Obesità e sindrome metabolica
Forme genetiche di iposurrenalismo primario

Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 12 ottobre 2021)
L’iposurrenalismo primario (PAI) è una condizione clinica rara, potenzialmente fatale, che necessita di un tempestivo approccio diagnostico e terapeutico. Tra le forme congenite (1), la più nota è l’iperplasia surrenalica congenita (CAH), dovuta a difetti nella steroidogenesi, che, in base al tipo e alla gravità, alterano la sintesi di mineralcorticoidi, glucocorticoidi e ormoni sessuali, sia a livello surrenalico sia gonadico. La causa più frequente di CAH è il deficit di 21-idrossilasi, che può dare tre diversi fenotipi in base al grado di attività enzimatica residua:
- forma con perdita di sali (attività enzimatica < 1%), caratterizzata da PAI a insorgenza nelle prime settimane di vita e iperandrogenismo, responsabile in entrambi i sessi di pubertà precoce, nelle femmine di ambiguità dei genitali alla nascita e successivo quadro simile al fenotipo classico della sindrome dell’ovaio policistico (PCOS), mentre nei maschi comporta infertilità e disturbi metabolici in età adulta;
- forma virilizzante semplice (attività enzimatica residua 1-2%), con manifestazioni da iperandrogenismo analoghe alla forma con perdita di sali; manca il quadro di iposurrenalismo clinico, anche se questo potrebbe essere slatentizzato in situazioni di stress;
- forma non classica (attività enzimatica residua del 20-50%), responsabile di diversi quadri di iperandrogenismo clinico a esordio in età adolescenziale-adulta (late-onset), con fenotipo simile alla PCOS nella femmina e spesso del tutto asintomatica nel maschio.
Le prime due forme vengono definite classiche e interessano circa 1:16.000 nati; la forma non classica, invece, ha prevalenza maggiore, attorno a 1:1000 nella popolazione caucasica.
Altre forme congenite di iposurrenalismo meno frequenti sono altri difetti enzimatici responsabili di iperplasia surrenalica congenita (deficit di 17α-idrossilasi/17,20 liasi, deficit di 11-idrossilasi, deficit di 3-ß-idrossi-steroido-deidrogenasi, deficit di citocromo P450 ossido-reduttasi), la sindrome di Allgrove o della tripla A (acalasia, Addison e alacrimia) (2), l’adrenoleucodistrofia X-linked (malattia perossisomale, caratterizzata dall'accumulo plasmatico e tissutale di acidi grassi a catena lunga e molto lunga, che interessa principalmente i maschi e provoca diversi disturbi neurologici, spesso associati o preceduti dall’insorgenza di PAI e a volte di ipogonadismo primario in età adulta) (3) e il deficit familiare di glucocorticoidi (associato a mutazioni nel gene del recettore per l'ACTH) (4). Inoltre, negli ultimi anni sono state scoperte molte altre cause genetiche rare di PAI, la cui diagnosi ha importanti implicazioni non solo sui familiari, ma anche sulla gestione e il follow-up del paziente affetto.
Deficit della proteina StAR
StAR, codificata dall’omonimo gene sul cromosoma 8, è la proteina chiave per l’inizio della sintesi di tutti gli ormoni steroidei, in quanto media il trasporto del colesterolo dalla superficie esterna della membrana mitocondriale a quella interna, dove inizia la steroidogenesi per azione di P450side-chain (P450scc). Deficit severi di StAR sono associati ad iperplasia surrenalica lipoidea congenita (LCAH), caratterizzata da PAI ad esordio peri-natale, fenotipo femminile nei maschi e ipogonadismo ipergonadotropo nelle femmine (5). Deficit parziali di StAR, invece, sono associati a forme di LCAH non classica, caratterizzata solo da PAI, anche a esordio tardivo o con solo deficit dei glucocorticoidi, mimando un quadro simile al deficit familiare di glucocorticoidi (familial glucocorticoid deficiency), altra rara sindrome genetica, non del tutto chiarita, associata a mutazioni nel gene del recettore per l'ACTH (4). Nei soggetti con deficit parziali viene comunque raccomandato un controllo a lungo termine dell’asse gonadico, sia in età puberale sia in età adulta, proponendo cautelativamente anche metodiche di crio-conservazione dei gameti, in particolare nei maschi, per possibili alterazioni della fertilità nel tempo.
