocr Metab Disord 2024, 25: 79-93.
- Mendelsohn AH, Elashoff DA, Abemayor E, St John MA. Surgery for papillary thyroid carcinoma. Is lobectomy enough? Arch Otolaryngol Head Neck Surg 2010, 136: 1055-61.
- Adam MA, Pura J, Gu L, et al. Extent of surgery for papillary thyroid cancer is not associated with survival. An analysis of 61,775 patients. Ann Surg 2014, 260: 601-5; discussion 605-7.
- Chen L, Wu YH, Lee CH, et al. Prophylactic central neck dissection for papillary thyroid carcinoma with clinically uninvolved central neck lymph nodes: a systematic review and meta-analysis. World J Surg 2018, 42: 2846-57.
- Eskander A, Merdad M, Freeman JL, Witterick IJ. Pattern of spread to the lateral neck in metastatic well-differentiated thyroid cancer: a systematic review and meta-analysis. Thyroid 2013, 23: 583-92.
- Lombardi D, Paderno A, Giordano D, et al. Therapeutic lateral neck dissection in well-differentiated thyroid cancer: analysis on factors predicting distribution of positive nodes and prognosis. Head Neck 2018, 40: 242-50.
- Avenia N, Vannucci J, Monacelli M, et al. Locally invasive thyroid cancer: options for a treatment. Updates Surg 2017, 69: 249-53.
- Russell MD, Kamani D, Randolph GW. Modern surgery for advanced thyroid cancer: a tailored approach. Gland Surg 2020, 9 suppl 2: S105-19.
- Filetti S, Durante C, Hartl D, et al. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2019, 30: 1856-83.
- Amin MB, Greene FL, Edge SB, et al. The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA Cancer J Clin 2017, 67: 93-9.
- Mitchell AL, Gandhi A, Scott-Coombes D, Perros P. Management of thyroid cancer: United Kingdom national multidisciplinary guidelines. J Laryngol Otol 2016, 130: S150-60.
- Silberstein EB, Alavi A, Balon HR, et al. The SNMMI practice guideline for therapy of thyroid disease with 131I 3.0. J Nucl Med 2012, 53: 1633-51.
- Pacini F, Basolo F, Bellantone R, et al. Italian consensus on diagnosis and treatment of differentiated thyroid cancer: joint statements of six Italian societies. J Endocrinol Invest 2018, 41: 849-76.
- Ruel E, Thomas S, Dinan M, et al. Adjuvant radioactive iodine therapy is associated with improved survival for patients with intermediate-risk papillary thyroid cancer. J Clin Endocrinol Metab 2015, 100: 1529-36.
- Gubbi S, Al-Jundi M, Foerster P, et al. The effect of thyrotropin suppression on survival outcomes in patients with differentiated thyroid cancer: a systematic review and meta-analysis. Thyroid 2024, 34: 674-86.
- Biondi B, Cooper DS. Benefits of thyrotropin suppression versus the risks of adverse effects in differentiated thyroid cancer. Thyroid 2010, 20: 135-46.
- Kiess AP, Agrawal N, Brierley JD, et al. External-beam radiotherapy for differentiated thyroid cancer locoregional control: a statement of the American Head and Neck Society. Head Neck 2016, 38: 493-8.
- Fussey JM, Crunkhorn R, Tedla M, et al. External beam radiotherapy in differentiated thyroid carcinoma: a systematic review. Head Neck 2016, 38 suppl 1: E2297-305.
- Blomain E, Berta S, Hug N, et al. Radiotherapy for brain metastases from thyroid cancer: an institutional and national retrospective cohort study. Thyroid 2022, 32: 781-8.
- Dudzinski SO, Cabanillas ME, Hamidi S, et al. Definitive radiotherapy for oligometastatic and oligoprogressive thyroid cancer: a potential strategy for systemic therapy deferral. J Natl Compr Canc Netw 2024, 23: e247072.
- Durante C, Costante G, Filetti S. Differentiated thyroid carcinoma: defining new paradigms for postoperative management. Endocr Relat Cancer 2013, 20: R141-54.
- Leenhardt L, Erdogan MF, Hegedus L, et al. 2013 European Thyroid Association guidelines for cervical ultrasound scan and ultrasound-guided techniques in the postoperative management of patients with thyroid cancer. Eur Thyroid J 2013, 2: 147-59.
- Miyauchi A, Kudo T, Miya A, et al. Prognostic impact of serum thyroglobulin doubling-time under thyrotropin suppression in patients with papillary thyroid carcinoma who underwent total thyroidectomy. Thyroid 2011, 21: 707-16.
- Castagna MG, Tala Jury HP, Cipri C, et al. The use of ultrasensitive thyroglobulin assays reduces but does not abolish the need for TSH stimulation in patients with differentiated thyroid carcinoma. J Endocrinol Invest 2011, 34: e219-23.
- Torlontano M, Crocetti U, Augello G, et al. Comparative evaluation of recombinant human thyrotropin-stimulated thyroglobulin levels, 131I whole-body scintigraphy, and neck ultrasonography in the follow-up of patients with papillary thyroid microcarcinoma who have not undergone radioiodine therapy. J Clin Endocrinol Metab 2006, 91: 60-3.
Linee Guida ATA sul carcinoma tiroideo differenziato: focus nei pazienti con DTC a basso rischio (ATA GUIDELINES 2025 UPDATE: Low-Risk DTC patients)
Marco Capezzone (Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.)
UOSD Endocrinologia, Ospedale Misericordia di Grosseto, USL Toscana sud-est Grosseto
(aggiornato al maggio 2026)
Le LG ATA 2025 per il carcinoma differenziato della tiroide (DTC) introducono un cambiamento sostanziale nella gestione diagnostico-terapeutica rispetto al documento ATA del 2015. Una delle novità più importanti riguarda la strategia terapeutica da adottare nei pazienti a basso rischio e la de-escalation del loro monitoraggio a lungo termine.
Da sottolineare tra le novità c’è il referto istopatologico, che deve indicare l’istotipo, la classificazione TNM, lo stato del margine di resezione, la presenza di invasione vascolare (compreso il numero di vasi interessati), il numero di linfonodi esaminati insieme al numero dei linfonodi positivi, le dimensioni della metastasi linfonodale maggiore e la presenza o assenza di estensione extra-nodale. Il patologo deve riportare inoltre la presenza dei sottotipi di PTC (carcinoma papillare della tiroide), di FTC/IEFVPTC (carcinoma follicolare/variante follicolare incapsulata invasiva del carcinoma papillare tiroideo) e OTC (carcinoma oncocitico tiroideo) a maggiore o minore aggressività, così come degli istotipi più frequentemente associati a sindromi familiari. Un referto istopatologico accurato è di importanza critica per guidare terapia e follow-up.
La categoria BASSO RISCHIO viene definita:
- nel PTC e sottotipi: tumore T1 e T2 (≤ 4 cm), uni-focale, pN0a, o cN0 e pN1a (≤ 5 LN+, tutti ≤ 2 mm), margini negativi o solo microscopicamente interessati + margine anteriore (R1);
- nei FTC/IEFVPTC: tumore T1 e T2 (≤ 4 cm), minimamente invasivo (solo invasione capsulare), pN0a, o cN0 e pN1a (≤ 5 LN+, tutti ≤ 2 mm; è comunque infrequente l’interessamento linfonodale), margini negativi o solo microscopicamente interessati + margine anteriore (R1);
- negli OTC: tumore T1 e T2 (≤ 4 cm), minimamente invasivo (solo invasione capsulare), pN0a, o cN0 e pN1a (≤ 5 LN+, tutti ≤ 2 mm; è comunque infrequente l’interessamento linfonodale), margini negativi o solo microscopicamente interessati + margine anteriore (R1).
La raccomandazione #11 riporta che la Sorveglianza Attiva può essere offerta come opzione di gestione appropriata per alcuni pazienti con PTC cT1aN0M0. Per i pazienti sottoposti a sorveglianza attiva, dovrebbe essere utilizzata l'ecografia del collo per monitorare la progressione di malattia (raccomandazione #12). Per i pazienti sottoposti a sorveglianza attiva, non è raccomandata la misurazione di routine dei livelli sierici di Tg e/o TgAb (raccomandazione #13).
In questi pazienti è indicata la resezione chirurgica in presenza di nuove metastasi linfonodali confermate da biopsia, crescita del tumore primario di almeno 3 mm, metastasi a distanza, evidenza di estensione extra-tiroidea, crescita posteriore, ansia del paziente, impossibilità di effettuare i controlli di follow-up e/o preferenza espressa per l'intervento chirurgico.
