Dipendenze da steroidi anabolizzanti

Salvatore Monti e Maria Grazia Deiana
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Facoltà di Medicina e Psicologia, Sapienza Università di Roma
(aggiornato al 21 febbraio 2017)
Gli steroidi anabolizzanti
Sono ormoni che includono il testosterone e i suoi analoghi sintetici. Questi sono modificati chimicamente in modo tale da potenziare gli effetti anabolizzanti e ridurre gli effetti androgenici. La terminologia corretta per queste sostanze è “steroidi anabolizzanti-androgenici” (AAS).
Gli steroidi anabolizzanti sono stati studiati a partire dalla fine degli anni ‘30 e inizialmente impiegati nel trattamento dell’ipogonadismo. Alcuni studi sperimentali sulle cavie mostrarono che gli steroidi favorivano la crescita dell’apparato muscolo-scheletrico; pertanto queste sostanze sono state utilizzate dai culturisti e, successivamente, il loro utilizzo si è diffuso nelle altre discipline sportive. Gli steroidi anabolizzanti sono utilizzati anche a scopo non sportivo: per esempio per aumentare la massa muscolare e migliorare l’aspetto fisico.
Diversi studi, soprattutto condotti in Nord America, hanno evidenziato che il 3-12% degli adolescenti maschi usano gli agenti anabolizzanti per questo scopo, mentre nelle donne l’uso è tra 1-2%. Attualmente gli steroidi anabolizzanti sono prescritti negli ipogonadismi, angioedema ereditario, disordini ematologici, condizioni di aumentato catabolismo (cachessia neoplastica), disfunzioni metaboliche causate da ustioni gravi, malattie infiammatorie polmonari, terapia radiante e malnutrizione AIDS-correlata.
Effetti
L’assunzione di tali sostanze ha una serie di effetti, tra i quali la crescita dell’apparato muscolo-scheletrico e la riduzione della massa grassa (effetti anabolizzanti) e lo sviluppo di caratteristiche sessuali maschili (effetti androgeni). L’incremento della massa muscolare indotto dal testosterone è dovuto a un aumento nella sintesi delle proteine muscolari. L’uso di dosi sovrafisiologiche permette l’aumento della forza e della velocità della contrazione muscolare e l’accelerata ripresa dopo un intenso esercizio fisico, stimolando inoltre l’eritropoiesi ed incrementando la sintesi di 2-3 difosfoglicerato (facilitando in questo modo il trasporto di ossigeno).
Gli effetti derivanti dall’assunzione in eccesso di AAS riguardano anche il comportamento: infatti, gli steroidi aumentano l’aggressività, diminuiscono la percezione della fatica, inducono uno stato di euforia ed incremento delle energie, aumentano il rischio di disordini psicotici e il rischio di suicidio/omicidio. Gli steroidi sembrano contribuire a indurre disturbi psichiatrici in soggetti predisposti.
Effetti collaterali
Malattia coronarica, ipertrofia ventricolare, eventi aritmici, danno epatico e epatite, insulino-resistenza. In tabella sono riportati gli effetti collaterali di elevate dosi di AAS sulla riproduzione e sulla funzione sessuale.
| Effetti collaterali endocrini degli anabolizzanti | ||
| Nel maschio | Nella femmina | In entrambi i sessi |
| Soppressione della spermatogenesi Atrofia testicolare Calvizie Ginecomastia Perdita della libido Disfunzione erettile Iperidrosi Ipogonadismo iatrogeno |
Anovulazione e amenorrea Dismenorrea Irsutismo e alopecia Atrofia mammaria Ipertrofia clitoridea Disfonia Approfondimento irreversibile della voce |
Soppressione delle gonadotropine Infertilità Striae distensae (o smagliature) Acne |
Secondo diversi studi, gli effetti indesiderati indotti da AAS sono reversibili alla sospensione, ma possono diventare irreversibili se l’abuso di AAS è di lunga durata.
Dipendenza
In letteratura non sono stati riportati dati riguardo la comparsa di dipendenza da AAS a dosi terapeutiche. L’uso per scopi non medici di AAS e, quindi, l’uso a dosi non terapeutiche, non causa un’intossicazione acuta (come accade per altre sostanze quali narcotici/allucinogeni/stimolanti), ma può determinare abuso e scatenare dipendenza. È stato recentemente calcolato che la prevalenza media di insorgenza di dipendenza da AAS è del 32.5-35%, nei soli studi americani.
Il motivo per cui alcuni uomini manifestino dipendenza da AAS non è ancora ben chiaro, sebbene l’ipotesi più accreditata sia una maggior suscettibilità individuale alla necessità di continuare l’uso degli steroidi. Il rischio di sviluppare dipendenza da steroidi è maggiore in coloro che presentano insoddisfazione della propria immagine corporea. La dismorfia muscolare è, infatti, una condizione nella quale l’individuo vede il proprio corpo debole e esile, nonostante sia invece normale e muscoloso, e rientra tra i criteri per la diagnosi del disordine dismorfico corporeo secondo il DSM5. Alcuni individui potrebbero inoltre essere maggiormente suscettibili agli effetti legati alla sospensione degli steroidi, quali l’ipogonadismo e i sintomi affettivi.
Nell’uomo la dipendenza da AAS è caratterizzata dalla comparsa della “sindrome da astinenza”, mediata dal sistema dei neurotrasmettitori neuroendocrini e corticali, che si caratterizza per la presenza di depressione, astenia, anoressia, insonnia, riduzione della libido e necessità di voler continuare l’uso nonostante vi siano effetti collaterali legati all’uso (“craving”); tali sintomi possono persistere per settimane o mesi.
La fase iniziale della sindrome da astinenza (durata 1 settimana) è simile a quella da oppiacei, mentre la seconda fase è caratterizzata prevalentemente da sintomi depressivi e “craving”. Vi è una forte associazione tra dipendenza da AAS e dipendenza da oppioidi. Infatti, nel 1989 è stato ipotizzato che la dipendenza da AAS segua un meccanismo oppioidergico, attraverso il quale gli AAS potrebbero incrementare l’attività degli oppioidi endogeni centrali; la sospensione degli AAS, riducendo questa attività, causerebbe una sindrome iperadrenergica acuta. Questa associazione è stata successivamente confermata: gli individui che abusano di AAS sono particolarmente a rischio di sviluppare abuso e dipendenza anche da oppioidi.
Gli AAS presentano importanti effetti modulatori su serotonina, norepinefrina, dopamina e GABA. Studi animali hanno inoltre mostrato che gli AAS modulano gli effetti di altre droghe d’abuso, come stimolanti del sistema nervoso centrale, cannabis e alcool.
Non ci sono evidenze riguardo il trattamento farmacologico della dipendenza da AAS, per cui la maggior parte dei pazienti è trattata sulla base dei sintomi presenti. Va favorito il ripristino della funzione dell’asse ipotalamo-ipofisi-gonade, magari avvalendosi della terapia con gonadotropina corionica o con clomifene. Il trattamento della dipendenza da AAS può avvalersi inoltre della psicoterapia cognitivo-comportamentale, degli anti-depressivi serotoninergici (inibitori selettivi del reuptake di serotonina come la fluoxetina), nel caso in cui la problematica più rilevante sia la dismorfia muscolare.
Bibliografia
- Jenssen IH, Johannessen KB. Aggression and body image concerns among anabolic androgenic steroid users, contemplators, and controls in Norway. Body Image 2015, 12: 6–13.
- Mhillaj E, et al. Effects of anabolic-androgens on brain reward function. Front Neurosci 2015, 9: 295.
- Pope HG Jr, et al. The lifetime prevalence of anabolic-androgenic steroid use and dependence in Americans: current best estimates. Am J Addict 2014, 23, 371–7.
- Kanayama G, et al. Treatment of anabolic-androgenic steroid dependence: emerging evidence and its implications. Drug Alcohol Depend 2010, 109: 6-13.
Gestione delle malattie intercorrenti e degli interventi chirurgici nel paziente endocrinopatico
Anestesia in pazienti con patologie endocrino-metaboliche
Neurochirurgia:
- Chirurgia della regione ipotalamo-ipofisaria
- Gestione peri-operatoria adenomi ipofisari
- Terapia neurochirurgica NFPA
- Terapia neurochirurgica prolattinomi
- Terapia neurochirurgica dell'acromegalia
- Terapia neurochirurgica del Cushing
- Terapia neurochirurgica del craniofaringioma
- Terapia chirurgica per l'orbitopatia di Graves
Chirurgia del collo:
- Terapia chirurgica tradizionale per la patologia tiroidea benigna
- Terapia chirurgica dei tumori tiroidei differenziati
- Terapia chirurgica mini-invasiva per la tiroide
- Chirurgia robotica per la patologia tiroidea
- Terapia chirurgica del carcinoma midollare
- Chirurgia radio/ecoguidata delle recidive locali di tumore tiroideo
- Terapia chirurgica dell'iperparatiroidismo primitivo
- Terapia chirurgica per l'iperparatiroidismo nell'ambito della MEN-1
- Terapia chirurgica per l'iperparatiroidismo nell'ambito della MEN-2
Chirurgia dell'addome e della pelvi:
- Chirurgia surrenalica
- Chirurgia bariatrica
- Gestione del diabete durante il ricovero in area chirurgica
- Terapia chirurgica per i NET
- Chirurgia per i NET duodeno-pancreatici nell'ambito della MEN-1
- Chirurgia per il feocromocitoma nell'ambito della MEN-2
- Iperplasia e carcinoma della prostata
- Terapia dei tumori ovarici
- Terapia dei tumori testicolari
- Attribuzione del genere e correzione delle ambiguità genitali
Varie:
- Terapia chirurgica per il piede diabetico
- La riattribuzione chirurgica del sesso nelle disforie di genere
- Terapia chirurgica per la disfunzione erettile
Modificazioni surrenaliche nel paziente critico
Ernesto De Menis, Francesco Frassoni
UO Medicina Interna, Dipartimento Medicina Clinica, Ospedale Generale, Montebelluna
Aspetti fisiopatologici
Le “malattie critiche” rappresentano situazioni cliniche in cui è richiesta un’estrema risposta adattativa alla condizione di “stress” e quindi è attivato l’asse CRH-ACTH-surreni (HPA). Le alterazioni surrenaliche sono diverse in relazione al tipo di “critical illness”, ad es. trauma, intervento chirurgico, eventi cardiovascolari, ma sicuramente risultano massime nella sepsi, in particolare nello shock settico (1). L’attivazione dell’asse HPA varia anche in relazione alla durata della condizione critica, come dimostrato da studi che hanno valutato sequenzialmente la risposta dell’asse HPA durante la degenza nelle unità di cure intensive (UCI); gli studi più estesi riguardano i pazienti ricoverati in UCI con sepsi.
Nelle fasi iniziali si ha un'attivazione del sistema HPA. Si ritiene che la secrezione di cortisolo aumenti per l’incremento della secrezione di ACTH sostenuta da un aumento di CRH e AVP. Tuttavia, la secrezione di cortisolo risulta essere sostenuta anche da stimoli ACTH-indipendenti, in modo particolare da citochine rilasciate durante la sepsi, e un ruolo importante sembrano avere i Toll-Like receptors stimolati da endotossine. In una fase successiva si osserva una riduzione delle concentrazioni di ACTH, ma le concentrazioni di cortisolo rimangono elevate: questa dissociazione è stata classicamente attribuita a meccanismi di stimolo surrenalico non ACTH-dipendenti (2). Tuttavia recentemente, utilizzando metodi di diluizione radioisotopica, è stato osservato che l’aumento della cortisolemia è sostenuto solo in minor parte dall’aumento della sintesi di cortisolo (aumento di due volte rispetto ai pazienti non critici), e per la maggior parte è dovuto all’aumento dell’emivita del cortisolo (circa cinque volte superiore) per una ridotta clearance (3). L’ipercortisolemia sarebbe comunque responsabile della riduzione della secrezione di ACTH. Se la degenza nelle UCI si prolunga, la riduzione delle concentrazioni di ACTH e quindi la minor stimolazione del surrene potrebbero determinare una condizione di iposurrenalismo tardivo (4). Nelle condizioni di sepsi si osservano anche alterazioni recettoriali (dei recettori glucocorticoidi) e post-recettoriali, che causano una relativa “resistenza” periferica ai glucocorticoidi (GC) (5).
L’attivazione fisiopatologica dell’asse HPA sembra conferire protezione rispetto alla malattia critica, per esempio garantendo la disponibilità di substrati energetici e la riduzione della flogosi, ma il protrarsi nel tempo di uno stato di iperattivazione glucocorticoide potrebbe determinare effetti negativi, quali aumento del catabolismo, miopatia e aumento di suscettibilità alle infezioni.
Valutazione funzione surrenalica
Nei pazienti con malattia critica l’insufficienza surrenalica assoluta può essere pre-esistente o può svilupparsi per la stessa malattia critica o le sue terapie. Fra le condizioni che possono causare insufficienza surrenalica de novo ricordiamo i traumi cranici e le emorragie subaracnoidee con insufficienza surrenalica secondaria, le sepsi per le alterazioni del microcircolo che possono causare emorragia surrenalica (come nella sepsi da meningococco) o trombosi surrenalica per lo stato ipercoagulativo, l’utilizzo di farmaci come etomidate.
L’argomento più controverso è l'esistenza di un'insufficienza surrenalica relativa, definita più recentemente come 'critical illness-related corticosteroid insufficiency' (CIRCI) (6). Il presupposto di tale concetto deriva da studi che avevano dimostrato che, nei pazienti critici, la sopravvivenza era inferiore nei soggetti che avevano una ridotta risposta all’ACTH test nonostante elevati livelli basali di cortisolemia. La condizione è caratterizzata da un difetto primariamente funzionale e transitorio della secrezione e/o dell’azione dei GC (1). Il sospetto di CIRCI nello shock settico deve essere primariamente formulato su base clinica, per la presenza di ipotensione nonostante l’espansione volemica e l'infusione di amine pressorie (7). Rimane invece controversa la definizione su base biochimica-ormonale, a causa delle varie interferenze sulle concentrazioni plasmatiche di cortisolo, dell’assenza di cut-off biochimici e dell’impossibilità pratica di valutare la resistenza periferica al cortisolo. Anche il test con ACTH, che era stato ampiamente utilizzato per identificare i pazienti con CIRCI, in genere utilizzando come cut-off di risposta l’aumento della cortisolemia di 10 µg/dL, non viene più considerato indispensabile nella diagnosi e selezione dei pazienti da trattare con GC (7); le più recenti linee guida suggeriscono di non eseguire più il test all’ACTH e di utilizzare un valore random di cortisolemia < 18 µg/dL come criterio per un eventuale utilizzo degli steroidi.
Terapia farmacologica con glucocorticoidi (alte dosi)
La terapia con dosi farmacologiche (alte dosi) di GC è stata ampiamente utilizzata in passato. Una serie di studi randomizzati e metanalisi (8) ha permesso di concludere che i GC si associano ad aumento della mortalità nella sepsi e probabilmente anche nello shock settico, per un aumento del rischio infettivo ed emorragico. Pertanto i GC ad alte dosi sono sconsigliati.
Terapia sostitutiva con glucocorticoidi
Il concetto di insufficienza surrenalica relativa nei pazienti con sepsi ha portato, dopo il fallimento della terapia ad alte dosi, all’introduzione della terapia con GC a dosi “sostitutive”: studi iniziali nello shock settico avevano suggerito che l’idrocortisone potesse ridurre il tempo di svezzamento dalle amine ed anche aumentare la sopravvivenza. Il primo studio randomizzato mirato (9), che utilizzava idrocortisone 150 mg/die (assieme a fludrocortisone), aveva dimostrato, nei pazienti con shock settico ed insufficienza surrenalica relativa (ACTH test), un aumento di sopravvivenza e una più rapida reversibilità dello shock. Tuttavia, il successivo studio randomizzato (CORTICUS) (10) non ha confermato tali risultati. In particolare, la sopravvivenza dei pazienti era sovrapponibile nei trattati e non trattati, ed anche i pazienti non responsivi all’ACTH test non mostravano miglioramenti. I due studi avevano differenze metodologiche, in particolare i pazienti di Annane (9) avevano un quadro più severo rispetto a quelli dello studio Corticus (10). Tuttavia, entrambi hanno contribuito a ridefinire le linee guida della sepsi e dello shock settico (7). Le linee guida suggeriscono che non si debbano utilizzare i GC nella semplice sepsi, mentre nello shock settico si suggerisce l’utilizzo di idrocortisone solo nei soggetti che non presentano risposta all'espansione volemica e alle amine pressorie. In questo caso la dose consigliata è idrocortisone 200 mg/die, e si deve utilizzare la via infusionale continua e non i boli. L'idrocortisone deve essere rapidamente sospeso quando non vi è più necessità di amine pressorie. Infine, per diagnosticare un'insufficienza surrenalica, non deve essere utilizzato il test all’ACTH ma è preferibile la cortisolemia con un cut-off di 18 µg/dL (7).
Conclusioni
I pazienti con malattia critica possono avere un'insufficienza surrenalica pre-esistente, ma bisogna sempre tenere presente che l’insufficienza surrenalica può svilupparsi in UCI per effetto della malattia critica stessa o dei suoi trattamenti farmacologici. Il concetto di ‘critical illness-related corticosteroid insufficiency’ è ancora ampiamente controverso. Gli studi clinici hanno condotto a linee guida che controindicano l’utilizzo di GC ad alte dosi nei pazienti settici, e riservano l’utilizzo di dosi “fisiologiche” di idrocortisone solo in pazienti con shock settico non responsivo ad espansione volemica e amine pressorie.
Bibliografia
- Boonen E, Van Den Berghe G. New concepts to further unreveal adrenal insufficiency during critical illness. Eur J Endocrinol 2016, 175: R1–9.
- Deak T. Cells and cytokine circuits: toward a working model for understanding direct immune-to-adrenal communication pathways. Endocrinology 2008, 149: 1433–5.
- Boonen E, et al. Reduced cortisol metabolism during critical illness. N Engl J Med 2013, 368: 1477-88.
- Boonen E, et al. Impact of duration of critical illness on the adrenal glands of human intensive care patients. J Clin Endocrinol Metabol 2014, 99: 4214–22.
- Molijn GJ, et al. Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. J Clin Endocrinol Metab 1995, 80: 1799–803.
- Marik PE, et al. Critical illness-related corticosteroid insufficiency. Chest 2009, 135: 181–93.
- Dellinger RP, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med 2013, 39: 165–228.
- Cronin DJ, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med 1995, 23: 1430–9.
- Annane D, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 2002, 288: 862-71.
- Sprung CL, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008, 358: 111-24.
Modificazioni metaboliche nel paziente critico
Luigi Magnani1 & Carmelo Sgarlata2
1UOC Medicina Interna, Ospedale di Voghera, ASST Pavia
2Istituto di Cura S. Margherita, Azienda di Servizi alla Persona di Pavia
IPERGLICEMIA
Prevalenza e fisiopatologia nel paziente critico
Il riscontro di iperglicemia nel paziente ricoverato con malattia critica è estremamente frequente: la prevalenza varia tra il 30 e il 40%, arrivando fino all’80% nelle unità di terapia intensiva (UTI), dove circa 8 su 10 pazienti iperglicemici non hanno una precedente diagnosi di diabete (1).
Una così elevata prevalenza di iperglicemia nei pazienti critici trova spiegazione in precise basi fisiopatologiche. Qualunque sia la patologia in atto, in questi pazienti si verifica una complessa attivazione neuro-ormonale (“stress response”), volta a preservare la sopravvivenza immediata dell’organismo, che innesca multipli meccanismi che conducono all’iperglicemia (2):
- rapido instaurarsi di uno stato di insulino-resistenza (principalmente in conseguenza dell’azione di citochine infiammatorie, quali TNF-α e IL-1 e 6);
- aumentata gluconeogenesi e glicogenolisi (per netto incremento nel paziente critico di ormoni quali cortisolo, catecolamine, GH, glucagone);
- concomitante riduzione della secrezione insulinica per inibizione delle β-cellule pancreatiche da parte della massiva attivazione α-adrenergica.
Inoltre, spessissimo questi pazienti vengono trattati con amine simpatico-mimetiche, steroidi, glucosio e supporti nutrizionali ev, che a loro volta tendono a indurre iperglicemia.
Significato prognostico
La presenza e l’entità dell’iperglicemia all’ingresso in ospedale costituiscono fattori prognostici sfavorevoli nel paziente critico e correlano strettamente con l’aumento della mortalità intra-ospedaliera e a lungo termine (3). In particolare, l’iperglicemia di nuovo riscontro (insorta cioè in pazienti non noti come diabetici) sembra correlare con un maggiore rischio di morte, non solo rispetto a quello dei soggetti euglicemici ma anche a quello dei diabetici noti. Tuttavia, la rilevanza clinica dell’iperglicemia nel paziente critico è stata a lungo ignorata, in quanto veniva in qualche modo considerata una risposta adattativa dell’organismo a fronte di un’aumentata richiesta energetica in condizioni estreme.
Il trattamento dell’iperglicemia nel paziente critico
Negli ultimi quindici anni il problema della gestione dell’iperglicemia nel paziente critico ha destato grande interesse nella comunità scientifica internazionale, come dimostrato dai numerosi studi pubblicati sull’argomento, talora dai risultati controversi.
Già nel 1997 lo studio DIGAMI (4) aveva dimostrato l’efficacia di un trattamento ipoglicemizzante intensivo (somministrazione di insulina in infusione continua in fase acuta, seguita nel follow-up da trattamento insulinico sottocutaneo intensivo) nel ridurre la mortalità in pazienti diabetici con infarto miocardico acuto. Nel 2001 la pubblicazione del “Leuven Surgical Trial” (5) accende un enorme interesse sull’argomento. In questo studio monocentrico in un’UTI chirurgica il mantenimento di uno stretto controllo glicemico (obiettivo glicemico 80-110 mg/dL) con trattamento insulinico intensivo ev determinava un’impressionante riduzione della mortalità relativa (-42% durante la degenza in UTI, -34% durante la degenza in ospedale), nonchè una netta riduzione dell’incidenza di gravi complicanze quali sepsi e insufficienza d’organo. Nonostante il grande entusiasmo destato dal Leuven trial, nessuno dei numerosi studi pubblicati negli anni successivi è stato in grado di replicare risultati così favorevoli.
Nel 2009 i risultati del vasto studio multicentrico internazionale “Nice-Sugar” (6), volto a valutare l’utilità di un controllo glicemico intensivo in pazienti ricoverati in UTI mediche e chirurgiche, non solo non hanno confermato i vantaggi di uno stretto controllo glicemico nel paziente critico, ma hanno evidenziato un significativo aumento di mortalità tra i pazienti sottoposti a terapia insulinica ev intensiva (obiettivo glicemico 81-108 mg/dL) rispetto a quelli trattati più permissivamente (≤ 180 mg/dL). Tra i pazienti trattati intensivamente si è evidenziato un notevole incremento del numero di ipoglicemie moderate e severe, possibile spiegazione del significativo aumento di mortalità. Va notato come esistano importanti differenze fra lo studio di Leuven e il Nice Sugar: la diversa tipologia di pazienti arruolati (solo chirurgici nel primo, chirurgici e medici nel secondo), le differenti modalità di rilevazione della glicemia (glicemia su prelievo arterioso vs reflettometria o prelievo venoso) e il diverso grado di addestramento degli operatori alla gestione del protocollo di infusione insulinica (elevato nel Leuven, standard nel Nice Sugar) potrebbero rendere ragione di risultati così differenti. In ogni caso, l’allarme provocato dai risultati del Nice Sugar ha spinto le principali linee guida internazionali a consigliare il mantenimento nel paziente critico di obiettivi glicemici meno stringenti rispetto a quanto inizialmente proposto.
Quali obiettivi glicemici, quali modalità di applicazione dei protocolli di infusione insulinica ev
Sebbene sia oggi unanimemente riconosciuto l’impatto prognostico sfavorevole dell’iperglicemia nel paziente con malattia critica e vi sia consenso generale nel considerare l’infusione insulinica ev continua quale terapia più appropriata per ottenere il controllo glicemico, vari aspetti sono ancora oggetto di discussione. Per quanto concerne gli obiettivi glicemici, gli standard 2016 per la cura del diabete dell’American Diabetes Association (7) e gli standard italiani per la cura del diabete mellito AMD-SID (8) raccomandano nel paziente critico l’utilizzo dell’infusione ev continua di insulina in presenza di persistente iperglicemia > 180 mg/dL, e confermano l’intervallo 140-180 mg/dL quale obiettivo glicemico appropriato nella maggior parte dei pazienti. In casi selezionati è consigliato un controllo glicemico più stretto (110-140 mg/dL), ma solo se ciò non comporta aumento del rischio d’ipoglicemia. Va però sottolineato come la scelta di questi obiettivi sia basata sul consenso degli esperti e su condivisibili preoccupazioni relative alla sicurezza del trattamento, ma non sia di fatto sostenuta da chiare evidenze.
Per ciò che riguarda le modalità di somministrazione dell’insulina, molti sono i protocolli validati presenti in letteratura, e non esistono studi clinici randomizzati di confronto tra essi. Un protocollo ideale deve (9):
- consentire di raggiungere l’intervallo glicemico desiderato rapidamente e in sicurezza (con una ridotta variabilità glicemica e un basso rischio d’ipoglicemia);
- prevedere un preciso algoritmo, che possa essere implementato in modo semplice e che comporti un ragionevole carico di lavoro (soprattutto infermieristico);
- prevedere una gestione quasi totalmente infermieristica con supervisione medica;
- includere un preliminare adeguato programma formativo degli operatori.
Tra i protocolli maggiormente utilizzati in area medica va ricordato quello di Yale (10), un algoritmo dinamico che considera nel calcolo della dose insulinica non soltanto il valore assoluto della glicemia ma anche l’andamento glicemico (direzione e velocità di modificazione dei valori di glicemia). In area chirurgica è invece molto utilizzato il protocollo di Markovitz (11), costituito da 4 algoritmi diversi alternativamente utilizzati per regolare la velocità di infusione dell’insulina sulla base dei valori glicemici.
Indipendentemente dal protocollo scelto, è sempre necessario:
- stimare attentamente il rischio di ipoglicemia, in base alle condizioni cliniche del singolo paziente, prima di decidere di iniziare la somministrazione ev di insulina;
- utilizzare insulina umana regolare, da somministrare mediante utilizzo di pompa-siringa previa diluizione in soluzione fisiologica (0.9% NaCl) nel rapporto di 1 UI/mL; qualsiasi altra modalità di somministrazione non è raccomandata, poiché non consente la necessaria flessibilità e la precisione posologica richiesta da tutti i protocolli;
- prevedere un protocollo di transizione dalla somministrazione di insulina ev alla terapia sottocutanea al termine della fase critica.
La scelta del protocollo per infusione insulinica è una decisione complessa, che non può prescindere dall’attenta analisi dello specifico setting clinico nel quale l’algoritmo dovrà essere utilizzato. L’implementazione di protocolli di questo tipo, infatti, non è ritenuta sicura e non è raccomandata in contesti in cui il supporto infermieristico non sia adeguato per eseguire con tempestività le numerose rilevazioni glicemiche previste dall’algoritmo e calcolare e attuare con precisione le conseguenti correzioni della velocità di infusione dell’insulina. Ciò va tenuto molto ben presente, in particolare al di fuori delle UTI, in reparti di tipo medico o chirurgico, ove l’aumento del carico di lavoro infermieristico necessario all’implementazione del protocollo può costituire un importante fattore limitante per un suo utilizzo in condizioni di sicurezza.
Notevoli vantaggi pratici sembrano emergere dalle iniziali esperienze di autori che hanno fatto ricorso all’utilizzo di sistemi automatici di rilevazione della glicemia (dispositivi sottocutanei per il monitoraggio glicemico continuo) e all’informatizzazione dei protocolli, che pare consentire, rispetto all’utilizzo dei protocolli cartacei, una notevole riduzione del rischio di errore nel calcolo della velocità di infusione dell’insulina e una significativa riduzione del carico di lavoro infermieristico (12).
IPOGLICEMIA
La più frequente alterazione della glicemia in corso di malattia critica è l’iperglicemia; con relativa frequenza però in questi pazienti si verificano anche episodi ipoglicemici, che talora fanno seguito a una prima fase iperglicemica.
Le cause di ipoglicemia nel malato critico sono numerose e coinvolgono plurimi meccanismi fisiopatologici. I depositi epatici di glicogeno sono sufficienti nella maggior parte dei pazienti critici a mantenere l’euglicemia per 8-12 ore, ma questo periodo è nettamente ridotto in casi di aumentato fabbisogno di glucosio o ridotta riserva di glicogeno, come nei soggetti con malattie croniche o malnutrizione. Nel paziente critico si ha un’esaltata glicogenolisi, con un rapido consumo delle scorte di glicogeno, mentre l’efficacia del processo di gluconeogenesi, energeticamente molto dispendioso, può risultare fortemente compromessa a causa di numerosi fattori (ipossia, acidosi, sviluppo di disfunzione renale e soprattutto epatica), risultando inadeguato al mantenimento dell’euglicemia. La sepsi, le polmoniti batteriche, lo scompenso cardiaco, l’insufficienza epatica, l’insufficienza renale acuta sono tra le patologie più comunemente associate a ipoglicemia nel paziente critico.
Il verificarsi di ipoglicemia ha un importante valore prognostico in questi pazienti, correlando significativamente con la mortalità e le complicanze. In uno studio su pazienti con polmonite acquisita in comunità, gli autori hanno osservato una significativa correlazione tra sviluppo di ipoglicemia e peggioramento degli esiti clinici, con aumento di circa 4 volte della mortalità a 30 giorni, più frequente insorgenza di complicanze gravi (come sepsi severa e shock settico) e incremento della necessità di ventilazione meccanica (29.8% vs 7.5%) o di supporto inotropo (21% vs 4.8%) (13). L’ipoglicemia che si verifica, seppur non frequentemente, nella sepsi, correla negativamente con la sopravvivenza. Nello scompenso cardiaco l’ipoglicemia è spesso dovuta a più fattori (aumento del dispendio energetico, sviluppo di ipossia tissutale, alterazioni della funzione epatica e renale) e correla con l’incremento dell’incidenza di fenomeni ischemici, aritmici e con la mortalità.
Nel paziente critico le manifestazioni cliniche dell’ipoglicemia, quali i sintomi adrenergici autonomici (palpitazioni, tremori, sudorazione, ecc) o neuroglicopenici (stato confusionale, convulsioni, perdita di coscienza, coma) possono apparire del tutto aspecifici e attribuibili ad altre cause. La loro comparsa o peggioramento impone l’immediata determinazione della glicemia, poiché in tale contesto clinico un eventuale episodio ipoglicemico intercorrente potrebbe altrimenti sfuggire. Nel monitoraggio dei pazienti con malattia critica, soprattutto in presenza di alterazioni significative dello stato di coscienza, appare quindi ragionevole consigliare determinazioni seriate dei valori glicemici, anche in presenza di precedente riscontro di euglicemia.
Trattamento dell’ipoglicemia
L’infusione endovenosa di un bolo glucosio alla dose iniziale di 25 g (soluzioni al 50% o al 33%) rappresenta il principale trattamento dell’ipoglicemia nel paziente critico. Nei casi più gravi o refrattari può essere associata la somministrazione endovenosa di glucagone, che però spesso determina nausea e vomito e successivo sviluppo di iperglicemia. Il glucagone, che agisce principalmente stimolando la glicogenolisi, non è indicato nei pazienti in cui si ipotizzi una deplezione di glicogeno come meccanismo patogenetico principale dell’ipoglicemia.
La risposta glicemica all’infusione di glucosio è spesso transitoria ed è pertanto talora necessario proseguire la somministrazione di destrosio mediante infusione di soluzioni al 5-10%, monitorando le variazioni glicemiche: ciò risulta di fondamentale importanza soprattutto in presenza di ipoglicemie di lunga durata (p.es da assunzione di sulfaniluree) (14).
BIBLIOGRAFIA
- Umpierrez GE, Isaacs SD, Bazargan N, et al. Hyperglycemia: an independent marker of in-hospital mortality in patients with undiagnosed diabetes. J Clin Endocrinol Metab 2002, 87: 978-82.
- McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin 2001, 17: 107-24.
- Falciglia M, Freyberg RW, Almenoff PL, et al. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 2009, 37: 3001-9.
- Malmberg K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. BMJ 1997, 314: 1512-5.
- van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in critically ill patients. N Engl J Med 2001, 345: 1359-67.
- NICE-SUGAR Study Investigators. Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009, 360: 1283-97.
- American Diabetes Association. Standards of Medical Care in Diabetes - 2016. Diabetes Care 2016, 39: S99–S104.
- Associazione Medici Diabetologi (AMD) - Società Italiana di Diabetologia (SID). Standard italiani per la cura del diabete mellito 2014: 231-44.
- Beltramello G, Manicardi V, Trevisan R. Trialogue. Managing hyperglycemia in internal medicine: instructions for use. Acta Diabetol 2013, 50: 465-73.
- Goldberg PA, Roussel MG, Inzucchi SE. Clinical results of an updated insulin infusion protocol in critically ill patients. Diabetes Spectrum 2005, 18: 188-91.
- Markovitz LJ, Wiechmann RJ, Harris N, et al. Description and evaluation of a glycemic management protocol for patients with diabetes undergoing heart surgery. Endocr Pract 2002, 8: 10-8.
- Beltramello G, Sgarlata C, Guerriero F, et al. Implementation of a Yale insulin infusion modified protocol in an Internal Medicine ward through the use of a digital application. Ital J Med 2015, 9 (s2): 6.
- Singanayagam A, Chalmers JD, Hill AT. Admission hypoglycaemia is associated with adverse outcome in community-acquired pneumonia. Eur Respir J 2009, 34: 932-9.
- Cryer P, Axelrod L, Grossman A, et al. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2009, 94: 709-28.
Il cuore come organo endocrino e bersaglio ormonale
Paolo Limone
SC Endocrinologia, Diabetologia e Malattie del Metabolismo, AO Ordine Mauriziano di Torino
INTRODUZIONE
Sono ben conosciuti da molti anni gli effetti sul cuore di vari ormoni “tradizionali”, quali ormoni tiroidei, catecolamine, GH, e le conseguenze di un loro eccesso o difetto. Vi sono peraltro numerosi altri ormoni, di più o meno recente individuazione, che possono esercitare importanti azioni sul cuore e sull’apparato cardio-vascolare: tra questi aldosterone, adrenomedullina, adiponectina, leptina, ghrelin. Per alcuni di questi sono stati bene accertati gli effetti sia fisiologici sia fisiopatologici sul cuore, e farmaci che interferiscono sulla loro azione sono correntemente utilizzati nel trattamento di alcune malattie cardio-vascolari per contrastare gli effetti negativi di un loro cronico eccesso. È peraltro allo studio la possibilità di utilizzare alcuni ormoni, quali GH o analoghi degli ormoni tiroidei, nel trattamento di condizioni cliniche quali lo scompenso cardiaco, per sfruttarne i potenziali effetti benefici. Per altri è stato individuato un ruolo in alcune condizioni patologiche, ma i risvolti diagnostici e terapeutici di queste conoscenze appaiono tuttora assai scarsi.
Oltre ad essere bersaglio di vari ormoni, il cuore è oggi considerato esso stesso un “organo endocrino”, in quanto le cellule muscolari cardiache sono in grado di sintetizzare e secernere varie sostanze ad azione paracrina o sistemica, per cui appare oggi ben evidente come il ruolo del cuore nella regolazione dell’omeostasi cardio-vascolare non sia limitato a una funzione di tipo meccanico, ma sia anche umorale.
IL CUORE COME ORGANO ENDOCRINO
Tra gli ormoni cardiaci, si distinguono:
- la famiglia dei peptidi natriuretici
- adrenomedullina/pro-adrenomedullina
- endotelina-1
1. Peptidi natriuretici
Si riconoscono: il Fattore Natriuretico Atriale (ANF) e il Brain Natriuretic Peptide (BNP), prodotti dai miocardiociti atriali, il Peptide Natriuretico di tipo C (CNP), riscontrato anche a livello del cuore ma prodotto prevalentemente in altre sedi (cellule muscolari lisce, endotelio, ecc).
ANF e BNP vengono secreti dagli atri in modo continuo, con velocità di secrezione aumentata dallo stiramento meccanico della muscolatura atriale. La secrezione è anche stimolata da adrenalina, endotelina-1 e, secondo recenti evidenze, glucagon-like-peptide (GLP-1).
Effetti fisiologici
- Rene: ANF e BNP esercitano effetto diuretico e natriuretico.
- Cuore e polmoni:
- riducono l’attività dei chemo- e barocettori, con conseguente riduzione dell’attività simpatica;
- aumentano l’attività parasimpatica, riducendo frequenza cardiaca e gittata cardiaca;
- riducono inoltre il tono della muscolatura liscia delle arteriole di resistenza;
- riducono crescita e proliferazione dei miocardiociti e dei fibroblasti cardiaci.
- Altri organi:
- riducono la secrezione di renina;
- inibiscono la sintesi di aldosterone e giocano un ruolo importante nel fenomeno dell’escape renale in presenza di ipersecrezione di mineralcorticoidi.
- ANF inibisce la secrezione di ADH;
Condizioni fisiopatologiche. Si osserva aumento della concentrazione dei peptidi natriuretici nelle seguenti condizioni patologiche: ipertensione arteriosa, insufficienza cardiaca, ipertrofia ventricolare sinistra, infarto miocardico, ipertensione polmonare.
Applicazioni cliniche. Il BNP ed il suo frammento NT-proBNP (dotato di maggior stabilità), sono marcatori utilizzati nella pratica clinica in casi di insufficienza cardiaca e rimodellamento ventricolare sinistro dopo infarto del miocardio (1).
2. Adrenomedullina
Appartiene alla superfamiglia della calcitonina, che comprende calcitonina, CGRP, amilina. È prodotta principalmente a livello della midollare surrenalica, mentre quantità minori sono prodotte dai miocardiociti e dai fibroblasti cardiaci e a livello arteriolare.
Possiede azioni vasodilatatrici, diuretiche e natriuretiche, nonchè effetti anti-proliferativi sui fibroblasti cardiaci e di riduzione della deposizione della matrice extra-cellulare.
Le concentrazioni plasmatiche aumentano nell’ipertensione arteriosa, nelle sindromi coronariche acute, nell’insufficienza cardiaca. Si ritiene che tale aumento abbia effetti protettivi sul cuore, riducendo sovraccarico pressorio, massa cardiaca, fibrosi.
Non vi sono al momento applicazioni cliniche del dosaggio dell’adrenomedullina (2).
3. Endotelina-1
Prodotta principalmente a livello endoteliale, viene secreta in misura minore anche dai miocardiociti.
L’endotelina-1 ha una potente e persistente azione vasocostrittrice, ha inoltre azione inotropa e cronotropa positiva sul cuore. Si pensa che l’endotelina-1 prodotta a livello cardiaco possa avere effetti locali di stimolo dell’ipertrofia nella fibrosi.
Le concentrazioni plasmatiche aumentano nell’insufficienza cardiaca, nell’infarto del miocardio, nell’ipertensione polmonare e nell’insufficienza renale.
L’antagonista bosentan è utilizzato nella pratica clinica e trova indicazione nel trattamento dell’ipertensione polmonare primitiva nei pazienti in classe funzionale III, nell’ipertensione polmonare secondaria a sclerodermia e a shunt sistemico-polmonari congeniti (3).
IL CUORE COME BERSAGLIO DI ORMONI
Ormoni tiroidei
Gli ormoni tiroidei regolano le funzioni cardio-vascolari, agendo direttamente sul cuore, sulla muscolatura liscia vascolare e sull’endotelio. Conseguenze indirette sono dovute inoltre agli effetti sulla termogenesi e sul rilascio/azione di altri ormoni, a loro volta implicati nel controllo cardio-vascolare.
Le azioni sul cuore sono svolte dalla T3 (la T4 non penetra nei miocardiociti), con azioni di tipo sia genomico sia non-genomico. La T3 aumenta la forza di contrazione del cuore e la velocità della contrazione sistolica e del rilasciamento diastolico e aumenta la frequenza cardiaca (sia come effetto diretto sia per interazione con le catecolamine); riduce inoltre le resistenze vascolari periferiche (compreso il circolo coronarico) e aumenta l’angiogenesi a livello delle arteriole coronariche. Gli ormoni tiroidei promuovono l’ipertrofia miocardica, sia fisiologica sia patologica (4).
Gli effetti genomici dipendono dall’interazione con i recettori nucleari e dalla conseguente modulazione dell’espressione di geni strutturali e regolatori, con effetto attivatore (sintesi di catene pesanti dell’α-miosina, sintesi dei recettori ß1-adrenergici, α-adrenergici, ecc) o repressore (sintesi di catene pesanti della ß-miosina, ecc); gli effetti non genomici sono mediati da recettori di superficie o extra-nucleari, si manifestano in tempi più brevi (secondi o minuti) e riguardano la modulazione diretta dei canali ionici di membrana, il contenuto intra-cellulare di calcio, la stimolazione della produzione di NO (5).
| Tabella 1 Effetti sull’apparato cardio-vascolare dell’eccesso o carenza di ormoni tiroidei |
||
| Parametro | Eccesso | Deficit |
| Frequenza/Inotropismo del cuore | ↑ | ↓ |
| Resistenze vascolari | ↓ | ↑ |
| Pressione diastolica | ↑ | ↑ |
| Post-carico | ↓ | ↑ |
| Gettata cardiaca | ↑ | ↓ |
| Termogenesi | ↑ | ↓ |
Asse GH-IGF-I
I miocardiociti esprimono recettori per GH e per IGF-1. Il GH ha importanti effetti sullo sviluppo del cuore e, nell’adulto, sul mantenimento della sua struttura e delle sue funzioni. Il GH è in grado di esercitare effetti sul metabolismo e sulla crescita delle cellule muscolari cardiache, anche indipendenti da IGF-1. IGF-1 stimola la sintesi proteica e l’ipertrofia dei miocardiociti, attivando geni muscolo-specifici.
L’ipertrofia miocardica indotta da GH-IGF-1 si accompagna anche a rimodellamento del cuore: l’IGF-1 stimola, infatti, la sintesi di collagene da parte dei fibroblasti e il GH ne aumenta la velocità di deposizione. GH e IGF-1 modulano inoltre la struttura del cuore, prevenendo l’apoptosi delle cellule miocardiche; questa azione potrebbe avere un effetto protettivo in condizioni di insulto ischemico.
Dati sperimentali suggeriscono che l’asse GH-IGF-1 aumenti anche la contrattilità miocardica, anche se le evidenze sono ancora deboli. L’IGF-1 influenza inoltre la funzione cardio-vascolare, aumentando la produzione vascolare di NO (6).
Un eccesso o un difetto di GH possono avere importanti conseguenze sul cuore. Un deficit di GH nell'adulto determina riduzione della massa ventricolare sinistra e della gittata cardiaca e ridotta capacità di esercizio, e la terapia sostitutiva con GH ha effetto anabolico sul cuore e migliora sia la funzione sistolica sia quella diastolica.
L’acromegalia si associa a cardiomegalia, ipertrofia ventricolare sinistra, sostituzione fibrosa e degenerazione dei miocardiociti, la cui entità si correla con la durata della malattia. Il trattamento dell’acromegalia con normalizzazione dei livelli di GH si associa a miglioramento delle alterazioni strutturali e funzionali (7).
Aldosterone
L’aldosterone viene prodotto non solo dalla glomerulare surrenale, ma anche da altri tessuti, tra cui il cuore stesso.
I più noti effetti dell’aldosterone avvengono tramite un processo più lento, di tipo genomico, mediato da recettori citosolici per i mineralcorticoidi presenti nei miocardiociti; secondo recenti evidenze, l’aldosterone esercita anche azioni attraverso meccanismi più rapidi di tipo non-genomico, mediati da un recettore che si ritiene essere una proteina accoppiata a G-protein.
L’aldosterone determina un ritardo nella depolarizzazione della membrana cellulare e un prolungamento del potenziale d’azione, attraverso un aumento della permeabilità della membrana al sodio; stimola inoltre la fibrosi miocardica, che si osserva in condizioni di eccesso di ormone, come si riscontra nell’iperaldosteronismo primario ed in quello secondario tipico dello scompenso cardiaco. La fibrosi è riconducibile ai seguenti meccanismi:
- infiammazione a livello delle pareti vascolari, con iperproduzione di citochine e chemochine, che partecipano al processo di fibrosi;
- stimolazione dell’attività dei macrofagi;
- regolazione della sintesi e della proliferazione delle fibre collagene (8).
Catecolamine
Sia l’adrenalina sia la noradrenalina agiscono sul cuore attraverso i recettori di tipo ß: i ß1 sono quelli più rappresentati nel cuore (oltre 4 volte i ß2); i ß3 sono poco espressi nel cuore sano, ma vengono iperespressi nell’insufficienza cardiaca (in cui diminuiscono i ß1). Le catecolamine determinano l’espressione di una serie di proteine a livello delle cellule muscolari cardiache, che determinano un aumento delle capacità di rilasciamento e un effetto inotropo positivo. La stimolazione della contrattilità miocardica è legata a un aumento del calcio intra-cellulare (9).
