Terapia farmacologica dell'obesità

Fabrizio Muratori
UOC Endocrinologia e Diabetologia, Ospedale Sant'Anna, Como
questo capitolo è in attesa di aggiornamento
RAZIONALE DELL’INTERVENTO FARMACOLOGICO NELLA TERAPIA INTEGRATA DELL’OBESITÀ
La gestione terapeutica dell’obesità è piuttosto complessa e richiede un approccio multidimensionale, con l’associazione di varie modalità terapeutiche integrate fra loro. Le linee guida dell’obesità indicano con molta chiarezza che il primo gradino della terapia è rappresentato dalla modificazione dello stile di vita attraverso l’educazione alimentare e l’esercizio fisico; il trattamento farmacologico dovrebbe quindi essere preso in considerazione solo dopo che è stata valutata l’efficacia della dieta, dell’esercizio fisico e, dove indicato, della terapia cognitivo-comportamentale e che tali approcci terapeutici si siano dimostrati inefficaci o nell’indurre perdita di peso o nel mantenimento del peso perso.
È comunque riportato in letteratura che la restrizione calorica, associata o meno a tecniche cognitivo-comportamentali, non è in grado di garantire, nella maggioranza dei casi, un calo ponderale adeguato e il mantenimento dei risultati raggiunti nel lungo periodo (1). Ormai è opinione comunemente consolidata che sia corretto utilizzare le varie terapie (dietologica, comportamentale, farmacologica e chirurgica) in modo integrato. È noto da molti anni che associare farmaci alla terapia del comportamento (educazione alimentare e terapia del movimento) induce un aumento della perdita di peso rispetto a quella ottenuta con il solo trattamento comportamentale (2,3). La decisione di iniziare il trattamento e la scelta del farmaco (quando fosse possibile) dovrebbero comunque avvenire dopo discussione con il paziente, sia dei potenziali benefici che dei limiti del farmaco, inclusi il suo meccanismo d’azione, gli effetti collaterali e il potenziale impatto sulla motivazione del paziente stesso. La terapia farmacologica dell’obesità richiede, infatti, una profonda conoscenza delle molecole a disposizione e dei loro meccanismi d’azione, ma soprattutto richiede esperienza nel campo terapeutico, per potere valutare al meglio le modalità e i tempi di somministrazione dei singoli farmaci (4).
Il ruolo del farmaco nell’obesità è di supportare gli altri presidi terapeutici ed è codificato dalle linee guida: secondo i National Institutes of Health, i farmaci anti-obesità sono indicati come parte di un programma globale che includa la dieta e l’attività fisica, in soggetti con BMI ≥ 30 kg/m2 oppure in soggetti con BMI ≥ 27 kg/m2 con altri fattori di rischio o altre patologie correlate all’obesità (tabella 1).
| Tabella 1 Guida per la scelta del trattamento integrato del paziente obeso |
|||||
| BMI | |||||
| Trattamento | 25–26.9 | 27–29.9 | 30–34.9 | 35–39.9 | > 40 |
| Dieta, esercizio fisico, terapia comportamentale | + | + | + | + | + |
| Terapia farmacologica | - | Con comorbilità | + | + | + |
| Terapia chirurgica | - | - | Con comorbilità | + | + |
A questo proposito si potrebbe obiettare che il limite di BMI di 27 è del tutto arbitrario, poiché se un paziente ha un BMI inferiore e presenta dei fattori di rischio correlati all’eccesso di peso, l’unica terapia efficace è la riduzione del peso stesso. Proprio per questo motivo l’uso di un eventuale farmaco ha in realtà una sola indicazione-controindicazione (che poi è la stessa di ogni terapia): il rapporto costo-beneficio, intendendosi per costo ogni elemento che può causare un danno al paziente.
Dopo la sospensione dal commercio di sibutramina nel gennaio 2010, in Italia è attualmente autorizzata una sola molecola per la terapia a lungo termine dell’obesità: orlistat. Vi sono studi controllati che valutano l’efficacia di questo farmaco per periodi che variano da due a quattro anni continuativi (5-7). Nella maggior parte degli studi che non utilizzano ausilio farmacologico il calo ponderale avviene nei primi sei mesi, a cui segue, nella maggior parte dei soggetti, un recupero ponderale. Anche con l’ausilio farmacologico si assiste a un calo ponderale più marcato nei primi sei mesi, seguito da un calo ponderale più lento nel periodo successivo. Alla sospensione del farmaco il peso viene generalmente recuperato, se il paziente non ha saputo modificare gli stili di vita in modo stabile. In questo contesto e alla luce delle attuali conoscenze, la terapia farmacologica ha lo scopo non tanto di aumentare il calo ponderale, ma di permettere a un maggior numero di soggetti obesi di raggiungere e mantenere gli obiettivi prefissati. Infatti, il ruolo dei farmaci nella terapia dell’obesità va inteso anche come un aiuto nel favorire l’adesione, da parte del paziente, all’approccio terapeutico globale, che comprende anche il trattamento nutrizionale, il cambiamento degli stili di vita e la terapia cognitivo-comportamentale (8,9).
Mantenimento del peso e supporto farmacologico
La posizione della comunità scientifica su questo aspetto della terapia dell’obesità è ormai consolidata. Il trattamento del paziente obeso (con le diverse modalità terapeutiche oggi a disposizione) deve proseguire anche dopo avere ottenuto un importante calo ponderale (10). Infatti, il mantenimento dei risultati ottenuti dopo riduzione del peso corporeo rappresenta uno degli aspetti più controversi e deludenti del trattamento del soggetto obeso, a causa dell’elevatissima frequenza di recidive a cui va incontro il paziente (11). Il successo, per il medico e il paziente, consiste nel raggiungere e mantenere l’obiettivo clinico stabilito. Per obiettivo clinico si intende raggiungere la riduzione ponderale necessaria a migliorare in modo sensibile i rischi legati all’obesità, specie quelli cardiovascolari. Sono stati condotti negli ultimi anni importanti studi per verificare se la terapia farmacologia (associata ovviamente a modifiche dello stile di vita, educazione alimentare e attività fisica) fosse in grado di modificare la tendenza al recupero di peso o addirittura fosse capace di fermarlo. I maggiori studi internazionali hanno dimostrato che l’aggiunta della terapia farmacologica (sibutramina, orlistat, lorcaserina e l’associazione fentermina/topiramato) induce un rallentamento consistente nel recupero di peso e, per quanto riguarda orlistat, è anche in grado di ridurre la comparsa di diabete mellito nei pazienti obesi seguiti per quattro anni (7-12).
In soggetti selezionati, si può ipotizzare il concetto di “ciclo terapeutico”, da prendere in considerazione a lungo termine. Se i risultati della precedente terapia sono stati soddisfacenti, negli anni successivi, quando compaiono situazioni o circostanze negative che comportino un recupero di peso, può essere presa in considerazione l’eventualità di nuovi cicli di terapia farmacologica per aiutare il paziente a gestire il momento difficile (9).
Bibliografia essenziale
- Douketis JD, Feightner JW, Attia J, Feldman WF. Canadian Task Force on Preventive Health Care.Periodic health examination, 1999 update: 1. Detection, prevention and treatment of obesity. CMAJ 1999, 160: 513-25.
- Weintraub M. Long-term weight control study: conclusions. Clin Pharmacol Ther 1992, 51: 642-6.
- James WP, Astrup A, Finer N, et al, for the STORM Study Group. Effect of sibutramine on weight maintenance after weight loss: a randomised trial. Lancet 2000, 356: 2119–25.
- Muratori F, Di Sacco G, Pellegrino D, Vignati F. La terapia farmacologica dell'obesità. In: Fatati G e Amerio ML (Eds), Dietetica e Nutrizione. Clinica, terapia e organizzazione, 2° Edizione, Il Pensiero Scientifico 2012: 491-538.
- Sjostrom L, Rissaanen A, Andersen t, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet 1998, 352: 167-72.
- Davidson MH, Hauptman J, Di Girolamo M, et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat. JAMA 1999, 281: 235-42.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of Diabetes in Obese Subjects (XENDOS) Study. Diabetes Care 2004, 27: 155–61.
- Weintraub M, Sundaresan PR, Schuster B, et al. Long term weight control: the National Heart, Lung and Blood Institute funded multimodal intervention study I-VII. Clin Pharmacol Ther 1992, 51: 581-646.
- Standard Italiani per la Cura dell'Obesità, SIO-ADI 2012-2013.
- Apfelbaum M, Vague P, Ziegler O, et al. Long-term maintenance of weight loss after a very-low-calorie diet: a randomized blinded trial of the efficacy and tolerability of sibutramine. Am J Med 1999, 106: 179-84.
- Wadden TA, Sternberg V, Letiziak KA, et al. Treatment of obesity by very-low-calorie diet, behavior therapy, and their combination: a five-year perspective. Int J Obes 1989, 13: 39-46.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012, 95: 297-308.
FARMACI CHE RIDUCONO L’ASSUNZIONE DI CIBO
È noto da molti decenni che l’ipotalamo ha una funzione importante nella regolazione dell’alimentazione. Infatti, negli animali da esperimento la stimolazione elettrica dell’ipotalamo laterale aumenta l’assunzione di cibo, mentre la stimolazione dell’ipotalamo ventro-mediale la diminuisce. Con l’avanzare della ricerca scientifica e delle conoscenze neurofisiologiche sulla regolazione del bilancio energetico, si è però osservato che nella regolazione dell’apporto alimentare il controllo del sistema che regola l’omeostasi del bilancio energetico è estremamente complesso, basandosi sull’integrazione, a livello di numerose aree cerebrali, tra le informazioni metaboliche provenienti dalla periferia e i segnali del sistema nervoso centrale. Lo studio di questi segnali rappresenta il più importante campo di ricerca nell’obesità ed è in rapida evoluzione. Da anni è comunque noto che noradrenalina, serotonina e dopamina, agendo su specifici siti a livello ipotalamico, determinano modificazioni del comportamento alimentare (1).
I farmaci ad azione centrale hanno quindi lo scopo di modulare l’assunzione del cibo e possono essere suddivisi in tre categorie.
- Farmaci che agiscono sulle vie centrali serotoninergiche: unico farmaco liberatore di serotonina è la lorcaserina, autorizzata in USA dal giugno 2012, un agonista selettivo dei recettori serotoninergici 5HT2C. Tale molecola non è attualmente autorizzata in Italia.
- Farmaci che agiscono sulle vie centrali catecolaminergiche: fentermina, mazindolo, dietilpropione, fendimetrazina (autorizzate come specialità medicinali negli USA da parte della FDA e non autorizzate in Italia)(2-8). Nel luglio del 2012 la FDA ha approvato, dopo lorcaserina sopra citata, anche l’uso di fentermina–topiramato, una combinazione di un farmaco ad azione noradrenergica e di un farmaco impiegato nel trattamento dell'epilessia e nella profilassi dell'emicrania, per il trattamento dell’eccesso ponderale in pazienti con BMI ≥ 30 kg/m2 o in pazienti con BMI ≥ 27 kg/m2 in presenza di comorbilità (9-13).
- Farmaci che agiscono sulle vie centrali catecolaminergiche e serotoninergiche: sibutramina. Sospesa dal mercato europeo e negli Stati Uniti nel 2010 (14-16).
Poiché le molecole di questa classe (farmaci ad azione centrale che riducono l’assunzione di cibo) non sono disponibili attualmente in Italia, l’esposizione di questo capitolo è prevalentemente di tipo informativo. In bibliografia vi sono molte voci utili ad approfondire l’argomento per chi fosse interessato.
Nella tabella 2 sono elencate le molecole anti-obesità autorizzate da FDA e AIFA per il trattamento dell’obesità. Il solo farmaco approvato per l’uso nell’obesità come specialità medicinale in Italia è Orlistat (farmaco ad azione periferica). Delle molecole di questa tabella, negli Stati Uniti la fentermina è ancora oggi il farmaco più utilizzato nel trattamento dell’obesità (dove è in commercio come specialità medicinale dal 1959). Il farmaco ad azione catecolaminergica più impiegato in Italia, nel recente passato, è stato la fendimetrazina. Questa era prescrivibile solo sotto forma galenica, unicamente da specialisti in endocrinologia, scienza dell’alimentazione, medicina interna, diabetologia, per un periodo non superiore a tre mesi consecutivi e solo in soggetti con BMI ≥ 30 kg/m2, ma dall’agosto 2011 ne è stata vietata da parte del Ministero della Salute la preparazione e la prescrizione.
| Tabella 2 Farmaci autorizzati per il trattamento dell’obesità (al 30/10/2013) |
||
| Autorizzato FDA | Autorizzato AIFA | |
| Dietilpropione | Sì | No |
| Fendimetrazina | Sì | No |
| Fentermina | Sì | No |
| Fentermina+Topiramato | Sì | No |
| Lorcaserina | Sì | No |
| Mazindolo | Sì | No |
| Orlistat | Sì | Sì |
| Sibutramina | No | No |
Bibliografia essenziale
- Parker JA, Bloom SR. Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology 2012, 63: 18-30.
- Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmunary hypertension. N Engl J Med 1996, 335: 609-16.
- Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med 1997, 337: 551-8.
- Hendrics EJ, Rothman RB, Greenway FL. How physician obesity specialists use drugs to treat obesity. Obesity (Silver Spring) 2009, 17: 1730-5.
- Cercato C, et al. A randomized double-blind placebo-controlled study of the long term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obesity 2009, 33: 857-65.
- Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev 2007, 59: 151-84.
- Bray GA. Medications for weight reduction. Med Clin North Am 2011, 95: 989-1008.
- Muratori F, Di Sacco G, Pellegrino D, Vignati F. La terapia farmacologica dell'obesità. In: Fatati G e Amerio ML (Eds), Dietetica e Nutrizione. Clinica, terapia e organizzazione, 2° Edizione, Il Pensiero Scientifico 2012: 491-538.
- Fidler MC, Sanchez M, Raether B, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011, 96: 3067-77.
- O’Neal PM, Smith SR, Weissman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity 2012, 20: 1426-36.
- Coleman E. FDA briefing document. NDA 22529 Lorquess (Lorcaserin hydrochloride). Tablets 10 mg Sponsor Arena Pharmaceuticals Advisory Committee (2010).
- Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377: 1341-52.
- Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr 2012, 95: 297-308.
- James WPT, et al. Effects of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Eng J Med 2010, 363: 905-17.
- Caterson ID, Finer N, Coutinho W, et al, on behalf of the SCOUT Investigators. Maintained intentional weight loss reduces cardiovascular outcomes: results from the Sibutramine Cardiovascular OUTcomes (SCOUT) trial. Diabetes, Obes Metab 2012, 14: 523-30.
- Bach DS, Rissanen AM, Mendel CM, et al. Absence of cardiac valve dysfunction in obese patients treated with sibutramine. Obes Res 1999, 7: 363-9.
FARMACI CHE RIDUCONO L’ASSORBIMENTO DEI NUTRIENTI
Orlistat appartiene a una nuova classe di farmaci per l’obesità, in quanto non agisce sopprimendo l’appetito, ma riducendo l’assorbimento dei grassi a livello del tratto gastrointestinale (1). L’azione farmacologica non si manifesta quindi per via sistemica, bensì nel tratto gastrointestinale. Orlistat è un potente inibitore irreversibile delle lipasi ed esplica la sua attività farmacologica nel lume dello stomaco e dell’intestino. La lipasi è l’enzima chiave coinvolto nella digestione dei lipidi alimentari. Le lipasi scindono i lipidi alimentari, in presenza di sali biliari, liberando acidi grassi dal glicerolo dei trigliceridi: gli acidi grassi liberi e il monogliceride sono successivamente disponibili per l’assorbimento attraverso la parete intestinale fino a giungere in circolo; l’anello ß-lattonico di orlistat forma un estere con i residui laterali della serina nel sito attivo delle lipasi, che è stabilizzato dai sali biliari. Il blocco delle lipasi determina una minore degradazione dei trigliceridi alimentari, con conseguente riduzione del loro assorbimento (circa il 30% in meno)(2). I trigliceridi non assorbiti passano attraverso il tratto intestinale e sono eliminati con le feci. Orlistat non ha effetti sulle altre funzioni pancreatiche, né sull’assorbimento dei carboidrati e delle proteine (3). Orlistat ha un assorbimento sistemico del tutto trascurabile (96% della dose totale).
Orlistat è stato valutato in studi clinici il cui disegno sperimentale prevedeva che i pazienti, a seguito di un periodo di run-in con il solo placebo, venissero randomizzati al placebo o al farmaco in esame. Dopo 12 mesi di trattamento, un cross-over randomizzava nuovamente i pazienti a placebo o Orlistat. La dieta è stata lievemente ipocalorica nel primo anno (circa 1500 Kcal/die), contenente approssimativamente 30% di calorie sotto forma di grassi, mentre nel secondo anno è stata assunta una dieta normocalorica. La perdita di peso nei 12 mesi è stata significativamente più elevata con orlistat, rispetto al placebo, in tutti gli studi clinici (10% di riduzione del peso iniziale rispetto al 6.1% del gruppo trattato con placebo). Vi è inoltre da aggiungere che la percentuale di pazienti che ha raggiunto una perdita di peso > 10% è stata significativamente più elevata nei pazienti trattati con orlistat (38.8% vs 17.6%). Altro dato significativo è che i pazienti trattati con orlistat durante il primo e il secondo anno, abbiano recuperato meno peso durante il secondo anno, rispetto ai pazienti che erano passati dalla terapia con orlistat al trattamento con placebo al termine del primo anno (4,5). La terapia con orlistat è inoltre associata a un significativo miglioramento di alcuni fattori di rischio per la patologia cardiovascolare, quali riduzione del colesterolo totale, del colesterolo LDL, dei trigliceridi e infine riduzione della pressione arteriosa (5). È importante notare che il miglioramento del quadro lipemico è maggiore nei soggetti che assumono il farmaco rispetto a quelli trattati con placebo, anche a parità di dimagrimento. Infatti, l’effetto di orlistat sul colesterolo LDL è indipendente dalla perdita di peso, come anche l’azione sull’assorbimento del colesterolo alimentare. Questi effetti sono legati al meccanismo d’azione del farmaco: il blocco delle lipasi induce una minore degradazione dei trigliceridi alimentari a formare acidi grassi e monogliceridi. Questa ridotta formazione di acidi grassi determina anche una minore solubilizzazione del colesterolo alimentare, con conseguente vantaggiosa riduzione del suo assorbimento.
La dose consigliata è di 120 mg (una capsula) prima, durante o fino ad un’ora dopo il termine dei pasti. Dal 2009 è in commercio la dose inferiore da 60 mg; in questo caso il farmaco non necessita di ricetta medica.
Gli effetti collaterali sono la diretta conseguenza dell’azione farmacologica di riduzione dell’assorbimento dei grassi alimentari e riguardano principalmente il tubo digerente con feci poltacee, oleose, crampi addominali, flatulenza, aumento della frequenza di defecazione, incontinenza fecale, disturbi tutti rilevati soprattutto in pazienti che non diminuivano la quota alimentare di grassi. Infatti, gli effetti collaterali sopra indicati non sono correlati alle dosi di farmaco utilizzate, bensì alla quantità di grasso eliminato con le feci.
Molti degli studi clinici condotti con orlistat hanno incluso una valutazione dei livelli delle vitamine liposolubili e del ß-carotene ed è stata notata una diminuzione significativa di dette vitamine, senza comunque effetti sfavorevoli sul metabolismo del calcio e sulle ossa (6). in pazienti trattati per lungo tempo con orlistat è consigliata la supplementazione con vitamine liposolubili: tale supplemento dovrebbe essere somministrato due ore prima o due ore dopo l’ultima somministrazione di orlistat (7). Orlistat, dato che può comportare un’aumentata eliminazione di ossalato urinario, è da usare con cautela in caso di nefrolitiasi da ossalato di calcio (8).
Il farmaco è praticamente privo di interazioni farmacologiche con altre molecole: non sono descritte interazioni sfavorevoli con alcool etilico assunto alla dose di 40 g/die, con contraccettivi orali, glibenclamide, pravastatina, atenololo, furosemide, nifedipina, digossina, fenintoina (9-15).
I criteri adottati in studi controllati sono la logica conseguenza dell’attività farmacologica della molecola, che, riducendo l’assorbimento dei grassi alimentari, è indicata soprattutto in quei pazienti che già sono aderenti a una dieta ipocalorica: in questi soggetti, infatti, riducendo ulteriormente l’introito calorico, si assiste a un maggiore calo ponderale. A nostro avviso, l’importanza di questo farmaco non è comunque data solo da questo aspetto, ma dagli effetti gastroenterici che insorgono quando si aumenta in modo eccessivo l’introito di grassi: questa sorta di “effetto antabuse” permette una maggiore aderenza alla dieta corretta. Ciò è probabilmente più evidente nei soggetti affetti da obesità morbigena (BMI > 40 kg/m2) (16) proprio perché in questi pazienti l’eccesso di alimenti grassi è notevolmente marcato. Queste considerazioni, unite alla scarsità di assorbimento sistemico e alla mancanza di interazione con altri farmaci, fanno ritenere orlistat un farmaco particolarmente indicato nelle fasi di mantenimento.
Effetto di orlistat nella prevenzione del diabete di tipo 2 in pazienti obesi
I dati più significativi sull’impiego di orlistat nei soggetti obesi diabetici o con ridotta tolleranza glicidica provengono dallo studio XENDOS, che ha valutato l’efficacia del farmaco, rispetto al placebo, nel ridurre la comparsa di diabete mellito di tipo 2 nell’arco di quattro anni (17). Il trattamento con farmaco o con placebo era accompagnato a modifiche dello stile di vita, dieta moderatamente ipocalorica e moderato esercizio fisico. Sono stati seguiti 3304 pazienti obesi (BMI > 30 kg/m2) con normale (79%) o ridotta (21%) tolleranza al glucosio, trattati per quattro anni con orlistat 120 mg o placebo 3 volte al dì. È stata valutata la progressione del peso e la progressione del diabete di tipo 2. Lo studio ha dimostrato che, rispetto al placebo, orlistat in associazione al cambiamento dello stile di vita determina a 4 anni un’evidente riduzione del peso e una significativa riduzione dell’incidenza di diabete mellito di tipo 2 (-37%) rispetto alle sole variazioni dello stile di vita e una significativa e sostanziale riduzione dei fattori di rischio cardiovascolare.
Un altro studio, condotto per un anno su 220 soggetti (18), ha dimostrato l’efficacia di orlistat rispetto al placebo, nel migliorare il controllo glicemico e ridurre la progressione del diabete, oltre a migliorare il profilo lipidico e i livelli pressori.
Orlistat e profilo lipidico
Orlistat è in grado di influire positivamente sul profilo lipidico anche grazie al suo meccanismo di azione. Gli effetti favorevoli di orlistat sul profilo lipidico sono stati documentati nello studio XENDOS (17), che ha dimostrato una riduzione significativa del colesterolo LDL a un anno (-11.4% vs placebo -1.6%) e a quattro anni (-12.8% vs -5.2%), associata a una riduzione significativa del rapporto colesterolo LDL/HDL: a un anno -0.5% vs placebo -0.3%, a quattro anni – 0.6% vs -0.4%.
Bibliografia essenziale
- Guerciolini R. Mode of action of orlistat. Int J Obes Relat Metab Disord 1997, 21: S12-23.
- Zhi J, Melia AT, Funk C, et al. Metabolic profiles of minimally absorbed orlistat in obese/overweight volunteers. J Clin Pharmacol 1996, 36: 1006-11.
- Zhi J, Melia AT, Eggers H, et al. Review of limited systemic absorption of orlistat, a lipase inhibitor, in healthy human volunteers. J Clin Pharmacol 1995, 35: 1103-8.
- Sjostrom L, Rissaanen A, Andersen t, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet 1998, 352: 167-72.
- Davidson MH, Hauptman J, Di Girolamo M, et al. Weight control and risk factor reduction in obese subjects treated for 2 years with orlistat. JAMA 1999, 281: 235-42.
- Zhi J, et al. The effect of orlistat, an inhibitor of dietary fat absorption on the pharmacokinetics of beta-carotene in healty volunteers. J Clin Pharmacol 1996, 36: 152-9.
- Bray GA. Medications for weight reduction. Med Clin North Am 2011, 95: 989-1008.
- Elliot WT, Chan J. Orlistat capsules (Xenical Roche Laboratoires).The physician’s therapeutic and drug alert. Amer Health Consultants 1999, 3: 90-2.
- Melia AT, et al. The interaction of the lipase inhibitor orlistat with ethanol in healthy 54 volunteers. Eur J Clin Pharmacol 1998, 54: 773-7.
- Zhi J, Melia AT, Koss-Twardy SG, et al. The influence of orlistat on the pharmacokinetics and pharmacodynamics of glyburide in healthy volunteers. J Clin Pharmacol 1995, 35: 521-5.
- Oo CY, Akbary B, Lee S, et al. Effect of orlistat, a novel anti obesity agent, on the pharmacokinetics and pharmacodynamics of pravastatin in patients with mild hypercolesterolemia. Clin Drug Invest 1999, 17: 217-23.
- Weber C, Tam YK, Schmidtke-Schrezenmeier G, et al. Effect of lipase inhibitor orlistat on the pharmacokinetics of four different anti-hypertensive drugs in healthy volunteers. Eur J Clin Pharmacol 1996, 51: 87-90.
- Melia AT, Mulligan TE, Zhy J. Lack of effect of orlistat on the bioavailability of a single dose of nifedipine extended-release tablets (Procardia XL) in healthy volunteers. J Clin Pharmacol 1996, 36: 352-5.
- Melia AT, Zhy J, Koss-Twardy SG, et al. The influence of reduced dietary fat absorption induced by orlistat on the pharmacokinetics of digoxin in healty volunteers. J Clin Pharmacol 1995, 35: 840-3.
- Melia AT, Mulligan TE, Zhy J. The effect of orlistat on the pharmacokinetics of phenytoin in healthy volunteers. J Clin Pharmacol 1996, 36: 654-8.
- Di Sacco G. Muratori F. Orlistat in estreme obesity. Int J Obes Relat Metab Disord 2000, 24 Suppl 1: 359.
- Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of Diabetes in Obese Subjects (XENDOS) Study. Diabetes Care 2004, 27: 155–61.
- Berne C, on behalf of the Orlistat Swedish Type 2 diabetes Study Group. A randomized study of orlistat in combination with a weight management programme in obese patients with Type 2 diabetes treated with metformin. Diab Med 2004, 22: 612–8.
Scheda orlistat
Beatrice Fazzalari
UOSD Endocrinologia e Diabetologia, AOU Sant’Andrea, Roma
Meccanismo d’azione
Orlistat riduce l’assorbimento dei grassi a livello del tratto gastro-intestinale, attraverso l’inibizione reversibile delle lipasi gastriche e pancreatiche. Forma un legame covalente con i residui di serina del sito attivo delle lipasi nel lume intestinale, inibendo di circa il 30% l'idrolisi dei trigliceridi alimentari in acidi grassi liberi e monogliceridi assorbibili. I trigliceridi non digeriti vengono così eliminati per via fecale, determinando un deficit calorico. L’azione farmacologica, pertanto, non si manifesta per via sistemica, bensì nel tratto digerente. Questo profilo farmaco-cinetico rende la molecola priva di effetti centrali sull'appetito o sul metabolismo.
Indicazioni
Orlistat è indicato, in associazione a un corretto stile di vita (dieta moderatamente ipocalorica e attività fisica), in pazienti adulti (≥ 18 anni) con BMI:
- ≥ 30 kg/m² (obesi);
- ≥ 27 kg/m² (sovrappeso), in presenza di altri fattori di rischio (es. ipertensione, diabete, dislipidemia).
È anche indicato nelle fasi di mantenimento per ridurre il rischio di recupero ponderale dopo precedente perdita di peso.
Popolazioni specifiche
Un utilizzo off-label prevede il trattamento con orlistat di pazienti obesi con scompenso cardiaco, come indicato dall’American Heart Association (6).
È approvato FDA per il trattamento a lungo termine dell’obesità pediatrica (≥ 12 anni) al dosaggio di 120 mg per tre volte al giorno (7), ma la scarsa tollerabilità della molecola ne limita l’utilizzo in questa popolazione. In italia non è autorizzato sotto i 18 anni.
Alcune metanalisi suggeriscono un miglioramento dei marcatori biochimici di danno epatico nei pazienti con MASLD/MASH, ma i dati sono eterogenei e non sufficienti a raccomandarne l’uso specifico per questa indicazione.
Controindicazioni
Gravidanza, allattamento, sindrome da malassorbimento cronico, colestasi, ipersensibilità a orlistat o a qualsiasi componente del prodotto.
