Overview sugli incidentalomi surrenalici

Giuseppe Reimondo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
Le masse surrenaliche sono tumori ad alta prevalenza e sono per lo più di riscontro occasionale, da cui la definizione di incidentalomi surrenalici. Negli studi radiologici la frequenza stimata è di circa il 4%, con un incremento fino al 10% nella popolazione anziana. Sebbene la lesione di più frequente riscontro sia l’adenoma, è clinicamente rilevante considerare sempre la possibilità che il riscontro occasionale di una massa surrenalica sia di natura maligna primitiva o secondaria.
Dal punto di vista diagnostico radiologico la TC e la RM mirate alle lesioni surrenaliche presentano un’accuratezza diagnostica sovrapponibile. La valutazione integrata delle dimensioni della lesione (< 40 mm), della densità sulla scansione basale (60% a 10 minuti dalla somministrazione del mezzo di contrasto alla TC, oppure la valutazione del segnale in opposizione di fase alla RM), permettono di effettuare una corretta diagnosi differenziale tra benigno e maligno nella quasi totalità dei casi.
Per quanto riguarda la diagnosi biochimica è clinicamente di primario rilievo escludere biochimicamente il sospetto di feocromocitoma e di sindrome di Cushing. Per quanto riguarda l’iperaldosteronismo, vi è concordanza fra le diverse linee guida che debba essere escluso solo nei pazienti con ipertensione e/o ipokaliemia.
Un capitolo a parte merita la sindrome di Cushing subclinica, la cui definizione prevede la presenza di uno stato ipercortisolemico in assenza di stigmate cliniche tipiche e altamente predittive di sindrome di Cushing manifesta. Nell’ambito dei pazienti con riscontro occasione di massa surrenalica, circa il 20% di essi presenta una sindrome di Cushing subclinica. Il valore clinico del riscontro di questa entità patologica risiede fondamentalmente nell’associazione con una maggiore frequenza di ipertensione, diabete, fratture vertebrali rispetto alla popolazione generale. Ancora oggi la vera difficoltà per questa sindrome è la definizione biochimica, sebbene la valutazione del cortisolo dopo soppressione con desametasone 1 mg rappresenti ormai l’indagine di riferimento. Nonostante non vi sia concordanza su questo argomento, la recente Position Statement AME ha adottato e raccomandato i seguenti criteri:
- valori di cortisolo dopo dex 1 mg inferiori a 1.8 µg/dL escludono la patologia;
- valori superiori a 5 µg/dL pongono un alto dubbio di sindrome di Cushing subclinica;
- valori intermedi sono considerati una condizione non definita, meritevole di successivi controlli e della valutazione di altri parametri, quali cortisoluria delle 24 ore e ACTH, in particolare in presenza di significative co-morbilità.
Follow-up
La tabella include le raccomandazioni delle linee guida.
| Raccomandazioni cliniche di position e consensus sulla gestione del paziente con incidentaloma surrenalico in corso di follow-up | |||||
| Follow-up | Valutazione ormonale | Imaging | |||
| Metodica | Frequenza | Metodica | Frequenza | ||
| NIH consensus conference (2002) | 4 anni | Dex 1 mg, metanefrine plasmatiche, PRA/aldosterone (se ipertensione o ipopotassiemia e solo alla diagnosi) | annuale | TC: monitoraggio per masse 60 mm | Ricontrollo a 6-12 mesi: non ulteriori controlli in assenza di incremento delle dimensioni |
| French Society of Endocrinology Consensus (2008) | 5 anni |
Dex 1 mg, metanefrine plasmatiche o urinarie (solo alla diagnosi), PRA/aldosterone (se ipertensione o ipopotassiemia e solo alla diagnosi) |
6 mesi, 2 anni e 5 anni | TC: monitoraggio per masse ≥ 40 mm | 6 mesi, 2 anni e 5 anni |
| AACE/AAES guidelines (2009) | 5 anni | Dex 1 mg + ACTH + DHEAS, metanefrine plasmatiche o urinarie, PRA/aldosterone (se ipertensione o ipopotassiemia) | annuale | TC: monitoraggio per masse ≥ 40 mm | 3-6 mesi, poi annualmente per 2 anni |
| AME position statement (2011) | Non determinato e da valutare su base individuale | 1 mg DST, metanefrine plasmatiche o urinarie, PRA/aldosterone (se ipertensione o ipopotassiemia) | non determinata e da valutare su base individuale | TC o RM | 3-6 mesi, ulteriori controlli su base individuale. Non raccomandato ricontrollo per masse < 20 mm e con HU < 10 |
Terapia
La scoperta occasionale di un carcinoma del corticosurrene, di un feocromocitoma o di un adenoma secernente cortisolo richiede come prima scelta terapeutica l’intervento chirurgico (prediligendo la via laparoscopica). Le lesioni che vengano etichettate radiologicamente compatibili con mielolipoma o cisti non richiedono alcun intervento e follow-up, a meno che non presentino dimensioni tali da determinare sintomatologia compressiva.
Le linee guida concordano anche nel candidare all’intervento chirurgico le masse surrenaliche con diametro superiore ai 40 mm, analogamente ad una lesione che presenta una crescita dimensionale maggiore di 10 mm al primo controllo di follow-up.
La figura propone un modello per la sindrome di Cushing subclinica, che tiene conto dei risultati fino ad ora disponibili e delle differenti posizioni a favore e contro l’indicazione chirurgica.
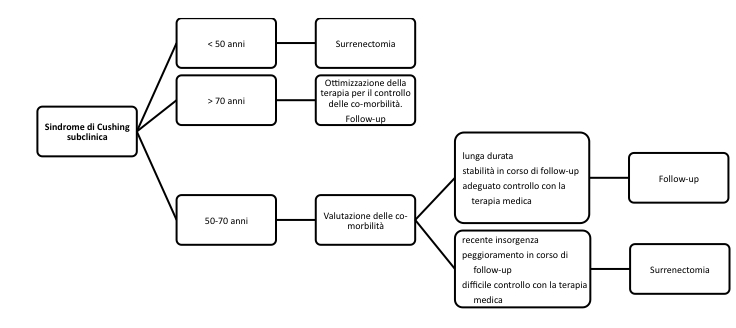
Proposta di gestione del paziente con sindrome di Cushing subclinica
Clinica e diagnostica incidentaloma surrenalico ed ipercortisolismo subclinico
Giuseppe Reimondo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
Le masse surrenaliche sono tumori ad alta prevalenza e sono per lo più di riscontro occasionale, da cui la definizione di incidentalomi surrenalici. L’orientamento comune, tuttavia, non è quello di considerare questa come la definizione di una sola entità, ma una sorta di comune denominatore di uno spettro di differenti patologie che presentano la stessa modalità di scoperta.
Epidemiologia
Negli studi radiologici la frequenza stimata è di circa il 4%, con un incremento fino al 10% nella popolazione anziana (1-5). Le frequenze delle diverse cause di incidentaloma surrenalico sono riportate nella tabella. Va sottolineato come sia clinicamente rilevante considerare la possibilità che il riscontro occasionale di una massa surrenalica sia di natura maligna, primitiva o secondaria.
| Frequenze dei differenti tipi di incidentaloma surrenalico | |||
| Tipo | Casistiche | ||
| mediche (%) | chirurgiche (%) | ||
| Adenoma | Totale | 80 | 55 |
| Non funzionante | 72 | 69 | |
| Cortisolo-secernente | 12 | 10 | |
| Aldosterone-secernente | 2.5 | 6 | |
| Feocromocitoma | 7 | 10 | |
| Carcinoma | 8 | 11 | |
| Metastasi | 5 | 7 | |
| Mielolipoma | - | 8 | |
| Cisti | - | 5 | |
| Ganglioneuroma | - | 4 | |
Diagnosi radiologica e FNAB
L’ecografia è una metodica per lo più operatore-dipendente, dal momento che non presenta la stessa sensibilità nella capacità di riscontrare masse surrenaliche rispetto a TC o RM (6, 7). Sono molto limitati gli studi che ne definiscono l’efficacia diagnostica, riportando una ottima sensibilità per masse surrenaliche di diametro > 30 mm (100%), ma non altrettanto accettabile per formazioni nodulari di dimensioni inferiori (circa 65%) (8). Peraltro, se da un lato non ha un ruolo nella differenziazione tra maligno e benigno, dall’altra rappresenta una buona tecnica alternativa nella valutazione in corso di follow-up (9).
La TC e la RM sono tecniche che permettono in ugual modo di distinguere le masse benigne dalle maligne.
Per quanto riguarda la TC, la tecnica di riferimento è quella a strato sottile, senza mezzo di contrasto. Se da un lato le dimensioni della lesione rappresentano un valido aiuto nell’ipotizzare un aumentato rischio di malignità della lesione (> 40 mm), più recenti studi hanno dimostrato come la misura della densità della lesione (indice di attenuazione espresso in unità Hounsfield, UH) abbia una maggiore accuratezza diagnostica, sebbene i due parametri possano essere considerati congiuntamente (10):
- valori di attenuazione ≤ 10 UH in condizioni basali hanno una sensibilità e una specificità elevata nel supportare l’ipotesi che la lesione sia benigna (10);
- valori > 10 UH sono considerati non conclusivi, poiché il 30% di queste lesioni sono adenomi a basso contenuto lipidico e richiedono un ulteriore approfondimento diagnostico (11, 12).
Un importante ausilio nella diagnosi differenziale è rappresentato dalla TC con mezzo di contrasto attraverso la valutazione della rapidità del wash-out:
- un wash-out assoluto ≥ 60% a 10 minuti risulta altamente suggestivo per adenoma;
- una percentuale di wash-out inferiore risulta sospetta per lesione maligna (13, 14).
Alla RM, in generale, gli adenomi appaiono ipo o iso-intensi in T1 rispetto al fegato e iper o iso–intensi in T2. La capacità di differenziazione fra lesioni benigne e maligne presenta la stessa accuratezza rispetto alla TC, in particolare con la valutazione della perdita di segnale in opposizione di fase (15, 16).
La scintigrafia con iodo-colesterolo di fatto non fornisce ulteriori caratterizzazioni alle lesioni (17). Recenti studi hanno invece valutato la PET-TC con FDG (fluorodesossiglucosio), dimostrando una buona sensibilità e specificità nel distinguere le lesioni. Non vi sono ancora precisi cut-off per quanto riguarda lo standardized uptake values (SUV), ma livelli < 1.5 sembrano essere altamente predittivi per una formazione benigna. La tecnica, tuttavia, al momento non è proponibile su ampia scala, per cui si la ritiene un valido ausilio per la valutazione delle lesioni che presentano caratteristiche dubbie a TC o RM (18-19). Verosimili ulteriori sviluppi potrebbero nascere con l’adozione di nuovi traccianti maggiormente specifici per la corteccia surrenalica.
Un cenno a parte deve essere fornito per quanto riguarda la FNAB delle lesioni surrenaliche. Tale tecnica nella patologia surrenalica ha un ruolo marginale, che va limitato a pazienti selezionati in cui vi sia il sospetto di lesioni metastatiche di tumori extra-surrenalici o nei rari casi in cui le valutazioni d’immagine risultino non conclusive (20). Prima di procedere alla manovra è indispensabile escludere biochimicamente la diagnosi di feocromocitoma (21).
