Differenziazione delle strutture cerebrali e del “pensiero”: aspetti neuropsicologici e strutturali dei DSD

Vickie Pasterski1, Franco D’Alberton2
1Dipartimento di Pediatria, Campus Biomedico, Università di Cambridge, UK, 1Università di Bologna, AOU Policlinico S.Orsola-Malpighi
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Nel pensiero comune, così come nel dibattito culturale, è quasi dato per scontato che gli aspetti psicologici e comportamentali legati alle differenze tra i sessi siano dovuti principalmente a fattori emotivi, relazionali, educativi, a loro volta influenzati dai modelli economici, culturali e sociali prevalenti. Nel tempo, invece, si è andata sempre più affermando l’evidenza che, come accade per le caratteristiche sessuali somatiche primarie, anche le differenze nel comportamento e nelle attitudini sessuali siano influenzate dal complesso processo biologico di differenziazione sessuale. Studi sugli esseri umani hanno, infatti, evidenziato il ruolo che gli ormoni gonadici pre- e post-natali, in particolare il testosterone, hanno nello sviluppo del comportamento dei bambini nei giochi e nelle scelte e negli interessi dei rispettivi sessi, nonché nell’orientamento sessuale e nell'identità di genere negli adulti. Studi sperimentali sugli animali avevano già dimostrato risultati simili per quanto riguarda i comportamenti che evidenziano differenze sessuali.
Sono state documentate le influenze dell’esposizione al testosterone perinatale sullo sviluppo neuro-comportamentale (1): la somministrazione di testosterone nelle cavie gravide produce nei cuccioli una maggiore mascolinizzazione e un’inferiore femminilizzazione nel comportamento sessuale. Ciò ha consentito di ipotizzare che questi effetti riflettessero cambiamenti nell’organizzazione dei sistemi neurali, come poi documentato da numerosissimi studi su varie specie (2,3).
Ricerche successive hanno stabilito diversi principi fondamentali riguardanti la differenziazione sessuale neuro-comportamentale:
- la differenziazione sessuale è dipendente dal testosterone, mentre l'estrogeno presumibilmente non femminilizza lo sviluppo neuro-comportamentale;
- gli effetti del testosterone sono graduati e lineari: un aumento di esposizione prenatale al testosterone si traduce in maggiore mascolinizzazione;
- la differenziazione sessuale neuro-comportamentale è multi-dimensionale: i comportamenti e i sistemi neurali che differiscono per maschi e femmine sono sensibili all'esposizione degli ormoni durante periodi critici leggermente diversi e possono variare nella loro sensibilità a diverse dosi di testosterone;
- gli effetti degli ormoni variano tra le specie;
- le caratteristiche maggiormente influenzate dagli ormoni, cioè quelle che evidenziano le differenze sessuali e la differenziazione sessuale neuro-comportamentale, potrebbero continuare a modificarsi nel periodo post-natale, fino a tre mesi dopo la nascita. È stato dimostrato che l'aumento del testosterone nei neonati maschi (chiamato mini-pubertà) ha ripercussioni sul comportamento tipico maschile nell’infanzia (4).
Le differenze sessuali comportamentali negli esseri umani sono già pronunciate nell'infanzia e includono preferenze di giocattoli, compagni di gioco e attività. Ad esempio: i maschi, abitualmente, preferiscono altri maschi come compagni di gioco, amano giocare con automobiline e armi e privilegiano uno stile di gioco più attivo e violento. Le bambine, prevalentemente, preferiscono giocare con altre femmine, con bambole e vestiti e mostrano uno stile di gioco più calmo (5).
Negli adulti le differenze sessuali più apparenti riguardano l'orientamento sessuale e l'identità di genere, con la maggior parte delle persone che dimostra attrazioni sessuali verso il sesso opposto e si identifica psicologicamente con il sesso cromosomico/gonadico/fenotipico (5).
Per quanto riguarda la personalità e gli aspetti cognitivi, le differenze sessuali sono molto più ridotte (6). Tuttavia, le bambine, in confronto ai bambini, tendono a mostrare livelli più alti di empatia e più bassi di aggressività fisica. Gli studi sulle attività cognitive che storicamente dimostravano differenze sessuali, ad esempio nelle abilità spaziali e matematiche a favore dei maschi e nelle attività verbali a favore delle femmine, hanno prodotto risultati non univoci, forse perché riflettono maggiormente i cambiamenti nelle influenze culturali e sociali.
Sono state individuate numerose differenze legate al sesso anche per quanto riguarda le strutture cerebrali. Innanzitutto, il volume totale del cervello è più grande negli uomini che nelle donne, anche se questo non significa che tutte le strutture cerebrali abbiano un maggiore volume nel maschio. Ad esempio, anche se l'amigdala è più grande negli uomini che nelle donne, le donne mostrano un maggiore spessore corticale in molte regioni (7), l’ippocampo è più grande (8) e vi è una maggior convoluzione in alcune parti del lobo frontale e corteccia parietale (9). Per quanto riguarda le funzioni cerebrali, gli studi hanno evidenziato schemi diversi di attivazione neurale durante l'esecuzione di una serie di operazioni cognitive. Tuttavia, nonostante le differenze di strutture e funzioni legate al sesso, poche differenze nel comportamento o nelle funzioni cognitive possono essere collegate in modo definitivo alla differenziazione sessuale. Ad oggi, l'unico risultato che è stato replicato in modo indipendente coinvolge il nucleo interstiziale dell'ipotalamo anteriore (INAH-3). Questa zona risulta più grande nei maschi che nelle femmine e nei maschi omosessuali (10,11). Solo prove indirette collegano con le esecuzioni cognitive regioni cerebrali differenziate a seconda del sesso. Ad esempio, l'area sagittale mediana delle regioni posteriori del corpo calloso correla negativamente con la lateralizzazione e positivamente con la fluenza verbale (3) e in entrambe le variabili sono presenti differenze rispetto al sesso.
È importante ricordare che lo sviluppo neuro-comportamentale è multidimensionale e vede implicati altri fattori, che le esperienze post-natali possono modificare la struttura cerebrale dei mammiferi per tutta la durata della vita e che negli esseri umani la neurogenesi continua anche nell'età adulta (12-13). In questa determinazione multidimensionale non possono venir trascurate la qualità delle relazioni genitori/bambini e del complesso gioco di identificazioni precoci che avviene tra i genitori e i loro figli, anche queste in grado di determinare mutamenti nell’organizzazione neuronale e nella stessa struttura del cervello (13). Inoltre, le influenze sociali e culturali, nonché lo sviluppo cognitivo, hanno dimostrato di influenzare in modo significativo i comportamenti sessuali (14-16).
Bibliografia
- Phoenix CH, Goy RW, et al. No title. Endocrinology 1959, 65: 163.
- Arnold AP. The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Horm Behav 2009, 55: 570–8.
- Hines M, Chiu L, McAdams LA, et al. Cognition and the corpus callosum: verbal fluency, visuospatial ability, and language lateralization related to midsagittal surface areas of callosal subregions. Behav Neurosci 1992, 106: 3–14.
- Pasterski V, Acerini CL, Dunger DB, et al. Postnatal penile growth concurrent with mini-puberty predicts later sex-typed play behavior: evidence for neurobehavioral effects of the postnatal androgen surge in typically developing boys. Horm Behav 2015, 69: 98–105.
- Hines M. Psychosexual development in individuals who have female pseudohermaphroditism. Child Adolesc Psychiatr Clin N Am 2004, 13: 641–56.
- Luders E, Narr KL, Thompson PM, et al. Gender effects on cortical thickness and the influence of scaling. Hum Brain Mapp 2006, 27: 314–24.
- Goldstein JM. Normal sexual dimorphism of the adult human brain assessed by in vivo magnetic resonance imaging. Cereb Cortex 2001, 11: 490–7.
- Gur RC, Turetsky BI, Matsui M, et al. Sex differences in brain gray and white matter in healthy young adults: correlations with cognitive performance. J Neurosci 1999, 19: 4065–72.
- Byne W. The medial preoptic and anterior hypothalamic regions of the rhesus monkey: cytoarchitectonic comparison with the human and evidence for sexual dimorphism. Brain Res 1998, 793: 346–50.
- LeVay S. A difference in hypothalamic structure between heterosexual and homosexual men. Science 1991, 253: 1034–7.
- Juraska JM. Neural plasticity and the development of sex differences. Annu Rev Sex Res 1998, 9: 20–38.
- Ming G, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci 2005, 28: 223–50.
- Balbernie R. Circuits and circumstances: the neurobiological consequences of early relationship experiences and how they shape later behaviour. J Child Psychother 2001, 27: 237-55.
- Fagot BI. The influence of sex of child on parental reactions to toddler children. Child Dev 1978, 49: 459-65.
- Pasterski V, Geffner ME, Brain C, et al. Prenatal hormones and postnatal socialization by parents as determinants of male-typical toy play in girls with congenital adrenal hyperplasia. Child Dev 2005, 76: 264-78.
- Perry DG, Bussey K. The social learning theory of sex differences: Imitation is alive and well. J Pers Soc Psychol 1979, 37: 1699-712.
Classificazione e nuova nomenclatura dei DSD
Silvano Bertelloni1, Giacinto Marrocco2, Antonio Balsamo3
1UO Pediatria 1, AOU Pisa; 2Chirurgia e Urologia pediatrica, Ospedale San Camillo, Roma; 3Università di Bologna, AOU Policlinico S.Orsola-Malpighi
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Secondo la Consensus di Chicago, con la dizione DSD si intendono tutte quelle condizioni congenite nelle quali il sesso cromosomico, gonadico o anatomico risulta non conforme rispetto agli usuali processi di sviluppo embrionario delle gonadi e/o dei genitali (1).
La tabella 1 riporta la nuova nomenclatura elaborata nella Consensus, mentre la tabella 2 sintetizza le principali forme cliniche di 46,XY DSD secondo la nuova classificazione.
| Tabella 1 DSD: la nomenclatura della Consensus di Chicago a confronto con la vecchia terminologia |
|
| Nuova | Vecchia |
| Disordini dello Sviluppo del Sesso | Intersessi |
| 46,XY DSD | Pseudo-ermafroditismo maschile Ipovirilizzazione o ipomascolinizzazione di un maschio 46,XY |
| 46,XX DSD | Pseudo-ermafroditismo femminile Virilizzazione o mascolinizzazione di una femmina XX |
| DSD ovo-testicolare | Ermafroditismo vero |
| DSD testicolare 46,XX | Maschio XX o Inversione sessuale XX |
| 46,XY con disgenesia gonadica completa | Inversione sessuale XY |
| Tabella 2 DSD: La nuova classificazione elaborata nella Consensus di Chicago |
||
| DSD con variazioni nei cromosomi sessuali | 45,X (sindrome di Turner e varianti) 47,XXY (Sindrome di Klinefelter e varianti) 45,X/46,XY (disgenesia gonadica mista, DSD ovo-testicolare) 46,XX/46,XY (DSD chimerico o ovo-testicolare |
|
| DSD 46,XY | Differenze dello sviluppo gonadico (testicolare) | Disgenesia gonadica completa (S. di Swyer) Disgenesia gonadica parziale Regressione testicolare DSD ovo-testicolare |
| Differenze della sintesi o dell’azione degli androgeni | Deficit della sintesi degli androgeni (es. deficit 17HSD3, deficit di 5α-reduttasi, mutazioni di StAR) Deficit di azione degli androgeni (es. CAIS, PAIS) Deficit del recettore LH (ipoplasia o aplasia delle cellule di Leydig) Deficit della sintesi o azione dell’AMH (s. da persistenza dei dotti di Muller) |
|
| Altre | Ipospadia severa Estrofia della cloaca Altre sindromi |
|
| DSD 46,XX | Differenze dello sviluppo gonadico (ovarico) | DSD ovo-testicolare DSD testicolare (es. SRY+, dup SOX9) Disgenesia gonadica |
| Eccesso di androgeni | Fetale (es. deficit 21-idrossilasi, deficit 11-idrossilasi) Feto-placentare (deficit di aromatasi, deficit POR) Materno (luteoma, arrenoblastoma, esogeno, ecc.) |
|
| Altre | Estrofia della cloaca Atresia vaginale Sindrome MURC Altre sindromi |
|
AMH: ormone anti-mulleriano; CAIS: sindrome da insensibilità completa agli androgeni; PAIS: sindrome da insensibilità parziale agli androgeni; MURCS: anomalie dei somiti mulleriani, renali, cervico-toracici; POR: citocromo P450 ossido-reduttasi
Benchè l’uso del cariotipo sia utile per la classificazione, dovrebbero essere evitati riferimenti non necessari ad esso; idealmente, quando possibile, dovrebbe essere usato un sistema basato su termini descrittivi (es. sindrome da insensibilità agli androgeni).
Gli aspetti maggiormente innovativi della nuova nomenclatura sono (2,3):
- la creazione di uno specifico acronimo (DSD), nuovo nella letteratura medica, usato come “contenitore” di un ampio gruppo di condizioni (a patogenesi ampiamente differente);
- la sostituzione di termini vaghi e discriminanti (es. pseudo-ermafroditismo o intersessi) con definizioni clinicamente o eziologicamente descrittive di una situazione genetica e clinica [es. insensibilità periferica agli androgeni (da mutazioni nel gene AR), deficit di 5α-reduttasi (da mutazioni nel gene SRD5A2), ecc.];
- la realizzazione di “etichette” diagnostiche più precise e uniformi, da utilizzare nell’ambito di studi scientifici (in modo da ottenere dati di esito più omogenei, da utilizzare come “guida” nel trattamento di nuove diagnosi);
- il ruolo chiave attribuito alla genetica (cariotipo, ecc.) nei percorsi patogenetici, diagnostici e terapeutici.
Tuttavia anche la nuova impostazione non è immune da critiche (4), tra cui:
- l’eccessiva enfasi data alla genetica (cariotipo, ecc.) che può risultare confondente, oltre che per i genitori e le persone affette, anche per i medici “non addetti ai lavori”;
- l’inserimento nell’ambito dei DSD di alcune forme cliniche che non presentano “disturbi nello sviluppo del sesso” (ad es. “vanishing testis syndrome”, criptorchidismo, ipospadia isolata, ipogonadismo ipogonadotropo), che può complicare la gestione clinica (e sociale) in alcuni soggetti, anche complicando l’accettazione della loro malattia;
- il termine “disordine” (“condizione patologica che richiede un trattamento”) può risultare male accetto in alcune culture (soprattutto se collegato al termine “sesso”) o da alcune persone.
Bibliografia
- Lee P, Houk CP, Ahmed SF, Hughes I. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics 2006, 118: e488-500.
- Hughes I. Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab 2008, 22: 119-34.
- Houk CP, Lee PA. Consensus statement on terminology and management: disorders of sex development. Sex Dev 2008, 2: 172-80.
- Barthold JS. Disorders of sex differentiation: a pediatric urologist’s perspective of new terminology and recommendations. J Urol 2011, 185: 393-400.
Il laboratorio di genetica per i DSD

Il laboratorio di endocrinologia per i DSD
Diagnostica per immagini per DSD
Santiago Vallasciani1, Cinzia Orazi2, Maria Angela Pavesi3, Giacinto Marrocco4
1UOSD Urologia Pediatrica, Ospedale Ca' Granda IRCCS, Milano; 2Dipartimento di Radiologia e Radiodiagnostica, Ospedale Pediatrico Bambino Gesù, Roma; 3Servizio di Radiologia Pediatrica, Clinica De Marchi, IRCCS Fondazione Ca’ Granda Ospedale Maggiore Policlinico, Milano; 4Chirurgia e Urologia pediatrica, Ospedale San Camillo, Roma
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
L’imaging riveste un ruolo chiave nella valutazione dei pazienti con DSD, che deve essere diversificata a seconda della fascia di età e della presentazione clinica.
Nel neonato e lattante con genitali ambigui l’obiettivo è di definire la condizione anatomica del paziente, in particolare dell’apparato genitale, al fine di porre una precoce e corretta diagnosi e definire l’iter terapeutico più indicato anche a lungo termine (1). L’indagine di prima istanza è rappresentata dall’ecografia, metodica non invasiva, affidabile e di agevole impiego, seguita in alcuni casi dalla genitografia e dalla risonanza magnetica (2,3).
L’esame ecografico deve essere condotto con scansioni per via trans-addominale e trans-perineale, in condizioni di replezione vescicale e deve mirare alla valutazione dettagliata degli organi genitali interni ed esterni, comprendendo la regione perineale, inguinale e gli spazi retro-vescicali (fig 1-5).
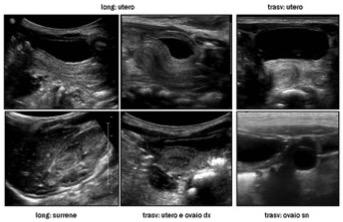
Figura 1: sindrome adreno-genitale a 3 mesi
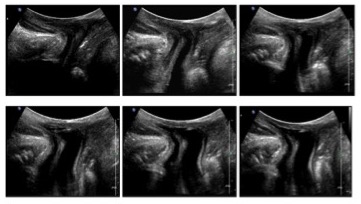
Figura 2: sindrome adreno-genitale a 3 mesi (scansioni perineali durante minzione: distensione uretra e vagina, seno uro-genitale)
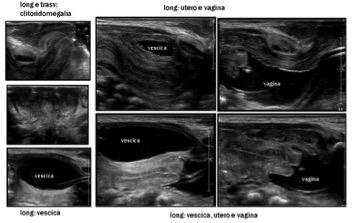
Figura 3: sindrome adreno-genitale a 2 mesi (genitografia ecografica: catetere in vescica e vagina)
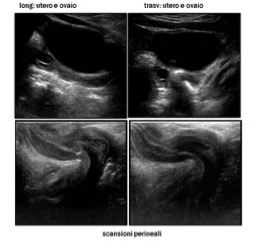
Figura 4: sindrome adreno-genitale a 8 mesi
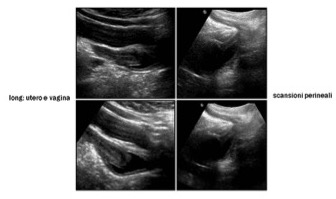
Figura 5: sindrome adreno-genitale a 9 mesi
Devono essere ricercati e definiti:
- l’utero e il canale vaginale (morfologia, dimensioni e struttura);
- eventuali strutture di derivazione mülleriana (utricolo prostatico);
- le gonadi: ovaie con presenza di follicoli, gonadi streak, testicoli con ecostruttura omogenea, oltre che in sede scrotale, anche con scansioni inguinali e addominali (più difficoltosa la valutazione dei testicoli in addome) (fig 6).