Preparazioni, via di somministrazione, posologia
Le formulazioni disponibili in Italia sono:
- Xenical/Beacita, capsule rigide orali (120 mg)
- Alli/Beacita, capsule rigide orali (60 mg)
La dose consigliata è di 120 mg (una capsula) per tre volte al giorno, con acqua, immediatamente prima, durante o entro un’ora dal termine dei tre pasti principali. La dose inferiore da 60 mg segue lo stesso schema, ma in questo caso il farmaco non necessita di ricetta medica.
Se un pasto viene saltato o non contiene grassi, la dose può essere omessa.
Effetti collaterali
Gli effetti gastro-intestinali (i più frequenti) sono direttamente legati al meccanismo d'azione e aumentano quando orlistat viene assunto con una dieta ricca di grassi (> 30% delle calorie totali giornaliere): feci poltacee e oleose, flatulenza, aumento della frequenza di defecazione, dolore addominale, nausea, vomito, incontinenza fecale (rara).
Effetti rari ma clinicamente rilevanti:
- epato-tossicità (segnalazioni post-marketing): epatite colestatica e colelitiasi; sono stati riportati rari casi di danno epatico grave con necrosi epato-cellulare o insufficienza epatica acuta. La FDA ha aggiunto un'avvertenza sull'etichetta riguardo al potenziale danno epatico grave;
- nefropatia da ossalato: orlistat può aumentare l’eliminazione urinaria di ossalato, con rischio di iperossaluria e nefrolitiasi da ossalato di calcio in pazienti con IRC pre-esistente;
- sanguinamento rettale.
Preparazioni, via di somministrazione, posologia
Capsule prima, durante o fino ad un’ora dopo il termine dei pasti:
- cp 120 mg (Beacita, Xenical)
- cp 60 mg (non necessita di ricetta medica)(Alli, Beacita)
Precauzioni d’uso
Non esistono dati di sicurezza oltre i 4 anni di trattamento continuativo.
Il trattamento deve essere interrotto in caso di mancato raggiungimento di almeno il 5% di calo ponderale a 12 settimane dall'inizio della terapia.
Raccomandazioni dietetiche: i pazienti devono seguire una dieta nutrizionalmente bilanciata e ipocalorica, moderatamente ipolipidica, con circa il 30% delle calorie derivanti dai grassi, distribuiti in modo equilibrato nei tre pasti principali. Un eccesso di grassi nel singolo pasto aggrava gli effetti gastro-intestinali (steatorrea, urgenza defecatoria). Consigliare abbondante assunzione di frutta e verdura.
Supplementazione vitaminica: in pazienti trattati per lungo tempo con orlistat è consigliata la supplementazione con vitamine liposolubili (A, D, E, K), assumendo quotidianamente un integratore multi-vitaminico contenente queste vitamine, poiché orlistat ne riduce l'assorbimento. Il supplemento deve essere assunto preferibilmente la sera prima di coricarsi o almeno 2 ore dopo l'orlistat.
Interazioni: orlistat può interagire con diversi farmaci che richiedono intervalli di somministrazione specifici e monitoraggio più frequente:
- ciclosporina: co-somministrazione non raccomandata (riduzione dei livelli plasmatici di ciclosporina e possibile diminuzione dell’efficacia immuno-soppressiva); se inevitabile, somministrare 3 ore dopo orlistat;
- acarbosio: evitare co-somministrazione per mancanza di studi di interazione;
- levo-tiroxina: somministrare almeno 4 ore prima o dopo Orlistat (meccanismo non del tutto chiarito: possibile riduzione dell’assorbimento di sali di iodio e/o LT4);
- warfarin e altri anti-coagulanti orali: può richiedere monitoraggio più stretto dell’INR (riduzione dell’assorbimento di vitamina K e potenziamento dell’effetto anti-coagulante);
- amiodarone: controllo più attento clinico e tramite ECG (lieve diminuzione nei livelli plasmatici di amiodarone);
- farmaci anti-epilettici liposolubili (es. valproato, lamotrigina): monitorare i livelli plasmatici dei farmaci (riduzione dell’assorbimento e rischio di convulsioni);
- altre possibili interazioni (ridotta efficacia): anti-retrovirali, anti-depressivi, litio, benzodiazepine.
Anziani (> 65 anni): non è necessario un aggiustamento posologico standard, ma utilizzare con cautela per maggior rischio di carenza di vitamine liposolubili e di interazioni farmacologiche.
Pazienti in terapia contraccettiva orale: in caso di diarrea severa orlistat-correlata, l'assorbimento dei contraccettivi orali può essere ridotto; raccomandare un metodo contraccettivo aggiuntivo.
Insufficienza renale lieve o epatica lieve-moderata: non sono necessari aggiustamenti posologici, tuttavia non è stata studiata la sicurezza in condizioni di grave insufficienza.
Modalità prescrittive
Orlistat, al dosaggio di 120 mg, è un medicinale disponibile in Italia in fascia C, non rimborsabile dal SSN, totalmente a carico del paziente.
Per le formulazioni da 60 mg non è richiesta la prescrizione medica.
Scheda liraglutide per obesità
Maria Chantal Ponziani
Commissione AME Obesità
SSD Diabetologia e Malattie Metaboliche ASL Novara
(aggiornata febbraio 2024)
Meccanismo d’azione
Liraglutide è un analogo del GLP-1, con un’omologia di sequenza del 97% rispetto al peptide endogeno. Le poche sostituzioni animoacidiche e il legame con un residuo di acido grasso permettono il legame del farmaco all'albumina, quindi una maggior permanenza in circolo, con aumento dell'emivita plasmatica del farmaco.
Liraglutide, in risposta alla introduzione di zuccheri, stimola la produzione di insulina e riduce la secrezione di glucagone: pertanto è un farmaco euglicemizzante. Rallenta, inoltre, lo svuotamento gastrico, riduce l’appetito, il peso corporeo e la massa grassa (1).
Farmacocinetica e farmacodinamica
L'assorbimento di liraglutide dopo iniezione sottocutanea è lento e i livelli massimi di concentrazione si raggiungono dopo 8-12 ore dalla somministrazione. L’ esposizione del farmaco aumenta proporzionalmente alla dose. Dopo somministrazione sottocutanea, la biodisponibilità assoluta di liraglutide è di circa il 55%. Il farmaco si lega per più del 98% alle proteine plasmatiche.
Studi condotti su soggetti sani nelle 24 ore successive alla somministrazione hanno dimostrato che il componente principale nel plasma era costituito da liraglutide intatta.. La liraglutide viene metabolizzata in modo simile alle proteine di grandi dimensioni; pertanto, non è stato identificato un organo principalmente responsabile della sua eliminazione. La clearance media dopo somministrazione sottocutanea di una singola dose è di circa 0.9-1.4 L/h, con emivita di eliminazione di circa 13 ore (2).
Indicazioni
Il farmaco è indicato, in aggiunta a una dieta ipocalorica e all’aumento dell’attività fisica, per la gestione del peso corporeo in pazienti adulti con BMI iniziale ≥ 30 kg/m2 (obesità), o ≥ 27 kg/m2 in presenza di comorbilità correlate al peso, come alterazioni del metabolismo glicidico, ipertensione arteriosa, dislipidemia o sindrome delle apnee notturne.
Il farmaco è indicato, sempre in aggiunta a una dieta ipocalorica e all’aumento dell’attività fisica, per la gestione del peso corporeo anche in adolescenti (> 12 anni) con obesità (corrispondente a BMI ≥30 kg/m2 per gli adulti secondo le tabelle internazionali, tabella 1) o con peso corporeo > 60 kg.
La terapia deve essere interrotta dopo 12 settimane se non si ottiene una riduzione del peso corporeo di almeno il 5% nell’adulto e 4% nell’adolescente (3).
| Tabella 1 Valori soglia di BMI per l'obesità tra i 12-18 anni in base al sesso secondo l’IOTF BMI corrispondente a 30 kg/m2 per gli adulti secondo i valori soglia internazionali (3) |
||
| Età | Maschi | Femmine |
| 12 | 26.02 | 26.67 |
| 12.5 | 26.43 | 27.24 |
| 13 | 26.84 | 27.76 |
| 13.5 | 27.25 | 28.20 |
| 14 | 27.63 | 28.57 |
| 14.5 | 27.98 | 28.87 |
| 15 | 28.30 | 29.11 |
| 15.5 | 28.60 | 29.29 |
| 16 | 28.88 | 29.43 |
| 16.5 | 29.14 | 29.56 |
| 17 | 29.41 | 29.69 |
| 17.5 | 29.70 | 29.84 |
| 18 | 30 | 30 |
Controindicazioni/non indicazioni
Gravidanza e allattamento: il trattamento deve essere interrotto in programmazione di gravidanza o se si verifica una gravidanza (4).
La sicurezza e l’efficacia di liraglutide per la gestione del peso corporeo non sono state stabilite e pertanto l’uso non è raccomandato in pazienti (3):
- di età ≥ 75 anni;
- trattati con altri prodotti per la gestione del peso;
- con obesità secondaria a disturbi endocrinologici o dell’alimentazione, oppure al trattamento con medicinali che possono causare un aumento di peso;
- con compromissione severa della funzionalità renale;
- con compromissione severa della funzionalità epatica;
- affetti da insufficienza cardiaca congestizia di classe IV NYHA;
- affetti da malattia infiammatoria intestinale o gastroparesi diabetica.
Precauzioni d'uso
I pazienti devono essere informati del possibile aumentato rischio delle condizioni sotto riportate ed istruiti a riconoscere la sintomatologia (5,6):
- pancreatiti;
- colelitiasi e colecistite;
- patologia tiroidea;
- aumento frequenza cardiaca;
- rischio ipoglicemico (riduzione del fabbisogno insulinico e/o di sulfoniluree per calo ponderale ed effetto euglicemizzante del farmaco);
- rischio iperglicemico (eccessiva riduzione/sospensione insulinica in soggetti insulino-trattati con scarsa secrezione insulinica residua).
Preparazioni, via di somministrazione, posologia
La dose iniziale del farmaco (Saxenda) è di 0.6 mg una volta al giorno al mattino prima di colazione. Per migliorare la tollerabilità gastro-enterica, la dose deve essere aumentata con incrementi di 0.6 mg a intervalli di almeno una settimana, fino a 3.0 mg una volta al giorno o alla dose massima tollerata. Non sono raccomandate dosi > 3.0 mg/die.
Negli adolescenti (12-18 anni) deve essere applicato un programma di aumento della dose simile a quello degli adulti.
| Tabella 2 Schema di titolazione del farmaco (3) |
|
| Dose (mg) | Settimane |
| 0.6 | 1 |
| 1.2 | 1 |
| 1.8 | 1 |
| 2.4 | 1 |
| 3.0 | Mantenimento |
Effetti collaterali
È generalmente ben tollerato, con effetti collaterali prevalentemente gastro-intestinali e raramente è necessaria la sospensione (7).
| Classificazione per sistemi e organi | Frequenza eventi avversi |
| Disturbi del metabolismo | Comune: ipoglicemia Non comune: disidratazione |
| Sistema nervoso | Molto comune: cefalea Comuni: vertigini, disgeusia |
| Patologie cardiache | Non comune: tachicardia |
| Patologie gastro-intestinali | Molto comuni: nausea, vomito, diarrea, stipsi Comuni: bocca secca, dispepsia, gastrite, malattia da reflusso gastro-esofageo, dolore addominale superiore, flatulenza, eruttazione, distensione dell’addome Non comune: pancreatite |
| Patologie epato-biliari | Comune: colelitiasi Non comune: colecistite |
| Patologie renali e urinarie | Rari: insufficienza renale acuta, compromissione della funzionalità renale |
| Patologie della cute e della sede di somministrazione | Comune: reazioni al sito di iniezione Non comune: orticaria |
| Patologie sistemiche | Comuni: astenia, affaticamento |
| Alterazione esami ematici | Comuni: aumento lipasi e amilasi |
Per ridurre l’incidenza degli eventi avversi, è necessario rispettare la titolazione suggerita e aumentare il dosaggio solo alla scomparsa di eventuali sintomi. Per prevenire o contrastare la nausea, evento avverso più comune, si consiglia di mangiare in piccole porzioni ed evitare cibi ricchi di grassi (8).
Relativamente alla possibile correlazione con il tumore pancreatico, un recentissimo studio storico di coorte con follow-up di 7 anni non ha documentato l’aumento dell’incidenza in pazienti trattati con analoghi GLP-1 (9).
Un’analisi dei dati provenienti dallo studio SCALE ha documentato che liraglutide 3 mg può determinare un aumento dose-indipendente di amilasi e lipasi, non correlato ad aumentato rischio di pancreatite acuta, Per tale ragione, gli autori concludono che non vi sono ragioni per proporre il monitoraggio di tali enzimi nei pazienti asintomatici durante la terapia con il farmaco (10).
Prescrivibilità
Prescrivibile in classe C per quanto attiene alla rimborsabilità (pertanto è a totale carico dell’assistito), ma con Ricetta Medica Ripetibile (RR). Cade il limite iniziale che ne aveva fatto un farmaco con prescrizione riservata allo specialista.
Bibliografia
- Knudsen LB, et al. The discovery and development of liraglutide and semaglutide. Front Endocrinol 2019,10: 155.
- Meece J. Pharmacokinetics and pharmacodynamics of liraglutide, a long-acting, potent glucagon-like peptide-1 analog. Pharmacotherapy 2009, 29: 33S-43S.
- AIFA. Riassunto delle caratteristiche del prodotto.
- Dion R, et al. Effects of GLP-1 agonists and SGLT2 inhibitors during pregnancy and lactation on offspring outcomes: a systematic review of the evidence. Front Endocrinol 2023,14: 1215356.
- Troels MJ, et al. Is there a link between liraglutide and pancreatitis? A post hoc review of pooled and patient-level data from completed liraglutide type 2 diabetes clinical trials. Diabetes Care 2015, 38: 1058-66.
- Long B, et al. GLP-1 agonists: a review for emergency clinicians. Am J Emerg Med 2024, 78: 89-94.
- Fujioka K. Safety and tolerability of medications approved for chronic weight management. Obesity 2015, 23 suppl 1: S7-11.
- AME, ADI, SIO, SICOB, SIGE. Linea guida per la terapia del sovrappeso e dell’obesità resistenti al trattamento comportamentale nella popolazione adulta con comorbilità metaboliche. LG ISS 193, 2023.
- Dankner R, et al. Glucagon-like peptide-1 receptor agonists and pancreatic cancer risk in patients with type 2 diabetes. JAMA Netw Open 2024, 7: e2350408.
- Steinberg WM, et al. Impact of liraglutide on amylase, lipase, and acute pancreatitis in participants with overweight/obesity and normoglycemia, prediabetes, or type 2 diabetes: secondary analyses of pooled data from the SCALE clinical development program. Diabetes Care 2017, 40: 839-48.
Scheda semaglutide ad alte dosi
Luisa Lener
Commissione AME Obesità
UO Endocrinologia, Università Cattolica del Sacro Cuore, Fondazione Policlinico Universitario “A. Gemelli”, IRCCS, Roma
(aggiornato febbraio 2024)
Meccanismo d’azione
Semaglutide è un analogo del GLP-1, con un’omologia di sequenza del 94% rispetto alla molecola endogena.
Controlla la glicemia, limita l’apporto calorico individuale determinando una significativa perdita di peso. Inoltre, ha anche un'azione specifica a livello centrale sui circuiti di ricompensa e interagendo con gli organi circum-ventricolari influisce sulle abitudini alimentari, favorendo una preferenza per cibi con minor contenuto lipidico (1).
Farmacocinetica e farmacodinamica
Il GLP-1 nativo viene degradato in pochi minuti ad opera della dipeptidil-difosfatasi 4 (DPP-4), per cui non può essere somministrato a scopi terapeutici. Grazie a una specifica modifica di 3 aminoacidi chiave, semaglutide è meno suscettibile alla degradazione da parte della DPP-4, il che le conferisce un'emivita di circa 165 ore, che nella pratica clinica consente la somministrazione settimanale (2).
Viene metabolizzata tramite clivaggio proteolitico, e solo minimamente a livello epatico e renale; i suoi metaboliti vengono escreti tramite urine e feci (2). Per tale motivo, non sono necessari aggiustamenti posologici in caso di insufficienza renale (fino a eGFR > 15 mL/min) (3) ed epatica lieve-moderata (Child-Pugh A) (4).
Indicazioni
Trattamento di seconda linea dell’obesità, al fallimento delle terapie comportamentali (come esercizio fisico e dieta) in pazienti > 18 anni con BMI > 30 kg/m2 o > 27 kg/m2 con almeno una complicanza correlata al peso, in combinazione con le terapie comportamentali (5,6).
Controindicazioni
Gravidanza (in caso di pianificazione di gravidanza, si consiglia di sospendere almeno 2 mesi prima), allattamento.
Non è raccomandato (per assenza di studi specifici) nelle seguenti popolazioni: diabete tipo 1, insufficienza renale terminale (eGFR < 15 mL/min), insufficienza epatica severa (Child-Pugh B e C), insufficienza cardiaca severa (NYHA classe IV).
Precauzioni d'uso
Usare con cautela in caso di anamnesi personale di carcinoma midollare della tiroide, MEN-2, pancreatite.
Preparazioni, via di somministrazione, posologia
Il farmaco (Wegovy) è disponibile in formulazione iniettiva, sotto forma di penne pre-riempite di dosaggio crescente (0.25 mg, 0.50 mg, 1 mg, 1.7 mg, 2.4 mg), da conservare in frigorifero (2–8°C) e somministrare sottocute una volta a settimana.
Si inizia dal dosaggio minimo di 0.25 mg e, se ben tollerato, si può aumentare al dosaggio superiore ogni quattro settimane, fino alla dose massima di 2.4 mg/settimana. In caso di effetti collaterali sarà necessario posticipare l’aumento della dose sino alla scomparsa, o alla riduzione, dei disturbi.
Una volta iniziato il trattamento e raggiunta la perdita di peso massima ottenibile (o il target personale), è necessario continuare il trattamento per mantenere gli effetti ottenuti. È possibile considerare l'impiego di dosi di mantenimento ridotte, ricorrendo alla minima dose efficace. In caso di parziale recupero del peso, è possibile incrementare nuovamente la dose di mantenimento.
Effetti collaterali
È generalmente ben tollerato, gli effetti collaterali sono prevalentemente gastro-intestinali e raramente è necessaria la sospensione (7).
| Effetto collaterale | Frequenza | Meccanismo |
| Nausea | Molto comune | Ridotto svuotamento gastrico e azione su SNC |
| Vomito | Comune | Ridotto svuotamento gastrico |
| Stipsi | Comune | Ridotta motilità intestinale |
| Diarrea | Molto comune | Possibile azione osmotica |
| Colelitiasi | Comune | Rapida perdita di peso e ridotta motilità colecistica |
| Iperamilasemia e iperlipasemia | Comune | Stimolo pancreas |
| Pancreatite acuta | Non comune | Stimolo pancreas |
Gli effetti collaterali sono parzialmente correlati al meccanismo d’azione del farmaco e dose-dipendenti. Per ridurne l’incidenza, è necessario rispettare la titolazione suggerita dal produttore e, comunque, aumentare il dosaggio solo alla scomparsa di eventuali sintomi. Per contrastare la nausea, il principale effetto avverso evidenziato dai pazienti, si consiglia di mangiare in piccole porzioni e di evitare cibi particolarmente grassi (8).
È stata esclusa la correlazione con lo sviluppo di carcinomi pancreatici (9). Studi su liraglutide, appartenente alla stessa famiglia, non hanno dimostrato correlazioni tra l’incremento dei livelli sierici di lipasi e amilasi e aumentata incidenza di pancreatite acuta (10); per tale motivo, AIFA non indica la sospensione del farmaco in caso di rialzo asintomatico di tali enzimi.
Prescrivibilità
Non limitazioni.
Bibliografia
- Gabery S, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 2020, 5: e133429.
- Jensen L, et al. Absorption, metabolism and excretion of the GLP-1 analogue semaglutide in humans and nonclinical species. Eur J Pharmac Sci 2017, 104: 31–41.
- Marbury TC, et al. Pharmacokinetics and tolerability of a single dose of semaglutide, a human glucagon-like peptide-1 analog, in subjects with and without renal impairment. Clin Pharmacokinet 2017, 56: 1381–90.
- Gangopadhyay KK, Singh P. Consensus statement on dose modifications of antidiabetic agents in patients with hepatic impairment. Indian J Endocrinol Metab 2017, 21: 341–54.
- Chao AM, et al. Semaglutide for the treatment of obesity. Trends Cardiovasc Med 2023, 33: 159–66.
- AME, ADI, SIO, SICOB, SIGE. Linea guida per la terapia del sovrappeso e dell’obesità resistenti al trattamento comportamentale nella popolazione adulta con comorbilità metaboliche. LG ISS 193, 2023.
- Wadden TA, et al. Effect of subcutaneous semaglutide vs placebo as an adjunct to intensive behavioral therapy on body weight in adults with overweight or obesity: the STEP 3 randomized clinical trial. JAMA 2021, 325: 1403–13.
- Shomali M. Optimizing the care of patients with type 2 diabetes using incretin-based therapy: focus on GLP-1 receptor agonists. Clin Diabetes 2014, 32: 32-43.
- Egan AG. Pancreatic safety of incretin-based drugs — FDA and EMA Assessment. N Engl J Med 2014, 370: 794-7.
- Steinberg WM, et al. Impact of liraglutide on amylase, lipase, and acute pancreatitis in participants with overweight/obesity and normoglycemia, prediabetes, or type 2 diabetes: secondary analyses of pooled data from the SCALE clinical development program. Diabetes Care 2017, 40: 839–48.
Valutazione multi-disciplinare del candidato all'intervento di chirurgia bariatrica
Giovanni De Pergola
Ambulatorio di Nutrizione Clinica, UOC di Oncologia Clinica, Dipartimento di Scienze Biomediche e Oncologia Umana, Università di Bari
La totalità delle più recenti linee guida internazionali e nazionali (SICOB 2008) prevede che il paziente obeso candidato alla chirurgia bariatrica venga sottoposto ad una approfondita valutazione pre-operatoria multidisciplinare. Questa dovrebbe essere affidata un team interdisciplinare, composto da esperti dedicati (chirurgo bariatrico, medico internista, anestesista, psicologo o psichiatra, nutrizionista e/o dietista, ecc.). Tale valutazione non deve essere necessariamente collegiale, purchè siano tenuti in giusta considerazione tutti i pareri espressi dai diversi componenti del team interdisciplinare.
Chirurgo bariatrico
Il chirurgo bariatrico deve avere competenze specifiche sulle tecniche operatorie, tali da potersi fare carico di tutte le fasi terapeutiche: selezione dei pazienti, scelta dell'intervento, fase peri-operatoria, assistenza post-operatoria, gestione delle eventuali complicanze, follow-up programmati (1). I Centri dove opera il chirurgo bariatrico dovrebbero assicurare un'attività continuativa, non inferiore ai 40 interventi per anno per quelli di 1° livello ed agli 80 interventi per anno per quelli di Riferimento regionale (1).
Internista
Una serie di patologie internistiche ha prevalenza maggiore nei pazienti obesi rispetto alla popolazione generale. In particolare, hanno maggiore prevalenza nell'obesità le patologie metaboliche (insulino-resistenza, diabete mellito tipo 2, dislipidemie, iperuricemia), cardiovascolari (ipertensione, cardiopatia ischemica, fibrillazione atriale, scompenso cardiaco, ictus, arteriopatia periferica, tromboflebiti, insufficienza del circolo linfatico degli arti inferiori), respiratorie (insufficienza respiratoria restrittiva, sindrome delle apnee notturne ostruttive, asma su base allergica), gastro-enterologiche (reflusso gastro-esofageo, colelitiasi, NAFLD, NASH), nefrologiche (sindrome nefrosica e insufficienza renale), osteoarticolari, cancro e anemia (prevalentemente sideropenica). Pertanto, è fondamentale la valutazione internistica dei pazienti candidati alla chirurgia bariatrica.
Endocrinologo
Le obesità secondarie a endocrinopatie (ipotiroidismo, Cushing, sindrome dell'ovaio policistico, deficit di GH, insulinoma) rappresentano complessivamente una percentuale inferiore al 10%, ma è imprescindibile una valutazione endocrinologica prima dell'intervento bariatrico, ad opera di un endocrinologo o di un internista con buone basi endocrinologiche.
Anestesista
L'obesità è associata a numerosi fattori di comorbilità che possono essere potenziali contro-indicazioni alla stessa chirurgia e, pertanto, la gestione dei pazienti obesi richiede una buona esperienza e una specifica competenza dell'anestesista.
Le alterazioni respiratorie rappresentano la prima causa di morbilità peri-operatoria nell'obeso. In particolare, una ipertensione arteriosa polmonare deve esortare alla prudenza e il grasso che infiltra i muscoli intercostali, il diaframma e l'addome riduce la compliance del torace, parietale e polmonare. La capacità funzionale residua diminuisce in modo esponenziale quando il BMI aumenta e questa riduzione si accentua in decubito dorsale.
Ancora, le conseguenze dell'obesità sulla cinetica dei farmaci sono molto variabili da un agente all'altro e due farmaci di una stessa classe terapeutica possono comportarsi in modo diverso. Pertanto, è lecito privilegiare l'utilizzo delle molecole meno liposolubili e con la durata d'azione più breve possibile.
Analisi psicologica
Il livello di competenza degli operatori che devono effettuare la valutazione dello stato mentale, psicologo e/o psichiatra, varia sulla base della tipologia e dell’entità del problema del paziente: sindromi psichiatriche maggiori, disturbi del comportamento alimentare (DCA), disturbi di personalità. Gli strumenti con cui si realizza la valutazione psichiatrica-psicologica sono il colloquio clinico e l’indagine psicometrica.
Il colloquio clinico serve ad indagare lo stato mentale del soggetto, la compliance all’intervento, l’anamnesi atta ad individuare condizioni psico-patologiche ostative all’intervento o che richiedono un trattamento specifico (farmacologico e/o psicoterapeutico) pre-operatorio e/o post-operatorio. L’indagine anamnestica è necessaria per riconoscere sindromi o eventi (abusi familiari, maltrattamenti) che possono interferire con il periodo post-operatorio, determinando un adattamento perturbato al cambiamento indotto dall’intervento o causare il fallimento del processo di cura o facilitare lo sviluppo di quadri psicopatologici di compenso.
L’indagine psicometrica mediante test, che soddisfa anche il criterio della riproducibilità, può fornire dati oggettivabili su personalità, condizioni sintomatiche relativamente al tono dell’umore, rapporto con il cibo, immagine corporea, presenza, intensità e qualità della componente impulsiva e qualità di vita del paziente candidato alla chirurgia bariatrica.
Il razionale e gli obiettivi della valutazione dello stato mentale nel paziente candidato alla chirurgia bariatrica possono essere riassunti in due punti principali.
- Esistono quadri psicopatologici i cui sintomi (per esempio il “binge eating”) condizionano l’aumento eccessivo di peso, ma esistono anche attitudini psicologiche e comportamentali che, pur non configurando un quadro clinico di rilevanza diagnostica, possono condizionare l’esito dell’intervento bariatrico e/o il benessere fisico e psichico del paziente operato, anche indipendentemente dal livello di riduzione ponderale raggiunto. Inoltre, la natura stessa di alcuni disturbi della sfera psichica non consente al paziente di costruire una motivazione e, quindi, la capacità di gestire il percorso bariatrico. In tutti questi casi, la valutazione psicologica può permettere sia di individuare fattori di rischio sia di fornire la base per programmare un trattamento mirato pre-operatorio e/o post-operatorio, migliorando l’outcome del paziente a lungo termine. Infine, l’assunzione di psicofarmaci può indurre un significativo incremento ponderale e rendere difficile ottenere un efficace dimagrimento. È quindi fondamentale la possibilità di proseguire con efficacia le terapie psicofarmacologiche del periodo pre-operatorio, particolarmente negli interventi di tipo prevalentemente malassorbitivo, che possono interferire con l’assorbimento dei farmaci psicotropi. In tali casi, prima di dare il consenso all’intervento, è opportuno considerare la farmacocinetica e la possibilità di sostituzione della via di somministrazione dei farmaci (per es. sostituzione della via orale con quella intramuscolare), al fine di evitare un peggioramento del disturbo mentale nel post-operatorio.
- Esistono stati psicopatologici che per gravità e qualità invalidante a lungo termine rappresentano delle reali controindicazioni alla chirurgia bariatrica. In questi casi, l’eventuale indicazione può essere posta solo individualmente per gravi motivi medici (prognosi quoad vitam infausta per motivi legati all’obesità), ma sempre con il formale consenso dello psichiatra di riferimento.