Diagnosi biochimica
La differenziazione fra una lesione benigna o maligna è di indubbia rilevanza clinica e gli strumenti a nostra disposizione ci permettono di raggiungere l’obiettivo nella quasi totalità dei casi. Molto più controversa è la questione che riguarda la valutazione ormonale. Due sono gli elementi principali che rendono questo campo molto discusso:
- il paziente per definizione non presenta segni e sintomi tipici di patologie surrenaliche, ma viene valutato in considerazione del riscontro occasionale di una massa surrenalica;
- i test di valutazione ormonale sono gli stessi previsti nei soggetti con alta probabilità clinica di avere la malattia e che comunque, anche in questi casi, presentano un’accuratezza diagnostica non sempre adeguata.
È clinicamente di primario rilievo escludere biochimicamente il sospetto di feocromocitoma e di sindrome di Cushing in tutti i pazienti con riscontro occasionale di massa surrenalica. Per quanto riguarda l’iperaldosteronismo, vi è concordanza fra le diverse linee guida che debba essere escluso solo nei pazienti con ipertensione e/o ipokaliemia. Per le procedure relative all’esclusione di queste patologie si rimanda ai capitoli di pertinenza.
Sindrome di Cushing subclinica
Il concetto di sindrome di Cushing subclinica include nella definizione la presenza di uno stato ipercortisolemico in assenza di stigmate cliniche tipiche e altamente predittive di sindrome di Cushing manifesta (1, 22). E’ applicabile anche al riscontro di incidentaloma ipofisario e alla terapia steroidea, ma è più diffusamente studiata e osservata in pazienti con riscontro occasionale di massa surrenalica, che presentano un ipercortisolismo ACTH-indipendente. Certamente ciascuno dei criteri citati presenta dei limiti, in particolare per quanto riguarda i segni e i sintomi di riconoscimento dell’eccesso di cortisolo, che sono per lo più dipendenti dall’esperienza personale del clinico che valuta il paziente. Clinici meno esperti potrebbero includere o non evidenziare segni e sintomi tipici della sindrome di Cushing conclamata (23), mentre la vera definizione di subclinico prevede che siano evidenziabili (qualora presenti) solo segni e sintomi meno specifici per ipercortisolismo e più tipici della sindrome metabolica (ad es. obesità centrale, ipertensione). Se queste condizioni vengono rispettate, nell’ambito dei pazienti con riscontro occasione di massa surrenalica, circa il 20% di essi presenta una sindrome di Cushing subclinica. La percentuale non è irrilevante, dal momento che l’incidentaloma surrenalico è presente in circa il 4% dei soggetti di media età e in più del 10% dei soggetti anziani (1, 3, 4, 24). Il valore clinico del riscontro di questa entità patologica risiede fondamentalmente nell’associazione con una maggiore frequenza di ipertensione, diabete, osteoporosi (in particolare fratture vertebrali) e aumentato rischio cardiovascolare rispetto alla popolazione generale (25, 26). Un recente studio (27) ha confermato, su una casistica molto ampia, come vi sia un aumentato rischio di eventi (diabete, osteoporosi, eventi cardiovascolari) progressivamente maggiore con l’aumentare dell’entità dell’ipercortisolismo subclinico. Peraltro, è stato anche dimostrato come l’ipercortisolismo subclinico sia un fattore di rischio indipendente rispetto a tutti gli altri co-fattori presi in considerazione a seconda della patologia (27).
La definizione biochimica è ad oggi ancora oggetto di discussione e non trova totale accordo nemmeno nelle linee guida o position delle società scientifiche. La valutazione del cortisolo dopo test di soppressione con desametasone 1 mg (dex 1 mg) dispone del maggior numero di studi ed è quella considerata più affidabile, sebbene la comparazione con altre indagini diagnostiche risulti molto eterogenea e la sensibilità e specificità diagnostica vari molto. Permane il problema della definizion dei cut-off. Nei pazienti con incidentaloma surrenalico sia il National Institute of Health, State-of-the-Science conference panel (24), che le linee guida di AACE/AAES (28) hanno definito il sospetto di sindrome di Cushing subclinica nel caso di mancata soppressione dei livelli di cortisolo < 5 µg/dL. Opinione differente è stata espressa dalla Società Francese di Endocrinologia che utilizza un cut-off di 1.8 µg/dL, direttamente trasferito dalle linee guida dell’Endocrine Society per la sindrome di Cushing manifesta (29). Tale cut-off presenta certamente una ottima sensibilità, ma una assai ridotta specificità, aumentando il rischio di falsi positivi e di indagini diagnostiche non necessarie. La Position Statement AME di recente pubblicazione ha espresso una via più flessibile: valori di cortisolo dopo dex 1 mg 5 µg/dL pongono un alto dubbio di sindrome di Cushing subclinica, mentre valori intermedi proposti anche da autorevoli gruppi, non hanno sufficienti evidenze, per cui si ritiene una condizione non definita, meritevole di successivi controlli e della valutazione di altri parametri, quali la cortisoluria 24 ore e l'ACTH, in particolare in presenza di significative co-morbilità (30). La valutazione del cortisolo salivare a mezzanotte nella ricerca della sindrome di Cushing subclinica non ha al momento fornito le stessa accuratezza diagnostica ormai riconosciuta nella sindrome di Cushing manifesta, per cui non rappresenta un test diagnostico validato e da considerare di primo livello (31,32).
Bibliografia
- Kloos RT, Gross MD, Francis IR, et al. Incidentally discovered adrenal masses. Endocr Rev 1995, 16: 460–84.
- Benitah N, Yeh BM, Qayyum A, et al. Minor morphologic abnormalities of adrenal glands at CT: prognostic importance in patients with lung cancer. Radiology 2005, 235: 517–22.
- Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. Study group on adrenal tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 2000, 85: 637–44.
- Barzon L, Sonino N, Fallo F, et al. Prevalence and natural history of adrenal incidentalomas. Eur J Endocrinol 2003, 149: 273–85.
- Bovio S, Cataldi A, Reimondo G, et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest 2006, 29: 298–302.
- Suzuki K, Fujita K, Ushiyama T, et al. Efficacy of an ultrasonic surgical system for laparoscopic adrenalectomy. J Urol 1995, 154: 484–6.
- Abrams HL, Siegelman SS, Adams DF, et al. Computed tomography versus ultrasound of the adrenal gland: a prospective study. Radiology 1982, 143: 121–8.
- Suzuki Y, Sasagawa I, Suzuki H, et al. The role of ultrasonography in the detection of adrenal masses: comparison with computed tomography and magnetic resonance imaging. Int Urol Nephrol 2001, 32: 303–6.
- Fontana D, Porpiglia F, Destefanis P, et al. What is the role of ultrasonography in the follow up of adrenal incidentalomas? Urology 1999, 54: 612–6.
- Hamrahian AH, Ioachimescu AG, Remer EM, et al. Clinical utility of noncontrast computed tomography attenuation value (Hounsfield units) to differentiate adrenal adenomas/hyperplasias from nonadenomas: Cleveland Clinic experience. J Clin Endocrinol Metab 2005, 90: 871–7.
- Lee MJ, Hahn PF, Papanicolaou N, et al. Benign and malignant adrenal masses: CT distinction with attenuation coefficients, size, and observer analysis. Radiology 1991, 179: 415–8.
- Pena CS, Boland GW, Hahn PF, et al. Characterization of indeterminate (lipid-poor) adrenal masses: use of washout characteristics at contrast enhanced CT. Radiology 2000, 217: 798–802.
- Caoili EM, Korobkin M, Francis IR, et al. Adrenal masses: characterization with combined unenhanced and delayed enhanced CT. Radiology 2002, 222: 629–33.
- Szolar DH, Kammerhuber FH. Adrenal adenomas and nonadenomas: assessment of washout at delayed contrast enhanced CT. Radiology 1998, 207: 369–75.
- Outwater EK, Siegelman ES, Radecki PD, et al. Distinction between benign and malignant adrenal masses: value of T1-weighted chemical-shift MR imaging. AJR Amer J Roentgenol 1995, 165: 579–83.
- Israel GM, Korobkin M, Wang C, et al. Comparison of unenhanced CT and chemical shift MRI in evaluating lipid-rich adrenal adenomas. AJR Amer J Roentgenol 2004, 183: 215–9.
- Falke TH, Sandler MP. Classification of silent adrenal masses: time to get practical. J Nucl Med 1994, 35: 1152–4.
- Groussin L, Bonardel G, Silvera S, et al. 18F-Fluorodeoxyglucose positron emission tomography for the diagnosis of adrenocortical tumors: a prospective study in 77 operated patients. J Clin Endocrinol Metab 2009, 94: 1713–22.
- Nunes MN, Rault A, Teynie J, et al. 18F-FDG PET for the identification of adrenocortical carcinomas among indeterminate adrenal tumors at computed tomography scanning. World J Surg 2010, 34: 1506–10.
- Berland LL, Silverman SG, Gore RM, et al. Managing incidental findings on abdominal CT: white paper of the ACR incidental findings committee. J Am Coll Radiol 2010, 7: 754–73.
- Casola G, Nicolet V, vanSonnenberg E, et al. Unsuspected pheochromocytoma: risk of blood-pressure alterations during percutaneous adrenal biopsy. Radiology 1986, 159: 733–5.
- Reincke M. Subclinical Cushing’s syndrome. Endocrinol Metab Clin North Amer 2000, 29: 43–56.
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008, 93: 1526-40.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Int Med 2003, 138: 424–9.
- Chiodini I. Clinical review: Diagnosis and treatment of subclinical hypercortisolism. J Clin Endocrinol Metab 2011, 96: 1223-36.
- Terzolo M, Pia A, Alì A, et al. Adrenal incidentaloma: a new cause of the metabolic syndrome? J Clin Endocrinol Metab 2002, 87: 998-1003.
- Di Dalmazi G, Vicennati V, Rinaldi E, et al. Progressively increased patterns of subclinical cortisol hypersecretion in adrenal incidentalomas differently predict major metabolic and cardiovascular outcomes: a large cross-sectional study. Eur J Endocrinol 2012, 166: 669-77.
- Zeiger MA, Thompson GB, Duh QY, et al; American Association of Clinical Endocrinologists; American Association of Endocrine Surgeons. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract 2009, 15: 450-3.
- Tabarin A, Bardet S, Bertherat J, et al; French Society of Endocrinology Consensus. Exploration and management of adrenal incidentalomas. French Society of Endocrinology Consensus. Ann Endocrinol (Paris) 2008, 69: 487-500.
- Terzolo M, Stigliano A, Chiodini I, et al; Italian Association of Clinical Endocrinologists. AME position statement on adrenal incidentaloma. Eur J Endocrinol 2011, 164: 851–70.
- Nunes ML, Vattaut S, Corcuff JB, et al. Late-night salivary cortisol for diagnosis of overt and subclinical Cushing’s syndrome in hospitalized and ambulatory patients. J Clin Endocrinol Metab 2009, 94: 456–62.
- Masserini B, Morelli V, Bergamaschi S, et al. The limited role of midnight salivary cortisol levels in the diagnosis of subclinical hypercortisolism in patients with adrenal incidentaloma. Eur J Endocrinol 2009, 160: 87–92.
Contributo della genetica per lo studio dei surreni
Nora Albiger
Divisione di Endocrinologia, Dipartimento di Scienze Mediche e Chirurgiche, Università di Padova
(aggiornato al 25 novembre 2021)
Le lesioni surrenaliche benigne sono relativamente frequenti nella popolazione generale e comprendono uno spettro di diverse entità, la cui diagnosi dipende dall’aspetto macroscopico dei surreni e dal profilo di secrezione ormonale.