L’esposizione cronica dei miocardiociti ad elevati livelli di catecolamine (feocromocitoma) provoca alterazioni strutturali e funzionali (verosimilmente legate al sovraccarico intra-cellulare di calcio e anche alla vasocostrizione α-mediata), che configurano una vera e propria miocardiopatia da catecolamine, caratterizzata da degenerazione e necrosi delle fibre miocardiche, foci di cellule infiammatorie, fibrosi ed edema miocardico diffuso (10).
Leptina
La leptina ha vari effetti metabolici sul cuore (stimolazione del metabolismo degli acidi grassi e del metabolismo glicidico) e di prevenzione dell’apoptosi.
La leptina può favorire ipertrofia miocardica, anche se non è chiaro se questo possa essere un effetto diretto o mediato dall’attivazione di altri sistemi neuro-ormonali (asse renina-angiotensina-aldosterone, sistema adrenergico). La leptina potrebbe anche proteggere il cuore dall’accumulo di grassi nell’obesità (11).
Ghrelin
È un ormone di origine gastrica, che, oltre agli effetti sulla secrezione di GH e ad azioni metaboliche, esercita significativi effetti cardio-vascolari. Recettori per Ghrelin sono ampiamente distribuiti nelle cellule muscolari cardiache e nei vasi.
Per quanto riguarda il cuore, Ghrelin aumenta la gittata cardiaca e la contrattilità miocardica, riduce la fibrosi miocardica e ha un effetto protettivo sul cuore legato a inibizione dell’apoptosi. A livello centrale riduce l’attività adrenergica, mentre a livello periferico aumenta la produzione di NO (12).
| Tabella 2 | |||||
| Azioni degli ormoni sulle attività del cuore | |||||
| Inotropa | Lusitropa | Cronotropa | Dromotropa | Batmotropa | |
| T3 | positiva | positiva | positiva | ||
| GH | positiva | ||||
| IGF-1 | positiva | ||||
| Aldosterone | negativa | negativa | |||
| Catecolamine | positiva | positiva | positiva | ||
| Endotelina | positiva | positiva | |||
| Ghrelin | positiva | negativa | |||
BIBLIOGRAFIA
- Ogawa T, De Bold AJ. The heart as an endocrine organ. Endocr Connect 2014, 3: R31-44.
- Kato J, Tsuruda T, Kitamura K, et al. Adrenomedullin: a possible autocrine or paracrine hormone in the cardiac ventricles. Hypertension Res 2003, 26: S113-9.
- Luscher TF, Burton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a new class of cardiovascular drugs. Circulation 2000, 102: 2434-40.
- Grais IM, Sowers JR. Thyroid and the heart. Am J Med 2014, 127: 691-8.
- Osmak-Tizon L, Poussier M, Cottin Y, et al. Non-genomic actions of thyroid hormones: molecular aspects. Arch Cardiovasc Dis 2014, 107: 207-11.
- Isgaard J, Arcopinto M, Karason K, et al. GH and the cardiovascular system: un update in a topic at heart. Endocrine 2015, 48: 25-35.
- Colao A. The GH-IGF-I axis and the cardiovascular system: clinical implications. Clin Endocrinol 2008, 69: 347-58.
- Kritis AA, Gouta CP, Liaretidou EI, et al. Latest aspects of aldosterone actions on the heart muscle. J Physiol Pharmacol 2016, 67: 21-30.
- Najafi A, Sequeira V, Kuster DWD, et al. ß-adrenergic receptor signalling and its functional consequences in the diseased heart. Eur J Clin Invest 2016, 46: 362-74.
- Ferreira VM, Marcelino M, Piechnik SK, et al. Pheochromocytoma is characterized by catecholamine-mediated myocarditis, focal and diffuse myocardial fibrosis, and myocardial dysfunction. J Am Coll Cardiol 2016, 67: 2364-74.
- Hall ME, Harmancey R, Stec DE. Lean heart: role of leptin in cardiac hypertrophy and metabolism. World J Cardiol 2015, 7: 511-24.
- Virdis A, Lerman LO, Regoli F, et al. Human ghrelin: a gastric hormone with cardiovascular properties. Curr Pharm Des 2016, 22: 52-8.
Meccanismi endocrini attivati in corso di scompenso cardiaco
Giulia Balbi
Medicina Interna, Ospedale S. Bortolo, Vicenza
Lo scompenso cardiaco è una sindrome in cui la pompa cardiaca, per un’anomalia strutturale e/o funzionale, non è in grado di garantire l’adeguata perfusione ai vari organi e tessuti.
Le cause di scompenso cardiaco vengono classificate in malattia ischemica, danno tossico, danno immuno-mediato e infiammatorio, patologie infiltrative, ormonali, nutrizionali e genetiche (1).
I segni e sintomi che caratterizzano questa sindrome sono attribuibili a una serie di meccanismi compensatori, che l’organismo mette in atto, nel tentativo di ripristinare un’adeguata gittata cardiaca (attraverso l’aumento della contrattilità miocardica, della frequenza cardiaca e l’espansione del liquido extra-cellulare) e di mantenere un’adeguata pressione sistemica (attraverso la vasocostrizione)(2). Alla base di questi meccanismi compensatori vi è un’attivazione neuroumorale/neuroendocrina (ormoni e neurotrasmettitori), che comporta effetti emodinamici e non emodinamici (infiammazione, stress ossidativo)(3). In acuto le risposte compensatorie controbilanciano il deficit di pompa cardiaca; in cronico, invece, questi meccanismi alimentano un circolo vizioso e comportano una progressiva tossicità a carico dei miociti, fino alla disfunzione cardiaca. Nelle prime fasi di malattia il paziente è asintomatico; man mano che la patologia progredisce e cronicizza, si assiste a una congestione di più organi e alla fibrosi, con comparsa dei sintomi (2).
I tre principali sistemi endocrini coinvolti sono:
- sistema nervoso adrenergico/simpatico;
- sistema renina-angiotensina-aldosterone;
- sistema dei peptidi natriuretici.
1. SISTEMA NERVOSO SIMPATICO (SNS)
In risposta al calo pressorio dovuto alla ridotta gittata cardiaca, si attiva il SNS, con aumentato rilascio e ridotta ricaptazione di noradrenalina a livello delle terminazioni nervose e rilascio di adrenalina dalla midollare del surrene (4). Le catecolamine determinano:
- incremento della contrattilità del ventricolo sinistro e aumento della frequenza cardiaca, al fine di mantenere un’adeguata gittata cardiaca;
- vasocostrizione polmonare e incremento del tono venoso, con aumento del pre-carico;
- vasocostrizione renale sull’arteriola efferente, per aumentare il filtrato glomerulare;
- riassorbimento di sodio nel tubulo prossimale, con conseguente sodio-ritenzione.
A lungo termine, l’iperstimolazione simpatica comporta down-regulation dei recettori ß-adrenergici, desensibilizzazione e perdita delle risposte inotropa e cronotropa (5). La stimolazione cronica dei recettori ß-adrenergici, inoltre, a livello cellulare porta a disfunzione del miocardio, con ri-espressione di isoforme proteiche fetali, perdita di cardiomiociti per apoptosi e necrosi (6).
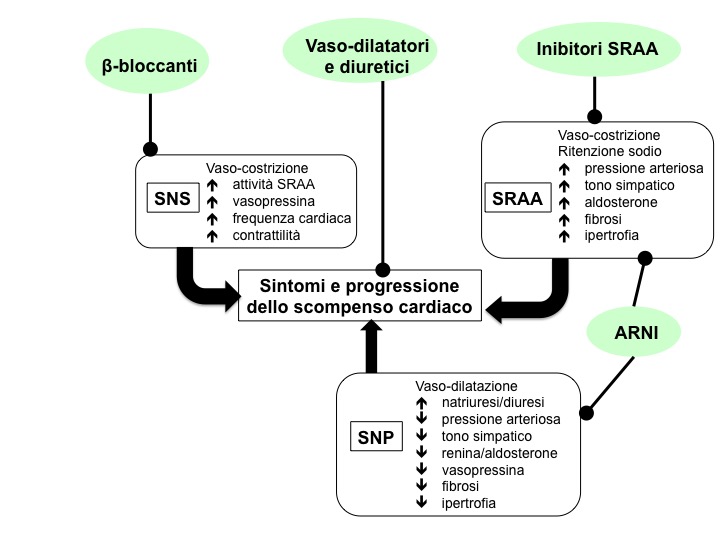
Terapia farmacologica mirata al SNS
È raccomandato l’utilizzo di ß-bloccanti, i quali proteggono il cuore dall’iperstimolazione cardiotossica delle catecolamine, riducono il rischio di ospedalizzazione, migliorano la sopravvivenza, riducono il rischio di aritmie, migliorano il flusso coronarico e inducono regressione del rimodellamento del ventricolo sinistro (4).
Linee guida ESC 2016 (1): nei pazienti con scompenso cardiaco e ridotta frazione di eiezione è raccomandato un ß-bloccante (in aggiunta a un ACE-inibitore) per ridurre il rischio di ricovero per scompenso e di morte (raccomandazione di grado IA).
2. SISTEMA RENINA-ANGIOTENSINA-ALDOSTERONE (SRAA)
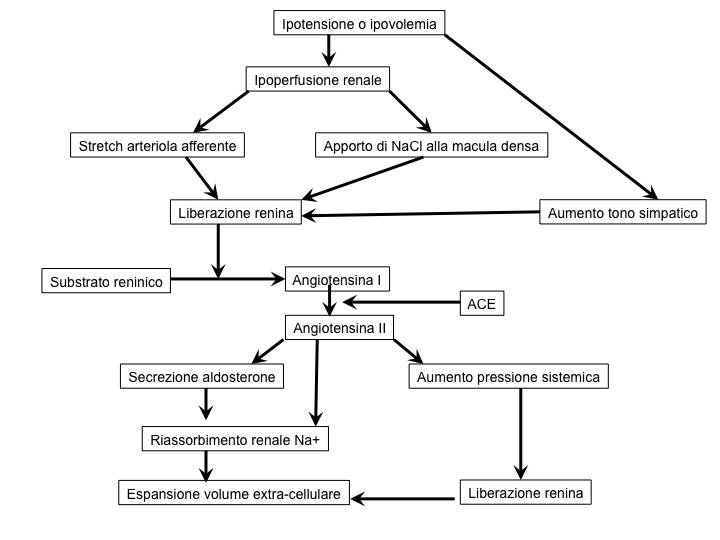
Renina
Esercita effetti emodinamici vascolari e renali, ma anche azione diretta sui tessuti, sui quali determina rimodellamento, disfunzione endoteliale e fibrosi. Il rilascio di renina è attivato dalle variazioni di liquidi extra-cellulari, attraverso l’azione su tre siti:
- barocettori della parete delle arteriole efferenti;
- barocettori cardiaci e arteriosi, che regolano l’attività nervosa simpatica e i livelli circolanti di catecolamine e aumentano la secrezione di renina attraverso i recettori ß1;
- cellule della macula densa del tubulo distale prossimale, stimolate dalla riduzione della concentrazione di sodio nell’ultrafiltrato.
L’introito di sodio costituisce il maggior determinante della secrezione di renina (7). Un’aumentata secrezione di renina determina un’aumentata produzione di angiotensina II e aldosterone, con riassorbimento di sodio ed espansione del fluido extra-cellulare.
Angiotensina II
Determina (7):
- vasocostrizione arteriolare, con incremento delle resistenze vascolari;
- riassorbimento di sodio e acqua a livello del tubulo prossimale e, indirettamente, nel tubulo distale, inducendo la secrezione surrenalica di aldosterone;
- vasocostrizione delle arteriole afferente ed efferente, con riduzione del flusso renale, aumento della pressione glomerulare (per mantenere il filtrato glomerulare quando il calo pressorio sistemico attiva il SRAA);
- azione infiammatoria su cellule del sangue (leucociti, cellule endoteliali, cellule muscolari lisce);
- proliferazione cellulare delle cellule muscolari lisce vasali (aterogenesi).
Aldosterone
Prodotto nel corticosurrene, viene stimolato principalmente dall’angiotensina II e dal potassio sierico. Determina un effetto sui valori pressori, inducendo un aumentato riassorbimento di sodio (nel tubulo renale). Inoltre, incrementa lo stress ossidativo, l’apoptosi, la fibrosi cardiaca e l’ipertrofia ventricolare sinistra (8).
Terapia farmacologica mirata al SRAA
I ß-bloccanti riducono il rilascio di renina indotto dalla stimolazione dei recettori ß1. Gli ACE-inibitori bloccano la conversione di angiotensina I in angiotensina II; i bloccanti dei recettori dell’angiotensina interferiscono con l’interazione tra angiotensina II e il suo recettore. È stato dimostrato che gli ACE-inibitori riducono mortalità e morbilità nei pazienti con scompenso cardiaco e ridotta funzione sistolica, pertanto sono raccomandati dalle linee guida ESC in tutti i pazienti sintomatici, se non controindicati o non tollerati, per inibire in maniera adeguata il SRAA. Sono, inoltre, raccomandati, nei pazienti asintomatici con disfunzione sistolica, per ridurre il rischio di sviluppare scompenso cardiaco, ospedalizzazione per scompenso cardiaco e morte. I ß-bloccanti sono raccomandati nei pazienti con storia di infarto del miocardio e disfunzione sistolica asintomatica per ridurre il rischio di morte. Spironolattone ed eplerenone sono raccomandati in tutti i pazienti sintomatici (anche se già in trattamento con ACE-inibitori e ß-bloccanti) con funzione sistolica ridotta, per ridurre mortalità e ospedalizzazione a causa dello scompenso cardiaco.
Linee guida ESC 2016 (1):
- nei pazienti sintomatici con scompenso cardiaco e ridotta frazione di eiezione è raccomandato un ACE-inibitore (in aggiunta a un ß-bloccante) per ridurre il rischio di ricovero per scompenso e di morte (raccomandazione di grado IA);
- nei pazienti sintomatici stabili con scompenso cardiaco e ridotta frazione di eiezione è raccomandato un ß-bloccante (in aggiunta a un ACE-inibitore) per ridurre il rischio di ricovero per scompenso e di morte (raccomandazione di grado IA);
- nei pazienti con scompenso cardiaco e ridotta frazione di eiezione, che rimangono sintomatici nonostante trattamento con un ACE-inibitore e un ß-bloccante, è raccomandato un anti-mineralcorticoide per ridurre il rischio di ricovero per scompenso e di morte (raccomandazione di grado IA).
3. SISTEMA DEI PEPTIDI NATIURETICI (SPN)
ll sistema dei peptidi natriuretici è costituito di 3 ormoni simili: ANP (atrial natriuretic peptide), BNP (B-type natriuretic peptide), prodotti entrambi dal cuore, e CNP (C-type natriuretic peptide), prodotto da endotelio e rene.
Le azioni che svolgono sono (9,10):
- soppressione della proliferazione cellulare;
- inibizione dell’infiammazione;
- riduzione dell’attivazione piastrinica;
- preservazione di funzione e struttura del miocardio;
- natriuretica (aumentando il filtrato glomerulare attraverso la dilatazione dell’arteriola afferente e la costrizione dell’efferente e inibendo il riassorbimento di sodio ed acqua);
- inibizione della secrezione di renina ed aldosterone;
- vasodilatatoria.
Il principale stimolo per la secrezione di ANP è lo stiramento delle pareti atriali, in risposta all’aumento di volume intra-vascolare o alla pressione transmurale cardiaca. Con la cronicizzazione dello scompenso cardiaco e l’aumento della pressione di riempimento ventricolare, vengono coinvolte anche le cellule miocardiche del ventricolo sinistro, che a loro volta producono ANP e BNP (8).
| Azioni fisiologiche dei peptidi natriuretici (10) | |
| Bersaglio | Effetti biologici |
| Rene | Aumento filtrazione glomerulare attraverso la vasodilatazione dell’arteriola afferente e la vasocostrizione dell’efferente Attivazione natriuresi attraverso l’inibizione dello scambio Na+/H+ nel tubulo prossimale, del co-trasporto Na+/Cl- nel tubulo distale e dei canali del Na nel dotto collettore Attivazione diuresi da inibizione dell’incorporazione stimolata da ADH nella membrana apicale del dotto collettore di acquaporina 2 |
| Cuore | Riduzione del pre-carico con caduta della gittata cardiaca |
| Vasi | Vasodilatazione Aumento della conduttività idraulica capillare Diminuzione del pre-carico e post-carico cardiaco |
| Ghiandole endocrine | Soppressione di SRAA, flusso simpatico, ADH, endotelina |
| Mitogenesi | Inibizione nelle cellule muscolari lisce Inibizione dell’ipertrofia fibroblastica mediata da fattori di crescita |
Terapia farmacologica mirata al SPN
Le più recenti novità farmacologiche riguardano questo ambito. Nelle ultime linee guida ESC, infatti, compare un nuovo farmaco, LCZ696 (valsartan (ARB)/sacubitril (NEP inhibitor)), della classe di farmaci ARNI (Ang receptor/NEP inhibitor), che combina un inibitore del recettore dell’angiotensina con un inibitore della neprilisina (11). La neprilisina è un’endopeptidasi, deputata alla degradazione di peptidi endogeni vasoattivi tra cui i PN. L’inibizione della neprilisina comporta un incremento del livello di questi peptidi, che controbilanciano l’iperattivazione neuroormonale che conduce a vasocostrizione, ritenzione di sodio e rimodellamento. Il razionale di questo tipo di terapia è l’azione simultanea su SRAA e SPN, con blocco del primo e potenziamento del secondo, che lo contrasta, senza aumentarne gli effetti collaterali (in particolare angioedema). I risultati emersi sono incoraggianti, in quanto nello studio PARADIGM HF il farmaco (confrontato con valsartan non in associazione) ha dimostrato di ridurre il rischio di morte e ospedalizzazione in pazienti con scompenso cardiaco cronico e frazione d’eiezione ridotta in classe NYHA II-IV (8,12,13).
Linee guida ESC 2016 (1): nei pazienti ambulatoriali con scompenso cardiaco e ridotta frazione di eiezione, che rimangono sintomatici nonostante un trattamento ottimizzato con ACE-inibitore, ß-bloccante e anti-mineralcorticoide, è raccomandato di sostituire l’ACE-inibitore con la combinazione sacubitril/valsartan, per ridurre ulteriormente il rischio di ricovero per scompenso e di morte (raccomandazione di grado IB).
CONCLUSIONI
La conoscenza dei meccanismi endocrini coinvolti nello scompenso cardiaco è utile per il clinico, al fine di impostarne una corretta gestione farmacologica; la terapia dello scompenso, infatti, è volta a contrastare l’iperattivazione neuroendocrina, principalmente attraverso la modulazione del SRAA (ACE-inibitori, sartani e anti-mineralcorticoidi) e del SNP (ß-bloccanti). Le linee guida 2016 per la diagnosi e cura dello scompenso cardiaco della European Society of Cardiology raccomandano, per tutti i pazienti sintomatici, con ridotta frazione di eiezione l’utilizzo di “antagonisti neuro-ormonali (ACE-inibitori, sartani e ß-bloccanti)”, in quanto “hanno dimostrato di migliorare la sopravvivenza” e, in aggiunta ai diuretici, in classe I B raccomandano anche gli ARNI, con la riserva di valutarne la sicurezza nel lungo termine (1).
BIBLIOGRAFIA
- 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur J Heart Fail 2016, 18: 891–975.
- Dumitru I. Heart failure. Medscape 2016.
- Colucci WS. Nitric oxide, other hormones, cytokines, and chemokines in heart failure. Uptodate 2016.
- Lymperopoulos A, et al. Adrenergic nervous system in heart failure. Pathophysiology and therapy. Circ Res 2013, 113: 739-53.
- Trispokiadis F, et al. The sympathetic nervous system in heart failure, physiology, pathophysiology and clinical implications. J Am Coll Cardiol 2009, 54: 1747-62.
- Colucci WS. Pathophysiology of heart failure: neurohumoral adaptations. Uptodate 2015.
- Fisher NDL. Overview of the renin-angiotensin system. Uptodate 2015.
- Hall ME, Yanes L, Long RC, Koch CA. Hormones of the cardiovascular system. Endotext 2015.
- Buglioni A, Burnett JC. Pathophysiology and the cardiorenal connection in heart failure. Circulating hormones: biomarkers or mediators. Clin Chim Acta 2015, 443: 3–8.
- Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis to treatment. Clin Sci 2016, 130: 57–77.
- Solomon SD, Zile M, Pieske B, et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 2012, 380: 1387-95.
- McMurray JJ, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014, 371: 993-1004.
- Ferrari L, Sada S, et al. Efficacy of angiotensin–neprilysin inhibition versus enalapril in patient with heart failure with a reduced ejection fraction. Intern Emerg Med 2015, 10: 369–71.
Ripercussioni endocrine dello scompenso cardiaco
Giovanni Scanelli e Alfredo De Giorgi*
UO Medicina 1, Ospedale “San Bortolo”, ULSS n. 6, Vicenza
*Scuola di specializzazione in Medicina Interna, Università di Ferrara
Definizione
Lo scompenso cardiaco o insufficienza cardiaca cronica (CHF) può essere definito come un’anomalia di struttura o funzione della pompa cardiaca, che porta all’incapacità del cuore di mantenere un apporto di ossigeno a livello dei tessuti periferici in quantità soddisfacente al loro fabbisogno metabolico (1). Tale incapacità si manifesta con sintomi e segni tipici, ad andamento ingravescente (dispnea e ridotta resistenza allo sforzo fisico, turgore giugulare, crepitazioni all’auscultazione del torace ed edemi declivi).
È una patologia molto frequente, presente in circa il 2% della popolazione adulta, per arrivare a percentuali cinque volte maggiori nei soggetti ultra80enni, con lieve prevalenza nel sesso femminile (13.5% vs 10.6%)(2). Tale condizione risulta in progressivo aumento, a causa sia dell’invecchiamento che dell’incremento delle comorbilità della popolazione generale.
| Cause di scompenso cardiaco | ||
| Malattie miocardiche | Cardiopatia ischemica | |
| Cardiopatia ipertensiva | ||
| Cardiomiopatia familiare | ||
| Cardiomiopatie acquisite | Infettive: batteriche, virali, da protozoi, da funghi, da parassiti, altro | |
| Autoimmuni: vaccini, farmaci, miocardiopatia a cellule giganti o da linfociti, sarcoidosi, miocardiopatia eosinofila | ||
| Da tossici esogeni: farmaci, sostanze d’abuso, metalli pesanti | ||
| Endocrine | ||
| Della gravidanza | ||
| Da infiltrazione di sostanze o cellule (amiloide, neoplasie) | ||
| Malattie valvolari | ||
| Malattie del pericardio | ||
| Malattie dell'endocardio | ||
| Scompenso cardiaco congenito | ||
| Aritmie e disturbi della conduzione cardiaca | ||
| Forme secondarie da aumentata gittata | ||
| Forme secondarie da eccesso di volume | ||
Per quanto riguarda le alterazioni periferiche nei pazienti con CHF, un interesse crescente stanno sviluppando le disfunzioni endocrine che vanno a definire il quadro della sindrome da deficit multiormonale, caratterizzata da modifiche del bilancio catabolismo/anabolismo nei pazienti con CHF, fino a quadri di cachessia cardiaca (3). Un recente studio condotto su una coorte di 202 pazienti con CHF (età media 64 ± 10 anni) ha rilevato che solo il 18% dei pazienti non presenta alcuna alterazione ormonale e il 75% mostra uno o due deficit ormonali (4).
Scompenso cardiaco e funzionalità tiroidea
Le alterazioni della funzionalità tiroidea sono particolarmente frequenti nei pazienti con CHF. Lo studio PROSPER ha mostrato come anche le forme subcliniche si associno a un aumentato rischio di CHF (5). L’effetto cardio-vascolare degli ormoni tiroidei è svolto principalmente dalla T3; questa agisce, infatti, sia sulla componente cardiaca muscolare, modificando la contrattilità miocardica e la gittata cardiaca, che sulle cellule pacemaker (aumento del rischio di aritmie), sia a livello vascolare periferico, con effetto pro-angiogenetico (mediato dal fattore di crescita vascolare) e con modifiche della resistenza vascolare (6). Sono noti anche effetti degli ormoni tiroidei sul volume ematico circolante e sul bilancio idroelettrolitico, dovuti in parte al danno renale indotto dalla disfunzione tiroidea (7). Tali modifiche del volume circolante possono essere alla base delle alterazioni funzionali che si verificano nello scompenso di cuore.
L’ipotiroidismo si associa a un aumento del 44% della mortalità per tutte le cause in pazienti con CHF e del 37% di morte o ospedalizzazione per eventi cardio-vascolari (8). Il trattamento della disfunzione tiroidea permette un miglioramento della resistenza all'esercizio fisico, aumenta la gittata cardiaca, la frazione di eiezione e riduce le resistenze vascolari periferiche (9).
Scompenso cardiaco e disfunzione renale
La connessione tra CHF e funzione renale è una condizione ben nota, rientrante nelle sindromi cardio-renali (10). Dal punto di vista endocrinologico, le correlazioni cardio-renali presenti in tali sindromi sono mediate da modifiche dell'asse renina-angiotensina-aldosterone (RAA) e dei peptidi natriuretici atriali (ANP, BNP e CNP). Il sistema RAA interviene nel controllo del bilancio idroelettrolitico e della pressione arteriosa, particolarmente importanti nei pazienti con CHF: l'angiotensina determina vasocostrizione arteriosa periferica e coronarica, riduzione della filtrazione glomerulare, ipertrofia dei miociti fino all'apoptosi, aumento dell'inotropismo cardiaco, con associato effetto pro-aritmico. Agisce inoltre indirettamente mediante l'aldosterone su riassorbimento tubulare di sodio e potassio, rimodellamento coronarico e reno-vascolare, disfunzione delle cellule endoteliali e dei barocettori carotidei, inibizione della captazione cardiaca di noradrenalina, con riduzione variabile della frazione di eiezione (11).
Altro sistema endocrino di interconnessione rene-cuore nei casi di CHF è rappresentato dal sistema dei peptidi natriuretici atriali (ANP) e ventricolari (BNP); questi, prodotti in seguito a stress di parete (da aumento delle pressioni intra-cavitarie cardiache), agiscono inducendo vasodilatazione periferica e natriuresi. Mediante inibizione del sistema RAA, permettono una parziale riduzione del pre-carico, con miglioramento delle prestazioni cardiache (12). Inibiscono inoltre l’attivazione del sistema nervoso simpatico, con soppressione della sintesi delle catecolamine, della vasopressina, del sistema RAA e dei meccanismi di ipertrofia e fibrosi del ventricolo sinistro (13). I livelli plasmatici dei peptidi natriuretici, in particolare il BNP, sembrerebbero correlati con la prognosi dei pazienti con CHF, anche se la loro maggiore utilità è attualmente nella diagnosi e come guida nel trattamento di CHF (14).
Le connessioni cuore-rene sono importanti anche per quanto riguarda l'approccio terapeutico nei pazienti con CHF: infatti, farmaci quali ACE-inibitori e diuretici, fondamentali nella terapia del CHF, possono essere responsabili di un ulteriore peggioramento della funzionalità renale, creando il circolo vizioso responsabile della cronicizzazione del quadro di scompenso.
Scompenso cardiaco e diabete mellito
La contemporanea presenza nei pazienti anziani di CHF e diabete tipo 2 (DM2) è abbastanza frequente: si tratta, infatti, di patologie a elevata prevalenza nella popolazione generale. Tale comorbilità, presente nel 25% dei pazienti con CHF (e nel 40% degli ospedalizzati per CHF)(15), non è comunque dovuta solo a un fattore epidemiologico: sono state identificate, infatti, diverse interconnessioni fisiopatologiche tra CHF e alterato metabolismo glucidico (16). Il DM predispone allo sviluppo di CHF, in quanto funge da fattore di rischio per diverse patologie, quali ipertensione, dislipidemie, alterazioni sia del sistema nervoso autonomo che del microcircolo vascolare periferico (17). Inoltre, l'insulino-resistenza, condizione alla base del DM2, è responsabile dell'alterato utilizzo cardiaco del glucosio, favorendo l'ossidazione degli acidi grassi, utilizzati come fonte energetica dal miocardio, con conseguenti lipotossicità cardiaca, alterazione della contrattilità miocardica, aumentata produzione di citochine infiammatorie e sintesi di specie reattive dell'ossigeno (ROS), che hanno l'effetto ultimo di indurre apoptosi miocitica e fibrosi: la cosiddetta cardiomiopatia diabetica (18).
Anche la terapia ipoglicemizzante si associa ad aumentato rischio di CHF; in particolare, la somministrazione di glitazonici si associa a un aumento di circa 2 volte del rischio di CHF, conseguente a fluido-ritenzione farmaco-indotta (19). Analogamente, anche gli inibitori DPP-4 sembrerebbero associarsi a un maggior rischio di CHF, ma il meccanismo patogenetico non è ancora del tutto definito (20) e i dati sono contraddittori.
Scompenso cardiaco e surrene
Nei pazienti con CHF è possibile riscontrare alterazioni funzionali e squilibri ormonali che riguardano tutte le componenti endocrine del surrene, in particolare, nella porzione corticale:
- zona glomerulare, che interviene nella sintesi dell'aldosterone, le cui alterazioni nella secrezione sono regolate dal sistema RAA e quindi interconnesse con la funzionalità renale;
- zona fascicolata, che regola la produzione dei glucocorticoidi, in particolare il cortisolo. Il suo ruolo nel CHF è correlato al suo effetto sui recettori mineralcorticoidi intra-citoplasmatici: sembrerebbe agire attivando tali recettori e aumentando i ROS conseguenti all'ipossia tissutale (21). In tali pazienti un aumento dei livelli di cortisolo sembra correlarsi a peggioramento della frazione d'eiezione, riduzione della tolleranza allo sforzo fisico, peggioramento della capacità ventilatoria e aumento della mortalità per eventi cardio-vascolari (22). Studi di correlazione tra cortisolo e CHF sono molto limitati e non sono disponibili dati sull'effetto cardio-vascolare del riequilibrio delle concentrazioni di tale ormone nei pazienti con CHF;
- zona reticolare, deputata alla sintesi degli ormoni sessuali: il testosterone e il deidroepiandrosterone (DHEA) sono associati a effetti protettivi, agendo sulla riduzione della lesione ischemica in seguito a infarto miocardico acuto (23), inducendo vasodilatazione periferica e, per azione sui canali del Ca2+, avendo anche un effetto anti-aritmico (riduzione del potenziale di depolarizzazione e della durata del QT corretto) (24). Ridotti livelli di testosterone e DHEA, presenti in circa il 43% dei pazienti con CHF, sono invece correlati a ridotte performance funzionali, aumento di eventi cardio-vascolari e mortalità (25). Inoltre, studi di intervento hanno dimostrato come la loro somministrazione si associ a miglioramento delle performance funzionali dei pazienti (26).
La midollare è deputata alla sintesi delle catecolamine: queste inducono vasocostrizione periferica, aumento della contrattilità e della frequenza cardiaca, con conseguente aumento del consumo di ossigeno miocardico. Nel CHF queste azioni da un lato rappresentano un meccanismo di compenso al ridotto apporto periferico di ossigeno, dall'altro risultano alla base del circolo vizioso che induce peggioramento del quadro clinico (27). Oltre all'effetto indiretto, esiste anche un effetto cardiotossico diretto delle catecolamine, che inducono sovraccarico miocitico di calcio, attivazione dei rettori α- e β-adrenergici, fibrosi interstiziale, apoptosi dei miocardiociti e disfunzione contrattile del ventricolo sinistro (28). È quindi fondamentale bloccare tale circolo vizioso con farmaci inibenti il sistema nervoso adrenergico (α- e β-bloccanti): questi determinano riduzione della mortalità e miglioramento dell'aspettativa di vita, particolarmente nei pazienti di età < 75 anni (29).
Scompenso cardiaco e disfunzione ipofisaria
Danni antero-ipofisari sembrerebbero correlarsi a quadri di scompenso cardiaco, non solo per alterazioni secondarie della funzione tiroidea e surrenalica, ma anche direttamente a seguito di inibizione della sintesi dell'ormone della crescita (GH) e del suo effettore periferico IGF-1, documentati in circa il 40-60% dei pazienti con CHF (30). L'IGF-1 agisce a livello cardio-vascolare, ostacolando il rimodellamento ventricolare post-infartuale e riducendo l'apoptosi miocitaria, inducendo vasodilatazione - con meccanismo dipendente dall'endotelina, dal NO e dal trasportatore Na+/K+ ATPasi - e a livello periferico migliorando le performance muscolari (31). Deficit di IGF-1 sembrerebbero avere importanti effetti prognostici in termini di sviluppo di CHF e di mortalità per eventi cardio-vascolari (32). Recenti studi confermano una riduzione degli eventi cardio-vascolari, un miglioramento della frazione di eiezione e migliori performance fisiche in seguito alla somministrazione esogena dell'ormone in pazienti con CHF (30,33).
Conclusione
Nei pazienti con CHF risulta fondamentale la valutazione della presenza di deficit ormonali singoli o multipli, che possono essere in alcuni casi la causa stessa del quadro di scompenso cardiaco. Tali deficit devono essere ricercati e trattati, per migliorare sia la qualità della vita che la prognosi dei pazienti. Sono necessari ulteriori studi atti a valutare sia l'impatto delle diverse disfunzioni endocrine sull'outcome dei pazienti con CHF, sia l'utilità clinica e su larga scala della terapia di reintegro del deficit ormonale, particolarmente nei pazienti anziani.
Bibliografia
- McMurray JJ, Adamopoulos S, Anker SD, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2012, 14: 803-69.
- Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 2016, 133: e38-60.
- Saccà L. Heart failure as a multiple hormonal deficiency syndrome. Circ Heart Fail 2009, 2: 151-6.
- Arcopinto M, Salzano A, Bossone E, et al. Multiple hormone deficiencies in chronic heart failure. Int J Cardiol 2015, 184: 421-3.
- Nanchen D, Gussekloo J, Westendorp RG, et al; PROSPER Group. Subclinical thyroid dysfunction and the risk of heart failure in older persons at high cardiovascular risk. J Clin Endocrinol Metab 2012, 97: 852-61.
- Klein I, Danzi S. Thyroid disease and the heart. Circulation 2007, 116: 1725-35.
- Iglesias P, Díez JJ. Thyroid dysfunction and kidney disease. Eur J Endocrinol 2009, 160: 503-15.
- Ning N, Gao D, Triggiani V, et al. Prognostic role of hypothyroidism in heart failure: a meta-analysis. Medicine (Baltimore) 2015, 94: e1159.
- Curotto Grasiosi J, Peressotti B, Machado RA, et al. Improvement in functional capacity after levothyroxine treatment in patients with chronic heart failure and subclinical hypothyroidism. Endocrinol Nutr 2013, 60: 427-32.
- Virzì G, Day S, de Cal M, e al. Heart-kidney crosstalk and role of humoral signaling in critical illness. Crit Care 2014, 18: 201.
- Sayer G, Bhat G. The renin-angiotensin-aldosterone system and heart failure. Cardiol Clin 2014, 32: 21-32.
- Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis to treatment. Clin Sci (Lond) 2016, 130: 57-77.
- Schlueter N, de Sterke A, Willmes DM, et al. Metabolic actions of natriuretic peptides and therapeutic potential in the metabolic syndrome. Pharmacol Ther 2014, 144: 12-27.
- Troughton R, Michael Felker G, Januzzi JL Jr. Natriuretic peptide-guided heart failure management. Eur Heart J 2014, 35: 16-24.
- Sarma S, Mentz RJ, Kwasny MJ, et al; EVEREST investigators. Association between diabetes mellitus and post-discharge outcomes in patients hospitalized with heart failure: findings from the EVEREST trial. Eur J Heart Fail 2013, 15: 194-202.
- Ingelsson E, Sundström J, Arnlöv J, et al. Insulin resistance and risk of congestive heart failure. JAMA 2005, 294: 334-41.
- Aroor AR, Mandavia CH, Sowers JR. Insulin resistance and heart failure: molecular mechanisms. Heart Fail Clin 2012, 8: 609-17.
- Dei Cas A, Khan SS, Butler J, et al. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Heart Fail 2015, 3: 136-45.
- Varas-Lorenzo C, Margulis AV, Pladevall M, et al. The risk of heart failure associated with the use of noninsulin blood glucose-lowering drugs: systematic review and meta-analysis of published observational studies. BMC Cardiovasc Disord 2014, 14: 129.
- Monami M, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and heart failure: a meta-analysis of randomized clinical trials. Nutr Metab Cardiovasc Dis 2014, 24: 689-97.
- Funder JW. Mineralocorticoid receptors: distribution and activation. Heart Fail Rev 2005, 10: 15-22.
- Tzanis G, Dimopoulos S, Agapitou V, Nanas S. Exercise intolerance in chronic heart failure: the role of cortisol and the catabolic state. Curr Heart Fail Rep 2014, 11: 70-9.
- Pongkan W, Chattipakorn SC, Chattipakorn N. Roles of testosterone replacement in cardiac ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther 2016, 21: 27-43.
- Herring MJ, Oskui PM, Hale SL, Kloner RA. Testosterone and the cardiovascular system: a comprehensive review of the basic science literature. J Am Heart Assoc 2013, 2: e000271.
- Santos MR, Sayegh AL, Groehs RV, et al. Testosterone deficiency increases hospital readmission and mortality rates in male patients with heart failure. Arq Bras Cardiol 2015, 105: 256-64.
- Mirdamadi A, Garakyaraghi M, Pourmoghaddas A, et al. Beneficial effects of testosterone therapy on functional capacity, cardiovascular parameters, and quality of life in patients with congestive heart failure. Biomed Res Int 2014, 2014: 392432.
- Triposkiadis F, Karayannis G, Giamouzis G, et al. The sympathetic nervous system in heart failure: physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009, 54: 1747-62.
- Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 2013, 113: 739-53.
- Chatterjee S, Biondi-Zoccai G, Abbate A, et al. Benefits of β blockers in patients with heart failure and reduced ejection fraction: network meta-analysis. BMJ 2013, 346: f55.
- Cittadini A, Saldamarco L, Marra AM, et al. Growth hormone deficiency in patients with chronic heart failure and beneficial effects of its correction. J Clin Endocrinol Metab 2009, 94: 3329-36.
- Napoli R, Guardasole V, Angelini V, et al. Acute effects of growth hormone on vascular function in human subjects. J Clin Endocrinol Metab 2003, 88: 2817-20.
- Arcopinto M, Isgaard J, Marra AM, et al. IGF-1 predicts survival in chronic heart failure. Insights from the T.O.S.CA. (Trattamento Ormonale Nello Scompenso CArdiaco) registry. Int J Cardiol 2014, 176: 1006-8.
- Cittadini A, Marra AM, Arcopinto M, et al. Growth hormone replacement delays the progression of chronic heart failure combined with growth hormone deficiency: an extension of a randomized controlled single-blind study. JACC Heart Fail 2013, 1: 325-30.
Ripercussioni sulla pressione arteriosa delle endocrinopatie e dei farmaci endocrini
Alberto Mazza
Centro Ipertensione Accreditato SIIA, Medicina Interna, Ospedale di Rovigo, Azienda ULSS 18
La regolazione e il mantenimento di livelli di pressione arteriosa (PA) tali da permettere un’adeguata perfusione degli organi vitali ed evitare i danni al sistema cardio-vascolare provocati dal patologico incremento dei valori pressori (ipertensione, PA clinica ≥ 140/90 mmHg) dipendono da numerosi sistemi, che interagiscono tra loro attraverso meccanismi di feed-back positivo e negativo (1). Tra questi, il sistema nervoso autonomo svolge un ruolo chiave, modulando la PA attraverso l’attivazione dei recettori adrenergici β1 e α, inducendo rispettivamente un aumento della frequenza e della gittata cardiaca e vasocostrizione arteriolare. Tuttavia, oltre a quella del sistema nervoso autonomo, la PA risente dell’azione di numerosi ormoni (2), come quelli steroidei (mineralcorticoidi, glucocorticoidi, estrogeni), tiroidei, delle catecolamine e degli ormoni peptidici (angiotensina II, ormone anti-diuretico e natriuretici atriali).
La grande maggioranza degli ipertesi (90-95%) non ha una causa nota dell’ipertensione: tale ipertensione, definita primitiva o essenziale, include una popolazione eterogenea nella quale, oltre ai suddetti meccanismi fisiopatologici, sono coinvolte alterazioni poligeniche e ambientali. Nel restante 5-10% dei casi l’ipertensione è secondaria a forme specifiche, che in casi ancora più rari (3%) sono endocrinopatie (3).
Seppur rare, le ipertensioni endocrine (IE) rappresentano un argomento di duplice interesse medico, sia biologico che clinico. Dal punto di vista biologico, le IE sono modelli sperimentali naturali di ipertensione, che contribuiscono alla conoscenza di come “l’iper-increzione ormonale sostenuta” sia in grado di indurre ipertensione. Dall’altra, il riconoscimento e la diagnosi precoce dell’IE (3) e la correzione della causa che ha generato l’ipertensione, permettono spesso di normalizzare l’incremento della PA, che di solito non risponde alla terapia anti-ipertensiva anche plurifarmacologica, tanto che alcune forme di IE - se non diagnosticate e trattate precocemente - possono presentare un decorso accelerato fino all’ipertensione maligna.
Nella pratica clinica non esiste una classificazione condivisa dell’IE, ma dal punto di vista fisiopatologico le IE si possono dividere in “primarie” e “secondarie” (tabella 1): nelle prime l’ormone modula direttamente i sopracitati determinanti fisiologici emodinamici della PA (es. iperaldosteronismi, feocromocitoma e iper-reninismi) e nelle altre l’ormone in specifiche endocrinopatie genera ipertensione indirettamente, in quanto recluta fattori che poi influenzano i determinanti emodinamici della PA (es. ipercortisolismi, distiroidismi, iperparatiroidismo e acromegalia).
| Tabella 1 Classificazione dell’ipertensione arteriosa endocrina |
|
| Primitiva | Iperaldosteronismo primario Iper-reninismo primitivo (reninoma) e secondario Feocromocitoma e altre neoplasie del sistema cromaffine |
| Secondaria | Sindrome di Cushing Acromegalia Ipotiroidismo Ipertiroidismo Iperparatiroidismo Diabete mellito tipo 2 Obesità Sindrome di Liddle Iperplasia surrenalica congenita Ipertensione in gravidanza Ipertensione indotta da estrogeni |
Sono qui di seguito elencate le principali forme di IE, per le quali viene proposto l’approccio terapeutico sul trattamento dell’ipertensione raccomandato dalle linee guida.
Ipertensione da iperaldosteronismo autonomo o primario
L’iperaldosteronismo autonomo (IAA), grazie al miglioramento del work-up diagnostico di screening biochimico e strumentale (4), è recentemente riconosciuto come la causa più frequente di IE (5). È caratterizzato da un’aumentata secrezione di aldosterone, svincolata dal controllo fisiologico esercitato dal sistema renina-angiotensina-aldosterone (RAAS).
L’adenoma corticale benigno secernente (o adenoma di Conn) è la forma più comune di IAA (65% dei casi), mentre l’iperplasia surrenalica bilaterale micro o macronodulare (iperaldosteronismo idiopatico) rappresenta la seconda causa di IAA (30% dei casi). Nell’IAA rientra una forma rara congenita, dove il fenotipo clinico (l’ipertensione) è controllabile con la somministrazione di glucocorticoidi (glucocorticoid-remediable aldosteronism).
Nell’IAA l’ipertensione arteriosa è principalmente indotta dalla ritenzione idrosalina secondaria all’azione renale dell’aldosterone, che clinicamente determina aumento della volemia e della portata cardiaca; tuttavia, spesso l’iperaldosteronismo anticipa gli effetti emodinamici attraverso un’azione di vasocostrizione sulle resistenze periferiche, mediata dall’angiotensina (AT) II.
L’ipertensione arteriosa tipica dell’IAA è spesso diastolica, da moderata (100 mmHg) a severa (110-120 mmHg), e di difficile controllo farmacologico. Nell’IAA da adenoma monolaterale la rimozione laparoscopica della neoformazione o del surrene riduce notevolmente la PA e normalizza l’ipertensione nel 50-60% dei casi (3). Le forme di IAA idiopatico vanno invece trattate con terapia medica con l’uso dello spironolattone, che alla dose di 25 mg è efficace nel controllare l’ipertensione e l’iperkaliemia ad essa associate. Tuttavia, a dosaggi superiori lo spironolattone può causare effetti collaterali che ne limitano l’impiego, come la ginecomastia nel maschio e disturbi mestruali nella donna, per blocco dei recettori per gli steroidi sessuali. In alternativa allo spironolattone, possono essere utilizzati altri diuretici risparmiatori di potassio, come canrenoato, amiloride e triamterene, ma è spesso necessaria la combinazione con calcioantagonisti diidropiridinici e inibitori del RAAS, come ACE-inibitori o sartani (5). Alcuni autori suggeriscono l’uso off-label dell’eplerenone, un antagonista selettivo dell’aldosterone privo degli effetti avversi dello spironolattone, il cui uso è indicato in Europa solo per la terapia dello scompenso cardiaco (6).