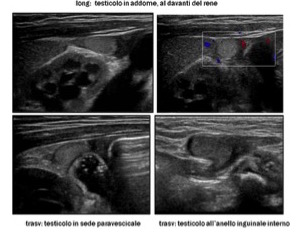
Figura 6: testicoli ritenuti
Le scansioni trans-perineali possono definire la morfologia dell’uretra, del canale vaginale, dell’eventuale seno uro-genitale, la possibile ostruzione vaginale e la distanza del livello di ostruzione dal piano perineale.
Qualora i reperti non siano sufficientemente caratterizzanti, l’introduzione di soluzione fisiologica mediante catetere in vagina, o nel seno uro-genitale, e in vescica, può consentire la visualizzazione della vagina stessa e della cervice (genitografia ecografica). La contemporanea distensione anche del retto può essere utile per la ricerca di eventuali fistole.
L’indagine deve essere sempre estesa alla valutazione dei reni (anomalie di numero, sede e fusione), dei surreni (ipertrofia) e, quando possibile, alla valutazione del midollo spinale (tethered cord).
L’indagine di livello successivo è la genitografia, che ha l’obiettivo di determinare la morfologia duttale interna (4). In particolare, consente di differenziare tra uretra maschile e femminile, di definirne i rapporti con lo sfintere esterno, di visualizzare una cavità vaginale e valutarne morfologia, dimensioni ed impressione cervicale sul fondo, l’eventuale confluenza della vagina e dell’uretra in un seno uro-genitale e la sua lunghezza, residui mülleriani (utricolo prostatico). Inoltre può essere riconosciuta la continuità del canale vaginale con la cavità uterina; possono essere visualizzate le salpingi, i dotti deferenti, tramiti fistolosi, e possono essere individuate anomalie urinarie associate (sbocchi ureterali ectopici, reflussi vescico-ureterali) (fig 7-8).
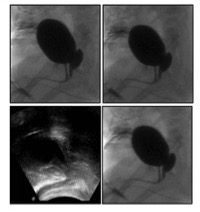
Figura 7: sindrome adreno-genitale a 9 mesi (genitografia: seno uro-genitale lungo)
Figura 8: ipospadia prossimale con residuo mulleriano
L’esame viene condotto in radioscopia pulsata, con bassa dose, a paziente in decubito laterale o laterale/obliquo, introducendo per pochi mm a livello dell’orifizio perineale un catetere tipo Foley (6-8 F) cuffiato esternamente a scopo occlusivo o, in alternativa, dispositivi specifici, iniettando per via retrograda una modesta quantità di mdc iodato sufficiente a delineare il seno uro-genitale, l’uretra e la vagina. Se il catetere entra in vagina, questo consente la dimostrazione dei rapporti con l’uretra, lo stato della cervice e la possibile visualizzazione della cavità uterina e delle salpingi. Se il catetere entra in vescica può essere retratto lentamente, continuando a introdurre mdc, al fine di delineare l’uretra, oppure può essere riempita la vescica valutando l’uretra in fase minzionale dopo rimozione del catetere stesso. Talvolta può essere necessaria l’introduzione di due cateteri, uno in vescica e uno in cavità vaginale, o più cateteri in caso di malformazioni complesse (cloaca). È importante comunque esaminare sempre tutti gli orifizi presenti sul perineo (5).
L’esame RM viene riservato ai casi in cui l’esame ecografico non risulta sufficiente alla definizione degli organi pelvici e/o delle gonadi. Tale indagine, che ha il vantaggio di non utilizzare radiazioni ionizzanti, è più sensibile nella valutazione degli organi pelvici, nella definizione dei rapporti spaziali e nella caratterizzazione tissutale. Consente la precisa valutazione dell’utero (presenza/assenza, morfologia, caratteri strutturali), la continuità con il canale vaginale, la presenza e il livello di un’eventuale ostruzione utero-vaginale, la differenziazione tra micropene e ipertrofia clitoridea (presenza/assenza del corpo spongioso), l’identificazione e la valutazione delle gonadi, con ottimale definizione della struttura ovarica e della struttura testicolare se in sede inguino-scrotale; risulta talora inefficace nel riconoscere testicoli intra-addominali e gonadi streak-like, rendendo necessaria, in casi selezionati, la valutazione laparoscopica. Consente inoltre la valutazione di tutte le anomalie associate (renali, scheletriche, midollari) e anche di possibili neoformazioni a origine dalle gonadi (fig 9).
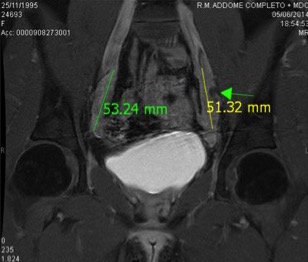
Figura 9: RM dimostrante gonadi endo-addominali in paziente affetta da insensibilità completa agi androgeni
Nei neonati e nei bambini più piccoli, rispetto ai bambini più grandi e agli adulti, tale indagine, comunque risulta limitata per la necessità di sedazione/anestesia generale, e in parte anche per una minore risoluzione tissutale (6).
Bibliografia
- Ahmed SF, Achermann JC, Arlt W, et al. UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol 2011, 75: 12–26.
- Al-Jurayyan NA, Al-Jurayyan RNA, Mohamed SH, Babiker AMI. Radiological imaging of disorders of sex development (DSD). Sudan J Paediatr 2013, 13: 10-6.
- Chavhan GB, Parra DA, Oudjhane K, et al. Imaging of ambiguous genitalia: classification and diagnostic approach. Radiographics 2008, 28: 1891–904.
- Riccabona M, Lobo ML, Willi U, et al; ESPR uroradiology task force and ESUR Paediatric Work Group. Imaging recommendations in paediatric uroradiology, part VI: childhood renal biopsy and imaging of neonatal and infant genital tract. Minutes from the task force session at the annual ESPR Meeting 2012 in Athens on childhood renal biopsy and imaging neonatal genitalia. Pediatr Radiol 2014, 44: 496–502.
- Vallasciani S, Ferro F, Atzori P, Martini L. Alternative homemade device for retrograde urethrogram in pediatric age. III ISHID World Congress 2009, Toronto, Canada.
- Orazi C, Cappa M, Schingo PMS, Tomà P. Ambiguous genitalia in imaging endocrine diseases in children. In: Imaging endocrine diseases in children (Avni F Editor), Springer 2012: 81-109.
Approccio psicologico ai DSD
Franco D’Alberton
Dipartimento Salute della Donna, del Bambino e dell’Adolescente, UO Pediatria, Programma Endocrinologia Pediatrica, AOU S. Orsola Malpighi, Bologna
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
L’approccio psicologico alle persone con DSD e ai loro familiari è molto cambiato negli ultimi vent’anni. L’opinione prevalente fin quasi all’inizio di questo secolo - e ancora presente in alcune realtà - era improntata alle idee di uno psicologo, John Money (1-3), che, sulla base del lavoro con i suoi colleghi endocrinologi e chirurghi del John Hopkins di Baltimora, aveva improntato una metodologia di lavoro che costruì il “paradigma tradizionale di trattamento” adottato come modello di riferimento per decenni dai clinici di tutto il mondo (4). Essa si basava su tre assunti fondamentali, cioè che:
- l’identità di genere sessuale fosse indifferenziata e plasmabile nei primi 18 mesi di vita sia in senso maschile che femminile;
- una “corretta” identità sessuale di genere presupponesse la presenza di genitali non ambigui, implicando con ciò la necessità di una chirurgia precoce, possibilmente entro la fine del secondo anno di vita;
- i genitori non dovessero avere dubbi nell’allevare il bambino/a secondo il sesso che era stato individuato e assegnato dal team di cura, generalmente in base all’apparenza dei genitali e alle possibilità di “ricostruzione” in senso maschile o femminile.
Questo approccio implicava la tendenza a non esplicitare le diagnosi ai pazienti, mantenendoli così all’oscuro delle loro condizioni. Il tenere le persone all’oscuro rispetto alle loro condizioni è un’idea che si è affermata fin dall’individuazione della sindrome di Morris. Infatti, quando nel 1953 Morris descrisse, con il linguaggio medico del tempo, la Sindrome da insensibilità completa agli androgeni (CAIS), nella parte psicologica affermò in modo categorico che non sarebbe stato saggio informare il paziente sulle sue condizioni “It goes without saying that it would be unwise to inform the patient of the true state of affairs” (5, p. 1209). A quei tempi, la consapevolezza di vivere con un DSD sembrava costituire un elemento inelaborabile per la psiche di una persona, configurabile di per sè come esperienza traumatica.
In realtà, da quando, verso la fine del ‘900, le persone direttamente interessate hanno incominciato a incontrarsi, condividere esperienze e far sentire la loro voce (aisuk.org, isna.org), un primo elemento rivendicato è stato il diritto delle persone ad essere informate sulle proprie condizioni. Ciò, lungi dal rappresentare un elemento traumatizzante, costituiva la base per un armonico rapporto con sé stessi e con i propri familiari, oltre che un inalienabile diritto della persona interessata.
Da quegli anni molte cose, pur faticosamente, hanno iniziato a cambiare e la Consensus di Chicago (6) ha costituito un primo momento di svolta nel percorso di presa in carico dei DSD. Tale svolta ha promosso un cambiamento nei protocolli di cura, favoriti dai migliori strumenti diagnostici e laboratoristici di cui oggi disponiamo e ispirati a una visione più aperta all’incertezza e al dubbio, in una visione meno normalizzante e più sensibile alla necessaria cautela nelle scelte di attribuzione di sesso al di fuori di un rigido binarismo sessuale. Oltre a ciò, ci si è resi conto inoltre che la complessità della situazione e l’intensità delle emozioni che l’incontro con il DSD suscita in chi se ne deve far carico necessita che non sia un unico individuo a prendersene cura, ma che nei centri che trattano queste condizioni vi sia la possibilità di contare su un gruppo di lavoro inter-disciplinare formato dai vari professionisti interessati (pediatri endocrinologi, neonatologi, genetisti, chirurghi, ginecologi, psicologi, ecc) (7). La condivisione delle idee, la circolazione del pensiero e la condivisione delle scelte con le persone interessate e i loro familiari dovrebbe ispirare ogni tappa del gruppo di lavoro (8,9).
Anche l’intervento psicologico è andato definendosi nel corso di questo processo, passando dal tentativo di curare qualcosa per sua natura patologico a un’ottica sostanzialmente preventiva e mirata a far circolare le informazioni e le emozioni ad esse collegate, evitando che queste si trasformino in disagi psicologici.
Le varie forme dell’intervento psicologico
Per ogni essere umano, il primo periodo della vita rappresenta la base di ogni futura stabilità emotiva e la qualità delle prime relazioni affettive e di cura segna il senso di benessere soggettivo e la fiducia che l’individuo sarà in grado di attribuire a sé stesso, agli altri e al mondo esterno.
Quando in una qualunque fase della vita si presenta un problema di natura organica, la sua accettazione nella rappresentazione che una persona ha di sé stessa dipende solo in parte dalla gravità oggettiva della difficoltà, ma soprattutto dall’equilibrio emotivo e dalla capacità di reazione della persona. Fondamentale a questo riguardo è la qualità del supporto sociale che il diretto interessato riceve: in primo luogo dai genitori e dalla famiglia allargata, successivamente dai professionisti che operano nelle strutture di cura a cui le persone si rivolgono. In molte condizioni di malattia organica, pratiche medico-chirurgiche necessarie al benessere del bambino possono lasciare segni nel corpo che solo il tempo e le relazioni emotive successive consentiranno di riassorbire all’interno di un discorso dotato di un senso condiviso.
Ogni elemento evitabile che può interferire con la qualità e la quantità delle relazioni affettive genitori/bambino, sia esso di natura sanitaria che emotiva, dovrebbe essere evitato nel periodo in cui si sviluppa l’apparato psichico del bambino, che prende forma all’interno della relazione con il corpo e con la mente dei genitori.
Quando la presenza di un DSD si affaccia nella vita di una famiglia, vi è un rischio oggettivo che la relazione genitori/bambino ne risenta in misura maggiore o minore e che pratiche mediche possano interferire in qualche modo con il fisiologico dipanarsi dello sviluppo psico-somatico. Una grande attenzione dovrebbe essere rivolta alla tutela della psiche del bambino, attraverso un intervento di natura preventiva, che prenda in carico le ansie e le preoccupazioni dei genitori che si trovano ad affrontare una realtà sconosciuta, spaventosa e che spesso evoca preoccupazioni irrealistiche.
Il primo intervento psicologico nel campo dei DSD non afferisce alla specificità del ruolo professionale dello psicologo, ma è quello svolto da un gruppo di lavoro affiatato ed esperto, composto dai vari specialisti che dei DSD si prendono cura. Essi, con la loro interazione contribuiscono a creare un solido ambiente di cura con interlocutori stabili e affidabili. Lo psicologo, in questa prima fase, contribuisce alla valutazione del caso attraverso la propria esperienza professionale nel campo dello sviluppo emotivo del bambino, sulle problematiche genitoriali e sulla gestione della ansie, oltre che sullo specifico psicologico dei DSD. La competenza dello psicologo è anche diretta a facilitare il lavoro all’interno del gruppo, affrontando eccessi di ansia che, anche nei curanti, possono portare a favorire che “l’azione” possa prendere il posto del pensiero.
Nel dipanarsi del lavoro di gruppo può rendersi necessario un intervento specifico e diretto dello psicologo di sostegno alla funzione genitoriale, soprattutto in situazioni familiari particolarmente complesse, intervento tanto più facilitato quanto più lo psicologo viene individuato e riconosciuto come un membro del team, “uno del gruppo”.
Questi incontri possono costituire la base di ulteriori sessioni nei casi in cui una maggior conoscenza del mondo interno e delle fantasie del bambino ormai cresciuto può contribuire a individuare con maggior accuratezza gli orientamenti rispetto all’identità di genere, spesso necessari per monitorare la crescita del bambino/a. Costituiscono anche la base per l’avvio di un confronto con i genitori e i medici su come affrontare la comunicazione della diagnosi e le comunicazioni riguardo a tutte le pratiche che il bambino deve sostenere. Si è visto nel tempo quanto sia importante per le persone poter essere informate e avvertire il senso di quello che sta loro capitando di vivere.
Da ultimo, lo psicologo clinico può portare il suo contributo specifico in situazioni in cui espressioni di sofferenza emotiva vengano a presentarsi nel corso dello sviluppo del bambino, dell’adolescente o dei suoi genitori.
Bibliografia
- Money J, Hampson JG, Hampson JL. Hermaphroditism: recommendations concerning assignment of sex, change of sex, and psychologic management. Bull Johns Hopkins Hosp 1955, 97: 284-300.
- Money J, Hampson JG, Hampson JL. An examinations of some basic sexual concepts: the evidence of human hermaphroditism. Bull Johns Hopkins Hosp 1955, 97: 301-19.
- Money J. Psychological evaluation of the child with intersex problems. Pediatrics 1965, 36: 51-5.
- Kanzakis K. Fixing sex. Duke University Press, Durham & London 2008.
- Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol 1953, 65: 1192-211.
- Hughes IA, Houk C, Ahmed SF, et al. Consensus statement on management of intersex disorders. Arch Dis Child 2006, 91: 554-63.
- Moran E, Karkazis C. Developing a multidisciplinary team for disorders of sex development: planning, implementation, and operation tools for care providers. Adv Urol 2012, 2012: 604135.
- D’Alberton F. Disclosing disorders of sex development and opening the doors. Sex Dev 2010, 4: 304-9.
- Liao LM, Green H, Creighton SM, et al. Service users’ experiences of obtaining and giving information about disorders of sex development. BJOG 2010, 117: 193–9.
DSD da alterazione del numero dei cromosomi sessuali
45,X DSD (sindrome di Turner e varianti)
45,X DSD (sindrome di Turner e varianti)
Edoardo Farinelli1, Clementina La Rosa2, Assunta Albanese3
1Clinica Pediatrica, Università di Perugia
2Adult Endocrine Unit, University College Hospital, London
3Paediatric Endocrine Unit, St George’s NHS Trust, London
INTRODUZIONE
La sindrome di Turner (TS) è la più frequente anomalia cromosomica nel sesso femminile. Le sue caratteristiche cliniche principali sono rappresentate da bassa statura, disgenesia gonadica con infantilismo sessuale ed amenorrea primaria, associate a un fenotipo caratteristico; a queste si accompagna un aumentato rischio di patologia cardiovascolare, endocrina, ossea e renale.
Questo articolo vuole fornire un riepilogo sulla TS, con particolare riferimento alla patologia d’organo associata per fascia d’età ed alla gestione clinica a lungo termine.
EPIDEMIOLOGIA E CENNI STORICI
La prima descrizione clinica di una femmina con bassa statura, mancato sviluppo delle ovaie, malformazione renale e altre caratteristiche dismorfiche, risale a un esame autoptico del 1768, ad opera del medico italiano Giovanni Morgagni (1). In seguito altri autori osservarono alcuni casi sporadici simili, tra i quali Funke (1902), Rossle (1922) ed in particolare il pediatra tedesco Otto Ullrich, che nel 1930 descrisse il caso di una bambina di 8 anni con bassa statura e pterigio del collo (2). Nel 1938, il medico americano Henry Hubertus Turner riportò ben 7 casi di giovani donne adulte con bassa statura e alcune caratteristiche dismorfiche già descritte da Ullrich, a cui si associava infantilismo sessuale (assenza o incompleto sviluppo dei caratteri sessuali secondari), descrivendo così quella condizione che oggi porta il nome di sindrome di Turner, o sindrome di Ullrich-Turner (3). Nel 1959 con il definirsi degli studi sulla cromatina sessuale, Charles Ford e altri chiarirono l’origine genetica della TS, osservando l’associazione tra l’assenza di uno dei due cromosomi X e le caratteristiche fenotipiche (4). È ormai noto che la TS è dovuta alla delezione completa o parziale del cromosoma X e che essa rappresenta l’anomalia cromosomica più comune nel sesso femminile, presentandosi approssimativamente con un’incidenza di 1:2500 nati di sesso femminile (5,6).