Le formazioni surrenaliche unilaterali sono frequenti. La maggior parte viene scoperta incidentalmente in studi di diagnostica per immagini. La prevalenza degli incidentalomi surrenalici va dall’1 al 7% della popolazione generale e aumenta con l’età (1). Gli incidentalomi che coinvolgono la corticale surrenalica sono nella maggior parte dei casi formazioni benigne, non secernenti, ma possono essere responsabili dell’ipersecrezione di cortisolo nel 5-47% dei casi e più raramente di aldosterone (1.6-3.3%).
Le lesioni surrenaliche bilaterali sono meno frequenti. Si possono distinguere due diverse entità, in relazione alle dimensioni e all’aspetto dei noduli:
- iperplasia surrenalica macro-nodulare bilaterale primaria (BPMAH per “primary bilateral macronodular adrenal hyperplasia”) caratterizzata dalla presenza di noduli > 10 mm;
- iperplasia surrenalica micro-nodulare (MiAH per “micronodular adrenal hyperplasia”), malattia rara nell’ambito della quale è più frequente la forma pigmentata primaria (PPNAD), caratterizzata da noduli < 10 mm, pigmentati, bilaterali e diffusi sulla corteccia surrenalica.
Nei tumori surrenalici, sono state descritte diverse alterazioni molecolari e cellulari. Fisiologicamente, nelle cellule della corticale del surrene l’ACTH si unisce al suo recettore, con conseguente attivazione della proteina Gs, che attiva a valle l’adenilato-ciclasi, con conseguente aumento della produzione di cAMP. La protein-chinasi A (PKA) è un tetramero, composto da due subunità catalitiche e 2 regolatorie. Il legame di 4 molecole di cAMP alle subunità regolatorie della PKA consente l’attivazione delle 2 subunità catalitiche, che fosforileranno diversi bersagli, responsabili dello stimolo alla trascrizione dei geni cAMP-dipendenti. Le fosfo-diesterasi coinvolte nella degradazione del cAMP agiscono come regolatori negativi (2).
Diversi meccanismi molecolari sono alla base della tumorigenesi surrenalica, molti dei quali coinvolgono geni onco-soppressori. Secondo l’ipotesi di Knudson, i tumori derivano dalla proliferazione clonale di una cellula a seguito di due distinti eventi mutazionali: perché si manifesti la malattia, devono verificarsi prima una mutazione germinale su uno dei due alleli di un gene onco-soppressore (che sarà ereditata) e poi una seconda mutazione a livello somatico sull’altro allele dello stesso gene (2).
Iperplasia surrenalica macro-nodulare bilaterale primaria (BPMAH)
PBMAH era inizialmente ritenuta una malattia sporadica, ma l’origine genetica è stata suggerita da diversi casi familiari con trasmissione autosomica dominante e sviluppo bilaterale delle lesioni. Inoltre, rari casi di PBMAH sono parte di quadri genetici più complessi, in cui la presenza delle alterazioni caratteristiche di ogni patologia contribuirà alla diagnosi finale. Ciò dimostra che la PBMAH è una malattia eterogenea, spesso associata a difetti in diversi geni (3). Il quadro di PBMAH può essere riscontrato in pazienti con diverse sindromi.
- MEN-1, che colpisce particolarmente le paratiroidi (80-100%), il pancreas (NET duodeno-pancreatici, 30-80%) e l’ipofisi (15-50%), causata da mutazioni inattivanti nel gene MEN1 (sul cromosoma 11q13), un onco-soppressore che codifica per la proteina menina. La frequenza di lesioni surrenaliche varia nei diversi studi, a seconda del metodo di screening utilizzato. Un’analisi retrospettiva multicentrica su 715 pazienti con MEN-1 segnalava ingrandimento surrenalico nel 20.4% dei casi.
- Leiomiomatosi ereditaria familiare e carcinoma delle cellule renali: si caratterizza per la presenza di leiomiomi cutanei multipli in diversi soggetti della stessa famiglia. La presentazione più frequente è con leiomiomi uterini e carcinoma renale. È una malattia autosomica dominante, provocata dalla presenza di mutazioni inattivanti del gene che codifica per fumarato-idratasi (sul cromosoma 1q43). La frequenza di lesioni surrenaliche è stimata del 7.8%, con forme multifocali e bilaterali nel 20% dei casi.
- Sindrome di McCune-Albright: caratterizzata dalla presenza di displasia fibrosa poli-ostotica, macchie cutanee caffè-latte e pubertà precoce. Nei primi anni di vita potrebbe verificarsi iperplasia surrenalica nodulare. In questi casi sono stati descritte mutazioni del gene GNAS, che codifica per la subunità alfa della proteina G, aumentando la produzione di cAMP.
- Poliposi adenomatosa familiare: i pazienti presentano poliposi colonica e predisposizione al cancro del colon e possono avere neoformazioni surrenaliche. È causata da mutazioni inattivanti del gene APC (sul cromosoma 5q22), un onco-soppressore che inibisce la via Wnt/β-catenina.
ARMC5 (Armadillo repeat containing 5) e PBMAH
Il sequenziamento diretto del DNA tumorale di pazienti con PBMAH sottoposti ad intervento chirurgico ha identificato la presenza di varianti del gene ARMC5 in 18/33 pazienti (4). In tutti i campioni di DNA tumorale erano presenti due alterazioni genetiche nel locus ARMC5, mentre il DNA leucocitario conteneva solo una delle 2 alterazioni identificate nel tumore, a conferma del ruolo di ARMC5 come onco-soppressore. Successivamente, altri studi nella PBMAH hanno confermato la frequente presenza di mutazioni di ARMC5, riportate in circa il 25% dei casi indice (5,6).
La funzione del gene ARMC5 non è ancora ben definita, anche se è noto che la sua sovra-espressione induce apoptosi in vitro e la sua inattivazione riduce la steroidogenesi.
Rispetto ai pazienti senza mutazioni, quelli con PBMAH che hanno mutazioni del gene ARMC5 presentano secrezione di cortisolo più marcata, con quadro clinico più grave, ghiandole surrenaliche più voluminose con un maggior numero di noduli (5,6).
Lo screening familiare dei parenti di primo grado, presumibilmente sani, dei casi indice di una coorte francese ha rivelato la presenza di mutazioni e iperplasia nodulare in diversi membri della famiglia (4). Questi risultati, insieme a quelli ottenuti in altre famiglie (7,8) suggeriscono l’esecuzione di test genetici precoci nei familiari dei pazienti affetti da PBMAH con mutazioni germinali di ARMC5, poiché la diagnosi precoce può evitare le complicanze della sindrome di Cushing clinica o subclinica.
Poiché ARMC5 è espresso in molti tessuti, è stata avanzata l’ipotesi di potenziali conseguenze proliferative delle mutazioni germinali in tessuti extra-surrenalici. Finora è stata trovata una variante somatica del gene ARMC5 in un meningioma intra-cranico di un membro di una famiglia con PBMAH e mutazioni del gene ARMC5. Successivamente, è stata riportata la presenza di meningiomi anche in altri membri di famiglie affette da PBMAH (9). Questi dati suggeriscono un ruolo di ARMC5 nella predisposizione allo sviluppo di meningiomi.
Sulla base di queste osservazioni, i portatori di mutazioni germinali di ARMC5 dovrebbero essere accuratamente monitorati per la presenza di lesioni surrenaliche funzionanti (cortisolo-secernenti) e meningiomi.
Espressione aberrante di recettori accoppiati alla proteina G in PBMAH
In alcuni casi di PBMAH la secrezione di cortisolo è in parte regolata dall’espressione aberrante di recettori accoppiati alla proteina G (GPCR). Questi GPCR portano all’attivazione della via cAMP/PKA, attivata dal recettore dell’ACTH, che porta alla trascrizione dei fattori della steroidogenesi. In circa l’80% dei pazienti con sindrome di Cushing da PBMAH sono stati identificati i GPCR per vasopressina, serotonina, LH, hCG, agonisti β-adrenergici, GIP-R (glucose-dependent insulinotropic polypeptide), glucagone e angiotensina II (i primi due sono più frequentemente coinvolti) (10). In metà di questi pazienti sono presenti risposte aberranti simultanee di cortisolo a diversi stimoli.
Nei pazienti con PBMAH e mutazioni del gene ARMC5 l’espressione aberrante di questi recettori è variabile. La forma GIP-dipendente è stata trovata solo in pazienti wild-type, a suggerire che mutazioni di ARMC5 possono essere associate a un particolare spettro di recettori illeciti.
Iperplasia surrenalica primitiva nodulare pigmentata (PPNAD)
È una rara forma d’ipercortisolismo ACTH-indipendente. Il 60% circa dei casi noti di PPNAD è espressione clinica del complesso di Carney, che comprende macchie cutanee pigmentate (lentiggini), mixomi cardiaci e cutanei, adenomi ipofisari GH- o PRL-secernenti, tumori a grandi cellule di Sertoli calcifici, noduli tiroidei benigni o maligni e schwannomi melanocitici psammomatosi (11).
Si trasmette con carattere autosomico dominante. Il gene responsabile PRKAR1A (sul cromosoma 17q22-24) è un potenziale onco-soppressore che codifica per la subunità regolatoria 1-alfa della PKA. Mutazioni inattivanti la subunità regolatoria della PKA causano un’attivazione costitutiva della via cAMP/PKA. Se l’ACTH stimola proliferazione delle cellule surrenaliche e secrezione di cortisolo, queste mutazioni possono causare tumorigenesi e ipercortisolismo (2,11).
Sono state identificate diverse mutazioni del gene PRKAR1A, ma solo per due è stata trovata un’alta prevalenza in uno studio di 353 pazienti con complesso di Carney e/o PPNAD: la c709-7del6 nell’introne 7, segnalata nei casi di PPNAD isolato, e la c.491-492delTG nell’esone 5, associata a mixomi cardiaci, lentiggini e tumori tiroidei. Mutazioni del gene PRKAR1A sono responsabili di più dei due terzi dei casi di complesso di Carney, del 37% delle forme sporadiche e del 60% delle forme familiari tipiche.
Nella patogenesi della PPNAD sono coinvolte anche mutazioni delle fosfo-diesterasi, in particolare di PDE11A e PDE8B. La prima probabilmente gioca un ruolo nello sviluppo della malattia per l’alterata degradazione di cAMP (2,12).
Mutazioni nella subunità catalitica della proteina PKA
Nel 2014 sono state descritte mutazioni somatiche attivanti del gene PRKACA, che codifica per la subunità catalitica alfa della proteina PKA. Quest’alterazione è stata identificata come causa frequente di adenomi surrenalici cortisolo-secernenti (circa 40% dei casi) (13). Non sono state descritte mutazioni germinali di questo gene nei pazienti con forme bilaterali. Tuttavia, in pazienti con PBMAH sono state descritte duplicazioni germinali in una regione del cromosoma 19 che include il locus PRKACA. Finora sono stati identificati solo 3 casi: una madre e suo figlio, che presentavano un quadro di PBMAH cortisolo-secernente diagnosticato, rispettivamente, nella 3° e 4° decade di vita, e un bambino di 2 anni operato per PBMAH (3).
Vedi anche:
Bibliografia
- Fassnacht M, Arlt W, Bancos I, et al. Management of adrenal incidentalomas: European Society of Endocrinology clinical practice guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016, 175: G1–34.