Ipertensione neuroendocrina
Il feocromocitoma e i paragangliomi sono neoplasie, spesso benigne, derivanti dalle cellule cromaffini, rispettivamente della midollare del surrene e dei gangli simpatici. Anche se sono una causa assai rara di ipertensione arteriosa, con una prevalenza dello 0.1-0.2% degli ipertesi, possono portare a gravi e potenziali effetti letali a seguito di crisi ipertensive indotte dal rilascio in circolo delle catecolamine. Tuttavia, nel 30% dei casi, in tali neoplasie la PA clinica è normale o addirittura vi è ipotensione ortostatica.
Il trattamento dell’ipertensione può essere difficile nelle forme diagnosticate in gravidanza (7) o nell’anziano (8), tanto che per la loro gestione è necessario ricorrere a un team multidisciplinare. Generalmente la rimozione chirurgica della neoplasia normalizza la PA, ma diventa fondamentale la gestione peri-operatoria dell’ipertensione (9). Infatti, questa prevede innanzi tutto il blocco farmacologico dei recettori adrenergici di tipo α e solo in seguito il blocco dei recettori adrenergici di tipo β. Infatti l’uso di β-bloccanti (atenololo, metoprololo, propranololo) prima degli α-bloccanti (doxazosina, prazosina o terazosina) va assolutamente evitato, perché può indurre un aumento paradosso della PA, a seguito della soppressione della componente vasodilatatoria sistemica mediata dai recettori β2-adrenergici. Anche se il labetalolo è tradizionalmente considerato l'agente ideale per il suo antagonismo α- e β-adrenergico, studi sperimentali non supportano il suo utilizzo in questo ambito clinico. In caso di inadeguato controllo della PA, è raccomandata la somministrazione di vasodilatatori come i calcioantagonisti (CA) diidropiridinici (amlodipina, nicardipina, nifedipina); è tuttavia non raccomandata la somministrazione orale o sublinguale di CA a breve durata d'azione come la nifedipina, potenzialmente pericolosa in quanto provoca una rapida e non modulabile riduzione della PA e non gestibile dai sistemi di autoregolazione cerebrale e coronarica. Nel caso invece di emergenze ipertensive con danno d’organo acuto, per il controllo della PA è raccomandato somministrare con cautela anti-ipertensivi per via parenterale (tabella 2).
| Tabella 2 Trattamento farmacologico delle emergenze ipertensive indotte da neoplasie neuroendocrine |
||
| Condizione | Farmaco | Contro-indicazioni/precauzioni |
| SCA | Nitroglicerina + β-bloccanti Nitroprussiato + β-bloccanti |
Idralazina |
| EPA | Nitroglicerina + diuretici dell’ansa Nitroprussiato + diuretici dell’ansa |
β-bloccanti Verapamil |
| Dissezione aneurisma aortico | Nitroprussiato + β-bloccanti | Vasodilatatori in generale |
| Ictus ischemico | Nitroglicerina Nitroprussiato Labetalolo |
Nifedipina |
| Encefalopatia ipertensiva | Nitroprussiato Labetalolo Nicardipina |
Clonidina α-metilDOPA |
| Ictus emorragico | Labetalolo Nicardipina |
Nitroprussiato (con precauzione) Nifedipina |
| ESA | Nimodipina | Nitroprussiato (con precauzione) |
| IRA | Fenoldopam Nicardipina |
Diuretici (con precauzione) |
| Eclampsia | α-metilDOPA Idralazina Solfato di magnesio |
Nitroprussiato |
| SCA: sindrome coronarica acuta; EPA: edema polmonare acuto; ESA: emorragia sub-aracnoidea; IRA: insufficienza renale acuta | ||
Ipertensione nell’iper-reninismo
La renina è un enzima (proteasi), che trasforma l’angiotensinogeno in AT I e assieme all’enzima di conversione (ACE) dell’AT I porta alla formazione dell’AT II, il più potente vasocostrittore endogeno dell’organismo. Pur non essendo un ormone e quindi non direttamente causa di IE, tutte le condizioni che determinano un aumento della renina circolante e tissutale determinano in realtà l’attivazione del RAAS, uno dei più importanti sistemi di regolazione della PA (1). L’aumento dei livelli di renina circolante può essere primitivo, come nei reninismi autonomi da neoplasie (3) di origine renale (emangiopericitoma e neuroblastoma), o extra-renale (tumori solidi), oppure secondario a ischemia renale. I reninismi primari sono molto rari e determinano un’ipertensione renina-dipendente curabile con l’asportazione della lesione.
Nei casi di ipertensione non controllata è raccomandato l’uso di ACE-inibitori e soprattutto degli antagonisti recettoriali tipo I dell’AT II, i sartani, che bloccano gli effetti vascolari dell’AT II (10).
Negli iper-reninismi secondari l’ischemia è prevalentemente secondaria a ipoperfusione del parenchima renale e in minor prevalenza (2-3% degli ipertesi) a stenosi dell’arteria renale (ipertensione renovascolare). La diagnosi precoce di quest’ultima è fondamentale, in quanto l’ipertensione, solitamente sistodiastolica da moderata a grave, può essere curata o migliorata mediante correzione chirurgica (generalmente percutanea) della stenosi (11). Nelle forme che non hanno tratto beneficio dalla rivascolarizzazione, il trattamento dell’ipertensione prevede l’uso di ACE-I e/o sartani e diuretici, eventualmente associati a CA e a bloccanti dei recettori β- e α-adrenergici.
Ipertensione da cortisolismi
Nelle diverse forme cliniche di ipercortisolismo, di cui la più nota è la sindrome di Cushing, l’ipertensione è usualmente moderata e solo in alcuni casi di grado severo ed è spesso (3) caratterizzata dalla perdita del fisiologico ritmo circadiano della PA, con evidenza di ipertensione notturna al monitoraggio ambulatoriale della PA nelle 24h (24h-ABPM).
Dal punto di vista fisiopatologico, nella genesi dell’ipertensione l’ormone (cortisolo) agisce indirettamente, reclutando numerosi meccanismi che regolano la PA (1), tanto che dal punto di vista emodinamico si distinguono un’ipertensione a elevata portata (ritenzione idrosalina -> aumento del volume circolante -> ipokaliemia e alcalosi metabolica) e una a resistenze elevate (alterazione del bilancio tra agenti vasocostrittori e vasodilatatori indotti dal cortisolo, con riduzione delle prostaglandine e aumento dell’AT II e del tono adrenergico).
La normalizzazione dell’ipertensione generalmente si ottiene con la rimozione/trattamento della causa dell’ipercortisolismo e nelle altre forme il controllo pressorio si ottiene con la somministrazione di diuretici, inibitori del RAAS e CA (10).
Ipertensione nei distiroidismi
Le variazioni dello stato di eutiroidismo interessano praticamente tutti i sistemi fisiologici, ma sono particolarmente pronunciati gli effetti su quello cardio-vascolare, in particolare sul controllo a medio e lungo termine della PA (1). Le patologie della tiroide inducono diversi cambiamenti emodinamici e influenzano la PA modulando la funzione endoteliale, la reattività vascolare, l’emodinamica renale e il RAAS (3). Tuttavia, nei distiroidismi i meccanismi emodinamici che regolano lo sviluppo e il mantenimento dell’ipertensione sono tra loro contrapposti. L'ipertiroidismo determina un aumento della risposta endotelio-dipendente, secondario allo stress di parete indotto dalla circolazione iperdinamica e contribuisce a ridurre la resistenza vascolare. Viceversa, l’ipotiroidismo è accompagnato a una marcata diminuzione della sensibilità agli agonisti simpatici, con conseguente aumento della resistenza vascolare periferica e della rigidità arteriosa. Inoltre, in modelli animali l’ipotiroidismo riduce la vasodilatazione endotelio-dipendente mediata dall’ossido nitrico (1).
L’ipertensione secondaria a disturbi della tiroide è di solito reversibile con il ripristino dello stato di eutiroidismo, ma in alcuni casi è necessario trattare farmacologicamente l’ipertensione. In particolare, nell’ipertiroidismo, caratterizzato da ipertensione prevalentemente sistolica e con elevata PA differenziale (12), i β-bloccanti sono il trattamento di prima scelta per il controllo sia della PA che della frequenza cardiaca, ma quando sono controindicati o non tollerati, è raccomandato l’uso di ACE-inibitori e CA (tabella 3).
L’ipotiroidismo è invece una tipica forma di ipertensione a bassa renina (27), caratterizzata da un aumento della PA diastolica, che mostra una migliore risposta ipotensiva a CA e diuretici; inoltre, nell’ipotiroidismo una dieta a basso contenuto di sodio sembra ulteriormente migliorare il controllo della PA (10). Tuttavia, non sono attualmente disponibili studi clinici randomizzati che abbiano confrontato l'efficacia delle diverse classi di farmaci anti-ipertensivi sul controllo pressorio in soggetti con distiroidismo; lo stesso dicasi per i singoli componenti delle diverse classi di anti-ipertensivi, dove a tutt’oggi non sono disponibili dati di efficacia.
| Tabella 3 Trattamento anti-ipertensivo raccomandato nei disturbi della tiroide |
||
| Farmaci anti-ipertensivi | Ipotiroidismo | Ipertiroidismo* |
| ACE-inibitori (o sartani se intolleranza) | - | ++ |
| Calcio-antagonisti diidro/non-diidropiridinici* | +++ | ++ |
| β-bloccanti | - | +++ |
| Diuretici | +++ | + |
| α-bloccanti | ++ | - |
Ipertensione nell’acromegalia
Si manifesta nel 20-40% dei pazienti acromegalici, specie di età avanzata e con storia di malattia di più lunga durata (3).
L’ipertensione, solitamente di entità moderata e non complicata, si instaura nelle fasi iniziali con un meccanismo volume-dipendente ad alta portata, per riduzione dell’escrezione sodica renale (stimolo diretto del GH sulla renina, che a sua volta attiva il RAAS); in seguito si ha aumento delle resistenze periferiche (danno d’organo endoteliale e aumento dell’attività adrenergica centrale forse indotto dall’iperinsulinismo). L’ipertensione può diventare complicata o resistente quando l’acromegalia si associa alla sindrome delle apnee notturne.
Il trattamento dell’ipertensione prevede l’uso dei comuni anti-ipertensivi e nelle prime fasi in particolare dei diuretici, associato alla restrizione dell’apporto di sodio (13). Nelle forme di ipertensione grave e complicata, il controllo pressorio avviene solo con il trattamento farmacologico/chirurgico dell’acromegalia.
Ipertensione nell’iperparatiroidismo
L’iperparatiroidismo primitivo è frequentemente associato all’ipertensione arteriosa (56-80%) e spesso assieme all’ipercalcemia ne caratterizza il quadro clinico all’esordio (3). L’ipertensione presenta spesso un profilo circadiano di tipo “non-dipping” (1), con ridotto calo pressorio notturno durante 24h-ABPM, che a sua volta, per aumento del carico pressorio globale, si associa a maggiore incidenza di danno d’organo cardiaco (ipertrofia ventricolare) e vascolare.
La diagnosi precoce e il conseguente trattamento chirurgico di paratiroidectomia (10) generalmente normalizza la PA. Non bisogna tuttavia dimenticare le forme di iperparatiroidismo iatrogeno, indotto paradossalmente da farmaci anti-ipertensivi come i diuretici (furosemide), molto frequente nella popolazione anziana (14).
Ipertensione da estro-progestinici
L’uso di estroprogestinici (EP) a scopo contraccettivo o in post-menopausa sembra esporre la donna ad aumentato rischio di ipertensione, tanto maggiore quanto maggiore è la durata del trattamento (10). Tuttavia, l’uso attuale di EP a scopo contraccettivo a basso dosaggio ha ridimensionato l’incidenza di ipertensione (1.2-2%).
Non è ancora del tutto chiaro il rapporto che lega menopausa, assetto ormonale, età e PA (37), e sono numerosi e non del tutto noti i meccanismi con i quali gli EP intervengono sulla modulazione della PA, anche se l’attivazione del RAAS sembra quello principale (1). In particolare, in modelli sperimentali l’attivazione del RAAS si associa a modifiche dell’emodinamica sistemica e soprattutto renale, per vasocostrizione delle arteriole renali, e ad aumento della frazione di filtrazione glomerulare, che regrediscono con la somministrazione di sartani. D’altra parte, è anche noto che gli estrogeni esercitano un’azione trofica sull’endotelio vasale, con rilascio di autacoidi vasodilatatori che controbilanciano l’attivazione vasocostrittrice del RAAS. Anche se non tutti gli studi sono concordi sugli effetti della terapia ormonale sostitutiva, la somministrazione di EP non sembra avere effetti negativi sulle donne normotese, e sembra essere sicura anche per le donne ipertese (10).
L’ipertensione non è da ritenersi una controindicazione alla prescrizione di terapia ormonale, anche se viene richiesto un attento monitoraggio nei primi mesi di somministrazione della stessa. Inoltre, come ormai uso comune, è raccomandato utilizzare formulazioni transdermiche e con un basso dosaggio di ormoni, e preferire i composti con caratteristiche più simili a estrogeno e progesterone naturale (40).
L’ipertensione da EP può tuttavia manifestarsi se coesistono altri gravi fattori di rischio di danno endoteliale - considerato il “primum movens” della patologia ipertensiva - come dislipidemia, tabagismo e forse fattori genetici individuali (15). In ogni caso l’uso dell’EP va sospeso se insorge ipertensione ed è controindicato se pre-esistono ipertensione giovanile, patologie cardiache o renali.
Bibliografia
- Dos Santos RL, da Silva FB, Ribeiro RF Jr, Stefanon I. Sex hormones in the cardiovascular system. Horm Mol Biol Clin Investig 2014, 18: 89-103.
- Carey RM. Overview of endocrine systems in primary hypertension. Endocrinol Metab Clin North Am 2011, 40: 265-77.
- Mazza A, Zamboni S, Armigliato M, et al. Endocrine arterial hypertension: diagnostic approach in clinical practice. Minerva Endocrinol 2008, 33: 127-46.
- Mazza A, Armigliato M, Zamboni S, et al. Endocrine arterial hypertension: therapeutic approach in clinical practice. Minerva Endocrinol 2008, 33: 297-312.
- Rossi GP, Bernini G, Caliumi C, et al, for the PAPY Study Investigators. A prospective study of the prevalence of primary aldosteronism in 1125 hypertensive patients. J Am Coll Cardiol 2006, 48: 2293–300.
- Takeda Y. Effects of eplerenone, a selective mineralocorticoid receptor antagonist, on clinical and experimental salt-sensitive hypertension. Hypertens Res 2009, 32: 321-4.
- Mazza A, Armigliato M, Ferretti A, et al. Gestational diastolic hypertension with gene mutation-related pheochromocytoma positive at 18F-DOPA PET/CT: diagnostic and therapeutic implications. Rev Esp Med Nucl Imagen Mol 2013, 32: 111-2.
- Mazza A, Rubello D. Malignant pheochromocytoma in the elderly: which is the best management in clinical practice? Nucl Med Commun 2015, 36: 1159-64.
- Mazza A, Armigliato M, Marzola MC, et al. Anti-hypertensive treatment in pheochromocytoma and paraganglioma: current management and therapeutic features. Endocrine 2014, 45: 469-78.
- ESH/ESC Task Force for the Management of Arterial Hypertension. 2013 Practice guidelines for the management of arterial hypertension of the European Society of Hypertension (ESH) and the European Society of Cardiology (ESC): ESH/ESC Task Force for the management of arterial hypertension. J Hypertens 2013, 31: 1925-38.
- Mazza A, Ravenni R, Armigliato M, et al. Mood disorders in uncontrolled hypertension despite multiple anti-hypertensive medications: searching for a link. High Blood Press Cardiovasc Prev 2016, 23: 41-6.
- Mazza A, Beltramello G, Armigliato M, et al. Arterial hypertension and thyroid disorders: what is important to know in clinical practice? Ann Endocrinol 2011, 72: 296-303.
- Bondanelli M, Ambrosio MR, degli Uberti EC. Pathogenesis and prevalence of hypertension in acromegaly. Pituitary 2001, 4: 239-49.
- Walker MD, Silverberg SJ. Cardiovascular aspects of primary hyperparathyroidism. J Endocrinol Invest 2008, 31: 925-31.
- Cacciatore B, Paakkari I, Hasselblatt R, et al. Randomized comparison between orally and transdermally administered hormone replacement therapy regimens of long-term effects on 24-hour ambulatory blood pressure in postmenopausal women. Am J Obstet Gynecol 2001, 184: 904-9.
Gestione delle aritmie cardiache in corso di ipertiroidismo
Michela Armigliato
UOS Endocrinologia e Reumatologia, Medicina Interna, Ospedale di Rovigo - Azienda ULSS 18
DATI EPIDEMIOLOGICI
La forme più frequenti di aritmia nell’ipertiroidismo in pazienti con cuore sano sono la tachicardia sinusale (a riposo o da sforzo), le ectopie sopraventricolari, forme non sostenute di tachiaritmia sopraventricolare e la ridotta variabilità della frequenza cardiaca; questi trigger elettrici contribuiscono allo sviluppo di parossismi di tachicardia atriale, fibrillazione atriale (FA) e flutter atriale. L’aritmia di maggior impatto clinico è la FA.
Fattori di rischio per lo sviluppo di FA nell’ipertiroidismo sono età, sesso maschile, ipertensione arteriosa, sottostanti patologie coronariche, scompenso cardiaco e malattie valvolari, T3-tossicosi. La FA è più frequente nei casi di T3-tossicosi e nel gozzo multinodulare tossico (GMN) rispetto al morbo di Graves, riflettendo l’età più avanzata dei pazienti con GMN (1).
Prevalenza della FA nei pazienti con ipertiroidismo
La prevalenza di FA e di altre forme di tachiaritmie atriali nei pazienti con ipertiroidismo clinico (IC) è circa del 13.8% vs il 2.3% della popolazione di controllo con normale funzione tiroidea. In uno studio su 13.000 pazienti ipertiroidei la prevalenza di FA è risultata < 2%, probabilmente a causa di una diagnosi precoce di ipertiroidismo: dopo suddivisione per età, la prevalenza si modifica progressivamente per ogni decade, raggiungendo il 15% nei pazienti ultra70enni (2).
In un altro studio l’incidenza cumulativa di FA nell’ipertiroidismo subclinico (IS) in pazienti di età > 60 anni era del 28% in un periodo di 10 anni (3). Questo dato conferma quello emerso dallo studio di 40.628 pazienti ipertiroidei del registro nazionale danese, in cui la prevalenza di FA è risultata dell’8.3% sia nell’IC che nell’IS, in quanto la forma subclinica si osserva nei pazienti più anziani (4).
Prevalenza dell’ipertiroidismo nei pazienti con FA
In uno studio in numerosi pazienti con FA di recente insorgenza, meno dell’1% presentava IC. Nonostante la bassa prevalenza, la possibilità di ripristinare il ritmo sinusale (RS), raggiungendo precocemente l’eutiroidismo, giustifica la determinazione del TSH in tutti i pazienti con FA o altre aritmie sopraventricolari non spiegabili diversamente.
GESTIONE DELLA FA IN CORSO DI IPERTIROIDISMO
La gestione delle aritmie cardiache indotte da amiodarone non è oggetto di questa trattazione.
Trattamento della tireotossicosi
Normalizzazione del livello di ormoni tiroidei plasmatici con terapia medica, radiometabolica o chirurgica, a seconda dell’eziologia di base.
Controllo della frequenza cardiaca
Il primo passo nel trattamento della FA nell’ipertiroidismo, indipendentemente dalla causa, è il controllo della frequenza cardiaca (FC). La scelta del farmaco dipende da quanto rapidamente si deve raggiungere lo scopo, dalla coesistenza di comorbilità e dall’evidenza di scompenso cardiaco acuto. Il controllo della FC si ottiene con l’uso di ß-bloccanti, calcioantagonisti non diidropiridinici e digossina (5).
ß-bloccanti (BB): in assenza di scompenso cardiaco, i BB (non selettivi e ß1-selettivi) sono i farmaci di prima scelta (classe 1, evidenza C). Controindicazioni assolute all’uso dei ß-bloccanti non selettivi sono asma bronchiale e BPCO severa, grave arteriopatia obliterante degli arti inferiori, fenomeno di Raynaud, ipoglicemia asintomatica del diabete mellito, bradicardia sinusale e blocco atrio-ventricolare di 2°-3° grado.
I BB riducono rapidamente i sintomi dovuti all’attivazione adrenergica, in quanto in molti tessuti, in corso di ipertiroidismo, vi è un’aumentata espressione di recettori ß-adrenergici. Nel paziente con ipertiroidismo in assenza di FA l’uso dei BB è off-label. Nel caso di FA associata a ipertiroidismo, la terapia con BB allo scopo di controllare la frequenza deve essere precoce ed efficace: uno studio ha dimostrato che in pazienti con cardiomiopatia dilatativa e ipertiroidismo, indipendentemente dalla presenza di FA, la reversibilità del danno correlava negativamente con durata di ipertiroidismo, sesso femminile, livelli di T3 e mancato impiego di BB.
I BB possono essere usati per via parenterale, per sfruttarne rapidamente l’azione nella crisi tireotossica, o per via orale. Il propranololo a dosi elevate (> 160 mg/die) riduce la produzione di T3 attraverso il blocco della 5’monodeiodinasi essendo molto liposolubile, ma il suo effetto è lento (7-10 giorni); atenololo e metoprololo sono BB ß1-selettivi e quindi possono essere usati in caso di controindicazioni relative, hanno azione più rapida e la mono o duplice somministrazione migliora la compliance del paziente.
L’atenololo per via parenterale, grazie alla lunga emivita, può essere utilizzato prima della tiroidectomia, garantendo copertura nella fase peri- e post-operatoria.
In gravidanza l’impiego di BB deve essere limitato a casi severi e per brevi periodo, in quanto sono stati segnalati ritardi di crescita e casi di aborto.
Per la scelta del tipo e relativo dosaggio dei BB utilizzati nella gestione delle aritmie nell’ipertiroidismo si rimanda alla tab 1.
| Tabella 1 ß-bloccanti raccomandati nella terapia dell’ipertiroidismo |
|||
| Principio attivo | Dose | Frequenza di somministrazione | Considerazioni |
| Propranololo | 10-40 mg/die | 3 o 4 volte/die | BB non selettivo, esiste molta esperienza |
| Atenololo | 25-100 mg/die per os 5 mg ev boli |
2 volte/die | ß1-selettivo, aumento compliance |
| Metoprololo | 25-50 mg/die per os 5 mg ev boli |
4 volte/die | ß1-selettivo |
| Nadololo | 40-160 mg/die | 4 volte/die | BB non selettivo, scarsa esperienza |
| Esmololo | Infusione venosa, 50-100 µg/kg/min | Reparti intensivi, crisi tireotossica | |
Calcio-antagonisti: quando i BB sono controindicati, possono essere utilizzati calcio-antagonisti non diidropiridinici (verapamil e diltiazem) per via parenterale (classe 1, evidenza C), ma prevedono cautela e monitoraggio pressorio ed elettrocardiografico, in quanto gravati dal rischio di ipotensione arteriosa e riduzione delle resistenze vascolari già ridotte nell’ipertiroidismo. Sono adatti per via orale nel controllo a lungo termine della frequenza cardiaca nei pazienti con controindicazione all’uso dei BB.
Digitale: la digossina (alle dosi di 0.25 mg in boli ripetuti) può essere utilizzata in caso di scompenso cardiaco, anche se spesso il paziente ipertiroideo è resistente alla sua azione per aumentata clearance del farmaco, aumento del volume di distribuzione, aumentato tono simpatico e ridotto tono vagale. Nell’anziano l’aumento della dose richiesta per raggiungere lo scopo comporta un aumentato rischio di tossicità.
Ripristino del ritmo sinusale e rischio cardio-embolico (RCE)
L’ipertiroidismo è considerato un fattore di rischio e causa reversibile di FA, ma solo 2/3 dei pazienti torna spontaneamente in RS entro 8-10 settimane dal raggiungimento dell’eutiroidismo; inoltre il RCE della FA è legato alla durata dell’aritmia. Per i pazienti che restano fibrillanti, resta l’opzione della cardioversione (CV) farmacologica o elettrica.
Quando è indicata la cardioversione?
Nella pratica clinica la si propone ai pazienti con FA:
- di durata < 12-24 mesi, in quanto le alterazioni atriali legate alla FA di lunga durata aumentano la probabilità di recidive;
- solo dopo almeno 3 mesi di persistente eutiroidismo.
Recenti studi stanno mettendo in discussione questi paradigmi: in uno studio retrospettivo su 106 pazienti con FA della durata di almeno 12 mesi e tireotossicosi, esenti da altre patologie cardiache, la CV elettrica seguita da 3 mesi di profilassi con disopiramide risultava efficace nel mantenere l’euritmia nel 67% dei casi con un follow-up di 80 ± 37 mesi (6). Questi autori affermano che come fattore predittivo di mantenimento del RS dopo CV è più importante l’assenza di anomalie strutturali cardiache rispetto alla durata della FA. È noto che il RCE correla con la durata della FA. Sul dilemma se sottoporre a CV i pazienti non ancora eutiroidei, con l’intento di abbreviare la durata della FA e il RCE, un recente studio poco numeroso non rilevava significative differenze nel rischio di recidiva di FA e sul RCE tra pazienti ancora ipertiroidei o eutiroidei dopo terapia (7). I risultati di questo studio discordano da quello di Siu et al (8), in cui si dimostrava che il tasso di recidiva di FA entro 24 mesi dalla CV risultava del 59% nei pazienti eutiroidei rispetto all’84% dei pazienti con disfunzioni tiroidee.
Quando sottoporre il paziente ad anti-coagulazione?
I pazienti con FA permanente hanno un RCE annuale di circa 3-4%, 5-7 volte maggiore dei soggetti in RS. Non è ancora assodato se i pazienti con FA e ipertiroidismo abbiano RCE maggiore. Il rischio di ictus nell’ipertiroidismo correla con l’età oltre che con la presenza di FA. Siu afferma che i pazienti ipertiroidei con FA hanno un rischio di sviluppare ictus del 9.4% entro 1 anno vs il 3% degli eutiroidei (9).
Nei pazienti più giovani, maschi, con FA con basso RCE (punteggio CHADS-VAS = 0), non sono certi i benefici dell’anti-coagulazione: in questa classe di pazienti può essere presa in considerazione la terapia anti-aggregante (10). È proprio in questa classe di pazienti che si impone il raggiungimento rapido dell’eutiroidismo, per abbreviare la durata della FA, e il trattamento in fase precoce con BB adeguatamente titolati, per ridurre il rischio di sviluppare tachi-cardiomiopatia. Nei pazienti ad elevato RCE, la scelta tra vecchi e nuovi anti-coagulanti non risente della coesistenza dell’ipertiroidismo; in caso di impiego di warfarin, potrebbero essere necessarie dosi più elevate, tenendo conto dell’aumentata clearance renale dell’anti-coagulante.
BIBLIOGRAFIA
- Klein I, Danzi S. Thyroid disease and the heart. Circulation 2007, 116: 1725-35.
- Shimizu T, Koide S, Noh JY, et al. Hyperthyroidism and the management of atrial fibrillation. Thyroid 2002, 12: 489-93.
- Sawin CT. Subclinical hyperthyroidism and atrial fibrillation. Thyroid 2002, 12: 501-3.
- Frost L, Vestergaard P, Mosekilde L. Hyperthyroidism and the risk of atrial fibrillation and flutter: a population based study. Arch Intern Med 2004, 164: 1675-8.
- Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid 2011, 21: 593-643.
- Nakazawa H, Lythall DA, Noh JY, et al. Is there a place for the late cardioversion of atrial fibrillation? A long-term follow-up study of patients with post thyrotoxic atrial fibrillation. Eur Heart J 2000, 21: 327-33.
- Gurdogan M, et al. Predictors of atrial fibrillation recurrence in hyperthyroid and euthyroid patients. Arq Bras Cardiol 2016, 106: 84-91.
- Siu CW, Jim MH, et al. Comparison of atrial fibrillation recurrence rates after successful electrical cardioversion in patients with hyperthyroidism–induced versus non hyperthyroidism–induced persistent atrial fibrillation. Am J Cardiol 2009, 103: 540-3.
- Siu CW, Pong V, et al. Risk of ischemic stroke after new onset atrial fibrillation in patients with hyperthyroidism. Heart Rhythm 2009, 62: 169-73.
- January CT, Wann LS, Alpert JS, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014, 64: e1-76.
Il rene come organo e bersaglio endocrino
Silvio Settembrini1 & Alessandra Fusco2
1Servizio di Endocrinologia, Diabetologia e Malattie Metaboliche, DS 26, Unità di Nefro-Diabetologia, UOC di Nefrologia e Dialisi, Ospedale dei Pellegrini, Napoli
2Ambulatorio di Endocrinologia e Diabetologia, Centro Acismom, Napoli
(aggiornato al 25 luglio 2017)
FUNZIONI DEL RENE
Il rene è considerato l’organo deputato a mantenere l’equilibrio dell’organismo e a regolare l’omeostasi dell’ambiente interno (plasma e liquido interstiziale).
È un organo a elevata specializzazione funzionale, le cui caratteristiche biochimiche sono contraddistinte da uno specifico network molecolare, variamente disposto nei diversi siti del nefrone, consistente in specifiche proteine strutturali ed enzimatiche, recettori e trasportatori, che mediano i segnali in arrivo (chimico-fisici, emodinamici e ormonali) in una corrispondente attività in uscita. Per tali motivi, il rene può essere considerato un organo di trasduzione bidirezionale: organo endocrino – bersaglio endocrino, con peculiarità autocrino-paracrine connesse alle finalità funzionali (1,2).
L’unità funzionale del rene è il nefrone; nell’uomo ci sono circa 1.2 milioni di nefroni per ogni rene. La tabella 1 riassume le fondamentali funzioni del rene.
| Tabella 1 Funzioni del rene |
|
| Emuntoria | Eliminazione dei prodotti finali del catabolismo azotato: urea, acido urico, creatinina, solfati, ecc. |
| Detossificazione dell’organismo da composti tossici e numerosi farmaci, con conseguente eliminazione. | |
| Regolazione del liquido extra-cellulare | Regolazione del volume (contenuto idrico dell’organismo), tramite ultra-filtrazione glomerulare, riassorbimento tubulare (passaggio selettivo di sostanze utili dall’ultra-filtrato al sangue) e secrezione tubulare (con passaggio di sostanze dal sangue nell’ultra-filtrato). Si modulano così recupero ed eliminazione di acqua (clearance dell'acqua libera), con conseguente escrezione di urina che, in base alle esigenze dell'equilibrio idrico ed elettrolitico, può essere ipertonica, isotonica o ipotonica (cioè con concentrazione di soluti maggiore, uguale o minore rispetto a quella del sangue). |
| Regolazione dell’osmolarità del liquido extra-cellulare, mediante riassorbimento di Na+ e acqua. | |
| Regolazione della concentrazione ematica di metaboliti e ioni | Regolazione del pH ematico e dell’equilibrio acido base, mediante riassorbimento e produzione ed eliminazione di bicarbonato e secrezione di idrogenioni. |
| Na+, K+, Cl-, PO43-, Ca2+, glucosio, aminoacidi, acido urico, urea, mediante integrazione tra processi di filtrazione e riassorbimento. | |
| Funzione metabolica | Produzione e degradazione di ormoni, citochine, autacoidi. |
| Gluconeogenesi. | |
| Controllo dell’attività emopoietica, con formazione, maturazione e produzione di globuli rossi. | |
Nel soggetto adulto a riposo il rene riceve circa il 20-25% della portata cardiaca (anche se la massa dei reni rappresenta solo lo 0.5% della massa corporea totale): ≈ 1200 mL/min; ≈ 1700 L/die; ≈ 63.000 L/anno; ≈ 44 milioni L in 70 anni.
Il flusso plasmatico renale è circa il 55% del flusso ematico renale, il volume del filtrato glomerulare è circa il 20% del flusso plasmatico renale: ≈ 180 L/die; ≈ 5 milioni L in 70 anni.
Il volume di urina prodotta è ≈ 1 mL/min, ≈ 1.5 L/die, ≈ 550 L/anno, ≈ 38500 litri in 70 anni.
Più del 99% del liquido filtrato dai reni deve tornare nel sangue (1,2).
IL RENE “METABOLICO”
Le attività metaboliche del rene variano in base all’area anatomo-funzionale.
L’area corticale, molto fornita di mitocondri, presenta uno spiccato metabolismo ossidativo tramite β-ossidazione degli acidi grassi (essenzialmente dell'acido palmitico), gluconeogenesi (a partire da lattato, glicerolo e aminoacidi gluconeogenici, soprattutto glutammina) e chetogenesi (in misura minore rispetto al fegato).
L’area midollare, scarsamente vascolarizzata (capta solo il 10% dell’ossigeno che arriva al rene) e poco fornita di mitocondri, energeticamente sviluppa solo la glicolisi, esclusivamente anaerobia, con produzione di acido lattico; il glucosio che viene utilizzato dalla midollare proviene o dal circolo ematico o dalla produzione corticale.
I reni, pur rappresentando insieme circa lo 0.5% del peso corporeo, consumano l’8–10% del metabolismo basale corporeo. Pertanto, l’elevato metabolismo energetico renale comporta un consistente consumo di O2, circa il 30-35% dell’O2 utilizzato/kg di peso renale, comparabile alle prestazioni energetiche del miocardio. Circa il 70-80% dell’energia sviluppata dal rene è adoperata nei meccanismi di trasporto attivo, finalizzati alla ricaptazione di molecole e ioni ultra-filtrati dal glomerulo (1,2).
I reni hanno anche importanti funzioni endocrine (3), secernendo diversi ormoni ad azione sistemica.
| Tabella 2 Sostanze ad azione ormonale e rene |
||
| Produzione | Azione | |
| Renina | x | |
| Angiotensina | x | |
| Aldosterone | x | |
| Eritropoietina | x | |
| Prostaglandine | x | x |
| Leucotrieni | x | |
| Sistema callicreina-chinine | x | |
| Fattore natriuretico renale | x | |
| Calcitriolo | x | x |
| ADH | x | |
| FGF-23 | x | |
| GH | x | |
| T3 | x | |
| PTH | x | |
| Catecolamine | x | |
| Endotelina | x | x |
| Insulina | x | |
| Glucagone, GLP-1 | x | |
Sistema renina-angiotensina (4,5)
La renina è un enzima proteolitico di 340 aminoacidi della classe delle idrolasi, secreto dalle cellule iuxta-glomerulari del rene, che trasforma l'angiotensinogeno (glicoproteina plasmatica di produzione soprattutto epatica ma rilasciata anche, costitutivamente, da cuore, vasi, reni e tessuto adiposo), in angiotensina I. Questa è un decapeptide, con scarsa attività biologica, che per opera di una chininasi II, l’ACE (solubile o localizzato su membrane cellulari, specie endoteliali, e a livello dei microvilli dell’orletto a spazzola del tubulo prossimale), viene idrolizzata con distacco dei 2 aminoacidi COOH- terminali, formando così l’angiotensina II. Questa ha una potente attività vasocostrittrice diretta, stimola il rilascio di aldosterone e ADH e a livello ipotalamico stimola il senso della sete. L’aldosterone agisce sul tubulo distale, favorendo il riassorbimento di Na+. Contemporaneamente, l'aumento di pressione sanguigna determinato dall'angiotensina II proprio a livello glomerulare fa aumentare l'ultra-filtrazione e la concentrazione di Na+ tubulare.
La regolazione della secrezione di renina è principalmente dipendente da tre fattori:
- fattori che riducono la pressione di perfusione renale, grazie a barocettori delle cellule juxta-glomerulari che percepiscono l'aumento o la diminuzione pressoria;
- riduzione della concentrazione di sodio nel tubulo distale, rilevata dalle cellule della macula densa, capaci di mandare alle cellule juxta-glomerulari segnali positivi attraverso le prostaglandine e negativi attraverso l'adenosina, stimolando o inibendo la secrezione di renina;
- liberazione di noradrenalina dalle terminazioni simpatiche, con stimolazione diretta dei recettori ß1 presenti nel rene; questi possono essere stimolati da tutti i farmaci ad azione simpatico-mimetica (come adrenalina e noradrenalina).
I valori di renina tenderanno quindi ad essere più elevati in caso di dieta iposodica, in gravidanza, in terapie con diuretici, anti-ipertensivi, estrogeni ed estro-progestinici.
Riduzioni di pressione e volemia determinano una cascata di eventi:
- ipertono simpato-adrenergico (per ridotte afferenze inibitorie baro-recettoriali), che, insieme alla diminuita perfusione renale e alla ridotta concentrazione luminale di sodio afferente alla macula densa (cellula sensing del sodio), stimola il rilascio di renina;
- la renina attiva l’angiotensina II, che riduce la filtrazione glomerulare e l’escrezione di acqua e sodio (insieme con l’aldosterone), determinando così il riequilibrio di volume ed elettroliti;
- l’ADH provoca riassorbimento di acqua nel tubulo collettore (solitamente impermeabile all’acqua).
| Tabella 3 Influenze su Angiotensinogeno e Renina |
||
| Angiotensinogeno | Renina | |
| Aumento | Glucocorticoidi Estrogeni Angiotensina II Nefrectomia Legatura degli ureteri Sindrome di Cushing Gravidanza |
Prostaglandina E2 Prostaciclina Callicreina Istamina Glucagone Paratormone Ipocalcemia Iposodiemia Gravidanza, estro-progestinici Diuretici |
| Diminuzione | Angiotensina Prostaglandine F Carico idrosalino |
Angiotensina II ADH Peptide natriuretico atriale Noradrenalina Endotelina Prostaglandine F Ipercalcemia Iperpotassiemia |
Eritropoietina
È una glicoproteina acida, costituita da una singola catena di 165 aminoacidi e da 4 catene oligosaccaridiche, con emivita di circa 3/4 ore.
Durante la vita fetale è prodotta dal fegato, mentre la produzione renale inizia durante gli ultimi mesi di gestazione e si completa alla nascita (resta comunque una minima attività epatica, che può arrivare al 10% in caso di anemia grave). La produzione renale avviene nei fibroblasti peri-tubulari della corticale e della midollare alta.
Lo stimolo alla produzione di EPO è legato a un sensore per l’ossigeno localizzato nelle cellule del tubulo prossimale e la cui azione è legata al consumo dell’O2 per il riassorbimento di Na. Alcune molecole partecipano alla trasmissione del segnale: prostaglandine, adenosina, androgeni, composti di ossigeno attivati, agonisti ß2-adrenergici, AMP-ciclico e NO. L’adenosina ha un ruolo fondamentale nella biosintesi dell’EPO, attraverso la stimolazione di protein-chinasi A, che porta alla fosforilazione di proteine nucleari utili per la trascrizione e/o la trasduzione del gene dell’EPO.
EPO è un importante regolatore dell’eritropoiesi: poiché la corticale del rene ha un elevato metabolismo aerobico, è molto sensibile all'apporto di ossigeno con il sangue. Se tale apporto diminuisce, le cellule della corticale immettono nel sangue EPO, che stimola il midollo osseo ad aumentare l'eritropoiesi: più eritrociti in circolo, infatti, significano maggiore possibilità di trasporto di ossigeno (6).
Prostaglandine
Sono acidi grassi insaturi ubiquitari, di cui esistono almeno 5 sedi di produzione a livello renale: parete vascolare di arterie e arteriole, glomerulo con capsula di Bowman, cellule epiteliali dei dotti collettori e dei tubuli corticali, cellule interstiziali della midollare.
Alcuni fattori agiscono sulla fosfolipasi stimolandone la sintesi: angiotensina II, ADH, bradichinina, noradrenalina, dieta ad alto contenuto sodico, iperosmolarità, infusione di soluzioni glucosate ipertoniche, di NaCl e mannitolo, furosemide.
Non sono noti i meccanismi di inibizione, ma alcuni farmaci intervengono a diversi livelli:
- inibiscono la fosfolipasi: corticosteroidi, calcio-antagonisti, mepacrina e sulfaniluree;
- inibiscono la ciclo-ossigenasi: alcuni FANS.
Le prostaglandine antagonizzano l’azione di vasocostrittori (noradrenalina e angiotensina II) e diuretici, determinando vasodilatazione renale e quindi intervenendo nei meccanismi di auto-regolazione renale.
Sono implicate nel controllo della secrezione di renina (ne stimolano la secrezione in risposta a ipovolemia e la loro stessa sintesi è incrementata dall’angiotensina II), nella regolazione del tono vascolare, nel controllo della funzione tubulare, inibiscono il riassorbimento di Na, si oppongono al riassorbimento di acqua indotto da ADH. Hanno quindi effetto diuretico e natriuretico. Anche l’azione dei diuretici dell’ansa è in parte mediata dallo stimolo alla sintesi delle prostaglandine (7,8).
Leucotrieni
Sono mediatori coinvolti in reazioni di ipersensibilità e di flogosi, responsabili di un potente effetto costrittore sulle cellule muscolari lisce di tutti i distretti, di un’azione vaso-permeabilizzante e di uno stimolo chemiotattico. I leucotrieni riducono l’osmolalità urinaria, aumentano il flusso urinario e svolgono un ruolo nell’escrezione di acqua.
Tra i leucotrieni, il 12-HETE è il principale prodotto delle cellule glomerulari, mesangiali ed epiteliali, responsabile di leucotassi e degranulazione leucocitaria (9).
Il sistema callicreina-chinine
Le callicreine sono proteasi che agiscono sul chininogeno, trasformandolo in chinina. La concentrazione della callicreina nel rene è maggiore nella corticale, decrescendo verso la midollare e le papille.
Le chinine attuano i loro effetti intra-renali attraverso lo stimolo alla sintesi di PGE-2 e di PGI-2. Il sistema agisce come vasodilatatore, soprattutto nelle zone juxta-midollari. Agiscono sull’equilibrio idrosodico, incrementando la diuresi e la natriuresi, grazie alla vasodilatazione, a un effetto tubulare diretto e a un’azione antagonista sull’ADH (8).
Endotelina
È la più potente sostanza endogena ad azione vasocostrittrice, sintetizzata nelle cellule endoteliali di tutto il macrocircolo, particolarmente a livello renale (nell’endotelio, nel mesangio e nelle cellule epiteliali della papilla).
Usualmente liberata in seguito a danno tissutale, la sua sintesi può essere stimolata da trombina, adrenalina, modificazioni della viscosità ematica, bradichinina, interleuchina 1, endotossina (anche in assenza di danno tissutale).
Ha azione costrittiva sulle cellule muscolari lisce e sui bronchi, cronotropa e inotropa positiva, interferisce con il trasporto ionico e la neurotrasmissione, regola la sintesi di prostaglandine, inibisce la renina, stimola la produzione di fattori natriuretici atriali.
A livello renale determina aumento delle resistenze (arteriola afferente > efferente), contrazione mesangiale e riduzione di flusso e filtrato. A questo si associa un aumento del volume urinario e dell’escrezione sodica per la liberazione dei fattori natriuretici atriali. Sul mesangio determina mitogenesi e aumento della matrice e sembra coinvolta nell’insufficienza renale acuta post-ischemica (10).
Fattore natriuretico renale
Sintetizzato nelle cellule corticali del nefrone distale, è strutturalmente identico al natriuretico atriale, con gli stessi effetti (11).
Vitamina D
Il PTH stimola il rene a produrre una 1-alfa-idrossilasi, che attiva la vitamina D prodotta dalla pelle e idrossilata in posizione 25 a livello epatico, trasformandola in calcitriolo, il suo metabolita attivo. Questo, a sua volta, stimola il riassorbimento di Ca2+ a livello del tubulo contorto distale (oltre a stimolare l’assorbimento intestinale di calcio e fosfato e ad avere effetti sull’osso) (12,13).
ADH/vasopressina
Sintetizzato nei nuclei ipotalamici sopra-ottico e para-ventricolare, modula il riassorbimento dell’acqua a livello del tubulo distale e soprattutto del dotto collettore.
Il rilascio di ADH è regolato da:
- iperosmolarità (> 280 mOsm/L);
- riduzione del volume extra-cellulare con soglia al 10%, tramite segnali stimolatori provenienti da barocettori cardiaci e vene polmonari, inibitori da aorta e seno carotideo;
- segnali stimolatori di angiotensina II, prostaglandine, acetilcolina, farmaci (alcaloidi della vinca, clofibrato, anti-depressivi triciclici, nicotina, isoproterenolo, carbamazepina, litio);
- segnali inibitori di peptidi oppioidi, fattore natriuretico atriale, farmaci (alcool, fenitoina, steroidi).
Agisce tramite recettori:
- V1, localizzati a livello di rene (cellule mesangiali, vasa recta, midollare), vasi, fegato, piastrine, accoppiati a PLC; a livello renale controllano filtrazione glomerulare, flusso ematico midollare e sintesi prostaglandine;
- V2 (con affinità molto maggiore dei V1), localizzati a livello di dotti collettori e porzione ascendente dell’ansa di Henle, accoppiati ad adenil-ciclasi; inseriscono acquaporina-2 (shuttle intra-cellulare per l’acqua) nella membrana cellulare dal compartimento intra-cellulare (14).