Deficit di P450scc
P450scc, codificato dal gene CYP11A1 sul cromosoma 15 e situato sulla parte interna della membrana mitocondriale, è l’altro fattore chiave assieme a StAR degli stadi iniziali della steroidogenesi, in quanto converte il colesterolo in pregnenolone. Mutazioni di P450scc, in base alla severità del deficit, provocano quadri simili a LCAH classica e non classica (6). Anche per i deficit parziali di P450scc, viene raccomandato il monitoraggio periodico della funzione sessuale soprattutto maschile, in quanto sono stati riportati casi di testicular adrenal rest tumors, soprattutto nei soggetti con scarso compenso surrenalico.
Mutazioni di recettori nucleari coinvolti nello sviluppo e nella funzione di surreni e gonadi
DAX-1: codificato dal gene NR0B1, localizzato sul braccio corto del cromosoma X, è stato descritto per la prima volta nel 1994 come responsabile di ipoplasia surrenalica congenita X-linked (7). Sono note numerose mutazioni genetiche, che, sempre sulla base del difetto di sintesi (completo o parziale), possono determinare la forma classica, caratterizzata da esordio precoce di PAI e ipogonadismo ipogonadotropo con infertilità, o non classica, caratterizzata da diversi fenotipi, a esordio tardivo, con ipogonadismo centrale parziale o anche con caratteristiche paradosse, come macropene e pubertà precoce. Il corretto inquadramento diagnostico di questi soggetti permette di ricercare anche eventuali forme di PAI subclinico nei familiari di sesso maschile della linea materna.
SF-1 (fattore steroidogenico 1): codificato dal gene NR5A1, localizzato sul cromosoma 9, è un altro fattore chiave che regola principalmente lo sviluppo testicolare e ovarico, mentre sembra meno determinante per lo sviluppo dei surreni. Infatti, alterazioni di SF-1 raramente provocano PAI, mentre risultano soprattutto associate a diversi spettri di disgenesia/disfunzione gonadica, descritti principalmente nel maschio: disturbi dello sviluppo sessuale, ipospadia, criptorchidismo, ipogonadismo e infertilità (8). Più recentemente sono state descritte mutazioni del gene NR5A1 anche in donne con insufficienza ovarica primaria sporadica o familiare. In tutti questi soggetti con disfunzioni gonadiche è raccomandato un follow-up a lungo-termine, perché resta ancora da chiarire se possano sviluppare nel tempo neoplasie gonadiche e PAI.
Disordini di crescita multi-sistemici
CDKN1C: inibitore della progressione del ciclo cellulare, viene espresso solamente dall’allele materno localizzato sul cromosoma 11, mimando una condizione X-linked. Mutazioni associate ad acquisizione di funzione di CDKN1C comportano una ridotta proliferazione cellulare associata alla sindrome IMAGe, caratterizzata da ritardo di crescita intra-uterino, displasia metafisaria (con arti corti), ipoplasia surrenalica congenita e anomalie genito-urinarie (9). Tale sindrome è stata descritta per la prima volta nel 1999 e risulta associata anche a tratti dismorfici (fronte prominente, setto nasale largo, orecchie basse) e diabete mellito. Di contro, mutazioni associate a perdita di funzione di CDKN1C sono state riscontrate nel 10% dei pazienti affetti da sindrome di Beckwith-Wiedemann, una malattia da iper-accrescimento, associata a malformazioni congenite e rischio di sviluppo di tumori, anche surrenalici.