GENETICA
La TS è citogeneticamente definita dal riscontro di un cariotipo anomalo, caratterizzato dall’assenza del secondo cromosoma X o di una parte di esso. Il cromosoma X assente/alterato può essere osservato in tutte le cellule corporee o solo in una certa percentuale di esse (mosaicismo). Lo stesso cromosoma X può inoltre presentare diverse anomalie strutturali, come ad esempio: isocromosoma, delezione, cromosoma ad anello o traslocazione. Nel 5% circa delle donne con TS è possibile anche ritrovare frammenti di cromosoma Y (7,8). Al caratteristico fenotipo della TS (bassa statura e infantilismo sessuale) corrisponde pertanto, non una singola anomalia del cariotipo ma, in realtà, un ampio spettro di anomalie genotipiche.
Determinare con esattezza il corredo genetico di una femmina con TS non è semplice e dipende, in primis, dal numero di cellule campione studiate. Mediante l’analisi del cariotipo “standard”, che si esegue mediamente in circa 20-30 cellule linfocitiche, si riscontra una monosomia (45,X-) in tutte le cellule analizzate (e pertanto definita “pura”) approssimativamente in circa il 50% dei casi; con l’analisi del cariotipo “ad alta risoluzione”, ovvero su un campione di almeno 100 cellule, prelevate su tessuti diversi (linfociti e fibroblasti, ad esempio), la percentuale di mosaicismo potrebbe risultare però molto più elevata, anche > 80% (9,10). In generale, secondo la maggior parte degli autori sugli aspetti genetici della TS, circa il 45% delle pazienti con TS diagnosticata dopo la nascita possiede un cariotipo con monosomia 45,X- “pura”, senza mosaicismo (11). I cariotipi a mosaico sono pertanto più della metà di tutti i casi di TS: in circa il 15% delle femmine con TS, la linea cellulare 45,X- coesiste con la normale linea 46,XX; altri tipi di mosaicismo sono caratterizzati dal riscontro di linee cellulari 47,XXX o, molto più frequentemente, nel 30% circa del totale dei casi, da linee cellulari in cui vi è un’anomalia strutturale della seconda X. L’anomalia cromosomica della seconda X più frequentemente riscontrata è l’isocromosoma X, formato da 2 bracci lunghi X e definito dalla dizione “46,X,i(Xq)” (12). Altre varianti strutturali della seconda X riscontrabili di frequente sono il cromosoma ad anello “r(X)” e la delezione del braccio lungo o del braccio corto della X “del(X)”.
Un’analisi approfondita del cariotipo, volta a ricercare/escludere con assoluta certezza la presenza di un mosaicismo, può comunque non essere sempre necessaria: ad esempio, il trovare o meno una linea cellulare normale (46,XX) in meno del 5% del numero totale di cellule corporee non cambia la prognosi, né la gestione clinica, di un caso di TS con cariotipo 45,X apparentemente “puro” (13). Molto più importante, dal punto di vista clinico e pratico, è invece ricercare e possibilmente escludere la presenza di linee cellulari con un intero cromosoma Y (ad esempio nel mosaicismo 45,X/46,XY) o con frammenti di esso, che possono trovarsi sia liberi che attaccati ad altri cromosomi (mediante un processo di traslocazione). Il riscontro di porzioni più o meno grandi di materiale genetico del cromosoma Y nel corredo genetico di una femmina con TS è correlato ad un elevato rischio di sviluppo di gonado-blastoma e pertanto, in tutti questi casi, viene in genere consigliato l’intervento di asportazione preventiva delle gonadi (12,13).
Il cromosoma X sano è di origine materna (XM) nel 60-80% dei casi e paterna (XP) nella restante parte; studi recenti hanno dimostrato che l’origine genitoriale della X potrebbe essere correlata al manifestarsi di determinati fenotipi (accessori) delle femmine con TS, come ad esempio la capacità di relazionarsi con altri individui (capacità cognitiva sociale, quoziente intellettivo verbale, comportamento di inibizione/disinibizione sociale) ed all’integrità funzionale o meno di altri organi ed apparati (14). In altre parole, sebbene ulteriori studi siano ancora necessari per confermare tale ipotesi, l’espressione o meno di numerosi geni del cromosoma X nelle femmine con TS sembra soggetta al fenomeno dell’imprinting.
PATOGENESI
L’eziopatogenesi della TS non è stata ancora chiarita. L’età materna avanzata al momento del concepimento non è un fattore di rischio per la TS, come invece accade nel caso di altre sindromi genetiche, tra le quali la sindrome di Down. A conferma di ciò, come già riportato in precedenza, studi di genetica molecolare hanno ampiamente dimostrato che il cromosoma X mancante o alterato è, nel 60-80% circa dei casi, di origine paterna, così come è di origine ovviamente paterna anche l’eventuale riscontro del cromosoma Y o di frammenti di esso (12,15).
La formazione di embrioni con TS è legata a un’anomala divisione cellulare, che può verificarsi o durante la gametogenesi (ipotesi pre-fertilizzazione) o durante le prime divisioni zigotiche (ipotesi post-fertilizzazione). Nello specifico, secondo l’ipotesi pre-fertilizzazione, un errore della divisione cellulare (meiotica) durante il processo di gametogenesi provoca la formazione di un gamete ipoaploide (con 22 cromosomi, per assenza del cromosoma sessuale, ad esempio di XP); successivamente, l’unione del gamete ipoaploide con un gamete normale del sesso opposto forma uno zigote con cariotipo aneuploide, di tipo monosomico (ad esempio: 45,XM-) che, se non abortito spontaneamente, attraverso le successive divisioni mitotiche, formerà un embrione le cui cellule avranno tutte un cariotipo identico allo zigote iniziale (45,X- “puro”). Recenti studi di genetica hanno però osservato che embrioni con cariotipo 45,X- si formano “fisiologicamente” in circa l’1-1.5% di tutte le gravidanze insorte spontaneamente e che essi vengono spontaneamente abortiti entro la 28° settimana di gestazione (10). In base ad analisi statistiche eseguite in questi studi, la frequenza di formazione di embrioni con corredo 45,X- (pari appunto all’1-1.5% di tutte le gravidanze) risulta troppo elevata per essere spiegata dalla possibilità che così tanti gameti anomali siano in grado di unirsi a gameti normali di sesso opposto.
Sembra allora più probabile che l’embrione con TS acquisisca il corredo monosomico 45,X- solo in una fase successiva alla formazione dello zigote e che quest’ultimo si sia formato dall’unione due gameti normali (10). Secondo l’ipotesi “post-fertilizzazione” dunque, a partire da uno zigote euploide, per via di un errore nella prima divisione mitotica, si formano due linee cellulari aneuploidi distinte (ad esempio una linea 45,X- ed una 47,XXX); successivamente, lo sviluppo della linea cellulare con il cariotipo 45,X- e la regressione della linea 47,XXX , porta alla formazione di un embrione il cui cariotipo è monosomico.
Va considerato però che, nella razza umana, il cariotipo 45,X- è l’unica monosomia compatibile con la vita; sembra molto difficile, in realtà, che un embrione con cariotipo di tipo 45,X- “puro” possa sopravvivere all’aborto spontaneo e addirittura riesca, completando i nove mesi di gestazione, a svilupparsi in una femmina che è pressoché perfetta, ad eccezione della bassa statura, dell’atresia ovarica e delle possibili malformazioni di altri organi, di solito non particolarmente gravi (10). Ciò premesso, appare allora ancora più probabile che l’errore di divisione cellulare che porta alla formazione dell’embrione con TS, avvenga nella seconda o nelle successive divisioni mitotiche, piuttosto che nella prima, e che l’embrione con TS vitale sviluppi comunque una seconda linea cellulare, oltre alla linea 45,X- (mosaicismo). Lo sviluppo di una seconda linea, di tipo 46,XX ad esempio, colmando l’aplo-insufficienza derivante dalla mancanza del secondo cromosoma X nella linea principale, potrebbe infatti essere il meccanismo che permette all’embrione con TS di sopravvivere e svilupparsi.
La maggioranza delle femmine con diagnosi di TS, anche se con cariotipo apparentemente di tipo 45,X- “puro”, potrebbe allora possedere una linea cellulare secondaria, detta “di salvataggio” (in inglese “rescue cell line”). Ipoteticamente ogni cariotipo apparentemente monosomico 45,X- “puro” e vitale potrebbe essere più probabilmente un mosaicismo che nel caso specifico è chiamato “mosaicismo nascosto o criptico”, perché non rilevato al momento della prima analisi del cariotipo (10).
DIAGNOSI
Prima dell’introduzione delle metodiche di studio del cariotipo, la diagnosi di TS avveniva sulla base delle evidenze fenotipiche. Attualmente, anche in presenza di fenotipo caratteristico, la conferma della diagnosi prevede l’esame del cariotipo che deve dimostrare l’assenza parziale o totale del secondo cromosoma X.
FENOTIPO E MANIFESTAZIONI CLINICHE
Le manifestazioni cliniche della TS possono essere diverse, variabilmente espresse da caso a caso, quindi non sempre facilmente identificabili. È riconosciuto un certo grado di correlazione genotipo-fenotipo e i quadri clinici più gravi appartengono di solito alle pazienti con monosomia pura, mentre le forme clinicamente più sfumate a soggetti con mosaicismi o con alterazioni strutturali della X (16). Nell’ultimo ventennio, importanti studi epidemiologici, condotti ad ampio spettro su popolazioni di pazienti con TS, hanno contribuito a precisare i dati sul rischio di morbilità e mortalità (17), nonché a chiarire la prevalenza delle caratteristiche cliniche per fascia d’età (18).
In epoca prenatale, i segni (ecografici) indicativi di una sospetta TS possono essere il riscontro, in un feto femmina, di edema generalizzato (igroma cistico), edema del dorso delle mani e dei piedi, aumento del numero delle pieghe cutanee della regione posteriore del collo, aumentata trans-lucenza nucale, presenza di malformazioni cardiache (tra le più frequenti: cuore sinistro ipoplasico, coartazione istmica dell’aorta, aorta bicuspide, ritorno venoso anomalo delle vene polmonari) e renali (rene a ferro di cavallo, duplicazioni della pelvi o degli ureteri) (19).
Alla nascita, il 20-33% dei soggetti affetti presenta linfedema delle mani e dei piedi e più raramente una piega cutanea caratteristica che si estende dal margine laterale del collo fino alle spalle (“pterigium colli”); possono essere presenti basso peso e/o diminuita lunghezza corporea e i segni della patologia cardiaca o renale.
Nelle età successive, fino alla pubertà, si manifesta il progressivo rallentamento della velocità di crescita e si possono rilevare altre caratteristiche fenotipiche tipiche delle TS. La bassa statura è senza dubbio la caratteristica più evidente e sempre presente. A partire dai 2-3 anni circa l’altezza delle TS appare inferiore rispetto alle altre bambine. Raggiunta l’età di 10 anni, ci sono in media 20 cm di differenza di altezza tra le TS e i controlli. Nel caso di mancata diagnosi fetale o perinatale, in età pediatrica la bassa statura è il segno che conduce alla diagnosi nella maggioranza dei casi (20). Le caratteristiche fenotipiche della TS diventano via via più evidenti con il progredire dell’età. Si rilevano soprattutto la facies tipica, con orecchie un po’ più grandi, leggermente ruotate posteriormente e a basso impianto, piega epicantica degli occhi, mandibola piccola, palato ogivale, denti affollati, abbondante cute nella regione posteriore del collo, dove il margine del cuoio capelluto arriva a coprire zone del collo normalmente prive di capelli. È frequente, inoltre, il riscontro di anomalie scheletriche (21), oltre alla bassa statura, quali:
- torace a scudo, ampio e largo, con un’apparente eccessiva distanza tra i capezzoli;
- cubito valgo: estendendo il braccio, si osserva che l’angolo formato tra l’asse del braccio e quello dell’avambraccio è più ampio di circa 15° rispetto alla norma;
- deformità di Madelung: anomalia scheletrica del polso, di solito presente bilateralmente, caratterizzata dall'accorciamento e dall'incurvamento del radio, che provoca una ben visibile tumefazione sul polso (che corrisponde alla testa dell’ulna) e una limitazione dei movimenti del polso;
- segno di Archibald (brevità del 4° metacarpo);
- scoliosi.
Altre caratteristiche corporee spesso presenti nelle bambine con TS sono: irsutismo, vitiligine, alopecia, nevi cutanei, displasia ungueale (unghie convesse “a cucchiaio”). In questa fascia d’età sono molto frequenti gli episodi di otite media ed esterna. Nello studio di Savendahl & Davemport del 2000 (18), la patologia a carico dell’orecchio si osservava nel 71% dei soggetti osservati, risultando così la patologia d’organo più frequente in assoluto nelle TS, subito dopo la bassa statura. Sono comuni anche le problematiche oculistiche, tra le quali miopia, ipermetropia, nistagmo e strabismo. È riportata anche un’aumentata incidenza di celiachia e patologia tiroidea autoimmune. A causa delle anomalie del palato, la patologia ortodontica è abbastanza tipica e pertanto è raccomandata la valutazione ortodontica di routine a tutte le bambine con TS a partire dai 7 anni.
Nel contesto psico-sociale, devono essere tenuti in considerazione eventuali difficoltà di apprendimento e frequenti problemi psicologici, emotivi e relazionali, dovuti in gran parte alla bassa autostima, derivante dalla presa di coscienza della condizione personale, dell’aspetto fisico e del riscontro di eventuali patologie. A ciò si aggiunge di frequente un’immaturità emotiva, rispetto alle coetanee, che può essere legata alla mancata/ritardata esposizione agli ormoni sessuali. Le ragazze con TS hanno un rischio aumentato di disturbi d’ansia e di tono dell’umore in senso depressivo; esse spesso denunciano poche amicizie, scarse relazioni con i pari, che nei casi più estremi possono determinare un certo grado di ritiro sociale (22).
In epoca adolescenziale-puberale si manifestano gli effetti della disgenesia ovarica (ipogonadismo). Nella stragrande maggioranza dei casi si ha mancanza di sviluppo dei caratteri sessuali, con conseguente infantilismo, assenza di scatto di crescita puberale ed amenorrea primaria. Solo nel 10-15% dei casi le ovaie mantengono una funzione sufficiente per avviare il processo di sviluppo puberale e solo in una percentuale ancora più bassa si ha il menarca spontaneo (23). Quando presenti, lo sviluppo puberale spontaneo e il menarca sono segni di una funzionalità ovarica sufficiente fino a quel momento, ma probabilmente destinata a un arresto prematuro, con amenorrea secondaria ed infertilità nella gran parte dei casi. Tradizionalmente le donne affette dalla TS sono, infatti, considerate non fertili. Tuttavia, recenti dati confermano che la possibilità di gravidanze spontanee è compresa tra il 2-7.5%, ma di queste gravidanze, meno del 30%-40% viene portato a termine (23,24).
In età adulta diventano frequenti le patologie cardiovascolari, tiroidea, ossea e quella del metabolismo glucidico. Per quanto riguarda le patologie endocrine, sembra che queste si presentino con un rischio relativo complessivo incrementato di quasi 5 volte rispetto ai controlli (25), in particolare con una significativa prevalenza della patologia tiroidea, del diabete mellito di tipo insulino-dipendente e non, assieme a una miscellanea di altri disturbi endocrini. È quindi evidente come l’insieme di questi problemi, soprattutto se non riconosciuti nel complesso della sindrome, determinino un progressivo deterioramento della qualità di vita, con aumento della morbilità e mortalità.
MORBILITÀ E MANAGEMENT
L’aspettativa di vita media dei soggetti affetti da TS è leggermente ridotta rispetto alla media della popolazione di riferimento, prevalentemente a causa della maggiore incidenza di patologie cardio-circolatorie e diabete (13,25). La TS non è associata ad alterate funzioni del sistema nervoso né, di solito, a ritardo mentale. Per il controllo a lungo termine di queste pazienti in età adulta e pediatrica, riportiamo in appendice il promemoria proposto dalla Turner Syndrome Support Society, UK.
Apparato cardio-circolatorio
La maggioranza della morbilità e mortalità nella TS è da attribuire alla patologia cardiaca congenita e acquisita. Sono molto frequenti, specialmente nei casi di monosomia pura, le malformazioni del cuore e dei grossi vasi: il 17-45% delle pazienti nasce, infatti, con malformazioni cardiache (26) e di queste, oltre il 75% presenta un difetto nel cono di efflusso del cuore sinistro, tipicamente coartazione dell’aorta, valvola aortica bicuspide o altra valvulopatia aortica e prolasso mitralico (27). Abbastanza frequente è anche lo sviluppo di ipertensione arteriosa sistemica, anche in assenza di malattia renale o aterosclerotica. Meno frequentemente è possibile osservare difetti a carico del cuore destro (per esempio ritorno venoso anomalo parziale) e dilatazione della radice aortica; quest’ultima è una condizione rara (< 5% dei casi di TS), ma può portare a rottura dell’aorta, potenzialmente fatale.