Per PTC cT1aN0M0, come alternativa alla sorveglianza attiva o alla resezione in pazienti selezionati, può essere considerata anche l’ablazione percutanea eco-guidata.
In entrambe queste opzioni non chirurgiche, è essenziale una decisione clinica condivisa tra il paziente e l'équipe medica in merito ai rischi e ai benefici di questo approccio.
Secondo la raccomandazione #15, quando si esegue la resezione per pazienti con carcinoma tiroideo di dimensioni < 2 cm senza estensione extra-tiroidea macroscopica (cT1) e senza metastasi (cN0M0), la procedura chirurgica iniziale dovrebbe essere la lobectomia tiroidea, a meno che non vi siano tumori bilaterali o altre indicazioni per rimuovere il lobo controlaterale. Per i pazienti con carcinoma tiroideo unilaterale a basso rischio > 2 e < 4 cm (cT2N0M0), la lobectomia tiroidea può essere il trattamento chirurgico iniziale preferibile, per il rischio e gli effetti collaterali significativamente inferiori.
Secondo la raccomandazione #19, per la maggior parte dei PTC piccoli, non invasivi, clinicamente negativi per metastasi linfonodali (cT1-T2, cN0) e per la maggior parte dei FTC non deve essere eseguita la dissezione profilattica dei linfonodi del compartimento centrale.
La raccomandazione #28 riporta che per determinare il rischio di persistenza/recidiva (localmente e/o a distanza) della malattia strutturale e/o la sopravvivenza nei pazienti con DTC, è raccomandato il sistema di stratificazione del rischio ATA 2025, che valuta le caratteristiche istopatologiche del tumore e il numero di linfonodi cervicali in combinazione con il sistema di stadiazione AJCC, le immagini post-operatorie e le misurazioni dei livelli sierici di Tg e TgAb (se appropriato).
La raccomandazione #30 suggerisce di misurare il livello sierico post-operatorio di Tg 6-12 settimane dopo la tiroidectomia totale, durante la terapia con ormoni tiroidei o dopo stimolazione del TSH. Tali misurazioni possono essere utili per ulteriori decisioni cliniche.
La raccomandazione #32 stabilisce che per il DTC a basso rischio NON è raccomandata di routine la terapia con radioiodio.
Secondo la raccomandazione #46, per i pazienti con malattia a rischio basso o intermedio, che non presentano evidenza di recidiva biochimica o strutturale, non è raccomandata la soppressione a lungo termine del TSH (i livelli di TSH vanno mantenuti nel range di normalità).
Per quanto riguarda il follow-up dei pazienti a basso rischio, le nuove LG ATA introducono una de-escalation del monitoraggio a lungo termine (raccomandazione #48):
- nei pazienti trattati con tiroidectomia totale e terapia con iodio radioattivo (RAI), che presentano una risposta eccellente, sostenuta per 5-8 anni dopo la terapia iniziale, può essere interrotta l'ecografia di routine e i pazienti possono essere seguiti successivamente con soli marcatori biochimici ogni 1-2 anni. Se poi presentano una risposta eccellente sostenuta per 10-15 anni, non necessitano più di monitoraggio biochimico di routine continuativo per il carcinoma tiroideo e dovrebbero essere considerati in remissione completa;
- i pazienti con DTC a basso rischio trattati con sola tiroidectomia totale che mantengono una risposta eccellente per 10-15 anni, non necessitano di monitoraggio biochimico di routine continuativo per il carcinoma tiroideo e dovrebbero essere considerati in remissione completa;
- nei pazienti con DTC a basso rischio trattati con lobectomia, in cui l'ecografia post-operatoria iniziale è negativa, le ecografie successive devono essere eseguite ogni 1-3 anni per 5-8 anni dopo la terapia iniziale. I noduli nel lobo residuo devono essere monitorati secondo le LG ATA sulla tiroide;
- nei pazienti con DTC a basso rischio trattati con lobectomia, se la Tg post-operatoria non è marcatamente elevata, non sono raccomandati di routine ulteriori dosaggi di Tg;
- i pazienti sottoposti a lobectomia o tiroidectomia totale senza successiva RAI non devono essere sottoposti a WBS con RAI nella sorveglianza;
- i pazienti con DTC a rischio di recidiva basso e basso-intermedio con risposta eccellente alla terapia non richiedono di routine il WBS diagnostico con RAI durante il follow-up.
| Raccomandazioni di de-escalation nei pazienti a basso rischio con risposta eccellente | |||
| Trattamento | Tireoglobulina non stimolata | Livelli di TSH da mantenere | Frequenza ecografia collo |
| Emitiroidectomia | 1 volta nel post-operatorio | Normali | Ogni 1-3 anni per 5-8 anni. |
| Tiroidectomia totale senza I-131 | Tg < 2.5 ng/mL e AbTg negativi | Normali | Ogni 1-3 anni per 5-8 anni, quindi interrompere, a meno che il livello di Tg non aumenti o gli AbTg non diventino rilevabili per la prima volta. |
| Tiroidectomia totale con I-131 | Tg < 0.2 ng/ml e AbTg negativi | Normali | |
Bibliografia
- Ringel MD, Sosa JA, Baloch Z, et al. 2025 American Thyroid Association management guidelines for adult patients with differentiated thyroid cancer. Thyroid 2025, 35: 841-985.
- LG ATA 2025 per carcinoma tiroideo differenziato: cosa cambia nella pratica clinica? AME Breaking News 20/2 – novembre 2025.
Introduzione alla classificazione della citologia tiroidea
Enrico Papini1 (Questo indirizzo email è protetto dagli spambots. È necessario abilitare JavaScript per vederlo.), Andrea Frasoldati2, Anna Crescenzi3 & Francesco Nardi4
1Endocrinologia & 3Patologia, Ospedale Regina Apostolorum, Albano Laziale (RM); 2Endocrinologia, Arcispedale S. Maria Nuova IRCCS, Reggio Emilia; 4Università di Roma Sapienza, Roma
L’agoaspirato con ago sottile (FNA) è lo strumento diagnostico più efficace per la valutazione della natura della patologia nodulare tiroidea e consente di selezionare i pazienti da seguire preferibilmente in modo conservativo rispetto a quelli da avviare alla exeresi chirurgica. L’impiego di una terminologia condivisa per l’inquadramento del nodulo in categorie diagnostiche a rischio crescente di malignità consente, considerati anche tutti gli altri criteri clinici e strumentali, di orientare nel modo più appropriato la gestione del paziente (1).
La nuova classificazione italiana della citologia tiroidea è stata ufficialmente pubblicata nel maggio 2014 sul Journal of Endocrinological Investigation (1). Il documento, messo a punto da un gruppo di esperti su mandato delle Società Italiane di Endocrinologia (AIT, AME e SIE) e di Anatomia Patologica e Citologia (SIAPEC-IAP), aggiorna la precedente classificazione del 2007 in base ai dati della letteratura, rendendola confrontabile con le altre classificazioni più utilizzate, quella americana, nota come “Bethesda” (2,3), e quella inglese, del Royal College of Pathologists of United Kingdom (UKRCP) (4), e fornisce a endocrinologi e citopatologi uno strumento di immediata utilità per la pratica clinica.
La tabella 1 riporta il confronto tra la classificazione italiana del 2014 e la precedente del 2007: è mantenuto lo schema con cinque categorie, associate al relativo atteso rischio di malignità e al suggerimento di un’azione clinica, ma sono presenti importanti modificazioni nelle categorie TIR 1 e TIR 3.
CONFRONTO FRA VECCHIA E NUOVA CLASSIFICAZIONE
Nella TIR 1 (“non diagnostico”) sono introdotte tre modifiche:
- in presenza di atipie citologiche significative, il campione viene incluso in una categoria di sospetto, indipendentemente dal numero delle cellule che lo compongono, come nelle classificazioni americana e inglese (2-4);
- i campioni ottenuti da lesioni cistiche nei quali non si raggiungono i limiti minimi di adeguatezza cellulare sono classificati come TIR 1C (cistico). Sono escluse le cisti colloidee, che sono incluse nella categoria TIR2;
- in caso di agoaspirati (FNA) ripetutamente non diagnostici, può essere eseguita una biopsia eco-guidata (‘core needle biopsy’) con ago 20-22 G per esame micro-istologico. La procedura si è dimostrata efficace nel ridurre il numero di casi “inadeguati” (5).
La TIR 3 (“indeterminato/proliferazione follicolare”) prevedeva nella classificazione 2007 l'intervento chirurgico come suggerimento terapeutico. È stata ora suddivisa in due sottoclassi:
- TIR 3A, che prevede follow-up con ripetizione di FNA;
- TIR 3B, che ha come opzione prioritaria l'exeresi chirurgica.