SAMD9: è un altro inibitore della proliferazione cellulare, localizzato sul braccio lungo del cromosoma 7 ed espresso durante lo sviluppo fetale. Mutazioni associate ad acquisizione di funzione si associano alla sindrome MIRAGE (mielodisplasia, infezioni, ritardo di crescita intra-uterino, ipoplasia surrenalica congenita, disgenesia gonadica ed enteropatia), descritta per la prima volta nel 2016, caratterizzata da un ampio spettro di manifestazioni cliniche, con diversa gravità, e associata a elevata mortalità, prevalentemente per infezioni gravi entro il secondo anno di vita (10). Una caratteristica peculiare di alcuni bimbi affetti da mutazioni di SAMD9 è che spesso sviluppano nelle cellule ematopoietiche monosomia del cromosoma 7 o delezioni del suo braccio lungo o mutazioni associate a perdita di funzione di SAMD9: tali meccanismi compensatori, eliminando l’allele mutato, conferiscono un vantaggio di crescita clonale, che può comportare lo sviluppo di sindromi mielodisplastiche o forme leucemiche. Infine, sono stati riportati anche bimbi con fenotipi più lievi, dovute a forme di mosaicismo per l’allele con mutazione di SAMD9.
POLE1: fattore chiave nella replicazione del DNA, localizzato sul cromosoma 12. Mutazioni associate alla sua perdita di funzione sono state riscontrate in bimbi con sindrome IMAGe-like, caratterizzata principalmente da deficit di crescita, ipoplasia surrenalica congenita (con diversi gradi di PAI), deficit del sistema immunitario e tratti dismorfici (11).
Una nuova sfingolipidosi: deficit di sfingosina-1-fosfato liasi tipo 1 (SGPL1)
SGPL1 è un enzima coinvolto nella degradazione delle ceramidi, il cui gene è localizzato sul cromosoma 10 e il cui deficit comporta una forma di sfingolipidosi, in cui l’accumulo di sfingolipidi e ceramidi a livello intra-cellulare comporta anche PAI, oltre a manifestazioni multi-sistemiche simili a quelle della malattia di Fabry o di Gaucher (disfunzioni neurologiche, linfocitopenia, sindrome nefrosica) (12). Il corretto inquadramento diagnostico del deficit di SGPL1 risulta, pertanto, fondamentale anche per individuare precocemente un possibile concomitante iposurrenalismo, che potrebbe essere mascherato dai cicli di terapia steroidea per la sindrome nefrosica.
Conclusioni
Le forme genetiche di PAI sono rare e di alcune, come quelle di recente scoperta, sono stati riportati solo un centinaio di casi; tuttavia, il loro corretto inquadramento diagnostico è molto utile per le importanti implicazioni epidemiologiche, terapeutiche e di follow-up per il paziente e i suoi familiari. La diagnosi e successiva valutazione genetica, pertanto, devono essere eseguite in centri esperti e specializzati.
Cosa fare di fronte a un bimbo o adolescente con recente diagnosi di PAI:
- ricercare attraverso una dettagliata anamnesi familiare possibili parenti affetti, individuando trasmissioni X-linked o aree geografiche di provenienza note per l’elevata ricorrenza di alcune mutazioni, come ad esempio le zone centrali della Turchia, ad alta prevalenza di varianti di CYP11A1;
- individuare altre manifestazioni cliniche eventualmente associate a PAI, che potrebbero orientare verso la diagnosi, come segni di iperandrogenismo, anomalie genito-urinarie, ritardo di crescita intra-uterino, tratti dismorfici, patologie autoimmunitarie;
- sulla base del quadro anamnestico e clinico, indagare i livelli di 17OH-progesterone, acidi grassi a catena molto lunga, la funzione gonadica, la presenza di anticorpi anti-corticale del surrene e anti-21-idrossilasi, per richiedere successivamente un eventuale test genetico mirato a un singolo gene;
- nelle forme congenite che rimangono non chiarite, proseguire con le tecniche di next-generation sequencing, oramai sempre più diffuse in alcuni laboratori di genetica dei centri di III livello, che permetteranno di individuare non solo nuove cause, prevalenza e distribuzione di tali forme, ma anche pannelli di geni o esoni target, che potranno rendere ancora più rapida e meno dispendiosa tale ricerca in futuro.