Klotho e FGF-23
L’FGF-23 prodotto dagli osteoblasti e osteoclasti ossei esercita le proprie attività biologiche attraverso l’interazione con un recettore di membrana (FGF-R). Vista la bassa affinità di FGF-23 per il suo recettore, è necessaria la presenza di cofattori. Klotho è una proteina prodotta a livello renale, necessaria per consentire l’attività ormonale di FGF-23. Questa interazione è di grande importanza per il metabolismo calcio-fosforo, in quanto l’FGF-23 riduce il riassorbimento tubulare dei fosfati, aumentandone l’escrezione urinaria, e Klotho inibisce la produzione di eccessive quantità di vitamina D attiva agendo sull'1-alfa-idrossilasi renale. Pertanto, il legame funzionale tra fosforemia, vitamina D ed FGF-23 è particolarmente sinergico. Elevati livelli sierici di fosfato e/o calcitriolo stimolano la produzione di FGF-23 da parte delle cellule ossee. L’interazione Klotho-FGF23 a livello renale incrementa l’endocitosi e inibisce la sintesi del co-trasportatore Na–Pi2a a livello tubulare. Di conseguenza, si riduce il riassorbimento renale di fosfato, con conseguente calo dei livelli sierici. Anche l’attivazione dell’FGF-R inibisce l’1-alfa-idrossilasi e stimola la sintesi della 24,25-idrossilasi, riducendo quindi la produzione di calcitriolo (15).
Insulina, GLP-1, glucagone
Insulina, GLP1 e glucagone svolgono un ruolo di grande importanza a livello renale, in quanto partecipano al controllo interattivo del riassorbimento di sodio e acqua.
L’insulina favorisce il riassorbimento di sodio, sia a livello tubulare prossimale, dove aumenta l’attività dello scambiatore sodio-idrogeno NHE3, sia a livello del tubulo distale, dove attraverso una chinasi glucocorticoide-dipendente SGK1 riduce l’ubiquinazione del canale epiteliale del sodio ENaC, fosforilando una proteina, NEDD 4-2. Pertanto, ENaC è up-regolata dall’aldosterone nella sua attività di riassorbimento del sodio, mentre l’insulina ne riduce la degradazione (16).
Il GLP-1, invece, ha attività contro-regolatorie rispetto all’insulina, in quanto inibisce lo scambiatore NHE3 nel tubulo prossimale, con una spiccata attività natriuretica (17,18) e riduce inoltre il riassorbimento di sodio e acqua indotto dall’ADH, bloccandone il trasporto di acqua con un meccanismo PGE2-mediato (19,20); inoltre, inibisce il signaling post-recettoriale mesangiale dell’angiotensina II (21).
Il glucagone, a sua volta, ha attività anti-diuretiche, con un’attività ADH-mimetica, aumentando il filtrato glomerulare e inducendo, in condizioni di inappropriata secrezione ormonale, espansione mesangiale con aumento di matrice (22,23).
BIBLIOGRAFIA
- Brenner and Rector’s. The Kidney, 11th Elsevier 2016.
- Renal Physiology, 5th Elsevier-Mosby Physiology Monograph Series 2013.
- Singh AK, Williams GH. Textbook of Nephro-Endocrinology. Academic Press-Elsevier 2009.
- Kobori H, et al. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007, 59: 251-87.
- Peach MJ. Renin-angiotensin system: biochemistry and mechanisms of action. Physiol Rev 1977, 57: 313–70.
- Jelkmann W. Erythropoietin. Front Horm Res 2016, 47: 115-27.
- Terragno NA, et al. Renal prostaglandins. Adv Prostaglandin Thromboxane Res 1976, 2: 561-71.
- McGiff JC. Interactions of prostaglandins with the kallikrein-kinin and renin-angiotensin systems. Clin Sci (Lond) 1980, 59 suppl 6: 105-16.
- Hartupee DA. Effect of leukotrienes of renal water excretion. Prostaglandins Leukot Essent Fatty Acids 1993, 48: 297-303.
- Kohan DE, et al. Physiology of endothelin and the kidney. Compr Physiol 2011, 1: 883-919.
- Wong PC, Guo J, Zhang A. The renal and cardiovascular effects of natriuretic peptides. Adv Physiol Educ 2017, 41: 179-85.
- Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Renal Physiol 2005, 289: F8-28.
- DeLuca HF. Regulation of the vitamin D endocrine system located in the kidney. Contrib Nephrol 1978, 13: 81-95.
- Boone M, Deen PM. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch 2008, 456: 1005–24.
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/Klotho endocrine pathways. Physiol Rev 2012, 92: 131-55.
- Brands MW, Manhiani MM. Sodium-retaining effect of insulin in diabetes. Am J Physiol Regul Integr Comp Physiol 2012, 303: R1101–9.
- Skov J. Effects of GLP-1 in the kidney. Rev Endocr Metab Disord 2014, 15: 197-207.
- Crajoinas RO, et al. Mechanisms mediating the diuretic and natriuretic actions of the incretin hormone glucagon-like peptide-1. Am J Physiol Renal Physiol 2011, 301: F355-63.
- Kutina AV, Golosova DV, Marina AS, et al. Role of vasopressin in the regulation of renal sodium excretion: interaction with glucagon-like peptide-1. J Neuroendocrinol 2016, DOI: 10.1111/jne.12367.
- Kutina AV, et al. Physiological mechanisms for the increase in renal solute-free water clearance by a glucagon-like peptide-1 mimetic. Clin Exp Pharmacol Physiol 2013, 40: 510-7.
- Mima A, et al. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCβ activation in diabetes. Diabetes 2012, 61: 2967-79.
- Bankir L, et al. Protein- and diabetes-induced glomerular hyperfiltration: role of glucagon, vasopressin, and urea. Am J Physiol Renal Physiol 2015, 309: F2–23.
- Li XC, Zhuo JL. Targeting glucagon receptor signalling in treating metabolic syndrome and renal injury in type 2 diabetes: theory versus promise. Clin Science 2007, 113, 183-93.
- Turner NN. Oxford Textbook of Clinical Nephrology, 4th Oxford University Press 2015.
Modificazioni ormonali in corso di insufficienza renale cronica e dialisi
Silvio Settembrini1 & Alessandra Fusco2
1Servizio di Endocrinologia, Diabetologia e Malattie Metaboliche, DS 26, Unità di Nefro-Diabetologia, UOC di Nefrologia e Dialisi, Ospedale dei Pellegrini, Napoli
2Ambulatorio di Endocrinologia e Diabetologia, Centro Acismom, Napoli
(aggiornato al 25 luglio 2017)
I reni sono organi complessi nei quali avvengono la sintesi e la degradazione di differenti ormoni. Le anomalie endocrine in pazienti con malattia cronica renale e in pazienti che ricevono un trattamento renale sostitutivo (TSR) possono dipendere da un numero di cause diverse. Inoltre, varie condizioni concomitanti, come infiammazione, acidosi metabolica e malnutrizione, possono partecipare alla patogenesi di molte alterazioni del sistema endocrino.
Il rene rappresenta insieme al fegato una delle principali sedi del metabolismo degli ormoni a struttura peptidica, rimuovendo dal 16 al 40% degli ormoni circolanti, intervenendo inoltre per il 20-80% sul loro metabolismo. I diversi ormoni peptidici (o i loro metaboliti attivi o inattivi) vengono filtrati a livello renale in base al flusso plasmatico renale e permeabilità della membrana glomerulare, dipendente da peso molecolare, forma, carica elettrica della molecola (1). Nelle cellule del tubulo prossimale, piccole molecole (come angiotensina o bradichinina) vengono idrolizzate sul versante luminale, mentre complessi più voluminosi (come insulina, PTH o glucagone) vengono assorbiti e quindi degradati per opera di enzimi lisosomiali (2). Per quanto riguarda gli ormoni steroidei, il rene svolge un ruolo fondamentale soprattutto sul metabolismo della vitamina D. Poiché il rene svolge un ruolo centrale nella regolazione dell’omeostasi ormonale, appare chiaro come la sua riduzione funzionale possa determinare una serie di modificazioni dell’equilibrio omeostatico del sistema endocrino (3). La conoscenza dei disordini endocrini in corso di insufficienza renale cronica (IRC) è di primaria importanza, perché la patologia endocrina altera la qualità di vita del paziente uremico e può comprometterne il pieno recupero psico-fisico.
È opportuno sottolineare che le misurazioni della concentrazione plasmatica di differenti ormoni hanno valore limitato nei pazienti con IRC e TSR: le concentrazioni ormonali possono essere inadeguate in un contesto di segnali di stimolo o di soppressione, il test può evidenziare isoforme inattive dell’ormone, la risposta degli organi bersaglio può essere amplificata o ridotta. Si ritiene, dunque, necessario interpretare le concentrazioni plasmatiche ormonali alla luce del contesto clinico (es. PTH in relazione alla calcemia o concentrazione insulinica in relazione alla glicemia). A seguire saranno riportate e discusse separatamente le alterazioni endocrine di più frequente riscontro nell’uremia cronica (tabella).
| Meccanismi delle alterazioni ormonali in corso di IRC | ||
| Catabolismo ormonale | Ridotta clearance renale | Insulina, PTH, leptina, adiponectina, gastrina |
| Alterata produzione | Riduzione della produzione degli ormoni nelle ghiandole endocrine | Testosterone, estrogeni |
| Riduzione della produzione ormonale da parte del rene | Calcitriolo, eritropoietina | |
| Iperproduzione ormonale reattiva per ristabilire l’omeostasi | PTH, FGF-23, eritropoietina | |
| Iperproduzione ormonale inappropriata dovuta ad anomalie del feed-back | ACTH, LH, PRL | |
| Pattern secretorio anomalo (pulsatilità, ritmo circadiano) | GH, LH | |
| Alterazione dell’attività ormonale | Aumento delle isoforme con minore attività biologica (per modifiche post-trascrizionali) | LH |
| Aumento della concentrazione delle proteine sieriche leganti gli ormoni e minore disponibilità della quota libera dell’ormone | IGF-I | |
| Riduzione della concentrazione delle proteine sieriche leganti gli ormoni e maggiore disponibilità della quota libera dell’ormone | Leptina | |
| Variazione della quantità e/o struttura del recettore ormonale | Recettore per la vitamina D | |
| Alterazione del signaling cellulare post-recettoriale | Insulina, GH | |
| Anomalie nell’attivazione dei pro-ormoni | Pro-insulina, tiroxina | |
Le anomalie endocrine più marcate nei pazienti con malattia cronica renale sono:
- carenza di: calcitriolo, testosterone, IGF-I ed eritropoietina (EPO);
- accumulo di: PRL, GH e insulina.
Le principali conseguenze cliniche delle sopracitate anomalie endocrine sono anemia, infertilità e malattie ossee.
| Classificazione funzionale dell'insufficienza renale | ||
| Stadio | Filtrazione glomerulare | VFG (mL/min) |
| G1 | Normale o aumentata | ≥ 90 |
| G2 | Lievemente diminuita | 60-89 |
| G3a | Da lievemente a moderatamente diminuita | 45-59 |
| G3b | Da moderatamente a severamente diminuita | 30-44 |
| G4 | Severamente diminuita | 15-29 |
| G5 | Insufficienza renale terminale | < 15 |
ERITROPOIETINA
Nell’adulto i reni sono responsabili della sintesi di circa l’85-90% dell’EPO in circolo, mentre il restante 10-15% deriva dal fegato (4,5).
Il principale stimolo per la sintesi di EPO nelle cellule peri-tubulari della corteccia renale è l’ipossia renale, che può essere causata da anemia o ipossiemia (6). L’ipossia stimola la sintesi del fattore inducibile dell’ipossia (HIF), responsabile dell’attivazione di diversi geni, tra cui quello per EPO (7). Un altro fattore stimolante la produzione renale di EPO è l’angiotensina II. Di contro, proteine tipicamente legate all’infiammazione (es. interleuchina–1 e TNF–α) inibiscono la secrezione di EPO, che può essere ridotta da fattori di infezione come CMV (8).
La concentrazione plasmatica di EPO in pazienti anemici con IRC e TSR è molto simile a quella di soggetti non anemici, ma è bassa nel contesto della concentrazione di emoglobina. Comunque, nei pazienti con IRC si verifica comunemente resistenza all’EPO (9). Nella pratica clinica nei pazienti con IRC e TSR non si esegue la valutazione della concentrazione plasmatica di EPO: le decisioni riguardanti il trattamento con agenti stimolanti l’eritropoiesi si basano sullo stato clinico e su misurazioni ripetute della concentrazione di emoglobina.
Nella popolazione generale, un basso livello di vitamina D è stato legato a un aumento di incidenza di ipertensione, malattia cardio-vascolare, sindrome metabolica, obesità, insulino-resistenza e albuminuria (10).
La prevalenza di deficit di 25-idrossivitamina D3 aumenta con la progressione dell’IRC e raggiunge l’80% in pazienti con IRC allo stadio 5. Inoltre, in pazienti con sindrome nefrotica, il 25(OH)D3 è escreto in eccesso con le urine. Nei pazienti trattati con dialisi peritoneale la vitamina D è eliminata con il liquido di dialisi peritoneale.
Nei pazienti con IRC si impiegano supplementi di ergocalciferolo, raccomandati se la concentrazione di 25(OH)D3 è < 25 ng/mL.
25(OH)D3 è ulteriormente idrossilato nel rene, con produzione del suo metabolita attivo -1,25(OH)D3 (calcitriolo). Con la riduzione del filtrato, si verifica il declino dell’attività dell’1α-idrossilasi. Inoltre, diminuisce la quantità di 25(OH)D3 inviata al rene (con meccanismo recettoriale che coinvolge la megalina) (11). In più, l’aumentata concentrazione di FGF-23 può inibire direttamente l’idrossilazione in posizione 1 e promuovere la sintesi di 24,25(OH)2D3, che sembra inattivo a livello metabolico. Così, in pazienti con IRC allo stadio 5 la concentrazione di calcitriolo è ridotta. Inoltre, in questi pazienti è stata descritta una riduzione nella densità dei recettori per il calcitriolo (VDR), che porta a una resistenza dell’organo bersaglio (11).
Il deficit di calcitriolo dei pazienti con IRC gioca un ruolo importante nello sviluppo di iperparatiroidismo secondario, ridotto assorbimento di calcio nell’intestino, ridotta mineralizzazione ossea e resistenza dello scheletro all’azione calcemica del PTH, così come miopatia e alterata crescita staturale.
Recenti studi hanno dimostrato che il deficit di calcitriolo aumenta la proteinuria nei pazienti con IRC e il trattamento con paracalcitolo sembra migliorare questa patologia. Inoltre, il trattamento con cinacalcet può dare benefici in questo gruppo di pazienti, poiché diminuisce la concentrazione di FGF-23, con minore degradazione di vitamina D3 e riduzione del rischio cardio-vascolare (12).
Alcuni studi suggeriscono che il deficit di calcitriolo sia responsabile di aumentata mortalità cardio-vascolare e generale nei pazienti con IRC. I risultati di piccoli studi di intervento suggeriscono che il trattamento con calcitriolo o altri agonisti di VDR può ridurre la mortalità in questi pazienti.
Alcuni studi hanno documentato una riduzione dei livelli di FGF-23 in pazienti uremici trattati con cinacalcet, con un miglioramento del metabolismo renale della vitamina D (13). Tuttavia questi dati richiedono ulteriore conferma su casistiche più ampie.
ALTERAZIONI DI INSULINA E GLUCAGONE
Diverse alterazioni contribuiscono all’alterato metabolismo dei carboidrati.
Secrezione e clearance insulinica
L’insulina è una proteina di 6 KDa, che viene liberamente filtrata nei glomeruli, escreta dai vasi peri-tubulari, riassorbita e metabolizzata in aminoacidi nel tubulo prossimale. Nei soggetti apparentemente sani la clearance dell’insulina è circa 200 mL/min, che supera la velocità di filtrazione glomerulare, a indicare che l’insulina viene anche captata a livello peri-tubulare. Si stima che in condizioni normali il rene elimini circa il 25% (6-8 unità/die) della quantità di insulina prodotta giornalmente dal pancreas (14).
Nell’IRC la secrezione insulinica è alterata, a causa soprattutto degli alti livelli di PTH e bassi livelli di calcitriolo. La clearance metabolica dell’insulina si riduce quando il GFR scende sotto 40 mL/min. Quando il filtrato renale è < 15-20 mL/min, l’emivita dell’insulina aumenta. Clinicamente queste modificazioni si traducono in una riduzione del fabbisogno insulinico nel paziente diabetico con IRC.
Il secondo fattore responsabile dell’iperinsulinemia nell’uremia è la resistenza periferica (vedi oltre).
Le alterazioni del metabolismo dell’insulina interferiscono sia sul metabolismo lipidico sia su quello delle proteine. Infatti, mentre da una parte l’iperinsulinemia può stimolare la sintesi delle VLDL ricche in trigliceridi, dall’altra anche la resistenza insulinica, riducendo l’attività della lipoprotein-lipasi, è in grado di determinare un incremento della trigliceridemia. L’insulina, inoltre, agisce sulla sintesi proteica, stimolando l’incorporazione degli aminoacidi a catena ramificata a livello tissutale.
Nell’uremia è stato dimostrato che il rilascio di insulina viene soppresso dall’acidosi metabolica. Infatti, il trattamento dialitico, correggendo l’equilibrio acido-base, migliora la risposta insulinica al glucosio (14). Il calcitriolo, deficitario in corso di IRC, quando somministrato in dialisi aumenta il rilascio di insulina e migliora la tolleranza glucidica, indipendentemente dalle modifiche delle concentrazioni di calcemia e PTH.
Insulino-resistenza
La resistenza periferica all’insulina, principalmente nei muscoli scheletrici che ne richiedono maggiori concentrazioni per aumentare la captazione del glucosio, compare nell’IRC precoce e tende ad aggravarsi con i vari stadi dell’IRC, fino a essere presente nella maggior parte dei pazienti con IRC avanzato (15). Dopo l’inizio di TSR, l’insulino-resistenza periferica si riduce marcatamente, ma solo dopo molte settimane di trattamento. Presumibilmente, una tossina uremica dializzabile indefinita è coinvolta nella patogenesi di un’attività impropria dell’insulina negli organi bersaglio. Tale molecola sarebbe specifica per l’IRC, non essendo stata riscontrata in pazienti non uremici con insulino-resistenza. Il difetto non riguarda solo il recettore insulinico, ma presumibilmente avviene anche a livello post-recettoriale, poiché è documentata la riduzione dell’attività della fosfatidil-inositol 3-chinasi (16).
Altri possibili meccanismi responsabili dell’insulino-resistenza sono l’aumento della gluconeogenesi epatica, la ridotta captazione epatica e muscolo-scheletrica del glucosio, la ridotta ossidazione del glucosio in anidride carbonica e acqua e soprattutto la diminuita sintesi di glicogeno (17).
Alcuni dei fattori coinvolti nell’insulino-resistenza sono modificabili. Per esempio, l’insulino-resistenza può essere migliorata da una dieta a basso contenuto proteico in pazienti pre-dializzati e dal trattamento con eritropoietina o calcitriolo in pazienti emodializzati.
Nei pazienti con IRC risultano frequentemente aumentate anche le concentrazioni di antagonisti dell’insulina, quali glucagone e GH, che parteciperebbero quindi alla patogenesi dell’insulino-resistenza insieme con acidosi metabolica, infiammazione cronica e aumentata attività del sistema renina-angiotensina (18).
Conseguenze cliniche dell’iperglicemia e dell’insulino-resistenza
Nei pazienti con IRC sia l’iperglicemia che l’insulino-resistenza aumentano la progressione dell’IRC e il rischio cardio-vascolare e contribuiscono allo sviluppo di ipertensione, dovuta a maggiore sensibilità al sale causata da aumentato riassorbimento tubulare di sodio. L’insulino-resistenza può anche partecipare allo sviluppo di malnutrizione, spesso riscontrata in questo gruppo di pazienti, e stimola il catabolismo del muscolo attraverso l’attivazione di una via proteolitica attraverso il sistema ubiquitina-proteasi (19).
Glucagone
Il glucagone è una proteina di basso peso molecolare, che viene filtrata a livello glomerulare, riassorbita e quindi degradata dalle cellule del tubulo prossimale.
In corso di IRC, la secrezione di glucagone risulta normale così come la soppressione in seguito alla somministrazione di glucosio e la stimolazione per infusione di arginina (18). I livelli plasmatici aumentano a causa della ridotta degradazione di glucagone e pro-glucagone.
L’accumulo di glucagone, causato dalla ridotta clearance renale, stimola la gluconeogenesi epatica e la conversione di alanina in glicogeno. Quest’ultimo meccanismo è ipotizzato essere alla base dell’ipercatabolismo proteico nell’uremia.
ALTERAZIONI DELLE ADIPOCHINE
Il tessuto adiposo può essere considerato come un organo endocrino, poiché produce una grande varietà di sostanze biologicamente attive, le adipochine. Nei pazienti con IRC si riscontra un’aumentata concentrazione di differenti adipochine.
La concentrazione plasmatica di leptina risulta aumentata, poiché la sua clearance è ridotta nel rene malato. La leptina stimola proliferazione e differenziazione di cellule ematopoietiche ed è verosimile che gli effetti della leptina e dell’eritropoietina siano sinergici. Inoltre, l’iperleptinemia stimola l’attività del sistema nervoso simpatico ed è perciò coinvolta in progressione dell’IRC, patogenesi dell’ipertensione e malattia cardio-vascolare (20,21).
Anche la concentrazione plasmatica di adiponectina è alta nei pazienti con IRC, a causa di alterazioni nella biodegradazione ed eliminazione dal rene malato (22). Le conseguenze cliniche dell’aumentata concentrazione di adiponectina non sono chiare: sembra che l’attività anti-aterosclerotica sia ridotta a causa della resistenza a livello recettoriale (23).
La concentrazione plasmatica di resistina risulta aumentata nei pazienti con IRC, soprattutto a causa di ridotta clearance renale (24). La resistina, alle concentrazioni tipiche dei pazienti con IRC, inibisce l’attività dei neutrofili. Potrebbe essere questa la causa di un’aumentata prevalenza di infezioni nei pazienti con IRC. Comunque, la resistina sembra avere un ruolo nella patogenesi della malattia cardio-vascolare in questi pazienti, perché pazienti emodializzati con bassa concentrazione sierica di resistina hanno bassa sopravvivenza senza ospedalizzazioni.
La concentrazione plasmatica di visfatina aumenta gradualmente con la riduzione del filtrato e si correla positivamente con la disfunzione endoteliale. Questa adipochina stimola l’adesione monocitaria alle cellule endoteliali. La visfatina può anche essere coinvolta nella patogenesi della malnutrizione nell’IRC. Inoltre, alte concentrazioni plasmatiche di visfatina sono predittive di mortalità cardio-vascolare (25).
ALTERAZIONI DELL’ASSE GH-IGF
Le alterazioni riscontrate nell’IRC possono avere importanti conseguenze cliniche: nei ragazzi la più importante è il ritardo di crescita con riduzione della statura definitiva, associata con aumento di morbilità e mortalità (26).
GH
Nel soggetto normale il GH viene filtrato dal glomerulo, fisiologicamente assorbito e metabolizzato a livello del tubulo prossimale.
Il GH aumenta nei pazienti uremici e la sua regolazione ipotalamo-ipofisaria è compromessa. La ridotta clearance renale rappresenta il fattore determinante l’aumento delle sue concentrazioni nell’IRC, sebbene si possa associare anche una maggiore secrezione (26). Contrariamente a quanto avviene nel soggetto sano, nell’IRC di grado avanzato, la somministrazione di glucosio induce un incremento paradosso delle concentrazioni di GH; sono da segnalare, inoltre, una risposta esagerata all’infusione di arginina e un’accentuata risposta secretoria dopo stimolazione con GHRH esogeno. I meccanismi dell’anomala regolazione del GH nell’IRC non sono stati completamente chiariti. Il primum movens sembra essere la compromissione del controllo ipotalamico: nonostante che nei modelli sperimentali di ratti uremici la risposta secretoria al GHRH sia sovrapponibile a quella dei controlli, la concentrazione di mRNA per GHRH risulta ridotta.
Tutte queste alterazioni hanno portato a ipotizzare una resistenza o insensibilità al GH in pazienti con IRC terminale (26). Questa può essere causata da ridotta densità dei recettori per GH negli organi bersaglio, poiché nei ragazzi e negli adulti con IRC la concentrazione della proteina legante il GH (frazione del recettore per GH che può essere usata per valutare la densità dei recettori) è ridotta in modo proporzionale alla riduzione della filtrazione glomerulare. La resistenza al GH può essere anche a livello post-recettoriale, con alterazione della trasduzione del segnale intra-cellulare JAK2-STAT. La trasduzione del segnale GH può essere soppressa anche dall’aumentata espressione dei geni SOCS2 e SOCS3. Altri fattori che contribuiscono alla resistenza al GH nell’IRC sono l’acidosi metabolica e l’infiammazione (27).
Durante la dialisi i livelli plasmatici di GH tornano per un breve periodo a valori normali, ma nel complesso rimangono elevati. Tuttavia, la crescita risulta egualmente compromessa nei bambini con uremia terminale. Per permettere una crescita ottimale, sono necessari apporto nutrizionale adeguato, prevenzione dell’osteodistrofia renale, correzione dell’acidosi metabolica, somministrazione di rhGH e dialisi efficiente se il paziente è in TSR (26).
IGF
La concentrazione di IGF-I tende a essere normale nella malattia renale pre-terminale (IRC 1-4). Le misurazioni con metodo RIA hanno dimostrato, in soggetti uremici, livelli di IGF-I normali o aumentati fino a 4 volte rispetto al soggetto sano, per la ridotta clearance renale (27). L’IGF-I libera sierica diminuisce con gli stadi dell’IRC: nello stadio 5 tale diminuzione è dovuta principalmente a elevate concentrazioni di proteine leganti (IGF-BP-1,2,4, e 6) (28). È stata anche riportata ridotta biodisponibilità di IGF-I negli organi bersaglio, come risultato dell’aumentata proteolisi di IGF-BP3, e l’attività circolante, misurata con i bioassay, è ridotta. L’apparente discrepanza può essere spiegata dall’accumulo, in corso di IRC, di peptidi circolanti ad attività inibitoria, non ancora caratterizzati. In ultimo, esiste una resistenza all’IGF-I, indipendente dalla presenza di fattori inibitori circolanti, causata da un difetto nella trasduzione del segnale intra-cellulare: nell’IRC è ridotta l’auto-fosforilazione del recettore tirosin-chinasico dell’IGF-I e la suscettibilità dell’IGF-IR all’IRS-1 (29).
La concentrazione sierica di IGF-II è nel range normale nella malattia renale pre-terminale e aumentata nell’IRC stadio 5.
Terapia con rhGH
La resistenza/insensibilità degli organi bersaglio al GH costituisce il razionale per il trattamento con rhGH nei ragazzi con ritardo della crescita causato da IRC. Tale trattamento è risultato sicuro ed efficace: non causa intolleranza glucidica né induce modificazioni del filtrato glomerulare; ottiene rapida crescita staturale (il 65% dei ragazzi trattati con rhGH può raggiungere un’altezza definitiva normale), accompagnata da aumento del peso corporeo e della circonferenza muscolare del braccio (26). La miglior risposta al trattamento con rhGH è stata riscontrata in pazienti in stadio pre-dialisi, probabilmente per una migliore sensibilità al GH. Il trattamento con rhGH è stato dimostrato essere efficace anche nel trattamento del ritardo di crescita staturale dopo trapianto di rene (causato soprattutto dalla somministrazione di glucorticoidi).
Nonostante i chiari benefici, la somministrazione di rhGH negli adulti con IRC non è esente da rischi, per cui servono studi che ne dimostrino efficacia e sicurezza prima di raccomandarlo in questo gruppo di pazienti.
Nell’individuo sano il rene contribuisce all’escrezione del cortisolo e dei suoi metaboliti idrosolubili. Con la riduzione del filtrato renale, aumenta l’emivita del cortisolo. In corso di IRC sono stati riportati livelli di cortisolemia nei limiti della norma o francamente elevati, tuttavia questi risultati contrastanti sono stati, in parte, attribuiti a problemi di metodologia di laboratorio (30). Infatti, i metodi immunologici sfruttano anti-sieri commerciali per il cortisolo che possono cross-reagire con i metaboliti steroidei, che nel paziente dializzato sono aumentati rispetto al soggetto normale. Pertanto, il riscontro di livelli elevati di cortisolemia, in corso di IRC, dovrebbe essere confermato con l’impiego di metodiche diagnostiche più specifiche.
Nel soggetto uremico, il legame del cortisolo alla CBG risulta normale, mentre il legame con l’albumina è ridotto. Il ritmo circadiano del cortisolo è conservato e la risposta all’infusione di ACTH adeguata, con un incremento delle concentrazioni di cortisolemia circolante. Inoltre, la somministrazione di CRH induce un incremento dei livelli di cortisolo ma non di ACTH. Non sono stati ancora spiegati i meccanismi, ma la terapia con eritropoietina ricombinante sembra migliorare la risposta dell’ACTH al CRH. Il soggetto uremico presenta una risposta attenuata al test di soppressione del cortisolo con desametasone, in particolar modo quando il farmaco viene introdotto per os. Sembrerebbero necessarie dosi superiori di desametasone nel test overnight per os (8 mg vs 1 mg del soggetto sano) per indurre una riduzione dei livelli a circa 2 µg/dL. Questo potrebbe dipendere da alterazione del feed-back ipofisi-surrene, da assorbimento ridotto oppure da elevato metabolismo del desametasone stesso. Dati contrastanti vengono riportati in uno studio che ha dimostrato concentrazioni plasmatiche adeguate di desametasone dopo somministrazione ad alte dosi sia per bocca sia per via ev, senza successiva soppressione della produzione di cortisolo.
Negli anni ’90 il cortisolo è stato proposto come marcatore di comorbilità. In pazienti con IRC pre-terminale, maggiori livelli plasmatici di cortisolemia correlavano con una maggiore richiesta di ospedalizzazione (31). L’ipercortisolemia con valori > 22 µg/dL si associava frequentemente a segni di malnutrizione, come ipoalbuminemia, elevato catabolismo proteico e riduzione della plica cutanea tricipitale ed è stato ipotizzato che l’ormone stesso potesse essere responsabile dello stato catabolico di questi pazienti.
I livelli plasmatici di cortisolo e ACTH aumentano durante il trattamento dialitico, rimangono elevati fino al termine della procedura e gradualmente tornano a livelli normali nelle 24 ore successive.
SISTEMA RENINA-ANGIOTENSINA-ALDOSTERONE
Il RAAS è un sistema sia endocrino circolatorio, che tissutale, ma prevalentemente tissutale e locale, che svolge in tutti gli organi, in situ, azioni sia paracrine che autocrine di regolazione delle funzioni d’organo (32,33). Oltre ai tradizionali meccanismi di controllo di volume e pressorio e di modulazione dell’handling del sodio e degli elettroliti, il RAAS tissutale svolge importanti funzioni sul differenziamento cellulare, tramite il signalling di angiotensina II, aldosterone e renina, che, sui loro recettori, regolano fattori trascrizionali quali il NF-κB, la via di segnale MAPK-ERK, e l’espressione genica di svariati fattori di crescita, quali TGF-β, citochine, chemochine, e di molecole di adesione e matrici extra-cellulari. In equilibrio, tramite l’enzima di conversione ACE-2 (34) il sistema RAAS può attivare la produzione di angiotensina 1-7 e 1-9, che svolgono attività contro-regolatorie sui loro rispettivi recettori MAS e AT2. Il RAAS tissutale svolge anche funzioni metaboliche sul controllo glicidico e lipidico (35-43).
Come in tutti gli organi, anche a livello renale sono presenti tutti gli elementi del RAAS (44,45), con una generazione intra-renale continua di angiotensina II, che viene compartimentalizzata tra tubulo prossimale e fluidi interstiziale. In corso di malattia renale cronica si attiva una disregolazione del RAAS (46), con un circolo vizioso di sovra-generazione di angiotensina II, che diventa un componente essenziale nel mantenimento di una elevata attività intra-renale di tutto il sistema RAAS, con conseguente alterazione del normale differenziamento delle strutture glomerulari e tubulari (mesangio, podociti, tubuli prossimali e distali, endoteli, membrane basali), loro apoptosi, chemiotassi macrofagica e linfocitaria e fibrogenesi evolutiva fino allo stato terminale di sovvertimento cito-architettonico renale, il rene grinzo (45).
L’angiotensina II stimola l’espressione intra-renale di angiotensinogeno (47), che a sua volta provoca aumentata sintesi di angiotensina II; inoltre, la generazione di specie reattive dell’ossigeno, dovuta all’aumentata quota di angiotensina II, aumenta l’espressione dell’angiotensinogeno a livello del tubulo prossimale. Questo loop è determinante nella progressione del danno renale, specie in presenza di diabete mellito, dove la glucotossicità acuta e cronica è un ulteriore elemento di amplificazione intra-renale del RAAS (40,48).
Angiotensina II e aldosterone inducono inoltre, quando sovra-regolati, un aumentato handling del sodio, agendo sui rispettivi trasportatori: NHE3 a livello del tubulo prossimale per angiotensina II ed ENaC a livello tubulare distale per aldosterone. Il sodio, a sua volta, genera un loop di amplificazione del RAAS intra-renale. L’aldosterone, inoltre, induce anch’esso, come l’angiotensina II, rimodellamento della rete vascolare renale, espansione mesangiale e senescenza delle cellule tubulari, con accumulo in situ di neutrofili e aumentata sintesi di collageno e fibrosi interstiziale (44,45).
Il rene è una delle sedi in cui avviene il metabolismo delle catecolamine: la catecol-O-metiltransferasi, enzima che catalizza la reazione di inattivazione delle catecolamine, è distribuita lungo le cellule del segmento S3 del tubulo prossimale e della porzione ascendente dell’ansa di Henle.
Nell’IRC di grado lieve i livelli plasmatici di catecolamine sono maggiori che nei sani, ma sono nella norma i livelli urinari di catecolamine e acido vanil-mandelico. All’aumento delle concentrazioni di noradrenalina dell’individuo uremico contribuiscono la ridotta captazione neuronale di catecolamine e la diminuita attività delle monoamino-ossidasi, che fisiologicamente ne determinano la degradazione a livello delle terminazioni sinaptiche. I livelli di catecolamine si normalizzano dopo il trapianto renale (51).
I pazienti in dialisi sembrano avere un’iperattività del sistema nervoso simpatico, valutabile a livello del nervo peroneale. Tuttavia, i livelli di adrenalina non sembrano aumentati. Poiché questa anomalia non è rilevabile nei soggetti anefrici, si è ipotizzato che il segnale afferente per l’iperattività simpatica sia inviato dal rene. I meccanismi non sono chiari, ma sembra verosimile che anche le tossine uremiche possano stimolare direttamente le vie nervose afferenti renali. Nell’IRC di grado più avanzato è stato dimostrato un aumento della produzione di noradrenalina (52). Sembra che adrenalina, noradrenalina e altri ormoni, come aldosterone, ADH, ANP, angiotensina II ed endotelina, rispondano in maniera appropriata alle modificazioni vascolari che si verificano durante la dialisi. Gli aumentati livelli di noradrenalina potrebbero essere responsabili dell’ipertensione arteriosa che si osserva in alcuni pazienti in dialisi nonostante il raggiungimento del peso secco mediante ultra-filtrazione adeguata. In questi soggetti è stata inoltre ipotizzata l’esistenza di una risposta pressoria esagerata alla noradrenalina.
Durante il trattamento emodialitico la clearance delle catecolamine può contribuire all’insorgenza degli episodi ipotensivi. Le perdite di noradrenalina sembrerebbero essere maggiori per diffusione rispetto alla convezione. Pertanto, quando viene praticata l’ultra-filtrazione come tecnica dialitica, le perdite di noradrenalina potrebbero essere inferiori e garantire una maggiore stabilità emodinamica. Durante l’ipotensione potrebbe anche esservi una mancata dismissione di adrenalina e noradrenalina in grado di contrastare l’evento, oppure coesistere una ridotta responsività d’organo alla noradrenalina (53).
Livelli elevati di adrenalina possono contribuire all’insulino-resistenza e a un incrementato catabolismo proteico nei pazienti con IRC. Resta da segnalare la difficoltà di porre diagnosi di feocromocitoma nel soggetto uremico a causa delle concentrazioni aumentate di catecolamine. Alcuni autori hanno stabilito che nei pazienti in TSR possa essere suggestivo di feocromocitoma un incremento di noradrenalina oltre 3.3 volte il valore normale.
ASSE IPOTALAMO-IPOFISI-TESTICOLO
L’IRC è la causa principale di molte alterazioni di questo asse, la maggior parte delle quali riguarda direttamente la funzione gonadica. Nei ragazzi uremici è stata documentata pubertà ritardata, che persiste anche con l’inizio della dialisi.
FSH
L’ormone normalmente stimola la crescita testicolare, la spermatogenesi e la sintesi di SHBG nelle cellule di Sertoli. Nei pazienti con IRC le concentrazioni sieriche di FSH possono essere sia elevate sia ai limiti superiori del range di normalità, ma la spermatogenesi è compromessa, per resistenza del testicolo all’FSH o per una disfunzione testicolare primaria (54,55).
LH
Nella maggior parte dei pazienti con IRC, le concentrazioni plasmatiche basali di LH sono elevate a causa del ridotto catabolismo e della perdita dell’inibizione del GnRH da parte dei ridotti livelli di testosterone (55).
PRL
Nella maggior parte dei maschi emodializzati le concentrazioni sieriche sono elevate ed è alterato il ritmo circadiano della secrezione (episodica durante il giorno con rari segnali secretori sonno-indotti). All’iperprolattinemia contribuiscono probabilmente sia il declino della clearance renale della PRL che l’aumentata velocità di produzione causata da un’inadeguata inibizione dopaminergica. L’accumulo di PRL causa l’inibizione della secrezione pulsatile di GnRH, così come la riduzione nella sintesi del testosterone, che si traduce in peggioramento delle funzioni sessuali e infertilità (54-56).
In alcuni pazienti con IRC trattati con bromocriptina sono stati descritti miglioramenti dell’iperprolattinemia e della funzione sessuale. Nei pazienti con IRC è stata anche descritta l’associazione tra iperprolattinemia e outcome cardio-vascolari negativi, probabilmente dipendente da disfunzione endoteliale. In un piccolo studio clinico si è riscontrata una riduzione della pressione e dell’ipertrofia ventricolare destra dopo somministrazione di bromocriptina in pazienti con IRC.
Androgeni e funzione sessuale
Nella maggior parte dei maschi emodializzati le concentrazioni sieriche di testosterone totale e libero sono basse, con ritmo circadiano conservato, sebbene risultino nella norma sia la capacità di legame che la concentrazione di SHBG. È stata riportata anche una ridotta concentrazione di androstenedione e deidro-epiandrosterone solfato. Non è ancora noto se la riduzione del testosterone sia causata da ridotta sintesi, da aumentato catabolismo o da una combinazione di entrambe. Inoltre, è ridotta e ritardata la risposta alla stimolazione con hCG (57-59). Anche la malnutrizione ha un ruolo: le concentrazioni di testosterone aumentano nei pazienti IRC in trattamento dietetico a basso contenuto proteico, con supplementi di amminoacidi essenziali e cheto-analoghi. Anche l’iperparatiroidismo secondario potrebbe contribuire alla riduzione dei livelli di testosterone e alle disfunzioni sessuali.
I pazienti uremici riferiscono comunemente diminuzione della libido e della potenza sessuale sia prima che dopo l’inizio della dialisi. Meno di metà degli uomini in dialisi ha una normale attività sessuale (58). Fattori neurologici, vascolari o ormonali possono essere responsabili della disfunzione sessuale. Nei pazienti dializzati la velocità di conduzione degli impulsi nervosi può essere ridotta, i riflessi bulbo-cavernosi appaiono assenti ed è presente una riduzione delle erezioni notturne. La funzione sessuale potrebbe migliorare in alcuni pazienti con l’inizio della dialisi, probabilmente per un miglioramento dello stato di salute generale, e non in particolare del sistema endocrino. In alcuni studi il miglioramento della funzione sessuale è stato messo in relazione con l’utilizzo dell’eritropoietina che sembra avere effetti positivi sull’asse ipofisi-surreni e ipofisi-gonadi.
Nei pazienti emodializzati non è rara la ginecomastia, fenomeno simile a quello osservato in pazienti malnutriti come nelle patologie croniche, dovuto alla diminuzione del rapporto testosterone/estrogeni (con ridotta sintesi di testosterone e aumento della sintesi degli estrogeni), ad aumento della sintesi di SHBG e a riduzione del testosterone libero (59).
Il deficit androgenico porta a incremento del tessuto adiposo e decremento della massa magra (soprattutto muscolare), sviluppo di patologie ossee con maggiore incidenza di fratture, anemia, riduzione della libido, danno della funzione sessuale e depressione. Infine, è stata recentemente descritta l’associazione di basse concentrazioni di testosterone con outcome cardio-vascolari peggiori (60).
La terapia con testosterone non è esente da rischi, è quindi necessaria una più ampia evidenza clinica di benefici prima che possa essere raccomandata nei pazienti con IRC e ipogonadismo (61,62).
Spermatogenesi
Nei maschi sottoposti a dialisi è spesso presente perdita della libido, della potenza sessuale e della spermatogenesi che, molto spesso, portano a infertilità (54,55). Lo spermiogramma evidenzia una riduzione del volume dell’eiaculato con oligo-azoospermia e bassa percentuale di spermatozoi mobili. A livello istologico testicolare, i tubuli seminiferi possono essere danneggiati, si può riscontrare riduzione del numero degli spermatociti maturi fino alla completa aplasia, e anche le cellule del Sertoli possono apparire atrofiche e può comparire fibrosi interstiziale con calcificazioni (58). Nei pazienti uremici anziani o dializzati da lungo tempo si osserva un progressivo peggioramento della spermatogenesi per incremento dei livelli di FSH. La causa del peggioramento sembrerebbe la presenza di “tossine uremiche” non rimosse dalla dialisi, ma si ipotizza anche un ruolo di alcuni materiali impiegati nella preparazione del circuito extra-corporeo di dialisi (59).
Nelle donne con IRC le irregolarità mestruali sono principalmente rappresentate dall’oligo-amenorrea in soggetti con filtrato glomerulare < 10-15 mL/min. Con filtrato glomerulare ulteriormente ridotto compare amenorrea, che solitamente si instaura già durante la fase di IRC pre-terminale.
Con la dialisi sono poche le segnalazioni di ripresa della funzione ovarica con ovulazione e cicli mestruali regolari, mentre nella maggior parte dei casi i cicli rimangono anovulatori.
LH
Nelle donne sane in età fertile la secrezione di LH avviene in maniera pulsatile e il feed-back dell’estradiolo riduce l’ampiezza dei polsi di LH. Nelle donne con IRC l’interruzione del rilascio ipotalamico ciclico di GnRH porta a perdita dell’appropriato rilascio pulsatile ipofisario di LH, il feed-back dell’estradiolo è ridotto e non riesce a diminuire i livelli di LH, che risultano aumentati nella maggior parte delle pazienti (54-56).
Queste alterazioni portano alla riduzione dell’ovulazione, causa diretta di infertilità in queste donne.
FSH
Contrariamente a quelle di LH, nella maggior parte delle donne in pre-menopausa con IRC, le concentrazioni sieriche di FSH sono normali (54-56) Questo fenomeno sembra in contrasto con l’assunto del fallimento ovarico primario nell’IRC e suggerisce una disregolazione dell’asse.
PRL
In donne con TSR le concentrazioni sono spesso aumentate, per alterato controllo ipotalamico della secrezione ipofisaria, con conseguente amenorrea (63).
Ormoni gonadici
A causa della disregolazione ipotalamica, le concentrazioni sieriche di estradiolo possono essere normali o basse, soprattutto se è presente iperprolattinemia, e nella seconda metà del ciclo le concentrazioni sieriche di progesterone sono diminuite a causa della luteinizzazione del follicolo difettoso (56). Le biopsie dell’endometrio dimostrano deplezione estrogenica anche a questo livello.
Una delle più importanti conseguenze della bassa concentrazione plasmatica di estrogeni è la malattia ossea. Le donne con amenorrea non solo hanno concentrazioni di estrogeni diminuite, ma anche una minor densità ossea a paragone con le pazienti dializzate con mestruazioni regolari.
Piccoli studi suggeriscono che il trattamento con estradiolo transdermico o con un SERM come il raloxifene può incrementare la densità ossea in donne post-menopausa in emodialisi. Specialmente alla luce di possibili eventi avversi cardio-vascolari, deve essere precisato che non sono disponibili studi di sicurezza a lungo termine relativi alla terapia ormonale sostitutiva o con SERM nelle donne con IRC (64,65).
Sessualità e gravidanza
Si hanno riduzione della libido e minor capacità di raggiungere l’orgasmo.
Pur essendo molto frequente la sterilità nelle pazienti uremiche, esistono casi di concepimento di bambini sani (54-56). La gravidanza portata a termine risulta rara, per l’elevata frequenza di morte fetale associata a polidramnios. L’aumentata segnalazione di nascite di bambini da parte di madri sottoposte a trattamento dialitico può essere messa in relazione all’uso della dialisi peritoneale o dell’emodialisi aggressiva giornaliera con membrane ad alto flusso e alla somministrazione di eritropoietina. Non esiste evidenza di superiorità della dialisi peritoneale rispetto all’emodialisi, ma si ritiene che si ottengano risultati migliori sottoponendo le pazienti a trattamenti più frequenti e duraturi.