Questa suddivisione (presente, con terminologia diversa, anche nelle classificazioni americana e inglese) ha lo scopo di ridurre il numero di pazienti inclusi nella categoria TIR 3 sottoposti a intervento chirurgico per patologia benigna.
Sono considerati “neoplasia follicolare” (TIR 3B) solo i campioni con elevata cellularità, scarsa o assente colloide e assoluta prevalenza di strutture microfollicolari/trabecolari.
Nei casi in cui, pur in presenza di elevata cellularità e scarsa colloide, non vi è un'assoluta prevalenza di strutture microfollicolari/trabecolari, la lesione è inclusa nella sottocategoria TIR 3A, con rischio atteso di malignità < 10% e suggerimento clinico conservativo.
Sono inclusi nella sottocategoria TIR 3B anche i campioni con alterazioni nucleari suggestive di carcinoma papillifero, troppo lievi o focali per includerli nella categoria TIR 4. Nelle classificazioni Bethesda (2,3) e UKRCP (4) queste lesioni sono incluse in categorie a rischio più basso che prevedono il follow-up con ripetizione della FNA. Casistiche recenti, tuttavia, riportano in queste lesioni un rischio di malignità > 25% (6), per cui questo gruppo è stato da noi incluso nella sottocategoria TIR 3B.
Non vi sono differenze sostanziali nelle altre categorie rispetto alla classificazione del 2007.
| Tabella 1 Confronto tra classificazione del 2007 e del 2014 |
|
| SIAPEC 2007 | SIAPEC-AIT-AME-SIE 2014 |
| Tir 1: non diagnostico |
TIR1: non diagnostico
|
| Tir 2: negatico per cellule maligne |
TIR 2: non maligno/benigno |
|
Tir 3: indeterminato (proliferazione follicolare |
TIR 3: indeterminato
|
|
Tir 4: sospetto di malignità |
TIR 4:sospetto di malignità |
| Tir 5: positivo per cellule maligne |
TIR 5: maligno |
CATEGORIE DI REFERTAZIONE CITOLOGICA 2014
Sono raccomandate cinque categorie diagnostiche principali. La tabella 2 sintetizza le categorie diagnostiche, il rischio di malignità atteso e le azioni cliniche suggerite.
TIR 1 (non diagnostica)
Comprende prelievi “inadeguati” (per contaminazione ematica o per artefatti) e “non rappresentativi” (assenza di almeno 6 gruppi di 10 tireociti ciascuno). In presenza di atipie citologiche significative, il campione viene incluso in una categoria di sospetto indipendentemente dal numero di cellule.
Se nei prelievi da lesioni cistiche sono presenti solo eritrociti, detriti e macrofagi con emosiderina, in assenza di colloide, la lesione è sottoclassificata come TIR 1C (cistica).
Azioni cliniche:
- TIR 1: non consente una diagnosi ed è quindi raccomandata la ripetizione di FNA eco-guidata, di regola dopo almeno un mese dalla precedente. In caso di FNA ripetutamente non diagnostiche, può essere eseguita una core-needle biopsy per esame micro-istologico;
- TIR 1C: in assenza di reperti clinici ed ecografici di sospetto, queste lesioni sono in genere benigne. In caso di sospetto clinico, ripetere la FNA per escludere un carcinoma papillifero cistico.
TIR 2 (non maligna/benigna)
Comprende la citologia da noduli colloidi o iperplastici, da tiroiditi autoimmuni o granulomatose e da altre, meno frequenti, condizioni non neoplastiche.
Azioni cliniche: follow-up clinico ed ecografico. Ripetere la FNA solo in caso di crescita o modificazioni ecografiche del nodulo e nei pazienti avviati a trattamenti ablativi non chirurgici.
TIR 3 (citologia indeterminata)
La maggioranza di questi casi è rappresentata da quadri microfollicolari, che corrispondono, sul piano istologico, a noduli adenomatosi iperplastici, adenomi o carcinomi follicolari o varianti follicolari del carcinoma papillifero. Sono distinte due sottoclassi a diverso rischio di malignità, caratterizzate in modo leggermente diverso rispetto al Bethesda System, per una prevista migliore definizione della probabilità di malignità (3):
- TIR 3A (lesione indeterminata a basso rischio di malignità): caratterizzata da accentuata cellularità, colloide scarsa e strutture microfollicolari numerose ma non sufficienti a far porre diagnosi di “neoplasia follicolare”. Sono inclusi in questa categoria anche strisci con alterazioni citologiche o architetturali, ma gravati da artefatti che ne limitano l’interpretazione;
- TIR 3B (lesione indeterminata a elevato rischio di malignità): elevata cellularità a disposizione monotona in strutture microfollicolari/trabecolari, con colloide scarsa/assente (quadro suggestivo di “neoplasia follicolare”). Questa categoria include anche campioni con alterazioni nucleari lievemente sospette per carcinoma papillifero.
Azioni cliniche:
- TIR 3A: stretto follow-up clinico ed ecografico. È raccomandata la ripetizione di FNA nel corso dei controlli successivi;
- TIR 3B: l’opzione prioritaria è l’exeresi chirurgica.
TIR 4 (sospetta per malignità)
Comprende campioni in cui la malignità è fortemente sospetta ma non certa. La maggioranza di questi casi corrisponde a un carcinoma papillifero, ma altre neoplasie sono occasionalmente incluse in questa categoria.
Azioni cliniche: exeresi chirurgica con eventuale esame intra-operatorio. Ripetizione di FNA nei soli casi pauci-cellulari o che richiedano una caratterizzazione pre-operatoria addizionale.
TIR 5 (maligna)
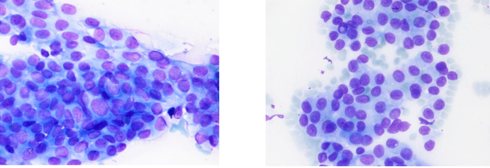
Comprende campioni con diagnosi citologica conclusiva di neoplasia maligna (papillifera, midollare, scarsamente differenziata, anaplastica, linfoma, tumore non epiteliale o metastatico). Ove possibile, deve essere formulata la diagnosi del tipo di tumore.
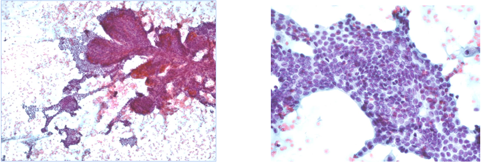
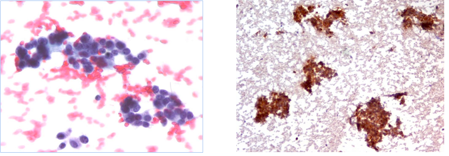
Azioni cliniche: resezione chirurgica con estensione basata su reperto citologico e quadro clinico. In caso di carcinoma anaplastico, linfoma o tumore metastatico, sono necessarie ulteriori procedure diagnostiche per definire la condotta terapeutica più appropriata.
| Tabella 2 Consenso Italiano 2014 |
|||
| Codice | Categoria diagnostica |
Rischio atteso di malignità (%) |
Azioni suggerite |
| TIR 1 |
Non diagnostico |
Non definito | Ripeti FNA eco-guidata dopo almeno 1 mese |
| TIR 1C | Non diagnostico-cistico |
Basso (variabile in base al quadro clinico) |
Valutare nel contesto clinico, eventualmente ripetere l'FNA |
| TIR 2 | Non maligno/benigno | < 3 |
Follow-up |
| TIR 3A |
Lesione indeterminata a basso rischio |
< 10 | Ripeti FNA/follow-up |
| TIR 3B |
Lesione indeterminata ad alto rischio |
15-30 | Exeresi chirurgica |
| TIR 4 | Sospetto per malignità | 60-80 |
Exeresi chirurgica con eventuale esame intraoperatorio |
| TIR 5 | Maligno | > 95 |
Exeresi chirurgica |
BIBLIOGRAFIA Essenziale
- Nardi F, Basolo F, Crescenzi A, et al. Italian Consensus for the classification and reporting of thyroid cytology. J Endocrinol Invest 2014, 37: 593-9.
- Baloch ZW, Livolsi VA, Asa SL, et al. Diagnostic terminology and morphologic criteria for cytologic diagnosis of thyroid lesions: a synopsis of the National Cancer Institute Thyroid Fine-Needle Aspiration State of the Science Conference. Diagn Cytopathol 2008, 36: 425-37.