- Bonnet-Serrano F, Bertherat J. Genetics of tumors of the adrenal cortex. Endocr Relat Cancer 2018, 25: 131–52.
- Drougat L, Espiard S, Bertherat J. Genetics of primary bilateral macronodular adrenal hyperplasia: a model for early diagnosis of Cushing’s syndrome? Eur J Endocrinol 2015, 173: M121–31.
- Assiè G, Libè R, Espiard S, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Engl J Med 2013, 369: 2105-14.
- Albiger NM, Regazzo D, Rubin B, et al. A multicenter experience on the prevalence of ARMC5 mutations in patients with primary bilateral macronodular adrenal hyperplasia: from genetic characterization to clinical phenotype. Endocrine 2017, 55: 959-68.
- Faucz FR, Zilbermint M, Lodish MB, et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab 2014, 99: E1113–9.
- Alencar GA, Lerario AM, Nishi MY, et al. ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab 2014, 99: E1501-9.
- Gagliardi L, Schreiber AW, Hahn CN, et al. ARMC5 mutations are common in familial bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab 2014, 99: E1784-92.
- Elbet U, Trovato A, Kloth M, et al. Molecular and clinical evidence for an ARMC5 tumor syndrome; concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J Clin Endocrinol Metab 2015, 100: E119-28.
- El Ghorayeb N, Bourdeau I, Lacroix A. Multiple aberrant hormone receptors in Cushing’s syndrome. Eur J Endocrinol 2015, 173: M45–60.
- Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: an overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol 2014, 386: 85–91.
- Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009, 94: 2085–91.
- Di Dalmazi G, Kisker C, Calebiro D, et al. Novel somatic mutations in the catalytic subunit of the protein kinase A as a cause of adrenal Cushing’s syndrome: a European multicentric study. J Clin Endocrinol Metab 2014, 99: E2093-100.
Terapia degli incidentalomi surrenalici
Giuseppe Reimondo
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
Management del paziente con incidentaloma surrenalico
La scoperta occasionale di un carcinoma del corticosurrene o di un feocromocitoma richiede come prima scelta terapeutica l’intervento chirurgico di surrenectomia (prediligendo la via laparoscopica), sebbene in entrambi i casi anche la radicalità non garantisca la guarigione del paziente. Analogo approccio vale per la diagnosi di adenoma surrenalico cortisolo-secernente. Al contrario, le lesioni che vengano etichettatE radiologicamente come compatibili con mielolipoma o cisti non richiedono alcun intervento e follow-up, a meno che non presentino dimensioni tali da determinare sintomatologia compressiva (1-5).
Nella maggior parte dei casi, tuttavia, le masse surrenaliche di riscontro occasionale non vengono sottoposte ad alcun trattamento e sono semplicemente avviate a un programma di follow-up.
Molti sono i fattori che intervengono nella decisione di sottoporre il paziente all’intervento chirurgico:
- la presenza di co-morbilità che controindichi l’intervento o, in alternativa, co-morbilità potenzialmente migliorabili dalla rimozione della lesione surrenalica;
- le dimensioni della lesione alla diagnosi e il riscontro di un accrescimento in corso di follow-up;
- l’attività secernente della lesione.
La possibilità che una massa surrenalica di riscontro occasionale sia una lesione maligna aumenta all'aumentare delle dimensioni:
- < 40 mm circa il 2%;
- tra 40 e 60 mm il rischio sale al 6%;
- > 60 mm sale a circa il 25% (1).
Pertanto le linee guida concordano nel candidare all’intervento chirurgico le masse surrenaliche con diametro > 40 mm, analogamente a una lesione che presenta una crescita dimensionale > 10 mm al primo controllo di follow-up (1-4). In genere, per riconoscere masse surrenaliche maligne che sono a rapida crescita si pone indicazione a un controllo radiologico (TC o RM) entro 6 mesi dalla diagnosi. Possono essere escluse le lesioni con dimensioni < 20 mm e con ipodensità. La sorveglianza radiologica ha un ruolo soprattutto nei primi anni dalla diagnosi, periodo nel quale la possibilità che una lesione (anche benigna) cresca fra 10 e 20 mm è di circa il 5-20% e, in tal caso, si può riconsiderare la possibilità di inviare il paziente all’intervento chirurgico (1-4).
Per quanto riguarda la possibilità che una lesione diventi secernente, è assai rara e per lo più si tratta di pazienti che sviluppano una secrezione autonoma di cortisolo quando già alla diagnosi presentavano una secrezione parzialmente autonoma(30 mm (1-4).
Non è ancora definito quale sia l’ottimale periodo di follow-up, ma può essere ragionevole seguire il paziente annualmente per circa 3-5 anni, periodo nel quale si possono identificare i pazienti con eventuale incremento dimensionale della lesione o variazione del quadro secretivo. Per quanto riguarda il controllo radiologico, come indicato da un recente lavoro (6), occorre evitare un eccesso di valutazione che possa determinare nel paziente ansie ingiustificate e costi addizionali, oltre a indurre un rischio di neoplasia correlato alle esposizioni radiologiche, sovrapponibile a quello di presentare una evoluzione maligna della lesione in corso di follow-up. Considerando tali presupposti è ipotizzabile un solo controllo entro 6-12 mesi dalla diagnosi per lesioni < 20 mm e con densità ridotta < 10 HU.
Nella tabella sono riportate le raccomandazioni cliniche per la gestione del follow-up fornite dalle consensus e position, fino ad ora disponibili.
| Raccomandazioni cliniche di position e consensus sulla gestione del paziente con incidentaloma surrenalico in corso di follow-up | |||||
| Follow-up | Valutazione ormonale | Imaging | |||
| Metodica | Frequenza | Metodica | Frequenza | ||
| NIH consensus conference (2002) | 4 anni | Dex 1 mg, metanefrine plasmatiche, PRA/aldosterone (se ipertensione o ipopotassiemia e solo alla diagnosi) | annuale | TC: monitoraggio per masse 60 mm | Ricontrollo a 6-12 mesi: non ulteriori controlli in assenza di incremento delle dimensioni |
| French Society of Endocrinology Consensus (2008) | 5 anni |
Dex 1 mg, metanefrine plasmatiche o urinarie (solo alla diagnosi), PRA/aldosterone (se ipertensione o ipopotassiemia e solo alla diagnosi) |
6 mesi, 2 anni e 5 anni | TC: monitoraggio per masse ≥ 40 mm | 6 mesi, 2 anni e 5 anni |
| AACE/AAES guidelines (2009) | 5 anni | Dex 1 mg + ACTH + DHEAS, metanefrine plasmatiche o urinarie, PRA/aldosterone (se ipertensione o ipopotassiemia) | annuale | TC: monitoraggio per masse ≥ 40 mm | 3-6 mesi, poi annualmente per 2 anni |
| AME position statement (2011) | Non determinato e da valutare su base individuale | 1 mg DST, metanefrine plasmatiche o urinarie, PRA/aldosterone (se ipertensione o ipopotassiemia) | non determinata e da valutare su base individuale | TC o RM | 3-6 mesi, ulteriori controlli su base individuale. Non raccomandato ricontrollo per masse < 20 mm e con HU < 10 |
Management e terapia dei pazienti con sindrome di Cushing subclinica
Se da un lato la diagnostica risulta complessa, non da meno è la ricerca di un accordo su come gestire i pazienti che presentino un ipercortisolismo subclinico. Vi sono alcuni studi che dimostrano come la surrenectomia possa migliorare le co-morbilità associate all’ipercortisolismo subclinico (7-10). Tuttavia, al di là del fatto che la maggior parte dei lavori è retrospettiva, vi sono alcuni limiti nei disegni degli studi, in particolare l’utilizzo di criteri disomogenei di inclusione dei pazienti, un periodo di follow-up post-intervento non sempre sufficientemente lungo da superare l’effetto del temporaneo iposurrenalismo realativo post-intervento e, non ultimo, la mancanza di una standardizzazione della terapia medica ottimale per il controllo delle co-morbilità, da comparare con i risultati dell’intervento chirurgico. Non vi è dubbio che vi siano comunque alcuni studi di maggiore rilievo. Il lavoro di Toniato e coll. (11) è l’unico randomizzato-controllato e, sebbene il trattamento medico pre- e post-operatorio non sia standardizzato e la descrizione dell’outcome non dettagliata, appare di interesse il risultato che circa il 25% dei pazienti sottoposti a intervento chirurgico abbiano normalizzato i livelli pressori e la glicemia. Chiodini e coll. (12) hanno confermato l’efficacia dell’intervento chirurgico, non solo nei soggetti con ipercortisolismo subclinico, ma anche nei pazienti che avevano masse non secernenti rispetto a soggetti che avevano rifiutato l’intervento chirurgico. Questo risultato pone ovviamente dei dubbi sul reale impatto della lieve ipersecrezione nel miglioramento del rischio cardiovascolare. Peraltro, vi sono anche esperienze che non riportano la superiorità della chirurgia rispetto al trattamento conservativo, sebbene anche in questo caso lo studio presenti dei limiti metodologici (13).
E’ opinione di chi scrive che al momento non vi siano studi sufficientemente solidi, in termini di selezione dei pazienti, valutazione prospettica, follow-up adeguato e, non ultimo, la comparazione con un ottimale regime di terapia medica, per dichiarare la superiorità della terapia chirurgica. Nella recente Position Statement AME raccomandiamo l’intervento chirurgico per i soggetti più giovani con sindrome di Cushing subclinica e per pazienti che presentano complicanze vascolari, metaboliche, ossee, potenzialmente correlate all’eccesso di cortisolo, di recente insorgenza, di difficile controllo o in peggioramento progressivo nonostante un adeguato regime terapeutico medico (4). Al contrario, l’età avanzata e la lunga durata delle co-morbilità sono fattori che rendono già di per sé poco efficace l’intervento chirurgico nei pazienti con ipercortisolismo manifesto (14,15). Va sottolineato come, in caso di intervento chirurgico, sia necessaria la terapia steroidea sostitutiva post-operatoria per compensare l’atteso iposurrenalismo relativo, associato al ricontrollo della funzionalità surrenalica residua.
La figura sintetizza una possibile proposta di gestione del paziente con sindrome di Cushing subclinica.
Proposta di gestione del paziente con sindrome di Cushing subclinica
Bibliografia
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Int Med 2003, 138: 424–9.
- Zeiger MA, Thompson GB, Duh QY, et al; American Association of Clinical Endocrinologists; American Association of Endocrine Surgeons. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract 2009, 15: 450-3.
- Tabarin A, Bardet S, Bertherat J, et al; French Society of Endocrinology Consensus. Exploration and management of adrenal incidentalomas. French Society of Endocrinology Consensus. Ann Endocrinol (Paris) 2008, 69: 487-500.
- Terzolo M, Stigliano A, Chiodini I, et al; Italian Association of Clinical Endocrinologists. AME position statement on adrenal incidentaloma. Eur J Endocrinol 2011, 164: 851–70.
- Chiodini I. Clinical review: Diagnosis and treatment of subclinical hypercortisolism. J Clin Endocrinol Metab 2011, 96: 1223-36.
- Cawood TJ, Hunt PJ, O'Shea D, et al. Recommended evaluation of adrenal incidentalomas is costly, has high false-positive rates and confers a risk of fatal cancer that is similar to the risk of the adrenal lesion becoming malignant; time for a rethink? Eur J Endocrinol 2009, 161: 513-27.