Bibliografia
- Buonocore F, Achermann JC. Primary adrenal insufficiency: new genetic causes and their long-term consequences. Clin Endocrinol 2020, 92: 11-20.
- Alhassoun M, Almakadma AH, Almustanyir S, et al. Triple A multisystem disorder: Allgrove syndrome. Cureus 2021, 13: e17476.
- Zhu J, Eichler F, Biffi A, et al. The changing face of adrenoleukodystrophy. Endocr Rev 2020, 41: 577-93.
- Meimaridou E, Hughes CR, Kowalczyk J, et al. Familial glucocorticoid deficiency: new genes and mechanisms. Mol Cell Endocrinol 2013, 371: 195-200.
- Bose HS, Sugawara T, Strauss JF, Miller WL. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med 2002, 335: 1870-9.
- Baker BY, Lin L, Kim CJ, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab 2006, 91: 4781-45.
- Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab 2015, 29: 607-19.
- Orekhova AS, Kalinchenko N, Morozov IA, et al. A novel mutation in the critical P-box residue of steroidogenic factor-1 presenting with XY sex reversal and transient adrenal failure. Horm Res Paediatr 2018, 89: 450-4.
- Vilain E, Le Merrer M, Lecointre C, et al. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J Clin Endocrinol Metab 1999, 84: 4335-40.
- Shima H, Hayashi M, Tachibana T, et al. MIRAGE syndrome is a rare cause of 46, XY DSD born SGA without adrenal insufficiency. PLoS One 2018, 13: e0206284.
- Logan CV, Murray JE, Parry DA, et al. DNA polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet 2018, 103: 1038-44.
- Prasad R, Hadjidemetriou I, Maharaj A, et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest 2017, 127: 942-53.
Overview sugli iposurrenalismi primari
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
(aggiornato al 10 settembre 2015)
L’iposurrenalismo primario o malattia di Addison è una condizione clinica descritta per la prima volta nel 1855 da Thomas Addison, determinata dalla carente produzione surrenalica di glicocorticoidi, mineralcorticoidi ed androgeni, mentre il difetto della midollare è clinicamente irrilevante (1).
Si tratta di una patologia piuttosto rara, con prevalenza di 93-140 casi/milione, incidenza di 4.7-6.2 casi/milione/anno (1,2), quest’ultima stimata in ulteriore aumento nella popolazione occidentale (2), e che compare prevalentemente nell’età adulta, con una maggiore frequenza nella quarta decade di vita e nel sesso femminile (1).
L’iposurrenalismo primitivo è classificabile dal punto di vista eziopatogenetico in varie forme, tra cui quella autoimmune risulta essere più frequente nei paesi industrializzati e quella infettiva nel resto del mondo (1,3).
Il quadro clinico si differenzia in una forma acuta, che rappresenta una vera emergenza clinica con le manifestazioni cliniche dello shock ipovolemico, e una forma cronica, in cui frequentemente i segni e i sintomi presentano un esordio graduale e sono aspecifici (1,3). Nella storia naturale della forma autoimmune sono state peraltro descritte due fasi pre-cliniche, prive di segni o sintomi, e caratterizzate soltanto da alterazioni auto-anticorpali o biochimiche/ormonali (4).
Nell’insufficienza surrenalica acuta, le condizioni di urgenza del quadro clinico, che richiedono un trattamento tempestivo, non consentono una valutazione accurata della funzione surrenalica e pertanto ci si dovrà limitare al dosaggio di cortisolemia e di ACTH (1,3).
Nell’insufficienza surrenalica cronica, la diagnosi di iposurrenalismo primario è difficile nelle fasi pre-cliniche della forma autoimmune, ma la presenza di ipotensione arteriosa, collasso, iperpigmentazione cutanea, iposodiemia, iperpotassiemia, ipercalcemia, acidosi ed ipoglicemia non diversamente spiegabili sono suggestivi per la presenza di iposurrenalismo (1,3). In particolare, l’iposodiemia può essere presente in oltre il 90% dei casi (3).