ALTERAZIONI TIROIDEE
In circa il 50% dei pazienti uremici sono riscontrabili disfunzioni tiroidee. L’uremia incide sul metabolismo periferico degli ormoni tiroidei.
Ormoni tiroidei
Negli studi sulla funzione tiroidea dei pazienti uremici, lo iodio legato alle proteine e la tireoglobulina sono risultati nella norma, mentre lo iodio inorganico plasmatico è generalmente aumentato. Le più importanti alterazioni degli ormoni tiroidei in corso di IRC sono la riduzione dei livelli di T3 totale (low T3 syndrome, per la ridotta attività della deiodinasi) e libera, con concentrazioni normali di rT3 e di FT4 (66). La riduzione delle concentrazioni di T3 dipende da tre fattori:
- ridotta conversione del T4 in T3 a livello periferico;
- acidosi metabolica;
- diminuito legame alle proteine trasportatrici, influenzato dagli elevati livelli di urea, creatinina, indoli e fenoli, come pure da ridotta clearance delle citochine infiammatorie TNF-alfa e IL-6.
In questo gruppo di pazienti sono comuni la sindrome da bassa T3 e l’ipotiroidismo subclinico. I pazienti IRC con concentrazioni sieriche più basse di T3 appaiono clinicamente eutiroidei, probabilmente poiché è aumentata l’espressione dell’RNA messaggero per i recettori nucleari α e β. Nonostante la riduzione nella clearance renale di rT3, le concentrazioni di questa molecola sono normali nell’IRC, in contrasto con il disturbo cronico non tiroideo, probabilmente per aumentata captazione cellulare di rT3 dallo spazio vascolare ed extra-vascolare (66).
Le ridotte concentrazioni degli ormoni tiroidei nei pazienti con IRC potrebbero non essere necessariamente indicative di disfunzione tiroidea, ma essere il riflesso della malattia cronica o della malnutrizione. Un basso livello di T3 nei pazienti con IRC è collegato con disfunzione endoteliale, aterosclerosi e alterazioni cardiache. Inoltre, un basso livello sierico di FT3 è stato collegato con aumentata mortalità cardio-vascolare nei pazienti in emodialisi (67).
L’influenza del trattamento dialitico sui test di funzionalità tiroidea è variabile. Le variazioni delle concentrazioni ormonali sono maggiori in corso di emodialisi rispetto alla dialisi peritoneale, dove i valori sono prossimi a quelli del soggetto sano. Durante il trattamento emodialitico, l’impiego di eparina come anti-coagulante, in grado di interferire sul legame di T4 alla tireoglobulina, in modo diretto competendo per i siti di legame delle proteine trasportatrici o indirettamente attraverso gli acidi grassi liberi, può spiegare l’aumento transitorio dei livelli di T4 (47). Le tossine uremiche (urea, creatinina, indoli, fenoli) inducono una disfunzione del recettore del TSH e del recettore nucleare di T3, alterando la risposta del TSH al TRH e la fisiologica risposta tissutale al T3 (68,69).
TSH
Le concentrazioni sieriche sono solitamente normali nonostante la tendenza a basse concentrazioni di T3 e T4, a suggerire una disregolazione dell’asse ipotalamo-ipofisi-tiroide. Nei pazienti con IRC la risposta del TSH al TRH è ridotta e lenta, a causa della ridotta clearance e del prolungato tempo di dimezzamento del TSH. È alterato anche il normale ritmo circadiano del TSH, con picco ridotto nel tardo pomeriggio o di prima mattina e diminuito rilascio notturno (70).
Ipotiroidismo primario
La diagnosi di ipotiroidismo in IRC può presentare difficoltà, perché i classici segni e sintomi dell’ipotiroidismo sono comuni al quadro clinico dell’IRC. Clinicamente nella maggior parte dei pazienti non si denotano alterazioni evidenti. Talora, si possono osservare segni suggestivi di ipotiroidismo, come intolleranza al freddo, cute secca, astenia, stipsi, sonnolenza, ma tale sintomatologia può ritrovarsi nel soggetto uremico indipendentemente dal quadro distiroideo. La presenza di raucedine è, invece, un sintomo sentinella, in quanto è particolarmente caratteristico di ipotiroidismo anche in dialisi. Tra le conseguenze cliniche dell’ipotiroidismo nell’IRC possono essere menzionati anemia, dolore muscolare e depressione. È noto che le ridotte concentrazioni di T3 sono predittori indipendenti di mortalità cardiovascolare e per tutte le cause nei pazienti con IRC.
La diagnosi di ipotiroidismo si basa sul riscontro di bassi livelli di FT4 con valori di TSH plasmatico nettamente > 20 mU/L (66).
L’IRC è associata a ridotta escrezione di ioduro, che causa l’aumento della concentrazione di ioduro inorganico e l’aumentato contenuto di ioduro nella tiroide, con iperplasia ghiandolare. L’eccesso intra-tiroideo di ioduro può contribuire all’ipotiroidismo attraverso il prolungato effetto Wolff-Chaikoff. In pazienti in emodialisi si verifica un transitorio aumento della concentrazione sierica di T4, causato dall’uso di eparina e anti-coagulanti. Poiché l’eparina compete con T4 per il sito di legame sulla TBG, con aumento delle concentrazioni sieriche di T4 per almeno 24 ore, i campioni ematici per la determinazione degli ormoni tiroidei dovrebbero essere raccolti prima della somministrazione di eparina che avviene prima della seduta dialitica (70).
Gozzo
Nei pazienti con IRC è riportata aumentata incidenza di gozzo ed esoftalmo. Peraltro, alcuni studi non confermano l’aumentata incidenza di gozzo, ma mostrano un aumento di volume della tiroide sia all’ecografia che all’istologia nel 50% dei pazienti (66).
BIBLIOGRAFIA
- Brenner and Rector’s. The Kidney, 11th Elsevier 2016.
- Turner NN. Oxford Textbook of Clinical Nephrology, 4th Oxford University Press 2015.
- Singh AK, Williams GH. Textbook of Nephro-Endocrinology. Academic Press-Elsevier 2009.
- Jelkmann W. Erythropoietin. Front Horm Res 2016, 47: 115-27.
- Souma T, Suzuki N, Yamamoto M. Renal erythropoietin-producing cells in health and disease. Front Physiol 2015, 6: 167.
- Heir P, et al. Oxygen-dependent regulation of erythropoietin receptor turnover and signaling. J Biol Chem 2016, 291: 7357-72.
- Souma T, Nezu M, Nakano D, et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J Am Soc Nephrol 2016, 27: 428-38.
- Eckardt KU, Kurtz A. Regulation of erythropoietin production. Eur J Clin Invest 2005, 35 suppl 3: 13-9.
- Fisher JW. Erythropoietin: physiology and pharmacology update. Exp Biol Med (Maywood) 2003, 228: 1-14.
- Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Renal Physiol 2005, 289: F8-28.
- DeLuca HF. Regulation of the vitamin D endocrine system located in the kidney. Contrib Nephrol 1978, 13: 81-95.
- Dusso AS, Tokumoto M. Defective renal maintenance of the vitamin D endocrine system impairs vitamin D renoprotection: a downward spiral in kidney disease. Kidney Int 2011, 79: 715-29.
- Martin A, David V, Quarles LD. Regulation and function of the FGF23/Klotho endocrine pathways. Physiol Rev 2012, 92: 131-55.
- Bellasi A, Di Micco L, Santoro D, et al, on behalf of UBI study investigators. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrology 2016, 17: 158.
- Leyking S, Fliser D. Insulin resistance in CKD. Clin J Am Soc Nephrol 2014, 9: 638–40.
- Spoto B, Pisano A, Zoccali C. Insulin resistance in chronic kidney disease: a systematic review. Am J Physiol Renal Physiol 2016, 311: F1087-108.
- Garibotto G, Sofia A, Russo R, et al. Insulin sensitivity of muscle protein metabolism is altered in patients with chronic kidney disease and metabolic acidosis. Kidney Int 2015, 88: 1419–26.
- Thomas SS, Zhang L, Mitch WE. Molecular mechanisms of insulin resistance in chronic kidney disease. Kidney Int 2015, 88: 1233–9.
- Liao MT, Sung CC, Hung KC, et al. Insulin resistance in patients with chronic kidney disease. J Biomed Biotechnol 2012, 2012: 691369.
- Schroth M, Kratzsch J, Groschl M, et al. Increased soluble leptin receptor in children with nephrotic syndrome. J Clin Endocrinol Metab 2003, 88: 5497–501.
- Cobo G, Cordeiro AC, Amparo FC, et al. Visceral adipose tissue and leptin hyperproduction are associated with hypogonadism in men with chronic kidney disease. J Ren Nutr 2017, 27: 243-8.
- Markaki A, Psylinakis E, Spyridaki A. Adiponectin and end-stage renal disease. Hormones (Athens) 2016, 15: 345-54.
- Yahya RS, Atwa MA, El-Sayed IH, et al. Adipocytokines in patients with chronic kidney disease stage 5. Clin Lab 2016, 62: 21-30.
- Marouga A, Dalamaga M, Kastania AN, et al. Circulating resistin is a significant predictor of mortality independently from cardiovascular comorbidities in elderly, non-diabetic subjects with chronic kidney disease. Biomarkers 2016, 21: 73-9.
- Kim HY, Bae EH, Ma SK, et al. Association of serum adiponectin level with albuminuria in chronic kidney disease patients. Clin Exp Nephrol 2016, 20: 443-9.
- Salas P, Pinto V, Zambrano MJ, Mericq V. Growth retardation in children with kidney disease. Int J Endocrinol 2013, 2013: 970946.
- Bach LA, Hale LJ. Insulin-like growth factors and kidney disease. Am J Kidney Dis 2015, 65: 327-36.
- Mak RH, Cheung WW, Roberts CT Jr. The growth hormone-insulin-like growth factor-I axis in chronic kidney disease. Growth Horm IGF Res 2008, 18: 17-25.
- Oh Y. The insulin-like growth factor system in chronic kidney disease: pathophysiology and therapeutic opportunities. Kidney Res Clin Pract 2012, 31: 26-3.
- N’Gankam V, Uehlinger D, Dick B, et al. Increased cortisol metabolites and reduced activity of 11beta-hydroxysteroid dehydrogenase in patients on hemodialysis. Kidney Int 2002, 61: 1859–66.
- Arregger AL, Cardoso EM, Zucchini A, et al. Adrenocortical function in hypotensive patients with end stage renal disease. Steroids 2014, 84: 57-63.
- Balakumar P, Anand-Srivastava MB, Jagadeesh G. Renin-angiotensin-aldosterone: an inclusive, an invigorative, an interactive and an interminable system. Pharmacol Res 2017, DOI: 10.1016/j.phrs.2017.07.003.
- Harris RC, Cheng HF. The intrarenal renin-angiotensin system: a paracrine system for the local control of renal function separate from the systemic axis. Exp Nephrol 1996, 4 suppl 1: 2-7.
- Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1–7): an evolving story in cardiovascular regulation. Hypertension 2006, 47: 515-21.
- Singh BM, Mehta JL. Interactions between the renin-angiotensin system and dyslipidemia: relevance in the therapy of hypertension and coronary heart disease. Arch Intern Med 2003, 163: 1296–304.
- Catar RA, Muller G, Heidler J, et al. Low-density lipoproteins induce the renin-angiotensin system and their receptors in human endothelial cells. Horm Metab Res 2007, 39: 801–5.
- Ma KL, et al. Interaction of RAS activation and lipid disorders accelerates the progression of glomerulosclerosis. Int J Med Sci 2013, 10: 1615–24.
- Carlsson PO. The renin-angiotensin system in the endocrine pancreas. JOP 2001, 2: 26-32.
- Cooper ME. The role of the renin-angiotensin-aldosterone system in diabetes and its vascular complications. Am J Hypertens 2004, 17: 16S-20.
- Hsueh WA, Wyne K. Renin-angiotensin-aldosterone system in diabetes and hypertension. J Clin Hypertens (Greenwich) 2011, 13: 224–37.
- Bindom SM, Lazartigues E. The sweeter side of ACE2: physiological evidence for a role in diabetes. Mol Cell Endocrinol 2009, 302: 193-202.
- Jandeleit-Dahm KA, Tikellis C, Reid CM, et al. Why blockade of the renin-angiotensin system reduces the incidence of new-onset diabetes. J Hypertens 2005, 23: 463-73.
- Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: a target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Amer J Physiol Heart Circul Physiol 2012, 302: H1219-30.
- Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am J Nephrol 2010, 31: 541–50.
- Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 2007, 59: 251–87.
- Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm 2007, 13 (8 suppl B): 9-20.
- Kobori H, Ozawa Y, Suzaki Y, et al. Young Scholars Award Lecture: Intratubular angiotensinogen in hypertension and kidney diseases. Am J Hypertens 2006, 19: 541–50.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993, 329: 1456-62.
- Zhuo JL, Li XC. New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides 2011, 32: 1551-65.
- Li XC, et al.The vasoprotective axes of the renin-angiotensin system: physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney disease. Pharmacol Res 2017, DOI: 10.1016/j.phrs.2017.06.005.
- Grassi G, Quarti-Trevano F, Seravalle G, et al. Early sympathetic activation in the initial clinical stages of chronic renal failure. Hypertension 2011, 57: 846-51.
- Iijima H, Okada Y, Tsunoda M, et al. Quantification of norepinephrine and its metabolites in the plasma of renal failure models. Nephron Physiol 2010, 116: 9-16.
- Hoch H, Stegbauer J, Potthoff SA, et al. Regulation of renal sympathetic neurotransmission by renal α(2A)-adrenoceptors is impaired in chronic renal failure. Br J Pharmacol 2011, 163: 438-46.
- Palmer BF. Sexual dysfunction in men and women with chronic kidney disease and end-stage kidney disease. Adv Ren Replace Ther 2003, 10: 48–60.
- Holley JL. The hypothalamic-pituitary axis in men and women with chronic kidney disease. Adv Chronic Kidney Dis 2004, 4: 337–41.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993, 22: 145-61.
- Edey MM. Male sexual dysfunction and chronic kidney disease. Front Med (Lausanne) 2017, 4: 32.
- Palmer BF, Clegg DJ. Gonadal dysfunction in chronic kidney disease. Rev Endocr Metab Disord 2017, 18: 117-30.
- Carrero JJ, Qureshi AR, Nakashima A, et al. Prevalence and clinical implications of testosterone deficiency in men with end-stage renal disease. Nephrol Dial Transplant 2011, 26: 184–90.
- Navaneethan SD, Vecchio M, Johnson DW, et al. Prevalence and correlates of self-reported sexual dysfunction in CKD: a meta-analysis of observational studies. Am J Kidney Dis 2010, 56: 670–85.
- Vecchio M, Navaneethan SD, Johnson DW, et al. Treatment options for sexual dysfunction in patients with male sexual dysfunction and chronic kidney disease: a systematic review of randomized controlled trials. Clin J Am Soc Nephrol 2010, 5: 985–95.
- Wang GC, Zheng JH, Xu LG, et al. Measurements of serum pituitary-gonadal hormones and investigation of sexual and reproductive functions in kidney transplant recipients. Int J Nephrol 2010, 2010: 612126.
- Gomez F, de la Cueva R, Wauters JP, Lemarchand-Beraud T. Endocrine abnormalities in patients undergoing long-term hemodialysis. The role of prolactin. Am J Med 1980, 68: 522–30.
- Ramesh S, Mann MC, Holroyd-Leduc JM, et al. The effect of hormone therapy on all-cause and cardiovascular mortality in women with chronic kidney disease: protocol for a systematic review and meta-analysis. Syst Rev 2015, 4: 44.
- Holley JL, Schmidt RJ. Changes in fertility and hormone replacement therapy in kidney disease. Adv Chronic Kidney Dis 2013, 20: 240–5.
- Iglesias P, Bajo MA, Selgas R, Díez JJ. Thyroid dysfunction and kidney disease: an update. Rev Endocr Metab Disord 2017, 18: 131-44.
- Rhee CM, Brent GA, Kovesdy CP, et al. Thyroid functional disease: an under-recognized cardiovascular risk factor in kidney disease patients. Nephrol Dial Transplant 2015, 30: 724-37.
- Zoccali C, Mallamaci F. Thyroid function and clinical outcomes in kidney failure. Clin J Am Soc Nephrol 2012, 7: 12–4.
- Santos GM, et al. Thyroid hormone receptor binding to DNA and T3-dependent transcriptional activation are inhibited by uremic toxin. Nuclear Receptor 2005, 3: 1.
- Mohamedali M, Reddy Maddika S, et al. Thyroid disorders and chronic kidney disease. Int J Nephrol 2014, 2014: 520281.
L’utilizzo dei farmaci endocrini secondo il grado di insufficienza renale
Silvio Settembrini1 & Alessandra Fusco2
1Servizio di Endocrinologia, Diabetologia e Malattie Metaboliche, DS 26, Unità di Nefro-Diabetologia, UOC di Nefrologia e Dialisi, Ospedale dei Pellegrini, Napoli
2Ambulatorio di Endocrinologia e Diabetologia, Centro Acismom, Napoli
(aggiornato al 25 luglio 2017) questo capitolo è in attesa di aggiornamento
| Classificazione funzionale dell'insufficienza renale | ||
| Stadio | Filtrazione glomerulare | VFG (mL/min) |
| G1 | Normale o aumentata | ≥ 90 |
| G2 | Lievemente diminuita | 60-89 |
| G3a | Da lievemente a moderatamente diminuita | 45-59 |
| G3b | Da moderatamente a severamente diminuita | 30-44 |
| G4 | Severamente diminuita | 15-29 |
| G5 | Insufficienza renale terminale | < 15 |
Acarbose: non è raccomandato in pazienti con grave insufficienza renale (clearance della creatinina < 25 mL/min).
Alendronato: non è raccomandato quando la clearance della creatinina è < 35 mL/min, in quanto non vi sono esperienze in proposito. Non è necessario aggiustare la dose nei pazienti con creatinina clearance > 35 mL/min.
Alirocumab: non sono necessari aggiustamenti della dose per i pazienti con compromissione renale lieve o moderata. Sono disponibili dati limitati in pazienti con compromissione renale severa, in cui l’esposizione ad alirocumab era circa 2 volte superiore rispetto ai soggetti con funzione renale normale.
Alogliptin: nei pazienti con eGFR < 60 mL/min somministrare al dosaggio di 12.5 mg/die, nei pazienti con eGFR < 30 mL/min usare al dosaggio di 6.25 mg/die.
Alprostadil: la farmacocinetica non è stata studiata nei pazienti con insufficienza renale, potrebbe essere necessario ridurre il dosaggio in pazienti con compromissione della funzionalità renale.
Anastrozolo: non si raccomandano aggiustamenti del dosaggio nelle pazienti con insufficienza renale lieve o moderata. Nelle pazienti con insufficienza renale grave, la somministrazione di anastrozolo deve essere eseguita con cautela.
Atorvastatina: non è richiesto nessun aggiustamento della dose.
Bezafibrato: è controindicato nei pazienti con ridotta funzionalità renale (clearance della creatinina < 60 mL/min).
Bromocriptina: nessuna segnalazione in RCP.
Buserelin: nessuna segnalazione in RCP.
Cabergolina: nessun segnalazione in RCP.
Canagliflozin: nei pazienti con eGFR < 60 mL/min è necessario ridurre la dose a 100 mg/die. Non è raccomandato nei pazienti con eGFR < 45 mL/min.
Cinacalcet: il profilo farmacocinetico nei pazienti con compromissione renale lieve, moderata e grave e in quelli in emodialisi o in dialisi peritoneale è simile a quello rilevato in volontari sani. Tuttavia, cinacalcet non è indicato nei pazienti con compromissione renale cronica non in dialisi, in cui si è evidenziato un aumento del rischio di ipocalcemia (< 8.4 mg/dL = 2.1 mmol/L) rispetto a pazienti con compromissione renale cronica in dialisi, che può essere dovuto a livelli sierici di calcio al basale inferiori e/o alla presenza di residua funzionalità renale.
Ciproterone: non è stato studiato in modo specifico in pazienti con funzionalità renale compromessa.
Clodronato: è eliminato principalmente per via renale, pertanto deve essere utilizzato con cautela in pazienti con insufficienza renale. Per il clodronato orale non sono disponibili dati di farmacocinetica in pazienti con insufficienza renale, con clearance della creatinina < 10 mL/min. L’uso in questi casi deve essere evitato, eccetto per trattamenti a breve termine in presenza di insufficienza renale puramente funzionale, causata da ipercalcemia. Si raccomanda di ridurre il dosaggio dell’infusione di clodronato secondo i valori di creatinina clearance come segue:
- 50-80 mL/min: riduzione del dosaggio del 25%;
- 12-50mL/min: riduzione del dosaggio del 25-50%;
- < 12 mL/min: riduzione del dosaggio del 50%. Si raccomanda che 300 mg di clodronato siano infusi prima dell’emodialisi, che la dose sia ridotta del 50% nei giorni liberi da dialisi e di limitare lo schema di trattamento a 5 giorni. É da notare che la dialisi peritoneale rimuove scarsamente il clodronato dalla circolazione.
Clomifene: nessuna segnalazione in RCP.
Colestiramina: nessuna segnalazione in RCP
Cortisone acetato: monitoraggio della risposta clinica nei pazienti con insufficienza renale.
Dapagliflozin: non è raccomandato nei pazienti con eGFR < 60 mL/min.
Dapoxetina: si raccomanda cautela nei pazienti affetti da disfunzione renale di grado lieve o moderato. L’uso di dapoxetina non è raccomandato in pazienti con disfunzione renale grave.
Denosumab: non è richiesto alcun aggiustamento della dose nei pazienti con compromissione renale. I pazienti con grave compromissione renale (clearance della creatinina < 30 mL/min) o sottoposti a dialisi hanno rischio maggiore di sviluppare ipocalcemia. Questo rischio, con il conseguente innalzamento dei livelli di PTH, aumenta con l’aumentare del grado di compromissione renale. In questi pazienti sono particolarmente importanti adeguato apporto di calcio e vitamina D e regolare monitoraggio della calcemia.
Desmopressina: controindicata in caso di insufficienza renale moderata o grave (clearance della creatinina < 50 mL/min).
Drospirenone: i livelli sierici nelle donne con lieve compromissione della funzionalità renale (clearance della creatinina 50-80 mL/min) sono paragonabili a quelli delle donne con funzionalità renale normale. I livelli sierici sono in media del 37% più alti nelle donne con compromissione moderata della funzionalità renale (30-50 mL/min). Il trattamento con drospirenone risulta anche ben tollerato dalle donne con funzionalità renale lievemente e moderatamente compromessa.
Dulaglutide: non è necessario alcun aggiustamento della dose in pazienti affetti da compromissione renale da lieve a moderata. L’esperienza in pazienti con compromissione renale grave (eGFR < 30 mL/min) o con malattia renale allo stadio terminale è molto limitata, perciò l’uso non è raccomandato in questi pazienti.
Empagliflozin: non è raccomandato nei pazienti con eGFR < 45 mL/min.
Estradiolo valerato/dienogest: la farmacocinetica non è stata studiata in pazienti con insufficienza renale.
Etinilestradiolo/Ciproterone acetato: non è stato studiato in modo specifico in pazienti con funzionalità renale compromessa.
Etinilestradiolo/Clormadinone: somministrare con cautela in caso di insufficienza renale.
Etinilestradiolo/Desogestrel: nessuna segnalazione in RCP.
Etinilestradiolo/Dienogest: nessuna segnalazione in RCP.
Etinilestradiolo/Gestodene: nessuna segnalazione in RCP.
Etinilestradiolo/Levonorgestrel: controindicato in caso di insufficienza renale grave.
Everolimus: non è necessario alcun aggiustamento della dose. In pazienti trattati con everolimus sono stati osservati casi di insufficienza renale (compresa insufficienza renale acuta), alcuni con esito fatale. La funzionalità renale deve essere monitorata in particolare quando i pazienti hanno fattori di rischio aggiuntivi che possono compromettere ulteriormente la funzionalità renale.
Evolocumab: non sono necessari aggiustamenti della dose in pazienti con compromissione renale da lieve a moderata. Non sono stati condotti studi su pazienti con compromissione renale grave (definita come eGFR < 30 mL/min), pertanto deve essere usato con cautela.
Exenatide: usare con cautela nei pazienti con eGFR < 50 mL/min, non è raccomandato nei pazienti con eGFR < 30 mL/min.
Ezetimibe: non è richiesto aggiustamento del dosaggio nel danno renale.
Fenofibrato: nei pazienti con danno renale è necessario ridurre la dose di fenofibrato, in base alla clearance della creatinina. Il trattamento deve essere interrotto in caso di aumento dei livelli di creatinina maggiori del 50% rispetto al limite superiore di normalità. Si raccomanda di controllare la creatinina nei primi 3 mesi successivi all’inizio del trattamento, e dopo periodicamente. Non è raccomandato in pazienti con grave malattia renale. In pazienti anziani con funzione renale compromessa deve essere presa in considerazione una riduzione di dose.
Finasteride: non sono necessari aggiustamenti del dosaggio in pazienti con insufficienza renale di vario grado (con clearance della creatinina al di sotto di 9 mL/min), dato che negli studi di farmacocinetica l’insufficienza renale non ha mostrato di influenzare l’eliminazione di finasteride. Finasteride non è stato studiato in pazienti in emodialisi.
Fludrocortisone: in caso di problemi renali è necessario aggiustare il dosaggio.
Flutamide: deve essere utilizzata con cautela in pazienti con disturbi della funzionalità renale.
Fluvastatina: nei pazienti con insufficienza renale da lieve a grave la farmacocinetica di fluvastatina rimane immodificata. Pertanto, in questi pazienti non sono necessari aggiustamenti della dose. Tuttavia, a causa di esperienza limitata con dosi > 40 mg/die, in caso di grave insufficienza renale (ClCr < 30 mL/min) queste dosi devono essere somministrate con cautela.
FSH ricombinante: la sicurezza nei pazienti con insufficienza renale non è stata testata.
Gemfibrozil: in pazienti con compromissione renale lieve-moderata (velocità di filtrazione glomerulare rispettivamente 50-80 e 30-50 mL/min) iniziare il trattamento alla dose di 900 mg/die e valutare la funzionalità renale prima di aumentare la dose. Non deve essere impiegato in pazienti con grave compromissione della funzionalità renale.
GH: non è stato osservato alcun accumulo in bambini affetti da IRC o da malattie renali allo stadio terminale.
Glibenclamide: non è raccomandato nei pazienti con IRC.
Gliclazide: non è necessario aggiustamento della dose nei pazienti con IRC da lieve a moderata, istituendo un attento monitoraggio; è controindicata nell’IRC grave.
Glimepiride: da usare con cautela in pazienti con eGFR < 60 mL/min; controindicata in caso di alterazione grave della funzionalità renale.
Gonadorelina: nessuna segnalazione in RCP.
Gonadotropina corionica ricombinante: la sicurezza nei pazienti con insufficienza renale non è stata testata.
Goserelin: non è necessario alcun adeguamento della posologia in pazienti con danno renale.
Ibandronato: in conseguenza della limitata esperienza clinica, non è raccomandato con clearance della creatinina < 30 mL/min, mentre non è necessario alcun aggiustamento di dose con insufficienza renale da lieve a moderata (≥ 30 mL/min).
Idrocortisone a rilascio modificato: non è necessario alcun aggiustamento della dose nei pazienti con insufficienza renale da lieve a moderata. Nei pazienti con insufficienza renale grave, si raccomanda il monitoraggio della risposta clinica e potrebbe essere necessario l’aggiustamento della dose.
IGF-I: non sono stati condotti studi su bambini con alterazione della funzionalità renale.
Insulina Aspart: non è necessario aggiustamento della dose.
Insulina Degludec: non è necessario aggiustamento della dose.
Insulina Detemir: non è necessario aggiustamento della dose.
Insulina Glargine: non è necessario aggiustamento della dose.
Insulina Glulisina: non è necessario aggiustamento della dose.
Insulina Lispro: non è necessario aggiustamento della dose.
Insulina NPH: non è necessario aggiustamento della dose.
Insulina umana regolare: non è necessario aggiustamento della dose.
Interferone: la funzione renale deve essere monitorata attentamente; controindicato nei pazienti con disfunzione renale grave.
Ketoconazolo: sebbene i dati siano limitati, la farmacocinetica non differisce tra pazienti con insufficienza renale e pazienti con funzione renale normale, e dunque non è raccomandato un aggiustamento del dosaggio.
Lanreotide: i soggetti con grave alterazione della funzionalità renale presentano una diminuzione di circa 2 volte della clearance sierica totale di lanreotide, con conseguente aumento dell’emivita e dell’AUC. Non è necessario tuttavia modificare la dose di inizio terapia nei pazienti con alterata funzionalità renale, poichè ci si aspetta che le concentrazioni sieriche di lanreotide in queste popolazioni siano ampiamente nel range di quelle tollerate con sicurezza nei soggetti sani.
Leuprorelina: non sono stati condotti studi clinici su pazienti con compromissione renale.
Levonorgestrel: controindicato in caso di insufficienza renale grave.
Levotiroxina sodica: nessuna segnalazione in RCP.
LH ricombinante: la sicurezza e la farmacocinetica non sono state testate nei pazienti con insufficienza renale.
Linagliptin: nessun aggiustamento della dose nei pazienti con IRC.
Liotironina sodica: nessuna segnalazione in RCP.
Liraglutide: non è richiesta correzione della dose per i pazienti con compromissione della funzionalità renale lieve o moderata (clearance della creatinina 60–90 mL/min e 30–59 mL/min, rispettivamente). Non vi è esperienza terapeutica in pazienti con grave compromissione della funzionalità renale (clearance della creatinina < 30 mL/min). Liraglutide attualmente non può essere raccomandato per l’uso in pazienti con grave compromissione della funzionalità renale, compresi i pazienti con malattia renale all’ultimo stadio.
Lovastatina: nei pazienti affetti da insufficienza renale grave (clearance di creatinina ≤ 30 mL/min) dosaggi > 20 mg/die devono essere attentamente valutati.
Medrossiprogesterone acetato: da usare con cautela nell’insufficienza renale.
Metformina: nei pazienti con eGFR < 60 mL/min somministrare con attenzione, facendo frequenti controlli della funzionalità renale (ogni 3-6 mesi). Nei pazienti con eGFR < 45 mL/min, non è raccomandato iniziare la terapia. Nei pazienti già in trattamento, continuare la terapia al dosaggio di 1000 mg/die o ridurre il dosaggio del 50%. Fare controlli frequenti della funzionalità renale (ogni 3 mesi). Nei pazienti con eGFR < 30 mL/min non è raccomandata.
Metimazolo: nessuna segnalazione in RCP.
Metirapone: nessuna segnalazione in RCP.
Mifepristone: in assenza di studi specifici, non è raccomandato nei pazienti con insufficienza renale.
Mitotane: non ci sono dati nei pazienti con insufficienza renale. Non è raccomandato l’uso in pazienti con insufficienza renale grave e andrebbe usato con cautela nel caso di insufficienza renale lieve o moderata. In questi pazienti è raccomandato il monitoraggio dei livelli plasmatici di mitotane.
Octreotide: l'alterata funzionalità renale non ha influenzato l'esposizione totale ad octreotide (AUC) somministrato come iniezione sottocutanea, pertanto non è necessario aggiustamento della dose nei pazienti con insufficienza renale.
Omega polienolici: sono disponibili solo informazioni limitate sull’uso in pazienti con compromissione renale.
Orlistat: gli effetti non sono stati studiati nell’insufficienza renale.
Pasireotide: anche se non stati effettuati studi clinici su pazienti con compromissione della funzione renale, la clearance renale ha un ruolo minore nell’eliminazione del pasireotide nell’uomo. Una moderata compromissione renale non dovrebbe avere un impatto significativo sui livelli di pasireotide in circolo, ma non si può escludere che l’esposizione sistemica aumenti nel caso di una grave compromissione renale.
Pegvisomant: non sono state stabilite la sicurezza e l’efficacia in pazienti con insufficienza renale.
Perclorato di potassio: nessuna segnalazione in RCP.
Pioglitazone: non è necessario alcun aggiustamento della dose nei pazienti con funzionalità renale compromessa (clearance della creatinina > 4 mL/min). Non sono disponibili informazioni su pazienti in dialisi, pertanto in tali pazienti non deve essere usato.
Pravastatina: nei pazienti con insufficienza renale moderata o grave, si raccomanda una dose iniziale di 10 mg/die. Il dosaggio deve essere aggiustato in base alla risposta dei parametri lipidici e sotto controllo medico.
Progesterone micronizzato: usare con cautela nell’insufficienza renale.
Propiltiouracile: non è richiesta alcuna modifica della dose per pazienti più anziani con funzionalità renale ridotta o obbligo di dialisi.
Raloxifene: meno del 6% della dose totale viene eliminata con le urine. In uno studio di farmacocinetica di popolazione, una riduzione del 47% della clearance della creatinina corretta per la massa magra ha determinato una riduzione della clearance del raloxifene e dei coniugati del 17% e 15%, rispettivamente. Raloxifene è controindicato nell’insufficienza renale grave.
Ranelato di stronzio: non è raccomandato nei pazienti con insufficienza renale grave (clearance della creatinina < 30 mL/min). Non è richiesto alcun adattamento posologico nei pazienti con insufficienza renale da lieve a moderata (30–70 mL/min).
Repaglinide: non è necessario aggiustamento del dosaggio, ma usare con cautela nei pazienti con eGFR < 30 mL/min.
Risedronato: non è necessario alcun aggiustamento della dose per i pazienti affetti da compromissione renale da lieve a moderata. È controindicato nei pazienti con grave danno renale (clearance della creatinina < 30 mL/min).
Rosuvastatina: non è necessario alcun aggiustamento della dose in pazienti con danno renale lieve o moderato. Nei pazienti con danno renale moderato (clearance della creatinina < 60 mL/min) la dose iniziale raccomandata è di 5 mg. La dose da 40 mg è controindicata nei pazienti con danno renale moderato. L’uso in pazienti con danno renale grave è controindicato a tutte le dosi.
Saxagliptin: non è necessario alcun aggiustamento della dose in pazienti con lieve insufficienza renale, mentre la dose deve essere ridotta a 2.5 mg una volta al giorno in pazienti con insufficienza renale moderata o grave. Poiché l’esperienza nei pazienti con insufficienza renale grave è molto limitata, saxagliptin deve essere usato con cautela in questa popolazione e non è raccomandato per i pazienti con malattia renale terminale (ESRD) che richiede emodialisi.
Sildenafil: nei volontari con compromissione renale da lieve a moderata (clearance della creatinina = 30-80 mL/min) la farmacocinetica non è alterata dopo l’assunzione di una dose singola orale di 50 mg, dunque non è necessario un aggiustamento del dosaggio nei pazienti con clearance della creatinina > 30 mL/min. Poiché la clearance è ridotta nei pazienti con compromissione renale grave (clearance della creatinina < 30 mL/min), deve essere presa in considerazione una dose di 25 mg. Sulla base dell’efficacia e della tollerabilità, la dose può essere aumentata gradualmente fino a 50 mg o 100 mg, secondo le necessità.
Simvastatina: non è necessario alcun aggiustamento della dosaggio in caso di insufficienza renale moderata. Nei pazienti con insufficienza renale grave (clearance della creatinina < 30 mL/min), dosaggi > 10 mg/die devono essere attentamente valutati e, se necessario, implementati con cautela.
Sitagliptin: viene escreta quasi immodificata nelle urine. Tuttavia, studi clinici hanno dimostrato che l’utilizzo di sitagliptin nell’IRC, a dosaggi ridotti, non ha causato ipoglicemie o eventi avversi. Pertanto il farmaco può essere utilizzato:
- nell’IRC lieve: a dose piena;
- nell’IRC moderata (VFG fino a 30 mL/min): al 50% in meno (50 mg/die);
- nell’IRC grave e in dialisi: al 75% in meno (25 mg/die).
Sunitinib: non è necessario un aggiustamento della dose inziale quando sunitinib è somministrato ai pazienti con compromissione renale (moderata-grave) o con malattia renale in fase terminale (ESRD) sottoposti a emodialisi. Gli aggiustamenti posologici successivi devono essere effettuati in base alla sicurezza e tollerabilità del singolo paziente. Le analisi di farmacocinetica di popolazione hanno indicato che la clearance apparente di sunitinib (CL/F) non è influenzata dalla clearance della creatinina nell’ambito del range preso in esame (42-347 mL/min). Le esposizioni sistemiche dopo una singola dose di sunitinib sono state simili in soggetti con compromissione renale grave (ClCr < 30 mL/min) e nei soggetti con funzionalità renale normale (ClCr > 80 mL/min). Sebbene sunitinib e il suo principale metabolita non siano eliminati attraverso emodialisi in soggetti con ESRD, le esposizioni sistemiche totali sono state inferiori del 47% per sunitinib e del 31% per il suo principale metabolita rispetto ai soggetti con funzionalità renale normale.
Tadalafil: la dose raccomandata è 10 mg nel paziente con compromissione della funzionalità renale. In uno studio clinico-farmacologico, dopo somministrazione di una dose di 10 mg, l’esposizione (AUC) al tadalafil è stata più alta nei soggetti con insufficienza renale lieve (clearance della creatinina da 51 a 80 mL/min) o moderata (clearance della creatinina da 31 a 50 mL/min) che nei soggetti sani. In un altro studio clinico-farmacologico, dopo un dosaggio di 10 mg, l’esposizione (AUC) al tadalafil in soggetti con insufficienza renale terminale sottoposti a dialisi è stata paragonabile all’esposizione nei soggetti sani. Non ci sono dati disponibili circa la somministrazione di tadalafil a dosi > 10 mg a pazienti con insufficienza renale. A causa dell’aumentata esposizione (AUC) al tadalafil, della limitata esperienza clinica e della mancanza di possibilità di influire sulla clearance con la dialisi, nei pazienti con grave insufficienza renale non è raccomandata la somministrazione di tadalafil una volta al giorno.
Tamoxifene: nessuna segnalazione in RCP.
Temozolomide: non sono disponibili dati sulla somministrazione in pazienti con danno renale. Sulla base delle proprietà farmacocinetiche, è improbabile che siano necessarie riduzioni della dose in pazienti con qualsiasi grado di danno renale. Tuttavia, occorre esercitare cautela quando viene somministrato in questi pazienti.
Teriparatide: non deve essere usato nei pazienti con insufficienza renale grave e deve essere usato con cautela in quelli con insufficienza renale di grado moderato.
Testosterone: non sono stati condotti studi formali in pazienti con funzionalità renale compromessa. Nei pazienti affetti da grave insufficienza renale, il trattamento può causare gravi complicanze, caratterizzate da edema, con o senza insufficienza cardiaca congestizia.
Tolvaptan: sulla base dei dati disponibili, non è necessario aggiustare la dose nei pazienti con compromissione renale da lieve a moderata, ma non è stato studiato in pazienti con grave compromissione renale ed è controindicato nei pazienti anurici.
Triptorelina: nessuna segnalazione in RCP.
Vardenafil: non è necessario modificare la dose nei pazienti con compromissione della funzionalità renale da lieve a moderata. In quelli con grave compromissione della funzionalità renale (clearance della creatinina < 30 mL/min) si deve prendere in considerazione una dose iniziale di 5 mg. In base alla tollerabilità e all’efficacia, la dose può essere aumentata a 10 mg e successivamente a 20 mg. La sicurezza non è stata studiata in pazienti con insufficienza renale terminale che richieda la dialisi, pertanto l’uso in questi pazienti è controindicato.
Vildagliptin: non è richiesto un aggiustamento della dose in pazienti con lieve compromissione della funzionalità renale (clearance della creatinina ≥ 50 mL/min). Nei pazienti con compromissione della funzionalità renale moderata o grave o con malattia renale allo stadio terminale, la dose raccomandata è 50 mg una volta al giorno.
Zoledronato: in pazienti con ipercalcemia neoplastica che manifestino anche una grave compromissione renale, il trattamento con acido zoledronico deve essere preso in considerazione solo dopo valutazione dei rischi e benefici del trattamento. Negli studi clinici sono stati esclusi i pazienti con valori di creatininemia > 400 µmol/L o > 4.5 mg/dL. Non è necessario alcun adattamento della dose in pazienti affetti da ipercalcemia neoplastica con valori di creatinina sierica < 400 µmol/L o < 4.5 mg/dL. Quando si inizia il trattamento nei pazienti con mieloma multiplo o con metastasi ossee da tumori solidi, devono essere determinate la creatininemia e la clearance della creatinina (ClCr), eseguendo un adeguamento posologico secondo il grado di compromissione renale:
- ClCr > 60 mL/min: 4.0 mg;
- ClCr 50-59 mL/min: 3.5 mg;
- ClCr 40-49 mL/min: 3.3 mg;
- ClCr 30-39 mL/min: 3 mg;
- ClCr < 30 mL/min: non raccomandato.
Dopo l'inizio della terapia, la creatininemia deve essere determinata prima di ciascuna somministrazione e il trattamento deve essere evitato in caso di peggioramento della funzionalità renale. Nei pazienti che mostrano segni di alterazione renale durante il trattamento, questo deve essere sospeso e ripristinato solo quando il valore della creatininemia ritorna entro il 10% del valore basale, con lo stesso dosaggio utilizzato prima dell'interruzione del trattamento.
| Classificazione funzionale dell'insufficienza renale | ||
| Stadio | Filtrazione glomerulare | VFG (mL/min) |
| G1 | Normale o aumentata | ≥ 90 |
| G2 | Lievemente diminuita | 60-89 |
| G3a | Da lievemente a moderatamente diminuita | 45-59 |
| G3b | Da moderatamente a severamente diminuita | 30-44 |
| G4 | Severamente diminuita | 15-29 |
| G5 | Insufficienza renale terminale | < 15 |
| Riassunto sull’utilizzo nei pazienti con diversi gradi di insufficienza renale (per specifiche e dettagli vedi testo) |
||||||
| Controindicato | Dati mancanti | Cautela/modifica dosi |
Utilizzabile |
|||
| Farmaco | G2 (60-89) |
G3a (45-59) |
G3b (30-44) |
G4 (15-29) |
G5 |
Dialisi |
| Acarbose | ||||||
| Alendronato | ||||||
| Alirocumab | ||||||
| Alogliptin | ||||||
| Alprostadil | ||||||
| Anastrazolo | ||||||
| Atorvastatina | ||||||
| Bezafibrato | ||||||
| Bromocriptina | ||||||
| Buserelin | ||||||
| Cabergolina | ||||||
| Canagliflozin | ||||||
| Cinacalcet | ||||||
| Ciproterone | ||||||
| Clodronato | ||||||
| Clomifene | ||||||
| Colestiramina | ||||||
| Cortisone acetato | ||||||
| Dapagliflozin | ||||||
| Dapoxetina | ||||||
| Denosumab | ||||||
| Desmopressina | ||||||
| Drospirenone | ||||||
| Dulaglutide | ||||||
| Empagliflozin | ||||||
| Estradiolo/Dienogest | ||||||
| EE/Ciproterone | ||||||
| EE/Clormadinone | ||||||
| EE/Desogestrel | ||||||
| EE/Dienogest | ||||||
| EE/Gestodene | ||||||
| EE/Levonorgestrel | ||||||
| Everolimus | ||||||
| Evolocumab | ||||||
| Exenatide | ||||||
| Ezetimibe | ||||||
| Fenofibrato | ||||||
| Finasteride | ||||||
| Fludrocortisone | ||||||
| Flutamide | ||||||
| Fluvastatina | ||||||
| FSH | ||||||
| Gemfibrozil | ||||||
| GH | ||||||
| Glibenclamide | ||||||
| Gliclazide | ||||||
| Glimepiride | ||||||
| Gonadorelina | ||||||
| Goserelin | ||||||
| hCG | ||||||
| Ibandronato | ||||||
| Idrocortisone a rilascio modificato | ||||||
| IGF-I | ||||||
| Insulina Aspart | ||||||
| Insulina Degludec | ||||||
| Insulina Detemir | ||||||
| Insulina Glargine | ||||||
| Insulina Glulisina | ||||||
| Insulina Lispro | ||||||
| Insulina NPH | ||||||
| Insulina umana regolare | ||||||
| Interferone | ||||||
| Ketoconazolo | ||||||
| Lanreotide | ||||||
| Leuprorelina | ||||||
| Levonorgestrel | ||||||
| Levotiroxina | ||||||
| LH | ||||||
| Linagliptin | ||||||
| Liotironina | ||||||
| Liraglutide | ||||||
| Lovastatina | ||||||
| Medrossiprogesterone acetato | ||||||
| Metformina | ||||||
| Metimazolo | ||||||
| Metirapone | ||||||
| Mifepristone | ||||||
| Mitotane | ||||||
| Octreotide | ||||||
| Omega polienolici | ||||||
| Orlistat | ||||||
| Pasireotide | ||||||
| Pegvisomant | ||||||
| Perclorato di potassio | ||||||
| Pioglitazone | ||||||
| Pravastatina | ||||||
| Progesterone | ||||||
| Propiltiouracile | ||||||
| Raloxifene | ||||||
| Ranelato di stronzio | ||||||
| Repaglinide | ||||||
| Risedronato | ||||||
| Rosuvastatina | ||||||
| Saxagliptin | ||||||
| Semaglutide | ||||||
| Sildenafil | ||||||
| Simvastatina | ||||||
| Sitagliptin | ||||||
| Sunitinib | ||||||
| Tadalafil | ||||||
| Tamoxifene | ||||||
| Temozolomide | ||||||
| Teriparatide | ||||||
| Testosterone | ||||||
| Tolvaptan | ||||||
| Triptorelina | ||||||
| Vardenafil | ||||||
| Vildagliptin | ||||||
| Zoledronato | ||||||
| Legenda | ||||||
| Controindicato | Dati mancanti | Cautela/modifica dosi | Utilizzabile | |||
Bibliografia
- AIFA. Banca dati farmaci.