- Ali SZ, Cibas ES. The Bethesda system for reporting thyroid cytopathology: definitions, criteria and explanatory notes. Springer, New York 2010.
- Cross PA, Chandra A, Giles T, et al. Guidance on the reporting of thyroid cytology specimens. 2009.
- Samir AE, Vij A, Seale MK, et al. Ultrasound-guided percutaneous thyroid nodule core biopsy: clinical utility in patients with prior non diagnostic fine-needle aspirate. Thyroid 2012, 22: 461-7.
- Renshaw AA. Should ”atypical follicular cells” in thyroid fine-needle aspirates be subclassified? Cancer Cytopathol 2010, 118: 186-9.
AGGIORNAMENTO 2026
a cura di Vincenzo Triggiani
Endocrinologia e Malattie Metaboliche, Dipartimento Interdisciplinare di Medicina, Università degli Studi di Bari “Aldo Moro”
Il Consenso Italiano per la Classificazione e la Refertazione della Citologia Tiroidea (ICCRTC), aggiornato al 2025, è stato elaborato come strumento per l'interpretazione e la comunicazione delle diagnosi citologiche dei noduli tiroidei e come supporto per le decisioni cliniche basate sui risultati morfologici. Questo documento costituisce l'edizione aggiornata del consenso pubblicato nel 2014.
Obiettivi principali:
- standardizzare la terminologia citologica per i risultati dell’ago-aspirazione tiroidea (FNA);
- fornire criteri morfologici chiari per l'inclusione in ciascuna categoria diagnostica;
- correlare i risultati citologici con la gestione clinica suggerita per i pazienti;
- migliorare la comunicazione tra gli specialisti coinvolti (patologi, endocrinologi, chirurghi).
Il consenso 2025 include gli avanzamenti scientifici recenti nel campo, come l'espansione delle tecniche di biologia molecolare, gli aggiornamenti della classificazione WHO 2022 (inclusa la ridefinizione di oncociti al posto di cellule di Hürthle) e l'inclusione di un ricco atlante citologico.
Il sistema ICCRTC mantiene cinque categorie principali, con due sottoclassi per la categoria indeterminata (TIR3A/TIR3B).
| Categoria | Definizione | ROM atteso* (%) | Azione clinica principale |
| TIR1 | Non-diagnostico: campioni inadeguati (quantitativamente o qualitativamente) o non rappresentativi. | 11.2% (stimato) | Ripetizione FNA. CNB se persistente inadeguatezza per noduli solidi. |
| TIR2 | Benigno/non-maligno: materiale adeguato e ben conservato, privo di atipie significative. | 3.8% (stimato) | Follow-up clinico ed ecografico periodico (1-3 anni). Ripetizione FNA solo per crescita significativa o successivo riscontro di caratteri ecografici sospetti. |
| TIR3A | Lesione indeterminata, probabilmente benigna: alterazioni architetturali e/o nucleari minime. | 12.4% | Ripetizione FNA. Possibile impiego delle indagini molecolari/immuno-citochimiche. Follow-up attivo per noduli a basso rischio ecografico. |
| TIR3B | Lesione indeterminata a rischio intermedio di malignità: atipia architetturale e/o nucleare più marcata della TIR3A. | 44.4% | Escissione chirurgica (prima scelta). Test molecolari da prendere in considerazione per affinare la stratificazione del rischio. |
| TIR4 | Sospetto per malignità: forte sospetto, ma diagnosi definitiva non stabilita (per scarsa cellularità, atipie incomplete). | 92.5% | Escissione chirurgica (prima scelta). Discussione multi-disciplinare. |
| TIR5 | Maligno: cellule con caratteristiche maligne distintive; spesso possibile diagnosi specifica di istotipo. | 99.7% | Trattamento chirurgico (prima linea). Sorveglianza attiva o termo-ablazione per piccole lesioni selezionate. |
| *I valori ROM (rischio di malignità) riportati derivano da meta-analisi su dati clinici reali. | |||
Note specifiche
- Categoria TIR1C (cistica): campioni fluidi da lesioni puramente cistiche. Vengono gestiti come cisti non sospette, a meno che non siano presenti fattori di rischio ecografico.
- Suddivisione della categoria TIR3: la distinzione in TIR3A/TIR3B è cruciale. L'ICCRTC posiziona l'atipia nucleare potenzialmente associata a carcinoma papillare nella categoria a rischio più alto (TIR3B), distinguendosi così dai sistemi The Bethesda System for Reporting Thyroid Cytopathology (TBSRTC 2023) e del UK Royal College of Pathologists (RCPath 2024) che tendono a collocare tale atipia nella categoria a rischio inferiore. Questo approccio si è dimostrato efficace nel separare il ROM, con un rischio di cancro molto più elevato nel TIR3B (44.4%) rispetto al TIR3A (12.4%).
Tecniche ausiliarie
L'uso di tecniche ancillari (immuno-citochimica e test molecolare) può essere importante specialmente per le categorie indeterminate (TIR3A e TIR3B) o per i tumori avanzati.
A. Immunocitochimica (ICC)
L'ICC è utilizzata per raffinare la diagnosi differenziale:
- marcatori tiroidei (tireoglobulina, TTF1 e PAX8) identificano in modo affidabile l'origine follicolare;
- calcitonina (Ct), cromogranina A e sinaptofisina diagnosticano il carcinoma midollare (MTC);
- metastasi: richiedono un panel di anticorpi specifico (es. CD10 e RCC per carcinoma renale, GATA-3 per paratiroidi).
B. Test molecolari
Le alterazioni molecolari (come quelle BRAF-like e RAS-like) sono associate a istotipi tumorali specifici e influenzano la gestione terapeutica:
- test rule-in: confermano la malattia, come il panel a 7 geni (che include BRAFV600E, RAS, RET/PTC, PAX8/PPARG) applicato ai noduli TIR3A e TIR3B;
- mutazione BRAFV600E: estremamente specifica per il carcinoma papillare (PTC); la sua rilevazione indica quasi certamente un cancro;
- alterazioni RAS-like: associate a ROM più variabile (19-85%) e si riscontrano anche in lesioni tiroidee benigne (adenoma follicolare).
Applicazioni cliniche:
- nei noduli TIR3A/TIR3B, l’esecuzione di test molecolari aiuta a stratificare il rischio;
- nelle categorie TIR4/TIR5 e nei tumori avanzati, l'analisi si concentra su marcatori prognostici e predittivi (TERT promoter, TP53, RET, NTRK) per guidare la terapia mirata.
Bibliografia
- Basolo F, et al. Italian Consensus for the classification and reporting of thyroid cytology - ICCRTC 2025 Update). Springer editore 2025.
Utilità della genetica nella gestione delle neoplasie tiroidee
Nadia Cremonini1 & Erica Solaroli2
1Libero-Professionista, Bologna
2UOSD Endocrinologia - Ospedale Maggiore-Bologna
(aggiornato ad aprile 2026)
Nelle ultime tre decadi i progressi nella biologia molecolare hanno contribuito in modo significativo alla comprensione delle alterazioni molecolari alla base della patogenesi dei carcinomi tiroidei.
Le indagini genetiche sono di supporto alla diagnostica, alla valutazione prognostica e più di recente al trattamento sistemico delle forme avanzate mediante terapie target. Vanno valutate sia le alterazioni della linea germinale (correlate alla sindrome MEN-2 per il carcinoma midollare della tiroide MTC, o altre sindromi ereditarie che includono carcinomi differenziati della tiroide), sia mutazioni somatiche, quindi presenti solo nel tessuto neoplastico.
CARCINOMA MIDOLLARE
Nei pazienti con MTC la ricerca di mutazioni germinali del proto-oncogene RET (identificato nel 1993) è fondamentale, e deve essere effettuata anche in assenza di anamnesi familiare positiva per la patologia (circa il 7% dei pazienti con MTC apparentemente sporadico risulta portatore di mutazione germinale RET). Anche la revisione delle linee guida (LG) dell’American Thyroid Association (ATA) del 2015 (1) raccomanda di proporre il test genetico, previo counseling, a:
- tutti i pazienti con presunto MTC sporadico;
- parenti di primo grado di pazienti con MTC ereditario;
- genitori di bimbi con fenotipo classico MEN-2B;
- pazienti con lichen cutaneo amiloidosico del dorso;
- bambini con malattia di Hirschprung.
Il test per le mutazioni di RET deve includere gli esoni 10 e 11 (da effettuarsi per primi, per maggiore frequenza di mutazione dei codoni in questi esoni) e, se negativi, gli esoni 8, 13, 14, 15 e 16.