- Tauchmanovà L, Rossi R, Biondi B, et al. Patients with subclinical Cushing’s syndrome due to adrenal adenoma have increased cardiovascular risk. J Clin Endocrinol Metab 2002, 87: 4872–8.
- Emral R, Uysal AR, Asik M, et al. Prevalence of subclinical Cushing’s syndrome in 70 patients with adrenal incidentaloma: clinical, biochemical and surgical outcomes. Endocr J 2003, 50: 399–408.
- Midorikawa S, Sanada H, Hashimoto S, et al. The improvement of insulin resistance in patients with adrenal incidentaloma by surgical resection. Clin Endocrinol (Oxford) 2001, 54: 797–804.
- Mitchell IC, Auchus RJ, Juneja K, et al. ‘Subclinical Cushing’s syndrome’ is not subclinical: improvement after adrenalectomy in 9 patients. Surgery 2007, 142: 900–5.
- Toniato A, Merante-Boschin I, Opocher G, et al. Surgical versus conservative management for subclinical Cushing syndrome in adrenal incidentalomas: a prospective randomized study. Ann Surg 2009, 249: 388–91.
- Chiodini I, Morelli V, Salcuni AS, et al. Beneficial metabolic effects of prompt surgical treatment in patients with an adrenal incidentaloma causing biochemical hypercortisolism. J Clin Endocrinol Metab 2010, 95: 2736–45.
- Sereg M, Szappanos A, Toke J, et al. Atherosclerotic risk factors and complications in patients with non-functioning adrenal adenomas treated with or without adrenalectomy: a long-term follow-up study. Eur J Endocrinol 2009, 160: 647–55.
- Iacobone M, Mantero F, Basso SM, et al. Results and long-term follow-up after unilateral adrenalectomy for ACTH independent hypercortisolism in a series of fifty patients. J Endocrinol Invest 2005, 28: 327–32.
- Fallo F, Sonino N, Barzon L, et al. Effect of surgical treatment on hypertension in Cushing’s syndrome. Am J Hypert 1996, 9: 77–80.
Overview sulle forme surrenaliche di Cushing
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 15 gennaio 2020)
Le forme di Cushing ACTH-indipendente costituiscono circa il 20% delle cause di ipercortisolismo endogeno. Sono dovute principalmente a tumori primitivi del cortico-surrene (80% adenomi e 20% carcinomi) e a forme di iperplasia surrenalica bilaterale.
Gli adenomi surrenalici presentano dimensioni generalmente < 4-5 cm, in alcuni casi possono secernere oltre al cortisolo anche androgeni e hanno una prognosi benigna.
I carcinomi surrenalici, invece, sono spesso grandi, radiologicamente molto disomogenei con aree necrotiche ed emorragiche, invasivi, hanno frquentemente una secrezione mista (cortisolo, androgeni, mineralcorticoidi) e si associano a prognosi infausta.
Le iperplasie surrenaliche ACTH-indipendenti rappresentano una malattia primitiva del cortico-surrene, caratterizzata da un ingrandimento bilaterale della corticale dei surreni, che può essere diffuso o nodulare (micro- o macro-nodulare, a seconda che le dimensioni dei nodi siano minori o maggiori di 1 cm). Sono forme molto complesse ed eterogenee, che possono determinare un’ipersecrezione autonoma di cortisolo o aldosterone, clinicamente manifesta o subclinica. Vi sono due forme principali di iperplasia ACTH-indipendente:
- le iperplasie pigmentate micro-nodulari (Primary Pigmented Nodular Adrenal Disease, PPNAD): si possono presentare in forma sporadica o associate al complesso di Carney (malattia rara, caratterizzata, oltre al PPNAD, da lesioni cutanee pigmentate e altre disendocrinopatie, mixomi cardiaci, ecc);
- le iperplasie surrenaliche macro-nodulari (primary bilateral macronodular adrenal hyperplasia, PBMAH): sono caratterizzate da diversi gradi di severità di ipercortisolismo, regolato dalla presenza di recettori aberranti responsivi a diversi stimoli (vasopressina, β-agonisti adrenergici, LH/βHCG, angiotensina II, GIP, ecc.) e in grado di attivare la via di trascrizione solitamente regolata dall’ACTH.
Queste forme di iperplasia vanno ben distinte dalle forme di iperplasia surrenalica ACTH-dipendente, come l’iperplasia surrenalica congenita da deficit enzimatici della steroidogenesi (il più frequente dei quali è quello della 21-idrossilasi) o alcune forme di Cushing ACTH-dipendente (soprattutto nell’anziano e di lunga durata), in cui lo stimolo cronico di ACTH può determinare un’iperplasia ghiandolare e l’insorgenza di nodi mono o bilaterali fino all’80% dei casi. Pertanto, va ricordato che la presenza di uno o più noduli surrenalici non rappresenta una prova sicura che l'ipercortisolismo sia primariamente causato dal surrene.
Bibliografia
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008, 93: 1526-40.
- Lacroix A, Bourdeau I. Bilateral adrenal Cushing's syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin North Am 2005, 34: 441-58.
Sindrome di Cushing da adenoma surrenalico
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 15 gennaio 2020)
Gli adenomi surrenalici secernenti cortisolo causano circa il 10% delle sindromi di Cushing. La diagnosi solitamente è prima clinica, caratterizzata da un quadro di ipercortisolismo florido, successivamente confermata biochimicamente e associata a valori di ACTH soppressi (1).
L’inquadramento diagnostico prevede lo studio dei surreni mediante TC o RM, che permettono la distinzione tra le forme benigne e maligne (tabella 1). Gli adenomi si presentano di dimensioni variabili, generalmente < 4 cm, rotondeggianti con margini regolari, omogeneamente ipodensi (< 10 unità Hounsfield, HU) e con rapido wash-out del mezzo di contrasto (2).
La buona caratterizzazione della massa surrenalica offerta da queste indagini radiologiche rende oggi molto meno utile la scintigrafia surrenalica con radio-colesterolo, mentre viene a volte utilizzato in seconda linea lo studio con PET con 18F-fluorodesossi-glucosio (18F-FDG), associato alla TAC per migliorare la risoluzione spaziale, in particolare nei pazienti con sospetta neoplasia primitiva o metastatica surrenalica.
| Tabella 1 Caratteristiche TC e/o RM orientative sulla natura di una massa surrenalica |
||
| Criteri | Benigna | Sospetta |
| Dimensioni | < 4 cm | > 4 cm |
| Margini | Regolari | Irregolari |
| Aspetto | Omogeneo | Disomogeneo |
| Wash-out del contrasto | Rapido | Tardivo |
| Attenuazione del segnale alla TC diretta | < 10 HU | > 10 HU |
| Intensità del segnale nelle sequenze T1 fuori fase alla RM | Abbattimento | Persistenza |
Anche se i meccanismi patogenetici responsabili dello sviluppo dei tumori del cortico-surrene sono poco conosciuti, negli ultimi anni le nostre conoscenze al riguardo sono molto aumentate (3). La progressione da adenoma a carcinoma surrenalico comporta una proliferazione cellulare di tipo monoclonale, con una progressiva alterazione del materiale cromosomico (amplificazioni e perdite). L’alterazione molecolare più frequentemente associata al fenotipo maligno comporta un’iperespressione del gene codificante IGF2 ed una perdita di funzione dei geni CDKN1C e H19 (4). Alcune forme genetiche familiari, inoltre, hanno predisposizione a sviluppare carcinomi surrenalici, tra cui la sindrome di Li-Fraumeni e la sindrome di Beckwith-Wiedemann (5).
Recentemente, si è focalizzata l'attenzione sul sistema dell'AMP ciclico e sul suo signaling (6). In alcuni casi, sono state identificate mutazioni somatiche di PRKAR1A e di alcune fosfo-diesterasi (11A e 8B), anche in casi di adenomi corticali sporadici del surrene e non solo nelle PPNAD (3,7). Analogamente, sono state identificate mutazioni genetiche coinvolte nella regolazione del signaling wnt/β-catenin in tumori surrenalici, sia benigni sia maligni (8). Tale via sembra essere responsabile anche del frequente riscontro di adenomi surrenalici o PBMAH nei pazienti con poliposi adenomatosa familiare affetti da mutazione germinale del gene APC (9).
Il trattamento dell’adenoma surrenalico cortisolo-secernente è la surrenectomia, solitamente per via laparoscopica, che permette la remissione praticamente nel 100% di casi (10).
Bibliografia
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2008, 93: 1526-40.
- Fassnacht M, Arlt W, Bancos I, et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016, 175: G1-34.
- Hernández-Ramírez LC, Stratakis Ca. Genetics of Cushing's syndrome. Endocrinol Metab Clin North Am 2018, 47: 275-97.
- Bertherat J, Bertagna X. Pathogenesis of adrenocortical cancer. Best Pract Res Clin Endocrinol Metab, 2009, 23: 261-71.
- Assie G, Giordano TJ, Bertherat J. Gene expression profiling in adrenocortical neoplasia. Mol Cell Endocrinol 2012, 351: 111-7.
- de Joussineau C, Sahut-Barnola I, Levy I, et al. The cAMP pathway and the control of adrenocortical development and growth. Mol Cell Endocrinol 2012, 351: 28-36.
- Vezzosi D, Bertherat J. Phosphodiesterases in endocrine physiology and disease. Eur J Endocrinol 2011, 165: 177-88.
- Berthon A, Martinez A, Bertherat J, Val P. Wnt/β-catenin signalling in adrenal physiology and tumour development. Mol Cell Endocrinol 2012, 351: 87-95.
- Gaujoux S, Pinson S, Gimenez-Roqueplo AP, et al. Inactivation of the APC gene is constant in adrenocortical tumors from patients with familial adenomatous polyposis but not frequent in sporadic adrenocortical cancers. Clin Cancer Res 2010, 16: 5133–41.
- Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2015, 100: 2807–31.
Primary Bilateral Macronodular Adrenal Hyperplasia, BPMAH
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 15 gennaio 2020)
L'iperplasia surrenalica macro-nodulare (primary bilateral macronodular adrenal hyperplasia, PBMAH) è una condizione complessa ed eterogenea, caratterizzata da surreni ingranditi bilateralmente, spesso asimmetricamente, per la presenza di multipli nodi > 1 cm. Rappresenta meno dell’1% di tutte le cause di sindrome di Cushing (1). Tale categoria, una volta denominata ACTH-independent macronodular adrenal hyperplasia (AIMAH), si associa a un’incompleta soppressione del cortisolo al test con 1 mg di desametasone, con diversi gradi di severità di ipercortisolismo. La secrezione di cortisolo in questi casi è regolata dalla presenza di recettori aberranti accoppiati a proteine G, responsivi a diversi stimoli (vasopressina, serotonina, LH/βHCG, β-agonisti adrenergici, angiotensina II, GIP e glucagone) e in grado di attivare la via di trascrizione solitamente regolata dall’ACTH. È stata, inoltre, riportata una certa secrezione di cortisolo stimolata in modo autocrino e paracrino da ACTH prodotto localmente, che ha portato alla più corretta denominazione di tali forme in PBMAH.