- Smyth B, et al. Prescribing for patients on dialysis. Aust Prescr 2016, 39: 21–4.
- Khanal A, et al. Dose adjustment guidelines for medications in patients with renal impairment: how consistent are drug information sources? Intern Med J 2014, 44: 77-85.
- Velenosi TJ, Urquhart BL. Pharmacokinetic considerations in chronic kidney disease and patients requiring dialysis. Expert Opin Drug Metab Toxicol 2014, 10: 1131-43.
- Richards S. Metformin and patients on dialysis. Aust Prescr 2016, 39: 74–5.
- Verbeeck RK, Musuamba FT. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur J Clin Pharmacol 2009, 65: 757-73.
- Flynn C, Bakris GL. Non insulin glucose-lowering agents for the treatment of patients on dialysis. Nat Rev Nephrol 2013, 9: 147-53.
- Levy J. Drug prescribing in patients on dialysis. Oxford Handbook of Dialysis (4 ed.) 2016.
- Baillie and Mason’s Dialysis of Drugs. 2016.
- The Renal Drug Handbook, 4th Radcliffe Publishing, London, 2014.
- Hahr AJ, Molitch ME. Management of diabetes mellitus in patients with chronic kidney disease. Clin Diabetes Endocrinol 2015, 1: 2.
Il fegato come organo e bersaglio endocrino
Flaminia Ferri & Stefano Corradini
Divisione di Gastroenterologia, Dipartimento di Medicina Traslazionale e di Precisione, Università "Sapienza" di Roma
(aggiornato al 15 luglio 2022)
Il fegato non è solo l'organo più grande del corpo, ma anche quello che svolge uno dei ruoli più importanti, in particolare ma non solo nella trasformazione di sostanze tossiche in elementi che l'organismo può eliminare. Il suo ruolo centrale è legato sia all’entità della vascolarizzazione ricevuta (25% della gittata cardiaca a riposo), sia alla peculiarità della sua vascolarizzazione (1). A differenza degli altri organi, la vascolarizzazione epatica in ingresso è costituita dalla sovrapposizione di due reti, l'arteria epatica e la vena porta (1,2): l'arteria epatica porta al fegato sangue ossigenato, mentre la vena porta fornisce sangue deossigenato proveniente da stomaco, intestino, pancreas, milza e colecisti (2,3).
Grazie a questa sua peculiarità, il fegato è un organo dinamico, che “comunica” con altri organi endocrini, tra cui l'ipofisi, il pancreas, l'intestino, la tiroide, le ghiandole surrenali e l'osso, attraverso la produzione di ormoni, proteine leganti ed epatochine, e svolge un ruolo di elaborazione e ridistribuzione dei combustibili metabolici (4-8). A loro volta gli ormoni prodotti dagli altri organi endocrini modulano le funzioni metaboliche e sintetiche del fegato (4-8). Per tali ragioni, le epatopatie si associano a numerose modificazioni endocrine, che possono manifestarsi con l’alterazione del metabolismo del glucosio, del metabolismo osseo, con l’ipogonadismo, oltre che interessare il GH e il cortisolo (4-8).
Ruolo del fegato nella produzione diretta di ormoni
25-idrossi vitamina D. È nota per il suo ruolo nell'omeostasi del calcio e delle ossa, ma anche per la sua attività immuno-modulatrice sulla risposta innata e adattativa. Il fegato svolge un ruolo importante nella sintesi della vitamina D, che proprio a livello epatico viene idrossilata dagli enzimi del citocromo P450 per produrre 25-idrossivitamina D, la principale forma di immagazzinamento della vitamina (9-12). La riduzione della sintesi che si osserva nella cirrosi epatica e nella malattia colestatica cronica rappresenta un importante fattore di rischio per la malattia osteoporotica (13).
IGF-1. È prodotto principalmente dal fegato e i suoi recettori sono espressi in maniera ubiquitaria (14). La sua funzione è promuovere la crescita e la differenziazione cellulare e regolare il metabolismo dei nutrienti (15). Nella cirrosi epatica il deficit di IGF-1 è associato ad un’alterazione del metabolismo glucidico e lipidico (14).
Angiotensinogeno. Prodotto principalmente dal fegato, è coinvolto nel sistema renina-angiotensina-aldosterone, e svolge quindi un ruolo chiave nel mantenimento dell’omeostasi della pressione arteriosa, attraverso la sua azione sull'equilibrio del sodio, sul volume intra- ed extra-vascolare e sulle resistenze vascolari sistemiche (16,17). Nel contesto della cirrosi epatica, il sistema renina-angiotensina-aldosterone risulta up-regolato, con conseguente riduzione della perfusione renale (18). I livelli circolanti di angiotensinogeno risultano pressoché nella norma fino alle fasi terminali dall’epatopatia (19).
Ruolo del fegato nel metabolismo ormonale
Il fegato svolge un ruolo nel metabolismo di numerosi ormoni, come, ad esempio, l’attivazione e disattivazione degli ormoni tiroidei (20) o come sede del metabolismo del GLP-1, con conseguente stimolazione della produzione di insulina, riduzione della motilità intestinale e dell’appetito (21).
A livello epatocitario avviene, inoltre, anche la biosintesi del colesterolo, che è alla base della formazione degli ormoni steroidei. Tali ormoni, una volta sintetizzati, subiscono la prima e la seconda fase del loro metabolismo proprio a livello delle cellule epatiche attraverso vari processi, tra cui l’idrossilazione, la riduzione, la glucurono-coniugazione, ecc (22,23). Il fegato svolge quindi un ruolo chiave nel mantenimento dell'equilibrio elettrolitico e della pressione arteriosa tramite il metabolismo dei mineralcorticoidi e dei glucocorticoidi (22). Il fegato, inoltre, è il sito principale per la conversione di estrogeni, progesterone e androgeni nei loro metaboliti, tramite gli enzimi delle monossigenasi citocromo P450 (24).
Produzione di binding-protein
Il fegato rappresenta la principale fonte di produzione delle proteine leganti gli ormoni, tra cui ormoni tiroidei (TBG), glucocorticoidi e mineralcorticoidi (CBG), vitamina D (DBP) e ormoni sessuali (SHBG), IGF-BP (5,25,26). Le binding-protein agiscono come reservoir degli ormoni circolanti e ne possono regolare la distribuzione tissutale e il target di destinazione (25,26). Tale funzione epatica è, a sua volta, regolata da diversi fattori (25,26).
Asse fegato-cellule alfa
Questo asse rappresenta il possibile legame tra l’iperglucagonemia e la steatosi epatica che si riscontrano tipicamente nella sindrome metabolica. È, infatti, ampiamente noto il ruolo svolto dal glucagone nella regolazione della produzione di glucosio a livello epatico e come i livelli di glucosio circolanti influenzino la secrezione di glucagone delle cellule alfa del pancreas (27).
Negli ultimi anni è emerso chiaramente anche il ruolo del glucagone nella regolazione del turn-over degli aminoacidi a livello epatico, aumentando l’ureagenesi. A loro volta, alcuni aminoacidi glucagonotropici, come l’alanina, stimolano la secrezione di glucagone dal pancreas e la proliferazione delle cellule alfa, completando così il feed-back (28,29). L’interruzione del feed-back, di conseguenza, si traduce in un quadro di iperglucagonemia e iperaminoacidemia. Tali quadri sono stati osservati nella NAFLD lieve anche prima dell’insorgenza del diabete mellito tipo 2, suggerendo che anche la lieve alterazione della funzionalità epatica che si verifica in tale contesto può ridurre la sensibilità al glucagone, con conseguente riduzione dell'ureagenesi (30).
Betatropina
In passato uno studio aveva dimostrato il ruolo della betatropina nella proliferazione delle ß-cellule pancreatiche. Tale scoperta sembrava aprire nuovi orizzonti nel trattamento del diabete tipo 2, ma successivamente i dati sono risultati discordanti. La funzione di tale ormone non è ancora chiarita, ma sicuramente svolge un ruolo sia nel metabolismo glucidico che lipidico, anche se i meccanismi alla base della sua azione devono essere ancora chiariti (28,31). Attualmente si ipotizza che l'insulina stimoli la secrezione di betatropina e che questa, a sua volta, vada a promuove sia l’adipogenesi che la regolazione del metabolismo dei trigliceridi (28,31).
Epatochine
Fetuina-A. È la prima epatochina scoperta. Agisce sia a livello osseo, regolando l’osteogenesi, ma anche a livello dei miociti e degli adipociti, inibendo la fosforilazione del recettore per l’insulina. Inoltre, a livello del tessuto adiposo inibisce l’espressione dell’adiponectina. Nei pazienti con insulino-resistenza la fetuina-A è elevata e rappresenta un fattore di rischio per il diabete tipo 2, mentre l’inibizione della produzione epatica di fetuina-A migliora la sensibilità all’insulina (28,32-38).
Tsukushi. Appartiene alla famiglia delle SLRP (small leucine-rich proteoglycans), che, oltre ad avere un ruolo nell’embriogenesi, svolgono un ruolo, ancora non del tutto chiaro, nella regolazione del tessuto adiposo, del peso e del dispendio energetico (39-42).
FGF21. È un’epatochina prodotta principalmente dal fegato, anche se la sua espressione avviene anche a livello del tessuto adiposo e del pancreas. La sua espressione a livello epatico è stimolata da numerosi fattori, tra cui fattori dietetici, come il digiuno prolungato (> 7 giorni) ma anche sovra-nutrizione e in particolare dieta ricca di carboidrati e grassi associata ad una ridotta assunzione di proteine. Anche fattori ormonali come glucagone e triiodiotironina ne aumentano l’espressione, mentre l’insulina la inibisce. L’FGF21 influenza l’appetito, il peso corporeo, il metabolismo lipidico e glucidico (43-54).
Activina E. È secreta come omodimero dell’inibina-ßE, che è principalmente espressa nel fegato. Nei modelli animali, la sovra-espressione epatica sembra avere un ruolo nella prevenzione del sovrappeso/obesità e dell’insulino-resistenza, attraverso la promozione dell’aumento del dispendio energetico mediante l’aumento dell’ossidazione dei grassi. Nonostante tali dati, però, il suo ruolo nella regolazione metabolica appare ancora controverso (55-57).
GPNMB (glycoprotein nonmetastatic melanoma protein B). È una proteina trans-membrana espressa dal fegato e da altri organi, che stimola la lipogenesi sia in vivo che in vitro. I suoi livelli di espressione e quelli circolanti sono aumentati sia nell’obesità che nell’insulino-resistenza. Inoltre, recentemente è stato evidenziato anche il suo ruolo nella regolazione dello stato infiammatorio a livello del tessuto adiposo bianco (58).
Fibroblast growth factor 19 (FGF19)
Il gene dell’FGF19 è espresso a livello degli enterociti ileali. Nella fase post-prandiale vengono rilasciati gli acidi biliari che si legano al farnesoid X receptor (FXR), provocando il rilascio dell’FGF19 nel circolo entero-epatico (59,60).
Il ruolo attualmente più noto dell’FGF19 è nella regolazione del riempimento della colecisti e nell’omeostasi degli acidi biliari, ma negli ultimi anni sta emergendo un ruolo chiave dell’FGF19 a livello metabolico (61,62). L’FGF19 stimola a livello epatico la sintesi proteica e del glicogeno e agisce come inibitore della gluconeogenesi (61,62). Per quanto riguarda il metabolismo lipidico, FGF19 ha un ruolo centrale a livello epatico nella produzione de novo di trigliceridi, nell’ossidazione degli acidi grassi, nella regolazione dello stress del reticolo endoplasmico (63,64).
In considerazione dei ruoli sovra-elencati l’FGF19 appare oggi uno dei target terapeutici più promettenti per il trattamento della NAFLD (65).
Conclusioni
La funzione endocrina del fegato è un campo in rapida evoluzione, che presenta ancora diverse controversie da chiarire, al fine di ottenere una corretta interpretazione dei risultati, evitando di semplificare eccessivamente un sistema estremamente complesso. Tuttavia, non c’è alcun dubbio che i dati disponibili aprono a un futuro in cui la funzione endocrina diventerà il target delle terapie non solo della steatosi ma anche dell’obesità e del diabete.
Bibliografia
- Lorente S, Hautefeuille M, Sanchez-Cedillo A. The liver, a functionalized vascular structure. Sci Reports 2020, 10: 16194.
- Svegliati-Baroni G, Patrício B, Lioci G, et al. Gut-pancreas-liver axis as a target for treatment of NAFLD/NASH. Int J Mol Sci 2020, 21: 5820.
- Eipel C. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World J Gastroenterol 2010,16: 6046.
- Watt MJ, Miotto PM, de Nardo W, Montgomery MK. The liver as an endocrine organ—Linking NAFLD and insulin resistance. Endocr Rev 2019, 40: 1367–93.
- Bikle DD. The free hormone hypothesis: when, why, and how to measure the free hormone levels to assess vitamin D, thyroid, sex hormone, and cortisol status. JBMR Plus 2021, 5: e10418.
- Malespin M, Nassri A. Endocrine diseases and the liver. Clin Liver Dis 2019, 23: 233–46.
- Rui L. Energy metabolism in the liver. Compr Physiol 2014, 4: 177–97.
- Grant DM. Detoxification pathways in the liver. J Inherit Metab Dis 1991, 14: 421–30.
- Konstantakis C, Tselekouni P, Kalafateli M, Triantos C. Vitamin D deficiency in patients with liver cirrhosis. Ann Gastroenterol 2016, 29: 297–306.
- Christakos S, Dhawan P, Verstuyf A, et al. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev 2016, 96: 365–408.
- Elangovan H, Chahal S, Gunton JE. Vitamin D in liver disease: current evidence and potential directions. Biochim Biophys Acta Mol Basis Dis 2017, 1863: 907–16.
- Smyk DS, Orfanidou T, Invernizzi P, et al. Vitamin D in autoimmune liver disease. Clin Res Hepatol Gastroenterol 2013, 37: 535–45.
- Nair S. Vitamin D deficiency and liver disease. Gastroenterol Hepatol (NY) 2010, 6: 491–3.
- Kineman RD, del Rio-Moreno M, Sarmento-Cabral A. 40 years of IGF1: understanding the tissue-specific roles of IGF1/IGF1R in regulating metabolism using the Cre/loxP system. J Mol Endocrinol 2018, 61: T187–98.
- Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer 2006, 13 suppl 1: S33-43.
- Patel S, Rauf A, Khan H, Abu-Izneid T. Renin-angiotensin-aldosterone (RAAS): the ubiquitous system for homeostasis and pathologies. Biomed Pharmacother 2017, 94: 317–25.
- Matsusaka T, Niimura F, Shimizu A, et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 2012, 23: 1181–9.
- Di Pascoli M, La Mura V. Renin-angiotensin-aldosterone system in cirrhosis: there’s room to try! Dig Liver Dis 2019, 51: 297–8.
- Sansoè G, Aragno M, Wong F. Pathways of hepatic and renal damage through non‐classical activation of the renin‐angiotensin system in chronic liver disease. Liver Int 2020, 40: 18–31.
- Luongo C, Dentice M, Salvatore D. Deiodinases and their intricate role in thyroid hormone homeostasis. Nature Rev Endocrinol 2019, 15: 479–88.
- Jin T, Weng J. Hepatic functions of GLP-1 and its based drugs: current disputes and perspectives. Amer J Physiol-Endocrinol Metab 2016, 311: E620–7.
- Schiffer L, Barnard L, Baranowski ES, et al. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: a comprehensive review. J Ster Biochem Molec Biol 2019, 194: 105439.
- Trapani L. Regulation and deregulation of cholesterol homeostasis: the liver as a metabolic “power station.” World J Hepatol 2012, 4: 184.
- Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett 2005, 227: 115–24.
- Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocrinol 2016, 230: R13-25.
- Hammond GL. Potential functions of plasma steroid-binding proteins. Trends Endocrinol Metab 1995, 6: 298–304.
- Gar C, Haschka SJ, Kern-Matschilles S, et al. The liver–alpha cell axis associates with liver fat and insulin resistance: a validation study in women with non-steatotic liver fat levels. Diabetologia 2021, 64: 512–20.
- Rhyu J, Yu R. Newly discovered endocrine functions of the liver. World J Hepatol 2021, 13: 1611–28.
- Wewer Albrechtsen NJ, Færch K, Jensen TM, et al. Evidence of a liver-alpha cell axis in humans: hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 2018, 61: 671–80.
- Pedersen JS, Rygg MO, Kristiansen VB, et al. Nonalcoholic fatty liver disease impairs the liver-alpha cell axis independent of hepatic inflammation and fibrosis. Hepatol Commun 2020, 4: 1610–23.
- Lu P, Chen X, Zhang Z, et al. Insulin upregulates betatrophin expression via PI3K/Akt pathway. Sci Rep 2017, 7: 5594.
- Bourebaba L, Marycz K. Pathophysiological implication of fetuin-A glycoprotein in the development of metabolic disorders: a concise review. J Clin Med 2019, 8: 2033.
- Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circul Res 2011, 108: 1494–509.
- Mathews ST, Rakhade S, Zhou X, et al. Fetuin-null mice are protected against obesity and insulin resistance associated with aging. Biochem Biophys Res Commun 2006, 350: 437–43.
- Ren G, Kim T, Papizan JB, et al. Phosphorylation status of fetuin-A is critical for inhibition of insulin action and is correlated with obesity and insulin resistance. Amer J Physiol-Endocrinol Metab 2019, 317: E250–60.
- Ramírez-Vélez R, García-Hermoso A, Hackney AC, Izquierdo M. Effects of exercise training on fetuin-A in obese, type 2 diabetes and cardiovascular disease in adults and elderly: a systematic review and meta-analysis. Lipids Health Dis 2019, 18: 23.
- Pan X, Wen SW, Bestman PL, et al. Fetuin-A in metabolic syndrome: a systematic review and meta-analysis. PLoS One 2020, 15: e0229776.
- Ochi A, Mori K, Emoto M, et al. Direct inhibitory effects of pioglitazone on hepatic fetuin-A expression. PLoS One 2014, 9: e88704.
- Dellett M, Hu W, Papadaki V, Ohnuma S. Small leucine rich proteoglycan family regulates multiple signalling pathways in neural development and maintenance. Dev Growth Differ 2012, 54: 327–40.
- Wang Q, Sharma VP, Shen H, et al. The hepatokine Tsukushi gates energy expenditure via brown fat sympathetic innervation. Nat Metab 2019, 1: 251–60.
- Liu D, Zhang P, Wei X, et al. Elevated serum Tsukushi levels in patients with hyperthyroidism. Front Endocrinol (Lausanne) 2020, 11: 580097.
- Mouchiroud M, Camiré É, Aldow M, et al. The hepatokine TSK does not affect brown fat thermogenic capacity, body weight gain, and glucose homeostasis. Mol Metab 2019, 30: 184–91.
- Fisher FM, Maratos-Flier E. Understanding the physiology of FGF21. Annu Rev Physiol 2016, 78: 223–41.
- Tezze C, Romanello V, Sandri M. FGF21 as modulator of metabolism in health and disease. Front Physiol 2019, 10: 419.
- Markan KR, Naber MC, Ameka MK, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 2014, 63: 4057–63.
- Erickson A, Moreau R. The regulation of FGF21 gene expression by metabolic factors and nutrients. Horm Mol Biol Clin Invest 2017, 30: 0016.
- Fazeli PK, Lun M, Kim SM, et al. FGF21 and the late adaptive response to starvation in humans. J Clin Invest 2015, 125: 4601–11.
- Zhang X, Yeung DCY, Karpisek M, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 2008, 57: 1246–53.
- Habegger KM, Stemmer K, Cheng C, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes 2013, 62: 1453–63.
- Adams AC, Astapova I, Fisher FM, et al. Thyroid hormone regulates hepatic expression of fibroblast growth factor 21 in a PPARα-dependent manner. J Biol Chem 2010, 285: 14078–82.
- Uebanso T, Taketani Y, Fukaya M, et al. Hypocaloric high-protein diet improves fatty liver and hypertriglyceridemia in sucrose-fed obese rats via two pathways. Amer J Physiol-Endocrinol Metab 2009, 297: E76–84.
- Martínez-Garza Ú, Torres-Oteros D, Yarritu-Gallego A, et al. Fibroblast growth factor 21 and the adaptive response to nutritional challenges. Int J Mol Sci 2019, 20: 4692.
- Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol 2020, 16: 654–67.
- Baruch A, Wong C, Chinn LW, et al. Antibody-mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proc Natl Acad Sci USA 2020, 117: 28992–29000.
- Sugiyama M, Kikuchi A, Misu H, et al. Inhibin βE (INHBE) is a possible insulin resistance-associated hepatokine identified by comprehensive gene expression analysis in human liver biopsy samples. PLoS One 2018, 13: e0194798.
- Namwanje M, Brown CW. Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb Perspect Biol 2016, 8: a021881.
- Fang J, Wang S-Q, Smiley E, Bonadio J. Genes coding for mouse activin βC and βE are closely linked and exhibit a liver-specific expression pattern in adult tissues. Biochem Biophys Res Commun 1997, 231: 655–61.
- Prabata A, Ikeda K, Rahardini EP, et al. GPNMB plays a protective role against obesity-related metabolic disorders by reducing macrophage inflammatory capacity. J Biol Chem 2021, 297: 101232.
- Walters JRF. Bile acid diarrhoea and FGF19: new views on diagnosis, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol 2014, 11: 426–34.
- Fon Tacer K, Bookout AL, Ding X, et al. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 2010, 24: 2050–64.
- Choi M, Moschetta A, Bookout AL, et al. Identification of a hormonal basis for gallbladder filling. Nat Med 2006, 12: 1253–5.
- Kir S, Kliewer SA, Mangelsdorf DJ. Roles of FGF19 in liver metabolism. Cold Spring Harb Symp Quant Biol 2011, 76: 139–44.
- Diraison F, Moulin P, Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab 2003, 29: 478–85.
- Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol 2016, 78: 181–205.
- Arab JP, Karpen SJ, Dawson PA, et al. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 2017, 65: 350–62.
Eziopatogenesi, clinica e criteri diagnostici della steatosi e steatoepatite
Flaminia Ferri & Stefano Corradini
Divisione di Gastroenterologia, Dipartimento di Medicina Traslazionale e di Precisione, Università "Sapienza" di Roma
(aggiornato al 15 dicembre 2022)
INTRODUZIONE
La malattia da fegato grasso non alcolica (NAFLD), dal termine inglese “non-alcoholic fatty liver disease”, si riferisce alla presenza di steatosi epatica in assenza di altre cause di accumulo secondario di grasso epatico (es. eccessivo consumo di alcol, positività per HCV, ecc.). La NAFLD può evolvere in cirrosi ed è probabilmente un'importante causa di cirrosi criptogenetica (1-4).
La NAFLD è suddivisa in (5,6):
- steatosi epatica non alcolica (NAFL), in cui la steatosi epatica è presente senza evidenza di infiammazione significativa;
- steato-epatite non alcolica (NASH), in cui la steatosi epatica è associata a infiammazione epatica. La NASH è associata a rischio di sviluppare fibrosi epatica, cirrosi ed epato-carcinoma.
Vista la grande prevalenza della NAFLD e della Sindrome Metabolica (MetS), fattore di rischio per la NAFLD, la gestione clinica di questi pazienti è appannaggio in gran parte anche degli endocrinologi, mentre l’invio allo specialista epatologo viene regolato da linee guida di pratica clinica (vedi sotto).
Epidemiologia, patogenesi e genetica
La NAFLD è la malattia epatica cronica più comune nelle nazioni industrializzate, con differenze di prevalenza secondo metodo diagnostico, età, sesso ed etnia, e rappresenta una delle principali indicazioni per il trapianto di fegato.
La prevalenza globale della NAFLD è stata del 27.9% nel periodo 2000-2010 e del 31.6% nel periodo 2011-2021 (7). La prevalenza globale della NASH nel periodo 2000-2021 è stata dell’8.3% e, in una coorte di mezza età degli USA, del 12-14% (7,8).
La NAFLD è spesso associata alle caratteristiche cliniche della MetS. La prevalenza globale della NAFLD è rispettivamente del 70% e del 75.3% nella popolazione in sovrappeso e in quella obesa. La prevalenza globale della NASH è rispettivamente del 33,5 e del 33,7% nella popolazione in sovrappeso e in quella obesa (9). La prevalenza globale della NAFLD e della NASH nei pazienti diabetici è, rispettivamente, del 55-70% e del 30-40% (10).
L’aumento di prevalenza e gravità della NAFLD, in correlazione con l'aumento dell'obesità e della MetS, evidenzia la necessità di un approccio razionale alla prevenzione, diagnosi e trattamento della malattia. La NAFLD è presente anche nel 7% delle persone normo-peso, più frequentemente nelle donne, in età più giovane e con normalità delle transaminasi (11). Anche in presenza di normalità delle transaminasi sieriche, e spesso in associazione con insulino-resistenza o diabete, la NAFLD può comunque essere progressiva (12).
La patogenesi della NAFLD non è stata completamente chiarita. Il pre-requisito a livello metabolico consiste in un sovraccarico di acidi grassi liberi che, per diversi motivi, gli epatociti riescono a gestire solo con un accumulo intra-citoplasmatico di gocciole lipidiche costituite prevalentemente da trigliceridi. La teoria ampiamente supportata implica l’insulino-resistenza come meccanismo chiave che porta allo sviluppo della steatosi epatica, e forse anche alla NASH. Altre teorie hanno proposto l’ipotesi di un "second hit", o di ulteriore danno ossidativo, che sarebbe necessario per manifestare la componente necro-infiammatoria della NASH (13). L’accumulo epatico di ferro, la leptina, la carenza di anti-ossidanti e il microbioma intestinale sono stati suggeriti come potenziali fattori di stress ossidativo.
Sono state identificate diverse varianti genetiche e alterazioni epigenetiche associate sia al rischio di steatosi epatica che alla progressione alla NASH fibrosante. Una variante di “patatin-like phospholipase domain-containing 3” (PNPLA3), molto diffusa nella popolazione generale, predispone alla malattia, sia per il rischio di steatosi che per la progressione di malattia. Al contrario, una variante di “Hydroxysteroid 17-β dehydrogenase 13” (HSD17B13) è protettiva per la progressione di malattia (14).
Aspetti clinici
Per la diagnosi e la gestione clinica della NAFLD in ambito non epatologico specialistico ed in particolare nel setting degli specialisti endocrinologi, sono disponibili le LG del 2016 della European Association for the Study of Diabetes (EASD) in collaborazione con la European Association for the Study of the Liver (EASL) e con la European Association for the Study of Obesity (EASO) (15), quelle del 2022 della Società Italiana di Diabetologia (SID) in collaborazione con l’Associazione Italiana per lo Studio del Fegato (AISF) e con la Società Italiana dell’Obesità (SIO) (16) e quelle del 2022 da parte della American Association of Clinical Endocrinology (AACE) in collaborazione con la American Association for the Study of Liver Diseases (17). Per quanto riguarda la diagnosi di NAFLD, le LG sono piuttosto omogenee.
La maggior parte dei pazienti con NAFLD è asintomatica. Nel raccogliere l’anamnesi familiare e personale, bisogna prestare particolare attenzione alle patologie associate, in particolare alle componenti che vanno a formare il quadro della MetS, e ad eventuali cause secondarie. Allo stesso modo, la presenza di diabete mellito di tipo 2, obesità o il riscontro accidentale di un’elevazione degli enzimi di cito-necrosi epatica nei pazienti con fattori di rischio metabolico necessita un approfondimento diagnostico mediante uno screening non invasivo per arrivare alla diagnosi di steatosi e predire il rischio di NASH e fibrosi (13).
L’esame obiettivo in questi pazienti non mostra alterazioni significative, tranne talvolta per la presenza di epatomegalia; solo i pazienti che hanno già sviluppato la cirrosi possono presentare all’esame obiettivo le stigmate tipiche della patologia, quali spider naevi e splenomegalia e, nei pazienti con malattia scompensata, ascite e/o ittero (18).
Gli esami di laboratorio nei pazienti con NAFLD e cirrosi epatica possono documentare aumenti lievi o moderati di AST e ALT, eventualmente associati a piastrinopenia, iperbilirubinemia e prolungamento dell’INR. Nei pazienti senza cirrosi epatica, sia con steatosi semplice che con NASH, le AST e le ALT possono essere lievemente o moderatamente aumentate (19). Generalmente, quando presente, l’elevazione va da due a cinque volte il limite superiore dei valori normali, con un rapporto AST/ALT < 1, a differenza della steatosi epatica alcolica, in cui in genere il rapporto AST/ALT risulta > 2 (19-23). È importante sottolineare che livelli normali di transaminasi non escludono la diagnosi di NAFLD o la presenza di un danno istologico rilevante con NASH e fibrosi fino alla cirrosi. Tuttavia, alti livelli di transaminasi si associano a esiti negativi (12,17,1924-26).
La diagnosi di NAFLD viene in genere posta in seguito al riscontro casuale di ipertransaminasemia e/o aumento della gamma-GT e/o di steatosi evidenziata incidentalmente mediante l’esame ecografico. La diagnosi di NAFLD si basa sulla presenza di steatosi epatica all’imaging radiologico, più frequentemente ecografia, o alla biopsia epatica, sulla mancanza di significativo consumo di alcool in corso o recente e sull’esclusione di altre malattie steatogene del fegato (steatosi secondarie). Inoltre, nei casi con ipertransaminasemia, vanno comunque escluse altre concomitanti eziologie di danno epatico cronico.
Esclusione di altre cause
Per porre la diagnosi di NAFLD è indispensabile escludere la presenza di cause secondarie di steatosi epatica:
- apporto alcolico: escludere attraverso l’anamnesi la presenza di valori a rischio, cioè > 294 g di alcool/settimana per gli uomini e > 196 g di alcool/settimana per le donne, secondo le LG Americane (17), oppure ≥ 30 g/die per gli uomini e ≥ 20 g/die per le donne secondo le LG Europee (15) e Italiane (16);
- virus delle epatiti: ricerca di HCV-Ab, HBsAg, HBsAb, HBcAb;
- malattie da accumulo: calcolare l’indice di saturazione della transferrina;
- malattie immunitarie: misurare i livelli di gamma-globuline totali, anticorpo anti-nucleo, anti-mitocondrio, anti-LKM;
- alfa-1-antitripsina;
- cupremia, ceruloplasminemia (se alto sospetto di M di Wilson, cupruria 24 ore);
- farmaci e droghe steatogeni: cocaina, estrogeni, tamoxifene, amiodarone, irinotecano, farmaci anti-retrovirali, lomitapide, metotrexate, naprossene, ibuprofene, diltiazem, verapamil, nifedipina, 5-fluorouracile, tetracicline, glucocorticoidi, valproato (30);
- se sospetto clinico, dovrebbero essere considerate altre cause più rare di steatosi epatica secondaria: lipodistrofia, malnutrizione, nutrizione parenterale, abetalipoproteinemia, S di Reye, deficit di lipasi acida lisosomiale, fibrosi cistica, steatosi acuta in gravidanza, sindrome HELLP (Hemolysis, Elevated Liver enzymes, low Platelet count) (15,29).
Nei pazienti con aumento delle transaminasi, vanno comunque escluse altre cause concomitanti di ipertransaminasemia eventualmente presenti, che di per sé, in genere, non causano steatosi:
- farmaci, vitamine e supplementi;
- colangite biliare primitiva;
- s. di Budd-Chiari;
- iper- o ipotiroidismo, s. di Cushing, ipogonadismo, deficit di GH, m. di Addison.
METODICHE DIAGNOSTICHE
Valutazione non-invasiva per paziente
L’ecografia è la metodica di prima linea per la diagnosi e il follow-up dei pazienti con NAFLD. Nei pazienti affetti il parenchima appare iperecogeno rispetto al parenchima del rene destro, per l’accumulo dei lipidi all’interno degli epatociti (25). Dal punto di vista semi-quantitativo l’ecografia può solo, in modo soggettivo, valutare se l’accumulo è presente in modo lieve, moderato o severo. Oltre alla diagnosi di steatosi epatica, l’ecografia ci permette di valutare i segni di un’eventuale presenza di cirrosi, di ipertensione portale e di lesioni occupanti spazio. Tuttavia, tale metodica risulta meno affidabile nei pazienti con BMI > 40 kg/m2 e quando la steatosi è appannaggio di meno del 20% degli epatociti, con il rischio, quindi, di sotto-stimare la prevalenza di NAFLD (26-28).
La metodica più performante per quantificare in maniera non invasiva sia la steatosi che la fibrosi epatica è la spettroscopia protonica di risonanza magnetica (H-MRS), in quanto consente di individuare la steatosi epatica quando è presente in ≥ 5% degli epatociti. Attualmente questa tecnica, per il costo e la scarsa diffusione sul territorio, è utilizzata solo a fini di ricerca, ma non trova ancora applicazione in ambito clinico (15).
Negli ultimi ha avuto un importante sviluppo in questo campo la “Controlled Attenuation Parameter” (CAP). La CAP è un mezzo alternativo per effettuare una diagnosi qualitativa e quantitativa della steatosi e si associa frequentemente alle tecniche elastometriche, che consentono di valutare la rigidità epatica e quindi di stimare in maniera indiretta il grado di fibrosi (17,29). Tale metodica non ha ancora un ruolo ben definito all’interno delle LG, ma negli ultimi anni ha dimostrato nella pratica clinica un ruolo sempre maggiore, sia nella fase diagnostica che nel follow-up, anche se sono ancora limitati gli studi di confronto con la biopsia o la H-MRS (15).
Una volta fatta la diagnosi di NAFLD, è necessario riconoscere se il paziente ha una o più delle caratteristiche cliniche della MetS, attraverso un’accurata anamnesi personale che includa anche i farmaci, l’esame obiettivo con la misurazione della pressione arteriosa e della circonferenza addominale e prescrivendo esami specifici di primo livello (trigliceridi, colesterolo totale, LDL e HDL, glicemia ed emoglobina glicata). Nei casi in cui non si pone diagnosi di diabete, vanno richiesti OGTT e HOMA per escludere uno stato pre-diabetico. Le singole caratteristiche della MetS vanno indagate a scopo terapeutico e prognostico. I soggetti con NAFLD e diabete mellito tipo 2 e con obesità sono infatti quelli in cui la malattia epatica progredisce più rapidamente (17).
Il ruolo della biopsia
La biopsia epatica rappresenta il gold-standard per distinguere la NAFL dalla NASH e per valutare presenza e gravità della fibrosi. La diagnosi di NASH richiede la presenza di steatosi, ballooning e infiammazione lobulare (31-33). Per quanto riguarda la fibrosi, i test non-invasivi hanno un’accuratezza accettabile solo per identificare i casi a basso rischio di fibrosi significativa (vedi sotto).
Per la sua natura invasiva e i costi associati, tutte le LG concordano su un utilizzo limitato, al di fuori di trial clinici, della biopsia epatica eseguita nei centri specialisti epatologici solo nei pazienti con NAFLD e sospetto di fibrosi significativa, in cui la conferma dell’ipotesi diagnostica avrebbe implicazioni prognostiche e condurrebbe a cambiamenti nella gestione dei pazienti (33).
Valutazione non-invasiva della fibrosi clinicamente significativa nei pazienti con NAFLD
È opportuno sottolineare come nei pazienti con NAFLD la presenza di fibrosi epatica, in particolare di quella clinicamente significativa (stadi F2-F4), sia associata a prognosi peggiore, dovuta a eccesso di mortalità per tutte le cause rispetto alla popolazione generale (15-17). L’eccesso di mortalità associato alla fibrosi epatica nei pazienti con NAFLD è indipendente dalla presenza di MetS o sue componenti ed è dovuto, in ordine di frequenza, a cancro extra-epatico, cirrosi, malattie cardio-vascolari ed epato-carcinoma (34).
Per questo motivo, sono stati sviluppati numerosi punteggi non-invasivi, allo scopo di individuare tra i pazienti affetti da NAFLD coloro che presentino un rischio elevato di avere una fibrosi clinicamente significativa. In realtà la miglior performance di questi test risiede nella capacità di escludere la fibrosi clinicamente significativa e sono:
- Fibrosis 4 calculator (FIB-4) = età * AST/piastrine * √ALT;
- NAFLD Fibrosis Score (NFS) = −1.675 + 0.037 – età + 0.094 – BMI + (1.13 * insulino-resistenza/diabete (sì = 1, no = 0)) + (0.99 * rapporto AST/ALT) – (0.013 * piastrine (109/L)) – (0.66 * albumina (g/dL)).
Per ogni paziente con NAFLD dovrebbero essere calcolati i marcatori surrogati di fibrosi (FIB-4 o NFS), al fine di escludere una fibrosi significativa (≥ F2) che rappresenta l’elemento con maggiore impatto prognostico (15-17). Inoltre, anche l’elastometria epatica, misurata mediante “vibration-controlled transient elastography” (FibroScan) oppure, in modo più accurato nei pazienti obesi, con “Acoustic radiation force impulse (ARFI) point shear wave elastography” (35) o con “Two-dimensional shear wave elastography” (36) permette di escludere una fibrosi significativa nei pazienti con NAFLD quando i valori siano < 8 KPa. Misure più accurate di elastometria si ottengono con l’elastografia in risonanza magnetica (MRE), il cui utilizzo è però limitato per la scarsa disponibilità e gli alti costi (15-17).
Rilevanza clinica della valutazione non-invasiva della fibrosi e invio allo specialista epatologo nei pazienti con diagnosi di NAFLD
Vista l’altissima prevalenza della NAFLD, le LG di pratica clinica prevedono che nei pazienti con questa diagnosi i MMG e gli altri specialisti come gli endocrinologi eseguano il calcolo del punteggio FIB-4 o, eventualmente, NFS (le LG europee, più vecchie prevedevano anche il dosaggio delle transaminasi) (15-17):
- nel caso in cui il paziente presenti punteggio FIB-4 < 1.30 e punteggio NFS < -1.455, è considerato a basso rischio di fibrosi significativa, non necessita di essere inviato presso un centro di epatologia e proseguirà il follow-up con la rivalutazione dei suddetti punteggi dopo 1-3 anni;
- se invece il paziente presenta punteggio FIB-4 ≥ 1.30 o punteggio NFS ≥ -1.455, necessita di valutazione elastometrica e, solo nel caso in cui si evidenzi un valore di rigidità epatica significativo (≥ 8 KPa), è indicato l’invio presso un centro di epatologia per l’approfondimento diagnostico, la stadiazione e il follow-up e, nel caso il paziente sia sottoposto a chirurgia bariatrica, deve eseguire la biopsia epatica intra-operatoria.
Screening delle popolazioni a rischio metabolico per NAFLD e riferimento specialistico epatologico
Vista l’altissima prevalenza della NAFLD e della MetS, le diverse LG di pratica clinica sovra-menzionate affrontano il problema dello screening per NAFLD nei pazienti con MetS, ma con diverse raccomandazioni.
Le LG europee genericamente raccomandano che i pazienti con insulino-resistenza e/o fattori di rischio metabolici vengano sottoposti a procedure diagnostiche per la diagnosi di eccesso di grasso nel fegato e che i soggetti con obesità o MetS eseguano di routine il dosaggio delle transaminasi e/o l’ecografia (15). Esse inoltre affermano che nei pazienti ad alto rischio (età > 50 anni, DM2, MetS) è consigliabile ricercare la fibrosi.
Le LG italiane specificano che, in caso di difficile accesso all’ecografia, lo screening nei pazienti con caratteristiche della MetS possa essere fatto direttamente con il dosaggio delle transaminasi e il calcolo non-invasivo dei punteggi di fibrosi (16).
Le LG americane sono più puntuali e prevedono che nei pazienti ad alto rischio per NAFLD, in cui non sia mai stata indagata la presenza di questa patologia (pre-diabete o DM2, obesità e/o ≥ 2 fattori di rischio cardio-metabolico della MetS, tra circonferenza addominale > 102 cm nei maschi e > 89 cm nelle femmine, trigliceridemia ≥ 150 mg/dL, colesterolo HDL < 40 mg/dL nei maschi e < 50 mg/dL nelle femmine, pressione arteriosa ≥ 130/≥ 85 mm Hg, glicemia a digiuno ≥ 100 mg/dL), sia i MMG che gli endocrinologi eseguano lo screening non invasivo della fibrosi con FIB-4 ed eventuale elastometria, seguendo poi il medesimo algoritmo delineato nel paragrafo precedente (17). Per quanto riguarda la diagnosi di steatosi epatica in queste popolazioni a rischio, le LG americane non prevedono di eseguire di routine l’ecografia epatica (verosimilmente per gli alti costi), bensì il calcolo di uno dei punteggi non-invasivi per la presenza di steatosi, come il “fatty liver index” (FLI). Il FLI ha un’accuratezza inferiore all’ecografia, ma si può calcolare partendo da (17) BMI, circonferenza addominale, trigliceridemia e gamma-GT:
FLI = (e0.953*loge(trigliceridi) + 0.139*BMI + 0.718*loge(gamma-GT) + 0.053*circonferenza vita −15.745)/(1+ e0.953*loge(trigliceridi) + 0.139*BMI + 0.718*loge(gamma-GT) + 0.053*circonferenza vita − 15.745) * 100.
Esiste anche un calcolatore online (https://www.mdcalc.com/calc/10001/fatty-liver-index).
Rilevanza clinica della diagnosi di steatosi epatica nei soggetti a rischio metabolico anche in assenza di fibrosi epatica
Anche se ancora non introdotta nelle LG, dovrebbe essere raccomandata la ricerca della steatosi epatica nei pazienti con MetS, indipendentemente dalla presenza e grado della fibrosi. Infatti, un grande studio recente, eseguito su 10 568 pazienti con NAFLD diagnosticata alla biopsia epatica e seguiti per più di 14 anni, ha dimostrato che anche la presenza di semplice steatosi, senza NASH né fibrosi epatica, si associa a un eccesso mortalità del 10.7% a 20 anni rispetto alla popolazione generale, indipendentemente dalla MetS (34). Quindi, nonostante le LG italiane nei pazienti con caratteristiche della MetS prevedano di non eseguire l’ecografia di screening in caso di difficile accesso a questa metodica e di passare direttamente alla valutazione non invasiva della fibrosi (16), è auspicabile, come previsto dalle LG americane, di eseguire almeno il FLI. Uno studio recente ha individuato dei cut-off di FLI differenti per fasce di BMI, sesso e circonferenza addominale, con discreta accuratezza per la diagnosi di steatosi epatica (37). Nei pazienti con valore di FLI oltre il cut-off per steatosi epatica, andrebbe eseguita l’ecografia per conferma. Nei casi con diagnosi di steatosi è importante spiegare al paziente che, a differenza di quanto ritenuto in passato, anche la steatosi semplice senza fibrosi non è una condizione benigna e, quindi, va trattata (38).
BIBLIOGRAFIA
- Caldwell SH, Oelsner DH, Iezzoni JC, et al. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology 1999, 29: 664–9.
- Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol 2004, 40: 578–84.
- Browning JD, Kumar KS, Saboorian MH, Thiele DL. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am J Gastroenterol 2004, 99: 292–8.
- Poonawala A, Nair SP, Thuluvath PJ. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: a case-control study. Hepatology 2000, 32: 689–92.
- Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980, 55: 434–8.
- Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Ann Intern Med 1997, 126: 137–45.
- Liu J, Tian Y, Fu X, et al. Estimating global prevalence, incidence, and outcomes of non-alcoholic fatty liver disease from 2000 to 2021: systematic review and meta-analysis. Chin Med J (Engl) 2022, 135: 1682-91.
- Harrison SA, Gawrieh S, Roberts K, et al. Prospective evaluation of the prevalence of non-alcoholic fatty liver disease and steatohepatitis in a large middle-aged US cohort. J Hepatol 2021, 75: 284-91.
- Quek J, Chan KE, Wong ZY, et al. Global prevalence of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in the overweight and obese population: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2023, 8: 20-30.
- Stefan N, Cusi K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol 2022, 10: 284-96.
- Younossi ZM, Stepanova M, Negro F, et al. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine 2012, 91: 319–27.
- Fracanzani AL, Valenti L, Bugianesi E, et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: a role for insulin resistance and diabetes. Hepatology 2008, 48: 792–8.
- Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018, 283: 356–70.
- Zhu X, Xia M, Gao X. Update on genetics and epigenetics in metabolic associated fatty liver disease. Ther Adv Endocrinol Metab 2022, 13: 20420188221132138.
- EASL–EASD–EASO. Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016, 64: 1388–402.
- Italian Association for the Study of the Liver (AISF), Italian Society of Diabetology (SID) and Italian Society of Obesity (SIO). Non-alcoholic fatty liver disease in adults 2021: A clinical practice guideline of the Italian Association for the Study of the Liver (AISF), the Italian Society of Diabetology (SID) and the Italian Society of Obesity (SIO). Dig Liver Dis 2022, 54: 170–82.