Per i pazienti con fenotipo MEN-2B, deve essere ricercata la mutazione del codone M918T (esone 16) e, se negativa, la mutazione del codone A883F (esone 15).
Se viene rilevata una mutazione germinale nel paziente, l’analisi genetica deve essere proposta ai familiari di primo grado, al fine di individuare i portatori della mutazione stessa, possibilmente prima dell’età raccomandata per la tiroidectomia profilattica.
I familiari non portatori di mutazione devono essere rassicurati e non devono essere sottoposti a screening biochimico per MTC, feocromocitoma o iperparatiroidismo.
In assenza di mutazioni RET, ma in presenza di sospetto elevato di MTC familiare, si deve verificare che sia stato effettuato uno screening genetico completo, prendere in considerazione la ricerca di nuove mutazioni e lo screening biochimico dei familiari a rischio dall’età di 5 anni.
Da anni è nota la stretta correlazione tra specifiche mutazioni germinali di RET, età di esordio e aggressività di MTC (2), e la correlazione genotipo-fenotipo nelle MEN-2A: l’identificazione di una specifica mutazione indica anche la tempistica per lo screening di feocromocitoma e iperparatiroidismo, poiché la loro incidenza ed età di esordio differisce in base alla mutazione (nei pazienti con mutazione RET C634 il feocromocitoma ha una incidenza del 50% nella V decade di vita e del 90% nell’VIII, l’iperparatiroidismo è presente in circa il 30% dei pazienti, mentre è nettamente inferiore nei pazienti con altre mutazioni).
In base alle LG ATA (1), la classificazione dell’MTC ereditario comprende tre livelli di rischio per MTC aggressivo:
- categoria “rischio più alto”: include pazienti con MEN-2B e mutazione RET M918T; per questi bambini il test RET, l'ecografia (US) tiroidea, il dosaggio di calcitonina (CT) e la tiroidectomia profilattica devono essere effettuati il prima possibile, preferibilmente entro il primo anno di vita;
- categoria “rischio alto”: include pazienti con mutazioni del codone C634 (MEN-2A) o mutazione del codone A883F (MEN-2B); il rischio di MTC aggressivo è inferiore, ma sempre elevato. La tiroidectomia profilattica è suggerita prima dei 5 anni o anticipata in caso di rilievo di livelli elevati di CT, con dissezione del comparto linfonodale centrale se CT > 40 pg/mL, o se evidenza di linfonodi metastatici;
- categoria “rischio moderato”: include pazienti con MTC ereditario e mutazioni diverse da M918T, C634 e A883F, che hanno un rischio inferiore rispetto alle altre due categorie; i bambini dovrebbero essere valutati clinicamente, con US del collo e dosaggio di CT a partire dai 5 anni; l’epoca della tiroidectomia andrebbe determinata in base al riscontro di valori elevati di CT, o prima se il monitoraggio a lungo termine risulta impossibile.
Per i pazienti con mutazioni RET rientranti nelle categorie di rischio ATA alto e moderato è importante considerare i livelli basali e stimolati di CT, al fine di proporre una maggiore personalizzazione del timing per la tiroidectomia nei portatori di mutazione: per tali categorie, si è dimostrato sicuro pianificare l’intervento nel momento in cui la CT stimolata diventa positiva, senza variazioni della percentuale di guarigione rispetto alla tiroidectomia profilattica classica (3,4). Requisito essenziale per questo approccio è la compliance del paziente e/o dei familiari.
Ricaduta clinica dello screening genetico per mutazioni germinali di RET
- Individua le forme ereditarie di MTC.
- Guida il clinico nella strategia di follow-up dei pazienti.
- Indica la tempistica di effettuazione dello screening genetico nei familiari, con conseguente precoce identificazione dei soggetti portatori della mutazione.
- Indica la tempistica della tiroidectomia totale profilattica nei portatori di mutazione.
Ogni qualvolta si deve proporre un test genetico, si raccomandano incontri pre-test tra clinico, genetista e paziente e/o familiari da sottoporre al test stesso, per illustrarne in modo semplice e comprensibile le motivazioni. Fondamentale anche il counseling post-test, per illustrare le decisioni di gestione clinica conseguenti al risultato.
Mutazioni somatiche di RET. In circa il 60% dei pazienti con forma sporadica di MTC (MTCs) si rilevano mutazioni somatiche di RET (solo nel tessuto neoplastico), percentuale che aumenta fino all’80% nelle forme metastatiche.
Un recente studio retrospettivo multi-centrico internazionale (USA, Australia,Francia, Italia) su 290 pazienti con MTCs (5) confermava che la mutazione somatica RET M918T era la la più frequente (53.6%), mentre in pochi casi venivano rilevate mutazioni nei codoni 634, 630, 620 e 883. Rispetto alle forme wild type (wtRET), i MTCs con mutazioni di RET erano associati sin dall’esordio a caratteri aggressivi, quali dimensioni maggiori, stadio IV AJCC, invasione vascolare e alto grado IMTCGS (International Medullary Thyroid Carcinoma Grade System). Analogamente al MTC in MEN-2B, nella forma somatica la mutazione RET M918T presentava rischio più elevato se comparata ad altre mutazioni somatiche di RET (età più giovane all’esordio di malattia, stadio IV AJCC, invasione vascolare ed extra-tiroidea, margini chirurgici positivi). Rispetto a wtRET, tutte le mutazioni somatiche di RET erano associate a minor sopravvivenza libera da malattia metastatica (DMFS), ma non a significativa differenza di sopravvivenza complessiva (OS) o malattia-specifica (DSS). Inoltre sono state rilevate mutazioni di RAS, TP53 e in minima percentuale di PIK3CA (fosfoinositide-3-chinasi), mutazione del promotore di TERT (telomerase reverse transcriptase). Da ricordare che le mutazioni RET e RAS sono mutualmente esclusive.
Gli MTCs RAS mutati presentavano un trend, non significativo, di associazione a migliore DMFS.
Anche in una casistica retrospettiva italiana (6) comprendente 141 casi di MTCs, la presenza di mutazioni di RET (in particolare la mutazione M918T) era significativamente più frequente nelle forme di alto grado vs le forme a basso grado (68.8% vs 29.4%) e nessun caso di MTC di alto grado presentava mutazione di RAS.
Quando effettuare lo studio molecolare su tessuto neoplastico in MTC sporadico
Sarebbe utile fare la ricerca di mutazioni somatiche di RET in tutti gli MTCs (ma anche per gli MTC con varianti germinali RET di significato non noto), data la loro correlazione con i caratteri della neoplasia; per le forme metastatiche e in progressione diventa raccomandata, in quanto sono oggi disponibili farmaci inibitori selettivi di RET, selpercatinib e pralsetinib, che hanno efficacia maggiore e minori effetti collaterali rispetto agli inibitori multi-kinasici, quali vandetanib e cabozantinib. Dal dicembre 2024 AIFA ha approvato l’utilizzo di selpercatinib come mono-terapia per il trattamento di adulti e adolescenti di età ≥ 12 anni con MTC avanzato con mutazione di RET (determina AIFA n. 781/2024).
NEOPLASIE DI ORIGINE FOLLICOLARE
I tumori tiroidei ad origine dalle cellule follicolari sono causati da eventi genomici somatici che comportano l’attivazione della via di segnale di protein-chinasi mitogeno-attivate. Le alterazioni genetiche comprendono eventi mutazionali “precoci” e “tardivi” (fig. 1).
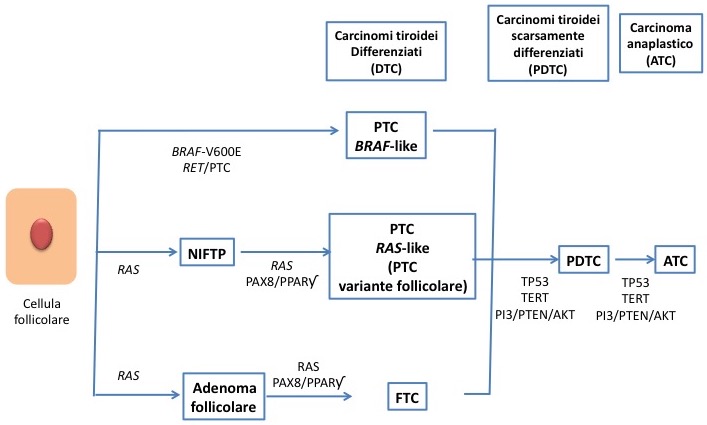
Figura 1 - Schema della tumorigenesi nei carcinomi tiroidei ad eziologia follicolare (modificato da 7)
L’attivazione iniziale in genere avviene tramite mutazioni interessanti BRAF e RAS, pertanto anche i tumori aggressivi mantengono caratteri BRAF-like, o RAS-like, mentre le variazioni “tardive” più frequenti, e associate a dedifferenziazione e progressione della neoplasia, sono rappresentate da mutazioni somatiche di TP53, del promotore di TERT e disregolazione di PI3K/PTEN/Akt. Inoltre si possono verificare fusioni geniche che coinvolgono RET, NTRK, ALK e BRAF.