In tutti i casi devono essere considerate le condizioni socio-economiche del paziente ed il livello di supporto familiare e sociale. Infatti, il follow-up richiede controlli clinici frequenti ed alcuni necessitano di terapie di supplementazione nutrizionale continuative (minerali, vitamine, aminoacidi) al fine di evitare stati carenziali. È quindi necessario accertarsi che il paziente possa essere seguito ed aiutato da persone a lui vicine e abbia la possibilità economica di sostenere il costo di tali terapie.
Dietista e/o Nutrizionista
Il ruolo del dietista (e/o nutrizionista) è quello di esaminare le comuni abitudini alimentari, come la colazione, la quantità e la frequenza di assunzione dei vari alimenti, eventualmente richiedendo al paziente di compilare un diario alimentare. Tali informazioni possono contribuire a comprendere l'origine della obesità, ma sono utili soprattutto per prospettare i cambiamenti delle abitudini alimentari che saranno indispensabili dopo l'intervento. Inoltre, a parte il ruolo specifico dello psicologo e/o dello psichiatra, anche il dietista (e/o nutrizionista) ha un ruolo importante nel valutare se il paziente possa fornire un consenso realmente consapevole ed informato ed abbia la giusta motivazione e disponibilità ad aderire a un programma dietologico di follow-up.
Altri specialisti che potrebbero essere importanti in una equipe multidisciplinare per specifiche tipologie di casi sono l'endoscopista, il chirurgo plastico, il cardiologo, il pneumologo e il fisiatra.
Bibliografia
Gli interventi di chirurgia bariatrica
Carlo Nagliati1 & Giuseppe M Marinari2
1Dipartimento di Chirurgia, Ospedale San Giovanni di Dio, Gorizia
2UO di Chirurgia Bariatrica, Humanitas Research Hospital, Rozzano (MI)
(aggiornato al 28 aprile 2022)
La chirurgia dell’obesità è una branca relativamente recente della chirurgia generale: i primi interventi, quasi aneddotici, risalgono agli anni ’50. Negli anni ’80 gli interventi bariatrici, tutti rigorosamente con accesso laparotomico, hanno cominciato ad essere praticati con numeri crescenti nel mondo occidentale. Negli anni ’90 due fenomeni contemporanei hanno contribuito all’esplosione della chirurgia bariatrica: l’imporsi dell’accesso laparoscopico e la contemporanea pandemia della malattia obesità. Il secondo dei due fenomeni ha portato ad un rapido sviluppo delle procedure anche in continenti (Medio ed Estremo Oriente, Sudamerica) dove fino a 20–25 anni fa la chirurgia dell’obesità praticamente non esisteva. Purtroppo, l’età ancora giovane fa sì che questa chirurgia non sia ad oggi regolata da una rigorosa EBM, sia perché continuano a nascere nuove proposte chirurgiche, sia perchè è molto difficile trovare studi RCT con follow-up prolungato (1).
Parlando di tecniche chirurgiche, è giusto puntualizzare che ormai tutti gli interventi si eseguono con accesso laparoscopico: solo in alcuni re-interventi, specie se successivi a plurime precedenti operazioni, è prudente utilizzare un accesso tradizionale laparotomico.
Le tecniche chirurgiche agiscono con differenti meccanismi di azione: restrittivo, malassorbitivo ed entero-ormonale, oppure — nella gran parte dei casi — con una combinazione di questi elementi. La perdita di peso si ottiene, quindi, con la riduzione forzata dell’introito di cibo, la riduzione dell’assorbimento intestinale e la modifica della secrezione di ghrelina, CCK, GLP-1 e PYY, responsabili di riduzione dell’appetito e sazietà precoce (2).
Secondo recenti indagini internazionali, gli interventi maggiormente eseguiti a livello mondiale sono rappresentati da Sleeve Gastrectomy (46-53%), Roux-en-Y Gastric Bypass (30-38%) e One-Anastomosis Gastric Bypass (5-7%) (3,4). Ancora presente, per quanto in forte calo, il Bendaggio Gastrico (3-5%) (3-5).
Negli ultimi anni oltre agli interventi chirurgici si stanno sviluppando le procedure endoscopiche (6,7), che costituiscono circa il 4% del totale (4). Al pallone intra-gastrico, si sono affiancate di recente diverse tecniche di sutura/plicatura gastrica e molteplici procedure di barriera o derivazione gastro-intestinale, quest'ultime perlopiù ancora sperimentali (6).
È opportuno segnalare che tutti gli interventi, se vengono rispettate le indicazioni e limitazioni, sono in convenzione SSN, comprese tutte le procedure di selezione pre-operatoria, di preparazione all'intervento e di follow-up post-operatorio.
PALLONE INTRA-GASTRICO
È una tecnica endoscopica, che si attua in sedazione profonda e non in anestesia generale: in corso di EGDS viene introdotto nello stomaco un pallone sgonfio, che viene poi gonfiato con aria o acqua e rilasciato nel fondo gastrico. La sua azione è transitoria, poiché dopo massimo 6-12 mesi di permanenza il pallone va rimosso con una nuova endoscopia, per solito un po’ più indaginosa della prima (in alcuni centri la rimozione viene attuata in anestesia generale).
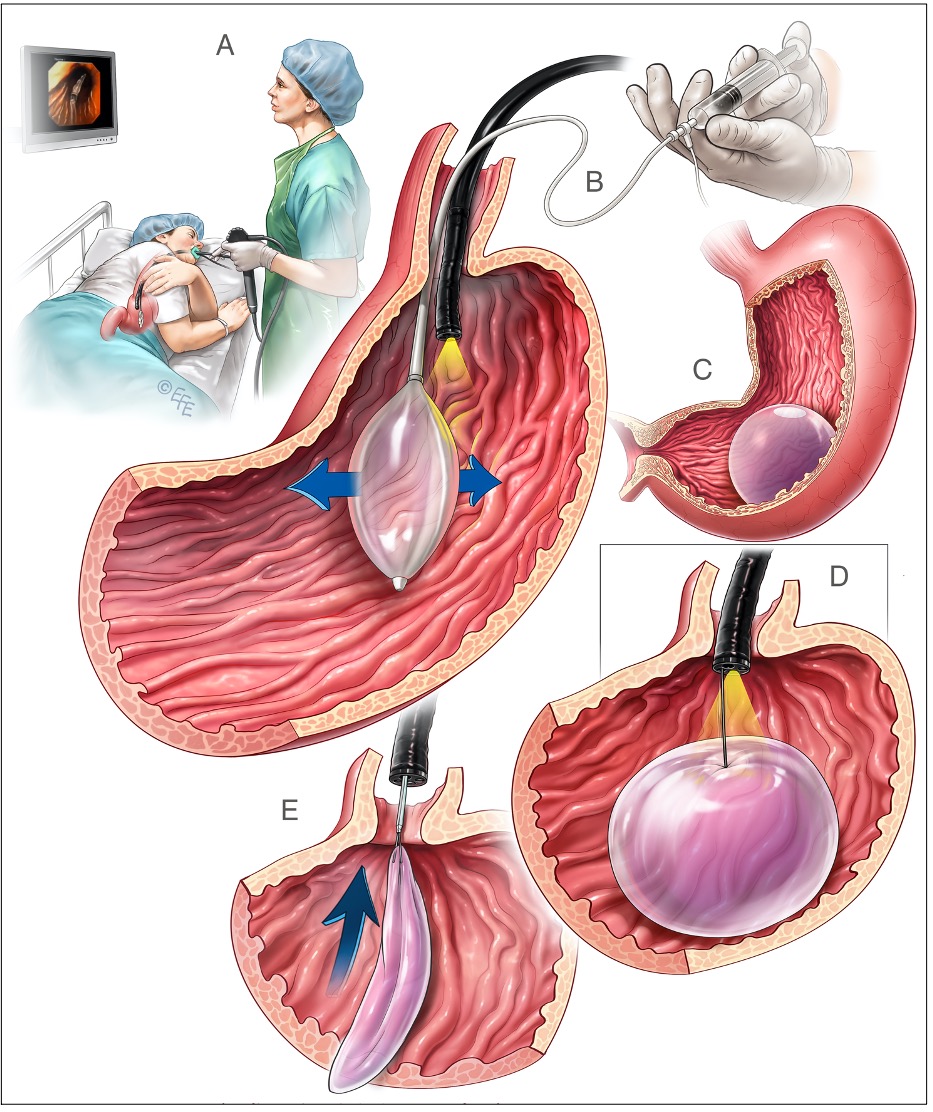
Figura 1: pallone intra-gastrico
BENDAGGIO GASTRICO (ASGB o LASGB, laparoscopic adjustable silicone gastric banding)
È stato a lungo l’intervento più diffuso in Italia e in Europa, sorpassato solo nel 2012 in Italia dalla sleeve gastrectomy (fonte: Registro Nazionale SICOB). L’intervento consiste nel posizionare un anello di silicone in regione immediatamente sotto-cardiale: si crea così una minima tasca gastrica prossimale (di circa 15 mL), che comunica con la restante porzione di stomaco distale attraverso un passaggio ristretto. Il bendaggio ha una parte pneumatica regolabile, collegata tramite un tubicino a un serbatoio posizionato nel sotto-cute: è quindi possibile, insufflando e desufflando, regolare il calibro della stenosi. Durante il pasto la piccola tasca gastrica prossimale si distende rapidamente, provocando un senso di sazietà (riferito a volte come fastidioso se non doloroso) e limitando quindi l’assunzione di cibo.
I vantaggi di questa procedura, che ha molto contribuito alla diffusione della chirurgia bariatrica, sono il minor rischio chirurgico rispetto alle altre procedure (in termini sia di mortalità sia di morbilità peri-operatoria), la reversibilità (l’anatomia del tubo digerente rimane intatta) e l'assenza di sequele metaboliche o nutrizionali a lungo termine. Tuttavia, va ricordato che i risultati sono molto legati al grado di collaborazione del paziente (capacità di modificare da subito e a lungo termine le abitudini alimentari) e che vi è un’elevata percentuale di complicazioni esofago-gastriche a medio-lungo termine (esofagite, mancata coordinazione peristaltica del 3° inferiore dell'esofago, dilatazione esofagea, dilatazione della tasca gastrica pre-bendaggio, stenosi gastrica), tali da obbligare a un'elevata percentuale di re-interventi (40% e oltre): in pratica il bendaggio è la procedura chirurgica meno pericolosa, ma al tempo stesso la meno efficace in termini di calo ponderale e di cura delle comorbilità, e quella che richiede più spesso un secondo intervento (1,8).
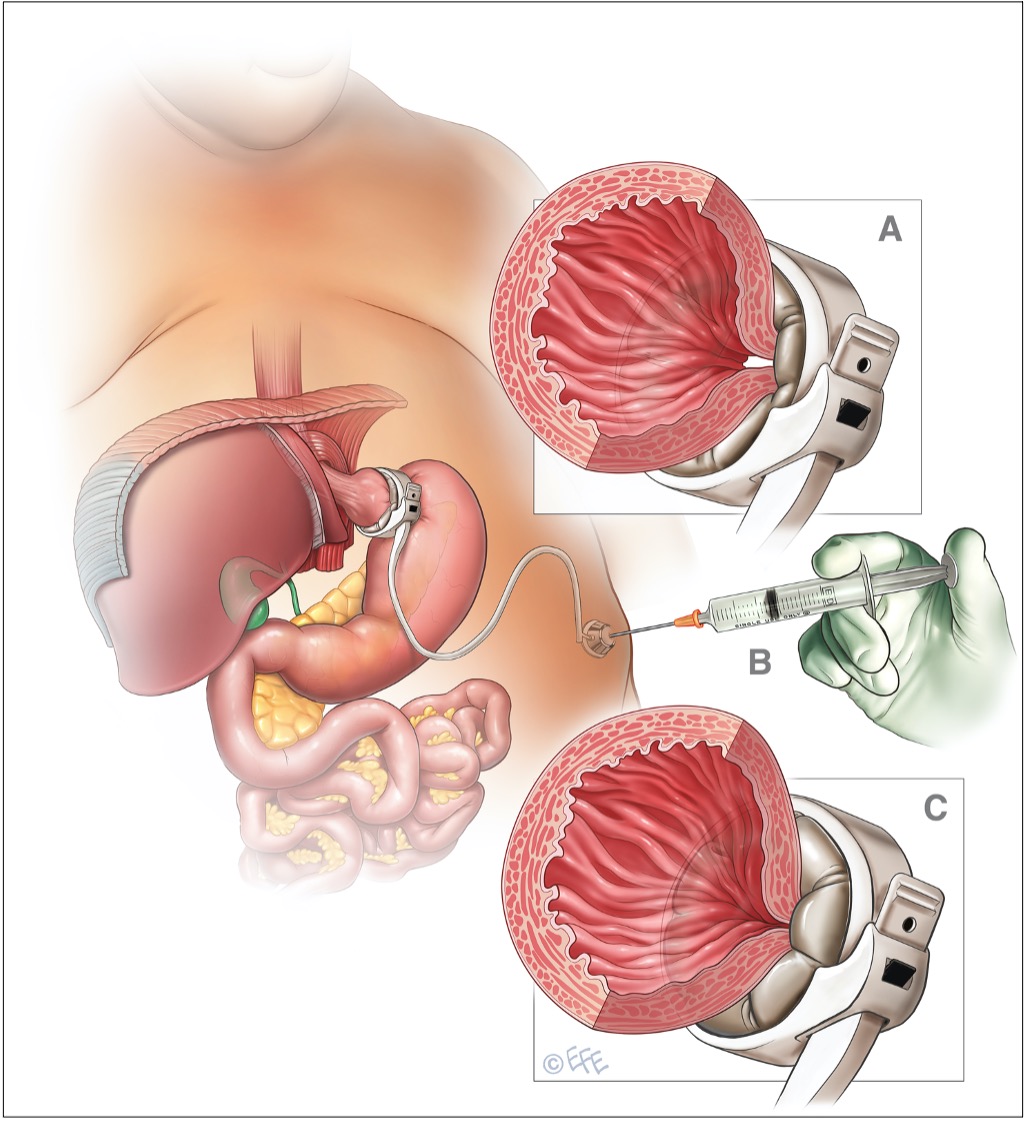
Figura 2: bendaggio gastrico regolabile
GASTROPLASTICA VERTICALE (VBG, vertical banded gastroplasty)
Funzionamento e risultati molto simili al Bendaggio Gastrico, ma rischio chirurgico decisamente maggiore, poiché la tasca gastrica prossimale si ottiene con una serie di suture e incisioni sullo stomaco. A causa di ciò è un intervento ormai pressochè scomparso, sia negli USA, dove la VBG era nata e dove nei primi anni ’90 era largamente eseguita, che nel resto del mondo.
SLEEVE GASTRECTOMY (SG)
Consiste in una resezione gastrica verticale, con tubulizzazione dello stomaco residuo. L’asportazione del fondo gastrico, con le sue cellule secernenti ghrelina, sembra il fulcro dell’intervento. La SG è un intervento abbastanza rapido, con un tempo di esecuzione medio < 60 minuti, ha minime complicazioni nutrizionali e soprattutto si segnala per una buona qualità di vita post-operatoria: per questi motivi è diventato l’intervento più eseguito e al tempo stesso più richiesto. Dal 2014 risulta l'intervento più eseguito al mondo (4,7).
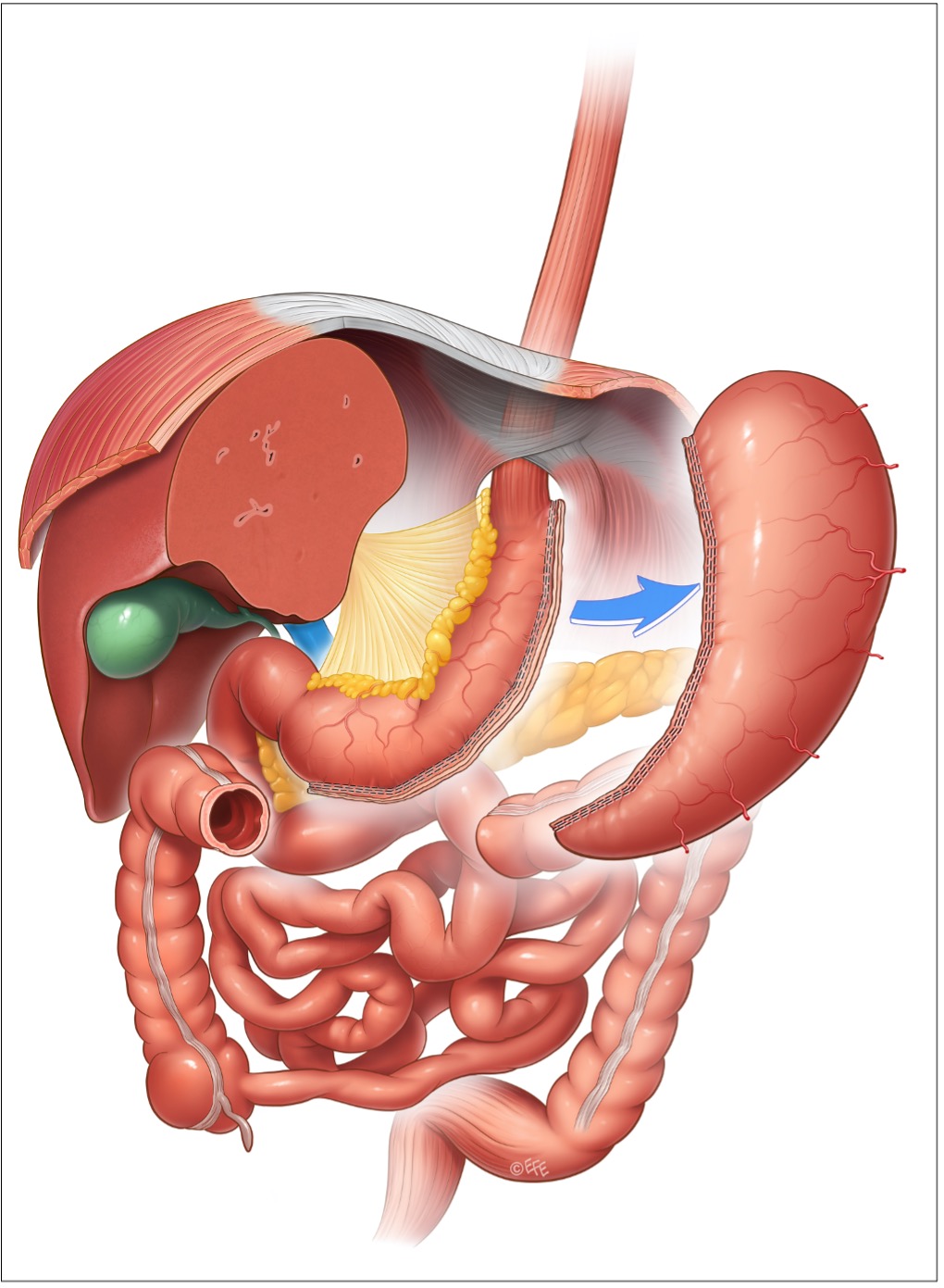
Figura 3: sleeve gastrectomy
BY-PASS GASTRICO (RYGB, Roux-en-Y gastric bypass)
È una procedura chirurgicamente più complessa: consiste nella creazione di una piccola tasca gastrica (volume pari a circa 30 mL), che viene completamente separata dal restante stomaco, che non viene asportato. Nella versione standard la continuità gastro-intestinale viene quindi ricostruita su un’ansa ad Y, dove il tratto intestinale anastomizzato allo stomaco (tratto alimentare) è lungo circa 100-150 cm, mentre il tratto intestinale, proveniente dal legamento di Treitz (tratto bilio-pancreatico, proveniente dallo stomaco escluso e dal duodeno) ed anastomizzato al tratto alimentare, è per solito lungo 70-100 cm: dall’anastomosi fra i 2 tratti intestinali l’intestino è unico e viene chiamato tratto comune.
Esistono numerose versioni di by-pass gastrico, che inducono a una certa confusione quando si parla del suo meccanismo di azione: poiché le piccole dimensioni della tasca gastrica conferiscono al RYGBP delle caratteristiche restrittive meccaniche, vi sono chirurghi che mettono un bendaggio non regolabile intorno all’anastomosi fra tasca gastrica e intestino, esaltando così le caratteristiche restrittive del RYGBP (banded RYGBP o by-pass gastrico di Fobi). Vi sono invece chirurghi che con l’aumentare del BMI spostano sempre più a valle verso la valvola ileo-cecale l’anastomosi fra tratto alimentare e tratto bilio-pancreatico, allungandoli entrambi a spese del tratto comune e spingendo il by-pass gastrico verso caratteristiche malassorbitive (Distal RYGBP o Long Limb RYGBP). In questo capitolo si fa riferimento al by-pass gastrico standard.
Il RYGBP è stata la procedura più eseguita negli ultimi 15 anni negli USA, ed è stato praticato in grossi numeri anche nel resto del mondo, al punto che oggi è considerato un gold-standard nella chirurgia dell’obesità: è molto efficace nella terapia del diabete tipo 2 e al contrario del bendaggio gastrico ha un tasso di re-interventi piuttosto basso. L’esclusione del duodeno dal transito alimentare comporta comunque un ridotto assorbimento di ferro e calcio.
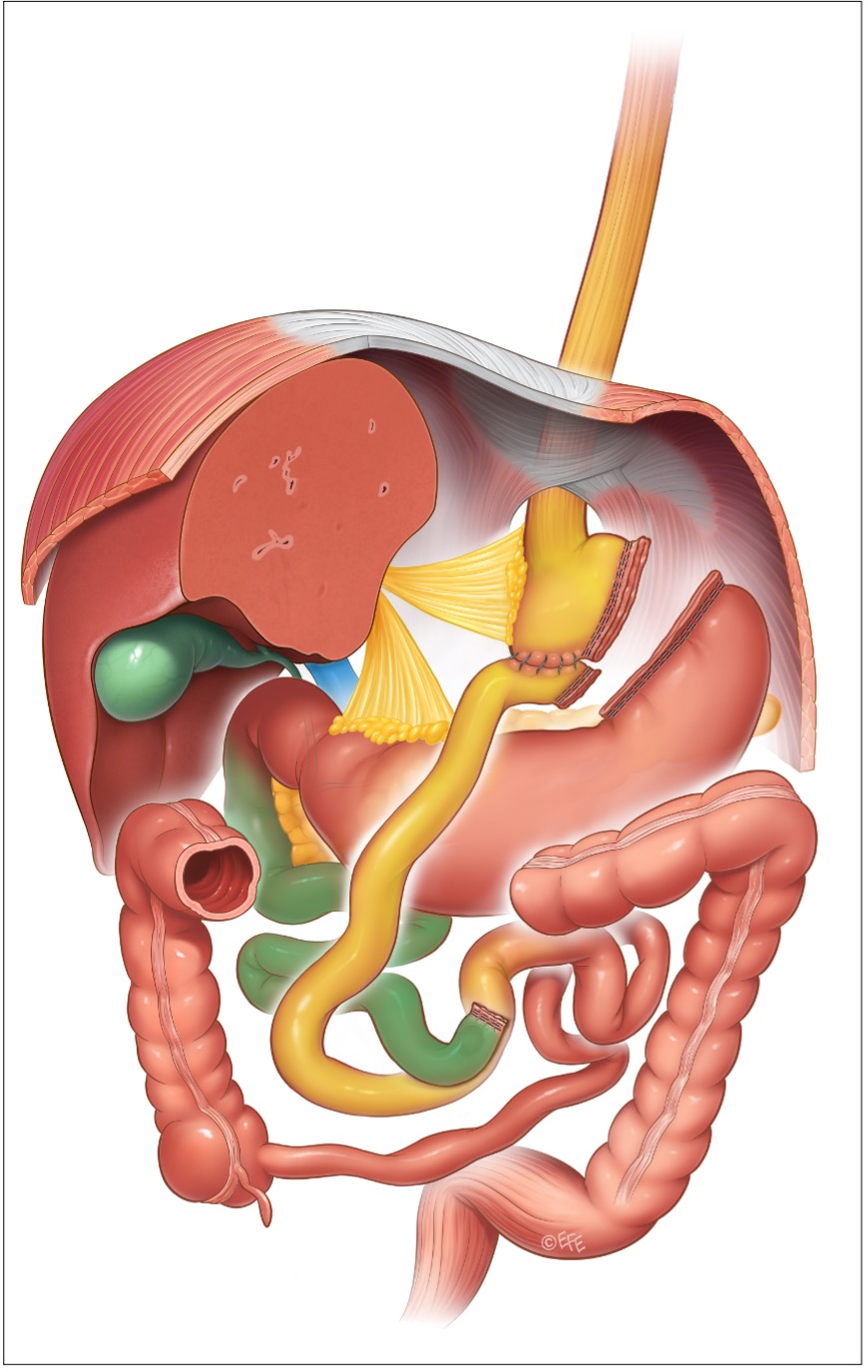
Figura 4: by-pass gastrico
ONE ANASTOMOSIS GASTRIC BY-PASS (OAGB)
Si tratta di una variante del by-pass gastrico standard, con significative differenze. Viene definito anche mini-gastric bypass (MGB) o by-pass gastrico ad omega. Ideato alla fine degli anni ’90, ricalca concettualmente i primissimi by-pass gastrici di 30 anni prima, basati sulle osservazioni di pazienti gastro-resecati, modificandone la tecnica per ovviare a importanti problematiche fra cui il reflusso biliare e l'esofagite da alcali che ne avevano determinato l’abbandono (9).
La tasca gastrica ottenuta dalla sezione dello stomaco è molto più lunga rispetto al by-pass standard ad Y e viene creata una sola anastomosi, laddove un’ansa intestinale (a circa 180-200 cm dal legamento di Treitz) viene anastomizzata allo stomaco (ricostruzione ad Ω).
L’intervento, come suggerito da diversi autori, ha meccanismo prevalentemente malassorbitivo (9). Sono state suggerite diverse tecniche per minimizzare la “sindrome da ansa afferente” e il reflusso biliare che possono presentarsi dopo l’intervento (10), il cui impatto oncogenico è ancora dibattuto (11).
I risultati sul calo di peso sono incoraggianti, con alcuni studi randomizzati che ne dimostrano la non inferiorità rispetto al by-pass gastrico standard a medio-breve termine (12). A fronte di una verosimile maggior incidenza di effetti avversi riferibili a quadri di malassorbimento, l’intervento, nelle sue varianti, sta guadagnando progressivamente popolarità (13), nonostante le perplessità di molti chirurghi.