- Cusi K, Isaacs S, Barb D, et al. American Association of Clinical Endocrinology clinical practice guideline for the diagnosis and management of nonalcoholic fatty liver disease in primary care and endocrinology clinical settings: co-sponsored by the American Association for the Study of Liver Diseases (AASLD). Endocr Pract 2022, 28: 528-62.
- Amarapurkar D, Kamani P, Patel N, et al. Prevalence of non-alcoholic fatty liver disease: population based study. Ann Hepatol 2007, 6: 161–3.
- Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107: 1103–9.
- Cohen JA, Kaplan MM. The SGOT/SGPT ratio? An indicator of alcoholic liver disease. Digest Dis Sci 1979, 24: 835–8.
- McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 2004, 8: 521–33.
- Sorbi D, Boynton J, Lindor KD. The ratio of aspartate aminotransferase to alanine aminotransferase: potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol 1999, 94: 1018–22.
- Falck-Ytter Y, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis 2001, 21: 17–26.
- Mofrad P, Contos MJ, Haque M, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003, 37: 1286–92.
- Hamaguchi M, Kojima T, Itoh Y, et al. The severity of ultrasonographic findings in nonalcoholic fatty liver disease reflects the metabolic syndrome and visceral fat accumulation. Am J Gastroenterol 2007, 102: 2708–15.
- Dasarathy S, Dasarathy J, Khiyami A, et al. Validity of real time ultrasound in the diagnosis of hepatic steatosis: a prospective study. J Hepatol 2009, 51: 1061–7.
- Ryan C. One hundred consecutive hepatic biopsies in the workup of living donors for right lobe liver transplantation. Liver Transpl 2002, 8: 1114–22.
- Saadeh S, Younossi ZM, Remer EM, et al. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology 2002, 123: 745–50.
- Ratziu V, Bellentani S, Cortez-Pinto H, et al. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol 2010, 53: 372–84.
- Cataldi M, Citro V, Resnati C, et al. New avenues for treatment and prevention of drug-induced steatosis and steatohepatitis: much more than antioxidants. Adv Ther 2021, 38: 2094–113.
- Kleiner D, Brunt E. Nonalcoholic fatty liver disease: pathologic patterns and biopsy evaluation in clinical research. Semin Liver Dis 2012, 32: 3–13.
- Kleiner DE, Brunt EM, van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41: 1313–21.
- Bedossa P. Utility and appropriateness of the fatty liver inhibition of progression (FLIP) algorithm and steatosis, activity, and fibrosis (SAF) score in the evaluation of biopsies of nonalcoholic fatty liver disease. Hepatology 2014, 60: 565–75.
- Simon TG, Roelstraete B, Khalili H, et al. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: results from a nationwide cohort. Gut 2021, 70: 1375-82.
- Attia D, Bantel H, Lenzen H, et al. Liver stiffness measurement using acoustic radiation force impulse elastography in overweight and obese patients. Aliment Pharmacol Ther 2016, 44: 366-79.
- Chimoriya R, Piya MK, Simmons D, et al. The use of two-dimensional shear wave elastography in people with obesity for the assessment of liver fibrosis in non-alcoholic fatty liver disease. J Clin Med 2020, 10: 95.
- Wu J, Tian S, Li H, et al. Population-specific cut-off points of fatty liver index: a study based on the National Health and Nutrition Examination Survey data. BMC Gastroenterol 2022, 22: 265.
- Tilg H, Targher G. NAFLD-related mortality: simple hepatic steatosis is not as 'benign' as thought. Gut 2021, 70: 1212-3.
Steatosi e steatoepatite in corso di obesità, dislipidemie e diabete mellito, loro gestione clinica ed impatto della nuova definizione di “Metabolic (dysfunction) associated fatty liver disease” (MAFLD)
Flaminia Ferri & Stefano Corradini
Divisione di Gastroenterologia, Dipartimento di Medicina Traslazionale e di Precisione, Università "Sapienza" di Roma
(aggiornato al 15 dicembre 2022)
INTRODUZIONE
La presenza di NAFLD è connessa con la diagnosi di sindrome metabolica (MetS), in quanto tutte le componenti della MetS correlano con l’accumulo di gocciole lipidiche nel citoplasma degli epatociti (1,2). Alla luce di tale dato, è indispensabile che ogni paziente in cui si pone la diagnosi di NAFLD sia sottoposto allo screening per le diverse componenti della MetS. Viceversa, nei pazienti in cui è stata posta diagnosi di MetS è indicata la valutazione per la NAFLD (cfr cap specifico).
Come già descritto nel capitolo precedente, la teoria più ampiamente supportata riguardante l’eziopatogenesi della NAFLD si basa sulla considerazione dell’insulino-resistenza (IR) come meccanismo chiave (3,4). La valutazione dell’IR può essere effettuata con i test dinamici, ma si ritiene che anche HOMA-IR possa essere sufficiente e più agevole per porre diagnosi di NAFLD correlata all’IR nei soggetti non-diabetici (5). L’HOMA-IR rappresenta, inoltre, un utile strumento per identificare i pazienti a rischio di sviluppare la steato-epatite/fibrosi avanzata e anche per il follow-up del paziente con NAFLD (5).
Un altro importante fattore per lo sviluppo della NAFLD è rappresentato dall’accumulo di tessuto adiposo a livello viscerale. Per tale ragione sia la misurazione del BMI che della circonferenza vita sono utili strumenti per identificare i pazienti affetti da NAFLD e predire il rischio di progressione di malattia (6,7).
Il trattamento della NAFLD, sia nei pazienti senza fibrosi che ancora di più in quelli con fibrosi, ha come scopo principale quello di ridurre la mortalità, sia per cause epatiche (cirrosi epatica ed epato-carcinoma) che per cancri extra-epatici (prima causa di morte in questi pazienti) e malattie cardio-vascolari (8). In considerazione dell’ezio-patogenesi della malattia, il trattamento della NAFLD include come primo passo quello dell’assunzione da parte del paziente di un corretto stile di vita.
STILE DI VITA
Dieta
In particolare, la riduzione del peso corporeo > 5% attraverso la dieta e l’esercizio fisico permette di migliorare la steatosi. La riduzione del peso corporeo ≥10% rappresenta la terapia primaria, sia nei pazienti obesi che sovrappeso, con la NASH e porta al miglioramento del quadro istologico anche della fibrosi, dell’insulino-resistenza e degli indici di necrosi epatocitaria (9,10). Anche se sono ancora pochi i dati di letteratura in tal senso, sembra che anche i soggetti con BMI normale e NAFLD rispondano bene alla perdita del 5% del peso corporeo (alcuni di questi avevano una circonferenza addominale aumentata) (11).
La dieta per i pazienti con NAFLD deve basarsi sulla distribuzione dei macro-nutrienti della dieta mediterranea in associazione con la riduzione dell’apporto calorico e l’eliminazione dei cibi maggiormente steatogeni, quali cibi pre-confezionati e bibite con alto contenuto di fruttosio aggiunto. La riduzione dell’apporto calorico deve essere di circa 500-1000 kcal/die, al fine di ottenere un calo ponderale pari a 0.5-1 kg/settimana (4).
Se non controindicato da altre comorbilità, si può suggerire al paziente la moderata assunzione di caffè, che ha dimostrato di svolgere un ruolo protettivo nei confronti dell’avanzamento dell’epatopatia nei soggetti con NAFLD (12).
Uso di alcool
Nei pazienti con NAFLD è indicata l’astensione dalle bevande alcoliche, sia nell’ottica della riduzione dell’apporto calorico, sia perché i dati presenti in letteratura indicano che in questi pazienti il consumo alcolico, sia regolare > 3 unità alcoliche/die nell’uomo o 2 unità alcoliche/die nelle donne, sia occasionale ma > 5 unità alcoliche nell’uomo o 4 unità alcoliche nelle donne in ciascuna occasione, è associato a maggiore incidenza di progressione della malattia e insorgenza delle sue complicanze (5). Inoltre, diversamente da quanto osservato nella popolazione generale, i soggetti con NAFLD non beneficiano dell’effetto dell’alcol sulla riduzione del rischio cardio-vascolare (13). Appaiono invece contrastanti i dati riguardanti gli effetti sul parenchima epatico di modeste quantità di alcol rispetto alla totale astinenza (5,14,15).
Attività fisica
Al fine di mantenere il cambiamento dello stile di vita non solo nel breve ma anche nel lungo termine, è consigliabile la personalizzazione dei programmi di attività fisica in base alle preferenze del paziente, in quanto è stato dimostrato che i pazienti con NAFLD beneficiano sia dell’attività aerobica che dell’allenamento di resistenza (5).
Si raccomanda di incrementare progressivamente l’intensità dell’attività, fino a raggiungere un’intensità che il paziente riuscirà a mantenere nel tempo, ad esempio una moderata attività aerobica può consistere in 3-5 sessioni/settimana di camminata veloce o cyclette della durata di 30 minuti (3).
Anche se gli effetti metabolici sono direttamente proporzionali alla quantità e intensità dell’esercizio, qualunque incremento dell’attività fisica apporta benefici ai pazienti con NAFLD (16-18).
TRATTAMENTI FARMACOLOGICI
Nei pazienti con fibrosi significativa (≥ F2 sec. Metavir) o NASH avanzata o NASH allo stadio iniziale ma in presenza di fattori di rischio per progressione di malattia (età > 50 anni, incremento delle ALT, MetS e/o DM2) (19), potrebbe essere indicato l’approccio farmacologico, ma finora nessun farmaco è stato approvato in Italia per il trattamento delle NASH e tra i farmaci in commercio al momento i dati a disposizione non permettono di formulare precise indicazioni terapeutiche, né sul farmaco da utilizzare né sulla durata della terapia. Per questi motivi, l’approccio terapeutico attuale a questi pazienti deve basarsi sul trattamento delle comorbilità della MetS associate alla NASH (5).
Alcuni farmaci già in commercio sono stata utilizzati off-label nei pazienti con NAFLD. Ad esempio, l’utilizzo di farmaci ad azione anti-ossidante, come la vitamina E, può essere preso in considerazione per il trattamento della NASH nelle persone senza DM, ma al momento non ci sono prove sufficienti per raccomandarla alle persone con DM e fibrosi avanzata (10). In alcuni studi si è osservato un parziale miglioramento del quadro istologico della NASH e la riduzione delle ALT, non confermati da altri studi. Oltre ai risultati discordanti, è stato anche sollevato un dubbio riguardante la sicurezza della terapia con vitamina E a lungo termine, in particolare in relazione all’aumento globale della mortalità, all’incremento dell’incidenza del carcinoma prostatico e di ictus emorragico.
Nell’ambito dei farmaci per il trattamento del DM2 sono stati testati diversi farmaci e, per offrire benefici cardio-metabolici nei pazienti con DM2 e NAFLD, devono essere presi in considerazione il trattamento con pioglitazone, o con gli agonisti del recettore del GLP-1 come liraglutide, o gli inibitori di SGLT-2, anche se con questi ultimi non ci sono prove di beneficio per il trattamento della NASH (10):
- pioglitazone: è stato documentato miglioramento dell’IR, riduzione/normalizzazione dei livelli di ALT e miglioramento del quadro istologico della NASH, ma senza ottenere regressione della fibrosi ove presente;
- liraglutide: l’efficacia nella NASH sembra essere anche a carico della fibrosi (10);
- metformina: non si è dimostrata efficace nell’indurre miglioramento istologico (20), ma può essere continuata secondo necessità per il trattamento dell'iperglicemia nelle persone con DM2 e NAFLD o NASH. È stata ipotizzata una sua azione sulla prevenzione dell’insorgenza dell’HCC e nel ridurre la mortalità per tutte le cause, ma solo in studi retrospettivi, per cui saranno necessari ulteriori studi per poterne confermare l’indicazione (21);
- nel caso dei pazienti in sovrappeso o obesità (BMI ≥ 27 kg/m2) con NAFLD o NASH è indicato l’utilizzo di semaglutide 2.4 mg/settimana (miglior evidenza) o liraglutide 3 mg/die (10). La semaglutide è stata approvata dall’EMA nel 2022 per il trattamento del sovrappeso complicato da comorbilità e dell’obesità, ma non è ancora entrata in commercio.
Per quanto riguarda i farmaci ipolipemizzanti, è stato valutato sia l’uso delle statine che degli omega-3. In passato, in particolar modo l’uso delle statine poteva apparire controverso, in quanto ritenute responsabili esse stesse di danno epatocitario. Le evidenze in letteratura dimostrano che sia le statine sia gli omega-3 possono essere utilizzati al fine di ridurre il rischio cardio-vascolare quando si verifica l’associazione della NASH con la dislipidemia (5). Per quanto riguarda le statine, una recente meta-analisi ha addirittura evidenziato come nella NAFLD migliorino le transaminasi, la gamma-GT, la steatosi e l’infiammazione epatica, senza però effetti sulla fibrosi epatica (22).
L’utilizzo degli acidi biliari è stato in tale contesto ampiamente testato. In passato diversi studi randomizzati hanno valutato l’efficacia dell’acido urso-dessicolico nel contesto nella NASH, dimostrando miglioramento solo transitorio degli indici di cito-necrosi epatica, che non si accompagnava a miglioramento significativo del quadro istologico (23-25).
Risultati molto incoraggianti vengono invece dagli studi riguardanti l’utilizzo dell’acido obeticolico, agonista del recettore farnesoide (FXR) e analogo dell’acido chenodessicolico. Il farmaco è già stato approvato per il trattamento della colangite biliare primitiva e negli studi effettuati nella NASH porterebbe a miglioramento dell’IR ma anche a regressione istologica, sia del quadro di NASH sia della fibrosi. I principali effetti collaterali che potrebbero limitare l’utilizzo di tale farmaco sono rappresentati dalla comparsa di prurito e dell’incremento delle LDL (26,27).
Dati promettenti sulla NASH emergono anche dall’uso della chirurgia bariatrica metabolica: se non controindicata da cirrosi scompensata, dovrebbe essere considerata per il trattamento della NAFLD e migliorare la salute cardio-metabolica se BMI ≥ 35 kg/m2, in particolare in presenza di DM2 (in casi selezionati con mancata risposta alla terapia anti-diabetica anche se BMI ≥ 30 kg/m2) (10). Studi recenti hanno, infatti, dimostrato come in particolare la sleeve gastrectomy abbia effetti positivi a breve-medio termine su calo ponderale, IR/DM2 e sul quadro istologico di NASH e fibrosi (28-30). Uno studio recentemente pubblicato ha documentato come la chirurgia bariatrica nei pazienti con NASH riduca nel lungo periodo il rischio di complicanze cardio-vascolari ed epatiche rispetto ai trattamenti non chirurgici (31).
IMPATTO DELLA NUOVA DEFINIZIONE DI “METABOLIC (DYSFUNCTION) ASSOCIATED FATTY LIVER DISEASE” (MAFLD)
È stato recentemente proposto (e accettato da molte, anche se non tutte, le maggiori società scientifiche) di utilizzare invece di NAFLD il termine MAFLD (32). La diagnosi di MAFLD prevede, oltre alla steatosi epatica, associata o meno a infiammazione e fibrosi epatica, la presenza di almeno una caratteristica tra DM2 e sovrappeso/obesità, oppure, nei pazienti in normopeso non diabetici, almeno due caratteristiche tra le seguenti (33):
- circonferenza vita ≥ 102 cm negli uomini e ≥ 88 cm nelle donne;
- pre-diabete, ossia emoglobina glicata (HbA1c) nel range 5.7-6.4% o glicemia a digiuno nel range 5.6-6.9 mmol/L (100-124 mg/dL), o livelli di glicemia post-carico a due ore nel range 7.8-11.0 mmol/L (140-200 mg/dL);
- pressione arteriosa ≥ 130/85 mm Hg o essere in terapia anti-ipertensiva;
- colesterolo HDL < 1.0 mmol/L (40 mg/dL) per i maschi e < 1.3 mmol/L (50 mg/dL) per le femmine o essere in terapia ipocolesterolemizzante;
- trigliceridemia ≥ 1.7 mmol/L (150 mg/dL) o essere in specifico trattamento farmacologico;
- HOMA-IR ≥ 2.5;
- proteina C-reattiva ad alta sensibilità (hs-CRP) > 2 mg/L.
La nuova terminologia MAFLD prevede la possibilità che il paziente possa avere altre eziologie di malattia cronica di fegato (alcol, virus, auto-immunità, ecc) e in tal caso la diagnosi del paziente sarà MAFLD con eziologia duplice o multipla. La prevalenza globale della MAFLD è del 38.8%, con prevalenza globale di MAFLD nei soggetti magri ed in quelli non obesi, spesso affetti da DM2 e/o ipertensione arteriosa, rispettivamente, del 5.4% e 29.8% (34). L’utilità della nuova terminologia MAFLD non risiede solo in un nome che definisce la malattia in senso positivo (e non più in senso negativo come nel caso del vecchio termine NAFLD “non-alcoholic fatty liver disease”), ma è molto rilevante per altri aspetti clinici:
- pur essendo l’80-85% dei pazienti coperto da entrambe le definizioni, circa il 15% dei pazienti che ha una diagnosi di MAFLD non rientra nella diagnosi di NAFLD (MAFLD NON-NAFLD) e circa il 4% dei pazienti con diagnosi di NAFLD non rientra nella diagnosi di MAFLD (NAFLD NON-MAFLD) (35,36). Questi gruppi di pazienti sono differenti dal punto di vista prognostico per quanto riguarda la mortalità cardio-vascolare e da diversi tipi di tumore, con il gruppo NAFLD NON-MAFLD associato a prognosi migliore rispetto agli altri due gruppi (37,38);
- dati recenti dimostrerebbero come, all’interno dei pazienti con diagnosi di MAFLD, la prognosi sia peggiore in alcuni sottogruppi, come quello dei pazienti diabetici o dei pazienti in normopeso, rispetto a quelli in sovrappeso o obesi (39,40);
- poiché la definizione di MAFLD non prevede più la differenza dicotomica tra NASH e NAFL, cioè solo steatosi senza infiammazione e possibile fibrosi, nei pazienti con diagnosi di MAFLD bisogna valutare la severità (vedi capitolo precedente) e monitorare la progressione della fibrosi epatica in modo non-invasivo ma anche trattare i pazienti senza fibrosi in quanto anch’essi ad aumentato rischio di mortalità (8);
- i pazienti con diagnosi di MAFLD necessiterebbero di una gestione più integrata da parte di diversi specialisti (gastroenterologi, endocrinologi, cardiologi, nutrizionisti, psicoterapeuti, ecc) in una struttura strettamente integrata (MAFLD clinic) (41).
Ultimo vantaggio della nuova diagnosi di MAFLD risiede in una miglior capacità di stratificare i pazienti in sottogruppi, allo scopo di bilanciare il gruppo placebo con il gruppo di studio negli studi clinici, rendendo i risultati di questi ultimi più applicabili al singolo paziente in un’ottica di medicina di precisione (33).
BIBLIOGRAFIA
- Alberti A, Vario A, Ferrari A, Pistis R. Chronic hepatitis C - natural history and cofactors. Aliment Pharmacol Ther 2005, 22: 74–8.
- Gaggini M, Morelli M, Buzzigoli E, et al. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5: 1544–60.
- Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018, 283: 356–70.
- Nobili V, Alisi A, Valenti L, et al. NAFLD in children: new genes, new diagnostic modalities and new drugs. Nat Rev Gastroenterol Hepatol 2019, 16: 517–30.
- EASL–EASD–EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016, 64: 1388–402.
- Frith J, Day CP, Robinson L, et al. Potential strategies to improve uptake of exercise interventions in non-alcoholic fatty liver disease. J Hepatol 2010, 52: 112–6.
- Bedogni G, Miglioli L, Masutti F, et al. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 2005, 42: 44–52.
- Simon TG, Roelstraete B, Khalili H, et al. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: results from a nationwide cohort. Gut 2021, 70: 1375-82.
- Petersen KF, Dufour S, Befroy D, et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54: 603–8.
- Cusi K, Isaacs S, Barb D, et al. American Association of Clinical Endocrinology clinical practice guideline for the diagnosis and management of nonalcoholic fatty liver disease in primary care and endocrinology clinical settings: co-sponsored by the American Association for the Study of Liver Diseases (AASLD). Endocr Pract 2022, 28: 528-62.
- DiStefano JK, Gerhard GS. NAFLD in normal weight individuals. Diabetol Metab Syndr 2022,14: 45.
- Saab S, Mallam D, Cox GA, Tong MJ. Impact of coffee on liver diseases: a systematic review. Liver Int 2014, 34: 495–504.
- Van Wagner LB, Ning H, Allen NB, et al. Alcohol use and cardiovascular disease risk in patients with nonalcoholic fatty liver disease. Gastroenterology 2017, 153: 1260-72.e3.
- Dunn W, Sanyal AJ, Brunt EM, et al. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). J Hepatol 2012, 57: 384–91.
- Moriya A, Iwasaki Y, Ohguchi S, et al. Roles of alcohol consumption in fatty liver: a longitudinal study. J Hepatol 2015, 62: 921–7.
- Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J Hepatol 2012, 56: 255–66.
- Keating SE, Hackett DA, George J, Johnson NA. Exercise and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol 2012, 57: 157–66.
- Bacchi E, Negri C, Targher G, et al. Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 randomized trial). Hepatology 2013, 58: 1287–95.
- Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol 2005, 42: 132–8.
- Vilar-Gomez E, Vuppalanchi R, Desai AP, et al. Long-term metformin use may improve clinical outcomes in diabetic patients with non-alcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Aliment Pharmacol Ther 2019, 50: 317-28.
- Zhang Z-J, Zheng Z-J, Shi R, et al. Metformin for liver cancer prevention in patients with type 2 diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab 2012, 97: 2347–53.
- Boutari C, Pappas PD, Anastasilakis D, et al. Statins' efficacy in non-alcoholic fatty liver disease: a systematic review and meta- Clin Nutr 2022, 41: 2195-206.
- Leuschner UFH, Lindenthal B, Herrmann G, et al. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology 2010, 52: 472–9.
- Dufour J, Oneta CM, Gonvers J, et al. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin E in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2006, 4: 1537-43.
- Lindor KD, Kowdley KV, Heathcote EJ, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology 2004, 39: 770–8.
- Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145: 574-82.e1.
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015, 385: 956–65.
- Lassailly G, Caiazzo R, Buob D, et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology 2015, 149: 379–88.
- Caiazzo R, Lassailly G, Leteurtre E, et al. Roux-en-Y gastric bypass versus adjustable gastric banding to reduce nonalcoholic fatty liver disease. Ann Surg 2014, 260: 893–9.
- Schauer PR, Bhatt DL, Kirwan JP, et al. Bariatric surgery versus intensive medical therapy for diabetes — 3-year outcomes. N Engl J Med 2014, 370: 2002–13.
- Aminian A, Al-Kurd A, Wilson R, et al. Association of bariatric surgery with major adverse liver and cardiovascular outcomes in patients with biopsy-proven nonalcoholic steatohepatitis. JAMA 2021, 326: 2031.
- Eslam M, Sanyal AJ, George J, et al. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158: 1999-2014.e1.
- Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020, 73: 202–9.
- Chan KE, Koh TJL, Tang ASP, et al. Global prevalence and clinical characteristics of metabolic-associated fatty liver disease: a meta-analysis and systematic review of 10 739 607 individuals. J Clin Endocrinol Metab 2022, 107: 2691-700.
- Ayada I, van Kleef LA, Alferink LJM, et al. Systematically comparing epidemiological and clinical features of MAFLD and NAFLD by meta‐analysis: focusing on the non‐overlap groups. Liver Int 2022, 42: 277–87.
- Lim GEH, Tang A, Ng CH, et al. An observational data meta-analysis on the differences in prevalence and risk factors between MAFLD vs NAFLD. Clin Gastroenterol Hepatol 2021, DOI: 10.1016/j.k.2021.11.038.
- Kang SH, Cho Y, Jeong SW, et al. From nonalcoholic fatty liver disease to metabolic-associated fatty liver disease: big wave or ripple? Clin Mol Hepatol 2021, 27: 257–69.
- Kim D, Konyn P, Sandhu KK, et al. Metabolic dysfunction-associated fatty liver disease is associated with increased all-cause mortality in the United States. J Hepatol 2021, 75: 1284–91.
- Huang J, Ou W, Wang M, et al. MAFLD criteria guide the subtyping of patients with fatty liver disease. Risk Manag Healthc Policy 2021, 14: 491–501.
- Chen X, Chen S, Pang J, et al. Are the different MAFLD subtypes based on the inclusion criteria correlated with all-cause mortality? J Hepatol 2021, 75: 987–9.
- Angelico F, Baratta F, Pastori D, del Ben M. Assessment of hepatic fibrosis in MAFLD: a new player in the evaluation of residual cardiovascular risk? Digest Liver Dis 2021, 53: 383–4.
Tossicità epatica dei farmaci endocrini e loro uso nei pazienti con epatopatia
Flaminia Ferri & Stefano Corradini
Divisione di Gastroenterologia, Dipartimento di Medicina Traslazionale e di Precisione, Università "Sapienza" di Roma
(aggiornato al 15 dicembre 2022)
INTRODUZIONE
L’utilizzo di alcuni dei farmaci endocrini nei pazienti con insufficienza epatica può essere normato dalla raccomandazione di non utilizzo per il rischio di tossicità epatica oppure di utilizzo a dosaggio ridotto per il rischio di accumulo del farmaco quando metabolizzato a livello epato/biliare. Di seguito vengono riportati per i vari farmaci la probabilità di provocare un danno epatico e, per quelli in cui il dato è disponibile nel sito https://www.drugs.com/disease-interactions/ (ultimo accesso 28 novembre 2022), l’eventuale raccomandazione di non utilizzo e/o utilizzo a dosaggio ridotto in caso di insufficienza epatica.
Il danno epatico indotto da farmaci (Drug Induced Liver Injury, DILI) può presentarsi in maniera estremamente eterogenea (tipo epato-cellulare o colestatico o misto). Sia nella gestione clinica che nella ricerca scientifica il danno epatico è definito in presenza di una delle seguenti opzioni (1):
- aumento di bilirubina totale > 2 volte il limite superiore del valore di normalità (ULN), associato a incremento di ALT ≥ 3 volte ULN;
- ALT ≥ 5 volte ULN;
- fosfatasi alcalina (ALP) ≥ 2 volte ULN.
Una volta definita la presenza del danno epatico, questo può essere classificato secondo il seguente valore di R, ottenuto dal rapporto tra ALT x ULN/ALP x ULN, in danno (1):
- epato-cellulare, se R è ≥ 5;
- misto, se R è compreso tra 2 e 5;
- colestatico, se R è ≤ 2.
A seconda del meccanismo patogenetico alla base del danno da farmaci, si può riconoscere un danno di tipo intrinseco e un danno di tipo idiosincrasico (1):
- la prima tipologia (danno intrinseco) si verifica in un numero elevato di individui che hanno assunto il farmaco, è dose/dipendente e l’insorgenza del danno è spesso ravvicinata all’assunzione del farmaco (1);
- il danno idiosincrasico si verifica solo in una piccola percentuale di individui, non è prevedibile, mostra un tempo di latenza più lungo e non è correlato alla dose. Si sospetta che questa tipologia di danno sia legata alla suscettibilità genetica dei singoli individui o di alcune popolazioni (1).
La probabilità che un farmaco possa causare un danno epatico è espressa dal likelihood score (2), riportato nel sito web livertox.nih.gov, e così classificato:
- A: farmaco per cui è ben nota (> 50 casi descritti) l’epato-tossicità su base intrinseca o idiosincrasica, con caratteristiche cliniche ben note;
- B: farmaco per cui sono già presenti tra 12 e 50 segnalazioni riguardanti la sua epato-tossicità o questa appare altamente probabile, anche qui con caratteristiche cliniche ben definite;
- C: farmaco che può indurre un danno epatico idiosincrasico probabile, con caratteristiche cliniche del danno non ben definite;
- D: farmaco per cui si sono verificate alcune segnalazioni (< 3) senza caratteristiche cliniche ben definite (epato-tossicità possibile ma rara);
- E: farmaco che è ampiamente utilizzato, ma per cui non è emerso un chiaro collegamento con il danno epatico, per cui si ritiene improbabile che il farmaco possa provocare la DILI;
- E*: il farmaco è una sospetta causa di danno epatico idiosincrasico, ma non ci sono sufficienti prove a riguardo;
- X: farmaci introdotti recentemente o usati raramente nella pratica clinica, per cui non si conoscono i rischi di sviluppare danno epatico.
SISTEMATICA
Acarbosio (2,3): la terapia con acarbosio è stata collegata a rari casi di danno epatico idiosincrasico, verificatosi da 2 a 8 mesi dopo l'inizio del trattamento. Generalmente il danno epatico acuto è moderato e la sospensione della terapia porta a risoluzione completa. Il danno epatico acuto indotto dall’acarbosio presenta caratteristiche simili anche nei pazienti con patologia epatica pre-esistente. In caso di pregressa epatite acuta indotta dal farmaco, la re-introduzione anche a distanza deve essere evitata, per l’elevato rischio di recidiva. Likelihood score: B.
Alirocumab (2,4): gli anticorpi monoclonali contro PCSK9 non sono stati associati né a ipertransaminasemia né ad iperbilirubinemia. Likelihood score: E.
Analoghi delle prostacicline (2,5): non sono stati associati a ipertransaminasemia e/o epatite acuta. Likelihood score: E.
Anastrozolo (2,6,7): il danno epatico correlato si manifesta con aumento transitorio e asintomatico delle transaminasi tra la prima e la quarta settimana dall’inizio della terapia, è generalmente lieve e auto-limitante. Sono stati segnalati casi di epatite acuta severa, con alterazione anche della funzione coagulativa. La risoluzione solitamente avviene rapidamente dopo la sospensione del farmaco. Il farmaco non è stato associato a casi di insufficienza epatica acuta o cronica, patologie delle vie biliari o sviluppo di steatosi epatica. Non sono necessari aggiustamenti del dosaggio di anastrozolo nei pazienti con compromissione epatica da lieve a moderata, ma il farmaco deve essere somministrato con cautela nei pazienti con grave compromissione epatica. Likelihood score: C.
Bisfosfonati (2,5,8-12): in letteratura vengono riportati casi di incremento delle transaminasi con comparsa di ittero, legati ai bisfosfonati più comunemente usati, quali zoledronato, alendronato, risedronato e ibandronato. Il danno epatico è di origine idiosincrasica ed è associato anche alla comparsa concomitante di nausea e dolori addominali. L’insorgenza della sintomatologia viene descritta solitamente nei primi 2-6 mesi dall’inizio del trattamento. Nella maggior parte dei casi descritti il quadro si presentava con gravità lieve-moderata, che andava incontro a risoluzione completa, seppur non sempre tempestiva, in seguito alla sospensione del farmaco. Sono stati descritti inoltre casi di aumento transitorio degli enzimi epatici in assenza di ittero, ma in associazione con lievi reazioni di ipersensibilità in caso di somministrazione di bisfosfonati per infusione endovenosa. Tale quadro è spesso associato alla dose iniziale e può attenuarsi o risolversi con le successive somministrazioni o tramite la pre-medicazione con glucocorticoidi o anti-istaminici. Non sono mai stati descritti casi di insufficienza epatica acuta o cronica legati all’uso di bisfosfonati, né sono stati descritti peggioramenti della funzione epatica nei pazienti con patologia epatica pre-esistente. In termini di rischio di induzione del danno epatico:
- alendronato e acido zoledronico rappresentano una causa rara ma probabile di danno epatico clinicamente evidente (likelihood score C);
- ibandronato e risedronato rappresentano una causa rara ma possibile di danno epatico clinicamente evidente (likelihood score D);
- etidronato e pamidronato sono considerati una causa rara non dimostrata ma sospetta di danno epatico clinicamente evidente (likelihood score E*).
Non sono disponibili dati sul riutilizzo di questa classe di farmaci in pazienti con precedente danno epatico indotto dai bisfosfonati.
Bromocriptina (2,5): può provocare aumenti lieve, asintomatici e auto-limitanti delle transaminasi. Raramente si verificano aumenti marcati, che necessitano di riduzione della dose o sospensione del farmaco. Non sono stati mai descritti casi di insufficienza epatica acuta o epatite cronica legate a questo farmaco. Likelihood score: D.
Chemioterapici e immuno-soppressori (2,13,14): i pazienti sottoposti a terapia immuno-soppressiva o chemioterapia, inclusi i farmaci biologici di nuova generazione, possono andare in contro a riattivazione dell’HBV (il rischio è definito basso se < 1%, moderato se compreso tra 1% e 10%, alto se > 10%). Si consiglia di effettuare preliminarmente i test HBsAg, anti-HBs Ab, anti-HBc Ab:
- tutti i pazienti che risultano HBsAg positivi devono effettuare la terapia anti-virale come trattamento (HBV-DNA positivi) o come profilassi (HBV-DNA negativi);
- in caso di HBsAg negatività e HBcAb positività, è necessario effettuare il test dell’HBV-DNA: se positivo, i pazienti devono essere trattati; se negativo, la terapia anti-virale è consigliata come profilassi solo nei soggetti con rischio elevato. Nei pazienti con rischio moderato e basso si effettua il monitoraggio di HBsAg e HBV-DNA ogni 1-3 mesi, sia durante che 12-18 mesi dopo la sospensione della terapia, e viene iniziata la terapia anti-virale immediatamente se si verifica la sieroconversione dell’HBsAg o si positivizza l’HBV-DNA. Nei pazienti HBcAb positivi, HBsAg/HBV-DNA negativi a rischio basso/moderato, si può consigliare comunque di iniziare la terapia anti-virale nel caso in cui sia necessaria una terapia di lunga durata, non sia possibile effettuare un adeguato monitoraggio oppure non sia prevedibile il rischio di riattivazione;
- nei pazienti sieronegativi è consigliata la vaccinazione contro l’HBV.
I pazienti che necessitano di terapia immuno-soppressiva o chemioterapia devono essere sottoposti anche al test dell’HCV-Ab; in caso di positività deve essere effettuato il dosaggio dell’HCV-RNA. Il trattamento con anti-virali è obbligatorio nei casi di HCV-RNA positivo. Il rischio di riattivazione dell’HCV è più basso rispetto all’HBV ma comunque presente, per cui in caso di negatività dell’HCV-RNA, si consiglia di effettuare il monitoraggio senza terapia di profilassi.
Per altre tematiche relative all’epato-tossicità non correlata all’HBV o HCV positività, si rimanda ai capitoli relativi ai singoli farmaci.
Ciproterone (2,5,15): in circa il 10-14% dei pazienti il ciproterone provoca un danno epato-cellulare associato talvolta ad aumento di ALP, che risulta lieve e transitorio e si risolve spontaneamente. La sintomatologia insorge nel primo semestre di trattamento. Talvolta è stato associato anche a casi di danno epatico clinicamente evidente con ittero. In questi casi si può evidenziare un danno grave associato a mortalità del 10%. Si consiglia quindi di interrompere il farmaco ai primi segni di danno epatico ed è sconsigliata la re-introduzione anche in caso di miglioramento. È stata segnalata anche la sensibilità crociata al danno epatico tra ciproterone e altri anti-androgeni. Si segnala inoltre che la terapia cronica ad alte dosi è associata anche a insorgenza di epato-carcinoma su fegato non cirrotico. Likelihood score: B.
Clomifene (2,16): le informazioni riguardanti l’effetto sui livelli di transaminasi sono scarse, anche perché solitamente il farmaco è somministrato in basse dosi e per un breve periodo. Più genericamente i farmaci utilizzati per il trattamento dell’infertilità possono provocare la sindrome da iperstimolazione ovarica, che può manifestarsi con dolore addominale e ascite, con contestuale elevazione lieve-moderata delle transaminasi e minima elevazione di bilirubina e ALP. Nel caso in cui si osservi un aumento delle transaminasi > 5 volte ULN, è consigliato ridurre la dose o sospendere il farmaco. L’uso del clomifene non è stato associato allo sviluppo di insufficienza epatica acuta o cronica. Il suo uso è controindicato nei pazienti con malattia epatica o anamnesi di disfunzione epatica. Likelihood score: C.
Colestiramina (2): ci sono scarse evidenze riguardo al collegamento con il danno epatico. Likelihood score: E.
Glucocorticoidi (2,17-19): a livello epatico il danno si evidenzia soprattutto in relazione alla terapia a lungo termine ed è un effetto dose-dipendente. I glucocorticoidi possono provocare l’insorgenza di NAFLD de novo o il peggioramento della NAFLD sottostante. Tale effetto collaterale è legato principalmente all’effetto diretto di tali farmaci sull’insulino-resistenza e sul metabolismo degli acidi grassi, ma è anche legato all’aumento ponderale che si osserva nel corso delle terapie corticosteroidee a lungo termine. Nei pazienti HBV+ e HCV+, l’epatopatia sottostante viene peggiorata dalla terapia cortisonica. In particolare, nei pazienti affetti da epatite cronica B i corticosteroidi possono indurre aumenti della replicazione virale, con contestuale riduzione delle transaminasi. Quando i livelli di corticosteroidi vengono riportati ai livelli fisiologici o il farmaco viene sospeso, portando alla normalizzazione delle difese immunitarie, si osserva un marcato incremento dei livelli di transaminasi (> 10-20 volte ULN). Tale condizione clinica può evolvere verso l’insufficienza epatica acuta su cronica o verso un’evoluzione più rapida dell’epatite cronica in cirrosi epatica. Per la gestione si consiglia di vedere il paragrafo riguardante i farmaci “Chemioterapici e Immunosoppressori”. In letteratura sono presenti anche casi di epatite acuta, con evoluzione talvolta in epatite severa o fulminante, comparsa successivamente a un ciclo breve ma ad alte dosi di metilprednisolone per via endovenosa, in cui non sono stati implicati virus epatotropi. Tali episodi si sono manifestati con l’insorgenza di ittero e aumento degli enzimi, espressione di danno epato-cellulare, dopo 1-6 settimane dalla sospensione della terapia con metilprednisolone. L’eziologia del danno epatocitario non è nota, ma potrebbe essere legata allo sviluppo di una grave epatite autoimmune innescata dalla terapia immuno-soppressiva e successiva normalizzazione delle difese immunitarie. In tale contesto, potrebbe essere appropriato riprendere la terapia con corticosteroidi, ma non ci sono sufficienti dati al riguardo. Non è chiaro se questo quadro clinico possa presentarsi anche con l’uso di alte dosi di prednisone o desametasone per via endovenosa. I pazienti con cirrosi devono essere monitorati più attentamente per gli effetti eccessivi degli steroidi. In questi pazienti possono essere necessari aggiustamenti del dosaggio. Likelihood score: A.
Denosumab (2): non ci sono dati che dimostrino che il farmaco sia intrinsecamente epato-tossico. Likelihood score: E*.
Dulaglutide (2): non ci sono dati in letteratura che ne documentino un’azione epato-tossica.
Estrogeni e contraccettivi orali (2,5,20-25): le prime formulazioni di contraccettivi orali sono state molto frequentemente associate a danno epato-colangiocitario, ma tale frequenza non è stata confermata con le nuove terapie anti-concezionali o con la terapia sostitutiva. La frequenza di danno epatocitario aumenta quando gli estrogeni sono associati ai progestinici. Estrogeni e contraccettivi orali posso provocare l’insorgenza di ittero, a causa della modesta inibizione dell'escrezione della bilirubina dall’epatocita in pazienti con patologie ereditarie che ne alterano il metabolismo (ad es. s. di Dubin-Johnson). Sono stati descritti casi in cui gli estrogeni e i contraccettivi orali nel corso dei primi 6 mesi di terapia possono indurre un danno misto o colestatico con ittero, astenia e prurito. Tale sintomatologia insorge tipicamente nei primi cicli di terapia. Le pazienti che sviluppano questi quadri hanno spesso una storia di colestasi gravidica, dato che induce a pensare che l’eziologia possa essere su base genetica. Il danno colestatico si risolve rapidamente con la sospensione della terapia, ma nelle pazienti sintomatiche può essere consigliato l’utilizzo di acido ursodesossicolico (12-15 mg/kg/die) fino alla risoluzione dei sintomi o alla risposta biochimica. L’utilizzo degli estrogeni e dei contraccettivi orali è stato associato all’insorgenza di tumori epatici, sia benigni (adenomi, angiomi, iperplasia nodulare focale, amartomi) che maligni. Il rischio di sviluppare un adenoma epatico nella popolazione che fa uso di contraccettivi orali è di circa lo 0.5%/anno, con potenziale evolutivo verso l’epato-carcinoma. La sospensione della terapia ormonale può portare talvolta a regressione dell’adenoma, in altri casi è necessario ricorrere al trattamento chirurgico. I contraccettivi orali sono stati anche associati ad aumentata incidenza delle trombosi venose e quindi anche a trombosi portale e/o delle vene sovra-epatiche (s. di Budd-Chiari). Tali quadri si evidenziano in particolar modo nei pazienti che presentano un quadro di trombofilia, ad es. carenza di proteina C o proteina S o mutazioni del fattore V di Leiden. L'uso di contraccettivi orali è stato associato anche a rari casi di peliosi epatica. Anche le patologie delle colecisti risultano più frequenti nelle donne che fanno uso di contraccettivi orali o della terapia ormonale sostitutiva. La colestasi associata ai contraccettivi contenenti estrogeni (OCC) è tipicamente lieve e si risolve rapidamente con l'interruzione. Alcuni casi, tuttavia, sono protratti e associati a prurito grave, con o senza ittero marcato. L'efficacia dell'ursodiolo nel trattamento della colestasi della gravidanza rende questo approccio appropriato nelle donne che sviluppano colestasi a causa di OCC, i cui sintomi sono problematici; il dosaggio è compreso tra 12 e 15 mg/kg/die e deve essere continuato fino alla risoluzione dei sintomi e delle principali anomalie degli esami biochimici. Non devono essere usati corticosteroidi. È tipica la recidiva di colestasi con la ripresa degli OCC, sebbene si possano trovare formulazioni a dose più bassa che non attivano la risposta. La gestione degli adenomi epatici correlati agli OCC è complessa. In molti casi, la semplice sospensione dei contraccettivi orali è seguita da regressione delle dimensioni del tumore, ma può essere necessario un intervento chirurgico per tumori più grandi e quelli in cui si ritiene che la trasformazione maligna rappresenti un rischio. La terapia con estrogeni deve essere somministrata con cautela nei pazienti con malattia epatica. Likelihood score: A.
Everolimus (2,26,27): in circa un quinto dei pazienti che lo assumono si verifica un aumento lieve, transitorio e auto-limitante delle transaminasi e in meno del 2% l’aumento risulta > 5 volte ULN. Non sono stati descritti casi di danno epatico clinicamente rilevante, ma questa terapia, come le altre immuno-soppressive e oncologiche, può portare a riacutizzatizione dell’epatite B (cfr. paragrafo “Chemioterapici e Immunosoppressori”). Nei pazienti con compromissione epatica lieve o moderata, si raccomanda la riduzione della dose. Nei pazienti con compromissione epatica grave, everolimus può essere utilizzato a dose ridotta se il beneficio desiderato supera il rischio. Likelihood score: E*.
Evolocumab (2): ad oggi non ci sono dati che suggeriscano che questa terapia possa provocare un danno epato-biliare. Likelihood score: E.
Exenatide (2): il farmaco non è stato associato a danno epato-biliare. È stato invece associato, seppur raramente, all’insorgenza di pancreatite acuta.
Ezetimibe (2, 28): se non è somministrata in combinazione con statine, l’elevazione di AST, ALT, gamma-GT e ALP si verifica tra il secondo e il decimo mese dall’inizio del trattamento in circa l’1% dei pazienti, è di grado lieve e auto-limitante e solitamente non si associa alla comparsa di ittero. Seppur rari, sono stati riportati casi di danno epatico acuto correlato all’uso di ezetimibe: nella maggior parte dei casi è auto-limitante ma in alcuni casi è stato associato a epatopatie e biliopatia cronica autoimmune o a insufficienza epatica acuta. La terapia con ezetimibe non è raccomandata nei pazienti con insufficienza epatica moderata o grave. Likelihood score: C.
Fibrati (2,5,29-32): l’uso di tutti i fibrati, in particolare fenofibrato, può essere associato a danno epatico, che può presentarsi come lieve-moderato fino a danno epatico acuto clinicamente evidente. Nella maggior parte dei casi si tratta comunque di incrementi transitori, asintomatici, per cui può non essere necessaria l’interruzione della terapia. La terapia con fenofibrato e con gemfibrozil deve essere somministrata con cautela nei pazienti con malattia epatica attiva. La terapia con clofibrato può essere considerata a dosaggio ridotto.
- Clofibrato: può provocare un aumento lieve e transitorio delle transaminasi e/o degli indici di colestasi, che si instaura più frequentemente dopo 2-3 mesi dall’inizio del trattamento. Tali alterazioni si risolvono spesso senza la necessità di sospendere il farmaco. L’uso del clofibrato non è stato associato allo sviluppo di insufficienza epatica acuta o cronica. Likelihood score: D.