Il numero delle mutazioni per singolo tumore aumenta a partire dalle forme ben differenziate fino ai tumori scarsamente differenziati (PDTC) e anaplastici (ATC) (8).
Mutazione BRAF-V600E. Oltre 20 anni fa veniva riportato questa mutazione in circa il 45% dei casi di carcinoma papillare della tiroide (PTC) (9). In seguito è stato riscontrato che la percentuale di tale mutazione è variabile nei diversi sottotipi di PTC: > 90% nella variante a cellule alte, 5-10% nel sottotipo follicolare, ma è presente anche nei sottotipi a cellule colonnari, e in molti casi del sottotipo hobnail (10).
BRAFV600E è anche un fattore determinante per il PDTC (circa il 30% dei casi) e per l’ATC (10-50% dei casi).
Ricerche concomitanti e successive rilevavano che le vie MAP (mitogen-activated protein)-chinasi e PI3K-Akt (phosphoinositide-3-kinase-protein B/Akt) sono le principali vie di trasduzione del segnale attivate nel carcinoma della tiroide e molti dei loro componenti sono mutati o attivati da ricombinazioni o alterazioni geniche:
- via MAP: BRAF -> MEK -> ERK;
- via PI3K-Akt: PI3K (PTEN) -> Akt -> mTOR.
Se molti studi hanno rilevato l’associazione tra la mutazione BRAF V600E e maggior rischio di metastasi linfonodali (N1), invasione extra-tiroidea e stadio TNM più elevato, con maggiore rischio di recidiva (11-14), altri studi non hanno attribuito valore prognostico a tale singola mutazione (15-17).
Mutazioni di PTEN. Il gene PTEN è un onco-soppressore, coinvolto nella via di segnale della fosfatidil-inositolo 3-chinasi (PI3K) e della protein-chinasi B (AKT), che regolano la crescita, la proliferazione e la sopravvivenza cellulare. In presenza di mutazioni del gene queste vie possono rimanere attivate in modo continuo, inducendo una crescita cellulare incontrollata e la formazione di tumori.
Le mutazioni di PTEN non rappresentano solo un evento precoce nei carcinomi tiroidei RAS-like (Ca follicolare - FTC) e nel Ca papillare variante follicolare incapsulato infiltrante (IEFVPTC), ma ulteriori alterazioni della via PTEN sono state rilevate in carcinomi tiroidei più avanzati, quali il carcinoma differenziato di alto grado (DHGTC), il PDTC e l’ATC (10). La sindrome di Cowden (CS) (parte della PTEN hamartoma tumor syndrome – PHTS), autosomica dominante, nel 25% dei casi dovuta a mutazioni germinali del gene PTEN, è oggi l’unica sindrome tumorale nota che comporta aumentato rischio di sviluppare nello stesso individuo carcinoma della tiroide e carcinoma della mammella (18).
Mutazioni di H-, K- ed N-RAS. Sono presenti nelle lesioni tiroidee a pattern follicolare: adenoma follicolare, FTC, sottotipo follicolare del PTC e NIFTP (neoplasia follicolare non invasiva con caratteri nucleari papillari) (19). La loro prevalenza è maggiore nelle aree con carenza iodica. Mutazioni di RAS sono state associate a comportamento clinico più aggressivo nei carcinomi differenziati della tiroide: sono di frequente riscontro nei PDTC (20-50% dei casi) e negli ATC (10-50% dei casi); N-RAS è la mutazione prevalente (8). RAS mutato pare attivare preferenzialmente la via PI3K-Akt: mutazioni o amplificazioni di PIK3CA (che codifica una subunità dell’enzima PIK3) comportano attivazione anomala di PI3K e sono più frequenti in FTC, PDTC e ATC vs PTC (20).
I PDTC e i carcinomi tiroidei differenziati di alto grado presentano mutazioni di BRAF, RAS o, meno di frequente, fusioni (in genere di RET e NTRK3); questi carcinomi possono anche esprimere mutazioni secondarie aggressive, quali mutazioni del promotore di TERT e in alcuni casi di TP53 e PIK3CA (8).
Mutazioni di TERT. Le due mutazioni C228T e C250T di TERT riscontrate in diversi istotipi di carcinoma tiroideo correlano con comportamento biologico più aggressivo della neoplasia e prognosi peggiore. La prevalenza è del 20-50% nei PDTC e del 30-70% negli ATC, mentre si riduce drasticamente nei PTC (circa 11%) e nei FTC (circa 17%); le mutazioni sono riscontrate soprattutto nei pazienti di età > 55 anni, con tumori di grandi dimensioni, spesso con estesa infiltrazione dei tessuti peri-tiroidei. Le mutazioni promuovono la progressione tumorale dei carcinomi tiroidei differenziati a tumore scarsamente differenziato e a carcinoma anaplastico, e sono significativamente associate a iodio-refrattarietà.
La coesistenza di una mutazione di TERT e di mutazioni BRAF o RAS potenzia il comportamento aggressivo dei DTC (possibile effetto sinergico), in particolare nei pazienti con carcinomi differenziati della tiroide ad alto rischio ATA e secondo il sistema TNM (21-23), e pertanto richiede grande attenzione nel follow-up.
Anche per i tumori follicolari a potenziale maligno incerto (forma rara con incidenza dello 0.5-3% di tutte le tiroidectomie, e con ottima prognosi), è bene ricercare la mutazione del promotore di TERT, in quanto tale mutazione è stata riscontrata in oltre il 30%, delle rarissime forme metastatiche di tale istotipo (24).
Mutazioni di TP53. TP53 è un gene soppressore maggiore nella regolazione del ciclo cellulare. Le sue mutazioni sono rare nei carcinomi differenziati della tiroide, mentre sono presenti nel 10-35% dei PDTC e nel 40-80% degli ATC.
Mutazioni somatiche di DICER 1. Nel 2001 DICER 1 è stato identificato come membro della famiglia delle nucleasi RNasi III, essenziale per la biogenesi di microRNA (miRNA) maturi. Mutazioni somatiche del dominio RNasi IIIb di DICER 1 ne compromettono la capacità di generare miRNA maturi, e si considera che siano alla base della tumorigenesi nei tumori associati a sindrome DICER 1 e sporadici.
Mutazioni somatiche di DICER 1 sono state rilevate in carcinomi tiroidei di origine follicolare, in particolare nell’FTC e nel PTC in età pediatrica, in assenza di altre mutazioni, mentre sono molto rare (< 1%) nel PTC dell’adulto (25). In genere i carcinomi tiroidei con una mutazione DICER 1 hanno andamento indolente, ma di recente (26) sono state identificate mutazioni somatiche di DICER 1 in cinque di sei bambini e adolescenti con PDTC, in assenza di mutazioni di BRAF, RAS, TERT, RET/PTC, mentre venivano riscontrate comutazioni “tardive”, quali TP53 e ATM, che si riscontrano in PDTC dell’adulto. In un paziente è stata riscontrata anche una mutazione germinale, permettendo di porre diagnosi di sindrome DICER 1.
Il PDTC in età infantile e adolescenziale è un’evenienza rara, aggressiva, che oltre allo studio genetico somatico richiede consulenza genetica per la ricerca di mutazione germinale, in quanto può essere la prima espressione clinica della sindrome DICER 1.
La sindrome DICER 1 è un disordine genetico raro, autosomico dominante, che predispone allo sviluppo di un ampio spettro di neoplasie benigne o maligne in età pediatrica e adulta: carcinoma embrionale del polmone, rabdomiosarcoma pleuro-polmonare, tumore di Wilms e tumori endocrini (tiroide, paratiroidi, ipofisi, pineale, pancreas endocrino, paragangliomi, tumori corticosurrenalici, ovarici e testicolari in particolare a cellule di Sertoli-Leydig).
Riarrangiamenti. Nell’ambito di riarrangiamenti genomici i PTC possono esprimere fusioni di geni cancro-relati, alcuni dei quali (RET, NTRK1-3, BRAF, ALK) rivestono grande importanza clinica, in quanto bersaglio di terapie target specifiche.