Oltre alle forme sporadiche, studi recenti hanno evidenziato anche la presenza di forme genetiche di PBMAH, nella maggioranza dei casi legate a mutazioni germinali di ARMC5, a trasmissione autosomica dominante, responsabile della tumorigenesi surrenalica, e più raramente legate a mutazioni dei geni PRKACA e GNAS, responsabili di un’attivazione costitutiva della via di segnale che regola la steroidogenesi (2). Tali forme genetiche sono spesso associate a quadri clinici più severi e i pazienti ARMC5-mutati tendono anche a presentare fenotipi multi- e macro-nodulari di maggiori dimensioni e possibile presenza di altri tumori, in particolare meningiomi, suggerendo una possibile nuova sindrome da neoplasie multiple (3,4).
I pazienti con PBMAH generalmente sono di età media o avanzata. Quando si manifesta nella prima decade di vita, è in genere espressione di una sindrome di McCune-Albright.
L’ipercortisolismo può essere manifesto, ma spesso è subclinico e questo è un elemento di importanza clinica, dal momento che le masse bilaterali rappresentano circa il 10% degli incidentalomi surrenalici e che la diagnosi è quindi spesso ritardata e successiva al riscontro radiologico. Sebbene la sindrome di Cushing sia la manifestazione clinica più frequente, sono riportati anche casi di concomitante secrezione di aldosterone, estrogeni o androgeni (5-7).
Dal punto di vista diagnostico, ci sono alcuni protocolli mirati alla ricerca di questi recettori anomali: si tratta di test ormonali complessi, in genere riservati a centri di terzo livello, che valutano le modifiche dei livelli di cortisolo e ACTH in condizioni basali e dopo alcuni stimoli farmacologici (GnRH, TRH, terlipressina) e non farmacologici (ortostatismo, pasto misto). Un test viene considerato positivo per la presenza di recettore illecito se si registra un incremento del cortisolo maggiore del 50% rispetto al basale, in assenza di variazioni dell’ACTH. Tali test possono avere importanti ripercussioni terapeutiche: per esempio, nel sospetto di una forma con recettori aberranti del GIP, rivelata da un test al pasto misto positivo, l'ipercortisolismo potrebbe essere controllato con l'impiego di analoghi della somatostatina. Anche i ß-bloccanti adrenergici e gli agonisti del GnRH si sono dimostrati efficaci nel trattamento medico di alcune forme di PBMAH, rispettivamente con espressione anomala dei recettori adrenergici e di LH. Tuttavia, l’efficacia di questi farmaci sembra essere solo temporanea (1).
Nella maggior parte dei casi, la surrenectomia bilaterale è raccomandata nelle forme associate a ipercortisolismo florido (con cortisoluria > 3-4 volte il valore massimo di norma), mentre la mono-surrenectomia con eventuale surrenectomia parziale contro-laterale viene solitamente considerata nelle forme di ipercortisolismo mild (con cortisoluria < 2-3 volte il valore massimo di norma), seppur studi crescenti ne evidenzino i benefici in termini di riduzione delle comorbilità correlate alle forme di sindrome di Cushing subclinico (8,9). La scelta di quale surrene asportare può dipendere dalle dimensioni, dalla diversa captazione alla scintigrafia con selenio o iodo-colesterolo o dal gradiente di secrezione di cortisolo valutato al cateterismo delle vene surrenaliche (utilizzando il dosaggio delle catecolamine come criterio di adeguata cateterizzazione). In questi casi, il periodico follow-up post chirurgico è fondamentale per la conferma della remissione dal Cushing e la valutazione di possibili recidive, ma anche per l’insorgenza di iposurrenalismo.
Bibliografia
- Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab 2009, 23: 245-59.
- Hernández-Ramírez LC, Stratakis Ca. Genetics of Cushing's Syndrome. Endocrinol Metab Clin North Am 2018, 47: 275-97.
- Espiard S, Drougat L, Libe R, et al. ARMC5 mutations in a large cohort of primary macronodular adrenal hyperplasia: clinical and functional consequences. J Clin Endocrinol Metab 2015,100: E926–35.
- Elbelt U, Trovato A, Kloth M, et al. Molecular and clinical evidence for an ARMC5 tumor syndrome: concurrent inactivating germline and somatic mutations are associated with both primary macronodular adrenal hyperplasia and meningioma. J Clin Endocrinol Metab 2015, 100: E119-28.
- Ghayee HK, Rege J,Watumull LM, et al. Clinical, biochemical, and molecular characterization of macronodular adrenocortical hyperplasia of the zona reticularis: a new syndrome. J Clin Endocrinol Metab 2011, 96: E243–50.
- Goodarzi MO, Dawson DW, Li X, et al. Virilization in bilateral macronodular adrenal hyperplasia controlled by luteinizing hormone. J Clin Endocrinol Metab 2003, 88: 73–7.
- Malchoff CD, Rosa J, DeBold CR, et al. Adrenocorticotropin-independent bilateral macronodular adrenal hyperplasia: an unusual cause of Cushing’s syndrome. J Clin Endocrinol Metab 1989, 68: 855–60.
- Albiger NM, Ceccato F, Zilio M, et al. An analysis of different therapeutic options in patients with Cushing's syndrome due to bilateral macronodular adrenal hyperplasia: a single-centre experience. Clin Endocrinol (Oxf) 2015, 82: 808-15.
- Perogamvros I, Vassiliadi DA, Karapanou O, et al. Biochemical and clinical benefits of unilateral adrenalectomy in patients with subclinical hypercortisolism and bilateral adrenal incidentalomas. Eur J Endocrinol 2015, 173: 719-25.
Primary Pigmented Nodular Adrenal Disease (PPNAD)
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 15 gennaio 2020)
L'iperplasia surrenalica primitiva nodulare pigmentata (Primary Pigmented Nodular Adrenal Disease, PPNAD) è una causa rara di sindrome di Cushing ACTH-indipendente.
Si può presentare isolata (50% dei casi) o associata al complesso di Carney, una sindrome sistemica rara a eredità autosomica dominante, caratterizzata dalla presenza di lesioni cutanee pigmentate, mixomi cardiaci e cutanei e diversi disturbi endocrinologici, tra cui i più comuni sono l'acromegalia, i tumori della tiroide e dei testicoli e la PPNAD. Molti di questi disturbi sono già presenti alla nascita, ma l’età media di diagnosi è attorno ai 20 anni (1).
In oltre la metà dei casi di PPNAD e/o complesso di Carney sono presenti mutazioni germinali del gene PRKAR1A, codificante l’α-subunità regolatrice cAMP-dipendente della proteina chinasi A, implicata nella tumorigenesi endocrina (2,3). Oltre alla PRKAR1A, sono coinvolte nella patogenesi della PPNAD anche mutazioni della fosfodiesterasi (in particolare i sottotipi 11A e 8B) (4).
Il quadro clinico è quello tipico di un ipercortisolismo florido e, presentandosi in giovane età, spesso si associa a un’importante compromissione ossea, che necessita di trattamenti specifici. Alcune volte l'ipercortisolismo può presentarsi in modo ciclico, ritardando e rendendo più difficile la diagnosi.
Dal punto di vista biochimico, caratteristicamente i pazienti con PPNAD presentano un aumento paradosso della secrezione di cortisolo durante test prolungato con desametasone (2 mg/die di desametasone per 2 giorni seguiti da 8 mg/die per altri 2 giorni): il cortisolo urinario aumenta progressivamente dal secondo giorno (> 50% rispetto al basale). Tale fenomeno, comunque, non è presente in tutti i casi di PPNAD e, si può osservare talvolta anche in presenza di adenomi surrenalici cortisolo-secernenti (5).
Dal punto di vista morfologico, i noduli surrenalici della PPNAD sono di piccole dimensioni (< 5 mm, solo raramente superano il centimetro) e ricchi di lipofuscina, che spiega la tipica colorazione scura, in un contesto di atrofia della restante corticale del surrene. Alla TC o RM spesso i surreni sono di piccole dimensioni e non si evidenziano noduli; a volte, però, sono presenti nodularità allineate, che causano un caratteristico aspetto a "collana" (2,3).
La bisurrenectomia è il trattamento di scelta, anche se in alcuni casi selezionati la surrenectomia monolaterale consente una normalizzazione prolungata della secrezione di cortisolo (6).
Bibliografia
- Vezzosi D, Vignaux O, Dupin N, Bertherat J. Carney complex: clinical and genetic 2010 update. Ann Endocrinol (Paris) 2010, 71: 486-93.
- Lacroix A, Bourdeau I. Bilateral adrenal Cushing's syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin North Am 2005, 34: 441-58.
- Horvath A, Stratakis C. Primary pigmented nodular adrenocortical disease and Cushing's syndrome. Arq Bras Endocrinol Metab 2007, 51: 1238-44.
- de Joussineau C, Sahut-Barnola I, Levy I, et al. The cAMP pathway and the control of adrenocortical development and growth. Mol Cell Endocrinol 2012, 351:28-36.
- Louiset E, Stratakis CA, Perraudin V, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. J Clin Endocrinol Metab 2009, 94: 2406-13.
- Kyrilli A, Lytrivi M, Bouquegneau MS, et al. Unilateral adrenalectomy could be a valid option for primary nodular adrenal disease: evidence from twins. J Endocr Soc 2018, 3: 129-34.
Overview sui carcinomi surrenalici
Pina Lardo & Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare, Ospedale Sant'Andrea, Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 18 gennaio 2021)
Epidemiologia e prognosi
Il carcinoma cortico-surrenalico è una rara forma di neoplasia endocrina, la cui incidenza è stimata intorno ad un caso ogni 1.5 milioni di individui all’anno. La distribuzione per età segue un andamento di tipo bimodale, con un primo picco in età pediatrica e un secondo nella quinta decade di vita. La neoplasia spesso si associa a una sindrome ormonale manifesta, rappresentando un modello complesso di malattia endocrino-oncologica. L’associazione della malattia neoplastica con quella ormonale complica l’evoluzione clinica, rendendo critica la gestione del paziente (1).
Eziopatogenesi
Generalmente di origine sporadica, il cancro della corticale surrenalica riconosce in alcuni loci genici sui cromosomi 11 e 17 e nei geni dell'IGF-II, p53 e ß-catenina i principali effettori del processo tumorigenico (2).
Clinica
Il quadro clinico è frequentemente condizionato dall'attività funzionale della neoplasia. L'esordio è caratterizzato dalla comparsa, progressivamente ingravescente, di segni e sintomi riconducibili all'ipercortisolismo, con e senza virilizzazione. Nella maggior parte dei casi prevalgono le forme con eccesso di glucocorticoidi e quelle a secrezione mista di glucocorticoidi e androgeni. I carcinomi secernenti estrogeni e mineraloattivi sono piuttosto rari. Le neoplasie “non secernenti” mancano spesso di una clinica peculiare, essendo la loro presenza rilevata casualmente da metodiche di imaging o da segni per lo più di tipo meccanico dipendenti dalla crescita della massa. Talora, nei casi di malattia ad evoluzione metastatica, la comparsa di manifestazioni ad essa correlata permette il riconoscimento della neoplasia (1).
Diagnostica
La valutazione biochimica ormonale è importante nella diagnosi delle diverse forme ad attività ormonale. La spettrometria di massa è una metodica promettente, in associazione alla diagnostica per immagini radiologica (TC, RM) e medico-nucleare (PET), per la diagnosi differenziale delle lesioni surrenaliche maligne da quelle benigne (3,4).
Terapia
L'asportazione chirurgica completa della neoplasia, affidata a un team chirurgico esperto in patologia surrenalica, rappresenta il gold standard della terapia.