In tutti i pazienti con sospetto clinico va eseguita una valutazione ormonale basale, con dosaggio al mattino (ore 7-9) di cortisolemia, ACTH, renina o attività reninica ed aldosterone; il dosaggio del DHEA-S non è attualmente riconosciuto come criterio diagnostico aggiuntivo (1,3-6).
In tutti i pazienti con forte sospetto clinico e valutazione ormonale basale di norma va comunque effettuata una valutazione ormonale dinamica, mediante test con ACTH sintetico alla dose di 250 µg con valutazione della cortisolemia (1,3-7). In pazienti con forme “mild” o subcliniche, è stato proposto da alcuni autori il test con ACTH sintetico alla dose di 1 µg, ma non è stato ancora universalmente accettato come test di provata superiorità diagnostica (8). Nel sospetto di una forma autoimmune vanno eseguiti specifici esami anticorpali (di non frequente disponibilità), quali anticorpi anti-corteccia surrenalica (ACA) e anticorpi anti-21-idrossilasi (3,4,9).
La terapia dell’insufficienza surrenalica acuta prevede l’impiego di idrocortisone per via e.v. e la correzione della disidratazione con un adeguato apporto idrico (3,10-13).
Nell’insufficienza surrenalica cronica il trattamento sostitutivo prevede l’impiego di glicocorticoidi, idrocortisone o cortisone acetato, quest'ultimo maggiormente impiegato nel nostro paese, e mineralcorticoidi, 9α-fluoro-idrocortisone. La terapia con DHEA è invece considerata opzionale (3,10-13).
Bibliografia
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003, 361: 1881-93.
- Løvås K, Husebye ES. High prevalence and increasing incidence of Addison's disease in western Norway. Clin Endocrinol (Oxf) 2002, 56: 787-91.
- Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014, 275: 104-15.
- Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndrome: autoantibodies, autoantigens, and the applicability in diagnosis and disease prediction. Endocr Rev 2002, 23: 327-64.
- Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab 1994, 79: 923-31.
- Dorin RI, Qualis CR, Crapo LM. Diagnosis of adrenal insufficiency. Ann Intern Med 2003, 139: 194-204.
- Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab 1992, 75: 259-64.
- Laureti S, Arvat E, Candeloro P, et al. Low dose (1 µg) ACTH test in the evaluation of adrenal dysfunction in pre-clinical Addison’s disease. Clin Endocrinol 2000, 53: 107-15.
- Falorni A, Nikoshkov A, Laureti S, et al. High diagnostic accuracy for idiopathic Addison's disease with a sensitive radiobinding assay for autoantibodies against recombinant human 21-hydroxylase. J Clin Endocrinol Metab 1995, 80: 2752-5.
- Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab 2009, 94: 1059-67.
- Crown A, Lightman S. Why is the management of glucocorticoid deficiency still controversial: a review of the literature. Clin Endocrinol 2005, 63: 483-92.
- Hahner S, Allolio B. Therapeutic management of adrenal insufficiency. Best Pract Res Clin Endocrinol Metab 2009, 23: 167–79.
- Quinkler M, Hahner S. What is the best long-term management strategy for patients with primary adrenal insufficiency? Clin Endocrinol 2012, 76: 21–5.
- Dettagli
Clinica e diagnostica iposurrenalismo primario
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
(aggiornato al 10 settembre 2015)
Clinica
Occorre distinguere il quadro clinico dell’insufficienza surrenalica acuta da quella cronica (1).
Nell’insufficienza surrenalica acuta, secondo uno studio tedesco (2) condizione più frequente di quanto si riteneva in passato (6.3 casi per 100 pazienti/anno), il quadro clinico è drammatico e, se non si realizza un intervento terapeutico immediato, porta rapidamente all’exitus. Spesso l’insufficienza surrenalica acuta si manifesta in conseguenza di stress intercorrenti (malattie febbrili, sepsi, traumi, interventi chirurgici) in un paziente affetto, talvolta inconsapevolmente, da insufficienza surrenalica cronica latente; più raramente è causata da un'emorragia surrenalica bilaterale in corso di sepsi, ritenuta oggi espressione di una variante particolarmente grave di coagulazione intravascolare disseminata (sindrome di Waterhouse-Friederichsen), oppure in seguito a vomito, diarrea o malattie febbrili intercorrenti in un paziente affetto da insufficienza surrenalica cronica che non ha modificato la propria terapia glucocorticoidea sostitutiva in maniera adeguata (1-3).