- Riarrangiamento PAX8/PPARγ: è presente nel 20-50% degli FTC e in circa il 10% degli adenomi follicolari. Negli FTC è stato associato a fenomeni di invasione vascolare e a pazienti più giovani, ma non a prognosi sfavorevole. Il riaarrangiamento è raramente presente nelle forme a scarsa differenziazione.
- Fusioni del gene RET (RET/PTC): si tratta di riarrangiamento cromosomico, che dà origine a geni di fusione chimerici, e nello specifico i riarrangiamenti somatici di RET consistono nella fusione del dominio chinasico di RET all’estremità in 3’ con la porzione all’estremità in 5’ di un altro gene partner. Nei PTC i riarrangiamenti di RET si riscontrano nelle forme sporadiche, e in oltre il 90% dei casi si tratta di fusioni CCDC6-RET (RET-PTC1) o NCOA4-RET (RET-PTC3)(27). I recettori RET attivati stimolano le vie di segnale MAPK e PI3K-AKT, promuovendo la tumorigenesi. RET- PTC1 è presente in circa il 7-15% dei PTC classici, mentre RET-PTC3 è associato a esposizione a radiazioni ionizzanti, sia accidentali (come nel caso dei PTC post-Chernobyl), sia a scopo terapeutico, con prevalenza del 30-60% nei PTC insorti dopo esposizione alle radiazioni.
- Fusioni di NTRK: la famiglia NTRK (Neurotrophic Tropomyosin Receptor Kinase) comprende tre geni (NTRK1, NTRK2, NTRK3), che codificano per tre diverse proteine TRK (rispettivamente, TRKA, TRKB e TRKC), con funzione di recettori per diverse neurotrofine, rivestendo un ruolo fondamentale nello sviluppo e nella differenziazione del sistema nervoso. Il legame recettori TrkA, TrKB e TrkC – neurotrofine attiva una serie di vie di segnale cellulare: TrkA si lega a NGF (Nerve Growth Factor) e TrkB a BDGF(Brain-Derived Growth Factor), causando entrambi l'attivazione deille vie di segnale MAPK/RAS/ERK, PLC-(phospholipase C-gamma) e PI3K. La formazione di geni di fusione, che rappresenta il meccanismo più diffuso attraverso cui si esplica l'attivazione costitutiva dei geni NTRK, è il risultato di riarrangiamenti intra- (in particolare nel caso di NTRK1) o inter-cromosomici. I geni partner di NTRK sono molteplici, e in tutti i casi il gene chimerico conserva la regione 3' di NTRK con il dominio ad attività tirosin-chinasica, unitamente alla regione 5' del gene partner (28). La frequenza di fusioni NTRK varia dall’1 al 6% dei PTC nell’adulto (con la prevalenza più elevata nel DTC iodio-refrattario e nel sesso femminile) e dal 4 al 26% nei PTC dell’età pediatrica. In un recentissimo lavoro su 65 pazienti (37 adulti e 28 pediatrici) con carcinoma tiroideo (54 PTC, 4 PDTC e 7 ATC) positivi per fusione NTRK (24 NTRK1 e 41 NTRK3), è stata riscontrata una netta prevalenza di fusione NTRK3 negli adulti, e una percentuale simile delle due fusioni nei bambini. Mentre nei bambini NTRK1 era associata a stadio più avanzato del tumore, ma non a metastasi a distanza, negli adulti non vi erano differenze cliniche tra NTRK1 e NTRK3 (29). L’identificazione delle neoplasie tiroidee NTRK-positive ha assunto grande importanza in seguito alla dimostrazione che inibitori selettivi di TRK inducono alte percentuali di risposta in diversi tipi di carcinoma NTRK-positivi. Tra questi, entrectinib, un inibitore sia dei recettori NTRK sia di ROS1 e ALK, e larotrectinib, un inibitore selettivo dei recettori NTRK, sono stati approvati nel 2019 da FDA e successivamente da EMA (30).
- Alterazioni di ALK: il gene ALK (anaplastic lymphoma kinase), che codifica per un recettore tirosin-chinasico trans-membrana, è espresso nel sistema nervoso durante l’embriogenesi e viene silenziato nei tessuti adulti. Oltre che nello sviluppo del sistema nervoso, ALK gioca un ruolo importante nella regolazione della crescita, della differenziazione e della trasformazione cellulare. Sono state identificate diverse alterazioni del gene ALK: le più comuni sono le fusioni tra la porzione 5’ di un gene partner contenente il promotore e la regione 3’ di ALK codificante per il dominio chinasico, con conseguente attivazione della trasduzione del segnale a valle, aumento della proliferazione e della sopravvivenza cellulare. In vari tipi di tumore sono stati osservati riarrangiamenti di ALK: nei PTC la fusione di ALK è stata segnalata nello 0-2.2% di pazienti non selezionati, nel 2-7.3% di pazienti pediatrici e giovani adulti e fino al 12.7% di pazienti esposti a radiazioni. Data la rarità dei PTC con riarrangiamento di ALK, il loro comportamento clinico non è ancora ben caratterizzato. La fusione di ALK può fungere da potenziale bersaglio terapeutico grazie agli inibitori mirati della tirosin-chinasi (31).
PD1 – PDL-1. Negli ultimi anni, con l'avvento delle immuno-terapie, in particolare il blocco dei check-point immunitari, abbiamo assistito a cambiamenti nel trattamento del cancro. Le molecole dei check-point immunitari, espresse su varie cellule immunitarie, come le cellule T attivate, i macrofagi e le cellule dendritiche, regolano l'attivazione immunitaria e prevengono risposte immunitarie eccessive che potrebbero portare a malattie autoimmuni. La morte programmata 1 (PD-1), un recettore dei check-point immunitari espresso prevalentemente sulle cellule T, interagisce con il suo ligando, PD-L1, per promuovere la tolleranza immunitaria e regolare l'infiammazione. L'espressione di PD-L1 rappresenta un pre-requisito per l'uso di inibitori dei check-point immunitari nel trattamento di alcuni tumori (melanoma, carcinoma renale, tumori della testa e del collo, carcinoma polmonare non a piccole cellule). Storicamente, il carcinoma tiroideo è stato classificato come tumore "immunologicamente freddo", suggerendo un potenziale limitato per l'applicazione efficace di trattamenti basati sul sistema immunitario, ma studi recenti sulle forme avanzate di carcinoma tiroideo, inclusi ATC e PDTC, suggeriscono che il sistema immunitario svolga un ruolo critico nella loro patogenesi e progressione. C’è un'ampia variabilità nell'espressione di PD-L1 nel carcinoma tiroideo, che va dal 5 all'87%: ciò può essere dovuto a differenze nella popolazione testata, nei metodi di rilevamento, nelle tecniche di analisi e nell'interpretazione (32). I tassi di negatività del PD-L1 potrebbero essere falsi negativi derivanti dall'utilizzo di un campione d'archivio con degradazione del PD-L1, che impedisce una valutazione attendibile. È quindi importante che l'espressione del PD-L1 venga studiata su campioni bioptici raccolti di recente o con una quantità adeguata di tessuto per effettuare un'analisi ragionevole (33). Una delle principali sfide nell'utilizzo di PD-L1 come biomarcatore predittivo nell'ATC è l'assenza di valori soglia standardizzati per il processo decisionale clinico.
CONSIDERAZIONI CLINICHE
Nei pazienti con diagnosi di carcinoma tiroideo di derivazione delle cellule follicolari, il test molecolare riveste importanza in due stadi della gestione:
- dopo la diagnosi di carcinoma fornisce informazioni utili sotto il profilo prognostico e aiuta nelle decisioni terapeutiche;
- nei pazienti con forme progressive locali inoperabili o metastatiche, e iodo-refrattarie, guida nella scelta della terapia sistemica in base ad alterazioni geniche che possono costituire un bersaglio; anche i pazienti con forme ad alto rischio di recidiva strutturale dovrebbero essere testati per mutazioni somatiche.
Se non è disponibile un pannello NGS (next generation sequencing), che valuta anche rare alterazioni geniche, il primo step di studio molecolare dovrebbe comprendere TERT (ai fini prognostici), e BRAF, NTRK, RET-PTC e ALK, al fine di individuare i pazienti che potenzialmente possono beneficiare di terapie sistemiche target (34):
- selpercatinib per i DTC con neoplasia positiva per fusione RET-PTC;
- larotrectinib e entrectinib per i carcinomi tiroidei con fusione di NTRK.