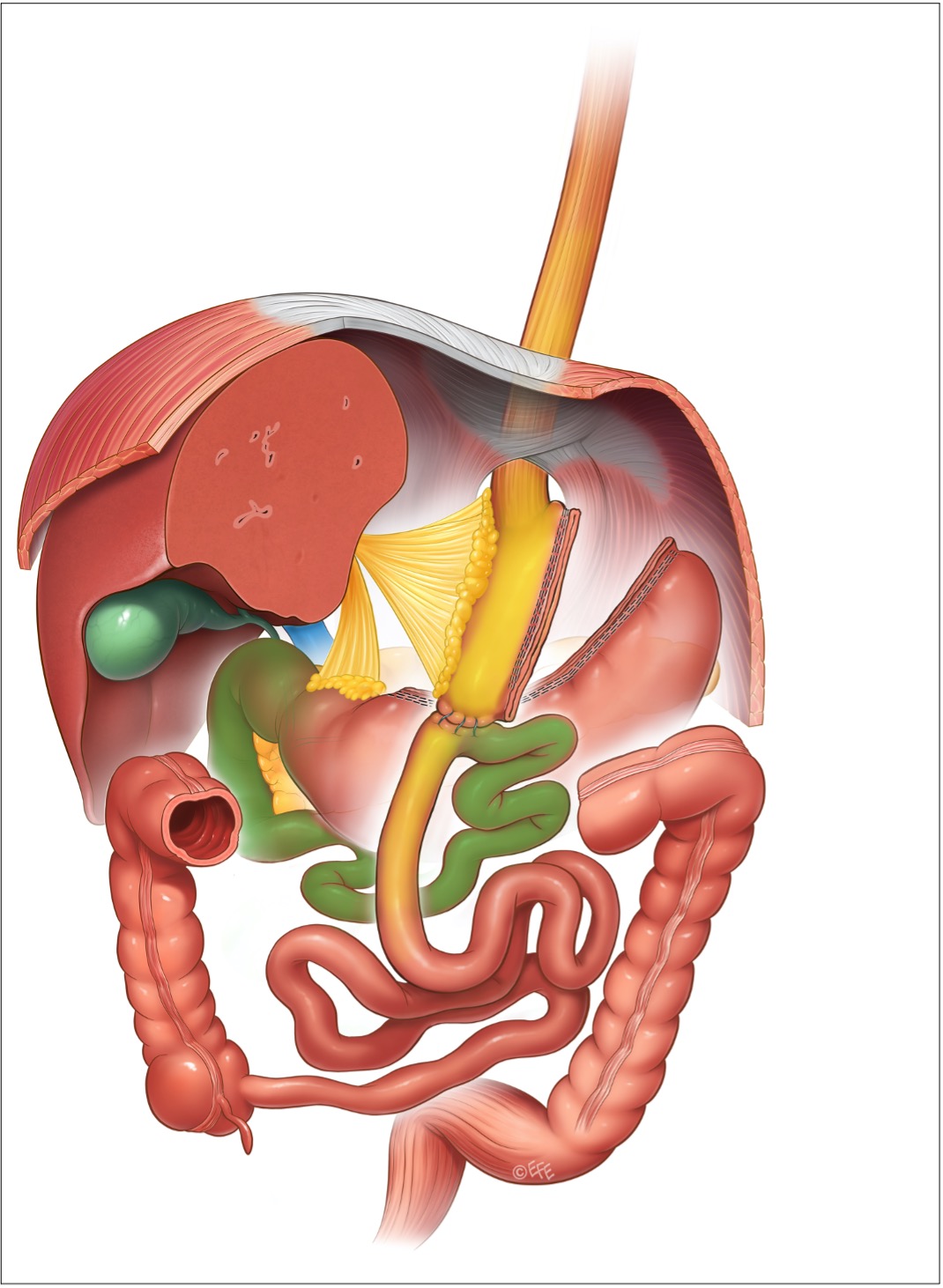
Figura 5: by-pass gastrico a singola anastomosi
DIVERSIONE BILIO-PANCREATICA (BPD, biliopancreatic diversion)
Nell’idea di creare una sindrome da intestino corto controllata, i primi interventi eseguiti negli anni ’50 e ‘60 furono di tipo malassorbitivo: prima il by-pass ileo-colico e successivamente il by-pass digiuno-ileale, che negli USA fu eseguito per più di 10 anni in oltre 100.000 casi. Entrambi ormai appartengono alla storia della chirurgia dell’obesità, a causa dell’alto tasso di complicazioni gravi (insufficienza epatica, insufficienza renale, artrite da by-pass: forma di artrite migrante, coinvolgente soprattutto le piccole articolazioni, legata al dismicrobismo intestinale).
Alla fine degli anni ’70 un chirurgo italiano (Nicola Scopinaro) volle riprendere la strada del ridotto assorbimento intestinale per la cura dell’obesità e creò così un nuovo intervento, la diversione bilio-pancreatica (BPD), che rispetto agli interventi malassorbitivi che l’avevano preceduta aveva il pregio di non avere anse intestinali escluse. Il ridotto assorbimento intestinale della BPD si ottiene con una resezione gastrica distale (comprendente quindi il piloro) e, come nel by-pass gastrico, con una ricostruzione su ansa ad Y, dove però, al contrario che nel RYGBP, il tratto alimentare e il tratto bilio-pancreatico sono lunghi (alimentare di 200 cm o poco più e bilio-pancreatico di lunghezza variabile ma non determinante) e il tratto comune è corto (nella versione originale il tratto comune era cm 50, ma molti chirurghi praticano un tratto comune più lungo – per solito cm 70/75 – per ridurre gli effetti collaterali e quindi le complicazioni della BPD).
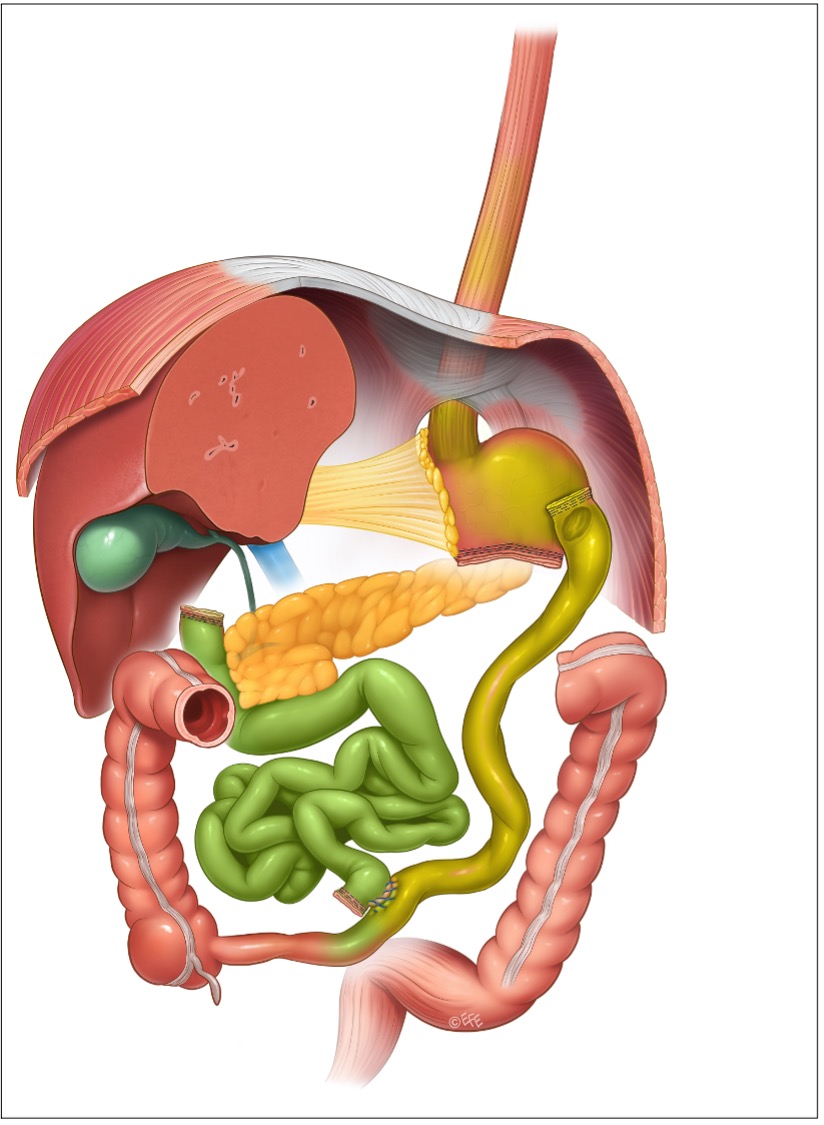
Figura 6: diversione bilio-pancreatica
Esiste anche una versione della BPD dove la resezione gastrica è verticale (con conservazione del piloro, come nella sleeve gastrectomy), mentre la ricostruzione intestinale è identica: si chiama duodenal switch o diversione bilio-pancreatica con duodenal switch (DS o BPD/DS). È un intervento praticato da molti anni negli USA e oggi diffuso ovunque nell’ambito di quella che viene chiamata terapia sequenziale, dove in un primo tempo si esegue una sleeve gastrectomy, e se negli anni si verifica un recupero di peso, poiché l’approccio restrittivo non è sufficiente, si esegue un secondo tempo chirurgico per aggiungere malassorbimento, convertendo la SG in un DS.
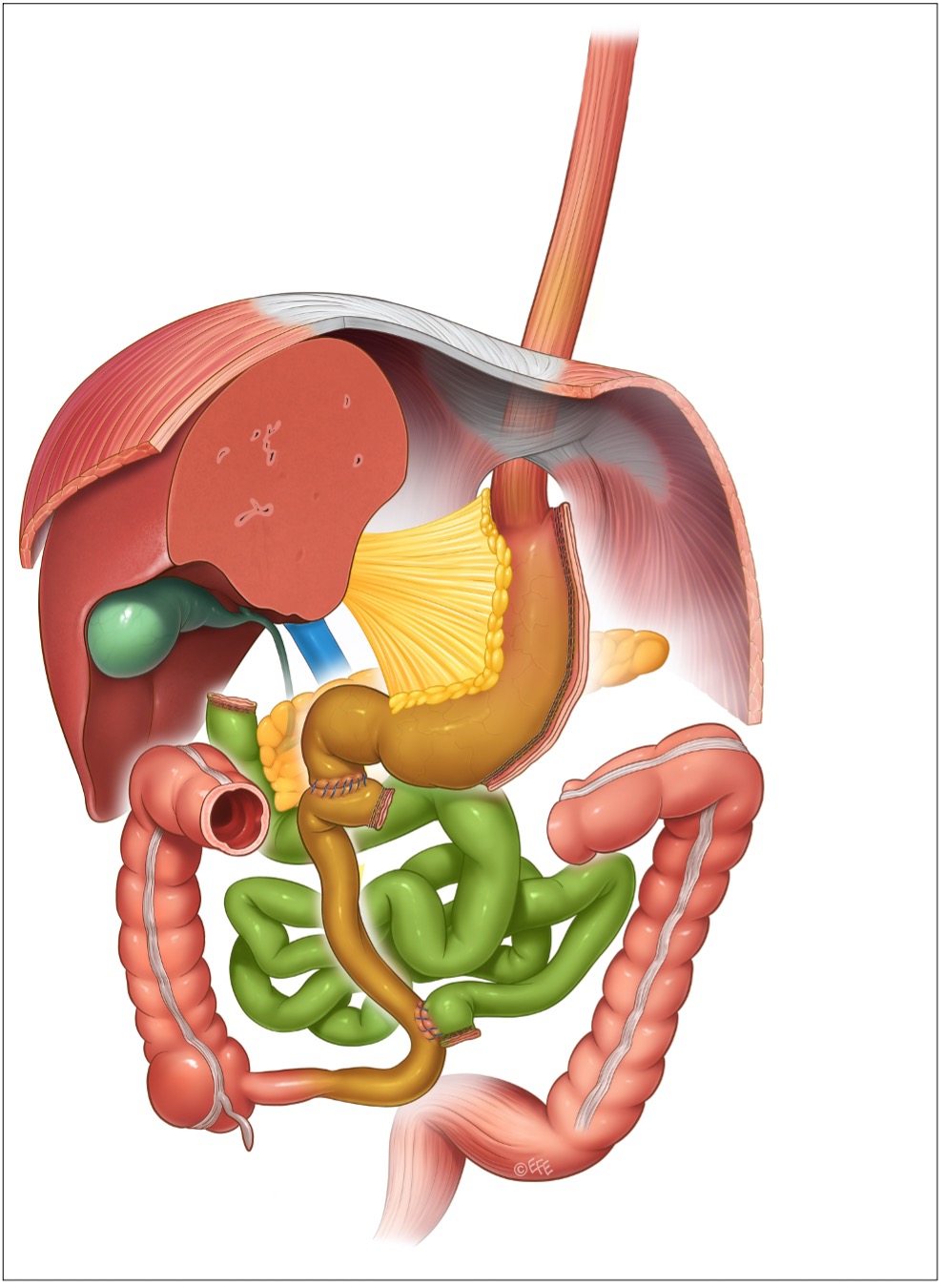
Figura 7: duodenal switch
Si tratta di procedure molto efficaci sia in termini di perdita di peso e mantenimento del peso perso, sia in termini di risoluzione delle comorbilità (14), ma con possibili conseguenze nutrizionali gravi e pesanti effetti collaterali indesiderati. La collaborazione del malato è sempre necessaria, ma, mentre nelle procedure restrittive è indirizzata alla modifica delle abitudini alimentari, nelle procedure malassorbitive la collaborazione va posta nell’assunzione degli integratori necessari e nella stretta frequenza del follow-up nutrizionale. Le procedure malassorbitive sono parte integrante e necessaria dello strumentario di un chirurgo bariatrico completo, ma vanno applicate in un ristretto numero di casi con grande attenzione alla selezione del malato (vedi capitolo successivo).
PROCEDURE ENDOSCOPICHE
È dimostrato come tutte le procedure endoscopiche (con qualche riserva per il pallone intra-gastrico riempito ad aria) siano più efficaci nel breve termine rispetto al solo intervento conservativo in termini di percentuale di perdita di peso (15). Le terapie bariatriche endoscopiche si sono evolute soprattutto come ponte tra la terapia conservativa e l’approccio chirurgico, data le loro caratteristiche di mini-invasività.
A parte il “classico” pallone intra-gastrico (di cui si è già discusso), popolarità sempre maggiore stanno acquisendo le plicature gastriche endoscopiche, che stanno aprendo le porte a nuove procedure restrittive (6). Alcune delle tecniche citate sono ancora sperimentali (16).
La restrizione gastrica può essere effettuata secondo modalità diverse.
Dispositivi che occupano spazio: pallone intra-gastrico. Una sfera in materiale biocompatibile viene introdotta nella cavità gastrica, riducendone la capacità e alterandone la motilità (vedi sopra).
Dispositivi che applicano suture: endoscopic sleeve gastroplasty. Viene effettuata una sutura a livello gastrico lungo la grande curvatura, riducendo le dimensioni dello stomaco e modificandone la motilità.
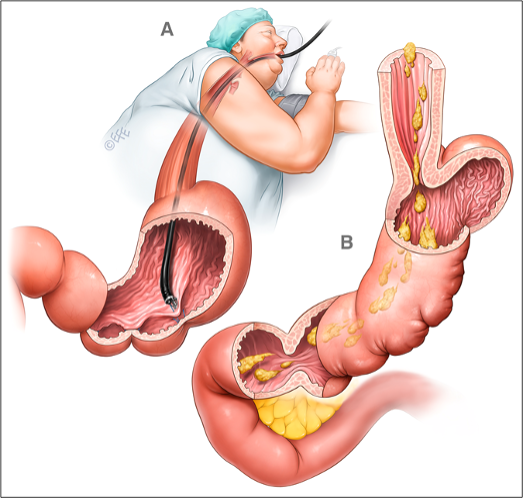
Figura 8: plicatura endoscopica
Tecniche che modificano l'assorbimento: duodenal-jejunal bypass liner endobarrier. Un dispositivo tubulare lungo 60 cm composto da fluoropolimero viene ancorato al bulbo duodenale e fatto progredire fino al digiuno prossimale, impedendo il contatto degli ingesti con la mucosa intestinale, provocando un by-pass dell’assorbimento da parte della prima porzione del tenue.
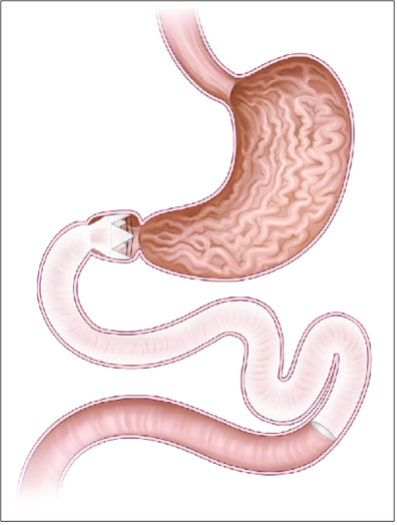
Figura 9: Endobarrier
Incisionless anastomosis system. Una serie di magneti viene introdotta endoscopicamente a livello di ileo terminale e digiuno prossimale, inducendo una fistola-anastomosi da decubito e provocando una diversione enterale.
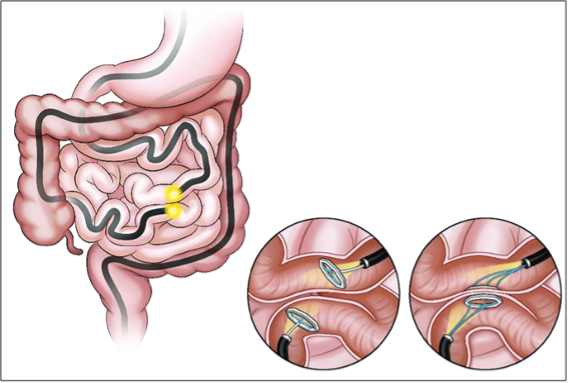
Figura 10: Incisionless Anastomosis System
Altre tecniche: aspireassist therapy. Viene confezionata una gastrostomia percutanea, collegata à la demande a un dispositivo aspirante finalizzato a drenare parte degli ingesti. Per quanto poco utilizzata, questa tecnica è troppo vicina alla bulimia nervosa per essere presa in considerazione nell’uso routinario.
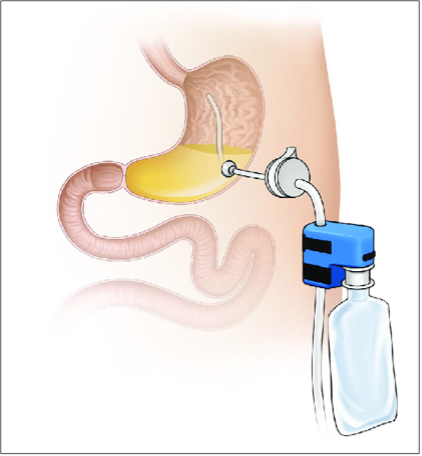
Figura 11: Aspireassist
Per i risultati delle diverse tipologie di intervento, vedi capitolo specifico.
BIBLIOGRAFIA
- Colquitt JL, Pickett K, Loveman E, Frampton GK. Surgery for weight loss in adults. Cochrane Database Syst Rev 2014, 8: CD003641.
- Miras AD, le Roux CW. Mechanisms underlying weight loss after bariatric surgery. Nat Rev Gastroenterol Hepatol 2013, 10: 575–84.
- Welbourn R, Hollyman M, Kinsman R, et al. Bariatric surgery worldwide: baseline demographic description and one-year outcomes from the fourth IFSO global registry report 2018. Obes Surg 2019, 29: 782-95.
- Angrisani L, Santonicola A, Iovino P, et al. IFSO worldwide survey 2016: primary, endoluminal, and revisional procedures. Obes Surg 2018, 28: 3783-94.
- Angrisani L, Santonicola A, Iovino P, et al. Bariatric surgery worldwide 2013. Obes Surg 2015, 25: 1822-32.
- Davis M, Kroh M. Novel endoscopic and surgical techniques for treatment of morbid obesity: a glimpse into the future. Surg Clin North Am 2016, 96: 857-73.
- Angrisani L, Santonicola A, Iovino P, et al. Bariatric surgery and endoluminal procedures: IFSO worldwide survey 2014. Obes Surg 2017, 27: 2279-89.
- Franco JV, Ruiz PA, Palermo M, et al. A review of studies comparing three laparoscopic procedures in bariatric surgery: sleeve gastrectomy, Roux-en-Y gastric bypass and adjustable gastric banding. Obes Surg 2011, 21: 1458-68.
- Rutledge R, Kular K, Manchanda N. The mini-gastric bypass original technique. N Int J Surg 2019, 61: 38-41.
- Deitel M, Kular KS, Musella M, Rheinwalt KP. Letter to the Editor: MGB and OAGB. Obes Surg 2018, 28: 2535-6.
- Rutledge R, Deitel M, Carbajo MA, et al. Commentary: Cancer after the OAGB-MGB. Obes Surg 2020, 30: 755-8.
- Robert M, Espalieu P, Pelascini E, et al. Efficacy and safety of one anastomosis gastric bypass versus Roux-en-Y gastric bypass for obesity (YOMEGA): a multicentre, randomised, open-label, non-inferiority trial. Lancet 2019, 393: 1299-309.
- Welbourn R, Hollyman M, Kinsman R, et al. Bariatric surgery worldwide: baseline demographic description and one-year outcomes from the fourth IFSO global registry report 2018. Obes Surg 2019, 29: 782-95.
- Buchwald H, Estok R, Fahrbach K, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med 2009, 122: 248-56.
- Jung SH, Jai HY, Hyuk SC, et al. Comparative efficacy of bariatric endoscopic procedures in the treatment of morbid obesity: a systematic review and network meta-analysis. Endoscopy 2020, 52: 940-54.
- Orlandini B, Gallo C, Boškoski I, et al. Procedures and devices for bariatric and metabolic endoscopy. Ther Adv Gastroint Endosc 2020, 13: 263177452092564.
Indicazioni e risultati dei diversi interventi di chirurgia bariatrica
Carlo Nagliati1 & Giuseppe M Marinari2
1Dipartimento di Chirurgia, Ospedale San Giovanni di Dio, Gorizia
2UO di Chirurgia Bariatrica, Humanitas Research Hospital, Rozzano (MI)
(aggiornato al 26 maggio 2023)
INTRODUZIONE
Parlare di risultati comporta parlare anche di complicanze, e quindi, prima di risultati ed indicazioni specifiche di ogni intervento, è doveroso riportare mortalità e morbilità della chirurgia dell’obesità.
La mortalità della chirurgia bariatrica negli ultimi 20 anni è crollata: i dati oggi disponibili mostrano una mortalità peri-operatoria variabile dallo 0.03% allo 0.2%. Parimenti anche la morbilità a 30 giorni (espressa in termini di re-interventi, necessità di lunghe degenze, embolia polmonare) si mantiene < 6%, variando un po’ a vantaggio della sleeve gastrectomy quando confrontata con il bypass gastrico (1,2). Diversi fattori contribuiscono a questo forte miglioramento: l’età media della popolazione bariatrica (40-45 anni), lo sviluppo di tecniche laparoscopiche, che senza dubbio permettono una precoce mobilità, con riduzione delle complicazioni polmonari e trombo-emboliche, e infine il fatto che la chirurgia bariatrica non è tempo–dipendente. I pazienti possono e devono essere ottimizzati prima di essere operati: l’adozione di protocolli ERAS (Enhanced Recovery After Surgery), sempre più utilizzati in Italia, spinge molto per la necessità di operare solo pazienti le cui comorbilità sono compensate al massimo (3-5).
I risultati della chirurgia bariatrica possono essere espressi in due modi: analizzando intervento per intervento risultati ed effetti collaterali, oppure prendere la chirurgia bariatrica nell’insieme e analizzare il suo effetto sulle principali comorbilità e sulla mortalità associata. Nel primo caso resta sempre valido quanto illustrato da Pichè (6), riassunto in tabella 1. Tuttavia, la letteratura più recente preferisce analizzare i risultati della bariatrica nell’insieme.
| Tabella 1 Risultati della chirurgia bariatrica |
||||
| BGR | SG | RYGBP | BPD/DS | |
| Calo ponderale | ||||
| 1 anno | 14-30% | 20-28% | 23-43% | 38-52% |
| > 6 anni | 13-14% | 22% | 25-28% | 36-55% |
| Remissione comorbilità | ||||
| Diabete | ||||
| 1 anno | 23-61% | 57-81% | 17-93% | 59-95% |
| > 6 anni | 20-74% | 14-86% | 50-84% | 90-100% |
| Dislipidemia | ||||
| 1 anno | 17% | 16-83% | 33-47% | 33-65% |
| 2-5 anni | 23-61% | 5-48% | 52-97% | 70-100% |
| Ipertensione | ||||
| 1 anno | 19-55% | 15-82% | 20-45% | 24-53% |
| 2-5 anni | 17-64% | 25-75% | 29-80% | 57-85% |
| Apnee notturne | ||||
| 1 anno | 78% | 52-100% | 33-100% | 100% |
| 2-5 anni | 33-90% | 39-91% | 67-80% | 74-92% |
| Legenda. BGR: bendaggio gastrico regolabile; SG: sleeve gastrectomy; RYGBP: bypass gastrico; BPD/DS: diversione bilio-pancreatica con o senza duodenal switch | ||||
EFFETTI DELLA CHIRURGIA BARIATRICA E METABOLICA SULLA MORTALITÀ A LUNGO TERMINE
La chirurgia bariatrica allunga l’attesa di vita, riducendo la mortalità generale e in particolare quella correlata a eventi cardio-vascolari (CV), diabete e cancro.
Una recente revisione (7) analizza gli effetti positivi della chirurgia dell’obesità su diabete, ipertensione, dislipidemie, apnee notturne, aggiungendo però all’analisi anche gli effetti su insufficienza cardiaca, infarto del miocardio, ictus, fibrillazione atriale e cancro: le conclusioni sono a favore della chirurgia, che riduce la mortalità sia nei soggetti a maggior rischio (per età e comorbilità) sia nei soggetti a rischio inferiore.
Una recente metanalisi (8) ha evidenziato il prolungamento dell’attesa di vita nei soggetti obesi operati di bariatrica rispetto alla popolazione di obesi non operati, dato più evidente in chi era affetto da diabete. Il dato di ridotta mortalità e ridotti eventi CV in soggetti obesi e diabetici, particolarmente se con BMI > 35, se di età > 55 anni e con diabete > 15 anni, è confermato da un altro studio di coorte su 6910 pazienti (9). Un altro recente studio retrospettivo (10) su un campione numerosissimo (13 900 sottoposti a sleeve gastrectomy, 17 258 a bypass gastrico e 87 965 obesi non operati) conferma la ridotta mortalità per qualunque causa nei soggetti obesi operati rispetto ai non operati. La riduzione della mortalità a 5 anni correlata a eventi CV o diabete negli operati di bypass è maggiore rispetto agli operati di sleeve gastrectomy, che tuttavia mostrano ridotto tasso di re-interventi e re-ingressi peri-operatori.
Alla chirurgia bariatrica è sempre stato scientificamente ed intuitivamente attribuito un effetto contro gli eventi CV (11), ma esiste ormai l’evidenza di come la chirurgia bariatrica riduca il rischio tumorale e la mortalità associata. Già nel 2009 il SOS Study (12) evidenziava come l’insorgenza di tumori fosse inferiore nelle donne operate per obesità rispetto alle non operate (ma non negli uomini). Nel 2011 Ashrafian (13) parlava di un’azione anti-tumorale della chirurgia dell’obesità, che si estrinseca attraverso diversi percorsi, sia ormonali sia anti-infiammatori. Feigelson (14,15) mostra l’effetto del calo ponderale ottenuto tramite la chirurgia nel ridurre l’incidenza di tumore della mammella, sia pre- sia post-menopausale, e mostra anche come ogni aumento di 5 punti di BMI dopo un primo tumore aumenti il rischio di un secondo cancro (16). Adams (17) in un recente studio retrospettivo a 40 anni ha analizzato mortalità e calo ponderale confrontando due gruppi di soggetti affetti da obesità, uno sottoposto a chirurgia e uno a terapia convenzionale (21 837 soggetti per gruppo). Nel gruppo trattato con chirurgia bariatrica la mortalità è stata inferiore del 29% per malattia CV, del 43% per cancro e del 72% per diabete.
RISULTATI DELLE DIVERSE PROCEDURE
Pallone intra-gastrico
Risultati: nei 6 mesi di permanenza offre un calo ponderale di circa 4-9 punti di BMI. È importante ricordare come il pallone sia un provvedimento temporaneo, a cui dovrebbe seguire un intervento bariatrico: in caso contrario, il recupero ponderale è quasi obbligatorio (5).
Complicanze: sono possibili nausea, vomito, reflusso gastro-esofageo; è possibile anche che il pallone si sgonfi spontaneamente, con perdita dell’effetto, ma soprattutto con rischio di occlusione intestinale da corpo estraneo. Alla rimozione del pallone è possibile constatare la presenza di un’ulcera gastrica da decubito (6,7).
Indicazione: ottenere un calo ponderale nei super-obesi, in preparazione a intervento bariatrico (in caso di alto rischio anestesiologico), oppure ridurre il peso di persone non candidabili alla chirurgia (per età, o BMI < 35), tenendo però presente il probabile recupero ponderale nel tempo. Esiste anche la proposta di utilizzare il pallone per la terapia del sovrappeso (8).
Bendaggio gastrico (ASGB o LASGB)
Risultati: a medio-lungo termine (> 5 anni) offre una perdita media pari al 40-50% del sovrappeso pre-operatorio, con bassa mortalità (0.05%) in paragone ad altre procedure, ma con elevato tasso di re-interventi (anche > 50% in numerose casistiche) per complicanze gastro-esofagee o per fallimento nella perdita di peso (9-11). Il LASGB sembra molto operatore-dipendente e addirittura nazione-dipendente: i risultati australiani sono ottimi, molto diversi da quelli ottenuti altrove (12). Per ottenere buoni risultati con il LASGB è fondamentale un’attenta selezione.
Indicazione: l’alto tasso di re-interventi e i deludenti risultati sul peso ne fanno ormai un intervento desueto.
Sleeve gastrectomy (SG) e by-pass gastrico (RYGBP)
SG è l’intervento più eseguito al mondo.