Il paziente presenta le manifestazioni cliniche dello shock ipovolemico, con profonda prostrazione, confusione, ipotensione arteriosa, tachicardia, nausea, vomito, disidratazione, talvolta dolori crampiformi all’addome (pseudo-addome acuto); la febbre può essere manifestazione di tale condizione o del processo morboso scatenante (1-3).
Nell’insufficienza surrenalica cronica frequentemente i segni e i sintomi presentano un esordio graduale, sono aspecifici, dovuti alla cronica carenza ormonale (1,3).
In particolare, tra i sintomi si annoverano astenia, anoressia, nausea, vomito, ricerca di cibi salati, epigastralgie e algie addominali, irritabilità e depressione, calo ponderale, polimialgie diffuse, vertigini, riduzione o perdita della libido (nel sesso femminile). Tali sintomi si aggravano col tempo. Inizialmente si ha infatti una fase di ridotta riserva surrenalica, con secrezione di glicocorticoidi conservata in condizioni basali, ma insufficiente in situazioni di stress; quando la perdita di tessuto corticale raggiunge circa il 90%, si ottiene il quadro completo di insufficienza surrenalica (1,3).
I segni clinici più tipici sono caratterizzati da iperpigmentazione cutanea o melanodermia (con localizzazione prevalentemente nelle zone esposte alla luce e allo sfregamento, quali volto, gomiti, ginocchia, pliche palmari, areole mammarie, cicatrici, e delle mucose della guancia, lingua, gengive, mucosa anale e vulvo-vaginale), ipotensione arteriosa, iposodiemia, iperpotassiemia, anemia, linfocitosi ed eosinofilia, TSH aumentato, ipercalcemia, ipoglicemia (più frequentemente si manifesta a digiuno, dopo attività fisica o dopo assunzione di alcolici), riduzione e successivamente scomparsa dei peli pubici e ascellari (nel sesso femminile), alterazioni mestruali di grado assai variabile (1,3).
Nella storia naturale della forma autoimmune sono state peraltro descritte due fasi pre-cliniche, prive di segni o sintomi, e caratterizzate da alterazioni auto-anticorpali (fase potenziale, stadio 0) o biochimiche/ormonali (fase subclinica, stadio 1, 2 e 3), come illustrato in figura 1 (4).
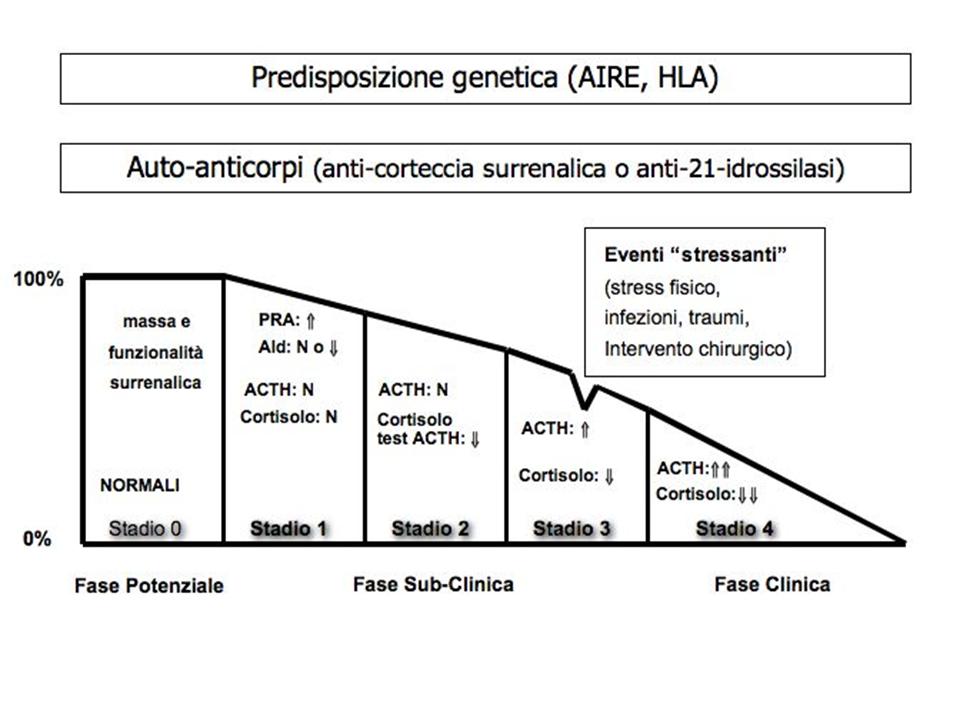
In alcune forme non autoimmuni (es. forma infettiva) può essere presente un deficit secretivo parziale della sola zona fascicolata, con preservata secrezione mineralcorticoide e mancanza di ipotensione arteriosa, iposodiemia ed iperpotassiemia (1,3).