Data l’elevata incidenza di difetti cardiaci associati, tutte le pazienti con TS devono essere attentamente valutate sul piano cardio-circolatorio; la prima visita cardiologica, completa di ECG, ecocardiografia bidimensionale ed eco-color-doppler, deve essere fatta non appena posta la diagnosi di TS. In caso di dubbi sull’eventuale presenza di difetti cardiaci o al fine di caratterizzare meglio gli stessi, è raccomandata la pronta esecuzione di una RM cardiaca (o eventualmente di una TC). I successivi controlli cardiologici andranno programmati in base alla situazione clinica e alle indicazioni del cardiologo. In particolare, tutte le bambine con TS, anche se con apparato cardiovascolare e pressione arteriosa normale, andranno attentamente rivalutate durante il periodo di passaggio tra l’età adolescenziale e l’età adulta e nell’eventuale momento in cui decidessero di programmare una gravidanza. Anche in caso di apparente normalità dell’apparato cardio-circolatorio, gli esperti (28) raccomandano comunque l’esecuzione di una RM cardiaca di controllo, da effettuare non appena l’età della paziente consenta la sua piena collaborazione allo svolgimento della procedura radiologica, e un monitoraggio ecocardiografico della valvola e della radice aortica ogni 3-4 anni. Inoltre, in considerazione dell’aumentato rischio di dilatazione della radice aortica e di dissecazione aortica, deve essere prontamente instaurato il trattamento dell’eventuale ipertensione arteriosa.
Apparato osteo-articolare
A partire dai 18 mesi di vita è possibile rilevare un rallentamento della velocità di crescita, che progressivamente porterà circa il 95% dei soggetti a una statura finale di circa 20 cm inferiore all’altezza media per il corrispondente gruppo etnico di appartenenza (20). La bassa statura viene vissuta sia dal soggetto affetto che dai genitori come uno dei problemi principali.
Sin dai primi anni ’80, numerosi studi hanno complessivamente dimostrato l’efficacia della terapia con rhGH nel migliorare la statura definitiva delle bambine con TS, nonostante nelle stesse non sia stata dimostrata una reale insufficienza di GH. In particolare, questi studi hanno provato che l’inizio precoce, la dose e la durata totale della terapia con rhGH sono tutti fattori che hanno una correlazione diretta con l’incremento della statura finale delle TS, che, secondo alcuni autori, può teoricamente arrivare anche a oltre 11 cm (13). Nel 2005, i risultati di uno studio randomizzato e controllato sull’altezza finale di 154 ragazze canadesi con TS hanno mostrato che il gruppo di ragazze in cui era stata effettuata la terapia con rhGH aveva raggiunto in media un’altezza finale di 7.2 cm superiore al gruppo di controllo in cui non era stata effettuata la terapia (29). I possibili effetti collaterali della terapia con rhGH sono complessivamente accettabili e non differiscono da quelli che possono verificarsi nei soggetti trattati per GHD, anche se il rischio di alcune complicanze metaboliche è maggiore nelle bambine con TS, per via della predisposizione verso diabete mellito ed insulino-resistenza.
Dopo aver discusso con i genitori i potenziali benefici e rischi della terapia con rhGH, l’endocrinologo pediatra può pertanto iniziare la terapia, non appena la paziente si trovi al di sotto delle due deviazioni standard (SD) di altezza per l’età (di solito dopo i 2 anni di vita) e senza necessità di eseguire test di stimolo per il dosaggio del GH (endogeno) (30). Il trattamento prevede somministrazioni singole quotidiane di rhGH, per via iniettiva sottocutanea, con posologia più elevata rispetto a quella usata nei pazienti GHD (ovvero circa 0.035-0.05 mg/kg/die). Una volta iniziata la terapia, le pazienti con TS vengono seguite mediante visite di controllo endocrinologiche periodiche, di solito ogni 4-6 mesi. Durante queste visite la dose di rhGH viene aggiustata, in base alla risposta individuale al farmaco, alle variazioni del peso corporeo, all’insorgenza di eventuali effetti collaterali ed anche in base alla concentrazione ematica di IGF-1, che di norma viene mantenuta entro il limite massimo delle 2 SD al di sopra della media. La terapia va proseguita di norma fino al completamento della crescita o eventualmente al raggiungimento di una statura soddisfacente.
Sebbene la terapia con rhGH sia efficace e relativamente sicura, non è in grado di far recuperare completamente la differenza di altezza finale delle pazienti con TS rispetto ai controlli. In particolare, se l’altezza all’inizio della terapia è già gravemente compromessa e/o la terapia viene comunque iniziata tardivamente o se la velocità di crescita rimane lenta nonostante una buona aderenza al trattamento o se vi è un precoce raggiungimento di età ossea puberale, le bambine con TS potrebbero ottenere solo modesti benefici dalla terapia con rhGH (31). Al fine di ottimizzare la crescita, sono stati effettuati vari studi su schemi di terapia combinata rhGH + oxandrolone (Ox), uno steroide di sintesi derivato del diidrotestosterone (DHT). Gli schemi di terapia combinata adottati (31) prevedono, oltre alla terapia convenzionale con rhGH, l’aggiunta a partire dagli 8-10 anni di età circa di Ox ad una dose molto bassa (0.03-0.06 mg/kg/die, fino a un massimo di circa 2.5 mg/die). I risultati di tre recenti studi prospettici, randomizzati e controllati, eseguiti in doppio cieco, hanno complessivamente mostrato un effettivo incremento medio della velocità di crescita e dell’altezza finale (pari a circa 2.3-4.6 cm) nei gruppi trattati con Ox, rispetto ai controlli. I possibili effetti collaterali dell’Ox sono stati dettagliatamente analizzati nei vari studi: dosi di Ox pari a 0.03-0.05 mg/kg/die sembrano essere sicure, solo raramente associate a modici effetti di virilizzazione, tra i quali irsutismo, acne, clitorido-megalia e approfondimento del tono della voce. In sintesi, sebbene gli effetti avversi a lungo termine non siano ancora stati determinati, la terapia combinata rhGH + Ox appare oggi una valida opzione per promuovere la crescita delle bambine con TS ed insufficienza ovarica, in particolare in determinati gruppi di pazienti, come ad esempio in quelli con diagnosi tardiva od in terapia con rhGH, ma con scarsa velocità di crescita (31,32).
Un'altra strategia per incrementare la statura finale delle TS può essere il ritardare l’induzione dello sviluppo puberale, mediante l’introduzione ritardata, di circa 2 anni, della terapia estrogenica sostitutiva, ad esempio a 14 anni anziché a 12. Rispetto a questa strategia, l’utilizzo della terapia combinata con Ox ha il vantaggio di promuovere la crescita e l’altezza finale senza però provocare ritardi dello sviluppo corporeo, evitando quindi le possibili conseguenze sul piano cardiovascolare, scheletrico, riproduttivo e psicologico della prolungata carenza estrogenica. Infine, schemi di terapia combinata rhGH + Ox che prevedano anche l’introduzione ritardata della terapia estrogenica sostitutiva, non hanno dimostrato effetti superiori, in termini di miglioramento della statura definitiva, rispetto alla sola terapia combinata rhGH + Ox e pertanto non sono raccomandati.
Probabilmente esiste un limite a cui può essere portata l’altezza finale delle bambine affette da TS (32). Negli ultimi anni, è stata posta molta enfasi sull’importanza di un’esposizione graduale a dosi fisiologiche di estrogeni sin dall’età infantile. Questa metodica sembra avere benefici nella risposta di crescita alla terapia con rhGH (contrariamente a quanto tradizionalmente ritenuto), nel normalizzare l’età d’insorgenza del telarca e dello sviluppo puberale e addirittura nel ridurre i problemi del comportamento e nel migliorare l’autostima e le abilità cognitive. Gli studi in questione (33-35) hanno suggerito l’uso di dosi crescenti di etinil-estradiolo, iniziando a partire dai 5 anni d’età (25 ng/kg/die per la fascia d’età compresa tra 5-8 anni; 50 ng/kg/die per la fascia 8-12 anni) fino ai 12 anni, quando potrà essere iniziato il regime standard per l’induzione alla pubertà (100 ng/kg/die per la fascia 12-14 anni; 200 ng/kg/die per l’età tra 14-15 anni; 400 ng/kg/die raggiunta l’età di 15-16 anni; 800 ng/kg/die dopo i 16 anni). Nel caso che nel corso di tale terapia si presentasse il rischio di un’accelerazione dello sviluppo puberale, gli stessi autori consigliano un dimezzamento della dose di estrogeni per sei mesi.
Al di là della bassa statura, le altre problematiche più frequentemente riscontrate nelle bambine con TS a carico dell’apparato osteo-articolare sono (36):
- bassa densità minerale ossea, con conseguente rischio di osteoporosi e fratture (specialmente a carico delle braccia);
- displasia congenita dell’anca;
- ipoplasia delle vertebre cervicali e deformità della colonna vertebrale (scoliosi, cifosi, lordosi);
- cubito valgo e deformità di Madelung;
- sproporzione corporea complessiva tra segmento superiore ed inferiore.
In età adulta, è raccomandabile effettuare un piano di controlli seriati per le problematiche inerenti la ridotta densità minerale ossea e l’insufficienza ovarica: potranno essere periodicamente misurati i livelli sierici di vitamina D (25-OH-D) e PTH e potrà eventualmente essere consigliata anche l’esecuzione della mineralometria ossea a raggi X (DEXA-scan). Ove indicato, potranno essere consigliati un’appropriata terapia con calcio (1.2-1.5 g/die), vitamina D e lo svolgimento di una regolare attività fisica.
Apparato riproduttivo
Nei soggetti con TS le gonadi si sviluppano regolarmente per le prime 14-16 settimane di vita fetale; successivamente, a partire dalla 18° settimana circa, inizia una progressiva perdita di ovociti e degenerazione ovarica caratterizzata da graduale sostituzione del tessuto ovarico con tessuto connettivale (disgenesia gonadica), che porterà le stesse ovaie ad assumere un aspetto “a banderella fibrosa” (“streak gonads” in inglese). Il processo di degenerazione ovarica si completa in oltre il 90% dei casi entro i primi anni di vita e di conseguenza, nella grande maggioranza delle TS, le ovaie non possiedono una funzionalità sufficiente a dare avvio o quantomeno a completare il processo di sviluppo puberale. In media, soltanto un terzo delle giovani con TS sviluppa una pubertà spontanea e solo una metà di queste ultime riuscirà a presentare un menarca spontaneo (23,37).
L’ipogonadismo determina il mancato/incompleto sviluppo dei caratteri sessuali (infantilismo sessuale), con i conseguenti problemi fisici quali amenorrea (primaria nel 90% dei casi), infertilità, mancanza dell’accelerazione accrescitiva puberale, ridotta mineralizzazione ossea e conseguente aumentato rischio di osteoporosi, complicanze metaboliche e cardiovascolari (38). Nelle bambine con TS è pertanto raccomandato, a partire dai 9-10 anni di età, effettuare un attento monitoraggio dello sviluppo puberale attraverso l’esame fisico e tramite il dosaggio dei livelli di LH ed FSH. Alle pazienti giovani adulte che vogliano considerare la riproduzione assistita in fase successiva, può essere offerta la crio-conservazione degli ovociti o di tessuto ovarico. In questo caso la misurazione dell’ormone anti-mulleriano (AMH) è considerata utile per la valutazione della riserva ovarica. Recenti studi hanno, infatti, evidenziato che nelle pazienti con TS questo parametro di laboratorio è inversamente correlato alla misurazione di LH, FSH e si presenta proporzionalmente più elevato nelle pazienti con mosaicismo e in quelle che hanno raggiunto uno sviluppo puberale spontaneo (39).
Raggiunta l’età di 12 anni circa, in assenza di sviluppo puberale spontaneo e in presenza di alti livelli sierici di gonadotropine, è generalmente opportuno iniziare una terapia ormonale sostitutiva, per permettere lo sviluppo dei caratteri sessuali secondari e per evitare le problematiche psico-fisiche connesse alla carenza di ormoni sessuali. Al fine di mimare il fisiologico sviluppo puberale, uno schema convenzionale di induzione della pubertà e di sostituzione ormonale prevede la somministrazione di preparazioni estrogeniche (etinil-estradiolo per os) a dosi inizialmente molto basse, ovvero pari a 0.1 µg/kg/die circa, per una durata di circa 12 mesi. Successivamente, la dose giornaliera di etinil-estradiolo va progressivamente incrementata, mediante la prescrizione di dosi di farmaco via via crescenti ogni 6-9 mesi circa (33). In alternativa all’etinil-estradiolo, può essere utilizzato il 17ß-estradiolo, somministrabile per via orale o per via trans-dermica: la dose iniziale è pari a 1/10-1/8 della dose sostitutiva per una donna adulta (2 mg/die per via orale e 0.1 mg/die per via trans-dermica), da incrementare in un arco di tempo di circa 2-4 anni (28). In generale, in base ai dati scientifici disponibili su altre condizioni di ipogonadismo, la somministrazione trans-dermica sembra la strategia preferibile per mimare la pubertà fisiologica, con il beneficio di essere associata a minor rischio di trombosi venosa profonda (40). Intorno ai 14 anni, ovvero dopo circa 2 anni dall’inizio della terapia estrogenica, o comunque al raggiungimento dei primi sanguinamenti uterinici o di uno stadio Tanner IV o V dello sviluppo del seno, va aggiunto in terapia il progestinico: inizialmente, quando la dose giornaliera di estrogeni è ancora relativamente bassa rispetto alla dose di un adulto, il progestinico può essere semplicemente addizionato alla terapia di base estrogenica, somministrandolo per 7-11 giorni al mese, di solito dal 15° al 25° giorno del ciclo (fase post-ovulatoria), a una dose giornaliera pari a 5-10 mg di noretisterone oppure 30 μg di levonorgestrel o 5–10 mg di medrossi-progesterone o 5-10 mg di didrogesterone o 200 mg di progesterone micronizzato. Complessivamente il graduale e lento incremento della dose di estrogeni permette di iniziare e di far progredire lo sviluppo dei caratteri sessuali secondari, senza provocare la prematura chiusura delle epifisi delle ossa lunghe, consentendo così alle bambine con TS di ottimizzare la crescita staturale sfruttando l’intero potenziale di crescita. Il graduale incremento di estrogeni, associato all’aggiunta del progestinico solo in una seconda fase, ovvero al raggiungimento dello stadio IV di Tanner di sviluppo mammario, previene inoltre l’anti-estetico sviluppo del “seno a tubo” (tubular breast), che talvolta si osserva nelle TS durante lo sviluppo (41).
Al raggiungimento della completa maturazione del seno e/o della piena dose di estrogeni, la terapia sostitutiva ormonale può essere semplicemente proseguita attraverso la prescrizione di pillole estro-progestiniche monofasiche (pillola contraccettiva orale). Secondo molti esperti comunque, poiché la posologia ormonale della pillola contraccettiva orale è sicuramente in eccesso rispetto alla reale necessità funzionale sistemica della donna con TS, è preferibile proseguire la terapia estrogenica sostitutiva mediante l’utilizzo di dosi appropriate ottenute con 17ß-estradiolo per via orale o trans-dermica (42). È opinione di molti specialisti che una tardiva esposizione agli estrogeni potrebbe portare a uno sviluppo incompleto della ghiandola mammaria e dell’utero e che viceversa esiste un intervallo di tempo ideale durante il quale il trattamento estrogenico sostitutivo può ottimizzare il loro sviluppo. Allo stesso modo, l’introduzione eccessivamente precoce di progestinici potrebbe avere un effetto avverso sullo sviluppo del seno. Questi concetti rimangono tuttavia teorici senza specifica evidenza scientifica. In ogni caso l’uso della pillola estro-progestinica (contraccettivo orale) è assolutamente sconsigliato durante l’induzione dello sviluppo puberale.
Il trattamento ormonale sostitutivo deve essere proseguito fino all’età della menopausa, adottando i dosaggi e le formulazioni più appropriate in base a età, bisogni specifici ed eventuali problemi medici associati.
Per quanto riguarda la fertilità, nel 5-10% delle donne con TS è possibile una gravidanza spontanea, in particolare nelle pazienti con sviluppo sessuale spontaneo. Ove indicato, deve essere pertanto offerta l’adeguata contraccezione e anche la valutazione della riserva ovarica per la crio-conservazione degli ovociti, per un’eventuale futura gravidanza. Le principali tecniche per la riproduzione assistita che possono essere offerte a una donna con TS che desideri un figlio sono rappresentate dalla fertilizzazione in vitro eterologa (con donazione di ovocita da una donatrice sana e successivo trasferimento embrionale nell’utero, a fresco o dopo crio-conservazione fino al momento desiderato per la gravidanza) od omologa (in cui l’ovocita può essere prelevato dalla madre e crio-conservato anche per molti anni, per scongiurare il rischio che al momento desiderato per la gravidanza non vi siano più follicoli sani) (43). In ogni caso, indipendentemente dalla metodica scelta per il concepimento, va sottolineato che una gravidanza in una donna con TS può essere rischiosa: possono infatti aggravarsi le eventuali problematiche cardiache (aumentato rischio di dilatazione, dissezione e rottura aortica), tiroidee, di pressione arteriosa (pre-eclampsia). Inoltre c’è un aumentato rischio di parto prematuro e di scarsa crescita del feto (intrauterine growth restriction, IUGR). Difetti cardiaci importanti, che hanno magari già necessitato di correzione chirurgica, così come la presenza di ipertensione e/o dilatazione aortica possono essere considerati di per sé controindicazioni ad una gravidanza. Al momento del parto, inoltre, possono verificarsi ulteriori problematiche (sproporzione feto-pelvica, aumento del carico di lavoro cardiaco) e pertanto può essere raccomandato il taglio cesareo. Nel caso di gravidanze spontanee o in vitro con ovocita crio-preservato, sebbene la limitatezza dei casi non permetta un’accurata stima dei rischi, bisogna tenere presente anche la possibilità di trasmissione dell’anomalia cromosomica da madre a figlia (44,45) e l’insorgenza di altre anomalie cromosomiche (46). In sintesi, la donna con TS che voglia intraprendere una gravidanza deve eseguire accertamenti e consulenze mediche approfondite multi-specialistiche, in particolar modo cardiologiche, sia a priori che durante tutto il periodo della gravidanza, al fine di valutare i possibili rischi per la sua salute e per la salute del proprio bambino (43). In considerazione del rischio d’insorgenza di anomalie cromosomiche in gravidanze spontanee od indotte con ovocita crio-preservato, è importante considerare l’esecuzione di una consulenza genetica e della diagnostica pre-impianto.