Risultati: in studi randomizzati sono di gran lunga migliori del bendaggio gastrico (13). Infatti, a fronte di un maggiore rischio peri-operatorio, è maggiore il calo ponderale (circa il 60% del sovrappeso), maggiore la percentuale di scomparsa delle comorbilità e minore il numero di re-interventi nel medio-lungo termine. Al contempo però, non avendo la SG caratteristiche malassorbitive, le complicanze nutrizionali nel follow-up sono trascurabili e di facile gestione (9,14-16). Nel confronto con RYGBP, la SG ha efficacia sovrapponibile in termini di calo ponderale e risoluzione delle comorbilità (18,19), tempo di esecuzione chirurgica inferiore e minor tasso di problematiche nutrizionali nel follow-up (per l’assenza di malassorbimento) (9,19). Il RYGBP è migliore di SG nei confronti della malattia da reflusso gastro-esofageo (19), così come nella percentuale di remissione in caso di diabete mellito di tipo 2 con insorgenza > 10 anni (20). Entrambi gli interventi hanno una percentuale di fallimento nel mantenimento del peso perso, che si accompagna anche a ricomparsa delle comorbilità (21). Tale percentuale dipende dal BMI di partenza (21), dato da non trascurare nella selezione dei malati:
- se il BMI è < 50, è intorno al 6% per entrambi gli interventi;
- se il BMI è > 50, è intorno al 15% per SG e del 15-20% in media per RYGBP.
Indicazione: la SG copre la maggior parte dei casi. Sono ottimi candidati i soggetti con BMI 40÷50, affetti o meno da comorbilità, ma la SG può essere proposta anche in caso di BMI superiore, come eventuale primo tempo di una terapia sequenziale (la percentuale di conversione in duodenal switch è dal 6 al 20%, a seconda delle diverse casistiche). Per la sua relativa minore aggressività e facilità di gestione, la SG è particolarmente indicata in caso di soggetti più fragili, come malati ad alto rischio, anziani, adolescenti, candidati a trapianto, cirrosi Child A, malattie infiammatorie intestinali. La preferenza va invece al RYGBP nei casi di grave malattia da reflusso GE e nel diabete tipo 2 di lunga data.
Mini bypass gastrico/one anastomosis gastric bypass (OAGB)
Risultati: ideato a fine anni ’90, è stato progressivamente adottato da diversi Centri in Europa e in Asia (23,24). I risultati sono comparabili o maggiori rispetto al bypass gastrico tradizionale, sia in termini di peso (30-40% di calo ponderale sul peso totale) che in riferimento al miglioramento delle comorbilità, in particolare del diabete mellito (25).
Effetti collaterali: al pari del bypass gastrico tradizionale, sono possibili stati carenziali, con maggior incidenza di malassorbimento di ferro e vitamine lipo-solubili, per cui è indicata l’integrazione di micro-nutrienti a tempo indeterminato. La persistenza di dieta ricca di grassi può portare a flatulenza e steatorrea, con significativo impatto negativo sulla qualità di vita (25). Una percentuale variabile a seconda delle casistiche presenta reflusso biliare, sintomatico o meno. Allo stato attuale non esistono evidenze circa gli effetti della procedura in termini di rischio di cancro gastro-esofageo (26).
lndicazione: ricalcano sostanzialmente quelle del bypass gastrico tradizionale, in termini di peso e comorbilità presenti. È dibattuto l’effetto della procedura in caso di reflusso gastro-esofageo, vista la possibilità di reflusso biliare post-operatorio. Nel complesso, il crescente numero di interventi eseguiti e gli studi effettuati sui pazienti operati dimostrano che tale procedura è tecnicamente relativamente semplice, sicura ed efficace (26).
Diversione bilio-pancreatica (BPD)
Risultati: in confronto con tutti gli altri interventi è la procedura più efficace a nostra disposizione, sia per perdita e mantenimento del peso (> 70% del sovrappeso) sia per risoluzione delle comorbilità (27).
Effetti collaterali: sgradevoli sull’alvo (evacuazioni frequenti, feci maleodoranti), ma il risultato globale percepito è stato riportato come buono o molto buono dalla maggioranza degli operati (27). Le complicanze nutrizionali (malnutrizione proteica, ipovitaminosi, osteoporosi) la rendono tuttavia un intervento pericoloso, anche se la modifica delle lunghezze intestinali e l’introduzione del duodenal switch hanno ridotto l’incidenza delle complicanze e migliorato la qualità di vita (28).
lndicazione: è un intervento ad alta complessità nella gestione del follow-up, con rischio di complicanze maggiori e molti effetti collaterali. In ogni caso la BPD può essere praticata solo in persone con alimentazione spontaneamente ricca in proteine e con reddito sufficiente a coprire le spese per integratori ed esami emato-chimici frequenti. Tutto questo ha fatto sì che negli ultimi anni la BPD sia stata eseguita sempre meno. Inoltre, è recentemente uscita una pubblicazione (29) della scuola di Genova (dove l’intervento era stato messo a punto) che rappresenta un passo importante verso il definitivo abbandono della metodica. In pratica viene riportato che nei pazienti obesi e diabetici, nonostante offra un miglior risultato in termini di perdita di peso e di compenso glucidico in confronto alla terapia conservativa o a altre procedure bariatriche, la mortalità a lungo termine è maggiore. In pratica, per quanto siano importanti gli effetti positivi, quelli negativi sono ancora maggiori, al punto da determinare una maggiore mortalità.
PROCEDURE ENDOSCOPICHE
Pallone intra-gastrico/Plicature gastriche/altre procedure
Risultati: pur in presenza di evidenze che riportano la superiorità di tali trattamenti rispetto all’approccio conservativo (terapia dietetica e comportamentale), non sono disponibili studi su vasta scala e i risultati sono nel complesso frammentari ed eterogenei (30). Viene riportato calo ponderale del 7-18% sul peso totale (31).
Effetti collaterali: frequenti nausea e vomito; alcuni report evidenziano il rischio di perforazione gastrica e (per il pallone intra-gastrico) di occlusione intestinale. Aneddotico il rischio di pancreatite. Non sembra significativo il rischio di carenze nutrizionali (32).
lndicazione: c’è una certa eterogeneità. Alcuni autori pongono le procedure endoscopiche fra la terapia farmacologica e la chirurgia bariatrica, quindi per lo più riservate a pazienti affetti da obesità di I o II grado in assenza di comorbilità, non candidabili quindi a trattamento chirurgico invasivo. Altre procedure, in particolare le tecniche che portino a parziale malassorbimento o aspirazione del contenuto gastrico, potrebbero trovare indicazione nelle classi di obesità superiore, soprattutto nel caso in cui il paziente rifiuti un intervento chirurgico (30).
BIBLIOGRAFIA
- Arterburn DE, Telem DA, Kushner RF, Courcoulas AP. Benefits and risks of bariatric surgery in adults: a review. JAMA 2020, 324: 879-87.
- Sundbom M, Näslund E, Vidarsson B, et al. Low overall mortality during 10 years of bariatric surgery: nationwide study on 63,469 procedures from the Scandinavian Obesity Registry. Surg Obes Relat Dis 2020, 16: 65-70.
- Stenberg E, Dos Reis Falcão LF, O'Kane M, et al. Guidelines for perioperative care in bariatric surgery: Enhanced Recovery After Surgery (ERAS) Society Recommendations: a 2021 update. World J Surg 2022, 46: 729-51.
- Trotta M, Ferrari C, D'Alessandro G, et al. Enhanced recovery after bariatric surgery (ERABS) in a high-volume bariatric center. Surg Obes Relat Dis 2019, 15: 1785-92.
- Nagliati C, Troian M, Pennisi D, Balani A. Enhanced recovery after bariatric surgery: 202 consecutive patients in an Italian bariatric center. Obes Surg 2019, 29: 3133-41.
- Pichè ME, Auclair A, Harvey J, et al. How to choose and use bariatric surgery in 2015. Can J Card 2015, 31: 153-66.
- Hua Y, Lou YX, Li C, et al. Clinical outcomes of bariatric surgery - Updated evidence. Obes Res Clin Pract 2021, 16: 1-9.
- Syn NL, Cummings DE, Wang LZ, et al. Association of metabolic-bariatric surgery with long-term survival in adults with and without diabetes: a one-stage meta-analysis of matched cohort and prospective controlled studies with 174 772 participants. Lancet 2021, 397: 1830-41.
- Doumouras AG, Lee Y, Paterson JM, et al. Association between bariatric surgery and major adverse diabetes outcomes in patients with diabetes and obesity. JAMA Netw Open 2021, 4: e216820.
- Courcoulas AP, Johnson E, Arterburn DE, et al. Reduction in long-term mortality after sleeve gastrectomy and gastric bypass compared to non-surgical patients with severe obesity. Ann Surg 2023, 277: 442-8.
- Kwok CS, Pradhan A, Khan MA, et al. Bariatric surgery and its impact on cardiovascular disease and mortality: a systematic review and meta-analysis. Int J Cardiol 2014, 173: 20-8.
- Sjöström L, Gummesson A, Sjöström CD, et al; Swedish Obese Subjects Study. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol 2009, 10: 653-62.
- Ashrafian H, Ahmed K, Rowland SP, et al. Metabolic surgery and cancer: protective effects of bariatric procedures. Cancer 2011, 117: 1788-99.
- Feigelson HS, Caan B, Weinmann S, et al. Bariatric surgery is associated with reduced risk of breast cancer in both premenopausal and postmenopausal women. Ann Surg 2020, 272: 1053-9.
- Feigelson HS, Bodelon C, Powers JD, et al. Body mass index and risk of second cancer among women with breast cancer. J Natl Cancer Inst 2021, 113: 1156-60.
- Khalid SI, Maasarani S, Wiegmann J, et al. Association of bariatric surgery and risk of cancer in patients with morbid obesity. Ann Surg 2022, 275: 1-6.
- Adams TD, Meeks H, Fraser A, et al. Long-term all-cause and cause-specific mortality for four bariatric surgery procedures. Obesity (Silver Spring) 2023, 31: 574-85.
- Franco JV, Ruiz PA, Palermo M, et al. A review of studies comparing three laparoscopic procedures in bariatric surgery: sleeve gastrectomy, Roux-en-Y gastric bypass and adjustable gastric banding. Obes Surg 2011, 21: 1458-68.
- Romy S, Donadini A, Giusti V, et al. Roux-en-Y gastric bypass vs gastric banding for morbid obesity. A case-matched study of 442 patients. Arch Surg 2012, 147: 460-6.
- Zhang N, Maffei A, Cerabona T, et al. Reduction in obesity-related comorbidities: is gastric bypass better than sleeve gastrectomy? Surg Endosc 2013, 27: 1273-80.
- Jiménez A, Casamitjana R, Flores L, et al. Long-term effects of sleeve gastrectomy and Roux-en-Y gastric bypass surgery on type 2 diabetes mellitus in morbidly obese subjects. Ann Surg 2012, 256: 1023-9.
- Vidal P, Ramón JM, Goday A, et al. Laparoscopic gastric bypass versus laparoscopic sleeve gastrectomy as a definitive surgical procedure for morbid obesity. Mid-term results. Obes Surg 2013, 23: 292-9.
- Georgiadou, D, Sergentanis TN, Nixon A, et al. Efficacy and safety of laparoscopic mini gastric bypass. A systematic review. Surg Obes Relat Dis 2014, 10: 984-91.
- Welbourn R, Hollyman M, Kinsman R, et al. Bariatric surgery worldwide: baseline demographic description and one-year outcomes from the fourth IFSO global registry report 2018. Obes Surg 2019, 29: 782-95.
- Robert M, Espalieu P, Pelascini E, et al. Efficicacy and safety of one anastomosis gastric bypass versus Roux-en-Y gastric bypass for obesity (YOMEGA): a multicentre, randomised, open-label, non-inferiority trial. Lancet 2019, 393: 1299-309.
- De Luca M, Tie T, Ooi G, et al. Mini Gastric Bypass-One Anastomosis Gastric Bypass (MGB-OAGB)-IFSO Position Statement. Obes Surg 2018, 28: 1188-206.
- Marinari GM, Murelli F, Camerini G, et al. A 15-year evaluation of biliopancreatic diversion according to the Bariatric Analysis Reporting Outcome System (BAROS). Obes Surg 2004, 14: 325-8.
- Dorman RB, Rasmus NF, al-Haddad BJ, et al. Benefits and complications of the duodenal switch/biliopancreatic diversion compared to the Roux-en-Y gastric bypass. Surgery 2012, 152: 758-65.
- Papadia F, Carlini F, Longo G, et al. Pyrrhic victory? Long-term results of biliopancreatic diversion on patients with type 2 diabetes and severe obesity. Surg Obes Relat Dis 2023: DOI: 10.1016/j.soard.2023.04.300.
- Glass J, Chaudhry A, Zeeshan MS, et al. New era: endoscopic treatment options in obesity - a paradigm shift. World J Gastroenterol 2019, 25: 4567–79.
- Stier C, Balonov I, Stier R, et al. Endoscopic management of clinically severe obesity: primary and secondary therapeutic procedures. Curr Obes Rep 2020, 9: 339-47.
- Král J, Machytka E, Horká V, et al. Endoscopic treatment of obesity and nutritional aspects of bariatric endoscopy. Nutrients 2021, 13: 4268.
Preparazione all'intervento di chirurgia bariatrica e follow-up post-operatorio
Giovanni De Pergola
Ambulatorio di Nutrizione Clinica, UOC di Oncologia Medica, Dipartimento di Scienze Biomediche e Oncologia Umana, Università degi Studi Aldo Moro di Bari
PREPARAZIONE ALL’INTERVENTO
Profilassi del trombo-embolismo venoso (1)
Il paziente candidato all’intervento di chirurgia bariatrica ha un rischio elevato di sviluppare tromboembolismo venoso e deve quindi ricevere misure preventive.
È necessario l’impiego di metodi meccanici, quali il bendaggio degli arti inferiori, le calze elastiche anti-trombo, la compressione pneumatica intermittente e la mobilizzazione precoce.
Non vi sono invece linee guida che definiscano la dose di eparina a basso peso molecolare, aggiustata per il peso del paziente, da somministrare in prevenzione. La sera prima dell'intervento, è consuetudine somministrare 4000 UI di enoxaparina sodica se il BMI del paziente è 50. Peraltro, l’adozione di tutte le misure, fisiche e farmacologiche, non elimina il rischio di tromboembolismo venoso in chirurgia bariatrica.
È importante distinguere i pazienti con anamnesi positiva per trombosi venosa profonda o eventi riconducibili a embolia polmonare, da quelli con anamnesi negativa: soltanto i primi dovrebbero effettuare un eco-doppler venoso degli arti inferiori, essere sottoposti a posizionamento di filtro cavale temporaneo ed eseguire terapia con eparine a basso peso molecolare, che comunque deve essere continuata nei 30 giorni successivi all’intervento. La mobilizzazione deve essere precoce e, possibilmente, deve avvenire entro 3 ore dalla fine dell'intervento.
Profilassi antibiotica (1)
L’obesità per sè favorisce le infezioni post-operatorie di ferita, sia per complicazioni di tipo meccanico sia per la possibilità che l’antibiotico non raggiunga livelli sierici e tissutali di efficacia. La dose per la profilassi non deve essere inferiore a quella terapeutica ed è invece preferibile che corrisponda alla più alta dose terapeutica media, modificando il dosaggio in funzione del peso. La scelta, le modalità ed i tempi di somministrazione dell’antibiotico seguono le comuni regole consigliate per la chirurgia gastro-intestinale maggiore.
FOLLOW-UP POST-OPERATORIO
Profilassi del trombo-embolismo venoso
Vedi sopra
Deficit nutrizionali
L’entità dei deficit nutrizionali deriva dal tipo di intervento di chirurgia bariatrica e le carenze sono una conseguenza quasi inevitabile degli interventi malassorbitivi, a causa della riduzione della superficie assorbente intestinale. In particolare, poiché l’intestino prossimale è la sede principale di assorbimento delle vitamine e dei sali minerali, il by-pass gastrico Roux-en-Y (RYGB) e la diversione bilio-pacreatica (DBP) si associano a deficit nutrizionali multipli.
Le principali alterazioni cliniche del deficit nutrizionale, assolutamente da ricercare, sono l’iperparatiroidismo secondario, l’osteoporosi, l’anemia, la neuropatia e l’iperomocisteinemia.
Per quanto attiene alle prime due alterazioni, la chirurgia bariatrica può avere un effetto significativo sul rischio di frattura dopo 3-5 anni nei pazienti che hanno perso più peso (2). Un elevato rischio di deficit di vitamina D esiste dopo gli interventi di bendaggio gastrico laparoscopico (LAGB), RYGB e DBP, ma il rischio di osteoporosi è particolarmente elevato dopo DBP, che conferisce anche un elevato rischio di ipocalcemia.
Un elevato rischio di carenza di ferro è presente dopo RYGB (nel 50% dei pazienti dopo 2 anni dall’intervento) e dopo DBP (nel 100% dei pazienti dopo 5 anni dall’intervento); il metabolismo del ferro può essere compromesso per riduzione dell’acidità gastrica e/o per esclusione del duodeno e del digiuno prossimale e/o per riduzione dell’assunzione alimentare.
La DBP induce anche un rischio medio (non elevato) di carenza di vitamina A, vitamina B12, vitamina K e zinco. Il RYGB può invece essere responsabile di un elevato rischio di deficit di vitamina B12, a sua volta responsabile di neuropatia, mielopatia e depressione, sino alla demenza. Anche la carenza di altre vitamine può favorire specifiche complicanze neurologiche (3): è comune il deficit di vitamina B1, che induce l’encefalopatia di Wernicke e la sindrome di Korsakoff; il deficit di vitamina B2 è presente nel 14% dei casi di chirurgia bariatrica e favorisce la sindrome del piede che brucia, mentre il deficit di vitamina B6 è presente nel 17% dei casi ed induce polineuropatia. Sono rari il deficit di vitamina E, che favorisce neuropatia periferica e miopatia, ed il deficit di rame, che favorisce mielopatia e atassia sensoriale.
Sintomi, segni e patologie da ricercare
I pazienti che hanno eseguito una RYBG possono presentare sintomi post-prandiali di ipoglicemia e in questi casi deve essere esaminata la possibilità di una dumping syndrome o di una inappropriata secrezione di insulina (4). Altre possibili manifestazioni indotte dalla chirurgia bariatrica sono il vomito e l’intolleranza ad alcuni alimenti, dovute prevalentemente alla carenza della pepsina e degli enzimi pancreatici e al breve tempo di contatto degli enzimi con il contenuto alimentare. Sono generalmente poco tollerate la carne rossa, la pasta, il pane e le verdure crude. I pazienti sottoposti a chirurgia bariatrica presentano un maggiore rischio di attacchi di gotta dopo una significativa perdita di peso (4) e non va dimenticata la possibilità di insufficienza renale acuta (6% dei casi).
Esami da eseguire nei 2 anni successivi all’intervento
Gli esami fondamentali, da eseguire dopo 1, 3, 6, 12, 18 e 24 mesi sono glicemia, creatininemia, elettroliti, emocromo completo e test di funzionalità epatica (4). Dopo 6, 12, 18 e 24 mesi risultano necessari sideremia, ferritina, vitamina B12, acido folico, calcio, paratormone, vitamina D e albuminemia (4). Dopo 1 e 2 anni è indicata la mineralometria ossea computerizzata. Esami opzionali sono vitamina B1 (dopo 3, 6, 12, 18 e 24 mesi), zinco (dopo 6, 12 e 24 mesi) e vitamina A (dopo 24 mesi) (4).
| Esami post-operatori (in parentesi quelli opzionali) | ||||||
| Esame | Mesi post-operatori | |||||
| 1 | 3 | 6 | 12 | 18 | 24 | |
| Glicemia | x | x | x | x | x | x |
| Creatininemia | x | x | x | x | x | x |
| Elettroliti | x | x | x | x | x | x |
| Emocromo completo | x | x | x | x | x | x |
| Test di funzionalità epatica | x | x | x | x | x | x |
| Sideremia | x | x | x | x | ||
| Ferritina | x | x | x | x | ||
| Vitamina B12 | x | x | x | x | ||
| Acido folico | x | x | x | x | ||
| Calcio | x | x | x | x | ||
| PTH | x | x | x | x | ||
| Vitamina D | x | x | x | x | ||
| Albuminemia | x | x | x | x | ||
| MOC | x | x | ||||
| Vitamina B1 | (x) | (x) | (x) | (x) | (x) | |
| Zinco | (x) | (x) | (x) | |||
| Vitamina A | (x) | |||||
Dieta dopo chirurgia bariatrica
La dieta dopo l’intervento deve essere progressiva:
- prime 1-2 settimane: liquida;
- per 3-4 settimane: semiliquida;
- per 4 settimane: semisolida;
- dal 3° mese: normale.
Per gli interventi di RYGB e DBP sono fondamentali supplementi multi-vitaminici e multi-minerali, in particolare di calcio e vitamina D. Per quanto concerne il calcio, si raccomanda 1 g per il RYGB e 2 g per la DBP. Per quanto attiene alla vitamina D, sono consigliabili 2000-2500 UI di colecalciferolo per bocca 1 volta alla settimana o, meglio, 100.000 UI di colecalciferolo i.m. 1 volta al mese nel caso della DBP.
Sia il RYGB sia la DBP richiedono specificatamente anche vitamina B12, ferro e vitamina C e DBP anche le vitamine liposolubili (A, E e K).
Un aspetto molto importante è il deficit proteico. In considerazione dell’intolleranza agli alimenti e del malassorbimento, può instaurarsi una condizione di malnutrizione proteica (albuminemia < 3.5 g/dL) e, quindi, l’apporto di proteine deve essere di 60-120 g/die, anche ricorrendo a supplementi proteici per via orale. È buona regola stabilire l’apporto proteico di base (1-1.5 g/Kg peso ideale) e aggiungere carboidrati e grassi sino ad ottenere l’introito energetico desiderato.
A lungo termine, il recupero del peso e i deficit nutrizionali possono dipendere da scarsa adesione alle raccomandazioni nutrizionali e di stile di vita, ridotto introito di alcuni alimenti e malassorbimento. Pertanto, la chiave del successo a lungo termine è rappresentata dall’educazione alimentare e dal follow-up del paziente da parte di un’equipe multidisciplinare. Il recupero ponderale può essere ricondotto a molteplici cause:progressivo aumento dell’apporto calorico, alterazione dei segnali neurali che influenzano l’asse fame-sazietà, meccanismi adattativi intestinali con incremento della capacità assorbitiva e dilatazione della tasca gastrica (negli interventi di natura restrittiva). Una dieta a basso indice glicemico, moderatamente ricca in proteine e con un elevato contenuto di fibre è più efficace nel prevenire il recupero del peso; l’introito di fibre può essere aumentato mediante supplementi di fibre solubili (5 g/die prima di ogni pasto) (5).
BIBLIOGRAFIA
- Società Italiana di Chirurgia dell’Obesità e delle malattie metaboliche (S.I.C.O.B.). Linee guida e stato dell’arte della chirurgia bariatrica e metabolica in Italia. EDISES s.r.l. – Napoli, 2008.
- Lamohamed A, de Vries F, Bazelier MT, et al. Risk of fracture after bariatric surgery in the United Kingdom: population based, retrospective cohort study. BMJ 2012, 345: e5085.
- Becker DA, Balcer LJ, Galetta SL. The neurological complications of nutritional deficiency following bariatric surgery. J Obes 2012: 608534.
- Heber D, Greenway FL, Kaplan LM, et al, and Endocrine Society. Endocrine and nutritional management of the post-bariatric surgery patient: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2010, 95: 4823-42.
- Faria SL, de Oliveira Kelly E, Lins RD, Faria OP. Nutritional management of weight regain after bariatric surgery. Obes Surg 2010, 20: 135-9.
Gravidanza post chirurgia bariatrica
Silvia Irina Briganti
Università Telematica San Raffaele
(aggiornato al 15 luglio 2025)
La chirurgia bariatrica è oggi una delle opzioni terapeutiche più frequenti in caso di obesità severa. Per essere candidati a questo tipo di chirurgia i pazienti devono soddisfare dei criteri specifici di selezione, tra cui l’indice di massa corporea (BMI) e l’eventuale presenza di comorbilità.
La fase pre-operatoria
È frequente la presenza di marcata irregolarità mestruale, in particolare oligo-amenorrea o amenorrea secondaria. La perdita di peso che segue l’intervento di chirurgia bariatrica è spesso in grado di determinare il ripristino della regolarità mestruale anche in tempi brevi (1). Inoltre, le modifiche anatomiche a carico del tratto gastro-intestinale (in particolare quelle relative all’acidità intra-gastrica) possono avere ripercussioni sulla capacità di assorbimento di alcuni farmaci, tra cui i contraccettivi orali (2). Ne consegue, quindi, quanto sia fondamentale in fase pre-operatoria una corretta informazione circa le tematiche relative alla fertilità, al fine di evitare gravidanze indesiderate. In particolare, è consigliabile suggerire di valutare terapie contraccettive alternative a quella orale, ad esempio mediante applicazione di cerotti trans-dermici o dispositivi intra-uterini, che possano by-passare il tratto digestivo e quindi non risentire delle modifiche di assorbimento. È altresì fondamentale informare adeguatamente le pazienti, ma anche i partner, che, per quanto le procedure di chirurgia bariatrica possano migliorare la funzione ovarica e la regolarità mestruale, non devono essere considerate terapie specifiche per l’infertilità di coppia, condizione che richiede comunque un inquadramento specialistico integrato multi-disciplinare (3).
La fase post-operatoria
Le linee guida raccomandano di non avviare una ricerca attiva di gravidanza prima che siano trascorsi almeno 12-18 mesi dall’intervento di chirurgia bariatrica. Le ragioni di tale attesa sono da ricercare principalmente nella stabilizzazione del peso e dello stato nutrizionale materno e nella prevenzione di eventuali carenze vitaminiche e minerali, potenzialmente dannose sia per la madre che per il nascituro. Nutrienti particolarmente a rischio di carenza per effetto delle modifiche anatomiche post-chirurgia bariatrica sono calcio, ferro, vitamina D, acido folico, vitamina A, vitamina K, vitamina B1 e vitamina B12 (3,4).
| Tabella 1 Carenze minerali e vitaminiche ed esiti materno-fetali in gravidanza |
|
| Calcio | Sviluppo osseo |
| Ferro | Anemia |
| Vitamina A | Micro-oftalmia fetale Malformazioni fetali congenite |
| Vitamina B1 | Encefalopatia |
| Vitamina B12 | Anemia |
| Vitamina D | Pre-eclampsia |
| Acido folico | Anemia Difetti di chiusura del tubo neurale |
| Vitamina K | Emorragia cerebrale fetale |
Dopo l’intervento di chirurgia bariatrica occorre sottoporre i pazienti a uno screening emato-chimico periodico, al fine di individuare eventuali carenze da supplementare: eseguire di routine (a 3-6 e 12 mesi per il primo anno e, successivamente, ogni 12 mesi o prima, secondo necessità) la valutazione dell’emocromo e il dosaggio dei livelli ematici di calcio, fosfato e magnesio, ferro, transferrina e ferritina, vitamina B12 e acido folico, sodio e potassio, vitamina D e PTH.
È consigliabile raccomandare l’assunzione di integratori multi-vitaminici e multi-minerali per almeno 12 mesi dopo procedure restrittive (ad esempio sleeve gastrectomy) e a vita dopo procedure malassorbitive (ad esempio by-pass gastrico) (2,3,5).
Per tutto il primo anno post-intervento è consigliabile abbinare una terapia con inibitore di pompa protonica (PPI), per ridurre il rischio di reflusso gastro-esofageo.
In caso di gravidanza, il monitoraggio laboratoristico può essere più o meno frequente, tuttavia è assolutamente fondamentale che la paziente metta al corrente il più precocemente possibile l’equipe di follow-up post-chirurgia bariatrica dello stato gravidico in atto. In corso di gravidanza è obbligatorio sospendere il PPI e tutti gli integratori la cui assunzione non sia chiaramente considerata sicura per mancanza di evidenze scientifiche e studi (rientrano in questa categoria i principali integratori consigliati in fase post-chirurgica) e affidarsi a formulazioni multi-vitaminiche e multi-minerali approvate in gravidanza, preferibilmente previo contatto con il ginecologo curante (2,3,5):
- vitamina D: formulazioni a base di colecalciferolo al dosaggio di 400 UI/die;
- acido folico: 800 µg/die se BMI all’inizio della gestazione < 30 kg/m2 oppure 5 mg/die se BMI > 30 kg/m2. La corretta supplementazione è particolarmente rilevante per evitare lo sviluppo di difetti di chiusura del tubo neurale;
- ferro: 65 mg/die;
- calcio: 1200 mg/die;
- vitamina B12: 1000 µg/settimana, preferibilmente in formulazione intramuscolare;
- vitamina A: non superare il dosaggio di 5000 UI/die, perchè l’eccesso può causare malformazioni fetali cerebrali e cardio-vascolari.