- Fenofibrato: in circa un quinto dei pazienti che lo assumono si verifica un aumento lieve e transitorio delle transaminasi e in meno del 5% l’aumento risulta > 3 ULN. Solo occasionalmente è necessario interrompere la terapia per ottenere una normalizzazione dei valori; è indicato, quindi, effettuare il monitoraggio dei valori di transaminasi nei pazienti che assumono tali terapie e si raccomanda l’interruzione del farmaco solo nei casi in cui tali valori superino di 3 volte l’ULN. Sono stati anche descritti casi di danno epatico clinicamente evidente, quasi sempre di tipo epato-cellulare e solo raramente di tipo colestatico o misto. Il quadro di danno acuto insorge solitamente nei primi mesi di terapia. Un quadro di danno cronico anche severo può comparire dopo mesi o talvolta anni nei pazienti che non hanno interrotto la terapia nonostante la presenza di danno epatico persistente e si presenta tipicamente con caratteristiche autoimmuni (iperglobulinemia associata a positività degli ANA o gli ASMA e/o “Vanishing Bile Buct Syndrome”). Nei casi in cui sia stato necessario sospendere il farmaco, non si raccomanda il re-inserimento, neanche a distanza di tempo. L’utilizzo del cortisone in tale contesto ha efficacia dubbia. Likelihood score: B.
- Gemfibrozil: in circa un quinto dei pazienti che lo assumono si può avere un aumento lieve e transitorio delle transaminasi ma in meno del 5% l’incremento è > 3 ULN. Si tratta spesso di aumenti asintomatici e transitori che non necessitano dell’interruzione del farmaco. Raramente si è osservato un danno epatico clinicamente evidente, la cui risoluzione comunque si verifica rapidamente dopo la sospensione. L’uso del gemfibrozil non è stato mai associato allo sviluppo di insufficienza epatica acuta o cronica. Likelihood score: C.
Finasteride (2,33): nel corso degli studi controllati la finasteride, sia 1 mg/die che 5 mg/die, è stata associata a un basso tasso di aumento delle transaminasi. Tali aumenti si sono dimostrati transitori e solo in casi sporadici hanno richiesto la modifica della posologia. La terapia deve essere somministrata con cautela nei pazienti con malattia epatica, in quanto l'esposizione potrebbe essere maggiore. Likelihood score: E.
Flutamide (2,34): in più del 60% dei pazienti che la assumono si verifica un aumento asintomatico e transitorio delle transaminasi. Solo nel 3-5% dei pazienti l’aumento risulta > 5 ULN e nello 0.1-1% si verifica un danno epatico acuto sintomatico, che può evolvere verso l’insufficienza epatica acuta. Il danno è solitamente di tipo epato-cellulare, è dose-dipendente e insorge mediamente dopo tre mesi dall’inizio dell’assunzione, ma talvolta può presentarsi più precocemente o più tardivamente. Non sono descritti casi di danno cronico. Si raccomanda, quindi, il monitoraggio attento e regolare dei livelli di ALT, soprattutto nei primi sei mesi di terapia. In caso di insorgenza del danno, il farmaco deve essere sospeso e dopo 1-2 mesi si inizia a osservare miglioramento. Non è raccomandata la re-introduzione del farmaco, in considerazione dell’elevato rischio di recidiva del danno. La flutamide deve essere somministrata con estrema cautela nei pazienti con ipertransaminasemia o con funzionalità epatica compromessa ed è controindicata nei pazienti con grave compromissione epatica. Likelihood score: A.
Gonadotropine (2): la somministrazione non è associata a epato-tossicità, ma il danno epatico può essere indotto dalla comparsa della sindrome da iperstimolazione ovarica. Likelihood score: E.
Goserelin (2,35): aumenti isolati e transitori delle transaminasi si verificano in meno del 5% dei casi e spesso non richiedono né l’interruzione del farmaco né l’aggiustamento della dose. Il danno epatico clinicamente evidente è un evento raro. Likelihood score: D.
Inibitori delle dipeptidil peptidasi-4 (2,36-40): il danno epatico è raro e l’eziopatogenesi non è nota. Il danno epato-cellulare si evidenzia tra la 2° e la 12° settimana con l’aumento delle transaminasi e regredisce rapidamente dopo la sospensione. Raramente si può verificare danno epatico acuto clinicamente evidente. Non è stata riportata (ma non si può escludere) reattività crociata tra le gliptine. Per il sitagliptin è stato descritto un caso di danno epatico clinicamente evidente in un soggetto HCV+. Linagliptin e sitagliptin: Likelihood score D; alogliptin e saxagliptin: Likelihood score E*.
Inibitori delle fosfodiesterasi tipo 5 (2,5,37)
- Sildenafil: in letteratura sono presenti almeno 5 casi di danno epatico acuto di tipo epato-cellulare, colestatico o misto, in cui il danno si è presentato tra il I e il III mese di terapia, senza evoluzione in insufficienza epatica. Il meccanismo non è chiaro, potrebbe essere tossico oppure ischemico, ma rappresenta comunque un evento raro. Likelihood score: C.
- Tadalafil: è stato collegato in rari casi a sviluppo di lieve ipertransaminasemia o a epatite colestatica insorta nei primi giorni dall’inizio del trattamento, in nessun caso con evoluzione verso l’insufficienza epatica. Non sono stati descritti casi di danno cronico epato-cellulare o biliare. Likelihood score: D.
- Vardenafil: non è associato a casi di danno epatico clinicamente evidente. Likelihood score: E*.
Inibitori del cotrasportatore sodio-glucosio tipo 2 (SGLT-2) (2,41,42): questa terapia è stata raramente associata a danno epatico, nella maggior parte dei casi si sono osservati invece miglioramenti nei livelli di ALT, legati probabilmente al miglior controllo glicemico con conseguente miglioramento della condizione di steatosi o steato-epatite spesso associata. Ertugliflozin e canagliflozin non sono raccomandati per l'uso in pazienti con compromissione epatica grave a causa della mancanza di dati clinici. Dapagliflozin non è stato studiato in pazienti con compromissione epatica grave e si raccomanda cautela. Non sono necessari aggiustamenti del dosaggio nei pazienti con compromissione epatica lieve o moderata. Dapagliflozin: Likelihood score D; canagliflozin, empagliflozin, ertugliflozin: Likelihood score E*.
Insulina (2,5,43,44): se somministrata a dosi elevate o intermittenti nei bambini e giovani adulti affetti da diabete mellito di tipo 1 scarsamente controllato o in soggetti con scompenso glicemico legato all’uso dei corticosteroidi, può portare all’insorgenza di epatopatia glicogenica, che si presenta clinicamente con epatomegalia, dolore addominale e aumenti delle transaminasi fino a 30 volte ULN. Il danno epatico migliora dopo aver ottenuto un buon controllo glicemico, ma il quadro può recidivare in caso di ripetuti episodi di scompenso glicemico. Nelle forme gravi si può presentare la sindrome di Mauriac. La patologia non provoca un danno epatico cronico. Non ci sono controindicazioni all’uso nelle patologie epatiche croniche e nella cirrosi epatica, mantenendo sempre un adeguato monitoraggio. Likelihood score: A.
Interferone (2,45): gli aumenti degli enzimi sierici durante la terapia sono generalmente auto-limitanti e benigni. Sono più frequenti nei pazienti con meno di un anno di età, in cui si consiglia il monitoraggio mensile in corso di terapia. Nei casi in cui l’elevazione dovesse superare di 5 volte l’ULN, è consigliata la riduzione della dose o la sospensione temporanea. Likelihood score: E.
Ketoconazolo (2,5,44,46): fino a un quinto dei pazienti può presentare incrementi lievi e transitori delle transaminasi. Il danno epato-tossico clinicamente evidente, con possibile evoluzione verso l’insufficienza epatica acuta, si verifica invece in un numero compreso tra 1: 2000 e 1:15000 e compare tra il 1° e il 6° mese dall’inizio del trattamento e si risolve dopo 1-3 mesi dalla sospensione. Il rischio di recidiva è elevato, per cui deve essere evitato il re-inserimento della terapia. È descritta reattività crociata con altri farmaci della stessa classe, ma è scarsa; si raccomanda comunque estrema cautela nei soggetti che hanno presentato un danno clinicamente rilevante. Nei soggetti con epatite cronica o cirrosi l’uso del ketoconazolo dovrebbe essere evitato. Likelihood score: A.
Lanreotide (2): non sono segnalate variazioni significative dei livelli di AST, ALT, gamma-GT o ALP, né in acuto né in cronico. Negli studi clinici è stata osservata ridotta clearance di lanreotide nei soggetti con compromissione epatica da moderata a grave, in cui il trattamento deve essere iniziato alla dose di 60 mg. Si deve usare cautela quando si usa questo agente in pazienti con insufficienza epatica moderata o grave per un intervallo di somministrazione prolungato. Come per gli altri analoghi della somatostatina, anche l’uso di lanreotide aumenta l’incidenza di calcoli della colecisti, probabilmente legata a riduzione della contrattilità della colecisti, come conseguenza dell’inibizione della secrezione di colecistochinina. L’uso prolungato si associa a colelitiasi in circa un terzo dei pazienti. Come si osserva nei casi di colelitiasi ad altra eziologia, anche in questo caso la maggior parte dei pazienti non manifesta sintomi e solo in una piccola percentuali di casi si verifica una colecistite sintomatica. Likelihood score: E*.
Leuprolide (35): è stata associata a lievi aumenti delle transaminasi nel 3-5% dei casi, ma in meno dell’1% l’aumento è risultato ≥ 3 volte l’ULN. Tali incrementi sono solitamente transitori e possono risolversi senza necessità di sospensione. Il farmaco non è mai stato associato a danno epatico clinicamente evidente. Ci sono dati a supporto di una sensibilità crociata al danno epatico tra gli analoghi del GnRH. Likelihood score: E.
Liraglutide (2): il farmaco non è associato ad epato-tossicità.
Metformina (2,5,37,44): lieve ipertransaminasemia in meno dell’1% dei pazienti in trattamento, mentre al contrario è frequente il miglioramento dei livelli di transaminasi nei soggetti con steatosi. Un danno epatico clinicamente evidente, di tipo epato-cellulare, colestatico o misto, è un evento estremamente raro: sono descritti poco più di una decina di casi in letteratura, nonostante l’ampia diffusione del farmaco, nessuno evoluto verso un quadro di epatite fulminante. La risoluzione del danno è rapida dopo l'interruzione e non è stata descritta reattività crociata per il danno epatico con altri farmaci anti-diabetici. Può essere utilizzata in sicurezza nei pazienti con epatite cronica o cirrosi compensata e si è mostrata particolarmente efficace nel trattamento dei pazienti con NAFLD associata a diabete mellito. È sconsigliato l’utilizzo nei pazienti con epatopatia associata a disturbo da uso di alcol o nei pazienti con cirrosi epatica scompensata, a causa dell’elevato rischio di insorgenza di acidosi lattica. Likelihood score: B.
Metimazolo (2,5,37,47): è una causa ben nota di danno epatico. Si tratta spesso di aumenti transitori e non significativi, che si verificano nei primi tre mesi dall’inizio della terapia ad alte dosi. Il farmaco è in grado però di provocare un danno epatico clinicamente evidente e sintomatico anche su base idiosincrasica: in questi casi il danno si verifica tra la 2° e la 12° settimana di terapia e si presenta con un quadro tipicamente colestatico prolungato, associato alla presenza di ittero per almeno 2-8 settimane. Il fattore confondente in questo contesto è il fatto che l’ipertiroidismo di per sè si può presentare con quadro clinico analogo. Il danno da metimazolo non evolve verso un quadro istologico di “Vanishing Bile Buct Syndrome”. La risoluzione del quadro clinico è solitamente rapida dopo la sospensione del farmaco, che deve avvenire immediatamente dopo il riscontro della malattia epatica. È indicato il passaggio a propiltiouracile, ma nei casi più severi può essere più appropriata la terapia chirurgica o con radioiodio. Likelihood score: A.
Mifepristone: l’impiego, in associazione al misoprostolo, legato all’aborto farmacologico non è associato a danno epatico, diversamente da quando invece è utilizzato in monoterapia a lungo termine con dosi più elevate. In tale contesto si può frequentemente osservare un aumento degli enzimi epatici, in particolare degli indici di colestasi, talvolta associato ad astenia, ittero e prurito. In questi casi è necessario interrompere definitivamente la terapia. Esistono informazioni limitate sulla sicurezza del mifepristone nei pazienti con compromissione epatica da lieve a moderata; pertanto, i produttori raccomandano che la dose massima non superi i 600 mg/die. L'uso del mifepristone non è raccomandato nei pazienti con grave malattia epatica, poiché la farmaco-cinetica in questi pazienti non è stata studiata. Likelihood score: D.
Mitotane (2): mentre è molto frequente un aumento lieve delle transaminasi (> 50% dei pazienti), un aumento > 5 volte l’ULN si verifica in meno dell’1% dei casi, mentre non sono descritti casi di sviluppo di danno epatico clinicamente evidente, ma è necessario considerare il suo impiego clinico limitato. Likelihood score: E*.
Octreotide (2,5): in una piccola percentuale di pazienti si verificano aumenti lievi, transitori e asintomatici dei livelli di transaminasi e degli indici di colestasi. In letteratura sono stati descritti diversi casi di danno epatico acuto, con esordio nei primi 6 mesi dall’inizio della terapia e rapida risoluzione dopo l’interruzione. La re-introduzione è sconsigliata, per l’elevato rischio di recidiva del danno anche in forma più severa: in caso di ripresa del farmaco, il monitoraggio deve essere molto attento. All’uso del farmaco non sono stati però associati casi di insufficienza epatica acuta o di “Vanishing Bile Buct Syndrome”. Nei pazienti con cirrosi epatica l'emivita del farmaco può essere aumentata, rendendo necessario l’aggiustamento del dosaggio, da titolare in base alla risposta clinica e alla velocità di risposta. È necessario inoltre segnalare che l'octreotide riduce la contrattilità della colecisti, che nei pazienti in terapia a lungo termine può provocare un aumento dell’incidenza della formazione dei calcoli biliari. Il trattamento profilattico con acido ursodesossicolico non sembra utile. Likelihood score: C.
Omega polienolici (2): possono provocare moderati aumenti delle ALT, ma non danno epatico clinicamente evidente. Si consiglia un monitoraggio più stretto nei pazienti con danno epatico pre-esistente. Likelihood score: E.
Orlistat (2,49,50): in letteratura diversi casi associano l’uso del farmaco a danno epatico epato-cellulare, con possibile evoluzione verso l’insufficienza epatica acuta e l’epatite fulminante. La causa del danno epatico non è nota, in quanto l’orlistat ha un’azione diretta sulla lipasi pancreatica e gastrica e solo l’1-3% viene assorbito. Pertanto, tali effetti non erano attesi né sono stati descritti da ampi studi clinici. L’ipotesi è che il danno epatico dipenda da un meccanismo di ipersensibilità. Likelihood score: C.
Ormoni tiroidei (2,5,51): le informazioni riguardo all’ipertransaminasemia con l’uso di ormoni tiroidei sono scarse, ma nonostante siano farmaci ampiamente prescritti non sono associati ad aumento degli enzimi epatici. Solo alte dosi di levotiroxina e altri preparati per la tiroide sono stati associati a danno epatico di entità lieve-moderata, con aspetto epato-cellulare o misto; in caso di danno epatico sottostante, possono rappresentare la causa del peggioramento della funzione epatica. Sono stati inoltre descritti rari casi di epatite su base autoimmune senza comparsa di auto-anticorpi ma caratterizzati da eosinofilia. I casi di danno epatico legati a levotiroxina e estratti tiroidei sono stati tutti segnalati in pazienti asiatici, suggerendo una possibile predisposizione genetica. In tale contesto il danno si risolve dopo la sospensione della terapia e la re-introduzione non è consigliata in considerazione dell’elevato numero di recidive. Likelihood score: C.
Pasireotide (2,52): in circa un terzo dei pazienti si verificano aumenti lievi, transitori e asintomatici degli enzimi epatici. L’uso di pasireotide non è stato associato a danno epatico clinicamente evidente. La molecola provoca l’inibizione della contrattilità della colecisti, con conseguente aumento dell’incidenza di calcoli di colesterolo nei soggetti trattati (20-30%); talvolta i pazienti hanno sviluppato sintomi che hanno richiesto il ricovero e il trattamento endoscopico o chirurgico. In questi pazienti è stata descritta la formazione di calcoli anche dopo la colecistectomia, sia nella via biliare principale che nelle vie biliari intra-epatiche. Il trattamento profilattico con acido ursodessossicolico non sembra utile. Likelihood score: E*.
Pioglitazone (2,5,37,53,54): non è associato ad aumento di AST e ALT. Nonostante la diffusione su larga scala del farmaco, in letteratura sono descritti poco più di una dozzina di casi di danno epatico clinicamente evidente, che esordisce nei primi sei mesi di utilizzo del farmaco, con quadro variabile tra epato-cellulare, colestatico e misto. Il danno in alcuni casi è evoluto verso l’insufficienza epatica acuta, ma nella maggioranza dei pazienti il recupero è stato completo nel corso dei 2-3 mesi successivi all’interruzione del trattamento. Non sono stati invece descritti casi di danno cronico epatico e/o biliare. Likelihood score: C.
Propiltiouracile (2,47,55): nei primi tre mesi di terapia si verifica frequentemente un aumento lieve, transitorio e asintomatico delle transaminasi, che si risolve anche continuando la terapia. Il propiltiouracile è anche associato a danno epatico clinicamente evidente, che si verifica in 1 caso su 1000, raramente con epatite fulminante e morte. Non è dose-dipendente, ma di tipo idiosincrasico, con esordio a distanza di 2-12 settimane dall’inizio della terapia. Il pattern è tipicamente di tipo epato-cellulare, in casi più rari misto o colestatico. L’interruzione della terapia non sempre garantisce la regressione del quadro clinico, che talvolta è evoluto in quadri di insufficienza epatica acuta. L’età adulta è meno soggetta a presentare l’evoluzione verso l’insufficienza epatica acuta rispetto ai bambini e agli adolescenti. Come per il metimazolo, anche per la tossicità da uso del propiltiouracile è necessario valutare la diagnosi differenziale con il danno epatico indotto dall’ipertiroidismo stesso, che può comunque rappresentare una concausa di gravità del danno. La terapia con derivati tioamidici deve essere somministrata con cautela nei pazienti con malattia epatica pre-esistente, anamnesi di abuso di alcool o epatite. Il trattamento deve essere interrotto se si verificano deterioramento della funzionalità epatica o altri segni di danno epatico e si può valutare l’uso del metimazolo in quanto la sua introduzione non è associata a recidiva del danno. Le ulteriori opzioni terapeutiche sono il radioiodio o l’intervento chirurgico. Likelihood score: A.
Repaglinide (2,44,56): il danno epatico in corso di terapia è possibile ma raro. La molecola può essere utilizzata nei pazienti con epatite cronica e cirrosi compensata, iniziando con la dose più bassa e procedendo con lenti incrementi sulla base di uno stretto monitoraggio. È consigliato invece evitarne l’uso nei pazienti con cirrosi scompensata. La repaglinide è quasi completamente metabolizzata nel fegato e i pazienti con funzionalità epatica compromessa possono essere esposti a concentrazioni più elevate del farmaco e dei suoi metaboliti, con aumentato rischio di gravi episodi ipoglicemici. Likelihood score: D.
Statine (2,5,44,57-63): sono associate ad aumento lieve, asintomatico e generalmente transitorio delle transaminasi, solitamente auto-limitante, spesso senza necessità di riduzione del dosaggio. Talvolta si può presentare anche un danno epatico clinicamente rilevante, sia nella forma epato-cellulare, sia colestatica, sia mista. La frequenza e l’epoca di insorgenza possono variare a seconda della molecola in esame. È indicato eseguire una valutazione delle transaminasi e degli indici di colestasi prima di iniziare la terapia e nel corso della terapia se clinicamente indicato. Nel caso in cui si verifichi un aumento anche sporadico di almeno 10 volte rispetto al l’ULN o persistente ≥ 5 ULN, è indicata la sospensione del farmaco; aumenti di minore severità spesso si risolvono spontaneamente, senza richiedere l’interruzione. Dovrebbe essere evitata la re-introduzione della stessa statina che aveva provocato il danno epatico, per l’elevato rischio di recidiva; il passaggio ad altra molecola è anch’esso a rischio: se effettuato, deve essere attentamente monitorato. L’utilizzo delle statine non è controindicato nei pazienti con epatite cronica, colangite biliare primitiva e cirrosi compensata, anzi può addirittura migliorare la fibrosi epatica. Una recente meta-analisi ha evidenziato come nella NAFLD le statine migliorino le transaminasi, la gamma-GT, la steatosi e l’infiammazione epatica, senza però effetti sulla fibrosi epatica. Si suggerisce comunque di iniziare il trattamento con il farmaco a bassa dose, incrementandola gradualmente in base alla risposta. In questi pazienti persiste la raccomandazione di monitoraggio delle ALT. Nei pazienti con cirrosi scompensata è raramente necessario l’uso di ipocolesterolemizzanti, in quanto questa fase della patologia epatica si associa solitamente a ipolipidemia anche in soggetti in passato dislipidemici. Nei pazienti in terapia con statine in cui si verifica uno scompenso della cirrosi epatica si può evidenziare una pronunciata risposta al farmaco per aumento delle concentrazioni plasmatiche.
- Atorvastina: la frequenza del danno epatico clinicamente evidente è circa 1:3000-5000 pazienti trattati. La latenza è estremamente variabile, da un mese a diversi anni, anche se il danno è maggiormente frequente nei primi 6 mesi. Clinicamente può presentare le caratteristiche di un’epatite autoimmune, con riscontro di ANA positività e aumento delle immunoglobuline. In questi casi può non essere sufficiente la sospensione del farmaco, ma può essere necessaria la terapia steroidea o immuno-soppressiva a lungo termine. Likelihood score: A.
- Fluvastatina: si associa ad aumento delle transaminasi, in genere lieve, asintomatico e transitorio. In meno dell’1% si verifica un aumento significativo al di sopra di 3 volte l’ULN, solitamente nei pazienti in trattamento con il dosaggio più alto. Il danno epatico clinicamente rilevante, con possibile evoluzione in insufficienza epatica acuta, rappresenta un evento piuttosto raro, di tipo misto o colestatico e si verifica tra il primo e il quarto mese dall’inizio del trattamento. Il farmaco non è stato associato allo sviluppo di insufficienza epatica cronica o da “Vanishing Bile Buct Syndrome”. Likelihood score: A.
- Lovastatina: è associata ad aumenti delle transaminasi, spesso lievi, asintomatici e generalmente transitori. L’epato-tossicità è dose-dipendente ed è indicata l’interruzione se si presentano valori > 10 volte ULN o per valori persistentemente > 5 volte ULN. È raro il danno epatico clinicamente evidente, che si presenta con pattern tipicamente colestatico, con insorgenza variabile tra le poche settimane dall’inizio della terapia fino a diversi anni dopo. È descritta la sensibilità crociata al danno epatico tra lovastatina e prodotti a base di riso rosso fermentato. Prima di iniziare la terapia, si raccomanda di effettuare gli esami ematochimici per escludere la presenza di danno epato-cellulare o colestatico, e la ripetizione dei test in corso di terapia. In caso di insorgenza, le lievi alterazioni delle transaminasi si risolvono spontaneamente entro poche settimane anche senza interruzione. Nel caso in cui l’elevazione sia più marcata o persistente, è necessario sospendere definitivamente il farmaco. Likelihood score: B.
- Pravastatina: la frequenza del danno epatico clinicamente evidente è ancor più rara rispetto alle altre statine (circa 1:100.000 pazienti trattati). La latenza è tra 2 e 9 mesi, la normalizzazione del quadro clinico si osserva dopo pochi mesi. Likelihood score: B.
- Rosuvastatina: si associa a lieve aumento delle transaminasi (1-3% dei casi), asintomatico e transitorio. In circa l’1% si verifica un aumento significativo, > 3 volte dell’ULN, solitamente nei pazienti in trattamento con il dosaggio più alto. La rosuvastatina è anche raramente (1:10.000 pazienti) associata a danno epatico clinicamente evidente, il cui esordio si verifica solitamente dopo 2-4 mesi e presenta un pattern prevalentemente epato-cellulare. Non si può escludere un’evoluzione verso l’insufficienza epatica acuta. Alcuni quadri possono risultare simili all’epatite autoimmune e, in caso di mancato miglioramento tempestivo dopo la sospensione del farmaco, può essere indicata la terapia con corticosteroidi, di durata minima e seguita da attento follow-up. Likelihood score: A.
- Simvastatina: nel 5% dei pazienti si riscontrano aumenti lievi, transitori e asintomatici delle ALT; aumenti moderati (> 3 volte l’ULN) nell'1-2% dei casi. Gli aumenti sono più frequenti nei soggetti che assumono dosi elevate in maniera cronica. Il danno epatico clinicamente rilevante è un evento estremamente raro, considerando anche l’enorme diffusione del farmaco; in tali contesti non si può comunque escludere un’evoluzione verso l’insufficienza epatica acuta. Il tempo di latenza può variare da pochi giorni a 3 anni, con una maggiore frequenza tra il primo e il sesto mese. Sono stati descritti anche quadri clinici simili all’epatite autoimmune, che hanno richiesto anche terapia immuno-soppressiva a lungo termine. Alla simvastatina sono stati attribuiti rari casi di insufficienza epatica acuta e morte. Seppur in studi con bassa numerosità, è stato valutato l’utilizzo della simvastina nel soggetto cirrotico: gli studi hanno dimostrato bassa tossicità epatica e muscolare con il dosaggio da 20 mg, e un netto aumento degli effetti collaterali con il dosaggio da 40 mg. Likelihood score: A.
Steroidi anabolizzanti androgeni (2,5,64,65): questa classe di farmaci è causa di danno epatico sotto diverse forme: ipertransaminasemia transitoria, sindrome colestatica acuta, danno vascolare, tumori primitivi epatici benigni e maligni. Quest’ultimi rappresentano la complicanza più severa e si manifestano in soggetti che li assumono da lungo tempo. In caso di danno epatico, è necessario raccomandare la sospensione totale, sconsigliando la riduzione della posologia o il passaggio a un'altra formulazione; la risoluzione del quadro appare comunque lenta. I pazienti con patologie epatiche pre-esistenti devono essere monitorati più attentamente: se la funzionalità epatica diminuisce o si verifica tossicità, la terapia deve essere sospesa. Likelihood score: A.
Sulfaniluree (2,5,44): l’uso di tutte le forme attualmente disponibili di questa classe di farmaci provoca raramente un danno epatico clinicamente rilevante. L’insorgenza del danno si verifica dopo 3-12 settimane dall'inizio della terapia, sotto forma di danno epato-cellulare, colestatico o misto, che si risolve rapidamente dopo l’interruzione. Questa categoria di farmaci è raramente associata a insufficienza epatica acuta. Nei pazienti con epatite cronica o cirrosi compensata le sulfoniluree possono essere prescritte, iniziando con la dose più bassa e procedendo con lenti incrementi sulla base di uno stretto monitoraggio. Questa categoria di farmaci deve essere evitata nei pazienti con cirrosi scompensata. Likelihood score: glibenclamide B; gliclazide D; glimepiride C; glipizide C.
Sunitinib (2,66,67): in circa il 40% dei soggetti trattati si osservano aumenti delle transaminasi, generalmente asintomatici e di grado lieve, e solo nel 2-3% il rialzo è > 5 volte l’ULN. In questi casi o quando il rialzo si associa anche ad aumento della bilirubina, è consigliato l’aggiustamento terapeutico o la sospensione transitoria del farmaco; la ripresa della terapia dovrà essere effettuata con dosaggio inferiore, eventualmente associato a prednisone (10-20 mg/die). In alcuni casi si può osservare anche iperbilirubinemia isolata di grado lieve-moderato, legata all’interazione tra il farmaco e l'UDP-glucuroniltransferasi epatica. Si possono osservare anche quadri di danno epatico clinicamente evidente, con pattern tipicamente epato-cellulare, e anche quadri clinici di encefalopatia epatica acuta, caratterizzati da confusione, irritabilità e aumento dell’ammoniemia, che regrediscono con la sospensione ma possono recidivare dopo la re-introduzione della terapia. Non sono descritti casi di reattività crociata con altri inibitori della tirosin-chinasi. In considerazione del rischio di danno epatico da sunitinib, è importante evitare l’uso di altri agenti epato-tossici. Likelihood score: B.
Tamoxifene (2,68): è stato associato a rari casi di danno epatico clinicamente evidente di tipo idiosincrasico, che si presenta solitamente entro i 6 mesi dall’inizio del trattamento e può avere un pattern prevalentemente colestatico ma anche misto o epato-cellulare. Nella maggior parte dei casi si tratta di un danno auto-limitante, ma sono stati anche descritti casi di insufficienza epatica. Il tamoxifene, se assunto per lunghi periodi, frequentemente, soprattutto nei soggetti con BMI elevato, induce steatosi semplice, talvolta steato-epatite e più raramente fibrosi e ipertensione portale. L’interruzione del farmaco determina un miglioramento, che però può essere molto lento. Il farmaco è stato associato anche ad aumentato rischio di trombosi venose, inclusa la trombosi portale. Si consiglia quindi il monitoraggio periodico delle ALT. In caso di valori persistentemente elevati, la prosecuzione della terapia deve essere valutata individualmente, misurando il rapporto rischi/benefici. In tale bilancio possono dare un contributo anche i sistemi non invasivi di valutazione delle fibrosi e talvolta anche la biopsia epatica. È consigliato comunque di eliminare i fattori che possono contribuire al danno, come alcool o BMI elevato. Il passaggio agli inibitori dell'aromatasi, come anastrozolo, letrozolo o exemestane, può rappresentare una valida alternativa. Likelihood score: B.
Temozolomide (2,7,70): in 1 caso su 10 si può verificare un aumento dell’AST, solitamente di modesta entità, che si auto-limita senza richiedere modiche della terapia. Più raramente si verifica epato-tossicità manifesta, con pattern misto nella fase iniziale e successivamente colestatico. Non sono stati descritti casi di re-introduzione della terapia dopo sospensione, mentre la sostituzione con altri agenti anti-neoplastici non ha portato a recidiva del danno epatico. Uno studio ha mostrato che la farmaco-cinetica della temozolomide in pazienti con cirrosi e compromissione epatica da lieve a moderata (classe Child-Pugh A - B) era simile a quella osservata nei pazienti con funzionalità epatica normale. Si deve usare cautela nella somministrazione a pazienti con grave compromissione epatica. Il farmaco si può associare anche a riattivazione dell’HBV nei soggetti con infezione occulta all’inizio della terapia. L’epatite HBV è risultata responsiva alla terapia anti-virale, che ha consentito talvolta anche di riprendere il temozolomide. Si raccomanda quindi lo screening per HBsAg, anti-HBc Ab e HBsAb in coloro che devono iniziare la terapia. Nei pazienti con evidenza sierologica di epatite B in corso o pregressa, deve essere monitorato l’HBV-DNA, iniziando la terapia anti-virale se positivo. Un’alternativa può essere somministrare la terapia anti-virale in profilassi durante il trattamento e nei 6 mesi successivi. Likelihood score: B.
Teriparatide (2): non è associato a epato-tossicità. Likelihood score: E.
Vaptani (1,2,71,72): in circa il 4-5% dei pazienti trattati è stato documentato a livello biochimico un danno epato-cellulare o misto, clinicamente evidente nello 0.1% dei casi, con tempo di latenza variabile tra 3 e 9 mesi. La sintomatologia era caratterizzata da ittero, astenia, nausea e dolori addominali. L’esame istologico documentava un quadro di epatite acuta con lieve colestasi. L’interruzione della terapia ha portato a risoluzione del quadro clinico nell’arco di 1-3-mesi. In un solo caso è stata documentata la necessità di trapianto epatico. La re-introduzione della terapia dovrebbe essere evitata, perché può comportare recidiva del danno. Non ci sono informazioni sulla sensibilità crociata. L'uso di tolvaptan nei pazienti con cirrosi va valutato da caso a caso a livello specialistico, ma andrebbe evitato, poiché la capacità di recupero dal danno epatico può essere compromessa. Likelihood score: C.
Triptorelina (2): nessuna segnalazione di epato-tossicità. Gli studi clinici hanno mostrato che i soggetti con insufficienza epatica avevano un'esposizione maggiore al farmaco. Si raccomanda cautela e monitoraggio degli eventi avversi. Likelihood score: E.
BIBLIOGRAFIA
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: drug-induced liver injury. J Hepatol 2019, 70: 1222–61.
- LiverTox. Clinical and Research Information on Drug-Induced Liver Injury. livertox.nih.gov (ultimo accesso 3 novembre 2022).
- Chao C-T, Wang J, Huang J-W, Chien K-L. Acarbose use and liver injury in diabetic patients with severe renal insufficiency and hepatic diseases: a propensity score-matched cohort study. Front Pharmacol 2018, 9: 860.
- el Shahawy M, Cannon CP, Blom DJ, et al. Efficacy and safety of alirocumab versus ezetimibe over 2 years (from ODYSSEY COMBO II). Am J Cardiol 2017, 120: 931–9.
- Chalasani N, Bonkovsky HL, Fontana R, et al. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology 2015, 148: 1340-52.e7.
- Tomao F, Spinelli G, Vici P, et al. Current role and safety profile of aromatase inhibitors in early breast cancer. Expert Rev Anticancer Ther 2011, 11: 1253–63.
- Zapata E, Zubiaurre L, Bujanda L, Pirola A. Anastrozole-induced hepatotoxicity. Eur J Gastroenterol Hepatol 2006, 18: 1233–4.
- Guañabens N, Monegal A, Cerdá D, et al. Randomized trial comparing monthly ibandronate and weekly alendronate for osteoporosis in patients with primary biliary cirrhosis. Hepatology 2013, 58: 2070–8.
- Tadrous M, Wong L, Mamdani MM, et al. Comparative gastrointestinal safety of bisphosphonates in primary osteoporosis: a network meta-analysis. Osteoporos Int 2014, 25: 1225–35.
- Reid IR. Short-term and long-term effects of osteoporosis therapies. Nat Rev Endocrinol 2015, 11: 418–28.
- Rossini M, Adami G, Adami S, et al. Safety issues and adverse reactions with osteoporosis management. Expert Opin Drug Saf 2016, 15: 321–32.
- Schneider JS, Montani M, Stickel F. Drug-induced autoimmune hepatitis following treatment with zoledronic acid. Case Rep Gastroenterol 2017, 11: 440–5.
- Lee HL, Bae SH, Jang B, et al. Reactivation of Hepatitis C Virus and its clinical outcomes in patients treated with systemic chemotherapy or immunosuppressive therapy. Gut Liver 2017, 11: 870–7.
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017, 67: 370–98.
- Bessone F, Lucena M, Roma MG, et al. Cyproterone acetate induces a wide spectrum of acute liver damage including corticosteroid-responsive hepatitis: report of 22 cases. Liver Int 2016, 36: 302–10.
- Figueiredo JBP, Nastri CO, Vieira ADD, Martins WP. Clomiphene combined with gonadotropins and GnRH antagonist versus conventional controlled ovarian hyperstimulation without clomiphene in women undergoing assisted reproductive techniques: systematic review and meta-analysis. Arch Gynecol Obstet 2013, 287: 779–90.
- Itoh S, Igarashi M, Tsukada Y, Ichinoe A. Nonalcoholic fatty liver with alcoholic hyalin after long-term glucocorticoid therapy. Acta Hepatogastroenterol (Stuttg) 1977, 24: 415–8.
- Wald J, Farr R. Abnormal liver-function tests associated with long-term systemic corticosteroid use in subjects with asthma. J Allergy Clinic Immunol 1991, 88: 277–8.
- Shiota G, Harada K, Oyama K, et al. Severe exacerbation of hepatitis after short-term corticosteroid therapy in a patient with “latent” chronic hepatitis B. Liver 2000, 20: 415–20.
- Etminan M, Delaney JAC, Bressler B, Brophy JM. Oral contraceptives and the risk of gallbladder disease: a comparative safety study. CMAJ 2011, 183: 899–904.
- Lieberman DA, Keeffe EB, Stenzel P. Severe and prolonged oral contraceptive jaundice. J Clin Gastroenterol 1984, 6: 145–8.
- Steinbrecher UP, Lisbona R, Huang SN, Mishkin S. Complete regression of hepatocellular adenoma after withdrawal of oral contraceptives. Dig Dis Sci 1981, 26: 1045–50.
- Boake WC. Intrahepatic cholestatic jaundice of pregnancy followed by enovid-induced cholestatic jaundice. Ann Intern Med 1965, 63: 302.
- Bourgeois AL, Auriche P, Palmaro A, et al. Risk of hormonotherapy in transgender people: literature review and data from the French Database of Pharmacovigilance. Ann Endocrinol (Paris) 2016, 77: 14–21.
- Stannov SU, Ries A, Bang UC. Hepatotoxicity induced by a second-generation combined oral contraceptive: case report and review of the literature. Eur J Contracept Reprod Health Care 2019, 24: 322–4.
- Abdel-Rahman O, Fouad M. Risk of fatigue and hepatic and metabolic toxicities in patients with solid tumors treated with everolimus: a meta-analysis. Future Oncol 2015, 11: 79–90.
- Mir O, Toulmonde M, Coriat R, et al. Hepatitis B reactivation during everolimus treatment. Acta Oncol (Madr) 2016, 55: 1505–6.
- Kanagalingam T, Lazarte J, Wong DKH, Hegele RA. Liver injury associated with ezetimibe monotherapy. CJC Open 2021, 3: 195–7.
- Ho C-Y, Kuo T-H, Chen T-S, et al. Fenofibrate-induced acute cholestatic hepatitis. J Chin Med Assoc 2004, 67: 245–7.
- Rigal J, Furet Y, Autret E, Breteau M. Hépatite mixte sévère au fénofibrate? Revue de la littérature à propos d’un cas. Rev Med Interne 1989, 10: 65–7.
- Geng Q, Ren J, Chen H, Lee C, Liang W. Adverse events following statin-fenofibrate therapy versus statin alone: a meta-analysis of randomized controlled trials. Clin Exp Pharmacol Physiol 2013, 40: 219–26.
- Domínguez Tordera P, Comellas Alabern JF, Ronda Rivero F. Gemfibrozil hepatotoxicity: a case report. Int J Clin Pharm 2011, 33: 730–2.
- Hirshburg JM, Kelsey PA, Therrien CA, et al. Adverse effects and safety of 5-alpha reductase inhibitors (finasteride, dutasteride): a systematic review. J Clin Aesthet Dermatol 2016, 9: 56–62.
- Castelo-Branco C, Hernández-Angeles C, Alvarez-Olivares L, Balasch J. Long-term satisfaction and tolerability with low-dose flutamide: a 20-year surveillance study on 120 hyperandrogenic women. Gynecol Endocrinol 2016, 32: 723–7.
- Bolton EM, Lynch T. Are all gonadotrophin-releasing hormone agonists equivalent for the treatment of prostate cancer? A systematic review. BJU Int 2018, 122: 371–83.
- Rehman MB, Tudrej BV, Soustre J, et al. Efficacy and safety of DPP-4 inhibitors in patients with type 2 diabetes: meta-analysis of placebo-controlled randomized clinical trials. Diabetes Metab 2017, 43: 48–58.
- Hernández N, Bessone F, Sánchez A, et al. Profile of idiosyncratic drug induced liver injury in Latin America: an analysis of published reports. Ann Hepatol 2014, 13: 231–9.
- White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013, 369: 1327–35.
- Asakawa M, Mitsui H, Akihisa M, et al. Efficacy and safety of sitagliptin for the treatment of diabetes mellitus complicated by chronic liver injury. Springerplus 2015, 4: 346.
- Toyoda-Akui M, Yokomori H, Kaneko F, et al. A case of drug-induced hepatic injury associated with sitagliptin. Int Med 2011, 50: 1015–20.
- Rosenstock J, Seman LJ, Jelaska A, et al. Efficacy and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, as add-on to metformin in type 2 diabetes with mild hyperglycaemia. Diabetes Obes Metab 2013, 15: 1154–60.
- Bajaj HS, Brown RE, Bhullar L, et al. SGLT2 inhibitors and incretin agents: associations with alanine aminotransferase activity in type 2 diabetes. Diabetes Metab 2018, 44: 493–9.
- Sherigar JM, Castro J de, Yin YM, et al. Glycogenic hepatopathy: a narrative review. World J Hepatol 2018, 10: 172–85.
- Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis - a practical guide. Aliment Pharmacol Ther 2013, 37: 1132–56.
- Richeldi L. Assessing the treatment effect from multiple trials in idiopathic pulmonary fibrosis. Eur Respir Rev 2012, 21: 147–51.
- Kyriakidis I, Tragiannidis A, Munchen S, Groll AH. Clinical hepatotoxicity associated with antifungal agents. Expert Opin Drug Saf 2016, 16: 1–17.
- Suzuki N, Noh JY, Hiruma M, et al. Analysis of antithyroid drug-induced severe liver injury in 18,558 newly diagnosed patients with Graves’ disease in Japan. Thyroid 2019, 29: 1390–8.
- Kapur A, Angomchanu R, Dey M. Efficacy of use of long-term, low-dose mifepristone for the treatment of fibroids. J Obstet Gynaecol India 2016, 66: 494–8.
- Khalil H, Ellwood L, Lord H, Fernandez R. Pharmacological treatment for obesity in adults: an umbrella review. Ann Pharmacother 2020, 54: 691–705.
- Montero JL, Muntané J, Fraga E, et al. Orlistat associated subacute hepatic failure. J Hepatol 2001, 34: 173.
- Mandel SJ. Levothyroxine therapy in patients with thyroid disease. Ann Intern Med 1993, 119: 492-502.
- Öberg K, Lamberts SWJ. Somatostatin analogues in acromegaly and gastroenteropancreatic neuroendocrine tumours: past, present and future. Endocr Relat Cancer 2016, 23: R551–66.
- Kung J, Henry RR. Thiazolidinedione safety. Expert Opin Drug Saf 2012, 11: 565–79.
- Drugs for type 2 diabetes. Med Lett Drugs Ther 2017, 59: 9–18.
- Yang J, Li L, Xu Q, et al. Analysis of 90 cases of antithyroid drug-induced severe hepatotoxicity over 13 years in China. Thyroid 2015, 25: 278–83.
- Marshall V, Wilton L, Shakir S. Safety profile of repaglinide as used in general practice in England: results of a prescription-event monitoring study. Acta Diabetol 2006, 43: 6–13.
- Cai T, Abel L, Langford O, et al. Associations between statins and adverse events in primary prevention of cardiovascular disease: systematic review with pairwise, network, and dose-response meta-analyses. BMJ 2021, 374: n1537.
- Hartleb M, Rymarczyk G, Januszewski K. Acute cholestatic hepatitis associated with pravastatin. Am J Gastroenterol 1999, 94: 1388–90.
- Simon TG. When less is more: dosing simvastatin in decompensated cirrhosis. Lancet Gastroenterol Hepatol 2020, 5: 3–5.
- Pose E, Napoleone L, Amin A, et al. Safety of two different doses of simvastatin plus rifaximin in decompensated cirrhosis (LIVERHOPE-SAFETY): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Gastroenterol Hepatol 2020, 5: 31–41.
- Sung S, Al-Karaghouli M, Kalainy S, et al. A systematic review on pharmacokinetics, cardiovascular outcomes and safety profiles of statins in cirrhosis. BMC Gastroenterol 2021, 21: 120.
- Marrache MK, Rockey DC. Statins for treatment of chronic liver disease. Curr Opin Gastroenterol 2021, 37: 200-7.
- Boutari C, Pappas PD, Anastasilakis D, et al. Statins' efficacy in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Clin Nutr 2022, 41: 2195-206.
- Hassan A, Fontana R. Liver injury associated with sporting activities. Semin Liver Dis 2018, 38: 357–65.
- Zheng E, Sandhu N, Navarro V. Drug-induced liver injury secondary to herbal and dietary supplements. Clin Liver Dis 2020, 24: 141–55.
- Shantakumar S, Nordstrom BL, Djousse L, et al. Occurrence of hepatotoxicity with pazopanib and other anti-VEGF treatments for renal cell carcinoma: an observational study utilizing a distributed database network. Cancer Chemother Pharmacol 2016, 78: 559–66.
- Bunchorntavakul C, Reddy KR. Drug hepatotoxicity. Clin Liver Dis 2017, 21: 115–34.
- Yoo J-J, Lim YS, Kim MS, et al. Risk of fatty liver after long-term use of tamoxifen in patients with breast cancer. PLoS One 2020, 15: e0236506.
- Bonkovsky HL, Kleiner DE, Gu J, et al. Clinical presentations and outcomes of bile duct loss caused by drugs and herbal and dietary supplements. Hepatology 2017, 65: 1267–77.
- Desai VCA, Quinlan SC, Deitz AC, et al. Risk of severe acute liver injury among patients with brain cancer treated with temozolomide: a nested case-control study using the healthcore integrated research database. J Neurooncol 2017, 134: 89–95.
- Raina R, Chakraborty R, DeCoy ME, Kline T. Autosomal-dominant polycystic kidney disease: tolvaptan use in adolescents and young adults with rapid progression. Pediatr Res 2021, 89: 894–9.
- Muto S, Okada T, Yasuda M, et al. Long-term safety profile of tolvaptan in autosomal dominant polycystic kidney disease patients: TEMPO Extension Japan Trial. Drug Healthc Patient Saf 2017, 9: 93–104.