La terapia medica per il carcinoma cortico-surrenalico in stadio avanzato è rappresentata dal mitotane, farmaco adrenolitico approvato per la terapia di questa neoplasia. Tuttavia, numerosi studi suggeriscono l’opportunità che la chirurgia sia seguita da terapia adiuvante rappresentata sempre dal mitotane (1,5).
La chemioterapia, che generalmente segue la monoterapia con mitotane in caso di progressione della malattia, prevede l'impiego di schemi terapeutici tra cui quello più efficace è rappresentato dalla combinazione di etoposide, doxorubicina e cisplatino in associazione con mitotane (5).
Studi retrospettivi supportano l’ausilio della radioterapia in adiuvante dopo la chirurgia (6).
Le targeted therapies, sviluppate sulla base delle nuove conoscenze molecolari relative alla tumorigenesi cortico-surrenalica, rappresentano un futuro promettente nella strategia terapeutica di questa neoplasia (7).
Bibliografia
- Fassnacht M, Dekkers OM, Else T, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018, 179: G1-46.
- Bonnet-Serrano F, Bertherat J. Genetics of tumors of the adrenal cortex. Endocr Relat Cancer 2018, 25: R131-52.
- Bancos I, Taylor AE, Chortis V, et al. Urine steroid metabolomics for the differential diagnosis of adrenal incidentalomas in the EURINE-ACT study: a prospective test validation study. Lancet Diabetes Endocrinol 2020, 8: 773–81.
- Hennings J, Lindhe O, Bergstrom M, et al. [11C]metomidate positron emission tomography of adrenocortical tumors in correlation with histopathological findings. J Clin Endocrinol Metab 2006, 91: 1410-4.
- Berruti A, Baudin E, Gelderblom H, et al, on behalf of the ESMO Guideline Working Group. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012, 23 suppl 7: 131–8.
- Sabolch A, Else T, Griffith KA, et al. Adjuvant radiation therapy improves local control after surgical resection in patients with localized adrenocortical carcinoma. Int J Radiat Oncol 2015, 92: 252–9.
- Creemers SG, Hofland LJ, Korpershoek E, et al. Future directions in the diagnosis and medical treatment of adrenocortical carcinoma. Endocr Relat Cancer 2016, 23: R43-69.
Clinica e diagnostica carcinoma surrenalico
Pina Lardo & Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare, Ospedale Sant'Andrea, Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 18 gennaio 2021)
Epidemiologia
Il carcinoma corticosurrenalico (CCS) è un tumore endocrino raro, con un’incidenza di 0.7-2 nuovi casi per milione di abitanti/anno. Può insorgere a qualsiasi età, ma più frequentemente è stata osservata una distribuzione per età bimodale, con un primo picco d’insorgenza nell’infanzia e un secondo picco nella 5° decade. In entrambe le fasce di popolazione vi è una predilezione per il sesso femminile (rapporto F/M = 1.5-2.5/1) (1).
La maggior parte dei CCS in età adulta è sporadica, ma occasionalmente si presenta nell’ambito di sindromi ereditarie (s. di Li-Fraumeni, s. di Beckwith-Wiedeman, MEN-1, iperplasia surrenalica congenita, poliposi adenomatosa familiare, sindromi associate a mutazioni del gene ß-catenina) (2,3). Circa il 2% dei CCS sporadici si riscontra tra gli incidentalomi surrenalici (4).
Patogenesi
La tumorigenesi surrenalica è sostenuta da meccanismi estremamente eterogenei, sia in termini di sviluppo che di metastatizzazione.
L’analisi delle modalità di inattivazione del cromosoma-X in tessuti eterozigoti ha dimostrato che la popolazione cellulare del CCS è monoclonale, mentre gli adenomi possono essere mono- o poli-clonali. La monoclonalità indica che lo sviluppo neoplastico è il risultato di mutazioni genetiche intrinseche, in grado di conferire un vantaggio di crescita alle cellule inizialmente coinvolte (5).
Attraverso studi su alterazioni molecolari, presenti a livello della linea germinale in rare malattie ereditarie e a livello somatico in tumori sporadici, sono state identificate alcune regioni cromosomiche e alcuni geni potenzialmente coinvolti nel processo di tumorigenesi (5-8).
È stata costantemente dimostrata nei CCS la perdita di eterozigosità (LOH) in 17p13 (> 85%), che correla con il criterio patologico adottato per definire la malignità di una massa surrenalica (punteggio di Weiss) e potrebbe essere usata come marcatore di malignità oltre che come parametro predittivo di recidiva dopo resezione tumorale (8). La LOH della regione 11p15 è più frequente nel CCS rispetto agli adenomi (78.5 vs 9.5%), è associata a maggior rischio di recidiva e correla con il punteggio di Weiss.
Oncogeni:
- alterazioni genetiche o epigenetiche dell’imprinting nella regione 11p15 (dove è localizzato il gene IGF-II) sono implicate nella sindrome di Beckwith-Wiedemann, caratterizzata da macrosomia, macroglossia, anomalie dello sviluppo (in particolare difetti della parete addominale con onfalocele) e aumentata incidenza di tumori embrionali, tra cui tumore di Wilms, neuroblastoma, epatoblastoma e nel 5-10% dei casi anche CCS;
- gene APC, la cui mutazione germinale inattivante, responsabile dell’aumento dei livelli di β-catenina (che codifica per una proteina della via di segnale Wnt, normalmente attivata durante lo sviluppo embrionale, essenziale nello sviluppo della corteccia surrenalica) è causa della sindrome di Gardner, altra patologia ereditaria caratterizzata dalla presenza di numerosi polipi del colon in età giovanile e CCS in rari casi.
Onco-soppressori: da segnalare il gene TP53 (localizzato sul cromosoma 17p13, che codifica per una proteina essenziale per la regolazione della crescita cellulare e dell’apoptosi), la cui mutazione germinale ha una prevalenza del 3-7% nel CCS dell’adulto (9).
Quadro clinico
I tumori surrenalici vengono suddivisi clinicamente in (9):
- funzionanti, quando segni e sintomi clinici sono riconducibili alla loro esaltata attività endocrina;
- non funzionanti, quando l’attività secretoria, di precursori e/o ormoni, è incapace di generare una sindrome clinica manifesta.
Il CCS si manifesta (10,11):
- nel 40-60% dei casi con segni e sintomi dovuti all’ipersecrezione ormonale tumorale;
- in 1/3 dei casi con sintomi aspecifici dovuti a fenomeni compressivi locali, che possono essere responsabili di una diagnosi tardiva;
- nel restante 20-30% dei casi incidentalmente, durante l’esecuzione di esami eseguiti per altre motivazioni cliniche;
- raramente con i classici sintomi neoplastici (perdita di peso, cachessia, febbre, sudorazioni notturne) e generalmente sono rare le sindromi paraneoplastiche.
L’ipercortisolismo è la presentazione clinica più comune (50-80% dei CCS secernenti) e si manifesta con i segni e i sintomi tipici della sindrome di Cushing rapidamente progressiva (tra le manifestazioni sono frequenti diabete mellito, ipertensione arteriosa e disturbi psichiatrici). L’ipertensione, con o senza ipopotassiemia, è dovuta al fatto che gli elevati livelli di cortisolo sono in grado di saturare l’enzima 11-β-idrossisteroido-deidrogenasi di tipo 2, che converte il cortisolo in cortisone inattivo, con conseguente azione mineralcorticoide mediata dai glucocorticoidi.
Nel 40-60% dei casi, invece, il CCS si manifesta con una sintomatologia dovuta all’ipersecrezione di androgeni, evidente soprattutto nelle donne (può causare irsutismo, acne, alopecia, atrofia mammaria, alterazioni del ciclo mestruale, infertilità) e in età pediatrica (segni e sintomi di virilizzazione), mentre non è clinicamente evidente nel maschio adulto. La produzione di estrogeni si verifica solo nell’1-3% dei maschi affetti, causando ginecomastia e atrofia testicolare mediante la soppressione dell’asse gonadico, mentre nella donna può determinare sanguinamenti uterini anomali e aumento del volume mammario. Nella valutazione delle lesioni surrenaliche la possibilità di forme maligne deve essere pertanto sospettata, oltre che per le dimensioni, per gli elevati livelli di androgeni o estrogeni (1).
In circa metà dei CCS secernenti è presente co-secrezione di androgeni e cortisolo; in questi casi i quadri clinici sono il prodotto della sommatoria degli effetti delle due classi di ormoni steroidei, ad eccezione degli effetti catabolici indotti dal cortisolo che contrastano con quelli anabolici indotti dagli androgeni, annullando il segno clinico dell’atrofia muscolare.
L’ipersecrezione di aldosterone è rara, mentre più comunemente l’ipertensione e/o l'ipopotassiemia sono causati dall’azione mineralcorticoide del cortisolo in eccesso o dall’azione di precursori steroidei, come l’11-desossicorticosterone; a sua volta l’ipokaliemia è responsabile dello sviluppo di ipertensione per un effetto di vaso-costrizione arteriolare periferica (12,13).
I carcinomi cosiddetti non funzionanti esordiscono con una sintomatologia riconducibile all’accrescimento della massa tumorale. I segni e sintomi sono di tipo gastro-intestinale, generalmente aspecifici: anoressia, dimagramento, dolore epigastrico, nausea, vomito, dolore lombare relativo all’effetto compressivo esercitato dalla massa. Meno frequentemente, compaiono manifestazioni sistemiche, quali mialgia diffusa, stanchezza, astenia e febbricola persistente. Manifestazioni cliniche meno frequenti comprendono: segni di compressione della vena cava inferiore, quali edemi declivi agli arti inferiori e varicocele, dolore addominale acuto, ostruzione delle vene sovra-epatiche con sindrome di Budd–Chiari, ematuria causata da infiltrazione del tessuto renale, ostruzione delle vie urinarie superiori, dolore pelvico, paraplegia dovuta a infiltrazione delle strutture nervose spinali, dispnea e dolore toracico in seguito a compressione del diaframma, rottura del tumore con sanguinamento intra-lesionale o retro-peritoneale.
Talvolta un CCS non funzionante può diventare sintomatico e arrivare alla diagnosi per le localizzazioni metastatiche: dolore osseo per fratture patologiche o diagnosi incidentale di localizzazioni secondarie polmonari, epatiche o scheletriche (8,10).
Diagnostica ormonale
Il sospetto di CCS può derivare dal quadro clinico (rapida comparsa di segni e sintomi di ipersecrezione ormonale), dal quadro ormonale o da un esame di diagnostica per immagini con risultato sospetto o indeterminato.
In tutti i pazienti portatori di una massa surrenalica è sempre raccomandata la valutazione pre -operatoria della funzionalità surrenalica, in quanto consente di stabilire l’origine cortico-surrenalica del tumore, sospettarne la malignità e quindi influenzarne la strategia chirurgica (14) (tab 1).
| Tabella 1 Indagini ormonali in pazienti con sospetto CCS |
|
| Eccesso di | Test |
| Glucocorticoidi | Test di soppressione con desametasone 1 mg o cortisolo libero urinario 24 ore o cortisolo salivare. Livelli basali di ACTH (determinazione non necessaria se già escluso ipercortisolismo). |
| Ormoni sessuali e precursori steroidei | DHEA–S. 17-OH progesterone. Androstenedione. Testosterone (solo nelle donne). 17-β-estradiolo (solo nei maschi e nelle donne in post-menopausa). 11-desossicortisolo (se disponibile). |
| Mineralcorticoidi | Potassiemia. Rapporto Aldosterone/Renina (solo in pazienti con ipertensione e/o ipokaliemia). |
| Catecolamine | Metanefrine frazionate urinarie delle 24 ore o metanefrine libere plasmatiche. |
Le prescrizioni riguardo alla diagnostica mirano a escludere l’eccesso di secrezione di glucocorticoidi, androgeni o mineralcorticoidi da parte della massa. Il miglior test per valutare l’eccesso di secrezione di cortisolo è il test di soppressione con 1 mg di desametasone. In caso di mancata soppressione del cortisolo, è indicato il dosaggio dell’ACTH per dimostrare la condizione di ACTH-indipendenza legata alla primitività della lesione surrenalica.