Infine, è fondamentale ricordare che la paziente sottoposta a intervento di chirurgia bariatrica non è candidabile a screening per diabete gestazionale (GDM) con curva da carico orale con 75 g di glucosio, per la possibilità di ipoglicemia reattiva secondaria a dumping syndrome, effetto collaterale molto comune soprattutto in caso di interventi malassorbitivi come il by-pass gastrico. In queste pazienti lo screening per GDM prevede il dosaggio della glicemia venosa a digiuno e il monitoraggio glicemico a digiuno, un’ora e due ore dopo i pasti mediante determinazione della glicemia capillare e/o tramite monitoraggio glicemico continuo per almeno una settimana. Il timing dello screening per GDM deve rispettare le linee guida per tutte le donne in gravidanza:
- screening precoce tra la 16°-18° settimana ed eventuale ripetizione alla 24°-28° settimana in caso di presenza di specifici fattori di rischio (glicemia a digiuno 100-125 mg/dL, BMI > 30 kg/m2 o pregresso GDM nelle prime settimane di gravidanza);
- 24°-28° settimana per tutte le altre pazienti (6).
In conclusione, la gestione della gravidanza post-chirurgia bariatrica rappresenta una situazione delicata da inquadrare in centri di riferimento, preferibilmente all’interno di un equipe multi-disciplinare che coinvolga, oltre all’endocrinologo, anche il ginecologo e il chirurgo.
Bibliografia
- Maggard MA, Yermilov I, Li Z, et al. pregnancy and fertility following bariatric surgery. A systematic review. JAMA 2008, 300: 2286-96.
- Miller AD, Smith KM. Medication and nutrient administration considerations after bariatric surgery. Am J Health Syst Pharm 2006, 63: 1852-7.
- American College of Obstetricians and Gynecologists. ACOG practice bulletin no. 105: bariatric surgery and pregnancy. Obstet Gynecol 2009, 113: 1405.
- Karmon A, Sheiner E. Pregnancy after bariatric surgery: a comprehensive review. Arch Gynecol Obstet 2008, 277: 381–8.
- Ciangura C, Coupaye M, Deruelle P, et al. Clinical practice guidelines for childbearing female candidates for bariatric surgery, pregnancy, and post-partum management after bariatric surgery. Obes Surg 2019, 29: 3722-34.
- AMD-SID. Standard Italiani per la cura del diabete mellito. 2018.
Aggiornamenti obesità
In attesa degli aggiornamenti di Endowiki, si possono trovare articoli aggiornati ai seguenti link:
Definizione, classificazione ed epidemiologia della sindrome metabolica
Giorgio Borretta
Endocrinologia, AO S Croce e Carle, Cuneo
Definizione e classificazione
Il termine sindrome metabolica (SM) definisce l'aggregazione nello stesso individuo di molteplici fattori di rischio per lo sviluppo del diabete tipo 2 (DMT2) e della malattia cardiovascolare: iperglicemia, ipertrigliceridemia, bassi livelli di HDL-colesterolo, ipertensione arteriosa e obesità centrale (1-4). Queste componenti riflettono principalmente una condizione di ipernutrizione, stile di vita sedentario e relativo eccesso di adiposità. Tale relazione spiega la sempre più frequente coesistenza di tali anomalie nella popolazione generale, anche se la loro prevalenza ed interazione sono spesso diverse nei due sessi, nelle varie fasce di età e anche tra diversi gruppi etnici.
Oggi la SM è riconosciuta come entità clinica autonoma, con specifico codice di identificazione nosologica nelle versioni aggiornate del nomenclatore internazionale (ICD-IX: 277.7); ciononostante restano aperti alcuni problemi riguardo alla sua definizione diagnostica e soprattutto al significato clinico e prognostico della SM (3,4).
Dal 1998 vari organismi internazionali ed autorevoli società scientifiche hanno fornito indicazioni sulle modalità di diagnosi (tab. 1). Tuttavia, la proposta di criteri diagnostici in parte divergenti o contrastanti e in una certa misura arbitrari ha suscitato negli ultimi anni un acceso dibattito tra esperti. Infine nel 2009, una commissione congiunta (2), promossa da autorevoli associazioni scientifiche internazionali, ha pubblicato una nuova consensus sulla definizione diagnostica della SM (tab. 2).
| Tabella 1 Criteri diagnostici della SM |
||||
| Alterazione | AH-NHBLI (qualsiasi combinazione di 3 alterazioni) |
IDF (obesità centrale + 2 alterazioni) |
NCEP-ATPIII (qualsiasi combinazione di 3 alterazioni) |
WHO (iperglicemia o insulino-resistenza + 2 alterazioni) |
| Iperglicemia | FPG > 5.6 mmol/L o diabete noto | FPG > 5.6 mmol/L o diabete noto | FPG > 5.6 mmol/L o diabete noto | FPG > 5.6 mmol/L o IGT o diabete noto |
| Insulino-resistenza (IR) | - | - | - | HOMA-IR nel quartile più alto della popolazione |
| Obesità centrale | Circonferenza vita > 102 cm in uomini e > 88 cm in donne | Circonferenza vita > 94 cm in uomini e > 80 cm in donne | Circonferenza vita > 102 cm in uomini e > 88 cm in donne | Rapporto vita/fianchi > 0.9 in uomini e > 0.85 in donne e/o BMI > 30 |
| Ipertensione | Sistolica > 130 e/o diastolica > 85 mmHg e/o trattamento | Sistolica > 130 e/o diastolica > 85 mmHg e/o trattamento | Sistolica > 130 e/o diastolica > 85 mmHg e/o trattamento | Sistolica > 140 e/o diastolica > 90 mmHg e/o trattamento |
| Ipertrigliceridemia | > 1.7 mmol/L o trattamento | > 1.7 mmol/L o trattamento | > 1.7 mmol/L o trattamento | - |
| Basso HDL-C | < 1.03 mmol/L negli uomini o < 1.29 mmol/L nelle donne o trattamento | < 1.03 mmol/L negli uomini o < 1.29 mmol/L nelle donne o trattamento | < 1.03 mmol/L negli uomini o < 1.29 mmol/L nelle donne o trattamento | - |
| Dislipidemia | - | - | - | Trigliceridi > 1.7 mmol/L e/o HDL-C < 1.03 negli uomini o < 1.29 mmol/L nelle donne o trattamento |
| Microalbuminuria | - | - | - | UAE > 20 µg/min o ACR > 30 mg/g |
| Tabella 2 Nuovi criteri di diagnosi della SM (2) |
|
| Criteri | Valori |
| Aumento circonferenza vita | Popolazione specifici |
| Ipertrigliceridemia o trattamento | > 150 mg/dL (1.7 mmol/L) |
| HDL-C ridotto o trattamento | < 40 mg/dL (1.0 mmol/L) nei maschi o < 50 mg/dL (1.3 mmol/L) nelle donne |
| Ipertensione o trattamento | Sistolica > 130 e/o diastolica > 85 mmHg |
| IFG o trattamento ipoglicemizzante | > 100 mg/dL |
Le principali critiche espresse nei confronti della SM sono:
- alcuni importanti fattori di rischio per diabete tipo 2 e malattia cardio-vascolare non sono inclusi nella definizione clinica;
- differenti combinazioni delle componenti della SM conferiscono diversi livelli di rischio;
- la glicemia può essere da sola un valido fattore predittivo per diabete tipo 2;
- la SM può non essere un fattore di rischio indipendente rispetto alle singole componenti;
- i valori di riferimento per obesità nei differenti gruppi etnici non sono basati su chiari criteri di evidenza.
L'incertezza sulla definizione dei criteri diagnostici, tra cui l'inclusione o meno del diabete, i dubbi sulla insulino-resistenza quale elemento patogenetico unificante le diverse componenti della SM, una limitata efficacia predittiva del rischio cardiovascolare, hanno indotto autorevoli studiosi ad avanzare seri dubbi sul vantaggio clinico di diagnosticare la SM. Tale perplessità è almeno in parte condivisibile se pensiamo, ad esempio, che in varie popolazioni l'applicazione dei criteri diagnostici proposti da IDF quasi raddoppia la prevalenza di malattia accertata secondo i criteri dell'ATP III.
Un ulteriore problema aperto è se la SM sia un valido strumento di predizione del rischio. Recenti metanalisi hanno rilevato che la SM risulta maggiormente predittiva di DMT2 (rischio relativo: 3.1-5.1) che di malattia cardiovascolare (rischio relativo: 1.7-1.9), almeno nel breve-medio periodo (< 15 anni). L'elevato valore predittivo del rischio di diabete è da attribuire alla stessa natura della SM, i cui componenti sono tutti predittori indipendenti di DM. Al contrario, i criteri diagnostici di SM escludono numerosi fattori di rischio cardiovascolare, quali età, fumo, familiarità ed ipercolesterolemia, che evidentemente ne indeboliscono il potere predittivo. Il rischio cardiovascolare a breve termine risulterebbe infatti meglio definito da altri strumenti, quali ad esempio il Framinghan Risk Score o il Prospective Cardiovasculare Munster (PROCAM) score. Va comunque sottolineato che il valore predittivo di rischio cardiovascolare della SM aumenta considerevolmente quando il diabete emerge.
Nonostante critiche ed elementi di incertezza, è comunque opinione diffusa che la SM possa costituire non solo un interessante costrutto fisiopatologico, ma altresì un utile strumento clinico per il medico pratico (5). Tra gli elementi positivi della SM come fattore predittivo di diabete tipo 2 e malattia cardiovascolare vanno evidenziati:
- escludendo l'età è in grado di identificare un elevato rischio anche nei soggetti giovani;
- è dicotomico;
- è utile per spiegare ai pazienti il legame patogenetico tra le sue varie componenti;
- è più semplice da calcolare rispetto ad una complessa equazione di calcolo del rischio;
- è correlata ad un rischio di 3-5 volte maggiore per diabete tipo 2 e di quasi 2 volte per malattia cardiovascolare;
- è utile come fattore predittivo di rischio a lungo termine.
Ulteriore aspetto di interesse clinico della SM è la frequente associazione con altre condizioni morbose, quali la steatosi epatica non alcoolica (NAFLD), la sindrome delle apnee notturne su base ostruttiva (OSAS) e la malattia renale.
Infine, di particolare interesse per l'endocrinologo è il rapporto tra SM e alcune endocrinopatie, in particolare quelle emergenti di tipo subclinico. La sindrome di Cushing, in particolare, si manifesta abitualmente con fenotipo sovrapponibile alla SM. Recentemente è stato ipotizzato che anche le forme subcliniche di malattia, molto più frequenti e spesso associate a incidentaloma surrenalico, possano rappresentare una causa comune di SM. Altri studi riportano un aumento di insulino-resistenza e di anomalie glicolipidiche e pressorie in varie endocrinopatie relativamente frequenti, come l'iperaldosteronismo primario, la PCOS, l'ipogonadismo maschile e l'iperparatiroidismo primario; ulteriori evidenze supportano anche una relazione tra ipovitaminosi D e SM.
Occorre però precisare che a tutt'oggi non sono disponibili studi epidemiologici consistenti e che mancano prove conclusive di un nesso causale tra alcune endocrinopatie e SM e degli effetti del loro trattamento sulle componenti della SM e, soprattutto, sugli esiti ad essa correlati.
In conclusione, il principale valore sul piano clinico della SM è quello di offrire un modello semplice e di facile applicazione per la selezione dei soggetti a rischio di diabete e malattie cardiovascolari. La consapevolezza della SM, infatti, costituisce uno stimolo a ricercare scrupolosamente nel singolo paziente l'aggregazione dei vari componenti della SM e, nel contempo, a valorizzare in modo appropriato singole alterazioni metaboliche altrimenti considerate di secondaria importanza, come IFG/IGT o ipertrigliceridemia. Da una prospettiva di salute pubblica, il maggior vantaggio di focalizzare l'attenzione sulla SM è sicuramente quello di promuovere stili di vita volti a combattere l'epidemia di obesità e di favorire interventi specifici di prevenzione e controllo sui fattori di rischio cardiovascolare.
Epidemiologia
Negli ultimi decenni la prevalenza della SM è aumentata progressivamente, raggiungendo proporzioni epidemiche nei paesi economicamente più avanzati (4). I principali fattori che influenzano la prevalenza della SM sono: età, sesso, etnia e soprattutto i criteri utilizzati per la diagnosi.
I criteri proposti da WHO ed EGIR sono più restrittivi di ATP III e IDF; questi ultimi sono i più inclusivi.
Ulteriori fattori epidemiologicamente influenti sono quello socio-economico, che correla inversamente con la prevalenza di SM, il fumo di sigaretta, il consumo di alcool e il grado di sedentarietà.
Negli USA la percentuale di soggetti ultrasessantenni con SM supera il 40%; in Europa e in America Latina la prevalenza di SM nella popolazione adulta è di circa il 25%. Anche nei paesi asiatici emergenti (6) la prevalenza di SM è in aumento e i dati più recenti riportano un tasso di 8-13% negli uomini e di 2-18% nelle donne, a seconda dei criteri utilizzati per la diagnosi.
Alcuni studi hanno peraltro dimostrato che un intervento sullo stile di vita è efficace nel ridurre significativamente la prevalenza di SM.
Bibliografia
- Bonora E. La sindrome metabolica fra controversie e dati di fatto. Quaderni di Diabetologia nella pratica clinica 2007, 1: 5-14.
- Alberti KGMM, Eckel RH, Grundy SM. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung and Blood Institute; America Heart Association; World Heart Federation; International Atherosclerosis Society and International Association for the study of obesity. Circulation 2009, 120: 1640-5.
- Kahn R. The metabolic syndrome: what is the clinical usefulness. Lancet 2008, 371: 1892-3.
- Cornier MA, Dabelea D, Hernandez L, et al. The metabolic syndrome. Endocr Rev 2008, 29: 777-822.
- Cameron A. The metabolic syndrome: validity and utility of clinical definitions for cardiovascular disease and diabetes risk prediction. Maturitas 2010, 65: 117-21.
- Cho LW. Metabolic syndrome. Singapore Med J 2011, 52: 779-85.
Diagnostica e terapia della sindrome metabolica
Maurizio Nizzoli
UO Endocrinologia e Malattie Metaboliche, Dipartimento di Medicina Specialistica, Ospedale GB Morgagni, Forlì
DEFINIZIONE E INQUADRAMENTO DIAGNOSTICO
Sono stati proposti numerosi criteri diagnostici della SM da parte delle principali organizzazioni scientifiche, espressione da una parte della difficoltà di identificare un'esatta definizione e dall’altra dal ragionevole dubbio che può insorgere circa la reale esistenza della SM come entità nosografica a sé stante. Comunque nel 2001 il Center for Disease Control americano ha attribuito uno specifico codice ICD-9_CM, il 277.7 che codifica per la Dysmetabolic Syndrome X; nonostante questo, il dibattito riguardo la definizione e i criteri diagnostici continua ancora oggi. Il contrasto riguarda in particolare due aspetti: l’aumento della glicemia e l’obesità. Il primo coinvolge sia la condizione di pre-diabete e quindi l’alterata tolleranza del glucosio a digiuno (IFG), la ridotta tolleranza al glucosio (IGT, per la quale è necessario effettuare una curva da carico di glucosio come test diagnostico), sia la diagnosi di DM che per molti deve essere considerata una malattia a sé stante. Il secondo aspetto riguarda l’obesità centrale, come misurarla e quindi quali limiti considerare. Probabilmente sono i molteplici fenotipi clinici della SM, che peraltro possono cambiare per fenomeni fisiologici come l’invecchiamento e la menopausa, a creare questa protratta confusione di inquadramento nosografico e diagnostico. L’aumento della pressione arteriosa (PA) e la dislipidemia aterosclerotica (intesa come aumento dei trigliceridi, della Apoproteina B, delle lipoproteine LDL piccole e dense, e diminuzione del colesterolo HDL), sono gli altri fattori di rischio metabolico che caratterizzano la SM e sui i quali c’è invece un unanime consenso.
Nel 2009 alcune tra le maggiori società scientifiche (Diabetes Federation Task Force on Epidemiology and Prevention, National Heart, Lung, and Blood Institute, American Heart Association, World Heart Federeration, International Atherosclerosis Society, International Association for The study of Obesity) hanno cercato di armonizzare i vari criteri diagnostici per la SM: la presenza di almeno tre fattori di rischio su 5 per fare diagnosi di SM (tabella 1)(1). Nessuno è ritenuto indispensabile, compreso l’obesità viscerale sul cui limite per definirla non c’è consenso (vedi tabella 2) (1).
| Tabella 1 Criteri per la diagnosi clinica di Sindrome Metabolica |
|
| Fattore di rischio | Limiti |
| Circonferenza addominale * | In relazione alla popolazione o all’etnia (tabella 2) |
| Trigliceridi (oppure terapia specifica) ** | ≤ 150 mg/dL (1.7 mmol/L) |
| Colesterolo HDL (oppure terapia specifica) ** | ≤ 40 mg/dL (1 mmol/L) ≤ 50 mg/dL (1.3 mmol/L) |
| Pressione Arteriosa (oppure terapia specifica in paziente con ipertensione anamnestica) | Sistolica ≤ 130 e/o Diastolica ≤ 85 mmHg |
| Aumento della Glicemia (oppure terapia specifica) *** | ≤ 100 mg/dL (5.6 mmol/L) |
| è necessaria la presenza di almeno tre fattori su cinque * E’ raccomandato l’utilizzo dei limiti indicati dall’IDF per la popolazione di origine non Europea, mentre per quella Europea possiamo fare riferimento ai limiti indicati dall’IDF o dal AHA/NHLBI ** I farmaci più comunemente utilizzati per l'ipertrigliceridemia (o per Colesterolo HDL ridotto) sono fibrati, acido nicotinico e acidi grassi omega-3 (solo per trigliceridi) *** La maggior parte dei pazienti con diabete mellito di tipo 2, in base a questi criteri, ha la Sindrome Metabolica |
|
| Tabella 2 Limiti raccomandati dalle principali organizzazioni scientifiche per definire l’obesità addominale |
|||
| Popolazione | Organizzazione | Uomini | Donne |
| Europei | IDF | ≤ 94 cm | ≤ 80 cm |
| Caucasici | WHO | ≤ 94 cm (rischio moderato) ≤ 102 cm (rischio alto) |
≤ 80 cm (rischio moderato) ≤ 88 cm (rischio alto) |
| USA | AHA/NHLBI (ATP III) | ≤ 94 cm (rischio moderato) ≤ 102 cm (rischio alto) |
≤ 80 cm (rischio moderato) ≤ 88 cm (rischio alto) |
| Europei | Health Canada | ≤ 102 cm | ≤ 88 cm |
| Asiatici (compresi giapponesi) | IDF | ≤ 90 cm | ≤ 80 cm |
| Asiatici | WHO | ≤ 90 cm | ≤ 80 cm |
| Giapponesi | Japanese Obesity Society | ≤ 85 cm | ≤ 80 cm |
| Cinesi | Cooperative Task Force | ≤ 85 cm | ≤ 80 cm |
| Mediterranei, medio orientali | IDF | ≤ 94 cm | ≤ 80 cm |
| Africa sub-sahariana | IDF | ≤ 94 cm | ≤ 80 cm |
| America centrale e del sud | IDF | ≤ 90 cm | ≤ 80 cm |
L’obiettivo del clinico è, da una parte identificare precocemente le persone a rischio per la SM in modo da impostare un precoce intervento sullo stile di vita, e dall’altra, per coloro che già hanno la sindrome, ridurre il rischio di malattia aterosclerotica, che è raddoppiato, e diabete (5). In particolare la SM senza IGT e IFG comporta un rischio di diabete di 5 volte, mentre per coloro che hanno già il pre-diabete il rischio aumenta di 5-7 volte rispetto a quelli senza (6).
Identificazione dei pazienti a rischio per la SM
Valutare ogni tre anni le persone con uno o più fattori di rischio, con la misurazione della circonferenza addominale, della PA, della glicemia a digiuno e dell’assetto lipidico (2).
Pazienti con SM
L’approccio primario è costituito dall’intervento sullo stile di vita in modo da ottenere un adeguato calo ponderale. Dopo un periodo di 3–6 mesi valuteremo in ogni paziente il rischio cardiovascolare assoluto, utilizzando algoritmi validati come la carta del rischio cardiovascolare del “il progetto cuore” dell’Istituto Superiore di Sanità, oppure, qualora sia già presente il diabete, lo UKPDS Risk score, perché, oltre ai fattori di rischio classici, tiene in considerazione anche la durata del diabete e il grado di controllo glicemico.
Il calcolo del rischio ci consentirà di individuare l'obiettivo per la terapia medica, se necessaria, per la PA e la dislipidemia. In una prospettiva clinica potremmo affermare che mentre la diagnosi di SM individua il rischio relativo (di malattia cardiovascolare e diabete), le carte del rischio ci consentono di determinare il rischio cardiovascolare assoluto del nostro paziente (tabella 3).
TERAPIA
| Tabella 3 Obiettivi terapeutici per il trattamento della Sindrome metabolica |
||
| Ambito | Obiettivo | |
| Modifica stile di vita | Ridurre |
peso corporeo del 7-10% in un anno con un minor apporto calorico quotidiano (500-1000 Kcal/die in meno rispetto all’abituale) |
| Aumentare il consumo di frutta (tre volte al dì), verdura (due volte al dì), legumi e pesce | ||
| Fare regolare attività fisica (camminare almeno 30-60 minuti al dì per 5 giorni alla settimana) | ||
| Abolire il fumo | ||
| Fattori di rischio metabolici | Colesterolo - LDL | Rischio Molto Alto (risk SCORE ≤ 15%): ≤ 70 mg/dL Rischio Alto (risk SCORE 10-14%): ≤ 100 mg/dL Rischio Moderato (risk SCORE 5-10%): ≤ 115 mg/dL Rischio Basso (risk SCORE < 5%): ≤ 130 mg/dL |
| Pressione Arteriosa | < 130/80 mmHg | |
| HbA1c | < 6% se è presente Diabete < 6.5-7% |
|
| Stato protrombotico | Può essere raccomandata una bassa dose di ASA (75-100 mg/die) nei pazienti a rischio alto e molto alto in prevenzione primaria | |
Terapia ipolipemizzante
Il farmaco di prima scelta è la statina, per la quale è sempre necessario ottimizzare il dosaggio prima di prendere in considerazione la sostituzione o l'associazione con altri farmaci.
In casi di intolleranza a dosaggi elevati oppure quando non abbiamo raggiunto l’obiettivo, possiamo associare ezetimibe.
Per i pazienti con dislipidemia aterogena il farmaco di seconda linea è il fenofibrato; gli acidi grassi omega-3 sono da considerare farmaci di 3° linea.
Per la rimborsabilità dei farmaci da parte del SSN dobbiamo fare riferimento alla nota AIFA 13.
Terapia ipoglicemizzante
La metformina e i glitazoni (attualmente è disponibile solo il pioglitazone) sono farmaci insulino-sensibilizzanti e sono in grado di prevenire il diabete, seppure in minor misura rispetto un adeguato intervento sullo stile di vita. Non abbiamo dati sulla loro efficacia sul rischio cardiovascolare in prevenzione primaria in pazienti con pre-diabete.
L’acarbosio, seppure con un meccanismo diverso, può essere una valida alternativa in relazione al rischio cardiovascolare.
Il loro utilizzo in prevenzione primaria non è rimborsabile dal SSN.
Terapia anti-ipertensiva
L'obiettivo pressorio è più basso rispetto alla popolazione senza SM e lo è ancor più in presenza di DM o nefropatia o proteinuria.
Non abbiamo un consenso su quale farmaco utilizzare nella SM, ma ragionevolmente il farmaco di prima scelta può essere un ACE-inibitore o un bloccante del recettore dell’angiotensina II, se il primo non fosse tollerato, per il favorevole profilo sui fattori di rischio metabolico.
Terapia anti-aggregante
Mentre abbiamo dati “solidi” in prevenzione secondaria, in quella primaria c’è al momento molta incertezza nel rapporto rischio/beneficio; pertanto una basso dosaggio di ASA può essere raccomandabile solo in pazienti con un rischio cardiovascolare alto o molto alto.
BIBLIOGRAFIA
- Alberti KG, et al. Harmonizing the Metabolic Syndrome: A Joint Interim Statement of the International Diabetes Federation Task Force on Epidemiology and Prevention. National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the study of Obesity. Circulation 2009, 120: 1640-5.
- Rosenzweig GL, et al. Primary prevention of cardiovascular disease and type 2 diabetes in patients at metabolic risk: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008, 93: 3671-89.
- Kahn R, et al. The metabolic syndrome: time for a critical appraisal. Joint statement from American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia 2005, 48: 1684-99.
- Grundy SM, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Circulation 2005, 112: 2735-52.
- Grundy SM. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol 2012, 59: 635-43.
Fisiopatologia della sindrome metabolica
Silvio Settembrini
Servizio di Malattie Metaboliche e Diabetologia, ASL Napoli 1 Centro
Le basi fisiopatologiche della Sindrome Metabolica (o meglio “Endocrino-Metabolica” per la compartecipazione di svariati ormoni) sono collegate alla sua multifattorialità ed eterogeneità e pertanto il loro determinismo, in termini di progressione patogenetica è da riferire ai singoli fenotipi, che possono variare per “primum movens” e crono-ritmo. Il filone interpretativo principale della sindrome (1) vede però i suoi determinanti fisiopatologici essenziali nell’obesità centrale, viscerale, e nell’insulino-resistenza (2,3), che in genere segue l’aumento di massa adiposa e il prodursi di una condizione di steatosi multi-sistemica, che configura un quadro di “adiposopatia” diffusa (4).
La deposizione ectopica di tessuto adiposo in tutti i parenchimi (fegato, pancreas, cuore, rene, ecc) e peri-organo, con una contiguità topografica che determina una relazione di tipo paracrino negativa tra l’adipe e l’organo stesso (5), successiva cronologicamente all’aumento complessivo di massa adiposa, specie in sede viscerale e retro-peritoneale (6-8), condiziona le funzioni dell’organo infiltrato, avviando le premesse patogenetiche per la disfunzione d’organo e le successive complicanze (7, 8). Specialmente il fegato va incontro a una progressione di danno funzionale e biochimico, che condiziona il metabolismo glico-lipidico (9) con un’evoluzione fibro-infiammatoria (10, 11). Inoltre, l’infiltrazione lipidica, soprattutto a livello inter- e intra–muscolare, ma anche peri-intra-vascolare (12-14), induce una condizione di insulino-resistenza muscolare e vascolare e successivamente d’organo e sistemica (2, 3, 8, 15, 16). Questa è direttamente collegata all’insorgenza di ipertensione arteriosa (17, 18-20), anche per l’iperattivazione del sistema renina-angiotensina (RAAS) intra-adiposo (21), con conseguente aumentata espressione di angiotensinogeno ed amplificazione dell’ipertono simpatico (22), con reclutamento negativo del segnale insulinico, i cui substrati vengono fosforilati su residui di serina-treonina competitivi per quelli fisiologici su tirosina (23), il cui effetto è l’innesco della disfunzione endoteliale (24).
Rilevante è il ruolo delle adipochine, con alterato pattern secretivo nell’obesità addominale: poichè il tessuto adiposo diventa sede di infiammazione cronica (25), con produzione di fattori chemiotattici, quali la proteina MCP-1, attivanti l’infiltrazione macrofagica, di conseguenza si realizzano riduzione di adiponectina ed aumento di leptina, e aumentata espressione nei macrofagi intra-adiposi di interleuchina-6 e TNF-alfa, attivanti a cascata il background immuno-infiammatorio (6, 7, 15, 25, 26) che caratterizza la sindrome. Il terminale molecolare è il NFkB (fattore nucleare ubiquitario di trascrizione kB), responsabile dell’attivazione di numerosi geni collegati al danno vascolare e d’organo e all’alterato equilibrio emocoagulativo e trombotico (27), derivante dalla ridotta fibrinolisi, dall’aumentata viscosità ematica, dall’aumento del fibrinogeno, dall’infiammazione vascolare (PCR aumentata) (28, 29) ed inoltre dallo stress ossidativo e dall’aumentata secrezione adipocitaria di PAI 1 (inibitore dell’attivatore del plasminogeno). Il NFkB può essere considerato il punto di partenza delle manifestazioni della sindrome metabolica (diabete, obesità, aterosclerosi, ipertensione arteriosa)(30), ma del tutto recentemente è stato documentato un ruolo anche dei microRNA nell’omeostasi glico-lipidica e nel controllo vascolare (31). L’aumento in circolo di acidi grassi liberi (FFA), derivanti dalla disfunzione adipocitaria, condiziona a sua volta l’insulino-resistenza epatica, muscolare e adiposa, con aumentata produzione di lipoproteine VLDL e successiva dislipidemia aterogena, i cui aspetti peculiari sono la riduzione delle HDL, l’aumento dei trigliceridi, e la presenza di LDL piccole e dense altamente ossidate, vettrici di elevate quantità di aldeidi; da sottolineare che le stesse HDL diventano disfunzionali, in quanto anch’esse ossidate, e come tali eliminate dal rene (1, 32).