Sistema immunitario
Le pazienti con TS hanno un’elevata probabilità (circa doppia o tripla rispetto alla popolazione di riferimento) di sviluppare nel corso della vita malattie su base autoimmunitaria, in particolare: malattie infiammatorie intestinali (rettocolite ulcerosa e morbo di Crohn), tiroidite di Hashimoto, malattia celiaca, artrite idiopatica giovanile, diabete mellito tipo 1 e dermopatie autoimmuni (vitiligine, psoriasi ed alopecia areata) (47,48). La tiroidite di Hashimoto, in particolare, si riscontra nel 30-40% dei casi (49). Per questo motivo, i programmi di sorveglianza a lungo termine includono test di screening per le patologie autoimmuni (funzionalità tiroidea, anticorpi anti-tireoperossidasi, anti-transglutaminasi ed emoglobina glicata), a partire dai 4 anni di età e con cadenza annuale, da proseguire anche nell’età adulta (28).
Apparato urinario
In circa il 30-40% delle bambine con TS sono presenti malformazioni congenite del tratto urinario, tra le quali, si riscontrano tipicamente: duplicazioni del distretto escretore renale, rene a ferro di cavallo, stenosi uretero-vescicale e reflusso vescico-ureterale. La maggior parte di tali malformazioni sono inizialmente asintomatiche, ma possono con il tempo facilitare l’insorgenza di complicanze, quali ad esempio ipertensione, idronefrosi ed infezioni delle vie urinarie. È pertanto raccomandato che ogni bambina venga sottoposta, subito dopo la diagnosi di TS, a un’accurata valutazione nefro-urologica, completa di ecografia o altra metodica appropriata di imaging (50).
Apparato uditivo
Circa il 60-80% delle pazienti con TS sviluppa nel corso della vita otiti ricorrenti e problematiche dell’udito (18). La perdita dell’udito è progressiva, ma può presentarsi molto rapidamente, soprattutto dopo i 35 anni, portando a presbiacusia. Il trattamento ottimale per le forme di otiti ricorrenti, oltre alla terapia antibiotica, può avvalersi del posizionamento di tubicini di drenaggio trans-timpanici (tubi di ventilazione) per consentire il deflusso di accumuli di liquido dall’orecchio medio verso l’esterno, nonché la ventilazione e compensazione della pressione endo-auricolare (51,52). Nei programmi di monitoraggio a lungo termine delle pazienti con TS dovrebbe essere sempre incluso l’esame otoiatrico e audiometrico, per valutare eventuali problemi di ridotta capacità uditiva.
Aspetti metabolici
Nella TS si osserva un aumento di incidenza della patologia metabolica, con aumentato rischio di sviluppare ipertensione arteriosa, intolleranza glucidica, diminuzione della prima fase della risposta insulinica (53), obesità con riduzione dell’indice di massa magra e della capacità di affrontare l’esercizio fisico (54) e steatosi epatica (55-56). Il diabete, inoltre, con le sue complicanze a lungo termine rappresenta una della prime cause di mortalità nella TS. È pertanto raccomandabile che venga eseguito un attento monitoraggio dell’indice di massa corporea (BMI), dei livelli ematici di glicemia ed insulina (sia a digiuno che mediante test di tolleranza orale al glucosio), del profilo lipidico, dell’emoglobina glicata (Hb1Ac) e della funzionalità epatica, quest’ultima valutata anche mediante ecografie addominali volte ad escludere la presenza di steatosi. Le ragazze con TS dovrebbero essere educate a mantenere quanto più possibile uno stile di vita corretto, mediante un’alimentazione equilibrata e un regolare esercizio fisico.
Aspetti neuro-cognitivi e psico-sociali
La maggior parte delle pazienti con TS ha intelligenza e sviluppo cognitivo normali: vi è tuttavia una discrepanza, in media, tra i punteggi del quoziente intellettivo verbale (QIV) e del quoziente di performance intellettiva (QIP), con una netta riduzione di questi ultimi, che sembra essere correlata ai deficit delle funzioni esecutive e ai deficit visuo-spaziali. In generale, si osserva buona conoscenza linguistica, buona abilità nel leggere e pronunciare parole di uso non comune, nella comprensione della lettura e nella comprensione del linguaggio recettivo, mentre le difficoltà maggiori si riscontrano nell’esecuzione di un compito sulla base di informazioni non verbali, nei processi di orientamento sinistra-destra e nei test in cui viene misurata l’attenzione a dettagli visivi e la capacità di contestualizzare gli stessi, ovvero di afferrare il significato dei dettagli ed interpretarli nel contesto dell’immagine (57).
Le ragazze con TS spesso hanno ridotta socializzazione, con poche amicizie, scarse relazioni con i pari e scarsa auto-stima. Gli aspetti fenotipici maggiormente visibili e le eventuali difficoltà in ambito scolastico o socio-relazionale sono una frequente fonte d’ansia e di disturbo del tono dell’umore per queste ragazze, che rischiano nei casi più estremi un certo grado di isolamento sociale, a partire soprattutto dall’età adolescenziale in poi. È perciò molto importante che la scuola venga informata sulle caratteristiche cognitive della ragazze con TS, in modo da riconoscere per tempo le eventuali difficoltà e offrire il giusto supporto, se necessario. Nell’ambito del monitoraggio a lungo termine per le problematiche fisico-patologiche, non dovrebbero escludersi i controlli inerenti le possibili problematiche psicologiche connesse alla sindrome, che spesso investono tutto il contesto familiare e non solo l’individuo affetto (58,59).
Aspetti oncologici
I risultati degli studi eseguiti per valutare l’incidenza delle patologie neoplastiche nelle donne con TS mostrano dati contrastanti: uno studio (60) effettuato in Danimarca su 597 pazienti ha mostrato un netto aumento di incidenza di cancro del colon, con rischio relativo pari a 6.9. La medesima associazione era già stata segnalata su pubblicazioni precedenti di casi clinici (61-63). Nel 2001, un gruppo di ricercatori inglesi ha invece pubblicato uno studio (64) su 400 donne affette da TS, seguite per 31 anni, i cui risultati non mostravano un rischio aumentato di sviluppare patologie neoplastiche rispetto a quello della popolazione di riferimento. Successivamente però, lo stesso gruppo di autori inglesi ha pubblicato i risultati di un altro studio eseguito su scala nazionale su 3425 donne inglesi con TS: rispetto alla popolazione generale, veniva riportato un aumentato rischio di sviluppo di tumori del sistema nervoso centrale (in particolare meningioma), del tratto vescico-ureterale, dell’occhio e del corpo dell’utero, ma anche un ridotto rischio di sviluppo di tumori della mammella (65). Un particolare ed elevato rischio neoplastico è infine associato al riscontro nel cariotipo di una femmina con TS di materiale genetico appartenente al cromosoma Y (incidenza pari al 40-60% dei soggetti con TS) (66); come accade anche per altre sindromi caratterizzate da disordini della differenziazione sessuale, la presenza del cromosoma Y o di una parte di esso è associata a un concreto rischio di sviluppo di displasia gonadica (gonado-blastoma), che, all’incirca nel 60% dei casi, può successivamente condurre alla formazione di disgerminoma o di altro tumore a cellule germinali altamente invasivo (ad esempio: carcinoma embriogenetico/embrionale, teratoma maturo/immaturo, tumore del sacco vitellino, coriocarcinoma) (8,67). In questo gruppo di soggetti è pertanto fortemente raccomandata la gonadectomia profilattica (68,69).
CONCLUSIONI
La sindrome di Turner è dovuta a un’alterazione cromosomica complessa ed è perfettamente compatibile con la vita. Un aspetto di questa condizione è rappresentato dai problemi legati alla ridotta altezza e al mancato sviluppo sessuale. Le patologie d’organo associate sono numerose e devono essere monitorate regolarmente. Tipicamente il deterioramento della salute di queste pazienti si osserva soprattutto in età adulta, con progressivo peggioramento della qualità di vita. Per assicurare la migliore prognosi possibile, è necessario che le pazienti con TS siano sottoposte a programmi di controllo a lungo termine operati da un gruppo sanitario multidisciplinare, a partire dall’età pediatrica e continuato nell’età adulta.
APPENDICE
Promemoria per il controllo a lungo termine delle pazienti con sindrome di Turner in età pediatrica e adulta. Turner Syndrome Support Society, UK.
Si raccomanda che il piano di cura complessivo sia gestito da un endocrinologo pediatra all’interno di un centro regionale di riferimento. Questi seguirà regolarmente la paziente per monitorarne la crescita, lo sviluppo puberale e la successiva transizione a un servizio di endocrinologia dell’adulto. L’endocrinologo dovrà inoltre farsi carico del ruolo di supervisore e coordinatore dei diversi aspetti della salute della paziente.
La seguente checklist non intende essere prescrittiva per aree in cui il dibattito è aperto, ma fornire raccomandazioni da sottoporre eventualmente a verifica. La checklist servirà inoltre ad assicurare continuità tra i dipartimenti che si occuperanno della paziente nelle diverse fasi.

| CHECKLIST PEDIATRICA | |||||||||||
| ANAGRAFICA | ISTRUZIONE | Sì/no | Commenti | ||||||||
| Nome: | Scolarizzazione | ||||||||||
| Indirizzo: | Necessità di sostegno (specificare) | ||||||||||
| Data di nascita: | Difficoltà specifiche (dettagliare) | ||||||||||
| DIAGNOSI | Data | Come è stata posta | VALUTAZIONE GENERALE | Frequenza | |||||||
| Pre-natale | Peso/altezza/BMI | A ogni visita ambulatoriale | |||||||||
| Post-natale | Pressione | ||||||||||
| Funzione tiroidea | Ogni 12-24 mesi | ||||||||||
| ALLA DIAGNOSI | Sì/no | Data | Ab anti-tiroide e anti-trans-glutaminasi | ||||||||
| Cariotipo (specificare) | Glicemia e HbA1c | Annuali | |||||||||
| materiale del cromosoma Y rilevato con FISH |
IGF-I |
||||||||||
| gonadectomia eseguita se presente materiale Y | Età ossea | ||||||||||
| Consulenza genetica | OGTT | Se necessario | |||||||||
| Consulenza cardiologica | Insulinemia | ||||||||||
| Ecografia reni/pelvi | Esami epatici | Prima dell’induzione della pubertà e del trasferimento al servizio adulti | |||||||||
| Indirizzata a gruppo di supporto | Ecografia reni/pelvi | Alla diagnosi e poi se necessario | |||||||||
| Cardiologo | |||||||||||
| CRESCITA | Data | Genetista | |||||||||
| Inizio | Fine | Gruppo di supporto | |||||||||
| Trattamento con GH | Audiologo | Se necessario | |||||||||
| Trattamento con oxandrolone |
Problemi del linguaggio:
|
||||||||||
| ORL | |||||||||||
| PUBERTÀ | Sì/no | Data | Oculista | ||||||||
| Spontanea | Podiatra | ||||||||||
| Se non è partita spontaneamente a 10 anni, dosaggio LH/FSH e ecografia pelvica | Dermatologo | ||||||||||
| Indotta | Ortopedico | ||||||||||
| Menarca | Ortodonzista | ||||||||||
| Terapia ormonale sostitutiva/contraccettiva (specificare) | Psicologo | ||||||||||
| CHECKLIST ADULTO | ||||
| TRANSIZIONE ALLE CURE DELL’ADULTO | TERAPIA ORMONALE SOSTITUTIVA | Quando | ||
| Invio a endocrinologo con interesse per TS | Adeguatezza dose | Ogni visita | ||
| Invio a clinica con specialisti per TS | Via di somministrazione ottimale | |||
| Invio a cardiologo | ||||
| Invio a MMG | ||||
| Invio a ginecologo | CARDIOLOGIA | Quando | ||
| Counseling generale | Pressione | Ogni visita | ||
Valutazione post GH di:
|
Ecocardiografia | Ogni 3-5 anni | ||
| RM | Pre-gravidanza | |||
| Consulenza cardiologica | Se necessario | |||
| AUDIOLOGO | Quando | |||
| Consapevolezza deficit uditivo | Ogni visita | |||
| Problemi ORL in atto | ||||
| Test audiometrico | Ogni 5 anni | |||
| Protesi acustiche | Se necessario | |||
| RIPRODUZIONE | Quando | |||
| Progetti per fertilità | Ogni visita | |||
| Funzione sessuale | ||||
| Ecografia uterina | Pre-gravidanza | |||
| OSSO | Quando | |||
| Anamnesi fratture | Ogni visita | |||
| VALUTAZIONE GENERALE | Quando | Densitometria | Ogni 5 anni | |
| Peso e BMI | Ogni visita | |||
| Funzione tiroidea | Annuale | SOCIALITÀ | Quando | |
| Funzione renale | Anamnesi lavorativa | Ogni visita | ||
| Funzione epatica | Relazioni | |||
| Lipidogramma | Umore | |||
| OGTT | Ogni 5 anni | Invio psicologo | Al bisogno | |
| Ab anti-tiroide e anti-transglutaminasi | ||||
| Qualità della vita | Se necessario | |||
BIBLIOGRAFIA
- Tesch LG, Rosenfeld R. Morgagni, Ullrich and Turner: the discovery of gonadal dysgenesis. The endocrinologist 1995, 5: 327-28.
- Ullrich O. Uber typische kombinationsbilder multipler abartungen. Z Kinderheilk 1930, 49: 271-6.
- Turner HH. A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology 1938, 23: 566–74.
- Ford CE, Jones KW, Polani PE, et al. A sex chromosomal anomaly in a case of gonadal dysgenesis (Turner's syndrome). Lancet 1959, 1: 711–3.
- Stochholm K, Juul S, et al. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab 2006, 91: 3897–902.
- Nielsen J, Wolhert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-years incidence study in Arhus, Denmark. Hum Genet 1991, 87: 81-3.
- Gravholt CH, Juul S, Naeraa RV, Hansen J. Prenatal and postnatal prevalence of Turner’s syndrome: a registry study. BMJ 1996, 312: 16-21.
- Oliveira RM, Verreschi IT, Lipay MV, et al. Y chromosome in Turner syndrome: review of the literature. Sao Paulo Med J 2009, 127: 373-8.
- Frías JL, Davenport ML; Committee on Genetics and Section on Endocrinology. Health supervision for children with Turner syndrome. Pediatrics 2003, 111: 692-702.
- Hook EB, Warburton D. Turner syndrome revisited: review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum Genet 2014, 133: 417–24.
- Wolff DJ, Van Dyke DL, Powell CM. Laboratory guideline for Turner syndrome. Genet Med 2010, 12: 52-5.
- Zhong Q, Layman LC. Genetic considerations in the patient with Turner syndrome--45,X with or without mosaicism. Fertil Steril 2012, 98: 775-9.
- Sybert VP, McCauley E. Turner's syndrome. N Engl J Med 2004, 351: 1227–38.
- Abramowitz LK, Olivier-Van Stichelen S and Hanover JA. Chromosome imbalance as a driver of sex disparity in disease. J Genomics 2014, 2: 77-88.
- Uematsu A, Yorifuji T, Muroi J, et al. Parental origin of normal X chromosomes in Turner syndrome patients with various karyotypes: implications for the mechanism leading to generation of a 45,X karyotype. Am J Med Genet 2002, 111: 134–9.
- Chacko E, et al. Update on Turner and Noonan syndromes. Endocrinol Metab Clin North Am 2012, 41: 713-34.
- Gravholt CH, Juul S, Naeraa RV, Hansen J. Morbidity in Turner syndrome. J Clin Epidemiol 1998, 51: 147-58.
- Savendahl L, Davemport ML. Delayed diagnosis of Turner’s syndrome: proposed guidelines for change. J Pediatr 2000, 137: 455-9.
- Morgan T. Turner syndrome: diagnosis and management. Am Fam Physician 2007, 76: 405-10.
- Saenger P, et al. Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrinol Metab 2001, 86: 3061-9.
- Ly-Pen D, Andreu JL. Madelung's deformity. Reumatol Clin 2014, 10: 125-6.
- Hong DS, Dunkin B, Reiss AL. Psychosocial functioning and social cognitive processing in girls with Turner syndrome. J Dev Behav Pediatr 2011, 32: 512-20.
- Wasserman JD, Asch A. Reproductive medicine and Turner syndrome: ethical issues. Fertil Steril 2012, 98: 792-7.
- Chakhtoura Z, Touraine P. Fertility on women with Turner syndrome. Presse Med 2013, 42: 1508-12.
- Gravholt CH. Epidemiological, endocrine and metabolic features in Turner syndrome. Arq Bras Endocrinol Metabol 2005, 49: 145-56.
- Sybert VP. Cardiovascular malformations and complications in Turner syndrome. Pediatrics 1995, 101: e11.
- Völkl TM, Degenhardt K, et al. Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome. The Erlangen experience. Clin Cardiol 2005, 28: 88-92.
- Bondi CA, and the Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of Turner syndrome study group. J Clin Endocrinol Metab 2007, 92: 10-25.
- Stephure DK, Canadian Growth Hormone Advisory Committee. Impact of growth hormone supplementation on adult height in Turner syndrome: results of the Canadian randomized controlled trial. J Clin Endocrinol Metab 2005, 90: 3360-6.
- Agenzia Italiana del Farmaco (AIFA). Nota 39– Determinazione n. 616/2014 modifica alla Nota AIFA 39 (Gazzetta Ufficiale 18 novembre 2010, n. 270).
- Sas TCJ, Gault EJ, et al. Safety and efficacy of oxandrolone in growth hormone-treated girls with Turner syndrome: evidence from recent studies and recommendations for use. Horm Res Paediatr 2014, 81: 289–97.
- Gault EJ, Perry RJ, Cole TJ, et al. Effect of oxandrolone and timing of pubertal induction on final height in Turner's syndrome: randomised, double blind, placebo controlled trial. BMJ 2011, 342: d1980.
- Quigley CA, Wan X, et al. Effects of low estrogen replacement during childhood on pubertal development and gonadotropin concentration in patients with Turner Syndrome: results of a randomized, double blind, placebo controlled clinical trial. J Clin Endocrinol Metab 2014, 99: E1754-64.
- Ross JL, Quigley CA, Cao D, et al. Growth hormone plus childhood low-dose estrogen in Turner syndrome. N Engl J Med 2011, 364: 1230–42.