Diagnostica
Nell’insufficienza surrenalica acuta, la condizioni di urgenza del quadro clinico, che richiede un trattamento tempestivo, non consente una valutazione accurata della funzione surrenalica e pertanto ci si dovrà limitare al dosaggio basale di cortisolemia e di ACTH (1-3,5).
Nell’insufficienza surrenalica cronica, la diagnosi di iposurrenalismo primario è difficile nelle fasi pre-cliniche della forma autoimmune, ma la melanodermia non diversamente spiegabile deve sempre suggerire la presenza di iposurrenalismo (1,3,5-7).
In tutti i pazienti con sospetto clinico va eseguita una valutazione ormonale basale, con dosaggio al mattino (ore 7-9) di cortisolemia, ACTH, renina o attività reninica ed aldosterone (1,3,5-7), anche se queste ultime valutazioni non sono universalmente eseguite per l’estrema variabilità dei risultati, sia legata al metodo di dosaggio che a fattori interferenti, e per una bassa predittività di malattia rispetto a cortisolemia e ACTH (3); il dosaggio del DHEA-S non è attualmente riconosciuto come criterio diagnostico aggiuntivo (1,3,5-7).
La diagnosi di iposurrenalismo primario viene posta in presenza di valori ridotti di cortisolemia ed aumentati di ACTH. In particolare secondo una recente Consensus Europea (3), valori di cortisolemia < 9 µg/dL (250 nmol/L) risultano diagnostici, mentre valori di cortisolemia < 14 µg/dL (400 nmol/L) risultano suggestivi per iposurrenalismo primario. Per quanto riguarda i valori di ACTH, si considerano aumentati se > 100 pg/mL (22 pmol/L) (1,5-7). Qualora si valutino anche renina o attività reninica ed aldosterone, sono da considerarsi diagnostici per iposurrenalismo primario valori aumentati di renina o attività reninica plasmatica (PRA > 3.0 ng/dL/h) e valori di aldosterone normali o ridotti (< 5 ng/dL) (1,5-7).
In tutti i pazienti con forte sospetto clinico, anche se la valutazione ormonale basale è normale, va comunque effettuata una valutazione ormonale dinamica mediante test con ACTH sintetico alla dose di 250 µg con valutazione della cortisolemia: un picco di cortisolemia < 500-550 nmol/L (18-22 µg/dL) va ritenuto indicativo di iposurrenalismo primitivo soltanto nell’ambito di un adeguato contesto clinico; è stato suggerito l’impiego di cut-off di picco di cortisolo più bassi (< 415 nmol/L, 15 µg/dL) per aumentare la sensibilità del test (1,3,5-7).
In pazienti con forme “mild” o subcliniche, è stato proposto da alcuni autori Il test con ACTH sintetico alla dose di 1 µg, ma non è stato ancora universalmente accettato come test di provata superiorità diagnostica (8).