Gli FFA determinano da un lato infiltrazione lipidica, aumentata ossidazione degli acidi grassi, aumentata produzione epatica di glucosio, ridotta captazione periferica di glucosio, condizionanti l’innesco di alterata glicemia a digiuno e ridotta tolleranza glicidica (5, 33), e dall’altro, soprattutto a livello cardiovascolare, l’up-regulation della NADPH-ossidasi di membrana (34), con aumentata generazione di ROS e quindi di disfunzione mitocondriale, che determina le condizioni per una lipo-apoptosi cellulare: infatti, l’aumento di ceramide derivante dal palmitato blocca la protein-chinasi B (AKT-PKB) e riduce l’attività delle chinasi di sopravvivenza, inducendo un ridotto differenziamento cellulare negli organi bersaglio, creando le premesse nel tempo per le insufficienze d’organo (35-37).
Inoltre l’attivazione pluri-tissutale del RAAS (21, 38-40) gioca un ruolo importante (specie l’interferenza sul signaling insulinico di angiotensina II (23) e aldosterone (41, 42)) nel determinismo della sindrome (24, 43, 44), unitamente all’aumento del cortisolo dovuto alla disregolazione della 11ß–idrossisteroido-deidrogenasi e all’attivazione dell’asse ipotalamo-ipofiso-surrene (45-48). Altrettanto importante è il ruolo di androgeni (49-51) ed estrogeni (52-55), per il loro ruolo protettivo nel metabolismo glico-lipidico e sulla funzione endoteliale. Gli ormoni tiroidei svolgono un ruolo predisponente alla sindrome metabolica in caso di ipotiroidismo, per la ridotta azione sul metabolismo glico-lipidico, sui mitocondri e le fosforilazioni ossidative (56-60).
La sequenza patogenetica, nella progressione della sindrome metabolica e nell’accumulo dei fattori di rischio, spiega la conseguente morbilità cardio-vascolare (5, 61-65).
Fisiopatologia della sindrome metabolica (modificato da 66)
Bibliografia
- Crepaldi G. Origine e sviluppo della Sindrome Metabolica. Sindr Metab Mal Cardiovasc 2008, 1: 3, - Kurtis Ed.
- Schmidt AM. Insulin resistance and metabolic syndrome: mechanisms and consequences. Arterioscler Thromb Vasc Biol 2012, 32: 1753.
- Rask-Madsen C, Kahn CR. Tissue–specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol 2012, 32: 2052-9.
- Bays HE. Adiposopathy. Is “Sick Fat” a Cardiovascular Disease? JACC 2011, 57: 2461–73.
- Clement K, et al. Weight of pericardial fat on coronaropathy. Arterioscler Thromb Vasc Biol 2009, 29: 615-61.
- Phillips LK, Prins JB. The link between abdominal obesity and the metabolic syndrome. Curr Hypertens Rep 2008, 10: 156-64.
- Després JP, Lemieux I, Bergeron J, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol2008, 28: 1039-49.
- Türkoglu C, Duman BS, Günay D, et al. Effect of abdominal obesity on insulin resistance and the components of the metabolic syndrome: evidence supporting obesity as the central feature. Obes Surg 2003, 13: 699-705.
- Serné EH, de Jongh RT, Eringa EC, et al. Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension 2007, 50: 204-11.
- Kotronen A, Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol 2008, 28: 27-38.
- Joy T, Lahiry P, Pollex RL, et al. Genetics of metabolic syndrome. Curr Diab Rep 2008, 8: 141-8.
- Houben AJ, et al. Perivascular fat and the microcirculation: relevance to insulin resistance, diabetes, and cardiovascular disease. Curr Cardiovasc Risk Rep 2012, 6: 80–90.
- Chaldakov GN, et al. Perivascular adipose tissue: a novel component of global cardiometabolic risk. CMR @ Journal 2010, 3: 19-22.
- Ouwens DM, et al. The role of epicardial and perivascular adipose tissue in the pathophysiology of cardiovascular disease. J Cell Mol Med 2010, 14: 2223-34.
- Romeo GR, et al. Metabolic syndrome, insulin resistance, and roles of inflammation – mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol 2012, 32: 1771-7.
- Lann D, LeRoith D. Insulin resistance as the underlying cause for the metabolic syndrome. Med Clin North Am 2007, 91: 1063-77.
- Cuspidi C, Sala C, Zanchetti A. Metabolic syndrome and target organ damage: role of blood pressure. Expert Rev Cardiovasc Ther 2008, 6: 731-43.
- Stehouwer CD, Henry RM, Ferreira I. Arterial stiffness in diabetes and the metabolic syndrome: a pathway to cardiovascular disease. Diabetologia 2008, 51: 527-39.
- Mendizábal Y, et al. Hypertension in metabolic syndrome: vascular pathophysiology. Int J Hypertens 2013 (2013), Article ID 230868.
- Suzuki T, Homma S. Treatment of hypertension and other cardiovascular risk factors in patients with metabolic syndrome. Med Clin North Am 2007, 91: 1211-23.
- Marcus Y, et al. Adipose tissue renin–angiotensin–aldosterone system (RAAS) and progression of insulin-resistance. Molec Cell Endocrinol 2013, 378: 1-14.
- Kennedy RL, et al. Free fatty acid receptors: emerging targets for treatment of diabetes and its complications. Ther Adv Endocrinol Metab 2010, 1: 165.
- Folli F, et al. Crosstalk between insulin and angiotensin II signalling systems. Exp Clin Endocrinol Diabetes 1999, 107: 133-9.
- Sarzani R, Salvi F, Dessì-Fulgheri P, Rappelli A. Renin-angiotensin system, natriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in humans. J Hypertens 2008, 26: 831-43.
- Faloia E, et al. Inflammation as a link between obesity and metabolic syndrome. J Nutrit Metab 2012, 2012: 476380.
- Gustafson B, Hammarstedt A, Andersson CX, et al. Inflamed adipose tissue: a culprit underlying the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol2007, 27: 2276-83.
- Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thromb Haemost 2008, 99: 995-1000.
- Pravenec M, et al. Effects of human C-reactive protein on pathogenesis of features of the metabolic syndrome. Hypertension 2011, 57: 731-7.
- Horiuchi M, Masaki M. C-reactive protein beyond biomarker of inflammation in metabolic syndrome Hypertension 2011, 57: 672-3.
- Baker RG, Hayden MS, Gosh S. NF-kB, inflammation, and metabolic disease. Cell Metab 2012, 13: 11-22.
- Fernández-Hernando C, et al. MicroRNAs in metabolic disease. Arterioscler Thromb Vasc Biol 2013, 33: 178-85.
- Olufadi R, Byrne CD. Clinical and laboratory diagnosis of the metabolic syndrome. J Clin Pathol 2008, 61: 697-706.
- Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab 2000, 11: 351-6.
- Ullevig S, et al. NADPH Oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arterioscler Thromb Vasc Biol 2012, 32: 415-26.
- Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem 2001, 276: 14890-5.
- Sharma S, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. Faseb J 2004, 18: 1692-700.
- Zhang Y, Ren J. Role of cardiac steatosis and lipotoxicity in obesity cardiomyopathy. Hypertension 2011, 57: 148-50.
- Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes Rev 2012, 13: 136-49.
- Zhou M-S, Schulman IH, Zeng Q. Link between the renin–angiotensin system and insulin resistance: Implications for cardiovascular disease. Vasc Med 2012, 17: 330-41.
- Stiefel P, Vallejo-Vaz AJ, García Morillo S, Villar J. Role of the renin-angiotensin system and aldosterone on cardiometabolic syndrome. Int J Hypertens 2011 (2011), Article ID 685238.
- Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med 2009, 150: 776-83.
- Fujita T, et al. Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension 2010, 55: 813-8.
- Thethi T, et al. The link between the renin-angiotensin-aldosterone system and renal injury in obesity and the metabolic syndrome. Curr Hypertens Rep 2012, 14: 160–9.
- de Kloet AD, Krause EG, Woods SC. The renin angiotensin system and the metabolic syndrome. Physiol Behav 2010, 100: 525-34.
- Walker BR, Andrew R. Tissue production of cortisol by 11beta-hydroxysteroid dehydrogenase type 1 and metabolic disease. Ann NY Acad Sci 2006, 1083: 165-84.
- Góth M, Hubina E, Korbonits M. Correlations between the hypothalamo-pituitary-adrenal axis and the metabolic syndrome. Orv Hetil 2005, 146: 51-5.
- Stalder T, et al. Cortisol in hair and the metabolic syndrome. J Clin Endocrinol Metab 2013, 98: 2573-80.
- Abraham SB, et al. Cortisol, obesity, and the metabolic syndrome: a cross-sectional study of obese subjects and review of the literature. Obesity (Silver Spring) 2013, 21: E105-17.
- Hong-Yo Kang. Beyond the male sex hormone: deciphering the metabolic and vascular actions of testosterone. J Endocrinol 2013, 217: C1-3.
- Kelly M, Jones TH. Testosterone: a metabolic hormone in health and disease. J Endocrinol 2013, 217: R25-45.
- Kelly M, Jones TH. Testosterone: a vascular hormone in health and disease. J Endocrinol 2013, 217: R47-71.
- Cussons AJ, Stuckey BG, Watts GF. Metabolic syndrome and cardiometabolic risk in PCOS. Curr Diab Rep2007, 7: 66-73.
- Matic M, et al. Estrogen signalling and the metabolic syndrome: targeting the hepatic estrogen receptor alpha action. PLoS One 2013, 8: e57458.
- Mauvais-Jarvis F, et al. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 2013, 34: 309-38.
- Finan B, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med 2012, 18: 1847-56.
- Kota SK, Meher LK, Krishna S, Modi K. Hypothyroidism in metabolic syndrome. Indian J Endocrinol Metab 2012, 16 (Suppl 2): S332-3.
- Oh JY, Sung YA, Lee HJ. Elevated thyroid stimulating hormone levels are associated with metabolic syndrome in euthyroid young women. Korean J Intern Med 2013, 28: 180-6.
- Chugh K, Goyal S, Shankar V, Chugh SN. Thyroid function tests in metabolic syndrome. Indian J Endocrinol Metab 2012, 16: 958-61.
- Heima NE, Eekhoff EM, Oosterwerff M, et al. Thyroid function and the metabolic syndrome in older persons: a population-based study. Eur J Endocrinol 2012, 168: 59-65.
- Song Y, Yao X, Ying H. Thyroid hormone action in metabolic regulation. Protein Cell 2011, 2: 358-68.
- Nilsson PM. Cardiovascular risk in the metabolic syndrome: fact or fiction? Curr Cardiol Rep 2007, 9: 479-85.
- Grundy SM. Metabolic syndrome: a multiplex cardiovascular risk factor. J Clin Endocrinol Metab 2007, 92: 399-404.
- Obunai K, Jani S, Dangas GD. Cardiovascular morbidity and mortality of the metabolic syndrome. Med Clin North Am 2007, 91: 1169-84.
- Air EL, Kissela BM. Diabetes, the metabolic syndrome, and ischemic stroke: epidemiology and possible mechanisms. Diabetes Care 2007, 30: 3131-40.
- Towfighi A, Ovbiagele B. Metabolic syndrome and stroke. Curr Diab Rep 2008, 8: 37-41.
- Kassi E. Metabolic syndrome: definitions and controversies. BMC Med 2011, 9: 48.
Lipodistrofie
Simona Ferri
SC di Endocrinologia - Ospedale Maggiore e Bellaria - Azienda USL di Bologna
(aggiornato al 5 agosto 2016) questo capitolo è in attesa di aggiornamento
DEFINIZIONE E FISIOPATOLOGIA
Sono sindromi eterogenee, caratterizzate dall’assenza parziale o completa di tessuto adiposo bianco, che determina livelli bassi o indosabili di leptina, citochina derivante dal tessuto adiposo stesso. La leptina regola numerosi processi metabolici, inclusa l’omeostasi glucidica, l’insulino-sensibilità e l’ossidazione degli acidi grassi, e ha un ruolo importante nella regolazione dell'appetito (effetto anoressizzante). I pazienti affetti da lipodistrofia presentano difetti della differenziazione degli adipociti, che causano una maggiore disponibilità di acidi grassi liberi. Questi si depositano selettivamente nel tessuto adiposo viscerale, con conseguenti alterazioni metaboliche peculiari, quali insulino-resistenza, diabete mellito e ipertrigliceridemia. Tali alterazioni causano secondariamente lo sviluppo di aterosclerosi, steatosi epatica non alcolica (NAFLD) e pancreatiti acute. Spesso le donne affette presentano sindrome dell'ovaio policistico. L’80% dei pazienti con lipodistrofia soddisfa i criteri diagnostici per la sindrome metabolica.
CLASSIFICAZIONE E PATOGENESI
Si distinguono forme congenite e acquisite (tabella); la patogenesi delle forme congenite dipende da mutazioni con deficit di molecole coinvolte nella differenziazione degli adipociti e nel metabolismo lipidico. Le forme acquisite si verificano frequentemente nei pazienti HIV in terapia anti-retrovirale (HAART); in altri casi le sindromi lipodistrofiche si associano ad altre patologie autoimmuni (1).
| Classificazione lipodistrofie | |
| Congenite | Lipodistrofia generalizzata congenita (s. di Berardinelli-Seip) |
|
Lipodistrofie parziali familiari:
Atri tipi |
|
| Acquisite |
Lipodistrofia generalizzata acquisita (s. di Lawrence) |
| Lipodistrofia parziale acquisita (s. di Barraquer-Simons) | |
| Lipodistrofia HIV-relata | |
|
Lipodistrofie localizzate:
|
|
Nei pazienti con lipodistrofia è stato inoltre dimostrato un aumento sierico dei livelli di apolipoproteina CIII (apoCIII), importante regolatrice della sintesi di trigliceridi, sintetizzata principalmente a livello epatico e in minor parte nell’intestino tenue. ApoCIII è un’inibitore non competitivo della lipoproteina lipasi (LPL), che idrolizza i trigliceridi in acidi grassi liberi e favorisce il catabolismo delle lipoproteine ricche di trigliceridi con successiva clearance epatica. Un aumento di apoCIII è associato quindi a ipertrigliceridemia (2).
LIPODISTROFIE CONGENITE
Lipodistrofia generalizzata congenita (CGL) – s. di Berardinelli-Seip
Epidemiologia. Patologia a trasmissione autosomica recessiva, la prevalenza è stimata a livello mondiale circa 1/10.000.000. La caratteristica determinante è la totale assenza di tessuto adiposo, già riconoscibile a 2 anni di età (ma talvolta anche dalla nascita), con conseguente aspetto muscoloso.
Clinica. L’infanzia è caratterizzata da rapida crescita staturale e aumento dell'età ossea. A partire dall’adolescenza si manifesta tipicamente acanthosis nigricans, soprattutto a livello di collo e ascelle, ma anche di inguine e tronco. Frequentissime sono epatomegalia e steatosi, che spesso portano a cirrosi epatica; molto frequente anche la splenomegalia. Il fenotipo può apparire spesso acromegalico, per la presenza di prognatismo e ispessimento di mani e piedi. Nelle donne in età post-puberale si sviluppa frequentemente una s. dell’ovaio micropolicistico, associata spesso a clitoridomegalia e frequente infertilità, non descritta nel maschio. Sono frequenti le lesioni ossee di tipo litico, mentre in alcuni pazienti è stata descritta cardiomiopatia ipertrofica e ritardo mentale. A livello metabolico l’insulino-resistenza è per la maggior parte dei casi severa e spesso esita in diabete mellito tipo 2 già dall’adolescenza; anche l’ipertrigliceridemia è maggiormente severa, con frequenti complicanze pancreatitiche.
Genetica e patogenesi. Gli AGPAT (1-acylglycerol-3-phosphate O-acyltransferase) sono enzimi coinvolti nella sintesi di trigliceridi e fosfolipidi; l’isoforma AGPAT2 è espressa a elevati livelli nel tessuto adiposo e una sua mutazione pare che determini lipodistrofia mediante una deplezione di trigliceridi nel tessuto adiposo, riducendo inoltre la disponibilità di fosfolipidi, importanti per i segnali intra-cellulari e di membrana. I pazienti affetti da tale forma (CGL di tipo 1) presentano un risparmio del tessuto adiposo a carico delle aree meccanicamente protettive o di cuscinetto, come le regioni peri-articolari, le orbite, la vulva, i palmi delle mani, le piante dei piedi, lo scalpo, la lingua, il cavo orale e la regione peri-caliceale dei reni. Il risparmio di tali aree adipose sta a indicare una verosimile iperespressione delle altre isoforme di AGPAT.
Nelle CGL di tipo 2 si ha il coinvolgimento del gene che codifica per la seipina (BSCL2), che pare avere un ruolo importante nell’adipogenesi; la sua espressione è inoltre elevata a livello del tessuto encefalico e bassa a livello del tessuto adiposo: questo potrebbe spiegare il deficit cognitivo e il quadro clinico più grave che si può riscontrare nella CGL di tipo 2.
Le mutazioni di AGPT2 e BSCL2 sono responsabili del 95% delle forme. Molto più rare sono la CGL 3, associata alla mutazione di CAV1, che codifica per la caveolina, e la CGL 4, associata alla mutazione di PTRF.
Alcuni pazienti non presentano nessuna delle mutazioni note, a indicare che probabilmente anche altri geni sono coinvolti (1,3,4).
Lipodistrofia parziale familiare - varietà Dunnigan
Patologie a trasmissione autosomica dominante molto eterogenee. La più frequente, descritta da Dunnigan, comprende circa 200 pazienti e si caratterizza per una normale distribuzione del tessuto adiposo nell'infanzia, con progressiva perdita di grasso dagli arti e successivamente da addome e torace a partire dalla pubertà. Molti pazienti, in particolare le donne, accumulano grasso a livello di collo, visceri e viso, con aspetto cushingoide. La RMN dimostra in questi pazienti la perdita di grasso sottocutaneo, con accumulo dello stesso nello spazio inter-muscolare di braccia e gambe, oltre a eccesso di grasso intra-addominale. Le complicanze metaboliche, come insulino-resistenza, ipertrigliceridemia, bassi livelli di HDL, sono molto più frequenti nelle donne; generalmente il diabete si sviluppa dalla II decade di vita. Il meccanismo patogenetico coinvolge il gene LMNA, il cui difetto causa la morte degli adipociti.
Lipodistrofia parziale familiare tipo Kobberling
Sindrome rara a trasmissione autosomica dominante, la cui diagnosi è resa difficile dalla mancanza di test di validazione genetica. È caratterizzata da perdita di tessuto adiposo dagli arti inferiori, accumulo di grasso sottocutaneo a livello del tronco e ipertensione arteriosa. Le complicanze metaboliche sono quelle tipiche delle altre forme di lipodistrofia.
Lipodistrofia parziale familiare associata a mutazione del gene PPAR-gamma
Rara forma di lipodistrofia parziale, caratterizzata da perdita di tessuto adiposo da arti e viso, associata a mutazione del gene PPAR-gamma, che normalmente interviene nella differenziazione dell'adipocita.
Sono state inoltre descritte pazienti femmine con deplezione adiposa da braccia e gambe, con risparmio del tessuto adiposo del viso ed eccessiva deposizione intra-addominale senza mutazioni del gene PPAR-gamma né di LMNA, il che suggerisce che intervengano altre mutazioni.
Lipodistrofia associata a displasia mandibolo-acrale
Patologia estremamente rara, descritta in 40 pazienti, caratterizzata da ipoplasia clavicolare, acro-osteolisi, contratture articolari, pigmentazione cutanea, facies da neonato, alopecia, atrofia cutanea, anormalità della dentizione e lipodistrofia.
I pazienti affetti vengono suddivisi in varianti:
- di tipo A: deplezione delle riserve adipose sottocutanee negli arti e normale deposizione o eccesso a viso e collo;
- di tipo B: perdita di grasso sottocutaneo più generalizzata. In alcuni pazienti sono state descritte inoltre le complicanze metaboliche tipiche della lipodistrofia.
Anche in questi pazienti è stato riconosciuto come meccanismo patogenetico una mutazione del gene LMNA.
Altre forme di lipodistrofie congenite comprendono la sindrome SHORT, caratterizzata da bassa statura, iperestensibilità delle articolazioni, difetti corneali e dell'iride, dove la distribuzione del grasso è pressochè assente nel sottocutaneo, eccetto che nella zona sacrale e glutea (1).
LIPODISTROFIE ACQUISITE
Sono più frequenti delle forme ereditarie.
Lipodistrofia generalizzata acquisita (AGL) o s. di Lawrence
Epidemiologia: la lipodistrofia generalizzata è molto rara, descritta in 80 pazienti con un rapporto M:F = 1:3 e di cui la stragrande maggioranza è di razza caucasica.
Clinica: la perdita di tessuto adiposo avviene generalmente già dall’età infantile o dalla pubertà, ma può esordire anche nell’adulto, coinvolgendo gran parte dei distretti corporei, in particolare viso e arti; spesso il tessuto adiposo viene perso anche dal sottocutaneo di palme delle mani e dorso dei piedi, mentre risultano preservati il grasso retro-orbitario e il grasso bruno. Tipicamente i bambini affetti presentano appetito vorace. Spesso sono presenti già dall’infanzia acanthosis nigricans e epato-steatosi. Nel 20% dei pazienti è stata descritta cirrosi epatica.
Patogenesi: sembra immuno-mediata, in quanto è stato dimostrato che nel 25% dei pazienti la perdita di tessuto adiposo è preceduta da una pannicolite, generalmente successiva a terapia iniettiva sottocutanea con reazione granulomatosa. Inizialmente tale lesione va incontro a guarigione, con una perdita di tessuto adiposo localizzata, ma successivamente si manifesta il quadro della lipodistrofia generalizzata. Un altro 25% dei pazienti presenta un’altra patologia autoimmune associata, in particolare la dermato-miosite. Il restante 50% dei pazienti sembra essere affetto da una lipodistrofia generalizzata idiopatica. I pazienti che presentano una pannicolite come evento iniziale presentano una perdita di tessuto adiposo inferiore e una minore prevalenza di diabete mellito e ipertrigliceridemia (1).
Lipodistrofia parziale acquisita – s. di Barraquer Simons
Patologia rara, descritti circa 250 casi di varie estrazioni etniche, con rapporto M:F = 1:4. La perdita di tessuto adiposo è localizzata a viso, collo, arti superiori, torace e parte superiore dell’addome e avviene a partire dall’infanzia o adolescenza. In contrasto con la perdita di tessuto adiposo dai distretti corporei superiori, esso risulta invece in eccesso su anche e arti inferiori, soprattutto nelle donne. Le complicanze metaboliche non sono frequenti. È stato osservato che nel 20% dei pazienti affetti si ha lo sviluppo di una glomerulonefrite membrano-proliferativa, dopo circa 5-10 anni dall’esordio della lipodistrofia. Si è inoltre osservato in alcuni pazienti lo sviluppo di patologie autoimmuni, come LES. La base patogenetica risulta immuno-mediata, in quanto è stata osservata la presenza di bassi livelli di C3 e risulta inoltre dosabile una IgG policlonale, definita fattore nefritico C3, che attivando l’enzima C3-convertasi (C3b,Bb) riduce i livelli di C3 stesso, causando un’incontrastata attivazione della via alternativa del complemento. Rimane tuttora poco chiaro il motivo per il quale venga risparmiato il tessuto adiposo degli arti inferiori (1,5).
Lipodistrofia parziale in pazienti affetti da HIV
Epidemiologia: rappresenta il tipo più frequente di lipodistrofia, osservata nel 40% dei soggetti HIV in corso di terapia anti-retrovirale (HAART) per più di 12 mesi.
Clinica: la perdita di tessuto adiposo avviene a carico di arti, volto (in alcuni casi fino alla scomparsa della bolla adiposa di Bichat), regione temporale. Per quanto riguarda il lipo-accumulo, la manifestazione più eclatante è la comparsa di una massa circoscritta di tessuto adiposo nella regione dorso-cervicale. Talvolta si ha deposizione di adipe nel collo e nel tronco. Anche in questi pazienti si ha un accumulo di tessuto adiposo a livello viscerale, in particolare a livello epatico, con secondaria ipertrigliceridemia, epato-steatosi, insulino-resistenza e bassi livelli di colesterolo HDL. Rara è l’insorgenza di diabete mellito.
Patogenesi: è discussa. Allo stato attuale delle conoscenze è stato dimostrato che:
- vi è uno stretto legame causale con HAART, in particolare con gli inibitori della proteasi e gli inibitori nucleosidici della trascriptasi inversa;
- esiste una predisposizione genetica e una diversità razziale (i pazienti afro-americani risultano più protetti rispetto ai caucasici);
- l’attività fisica costante e il controllo dietetico svolgono un effetto protettivo e contenitivo del fenomeno.
I meccanismi con i quali gli HAART determinano lipodistrofia non sono ancora del tutto chiariti: gli inibitori della proteasi inibiscono l’adipogenesi, causando un’inibizione nella differenziazione dei pre-adipociti e inducono l’apoptosi degli adipociti maturi; gli inibitori nucleosidici della trascrittasi inversa determinano un danno mitocondriale. Altri meccanismi indiretti sono determinati dai cambiamenti immunologici secondari a HAART e infezione da HIV, che determinano riduzione della sintesi di colesterolo HDL.
Pazienti sottoposti allo stesso regime farmacologico possono rispondere, dal punto di vista metabolico, in maniera del tutto differente, a indicare una componente genetica. Sono in corso studi che sembrano evidenziare quadri genetici (su alcuni geni che codificano per proteine e ormoni implicati nel metabolismo lipidico, polimorfismi che riguardano geni implicati nell’apoptosi degli adipociti), che possono conferire protezione o propensione nei confronti della sindrome lipodistrofica (6).
Lipodistrofie localizzate
Caratterizzate dalla perdita di tessuto adiposo da piccole aree, con realizzazione di un’impronta. In alcuni pazienti vi può essere un’importante coinvolgimento del tronco o degli arti. Le cause possono essere molto varie, ma nella stragrande maggioranza dei casi sono determinate dalla terapia iniettiva insulinica o corticosteroidea, in rari casi da pegvisomant, oltre a pannicolite o sollecitazione pressoria localizzata ripetuta (1).
DIAGNOSI
Per porre diagnosi di lipodistrofia possono essere sufficienti le caratteristiche cliniche peculiari, le alterazioni biochimiche, la scarsità di grasso corporeo (valutato con DEXA o RMN) e bassi livelli di leptina, anche se sarebbe comunque necessario effettuare la ricerca del tipo di mutazione, che può orientare sul tipo di evoluzione clinica.
TERAPIA
Non vi è un’unica terapia per la lipodistrofia, ma ogni paziente deve ricevere una terapia personalizzata, finalizzata alle complicanze metaboliche che esso presenta.
Gestione della dislipidemia: dieta ipolipidica (< 15% di lipidi) ed esercizio fisico, evitando l’alcool. In caso di persistenza di alterato assetto lipidico nonostante dieta, esercizio fisico e mantenimento dell’euglicemia, è possibile utilizzare fibrati e omega-3 ad elevate dosi. In alternativa ai fibrati, possono essere prescritte statine, facendo attenzione nei pazienti HIV: per interazione con gli inibitori delle proteasi, sono preferibili rosuvastatina, pravastatina e fluvastatina. Evitare estrogeni per il trattamento della PCOS, in quanto potrebbero aggravare la dislipidemia.
Gestione dell’iperglicemia: si possono utilizzare insulino-sensibilizzanti (metformina o glitazonici). Particolarmente indicata risulta la metformina per l’effetto su peso, PCOS e steatosi. Indicata anche la terapia insulinica, che spesso deve essere effettuata ad alte dosi per la presenza di marcata insulino-resistenza.
Pazienti HIV: in quelli con dislipidemia non controllata potrebbe essere indicato il passaggio a una terapia anti-retrovirale differente. Anche se la perdita di tessuto adiposo sottocutaneo spesso può essere irreversibile, in alcuni casi si è assistito a un ripristino della normale distribuzione del grasso dopo mesi dal cambio di terapia. Sono stati inoltre condotti studi con tesamorelina, un GH-releasing hormone, che ha portato a riduzione della circonferenza addominale e del grasso addominale, della pressione e ad aumento dei livelli di adiponectina senza alterazioni dell’omeostasi glucidica. Purtroppo tale terapia risulta costosa e spesso i pazienti non sono complianti perchè è iniettiva sc; inoltre gli effetti favorevoli della terapia scompaiono alla sua sospensione.