Merita un cenno la valutazione della biologia molecolare nei carcinomi tiroidei inoperabili o con estensione locale ad elevatissimo rischio chirurgico: la rilevazione di mutazioni potenzialmente bersaglio consente il trattamento con terapie target specifiche a scopo neoadiuvante, con successiva possibilità di procedere alla chirurgia se si ottiene una buona risposta (35).
Nei pazienti con diagnosi di ATC deve essere ricercata “in urgenza” la mutazione BRAF V600E con test rapido PCR (Polymerase Chain Reaction) o IHC (immuno-istochimica) e nel contempo va avviata la valutazione con NGS, che oltre a confermare il risultato BRAF V600E positivo ottenuto con le tecniche rapide (con conseguente possibilità di terapia con dabrafenib, inibitore di BRAF, associato a trametinib, inibitore di MEK), nei pazienti con ATC BRAFV600E wild-type permette di rilevare eventuali altre alterazioni targettabili, inclusi NTRK, ALK e RET (36).
Inoltre va considerata anche la ricerca di espressione di PD-L1 mediante IHC: se positiva, considerare terapia con pembrolizumab (inibitore del check-point immunitario) e lenvatinib (inibitore tirosin-chinasico).
Per rilevare mutazioni marker nei tumori tiroidei, a parte la ricerca di espressione di PD-L1 che si effettua con immuno-istochimica (IHC), per le altre mutazioni o si dispone di pannelli NGS o si può ricorrere a diversi metodi, che presentano utilità clinica differente per le varie mutazioni:
- mutazioni di RET: il metodo NGS DNA-based è il più utile, seguito da PCR (polymerase chain reaction);
- fusioni di RET: oltre al metodo NGS, si possono ultilizzare i metodi FISH (fluorescence in-situ hybridization) e PCR (quest’ultimo ha minore utilità clinica);
- mutazione BRAF V600E: NGS, PCR e IHC presentano tutti elevata utilità clinica;
- fusioni ALK e fusioni NTRK: la maggiore utilità clinica nel loro rilevamento è data dalle metodiche NGS e FISH, mentre è inferiore per i metodi IHC e PCR (34).
BIBLIOGRAFIA
- Wells SA Jr, Asa SA, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. The American Thyroid Association guidelines task force on medullary thyroid carcinoma. Thyroid 2015, 25: 567-610.
- Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 2003, 349: 1517-25.
- Rohmer V, Vidal-Trecan G, Bourdelot A, et al. Prognostic factors of disease-free survival after thyroidectomy in 170 young patients with a RET germline mutation: a multicenter study of the Groupe Francais d’Etude des Tumeurs Endocrines. J Clin Endocrinol Metab 2011, 96: E509-18.
- Elisei R, Romei C, Renzini G, et al. The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab 2012, 97: 426-35.
- Xu B, Viswanathan K, Ahadi MS, et al. Association of genomic profile of medullary thyroid carcinioma with tumor characteristics and clinicale outcomes in an international multicenterstudy. Thyroid 2024, 34: 167-76.
- Censi S, Galuppini F, Clausi C,et al. Tumor grade and molcular characteristics associated with survival in sporadic medullary thyroid carcinoma. Thyroid 2024, 34: 177-85.
- Acquaviva G, Visani M, Repaci A, et al. Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathol 2018, 72: 6-31.
- Volante M, Lam AK, Papotti M, Tallini G. Molecular pathology of poorly differentiated and anaplastic thyroid cancer: what do pathologists need to know? Endocr Pathol 2021, 32: 63-76.
- Cohen Y, Xing M, Mambo E, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst 2003, 95: 625-7.
- Baloch ZW, Asa SL, Barletta JA, et al. Overview of the 2022 WHO classification of thyroid neoplasm. Endocr Pathol 2022, 33: 27-66.
- Xing M, Westra WH, Tufano RP, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab 2005, 90: 6373-9.
- Elisei R, Viola D, Torregrossa I, et al. The BRAFV600E mutation is an independent, poor prognostic factor for the outcome of patients with low-risk intrathyroid papillary thyroid carcinoma: single-institute results from a large cohort study. J Clin Endocrinol Metab 2012, 97: 4390-8
- Kim TH, Park YJ, Lim JA, et al. The association of the BRAFV600E mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer. A meta-analysis. Cancer 2012, 118: 1764-73.
- Xing M, Alzahrani AS, Carson KA, et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. J Clin Oncol 2015, 33: 42-50.
- Trovisco V, Soares P, Preto A, et al. Type and prevalence of BRAF mutations are closely associated with papillary thyroid carcinoma histotype and patients’ age but not with tumour aggressiveness. Virchows Arch 2005, 446: 589-95.
- Ito Y, Yoshida H, Maruo R, et al. BRAF mutation in papillary thyroid carcinoma in a Japanese population: its lack correlation with high-risk clinicopathological features and disease-free survival of patients. Endocr J 2009, 56: 89-97.
- Russo M, Malandrino P, Nicolosi ML, et al. The BRAFV600E mutation influences the short- and medium-term outcomes of classic papillary thyroid cancer, but is not an independent predictor of unfavorable outcome. Thyroid 2014, 24: 1267-74.
- Lu M, Liu H, Zheng B, et al. Links between breast and thyroid cancer: hormones, genetic susceptibility and medical interventions. Cancers (Basel) 2022, 14: 5117.
- Nikiforov YE, Seethala RR, Tallini G, et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma: a paradigm shift to reduce overtreatment of indolent tumors. JAMA Oncol 2016, 2: 1023–9.
- Alzahrani AL. Clinical use of molecular data in thyroid nodules and cancer. J Clin Endocrinol Metab 2023, 108: 2759-71.
- Song YS, Lim JA, Choi H, et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 2016, 122: 1370-9.
- Melo M, Gapar da Rocha A, Batista R, et al. TERT, BRAF, and NRAS in primary thyroid cancer and metastatic disease. J Clin Endocrinol Metab 2017, 102: 1898-907.
- Moon S, Song YS, Kim YA, et al. Effects of coexistent RAFV600E and TERT promoter mutations on poor clinical outcomes in papillary thyroid cancer: a meta-analysis. Thyroid 2017, 27: 651-60
- Hysek M, Jatt K, Hellgren S, et al. Spatial distribution patterns of clinically relevant TERT promoter mutations in follicularthyroid tumors of uncertain malignant potential. Advantages of the digital droplet PCR technique. J Mol Diagn 2021, 23: 212-22.
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014,159: 676–90.
- Chernock RD, Rivera B, Borrelli N, et al. Poorly differentiated thyroid carcinoma of childhood and adolescence: a distinct entity characterized by DICER1 Mod Pathol 2020, 33: 1264-74.
- Santoro M, Moccia M, Federico G, et al. RET gene fusions in malignancies of the thyroid and the other tissues. Genes (Basel) 2020, 11: 424.
- Wong D, Yip S, Sorensen PH. Methods for identifying patients with tropomyosin receptor kinase (TRK) fusion Pathol Oncol Res 2020, 26: 1385-99.
- Marczyk VR, Fazeli S, Dadu R, et al. NTRK fusion-positive thyroid carcinoma: from diagnosis to target therapy. JCO Precision Oncology 2025, DOI: https://doi.org/10.1200/PO.24.00321.
- Koehler VF, Achterfeld J, Sandner N, et al. NTRK fusion events and targeted treatment of advanced radioiodine refractory thyroid cancer. J Cancer Res Clin Oncol 2023, 149: 14035-43.
- Shih KP, Lee YC, Tsai JJ, et al. Clinicopathologic features and cytologic correlation of ALK-rearranged papillary thyroid carcinoma: a series of eight cases. Endocr Pathol 2024, 35: 134–46.
- Shobab L, Al-Souri D, Mathews-Kim L, et al. PD-L1 expression varies in thyroid cancer types and is associated with decreased progression free survival (PFS) in patients with anaplastic thyroid cancer. Cancers (Basel) 2024, 16: 3632.
- Chern B, Pinto D, Hy Lum J, et al. Nearly half of patients with anaplastic thyroid cancer may be amenable to immunotherapy. Biomedicines 2024, 12: 1304.
- Mete O, Boucher A, Shrader KA, et al. Consensus statement: recommendations on actionable biomarker testing for thyroid cancer management. Endocr Pathol 2024, 35: 293-308.
- Huang N-S, Wang Y, Wei W-J, et al. A systemic review of neo-adjuvant target therapy in locally advanced thyroid cancer. Holistic Integr Oncol 2022, 1: 16.
- Filetti S, Durante C, Hartl DM, et al. ESMO clinical practice guideline update on the use of systemic therapy in advanced thyroid cancer. Ann Oncol 2022, 33: 674-84.