Nel sospetto di una forma autoimmune vanno eseguiti specifici esami anticorpali (di non frequente disponibilità), quali anticorpi anti-corteccia surrenalica (ACA) e anticorpi anti-21-idrossilasi (3,4,9). Gli ACA e gli anti-21-idrossilasi sono immunoglobuline organo-specifiche, dimostrabili rispettivamente mediante tecniche di immunofluorescenza indiretta e di immunoprecipitazione o, più recentemente per gli anti-21-idrossilasi, mediante tecniche radioimmunologiche. Entrambi gli anticorpi riconoscono il loro auto-antigene nell’enzima 21-idrossilasi. Mentre la sensibilità dei due anticorpi è pressoché identica all’esordio della malattia, la sensibilità degli anti-21-idrossilasi è superiore in pazienti con lunga durata di malattia. La specificità diagnostica degli anti-21-idrossilasi è estremamente elevata e la loro presenza costituisce un fattore di rischio per lo sviluppo di insufficienza cortico-surrenalica clinica, in funzione del titolo anticorpale, dell’età del paziente (se presenti in età infantile il rischio a 15 anni è del 100%), del contesto clinico (presenza di altre patologie autoimmuni) (3,4,9).
Nella diagnostica dell’iposurrenalismo primario le indagini strumentali non sono generalmente necessarie nella forma ad eziologia autoimmune, possono essere invece indicate (TAC o RMN addome senza mdc) nel sospetto di forme infiltrative, emorragiche o neoplastiche (1,3).
Bibliografia
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003, 361: 1881-93.
- Hahner S, Loeffler M, Bleicken B, et al. Epidemiology of adrenal crisis in chronic adrenal insufficiency: the need for new prevention strategies. Eur J Endocrinol 2010, 162: 597–602.
- Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014, 275: 104-15.
- Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndrome: autoantibodies, autoantigens, and the applicability in diagnosis and disease prediction. Endocr Rev 2002, 23: 327-64.
- Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab 1994, 79: 923-31.
- Dorin RI, Qualis CR, Crapo LM. Diagnosis of adrenal insufficiency. Ann Intern Med 2003, 139: 194-204.
- Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab 1992, 75: 259-64.
- Laureti S, Arvat E, Candeloro P, et al. Low dose (1 µg) ACTH test in the evaluation of adrenal dysfunction in pre-clinical Addison’s disease. Clin Endocrinol 2000, 53: 107-15.
- Falorni A, Nikoshkov A, Laureti S, et al. High diagnostic accuracy for idiopathic Addison's disease with a sensitive radiobinding assay for autoantibodies against recombinant human 21-hydroxylase. J Clin Endocrinol Metab 1995, 80: 2752-4.
- Dettagli
Classificazione degli iposurrenalismi primari
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
Gli iposurrenalismi primitivi sono classificabili dal punto di vista eziopatogenetico nelle seguenti forme (1,2):
- autoimmune (80-90% dei casi, forma più frequente nei paesi industrializzati): isolata (40%, più frequentemente nel sesso maschile) o parte di una sindrome poliendocrino-autoimmune (60%, più frequentemente nel sesso femminile, tipi 1 o 2);
- infettiva (TBC, HIV, CMV, miceti);
- infiltrativa (sarcoidosi, amiloidosi, emocromatosi, istiocitosi);
- emorragica (S. di Waterhouse-Friederichsen, terapia anti-coagulante, traumi);
- trombotica (LES, panarterite nodosa, sindrome da anticorpi anti-fosfolipidi, traumi);
- neoplastica (carcinoma surrenalico, metastasi);
- congenite (adrenoleucodistrofia, iperplasia surrenalica congenita, ipoplasia surrenalica congenita, sindromi familiari da resistenza all’ACTH);
- da farmaci (mitotane, aminoglutetimide, chetoconazolo, mifepristone);
- iatrogena (interventi di surrenectomia bilaterale).
Bibliografia
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003, 361: 1881-93.
- Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndrome: autoantibodies, autoantigens, and the applicability in diagnosis and disease prediction. Endocr Rev 2002, 23: 327-64.