La terapia d’elezione delle lipodistrofie generalizzate, congenite o acquisite, è la Metreleptina, analogo ricombinante della leptina. Questa è somministrata uno o due volte al giorno sc, preferibilmente allo stesso orario per mimare il ritmo circadiano della leptina. La dose iniziale raccomandata è calcolata in base al peso corporeo (dose massima giornaliera per peso < 40 kg = 0.13 mg/kg; per peso > 40 kg = 10 mg). L’eliminazione avviene quasi esclusivamente per via renale. Gli effetti collaterali più comuni, riscontrati nel 10% dei pazienti, sono cefalea, ipoglicemia, dolore addominale e calo ponderale, ma complessivamente la terapia è ben tollerata. È stata descritta un’associazione tra terapia con metreleptina e linfoma a cellule T, ma non è stata stabilita una reale relazione causale. In alcuni pazienti affetti da patologia autoimmune si è verificato un’esacerbazione della patologia stessa (epatite autoimmune e glomerulonefrite membrano-proliferativa). Le controindicazioni alla terapia con metreleptina sono obesità comune (obesità leptino-resistente) e ipersensibilità al principio attivo. Il farmaco non è approvato per l'uso in pazienti con lipodistrofia correlata a HIV o in pazienti con malattie metaboliche, compreso il diabete mellito e l’ipertrigliceridemia, senza evidenza concomitante di lipodistrofia generalizzata. Sicurezza ed efficacia di metreleptina sono state valutate in uno studio in aperto a braccio singolo, che ha incluso 48 pazienti con lipodistrofia generalizzata congenita o acquisita e diabete mellito, ipertrigliceridemia e/o livelli elevati di insulina a digiuno. Lo studio ha mostrato riduzioni di HbA1c, glicemia a digiuno e trigliceridi. Possono svilupparsi anticorpi con attività neutralizzante verso leptina e/o metreleptina, che potrebbero causare infezioni gravi o perdita di efficacia del trattamento. In gravidanza l’uso di tale terapia non è raccomandato. Non vi sono informazioni sull’effetto della terapia con metreleptina sull’anziano, per cui anche in pazienti con funzione renale conservata occorre prestare attenzione soprattutto alla scelta della dose iniziale, iniziando con la dose minima efficace. Poichè la metreleptina può potenzialmente alterare l’espressione di CYP450, deve essere utilizzata con cautela nei pazienti in terapia con farmaci metabolizzanti CYP450. A livello molecolare la metreleptina presenta un’elevata analogia strutturale con IL6, IL11, IL12 e oncostatina M, tutte facenti parte della famiglia delle citochine a catena lunga. La metreleptina causa inoltre un effetto insulino-sensibilizzante, aumentando il consumo di glucosio da parte del muscolo scheletrico con secondario effetto ipoglicemizzante. Riduce inoltre il livello di trigliceridi, migliorando secondariamente la steatosi, incrementa i livelli di colesterolo HDL e riduce l’insulinemia. Inoltre la metreleptina ha la capacità di regolare numerosi assi ormonali, come il somatotropo, corticotropo, tireotropo e gonadotropo, oltre ad avere effetti su osteogenesi, angiogenesi, ematopoiesi. La terapia sostitutiva con leptina in pazienti con lipodistrofia riduce i livelli sierici di trigliceridi, con effetto apparentemente indipendente da una riduzione dei livelli di apoCIII, ma il meccanismo responsabile rimane a tutt’oggi sconosciuto (2). L’efficacia della terapia con metreleptina risulta maggiore nelle forme generalizzate rispetto alle localizzate: poichè i pazienti affetti da lipodistrofia generalizzata presentano mortalità e morbilità maggiori a causa delle suddette anomalie metaboliche, risulta intuitivo il maggiore effetto positivo della terapia dal punto di vista metabolico (7).
BIBLIOGRAFIA
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004, 350: 1220-34.
- Kassai A, Ranganath M, et al. Effect of leptin administration on circulating apolipoprotein CIII levels in patients with lipodystrophy. J Clin Endocrinol Metab 2016, 101: 1790-7.
- Lima J, et al. Clinical and laboratory data of a large series of patients with congenital generalized lipodystrophy. Diabetol Metab Syndr 2016, 8: 23.
- Akinci B, Onay H, et al. Natural history of congenital generalized lipodystrophy: a nationwide study from Turkey. J Clin Endocrinol Metab 2016, 101: 2759-67.
- Oliveira J, Freitas P, et al. Barraquer-Simons syndrome: a rare form of acquired lipodystrophy. BMC Res Notes 2016, 9: 175.
- Da Cunha J, Morganti Ferreira Maselli L, et al. Impact of antiretroviral therapy on lipid metabolism of human immunodeficiency virus-infected patients: old and new drugs. World J Virol 2015, 4: 56-77.
- Rodriguez A, Mastronardi C, et al. New advances in the treatment of generalized lipodystrophy: role of metreleptin. Ther Clin Risk Manag 2015, 11: 1391-400.
Valutazione dello stato nutrizionale
Marco Cipolat
SC Dietetica e Nutrizione Clinica, AO S. Croce e Carle, Cuneo
(aggiornato al 3 febbraio 2020)
L’Organizzazione Mondiale della Sanità nel 1973 affermò che lo stato nutrizionale è la condizione dell’organismo che risulta dall’assunzione, l’assorbimento e l’utilizzo degli alimenti, così come da fattori di significato patologico. Di solito la valutazione dello stato nutrizionale include misure antropometriche, misure biochimiche e dietetiche, anamnesi clinica e fisiologica e altri dati. Ne consegue che la valutazione dello stato nutrizionale può essere un processo molto complesso. Qui prenderemo in considerazione alcune tra le principali e più diffuse metodiche, sufficienti a fornire un’adeguata determinazione dello stato nutrizionale nell’uomo.
Dall’esame obiettivo possono essere già rilevati molti dati (tab 1).
| Tabella 1 Principali segni clinici di malnutrizione |
||
| Organo | Quadro clinico | Probabile carenza |
| Condizioni generali | Calo ponderale, aspetto emaciato | Proteine ed energia |
| Cute | Dermatite | Proteine, Zn, vitamina A |
| Sottocute | Assottigliato Edema |
Proteine ed energia Proteine e tiamina |
| Mucose | Pallide | Ferro, vitamina E |
| Unghie | Coilonichia Fragilità, striature |
Ferro Aspecifica |
| Capelli | Alterazioni colore e struttura Perdita |
Proteine ed energia Zn, acidi grassi essenziali, Fe |
| Labbra | Lesioni angolari bilaterali, cheilosi | Proteine, Fe, cianocobalamina, acido folico, niacina, piridossina, riboflavina |
| Muscoli | Atrofia | Proteine ed energia |
Il dato più importante da tenere in considerazione resta tuttavia il peso del paziente e conseguentemente l’indice di massa corporea, e il loro andamento nel tempo. Nel paziente malnutrito sono certamente i dati più significativi ed immediati di deplezione di massa corporea, in particolare di massa muscolare.
| Tabella 2 Valutazione malnutrizione |
|||
| Parametro | Lieve | Moderata | Grave |
|
Calo ponderale:
|
5-10% |
11-20% 21-40% |
>20% >40% |
| BMI | 17-18.4 | 16-16.9 | < 16 |
| Indice creatinina/altezza | 99-80 | 79-60 | < 60 |
| Albumina (g/dL) | 3.5-3.0 | 2.9-2.5 | < 2.5 |
| Transferrina (mg/dL) | 200-150 | 149-100 | < 100 |
| Pre-albumina (mg/dL) | 18-22 | 17-10 | < 10 |
| Retinol-binding protein (mg/dL) | 2.9-2.5 | 2.4-2.1 | < 2.1 |
| Linfociti/mmc | 2000-1500 | 1199-800 | < 800 |
Parametri biochimici
La loro valutazione approfondisce e rende completa la valutazione dello stato nutrizionale. Nel 2002 la SINPE nell’ambito delle Linee Guida per la Malnutrizione Ospedaliera ha suggerito quanto riportato in tabella 2 (1). In modo particolare si sottolinea il significato del dosaggio delle proteine.
Albuminemia: ha valori normali di 3.4-4.5 g/dL, emivita di 20 giorni e dà conto della sintesi proteica a medio-lungo termine. Fornisce sufficienti informazioni sulla malnutrizione proteica viscerale. È il parametro che ha più dati in letteratura ed è il maggiore indice prognostico in relazione allo stato malnutritivo. Limitazioni: iposintesi epatica, perdita proteica nefrosica, stati vari di malattia acuta.
Transferrinemia: ha valori normali di 220-350 mg/dl, emivita di 8-10 giorni e dà conto della sintesi proteica a breve-medio termine. Il patrimonio totale corporeo è piccolo e pertanto può ben monitorare gli andamenti della sintesi proteica. Viene a ridursi negli stati morbosi acuti e cronici, in quanto va incontro a più rapida degradazione. Limitazioni: si riduce in caso di anemia carenziale B12, anemia da malattia cronica, sovraccarico marziale, sindrome nefrosica, terapia steroidea e aumenta nella carenza marziale, nell’ipossia e nella terapia estrogenica.
Pre-albumina: ha valori normali di 15-30 mg/dl, emivita di 2-4 giorni e dà conto della sintesi proteica a breve termine. Viene a ridursi negli stati morbosi acuti e cronici in quanto va incontro a minore sintesi. Limitazioni: iposintesi epatica, sindrome nefrosica (per perdita) e patologie genetiche a carico della sintesi proteica.
Tra gli altri valori di laboratorio non vanno dimenticati i dosaggi delle vitamine e degli elettroliti e l’emocromo, utile (oltre che per la valutazione dello stato dell’emopoiesi e del metabolismo marziale) anche per la conta linfocitaria, che dà ragione della capacità delle difese immunitarie che dipendono dalla disponibilità di diversi macro- e micro-nutrienti.
Dati antropometrici
I parametri più frequentemente usati sono (2):
- per valutare la componente muscolare: circonferenza di coscia e braccio, area muscolare del braccio;
- per valutare la componente adiposa: circonferenza della vita (componente viscerale) e pliche cutanee (componente sotto-cutanea le seconde);
- per valutare la distribuzione dell’adipe: rapporto tra circonferenza della vita e dei fianchi, che può essere tipo androide (prevalentemente viscerale) o ginoide (prevalentemente sotto-cutanea).
L’area muscolare del braccio è una misura che si ottiene mediante una formula matematica che comprende la misurazione (in cm) della circonferenza del braccio e della plica tricipitale del braccio sinistro (2,3):
((((circonferenza – (π x plica))²)/(4π)) – 10 (se maschio) o - 6.5 (se femmina).
La rilevazione dello spessore delle pliche cutanee viene eseguita sollevando le pliche e applicando un calibro a molla per una pressione di 10 g/mm², generalmente in quattro sedi:
- tricipitale: a metà omero con braccio piegato a 90°;
- bicipitale: al braccio (usualmente sinistro);
- sovra-iliaca: a 2 cm dalla cresta iliaca sulla linea ascellare media;
- sotto-scapolare: 1 cm al di sotto dell’angolo della scapola).
La percentuale di grasso corporeo in relazione a età e sesso può essere stimata, in soggetti di età > 17 anni, mediante la somma delle quattro pliche secondo le tabelle pubblicate nel 1974 da Durnin e Womersley (4) (tab 3).
| Tabella 3 Contenuto di grasso, come percentuale del peso corporeo, in relazione alla somma delle 4 pliche cutanee (bicipitale, tricipitale, sotto-scapolare e sovra-iliaca) secondo età e sesso |
||||||||
| Pliche (mm) | Maschi | Femmine | ||||||
| 17-29 aa | 30-39 aa | 40-49 aa | > 50 aa | 17-29 aa | 30-39 aa | 40-49 aa | > 50 aa | |
| 15 | 4.8 | - | - | - | 10.5 | - | - | - |
| 20 | 8.1 | 12.2 | 12.2 | 12.6 | 14.1 | 17.0 | 19.8 | 21.4 |
| 25 | 10.5 | 14.2 | 15.0 | 15.6 | 16.8 | 19.4 | 22.2 | 24.0 |
| 30 | 12.9 | 16.2 | 17.7 | 18.6 | 19.5 | 21.8 | 24.5 | 26.6 |
| 35 | 14.7 | 17.7 | 19.6 | 20.8 | 21.5 | 23.7 | 26.4 | 28.5 |
| 40 | 16.4 | 19.2 | 21.4 | 22.9 | 23.4 | 25.5 | 28.2 | 30.3 |
| 45 | 17.7 | 20.4 | 23.9 | 24.7 | 25.0 | 26.9 | 29.6 | 31.9 |
| 50 | 19.0 | 21.5 | 24.6 | 26.5 | 26.5 | 28.2 | 31.9 | 33.4 |
| 55 | 20.1 | 22.5 | 25.9 | 27.9 | 27.8 | 29.4 | 32.1 | 34.6 |
| 60 | 21.2 | 235 | 27.1 | 29.2 | 29.1 | 30.6 | 33.2 | 35.7 |
| 65 | 22.2 | 24.3 | 28.2 | 30.4 | 30.2 | 31.6 | 34.1 | 36.7 |
| 70 | 23.1 | 25.1 | 29.3 | 31.6 | 31.2 | 32.5 | 35.0 | 37.7 |
| 75 | 24.0 | 25.9 | 30.3 | 32.7 | 32.2 | 33.4 | 35.9 | 38.7 |
| 80 | 24.8 | 26.6 | 31.2 | 33.8 | 33.1 | 34.3 | 36.7 | 39.6 |
| 85 | 25.5 | 27.2 | 32.1 | 34.8 | 34.0 | 35.1 | 37.5 | 40.4 |
| 90 | 26.2 | 27.8 | 33.0 | 35.8 | 34.8 | 35.8 | 38.3 | 41.2 |
| 95 | 26.9 | 28.4 | 33.7 | 36.6 | 35.6 | 36.5 | 39.0 | 41.9 |
| 100 | 27.6 | 29.0 | 34.4 | 37.4 | 36.4 | 37.2 | 39.7 | 42.6 |
| In 2/3 dei casi l’errore era entro 3.5% del peso corporeo di grasso per le donne ed entro 5% per gli uomini | ||||||||
Va sottolineato che in considerazione del “trend secolare” che si osserva sui parametri antropometrici, cioè alla tendenza nel corso degli anni all’aumento dell’altezza, dello spessore delle pliche, della circonferenza del braccio e dell’area muscolare del braccio, attribuibili al miglioramento globale dello stato nutrizionale della popolazione, sarebbe auspicabile che detti valori di riferimento fossero rivisti periodicamente mediante adeguati studi (5).
Analisi della composizione corporea
Il modello bicompartimentale di composizione corporea può essere analizzato mediante la metodica dell’analisi corporea bioimpedenziometrica (impedenziometria o BIA). Questa metodica si avvale del principio per cui i tessuti magri, contenendo la mggior parte dell’acqua e degli elettroliti, sono buoni conduttori di corrente elettrica, mentre il tessuto grasso, al contrario, funge da isolante elettrico.
L’impedenza è l’ostacolo elettrico di un conduttore al passaggio di una corrente alternata e sottostà alla legge di Ohm. Usando sensori di fase, il vettore dell’impedenza Z è scomponibile in vettore di impedenza resistiva (o resistenza R) e vettore di resistenza capacitativa (o reattanza Xc) secondo la formula:
Z = (R² + Xc²)0.5
I vettori in fase hanno direzione parallela al flusso di corrente e sono determinati dall’ostacolo al passaggio di corrente di un conduttore, mentre quelli in contro-fase sono perpendicolari al passaggio di corrente e sono determinati dalla resistenza capacitativa che offre un condensatore al passaggio di corrente. L’angolo di fase descrive il rapporto che c’è tra resistenza al passaggio di corrente e resistenza capacitativa. Un circuito completamente resistivo darà angolo zero, uno totalmente capacitativo avrà valore di 90°. La massa magra costituisce, per quanto detto sopra, un buon conduttore, mentre la massa data dall’adipe, dall’osso e dal connettivo un buon condensatore, responsabile della resistenza al passaggio di corrente. A basse frequenze (< 5 kHz) la corrente si distribuisce solo negli spazi extra-cellulari e non è registrabile alcuno sfasamento vettoriale, in quanto si ha solo componente resistiva. Le correnti ad alta frequenza (10-100 kHz) penetrano invece nelle cellule, le quali agiscono, mediante le membrane, come condensatori e in tal modo compare lo sfasamento tra i sinusoidi delle correnti in entrata ed uscita e sono misurabili valori di reattanza capacitativa (6). Con queste premesse viene introdotta nel corpo un corrente elettrica di bassa intensità (800 µA) e frequenza 50 kHz, mediante 2 elettrodi posti al dorso della mano e a livello malleolare. Altri 2 elettrodi posti prossimalmente a questi rilevano la caduta del potenziale elettrico.
I valori di resistenza bio-elettrica riflettono le dimensioni della massa magra, o meglio, priva di grasso o fat free mass (essendo compresa anche l’acqua e gli elettroliti inevitabilmente contenuti negli adipociti). Quindi la massa grassa è ottenuta per differenza tra il valore di massa corporea totale e il valore di fat free mass. La reattanza e l’angolo di fase di cui sono responsabili le membrane e la loro caratteristica di comportarsi come un condensatore, è espressione della massa cellulare. La massa extra-cellulare è quindi ricavata dalla differenza tra fat free mass e massa cellulare. Dall’angolo di fase, che indica quanto l’impedenza dell’organismo sia di tipo capacitativo piuttosto resistivo, è possibile ricavare dati quantitativi su tutti i settori corporei che rivestono un interesse in nutrizione clinica. Con questa metodica è pertanto possibile avere dati su:
- acqua corporea totale (TBW - total body water);
- acqua extra-cellulare (ECW - extra cell water);
- acqua intra-cellulare (ICW - intra cell water);
- massa cellulare (BCM - body cell mass);
- massa magra (FFM - fat free mass);
- massa grassa (FM - fat mass);
- massa muscolare (MM - muscle mass);
- metabolismo basale correlato alla massa cellulare.
Il vettore che si ottiene mettendo in relazione reattanza, resistenza ed angolo di fase è in grado di quantificare i liquidi corporei e la loro distribuzione tissutale. È pertanto possibile ottenere il diagramma sotto-riportato, che dà conto dello stato nutrizionale e dello stato di idratazione.
Il posizionamento del vettore nella parte alta del diagramma mette in evidenza la disidratazione, quello in basso l’iperidratazione, a sinistra il buono stato nutrizionale/muscolare, a destra la malnutrizione/scarso patrimonio muscolare. I punti che ricadono all’interno del secondo anello del diagramma sono compresi nel 75% percentile di distribuzione e dunque all’interno dell’intervallo di normalità (7).
Valutazione funzionale muscolare
La massa muscolare scheletrica è la principale riserva proteica dell’organismo. La forza muscolare correla con il trofismo muscolare e l’area muscolare del braccio. Tra i metodi di valutazione funzionale più facilmente effettuabili e diffusi, vi è la handrgrip strength (8), cioè la prova della stretta di mano, che viene effettuata mediante dinamometri a molla o di tipo pneumatico, utilizzando per lo più l’arto non dominante. Si effettuano 3 misurazioni, tenendo conto del valore più alto, e i risultati possono essere espressi come percentuale dello standard (9).
Negli ultimi anni questa metodica è stata proposta per la definizione dello stato di fragilità del paziente anziano e notevoli dati sono stati rilevati anche di recente in popolazioni di pazienti affetti da neoplasie. Limite della metodica è ancora la standardizzazione: non vi è ancora accordo in merito ai metodi di misurazione (diversi tipi di strumenti in uso e diverse modalità di rilevazione) e non vi è ancora un cut-off di normalità condiviso (10, 11). In particolare il cut-off di normalità nell’anziano viene posto a 17.5 kg per il soggetto di sesso femminile e a 30 kg per il soggetto di sesso maschile (12), mentre il gruppo ESGWOP lo pone a 20 kg per la donna e a 30 kg per l’uomo (13,14).
Bibliografia
- Linee guida per la Nutrizione Artificiale Ospedaliera. Rivista Italiana di Nutrizione Parenterale ed Enterale 2002, 20 S5: S9-11.
- Frisancho AR. Anthropometric standards for the measurement of growth and nutritional status. University of Michigan Press, Ann Arbor, 1990.
- Heymsfield SB, et al. Anthropometric measurement of muscle mass: revised equation for calculating bone free arm muscle area. Am J Clin Nutr 1982, 36: 680–90.
- Durnin JV, Womersley J. Body fat assessed from total body density and its estimation from skinfold thickness: measurements on 481 men and women aged from 16 to 72 years. Br J Nutr 1974, 32: 77-97.
- Shah M, Hannan PJ, Jeffery RW. Secular trend in body mass index in the adult population of three communities from the upper mid-western part of the USA: the Minnesota Heart Health Program. Int J Obes 1991, 15: 499-503.
- Baumgartner RN, Chumlea WC, Roche AF. Bioelectric impedance phase angle and body composition. Am J Clin Nutr 1988, 48: 16-23.
- Piccoli A, Rossi B, et al. A new method for monitoring body fluid variation by bioimpedance analysis: the RXc graph. Kidney Int 1994, 46: 534-9.
- Martin S, Neale G, Elia M. Factors affecting maximal momentary grip strength. Hum Nutr Clin Nutr 1985, 39: 137-47.
- Webb AR, Newman LA, et al. Handgrip dynamometry as a predictor of postoperative complications: reappraisal using age standardized grip strength. JPEN J Parenter Enteral Nutr 1989, 13: 30-3.
- Moreau J, Ordan MA, et al. Correlation between muscle mass and handgrip strength in digestive cancer patients undergoing chemotherapy. Cancer Med 2019, 8: 3677–84.
- Roberts HC, Denison HJ. A review of the measurement of grip strength in clinical and epidemiological studies: towards a standardised approach. Age Ageing 2011, 40: 423–9.
- Fried LP, Tangen CM, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001, 56: M146-56.
- Cruz‐Jentoft AJ, Baeyens JP, Bauer JM, et al. Sarcopenia: European consensus on definition and diagnosis. Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39: 412–23.
- Laurentani F, Russo C, Bandinelli S, et al. Age-associated changes in skeletal muscles and their effect on mobility: an operational diagnosis of sarcopenia. J Appl Physiol 2003, 95: 1851–60.
Principi di nutrizione artificiale
Marco Cipolat
SC Dietetica e Nutrizione Clinica, AO S. Croce e Carle, Cuneo
(aggiornato al 23 gennaio 2020)
Qualora il paziente non sia in grado di soddisfare i fabbisogni calorico-proteici con l’alimentazione naturale, è possibile farlo mediante la nutrizione artificiale. Questa è attuabile mediante tre vie di somministrazione:
- orale mediante integratori calorico-proteici (ONS);
- enterale (da qui in poi NE);
- parenterale (da qui in poi NP).
Scelta della via di somministrazione
È legata fondamentalmente all’evidenza, per cui più questa si avvicina alle modalità naturali e meno esporrà il paziente a complicanze metaboliche (iperglicemia, dislipidemia, ecc.) o legate alla via di somministrazione (es. calcolosi colecisti nei casi di lunga durata, dislocazione del dispositivo, infezione dello stesso, sino alla sepsi nel caso della NP). L’utilizzo del tratto gastro-enterico permette, inoltre, il mantenimento del trofismo entero-colico, assicurandone la funzionalità e ostacolando la traslocazione batterica verso il letto vascolare, che è causa frequente di sepsi e di insufficienza multi-organo. Va inoltre considerato che l’efficacia nel correggere o prevenire la malnutrizione non è legata alla modalità di somministrazione, in quanto è ormai ampiamente dimostrato che questo risultato è ottenibile indifferentemente mediante le tre metodiche. Ne consegue che il principio che ci deve guidare nella scelta della via di somministrazione potrebbe tradursi in “oral/enteral first”. In altre parole:
- ogni qualvolta il paziente soddisfi i propri fabbisogni per via orale almeno per il 50-70%, è di prima scelta la prescrizione di ONS;
- qualora il paziente non sia in grado di farlo e abbia una buona funzione del tratto gastro-enterico, va sempre scelta la NE;
- la NP va riservata per i casi in cui vi sia l’impossibilità a utilizzare il tratto gastro-enterico per gravi situazioni cliniche, tra cui gravi sindromi malassorbitive, fistole ad alta gittata, stati occlusivi, dolore post-prandiale non controllato, sindrome ischemica addominale, ecc.
È possibile inoltre somministrare gli apporti utilizzando contemporaneamente più vie, qualora il paziente non sia in grado di completare i fabbisogni mediante la via maggiormente fisiologica. È cioè possibile affiancare una quota di apporti per via parenterale a pazienti che stiano assumendo alimentazione naturale, ONS o NE, oppure è possibile effettuare NE in pazienti che assumono alimenti per os o ONS.
Scelta del dispositivo
Va scelto considerando i tempi per cui prevediamo di usare la via artificiale (1):
- per periodi brevi, sonda naso-gastrica o catetere venoso periferico;
- per tempi di attuazione di durata medio-lunga, PEG, digiunostomia o accesso venoso centrale.
Nutrienti
Oggi gli ONS, le miscele enterali e parenterali sono molteplici e legate allo stato clinico del paziente, alle patologie da cui è affetto, allo stato di maggiore o minore malnutrizione.
In particolare, vi sono ONS e miscele enterali monomeriche (cioè contenenti un solo tipo di nutrienti come malto-destrine o proteine e solitamente in polvere) o polimeriche (cioè contenenti più nutrienti), prive o ricche in fibre (e queste di varia tipologia), equilibrate o destinate alle varie patologie. Tra queste ricordo quelle semi-elementari (contenenti nutrienti pre-digeriti per pancreopatie o difficoltà digestivo-assorbitive), per diabete (in grado di dare un maggiore controllo delle glicemie), insufficienza renale, miscele da utilizzarsi nel paziente catabolico, miscele con scopi farmaco-nutrizionali (immuno-modulanti, indicate per portatori di lesioni da pressione, ecc), miscele atte a preparare il paziente alla chirurgia, come avviene nei protocolli ERAS/Fast track. Per ciascuna miscela rimando, per quanto attiene la composizione, alle varie schede tecniche reperibili presso i produttori e, per quanto attiene al loro impiego, alle linee guida delle varie società di nutrizione artificiale, reperibili su Pubmed.
Le miscele parenterali sono divisibili in sacche a bassa osmolarità (max 850 m Osm/L, circa), utilizzabili anche per vena periferica, e ad alta densità calorica e alta osmolarità, utilizzabili solo per vena periferica, ternarie (contenenti i tre macro-nutrienti) o binarie, in cui i lipidi non sono contenuti. In particolare, negli ultimi anni la disponibilità di substrati sempre più specifici e tecnologicamente avanzati ha permesso di modulare anche le miscele per NP, in modo da ottenere un effetto anti-infiammatorio (utilizzando in particolare i lipidi strutturati, in grado di fungere da precursori per trombossani e prostaglandine ad attività anti-infiammatoria) e anti-catabolico (con un particolare contenuto in oligo-elementi e amminoacidi). Anche per le composizioni delle sacche per NP rimando alle schede tecniche relative.
Infine, va ovviamente ricordato che, a differenza delle miscele enterali, che vengono digerite assorbite e veicolate nel torrente circolatorio in maniera analoga agli alimenti naturali, le miscele parenterali, essendo infuse direttamente nel torrente circolatorio, sono costituite solo da molecole elementari in grado di mantenersi in soluzione nella fase liquida del sangue. Le miscele enterali non sono quindi assolutamente utilizzabili per via parenterale. Per questo, da circa 5 anni è stato introdotto nella pratica clinica un sistema di linee di deflussione con raccordi utilizzabili solo per la via enterale, chiamato EN-fit (2). Tale sistema è stato progettato con un apposito protocollo ISO. Attualmente teoricamente non è dunque più tecnicamente possibile infondere nutrizione enterale per via venosa, con le gravi conseguenze ben immaginabili. È stato internazionalmente convenuto che sia i dispositivi di accesso (sonde, PEG, digiuno-stomie) che i deflussori, che il materiale di nursing (siringhe e schizzettoni) per NE siano prodotti solo con tali raccordi e che siano caratterizzati dal colore viola/lilla che li evidenziano rispetto al restante materiale sanitario.
Bibliografia
- Linee guida SINPE per la Nutrizione Artificiale Ospedaliera. Rivista Italiana di Nutrizione Parenterale ed Enterale 2002, 20 S5: S9-S11.
- Guenter P, Lyman B. ENFit enteral nutrition connectors. Nutr Clin Pract 2016, 31: 769-72.