- Ross JL, Cassorla FG, Skerda MC, et al. A preliminary study of the effect of estrogen dose on growth in Turner’s syndrome. N Engl J Med 1983, 309: 1104–6.
- Santos V, Marçal M, et al. Turner syndrome. From child to adult. A multidisciplinary approach. Acta Med Port 2010, 23: 873-82.
- Davemport ML. Approach to the patient with Turner Syndrome. J Clin Endocrinol Metab 2010, 95: 1487-95.
- Gonzalez L, Witchel SF. The patient with Turner syndrome: puberty and medical management concerns. Fertil Steril 2012, 98: 780-6.
- Visser JA, Hokken-Koelega AC, Zandwijken GR, et al. Anti-Mullerian hormone levels in girls and adolescents with Turner syndrome are related to karyotype, pubertal development and growth hormone treatment. Hum Reprod 2013, 28: 1899–907.
- Ankarberg-Lindgren C, Kristrom B, Norjavaara E. Physiological oestrogen replacement for puberty induction in girls, a clinical observational study. Horm Res Paediatr 2014, 81: 239–44.
- Beckmann CRB, et al. Lippincott Williams & Wilkins - Wolters Kluwer Health. 2010. Obstetrics and Gynecology, 6th edition: pag 312.
- Collett-Solberg PF, Gallicchio CT, et al. Endocrine diseases, perspectives and care in Turner syndrome. Arq Bras Endocrinol Metabol 2011, 55: 550-8.
- Hewitt JK, Jayasinghe Y, et al. Fertility in Turner syndrome. Clin Endocrinol (Oxf) 2013, 79: 606-14.
- Varela M, Shapira E, Hyman DB. Ullrich-Turner syndrome in mother and daughter: prenatal diagnosis of a 46,X,del(X)(p21) offspring from a 45,X mother with low-level mosaicism for the del (X)(p21) in one ovary. Am J Med Genet 1991, 39: 411–2.
- Aller V, Gargallo M, Abrisqueta JA. Familial transmission of a duplication-deficiency X chromosome associated with partial Turner syndrome. Clin Genet 1995, 48: 317–20.
- Tarani L, Lampariello S, Raguso G, et al. Pregnancy in patients with Turner’s syndrome: six new cases and review of literature. Gynecol Endocrinol 1998, 12: 83–7.
- Jorgensen KT, Rostgaard K, Bache I, et al. Autoimmune diseases in women with Turner's syndrome. Arthritis Rheum 2010, 62: 658–66.
- Lleo A, Moroni L, Caliari L, Invernizzi P. Autoimmunity and Turner's syndrome. Autoimmun Rev 2012, 11: A538-43.
- Grossi A, Crinò A, et al. Endocrine autoimmunity in Turner syndrome. Ital J Pediatr 2013, 39: 79.
- Carvalho AB, et al. Cardiovascular and renal anomalies in Turner syndrome. Rev Assoc Med Bras 2010, 56: 655-9.
- Verver EJ, et al. Ear and hearing problems in relation to karyotype in children with Turner syndrome. Hear Res 2011, 275: 81-8.
- Parkin M, Walker P. Hearing loss in Turner Syndrome. Int J Pediatr Otorhinolaryngol 2009, 73: 243-7.
- Bakalov VK, Cooley MM, Quon MJ, et al. Impaired insulin secretion in the Turner metabolic syndrome. J Clin Endocrinol Met 2004, 89: 3516-20.
- Gravholt CH, Naeraa RW, Nyholm B, et al. Glucose metabolism, lipid metabolism, and cardiovascular risk factors in adult Turner's syndrome. The impact of sex hormone replacement. Diabetes Care 1998, 21: 1062-70.
- Calcaterra V, Brambilla P, et al. Metabolic syndrome in Turner syndrome and relation between body composition and clinical, genetic, and ultrasonographic characteristics. Metab Syndr Relat Disord 2014, 12: 159-64.
- O'Gorman CS, Syme C, et al. An evaluation of early cardiometabolic risk factors in children and adolescents with Turner syndrome. Clin Endocrinol (Oxf) 2013, 78: 907-13.
- Lepage JF, et al. Contribution of executive functions to visuospatial difficulties in prepubertal girls with Turner syndrome. Dev Neuropsychol 2011, 36: 988-1002.
- Lepage JF, et al. Impact of cognitive profile on social functioning in prepubescent females with Turner syndrome. Child Neuropsychol 2013, 19: 161-72.
- D’Aprile R, Galasso C. La sindrome di Turner: guida pratica per la famiglia, IV edizione. 2014: 25-34.
- Hasle H, Olsen JH, et al. Occurrence of cancer in women with Turner syndrome. Br J Cancer 1996, 73: 1156-9.
- Herrera-Ornelas L, Ochi H, Petrelli N, et al. Nonfamilial Turcot's syndrome associated with Turner's syndrome, multiple carcinomas of the tongue, and cancer of the colon. J Surg Oncol 1984, 27: 251-4.
- Ochi H, Takeuchi J, Sandberg AA. Multiple cancers in a Turner's syndrome with 45,X/46,XXp-/46,XX/47,XXX karyotype. Cancer Genet Cytogenet 1985, 16: 335-9.
- Cheng HM, Tsai MC. Turner's syndrome associated with sigmoid polyp and colon cancer: report of a case. J Formos Med Assoc 1993, 92: 580-2.
- Swerdlow AJ, Hermon C, Jacobs PA, et al. Mortality and cancer incidence in persons with numerical sex chromosome abnormalities: a cohort study. Ann Hum Genet 2001, 65: 177-88.
- Schoemaker MJ, Swerdlow AJ, et al. Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. Lancet Oncol 2008, 9: 239-46.
- Hersmus R, Stoop H, et al. SRY mutation analysis by next generation (deep) sequencing in a cohort of chromosomal Disorders of Sex Development (DSD) patients with a mosaic karyotype. BMC Med Genet 2012, 13: 108.
- McDonough PG, Tho SPT. Clinical implications of overt and cryptic Y mosaicism in individuals with dysgenetic gonads. Wellness for girls and women with Turner Syndrome. Proceedings of the Consensus Conference – April 6-9, 2006, National Institute of Child Health and Human Development, National Institute of Health, Bethesda, Washington DC, USA International Congress Series 2006, 1298: 13-20.
- Matsumoto F, Shimada K, Ida S. Tumors of bilateral streak gonads in patients with disorders of sex development containing Y chromosome material. Clin Pediatr Endocrinol 2014, 23: 93-7.
- Brant WO, et al. Gonadoblastoma and Turner syndrome. J Urol 2006, 175: 1858-60.
Sindrome di Klinefelter
Vito Angelo Giagulli1, Marianna Bono2 & Piernicola Garofalo2
1Unità Territoriale di Endocrinologia e Malattie Metaboliche, Presidio di Assistenza Territoriale “F Jaia”, Conversano AUL/Bari
2UO Endocrinologia, AO Ospedali Riuniti Villa Sofia – Cervello, Palermo
(aggiornato al 21 febbraio 2019)
Definizione ed epidemiologia
La sindrome di Klinefelter (SK) è una malattia genetica, con uno spettro di quadri caratterizzati dalla presenza di almeno un cromosoma X soprannumerario rispetto a quello presente in un soggetto con cariotipo normale 46 XY.
I quadri fenotipici e clinici, quindi, possono variare a seconda che si tratti di soggetti 47 XXY, 48 XXXY, 49 XXXXY oppure 49 XXXYY, oltre ai più complessi mosaicismi, cioè quadri clinici e fenotipici misti, composti dalla presenza di linee cellulari diverse con diverse alterazioni genetiche (ad esempio: 46XY/47XXY, 47XXY/48XXXY, e così via). La gravità del quadro clinico dipende dal numero di cromosomi X in più (1).
Questa premessa potrebbe far pensare che le manifestazioni cliniche di questa sindrome siano così evidenti da renderla facilmente riconoscibile, ma così non è. Sappiamo oggi con certezza che la SK è sottostimata, in quanto soprattutto i mosaicismi danno molto spesso luogo a espressioni fenotipiche molto sfumate, che arrivano alla diagnosi in età adulta, quando il soggetto viene studiato per disturbi della sfera sessuale o per l’infertilità di coppia.
Dal punto di vista epidemiologico, la prevalenza stimata della SK è di 1 su 500 nati, per cui non è certo una patologia genetica rara, come era considerata fino a poco tempo fa. Solo nel 40% dei soggetti viene fatta una diagnosi precoce, mentre il 60% di questi pazienti giunge alla diagnosi in età adulta.
Clinica
Le manifestazioni possono essere svariate. Nelle forme più lievi i soggetti si presentano di alta statura, tendenti all’obesità, presentano ginecomastia (cioè presenza di tessuto mammario più sviluppato rispetto ai soggetti di sesso maschile non affetti), testicoli piccoli e di consistenza aumentata, piccole disabilità neuro-psico-motorie. Buona parte dei soggetti con SK giunge alla diagnosi in seguito ad indagini eseguite per scoprire le cause di infertilità di coppia: dopo avere eseguito un cariotipo o dopo l’esame del liquido seminale, che mostra assenza o forte riduzione del numero degli spermatozoi.
Diagnosi
La diagnosi di SK è oggi divenuta più precoce, addirittura spesso in epoca fetale, grazie a una maggiore sensibilizzazione e conoscenza della sindrome, e perchè molte coppie procreano in età più avanzata, motivo che porta a richiedere sempre più frequentemente l’amniocentesi (2). La diagnosi clinica o sui dati di laboratorio (deficit di testosterone con gonadotropine aumentate) deve comunque essere confermata dal cariotipo.
Terapia
Nei soggetti in cui la diagnosi viene posta in epoca prenatale, i soggetti vengono seguiti soprattutto per la ricerca di disabilità neuro-psicomotorie ed eventualmente supportati. In età adolescenziale ed adulta la terapia è sostitutiva con il testosterone, utilizzato in varie formulazioni (intramuscolare, transdermica), che serve a consentire un adeguato sviluppo dei caratteri sessuali secondari, a mantenere il desiderio sessuale, a prevenire l’osteoporosi (3).
Fertilità
Per diversi anni il paziente con la SK è stato ritenuto infertile (4). Questi dati risultavano, tuttavia, in parziale contrasto con gli studi osservazionali, che riportano sia gravidanze spontanee in coppie il cui partner è affetto da KS, sia la presenza di spermatozoi nell’eiaculato di circa il 10% dei soggetti con forme mosaiche (46XY/47XXY) e di almeno l’8% dei quelli con la forma classica (47XXY) (5). Queste evidenze hanno suggerito la possibilità che alcuni soggetti con KS abbiano alcune isole di spermatogenesi normale, rendendo possibile il recupero di spermatozoi dai testicoli (TESE) nell’ambito di un percorso altamente tecnologico di procreazione medicalmente assistita (ICSI: intracytoplasmatic sperm injection). Tuttavia, è necessario sottolineare l’opportunità di uno studio genetico pre-impianto per le coppie il cui partner è affetto da KS, al fine di escludere la presenza di spermatozoi aneuploidi tra quelli da utilizzare per la ICSI (6).
Sono a tutt’oggi poco noti i fattori che possono influenzare il tasso di ritrovamento di spermatozoi (SSR), il tasso di gravidanze (PR) e il tasso di nati vivi (LBR) dopo TESE in adulti con KS e successiva ICSI. Una meta-analisi suggerisce che, indipendentemente dalla tecnica utilizzata (microTESE o macroTESE), nel soggetto KS l’SSR è pari al 50%, così come PR e LBR dopo ICSI. Questi risultati sono indipendenti dai parametri presi in considerazione nello studio metanalitico (testosterone, FSH, LH, età e volume testicolare) (7).
Oggi queste tecniche sono utilizzate anche nei soggetti in età adolescenziale, per recuperare spermatozoi da crioconservare per consentirne un utilizzo futuro, visto che nel corso degli anni il numero degli spermatozoi si riduce inevitabilmente (8).
Sessuologia
Se l’associazione della KS con l’infertilità è ben nota, pochi sono gli studi riguardanti la sessualità di questi soggetti. Gli studi fin qui condotti hanno rilevato che i soggetti con KS lamentano prevalentemente desiderio ipoattivo e alcune forme non severe di disfunzione erettile, la cui genesi non va considerata specifica della sindrome ma squisitamente endocrina, cioè dovuta al deficit di testosterone (9,10). Infatti, uno studio controllato con placebo, sebbene di piccole dimensioni, ha documentato che il trattamento sostitutivo con testosterone è in grado di migliorare significativamente i disturbi lamentati (11). Al contrario, un recente studio osservazionale ha evidenziato che esistono caratteristiche comportamentali che vanno considerate specifiche della sindrome, in particolare dovute alla presenza di un cromosoma X extra. I pazienti con KS, infatti, presentano un rischio più elevato rispetto alla popolazione di controllo di tratti autistici, comportamenti parafiliaci e disforia di genere, sostenuti da una personalità ossessiva-compulsiva (12).
Co-morbilità metaboliche e cardio-vascolari
È generalmente noto che i soggetti con KS hanno un elevato rischio (fino al 70%) di ospedalizzazione, presentando elevata incidenza di malformazioni, rischio cardio-vascolare (CV), disturbi psichiatrici e malattie metaboliche rispetto a soggetti della stessa età. Nel soggetto con KS, infatti, il tasso standard di mortalità (SMR) è aumentato:
- per le patologie CV (disfunzione ventricolare diastolica, cardiopatie congenite, trombosi degli arti inferiori, ecc): SMR 1.3 (IC95% 1.1-1.5);
- per le malattie cerebro-vascolari (emorragie subaracnoidee, malformazioni artero-venose, ictus ischemico, ecc): SMR 2.2 (IC95% 1.6-3.0);
- per le malattie metaboliche (obesità, sindrome metabolica, diabete mellito tipo 2 conclamato, ecc): 4.8 (IC95% 2.9-7.4).
Tenendo conto di questi dati, si può affermare che la spettanza di vita di questi pazienti è ridotta rispetto alle popolazioni sane (13).
Fra i fattori patogenetici di queste patologie vanno considerati quelli legati a malformazioni congenite (per es. vascolari), quelli dovuti a uno stato trombofilico (elevati tassi di PAI 1, di fattori della coagulazione, iperreattività primitiva delle piastrine) e, in particolare, quelli dovuti a disturbi metabolici (obesità, dislipidemie, diabete mellito tipo 2, ecc) (14). Poiché per questi ultimi l’SMR risulta più elevato in questi pazienti e poiché è documentato un legame fisiopatologico tra il deficit di testosterone e le malattie metaboliche, si è focalizzato un crescente interesse per studi clinici e fisiopatologici sui disturbi metabolici e la SK. Le malattie metaboliche sono, infatti, veramente frequenti sia negli adolescenti con KS che soprattutto negli adulti. I ragazzi e gli adolescenti con KS presentano riduzione della massa magra, con significativo incremento del grasso localizzato prevalentemente a livello del tronco, che, come è noto, costituisce un fattore favorente l’insulino-resistenza e lo sviluppo della sindrome metabolica (15). Nonostante il trattamento sostitutivo con testosterone venga intrapreso tempestivamente nei KS, spesso non si riesce a superare la condizione sfavorevole di insulino-resistenza e a ottenere un miglioramento della composizione corporea e del profilo metabolico (9,16). Questo suggerisce una patogenesi genetica piuttosto che un ruolo esclusivamente metabolico (ipogonadismo). Viene ipotizzato che geni presenti sull’extra cromosoma X siano espressi in eccesso, causando alterazioni del metabolismo del glucosio o dell’infiammazione, che possono essere alla base dei disturbi metabolici (17).
Bibliografia
- Skakkeback A, et al. Quality of life in men with a Klinefelter syndrome. The impact of genotype, health, socioeconomics and sexual function. Gen Med 2018, 20: 214-22.
- Close S, et al. Complexities of care in Klinefelter Syndrome. An APRN perpective. Pediatr Endocrinol Rev 2017, 14 suppl 2: 462-71.
- Flannigan R, et al. Klinefelter syndrome. The effects of early androgen therapy on competence and behavioral phenotype. Sex Med Rev 2018, 4: 595-606.
- Fullerton G, et al. Should non mosaic Klinefelter syndrome men be labelled as infertile in 2009? Human Reprod 2010, 25: 588-97.
- Lanfranco F, et al. Klinefelter’s syndrome. Lancet 2004, 364: 272-83.
- Vailard F, et al. The high frequency of sperm aneuploidy in Klinefelter patients and nonobstructive azoospermia is due to meiotic errors in euploid spermatocytes. J Androl 2012, 33: 1352-59.
- Corona G, et al. Sperm recovery and ICSI outcomes in Klinefelter syndrome: a systematic review and meta-analysis. Hum Reprod Update 2017, 23: 265-75.
- Rives N, et al. Fertility preservation in Klinefelter Syndrome patients during the transition period. Endocr Dev 2018, 33: 149-57.
- Corona G, et al. Sexual dysfunction in subjects with Klinefelter’s syndrome. Int J Androl 2010, 33: 574-80.
- Giagulli VA, et al. Adding liraglutide to lifestyle changes, metformin and testosterone therapy boosts erectile function in diabetic obese men with overt hypogonadism. Andrology 2015, 3: 1094-103.
- Wu FC, et al. The behavioural effects of testosterone undecanoate in adult men with Klinefelter’s syndrome: a controlled study. Clin Endocrinol (Oxf) 1982, 16: 489–97.
- Fisher AD, et al. Hyperxuality, paraphilic behaviour, and gender dysphoria in individuals with Klinefelter’s syndrome. J Sex Med 2015, 12: 2413-24.
- Bojesen A, et al. Morbidity in Klinefelter’s syndrome: a Danish register study based on hospital discharge diagnoses. J Clin Endocrinol Metab 2006, 91: 1254–60.
- Calogero A, et al. Klinefelter syndrome: cardiovascular abnormalities and metabolic diseases. J Endocrinol Invest 2017, 40: 705-12.
- Bardsley MZ, et al. Insulin resistance and metabolic syndrome in prepubertal boys with Klinefelter syndrome. Acta Paediatr 2011, 100: 866–70.
- Pasquali D, et al. Cardiovascular abnormalities in Klinefelter syndrome. Int J Cardiol 2013, 168: 754–9.
- Zitzmann M, et al. Gene expression patterns in relation to the clinical phenotype in Klinefelter syndrome. J Clin Endocrinol Metab 2015, 100: E518–23.
47,XYY DSD
45,X/46,XY (disgenesia gonadica mista)
Gianni Russo, Silvia Meroni
UO Pediatria e Medicina dell’adolescenza, Università “Vita e Salute”, Ospedale San Raffaele, Milano
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Le disgenesie gonadiche (DG) rappresentano un gruppo eterogeneo di condizioni cliniche in cui lo sviluppo fetale delle gonadi è anomalo, talvolta asimmetrico; tale situazione è causata sia da anomalie strutturali o numeriche dei cromosomi sessuali, sia da mutazioni nei geni coinvolti nello sviluppo della gonade. La gonade può essere costituita solamente da tessuto fibroso, in assenza di tessuto endocrinologicamente attivo e gameti (streak gonadico), oppure svilupparsi in modo incompleto (gonade disgenetica). Il fenotipo dei pazienti con DG è pertanto molto variabile e dipende dall’entità di ormoni prodotti dalla gonade disgenetica, che a sua volta riflette la quantità di tessuto gonadico funzionante (1).
Nella pratica clinica, in accordo con la nuova classificazione proposta dal “Consensus Statement”, il termine disgenesia gonadica mista (DGm) è riservato ai casi di disgenesia gonadica in soggetti con mosaicismo 45,X/46,XY o sue varianti. Il cariotipo è quindi caratterizzato dalla presenza simultanea di una linea cellulare con monosomia del cromosoma X e una o più linee cellulari con un cromosoma Y strutturalmente normale o anomalo (isocromosoma, delezione, cromosoma ad anello, traslocazione, …). Le alterazioni strutturali del cromosoma nella linea cellulare 46,XY possono determinare fenotipi differenti: la delezione del braccio corto del cromosoma Y, contenente SRY, interferisce direttamente con lo sviluppo del testicolo; la delezione del braccio lungo del cromosoma Y, contenente le regioni “Azoospermia Factor regions”, determina infertilità maschile.
È una condizione rara (incidenza 1.5 per 10.000 neonati), ma probabilmente sotto-diagnosticata. Tali persone possono presentare un fenotipo estremamente eterogeneo: individui con fenotipo maschile normale, individui con ambiguità genitale (ipospadia e criptorchidismo mono/bilaterale) e individui con fenotipo femminile e caratteristiche turneriane. Lo sviluppo degli organi genitali interni è anch’esso eterogeneo: le strutture mulleriane possono essere normalmente sviluppate, parzialmente sviluppate o assenti, in base al grado di disgenesia testicolare. Questa ampia eterogeneità clinica, ormonale e gonadica potrebbe riflettere la diversa distribuzione delle linee cellulari 45,X e 46,XY nei diversi tessuti del singolo individuo (2).
La diagnosi può avvenire:
- in epoca pre-natale: esecuzione di esami diagnostici pre-natali per fattori fetali o materni;
- in epoca neonatale: presenza di genitali ambigui;
- in età pediatrica: presenza di bassa statura o della combinazione bassa statura e ritardo puberale;
- in età adulta: presenza d’infertilità.
I pazienti maschi con mosaicismo 45,X/46,XY possono presentare caratteristiche turneriane e bassa statura (3). Devono essere quindi sottoposti a screening malformativo cardiologico, renale e audiologico e, se presente bassa statura, possono beneficiare del trattamento con ormone della crescita ricombinante (4). Un normale fenotipo dei genitali in senso maschile alla nascita è predittivo di una buona funzione testicolare, tale da permettere un normale sviluppo puberale spontaneo.
I soggetti con cromosoma Y (o sue porzioni) e disgenesia gonadica presentano un rischio aumentato di sviluppare tumori a cellule germinali (intorno al 10-15%), come il gonadoblastoma o il carcinoma in situ (CIS), con la possibilità di trasformazione maligna. Lo sviluppo di tumori avviene prevalentemente dalla seconda decade di vita. In letteratura non c’è un consenso univoco in merito ai tempi di esecuzione della gonadectomia. In passato era raccomandata la gonadectomia precoce con possibilità di attendere fino all’epoca puberale, essendo il rischio di neoplasia accettabilmente basso in epoca pre-puberale. Gli studi più recenti suggeriscono un approccio più individualizzato e conservatore che tenga in considerazione diversi fattori, quali la localizzazione delle gonadi (addominale, inguinale o scrotale), il fenotipo dei genitali interni ed esterni e il sesso assegnato. La localizzazione addominale delle gonadi così come la scarsa virilizzazione dei genitali esterni si associano a maggior rischio neoplastico. Pertanto, nei soggetti con fenotipo maschile e gonadi in sede scrotale è possibile optare per un attento monitoraggio mediante un’auto-palpazione testicolare regolare e un’ecografia testicolare annuale. Al contrario, nelle femmine con sindrome di Turner, anche in considerazione dell’insufficienza gonadica, è indicata la gonadectomia.
Bibliografia
- Lindhardt Johansen M, et al. 45,X/46,XY mosaicism: phenotypic characteristics, growth, and reproductive function – a retrospective longitudinal study. J Clin Endocrinol Metab 2012, 97: E1540-9.
- McCann-Crosby B, Mansouri R, et al. State of the art review in gonadal dysgenesis: challenges in diagnosis and management. Int J Pediatr Endocrinol 2014, 2014: 4.
- Martinerie L, Morel Y, Gay CL, et al. Impaired puberty, fertility, and final stature in 45,X/46,XY mixed gonadal dysgenetic patients raised as boys. Eur J Endocrinol 2012, 166: 687-94.
- Richter-Unruh A, Knauer-Fischer S, Kasper S, et al. Short stature in children with an apparently normal male phenotype can be caused by 45,X/46,XY mosaicism and is susceptible to growth hormone treatment. Eur J Pediatr 2004, 163: 251–6.
46,XY DSD
Disordini nello sviluppo del testicolo
Disordini produzione o azione degli androgeni
- mutazioni di StAR, 20-22 desmolasi
- deficit di SF1
- deficit di 17βOH-steroido-DH
- deficit di 5-alfa-reduttasi
- insensibilità periferica agli androgeni
Disordini da alterata secrezione o azione dell’AMH
| Tabella 1 Diagnosi differenziale dei principali 46,XY DSD (senza deficit surrenalico) |
|||||
| Deficit di SF1^ | Ipoplasia del Leydig | Deficit di 5α-reduttasi | Insensibilità agli androgeni | ||
| Prevalenza | ? | ? | 1: 147.000* | ? | 1: 20.000/1: 99.000 |
| Ereditarietà | AD (AR) | AR | AR | AR | X-linked recessiva |
| Gene mutato | NR5A1 | LHGCR | 17β-HSD3 | SRD5A2 | Recettore androgeni |
| Cromosoma | 9q33.3 | 2p21 | 9q22 | 2p23 | Xq11-12 |
| N. mutazioni | ~30 | - | ~40 | ~50 | > 500 |
| Patogenesi | Alterato sviluppo del testicolo/ alterazione della steroido-genesi | Alterazione recettore per LH (mancata risposta a LH/hCG) | Alterata conversione di Δ4-A in T | Alterata conversione di T in DHT | Mancata risposta (completa o parziale) dei tessuti bersaglio agli androgeni |
| Gonadi | Streak o testicoli (variamente sviluppati) | Testicoli intra- o extra-addominali* | Testicoli extra- addominali (~90%) | Testicoli extra- addominali (~90%) | Testicoli intra-addominali (~70%) |
| Fenotipo alla nascita | Da femminile a maschile | Femminile o ambiguo | Prevalentemente femminile | Femminile o ambiguo | Da femminile a maschile |
| Vagina | Presente | Presente (70-80%) | Presente (80%) | Presente (50%) | Presente (maggioranza) |
| Strutture wolffiane | Da assenti a presenti | Maschili (ipoplasiche) | Usualmente maschili | Usualmente maschili | Assenti o maschili (ipoplasiche) |
| Strutture mülleriane | Presenti (~30-50%) | Assenti | Assenti | Assenti | Assenti (o rudimentali) |
| Fenotipo alla pubertà | Infantilismo sessuale/ virilizzazione | Infantilismo sessuale/ virilizzazione | Virilizzazione | Virilizzazione | Da femminilizzazione a virilizzazione |
| Sviluppo mammario | Assente o minimo | Assente | Variabile | Assente | Presente |
| Peluria androgenica | Variabile | Variabile | Maschile normale | Maschile normale | Assente o scarsa |
| Androgeniz-zazione SNC | Variabile | Variabile | Variabile | Usualmente presente | Variabile |
| Profilo steroideo | Dimin variabile di tutti gli androgeni | Dimin variabile di tutti gli androgeni | Δ4-A aument, T dimin, T/Δ4-A dimin | T N o aument, DHT dimin, T/DHT aument | Δ4-A N, T N o aument, DHT N o dimin, E2 N o aument |
| Risposta hCG test | Incremento assente o subnormale di tutti gli androgeni | Incremento assente o subnormale di tutti gli androgeni | Incremento Δ4-A > T (rapporto T/Δ4-A < 0.8-1.0) | Normale incremento T con dimin DHT | Incremento di tutti gli androgeni |
| AMH/inibina B | Dimin o normale | Normale o dimin | Normale | Normale | Normale o aument |
| Cambiamento sesso | Raro | Assente | Presente (30-50%) | Presente (~75%) | Usualmente non presente |
^possibile deficit surrenalico nelle forme in omozigosi (rare)
*1:300 - 1:150 maschi nella popolazione araba della striscia di Gaza; relativamente frequente nella Repubblica Domenicana, in Libano e Nuova Guinea
AD = autosomica dominante; AR = autosomica recessiva
T = testosterone; Δ4-A = D4-Androstenedione, DHT = diidrotestosterone
N = normale; Aument = aumentato, Dimin = ridotto
Disgenesia gonadica completa e parziale
Regressione testicolare (agenesia)
Paolo Ghirri1, Francesca Dini2, Nella Augusta Greggio2, Maria Carolina Salerno3
1UO Neonatologia, AOU Pisana, Pisa, Italy; 2UOS di Endocrinologia Pediatrica e Adolescentologia, DAIS per la Salute della Donna e del Bambino, AO Università di Padova; 3Sezione Pediatrica, Dipartimento di Scienze Mediche Traslazionali, Università Federico II, Napoli
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
La sindrome da regressione testicolare (TRS) (o “vanishing testis syndrome” nella letteratura urologica) è una condizione clinica caratterizzata dall’assenza o dall’incompleto sviluppo di un testicolo in pazienti con cariotipo 46,XY e genitali esterni generalmente maschili normali. Si considera che sia generalmente dovuta all’atrofia e successiva scomparsa, durante la vita fetale, di un testicolo inizialmente normale (1) ed è caratterizzata dalla presenza di un funicolo spermatico rudimentale in assenza di tessuto testicolare macroscopicamente visibile (2,3).
Interessa meno del 5% dei casi di criptorchidismo, ma è stata diagnosticata nel 35-60% dei casi con testicoli non palpabili (1, 4-6).
Eziologia
La TRS sembra essere determinata principalmente da fattori meccanici, come torsioni o infarti testicolari, avvenuti durante la vita fetale o perinatale; meno frequenti sembrano invece le patologie genetiche ed endocrine (7,8). A sostegno di tale ipotesi è stato analizzato il tessuto testicolare del testicolo contro-laterale in soggetti con vanishing testis monolaterale (9), non riscontrando alcuna anomalia, né un basso numero di cellule germinali.
Tra i fattori di rischio predisponenti, è stata evidenziata una correlazione tra incompleta discesa dei testicoli e torsione durante la vita intra-uterina e tra traumi a carico dei testicoli localizzati in sede scrotale e danno infartuale. Ciò giustificherebbe il maggiore coinvolgimento del testicolo sinistro, per la sua più precoce discesa rispetto al destro (tab).
La TRS è stata osservata anche in associazione ad anomalie genetiche, come micro-delezioni del cromosoma Y e persistenza dei dotti di Mülleriani. Inoltre, una regressione testicolare può essere parte dello spettro clinico della disgenesia gonadica parziale 46, XY, in cui i pazienti presentano genitali esterni malformati o micropene in associazione alla regressione completa di uno od entrambi i testicoli (4,10,11). Comunque nella maggior parte dei casi la TRS appare come una condizione sporadica e i pazienti risultano per il resto normali, con storia familiare silente.
Clinica
La regressione testicolare è per lo più unilaterale, con parziale o completa assenza di tessuto testicolare e per lo più genitali esterni maschili normali, sebbene più raramente si possano osservare spettri fenotipici variabili, dipendenti dall’entità e dal periodo prenatale in cui si sviluppa il danno testicolare in relazione allo sviluppo sessuale (8). Se la regressione testicolare fetale si realizza precocemente, tra l'8° e la 10° settimana di gestazione, i pazienti possono avere genitali esterni quasi femminili, con utero ipoplasico e dotti genitali rudimentali. La regressione che avviene dopo la 12°-14° settimana, invece, si associa a fenotipo maschile normale, con anorchia o gonadi atrofiche (testicoli rudimentali). I fenotipi intermedi si manifestano con genitali esterni parzialmente virilizzati e sviluppo variabile dei genitali interni (12).
Diagnosi e caratteristiche isto-patologiche
In presenza di un testicolo criptorchide non palpabile è indicata una valutazione ecografica e, nel caso di mancata visualizzazione del testicolo, un’esplorazione laparoscopica. La diagnosi di TRS in genere si basa sulla visualizzazione di elementi del funicolo spermatico (arterie testicolari, plesso pampiniforme, nervi e vasi deferenti) che terminano a fondo cieco, di solito con un piccolo nodulo fibroso, nel retro-peritoneo o vicino all’anello inguinale interno. Fanno parte del quadro istopatologico della TRS la presenza di fibrosi, calcificazioni, depositi di emosiderina, in associazione con strutture testicolari e para-testicolari riconoscibili (8,12-14).
La valutazione dei livelli plasmatici di ormone anti-mulleriano (AMH), inibina B e testosterone prima e dopo somministrazione di hCG per valutare la presenza di tessuto testicolare (cellule del Sertoli e cellule di Leydig) potrà essere riservata ai casi con entrambi i testicoli non palpabili e non visualizzati all’esame ecografico.
All’interno del residuo testicolare si possono trovare con prevalenza molto variabile (0-24% dei casi)(tab) residui dei tubuli seminiferi e delle cellule germinali, che, di per sé, determinano un maggior rischio di degenerazione maligna (8). Questo rischio sembra inoltre essere associato alla localizzazione del residuo testicolare: quelli intra-addominali hanno un rischio 6 volte maggiore rispetto a quelli localizzati in sede inguinale (5).
| Riepilogo identificazione e valutazione istologica dei residui testicolari | ||||||
| Ref | N° pazienti | N° abbozzi testicolari esaminati | Età media (range) | Tubuli seminiferi (%) | Cellule germinali (%) | Dx/Sin |
| Nataraja 2015 | 140 | 140 | 0.2-17 | 24 | 10 | 48/92 |
| Dhandore 2014 | 2 | 2 | 6-11 | 0 | 0 | 2/1 |
| Antic 2011 | 29 | 30 | 1-13 | 40 | 0 | 14/16 |
| Bader 2010 | 206 | 208 | 1.4-3 | 15 | 11 | 47/159 |
| Hegarty 2007 | 117 | 25 | 0-12 | 0 | 0 | -/- |
| Belman 2007 | 54 | 52 | 0-12 | 3 | 0 | 18/36 |
| Cendron 1998 | 29 | 29 | 2-4 | 0 | 0 | -/- |
Terapia
L’approccio terapeutico al residuo testicolare è molto controverso, principalmente per la variabile prevalenza, riportata in letteratura, di cellule germinali nel residuo testicolare. I due studi con le casistiche più ampie (2,15), 346 casi complessivamente, hanno messo in evidenza la presenza di cellule germinali nel 10-11% dei casi. Questo dato, insieme al fatto che l’escissione del residuo testicolare non comporta un allungamento dei tempi chirurgici, indica come possa essere preferibile l’asportazione dei residui testicolari per prevenire ogni rischio potenziale di malignità. L’asportazione del residuo testicolare potrà essere fatta anche successivamente, al momento del posizionamento della protesi testicolare. Molti autori inoltre raccomandano la fissazione del testicolo contro-laterale per prevenire eventuali torsioni (8).
Bibliografia
- Hegarty PK, Mushtaq I, Sebire NJ. Natural history of testicular regression syndrome and consequences for clinical management. J Pediatr Urol 2007, 3: 206-8.
- Bader MI, Peeraully R, Ba’ath M, et al. The testicular regression syndrome – do remnant require routine excision? J Pediatric Surg 2011, 46: 384-6.
- Spires SE, Woolums C, Pulito AR, Spires SM. Testicular regression syndrome. A clinical and pathologic study of 11 cases. Arch Pathol Lab Med 2000, 124: 694-8.
- Philibert P, Zenaty D, Audran F, et al. Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateral anorchia: a French collaborative study. Hum Reprod 2007, 22: 3255–61.
- Emir H, Ayik B, Elicevik M, et al. Histological evaluation of the testicular nubbins in patients with nonpalpable testis: assessment of etiology and surgical approach. Pediatr Surg Int 2007, 23: 41–4.
- Vinci G, Anjot MN, Trivin C, et al. An analysis of the genetic factors involved in testicular descent in a cohort of 14 male patients with anorchia. J Clin Endocrinol Metab 2004, 89: 6282-5.
- Rozanski TA, Wojno KJ, Bloom DA. The remnant orchiectomy. J Urol 1996, 155: 712-4.
- Pirgon Ö, Dündar BN. Vanishing testes: a literature review. J Clin Pediatr Endocrinol 2012, 4: 116-20.
- Huff DS, Wu HY, Snyder HM 3rd, et al. Evidence in favor of the mechanical (intrauterin torsion) theory over the endocrinopathy (cryptorchidism) theory in the pathogenesis of testicular agenesis. J Urol 1991, 146: 630–1.
- Berkovitz GD. Clinical and pathologic spectrum of 46, XY gonadal dysgenesia: its relevance to the understanding of sex differentiation. Medicine (Baltimore) 1991, 70: 375-83.
- Parigi GB, Bardoni B, Avoltini V, et al. Is bilateral congenital anorchia genetically determined? Eur J Pediatr Surg 1999, 9: 312-5.
- Sevin C. Sindrome da regressione testicolare. Orphanet Febbraio 2005. OMIN 273250.
- Aynsley-Green A, Zachmann M, Illig R, et al. Congenital bilateral anorchia in childhood: a clinical, endocrine and therapeutic evaluation of twenty-one cases. Clin Endocrinol (Oxf) 1976, 5: 381-91.
- De Freitas Vinhasa C, Felipe-Silvab A, Da Rochac RFC. Testicular regression syndrome: a case report. Autopsy and Case Reports 2012, 2: 65-8.
- Nataraja RM, Asher CM, Nash R, Murphy FL. Is routine exicision of testicular remnants in testicular regression syndrome indicated? J Pediatr Urol 2015, 11: 151.e1-5.
- Dhandore P, Hombalkar NN, Gurav PD, Ahmed MH. Vanishing testis syndrome: report of two cases. J Clin Diagn Res 2014, 8: ND03-4.
- Cendron M, Schned AR, Ellsworth PI. Histological evaluation of the testicular nubbin in the vanishing testis syndrome. J Urol 1998, 160: 1161–2.
Deficit di SF1
Lilia Baldazzi1, Rita Ortolano2, Soara Menabò1, Antonio Balsamo2
1Laboratorio di Genetica Molecolare, Dipartimento Salute della Donna, del Bambino e dell’Adolescente, Programma Endocrinologia Pediatrica, AOU S. Orsola-Malpighi, Bologna; 2Dipartimento di Scienze Mediche e Chirurgiche, UO Pediatria, Programma di Endocrinologia, AOU Bologna
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Lo Steroidogenic Factor 1 (SF-1), codificato dal gene NR5A1 sul cromosoma 9q33, è uno dei principali regolatori trascrizionali dello sviluppo surrenale/gonadico, della funzione riproduttiva, dell’espressione tessuto-specifica degli enzimi steroidogenici e del metabolismo energetico nei mammiferi; è espresso anche durante lo sviluppo dell’ipotalamo ventro-mediale, delle cellule gonadotrope dell’ipofisi e della milza (1,2).
Presente nella gonade indifferenziata, SF1 mostra un pattern di espressione dimorfico durante lo sviluppo testicolare o ovarico: persiste nel primo, dove stimola l’espressione di SOX9/AMH nelle cellule del Sertoli e degli enzimi steroidogenici nelle cellule del Leydig; sparisce nelle ovaie per riapparire all’inizio della follicologenesi ed esprimersi nell’ovaio adulto nelle cellule della granulosa, della teca e del corpo luteo (3). Nella specie umana il gene NR5A1 venne inizialmente studiato nei 46,XY DSD con disgenesia gonadica (GD) e le prime mutazioni furono descritte in casi con GD e insufficienza surrenale (4). Successivamente diverse mutazioni di NR5A1 sono state associate ad un ampio spettro fenotipico di casi 46,XY DSD con/senza insufficienza surrenalica, che varia da fenotipi PAIS-like (spesso femminile alla nascita con clitoride ipertrofico e segni suggestivi quali seno uro-genitale ed eventuale virilizzazione progressiva), a forme lievi di disgenesia gonadica e alterata androgenizzazione, a forme di ipospadia grave, sino a maschi fenotipicamente “normali”, ma sterili (5).
Nelle donne 46,XX il deficit di SF1 si può associare a Premature ovarian failure (POF), confermando il ruolo di questo fattore nella follicologenesi ovarica (6).
L’inquadramento diagnostico dei pazienti con sospetto deficit di SF1 include l’anamnesi (familiare, gravidica e personale) e la ricerca di eventuali segni dismorfici. All’esame obiettivo dei genitali esterni, oltre all’ispezione, è importante la ricerca di eventuali gonadi palpabili e l’attribuzione dell’external masculinization score (EMS) per il grado di ipovirilizzazione (tab).
| Punteggio EMS (mascolinizzazione esterna) (15) | |||||
| Score | Fusione scrotale | Micropene | Meato uretrale | Gonade destra | Gonade sinistra |
| 3 | Sì | No | Normale | ||
| 2 | Glande | ||||
| 1.5 | Scroto | Scroto | |||
| 1 | Pene | Inguine | Inguine | ||
| 0.5 | Addome | Addome | |||
| 0 | No | Sì | Perineo | Assente | Assente |
Punteggio: > 11 = virilizzazione normale; da 0 a 11 ipovirilizzazione
Contestualmente dovrebbero essere eseguiti:
- ecografia pelvica per la valutazione dei genitali interni con eventuali residui Mülleriani (le gonadi sono difficilmente visualizzabili);
- cariotipo e PCR per SRY;
- dosaggio di testosterone, cortisolo, Δ4-androstenedione, FSH/LH, AMH, inibina B (androgeni generalmente ridotti, AMH e inibina B ridotti/normali, cortisolo ridotto se c’è insufficienza surrenalica);
- test di funzionalità gonadica (hCG test) per verificare la funzione delle cellule di Leydig (la risposta può essere normale/subnormale o assente).
Gli esami di secondo livello includono:
- cisto-uretrografia minzionale per l’eventuale presenza di seno uro-genitale;
- RM per migliorare la visualizzazione di gonadi/genitali interni;
- eventuale laparoscopia esplorativa e biopsia gonadica (tubuli seminiferi ipoplasici, rari spermatogoni, cellule di Leydig con citoplasma vacuolare);
- test genetici: sequenziamento di NR5A1 e MLPA (identifica delezioni/duplicazioni in eterozigosi anche di altri geni per GD inclusi nel kit); eventuale sequenziamento dei geni SR5A2 ed AR in assenza di GD.
La prevalenza di mutazioni NR5A1 nei 46, XY DSD non CAIS varia dal 5 al 20%. Le mutazioni identificate includono tutte le tipologie (da missenso a larghe delezioni), distribuite su tutto il gene (5) e riscontrate prevalentemente in eterozigosi. In diversi studi si è confermato che l’aplo-insufficienza di SF1 è, nella maggior parte dei casi, sufficiente per alterare lo sviluppo genitale dei feti 46, XY (7) e rendere le donne 46,XX a rischio di POF (alcune mamme portatrici presentano poli-abortività). Il 90% circa delle mutazioni ha origine de novo o materna, ma sono descritti anche casi di ereditarietà paterna (penetranza incompleta/mosaicismo non identificato), per cui la ricerca delle mutazioni identificate è da effettuarsi in entrambi i genitori. Sono rare famiglie con alleli a bassa penetranza, in cui il fenotipo si manifesta solo nei probandi omozigoti e non nei genitori portatori. Le mutazioni di NR5A1 mostrano perciò espressività fenotipica complessa con diversa penetranza, che non consente di stabilire una correlazione genotipo-fenotipo diretta (8). Una migliore comprensione del meccanismo patogenetico di SF1 potrà venire dall’identificazione sia di nuovi geni target (9) sia della regolazione epigenetica (10), nonché dalle evidenze che il legame con specie chimicamente distinte di fosfolipidi gli consente di essere accoppiato/disaccoppiato al signaling di diverse molecole (11).
Il management dei pazienti 46,XY con mutazione del gene NR5A1 risulta complessa, in quanto in passato molti pazienti, considerati PAIS nonostante gene AR normale, venivano gonadectomizzati ed allevati come femmine, per cui sono esigui i dati sullo sviluppo della funzionalità gonadica nonché sul rischio tumorale. Tale rischio, intrinseco alla GD, è maggiore in gonadi ritenute in sede addominale e inguinale (5), pertanto è necessaria la valutazione dell’istologia gonadica con marcatori tumorali (OCT3/4; TSPY) (12) e un lungo follow-up.
L’assegnazione del sesso di allevamento è controversa e tiene conto, oltre al già citato rischio, di numerosi fattori, tra cui il potenziale sviluppo puberale, le problematiche chirurgiche, l’espressione dell’orientamento individuale e le aspettative dei genitori. Alcuni casi descritti allevati come maschi (13-15) hanno mostrato sufficiente produzione di testosterone con sviluppo puberale maschile normale, ma successivo ipogonadismo ipogonadotropo e oligo-azoospermia: è pertanto necessario monitorare la funzionalità dell’asse ipotalamo-ipofisi-gonadi. Per il rischio di insufficienza surrenalica è necessario inoltre eseguire un ACTH test.
Bibliografia
- Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994, 77: 481–90.
- Hoivik EA, Lewis AE, Aumo L, Bakke M. Molecular aspects of steroidogenic factor 1 (SF1). Mol Cell Endocrinol 2010, 315: 27-39.
- Buaas FW, Gardiner JR, Clayton S, et al. In vivo evidence for the crucial role of SF1 in steroid-producing cells of the testis, ovary and adrenal gland. Development 2012, 139: 4561-70.
- Achermann JC, Ito M, Ito M, et al. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nature Genet 1999, 22: 125-6.
- El-Khairi R, Achermann JC. Steroidogenic Factor-1 and human diseases. Semin Reprod Med 2012, 30: 374-81.
- Lourenco D, Brauner R, Lin L, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med 2009, 360: 1-11.
- Achermann J, Ozisik G, Ito M, et al. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor 1 in a dose-dependent manner. J Clin Endocrinol Metab 2002, 87: 1829-33.
- Ostrer H. Disorders of sex development (DSDs). An update. J Clin Endocrinol Metab 2014, 99: 1503-9.
- Mizutani T, Kawabe S, Ishikane S, et al. Identification of novel steroidogenic factor 1 (SF1)-target genes and components of the SF1 nuclear complex. Mol Cell Endocrinol 2015, 408: 133-7.
- Hoivik EA, Bjanesoy TE, Bakke M. Epigenetic regulation of the gene encoding steroidogenic factor-1. Mol Cell Endocrinol 2013, 371: 133-9.
- Blind RD. Disentangling biological signaling networks by dynamic coupling of signaling lipids to modifying enzymes. Adv Biol Regul 2014, 54: 25-38.
- Loojienga LHJ, Stoop H, Biermann K. Testicular cancer: biology and biomarkers. Virchows Arch 2014, 464: 301–13.
- Tantawy S, Lin L, Akkurt I, et al. Testosterone production during puberty in two 46,XY patients with disorders of sex development and novel NR5A1 (SF-1) mutations. Eur J Endocrinol 2012, 167: 125–30.
- Ciaccio M, Costanzo M, Guercio G, et al. Preserved fertility in a patient with a 46,XY disorder of sex development due to a new heterozygous mutation in the NR5A1/SF-1 gene: evidence of 46,XY and 46,XX gonadal dysgenesis phenotype variability in multiple members of an affected kindred. Horm Res Paediatr 2012, 78: 119–26.
- Philibert P, Polak M, Colmenares A, et al. Predominant Sertoli cell deficiency in a 46,XY disorders of sex development patient with a new NR5A1/SF-1 mutation transmitted by unaffected father. Fertil Steril 2011, 95: 1788.e5-9.
- Ahmed SF, Khwaja O, Hughes IA. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int 2000, 85: 120-4.
Deficit di 17β-idrossisteroido-deidrogenasi tipo 3
Silvano Bertelloni1, Antonio Balsamo2
1Dipartimento Materno-infantile, UO Pediatria Universitaria, AOU Pisana, Pisa; 2Dipartimento di Scienze Mediche e Chirurgiche, UO Pediatria, Programma di Endocrinologia, AOU di Bologna, Bologna
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Il deficit di 17β-idrossisteroido-deidrogenasi tipo 3 [17β-HSD3 (OMIM n. 264300), in passato riportato anche come deficit di 17-chetosteroido-reduttasi] viene trasmesso con modalità autosomica recessiva e rappresenta il più frequente dei 46,XY DSD da alterata sintesi del testosterone (1). Il deficit enzimatico altera infatti il passaggio critico da Δ4-androstenedione a testosterone (figura).
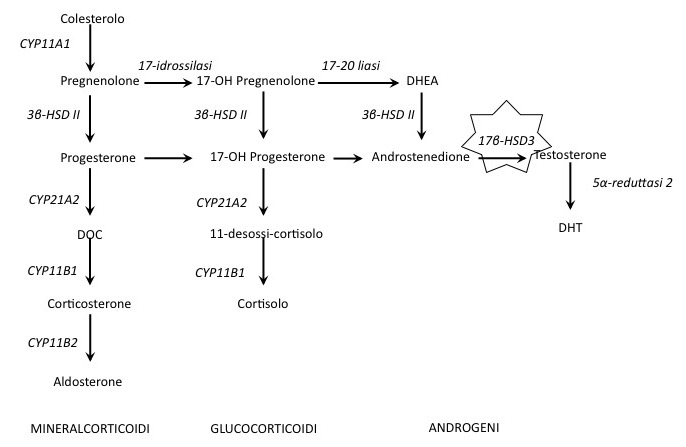
Via biosintetica degli androgeni, con deficit di 17βHSD-3.
La produzione di androgeni è controllata da vari enzimi: 17βHSD-3 converte il Δ4-androstenedione in testosterone con un processo di riduzione (1)
Sebbene l’esatta frequenza del deficit di 17β-HSD3 non sia nota, è stata descritta una diversa incidenza in rapporto alle varie popolazioni: in Olanda, è molto raro (incidenza stimata ~1: 147.000), mentre nelle regioni con alto livello di consanguineità risulta relativamente frequente (es. popolazioni araba della striscia di Gaza: ~1: 100-300). Non sono rari errori diagnostici, in quanto persone con questo DSD vengono spesso diagnosticate come affette da AIS (2).
Clinica
Alla nascita, i neonati presentano usualmente un fenotipo esterno femminile o modesta clitorido-megalia e fusione posteriore delle grandi labbra; i testicoli sono endo-addominali o a volte localizzati nelle grandi labbra. Date le caratteristiche fenotipiche, usualmente viene assegnato un sesso femminile. Meno frequentemente, si ha un’evidente ambiguità dei genitali con ipospadia più o meno importante e/o micropene.
Alla pubertà – nelle bambine non diagnosticate e con gonadi intatte - si ha usualmente una virilizzazione del fenotipo (crescita di peluria androgeno-dipendente, abbassamento del timbro della voce, crescita delle masse muscolari di tipo maschile, incremento delle dimensioni del clitoride fino a 5-10 cm) in conseguenza di aumento dei livelli di testosterone prodotto probabilmente da altri isoenzimi sotto lo stimolo dell’LH. Tuttavia, le caratteristiche fenotipiche sono non specifiche, risultando largamente sovrapponibili con quelle di altri 46,XY DSD da alterata sintesi o azione degli androgeni.
Dopo la pubertà, si può avere un cambiamento di sesso da femminile a maschile (tale evento è riportato maggiormente nei soggetti che vivono nelle aree ad alta frequenza di questo DSD) (tabella 1).
| Tabella 1 Deficit di 17β-HSD3: sesso assegnato alla nascita e successive ri-assegnazioni in regioni ad alta incidenza e nei paesi occidentali |
||||
| Epoca della vita | Sesso assegnato | |||
| Striscia di Gaza | Paesi Occidentali | |||
| Maschile | Femminile | Maschile | Femminile | |
| Alla nascita | 7 | 10 | 2 | 29 |
| Riassegnato in prepubertà | 15 | 2 | 0 | 0 |
| Riassegnato dopo pubertà | 11 | 0 | 3 | 0 |
| Totale | 33 | 12 | 5 | 29 |
Diagnosi
Dal punto di vista endocrino, ridotti livelli di testosterone con incremento dei valori di D4-androstenedione determinano un rapporto tra i due steroidi < 0.8, che permette la diagnosi. In età prepubere, deve essere effettuato un hCG test per la bassa sensibilità dei valori basali (tabella 2).
| Tabella 2 Deficit di 17β-HSD3: rapporto T/Δ4-A (cut-off 0.8) in rapporto all’età |
||||||
| Età | < 6 mesi | 1-10 anni | 11-18 anni | |||
| basale | picco | basale | picco | basale | picco | |
| Soggetti (n) | 9 | 9 | 7 | 20 | 24 | 14 |
| Sensibilità (%) | 100 | 89 | 57 | 90 | 92 | 93 |
L’analisi molecolare del gene HSD17/B3 permette la diagnosi di certezza. Sono state riportate in letteratura circa 30 mutazioni, senza una chiara relazione genotipo-fenotipo (2).
Trattamento
Per un trattamento ottimale, è indispensabile una diagnosi di certezza. In linea generale, una diagnosi molto precoce può indirizzare verso un’assegnazione di sesso di tipo maschile (3). Le bambine con fenotipo quasi o completamente femminile possono essere allevate come femmine, anche per la probabile mancata androgenizzazione del sistema nervoso centrale, dati i livelli molto bassi di testosterone prima della pubertà.
Nelle bambine con diagnosi molto tardiva in epoca puberale, la decisione finale di assegnazione del sesso richiede un percorso molto complesso anche dal punto di vista psicologico. Nei soggetti con maggiore virilizzazione, è indicata l’assegnazione al sesso maschile. Alla pubertà, si dovrà effettuare, se necessario, un trattamento sostitutivo con steroidi sessuali.
Bibliografia
- George MM, New MI, Ten S, et al. The clinical and molecular heterogeneity of 17-HSD-3 enzyme deficiency. Horm Res Paediatr 2010, 74: 229–40.
- Bertelloni S, Balsamo A, Giordani L, et al. 17-Hydroxysteroid dehydrogenase-3 deficiency: from pregnancy to adolescence. J Endocrinol Invest 2009, 32: 666-70.
- Jürgensen M, Hampel E, Hiort O, Thyen U.“Any decision is better than none” Decision-making about sex of rearing for siblings with 17â-Hydroxysteroid-dehydrogenase-3 deficiency. Arch Sex Behav 2006, 35: 358–70.