Anche in assenza di evidente ipersecrezione ormonale, in tutti i soggetti è raccomandata la valutazione dei precursori steroidei, che possono risultare aumentati per un’alterata regolazione della steroidogenesi nei CCS: 17OH-progesterone e DHEAS, quest’ultimo indicativo della produzione androgenica surrenalica, il cui valore aumentato può essere indicativo ma non esclusivo di malignità. A tal proposito, uno studio del 2000 (15) ne ha valutato l’efficacia diagnostica nel discriminare le lesioni benigne dalle maligne, evidenziandone frequentemente bassi livelli nei pazienti con incidentaloma surrenalico (sensibilità e specificità di bassi livelli di DHEAS nell’identificazione delle lesioni benigne, rispettivamente, del 41% e 100%) e alti livelli nei CCS. Purtroppo, la determinazione dei livelli di DHEAS è suscettibile di errore per la scarsa sensibilità delle attuali metodiche di dosaggio, prevalentemente in età avanzata in cui il bias è più evidente tra valori bassi e medio-bassi.
È inoltre raccomandato il dosaggio degli steroidi sessuali: testosterone e androstenedione nelle donne, 17ß-estradiolo nei maschi e nelle donne in post-menopausa (16).
La valutazione degli steroidi urinari con metodica gascromatografica è risultata altamente sensibile nel discriminare le lesioni benigne da quelle maligne. Questo tipo di analisi rileva nove steroidi (11desossi-corticosterone, corticosterone, progesterone, 17OH-progesterone, testosterone, DHEA, DHEAS, androstenedione, 17ß-estradiolo), con un pattern indicativo in entrambi i sessi dell’attivazione precoce della steroidogenesi (particolarmente della linea androgenica) nei CCS. Con questa tecnica, non ancora in uso clinico, è possibile evidenziare una secrezione ormonale “atipica” in circa l’80% dei CCS, anche in casi definiti non secernenti con le metodiche di dosaggio usuali, con evidenti vantaggi nell’identificazione precoce delle recidive e nella valutazione della risposta al trattamento, anche nei casi con imaging dubbio (17).
È sempre raccomandato escludere la presenza di un feocromocitoma, mediante il dosaggio delle metanefrine nelle urine delle 24 ore o nel plasma (nei centri che eseguono il dosaggio) ed eseguire lo screening per iperaldosteronismo, considerando che si riscontra spesso una soppressione isolata di renina anche in assenza di elevati valori di aldosterone, che potrebbe essere causata dall’azione mineralcorticoide del cortisolo o dalla secrezione di precursori steroidei con attività mineralcorticoide (1). In ogni caso, anche se raramente, è possibile che un CCS secerna aldosterone.
Imaging
Ha un ruolo fondamentale nella diagnosi del CCS, nella stadiazione mediante la valutazione del coinvolgimento di organi e strutture vascolari e linfonodali coinvolte, nella pianificazione del tipo di intervento chirurgico e durante il follow-up per monitorare la risposta al trattamento. (2). TC e RM senza mezzo di contrasto sono esami di prima scelta per l’identificazione delle lesioni surrenaliche e la distinzione tra forme benigne e maligne. La FDG-PET/TC trova oggi indicazione nella conferma della malignità di una lesione precedentemente identificata con le metodiche radiologiche.
La TC senza mdc valuta la densità tissutale mediante la misurazione dell’assorbimento dei raggi X; ciò permette di calcolare il valore di attenuazione tissutale misurato in unità Hounsfield (HU) e di quantificare l’assorbimento dei raggi X dei tessuti comparandolo con quello dell’acqua (pari a 0 HU). Vi è una correlazione inversa tra contenuto di lipidi e valore di attenuazione: maggiore è il contenuto lipidico, più basso sarà il valore HU (18). In particolare, un valore ≤ 10 HU è il valore soglia per la diagnosi di adenoma “lipid rich”, con sensibilità del 96-100% e specificità del 50-100% nel differenziare lesioni benigne da quelle maligne (19,20); un valore > 10 HU è considerato indeterminato, e circa il 30% degli adenomi viene ascritto alla categoria “lipid poor”, sovrapponibile, in alcuni casi, a quello dei feocromocitomi e delle lesioni maligne (21). Altri parametri da considerare nello studio TC sono:
- dimensioni: il rischio di malignità correla con il diametro tumorale, maggiore per tumori > 4 cm (sensibilità 97% e specificità 52%) e > 6 cm (sensibilità 91% e specificità 80%) (1). Nonostante le dimensioni > 6 cm siano suggestive di malignità, ciò non costituisce in assoluto un criterio discriminante, vista la sovrapposizione di dimensioni tra adenomi, CCS, metastasi e feocromocitomi, e infatti, ciascun valore soglia proposto come predittore di malignità ha mostrato un’inadeguata accuratezza diagnostica (22,23);
- omogeneità della lesione: il surrene normale e gli adenomi appaiono omogenei e simmetrici, con densità simile a quella dei reni; al contrario, i CCS presentano aspetto disomogeneo per la presenza di aree di necrosi, di emorragie e talvolta di calcificazioni. Queste ultime, presenti nel 30% dei CCS, sono raramente presenti negli adenomi, mentre si riscontrano nel 10% dei feocromocitomi (24);
- forma e margini: gli adenomi, diversamente dai carcinomi, hanno forma rotondeggiante e margini regolari che li separano dalle strutture circostanti.
La TC con mdc è in grado di fornire informazioni aggiuntive, permettendo una migliore distinzione degli adenomi lipid poor dai non adenomi (metastasi, feocromocitoma e carcinoma). Le lesioni maligne mostrano rapido enhancement ma più lento wash-out (< 60%) a causa della loro vascolarizzazione e avidità (25).
La RM senza mdc è in grado di acquisire scansioni a strato sottile secondo piani trasversali e coronali nelle sequenze T1- e T2-pesate. La ghiandola normale ha un’intensità di segnale in T1 e T2 uguale o leggermente inferiore a quella del fegato. Nella diagnostica surrenalica, l’esame è talora completato da sequenze T1-pesate acquisite dopo somministrazione ev di Gadolinio, che permette di sopprimere il segnale del grasso. Infatti, come per la TC, il contenuto lipidico contribuisce a distinguere le lesioni benigne da quelle maligne (26,27). I CCS appaiono eterogenei per la presenza di aree emorragiche e/o di necrosi, l’intensità di segnale è medio/alta nelle sequenze T1- e T2-pesate, l’enhancement dopo Gd è variabile, mentre il wash-out è generalmente lento. Poichè all’interno di un campo magnetico i protoni vibrano con una frequenza leggermente diversa se sono contenuti nell’acqua o nel grasso, mediante la selezione di sequenze specifiche si possono generare immagini diverse. Questa tecnica di chemical shift imaging consente di ottenere immagini “in fase” e “in opposizione di fase” (acqua/lipidi), fornendo informazioni spesso indispensabili per differenziare:
- adenomi lipid rich (contenenti materiale lipidico e acqua), che tipicamente perdono intensità di segnale nelle immagini in opposizione di fase rispetto a quelle in fase; la perdita di segnale rispetto agli organi vicini (generalmente la milza, per evitare effetti confondenti da steatosi epatica) è un indicatore accurato della presenza di grasso intra-cellulare;
- metastasi, carcinomi e feocromocitomi (non contenenti materiale lipidico) e adenomi lipid poor, che non presentano variazioni dell’intensità di segnale nelle immagini in opposizione di fase.
Nel differenziare gli adenomi dai non-adenomi tale metodica ha sensibilità dell’84-100% e specificità del 92-100%, simile alla TC senza mdc.
La 18F-FDG-PET è considerato il migliore esame per caratterizzare lesioni risultate “indeterminate” con le altre metodiche. Si basa sull’aumentata captazione di glucosio radio-marcato da parte dei tumori rispetto al tessuto normale; la misurazione quantitativa della concentrazione di 18F nei tessuti viene espressa mediante un indice, detto SUV (standard uptake value), valutato rispetto alla captazione a livello epatico: SUV compresi tra 1.45 e 1.8 sono indicativi di lesione benigna (28-30).
Tuttavia, queste tre metodiche, da sole, possono non rivelarsi sufficienti a definire una diagnosi di certezza. Anche nella stadiazione e nella valutazione delle recidive è raccomandabile la loro integrazione. Nei casi fortemente sospetti per CCS è importante una valutazione pre-operatoria anche del distretto toraco-addomino-pelvico, poichè nella maggior parte dei casi eventuali metastasi si localizzano a questo livello.
L’11C-metomidate, marcatore dell’11β-idrossilasi, è un tracciante PET in grado di differenziare le lesioni cortico-surrenaliche dai feocromocitomi e dalle metastasi riconducibili ad altre neoplasie, con sensibilità e specificità dell’89% e 96%, rispettivamente. Purtroppo, l’11C-metomidate non è in grado di discriminare le lesioni benigne da quelle maligne della corticale (31).
Istologia e fattori prognostici
La diagnosi istopatologica è spesso complicata a causa dell’eterogeneità di tali tumori, pertanto, nei casi dubbi, dovrebbe essere presa in considerazione una revisione da parte di anatomo-patologi esperti.
L’analisi immuno-istochimica di SF-1 (steroidogenic factor-1), fattore di crescita coinvolto nello sviluppo del tessuto surrenalico e nella regolazione della steroidogenesi, è il marcatore più sensibile e specifico, in grado di stabilire l’origine surrenalica del tumore con sensibilità del 98% e specificità del 100% (32); se non disponibile, possono essere utilizzati altri marcatori, come alfa-inibina, melan-A e calretinina.
È fondamentale che il referto istologico comprenda: il punteggio di Weiss con l’esatta conta delle mitosi, la positività per Ki67, lo stato dei margini di resezione chirurgica, la stadiazione patologica (invasione dei tessuti circostanti) e il coinvolgimento linfonodale.
Per la diagnosi anatomo-patologica delle lesioni surrenaliche è attualmente raccomandato il punteggio di Weiss (tab 2), basato sulla combinazione di nove criteri istologici patognomonici di malignità: un punteggio > 3 è indicativo di CCS, se è uguale a 3 è indicativo di tumore indeterminato. L’applicazione di tale punteggio richiede esperienza da parte del patologo e in alcune varianti è difficilmente riproducibile: sono stati evidenziati falsi negativi nella variante mixoide (per la difficoltà nell’identificare alcuni parametri) e falsi positivi nella variante oncocitica (per la presenza intrinseca di tre parametri, citoplasma eosinofilo, crescita diffusa ed atipie).
| Tabella 2 Criteri di Weiss |
||
| Criterio | Punteggio | |
| Struttura tumorale | Architettura con pattern diffuso > 33% | 1 |
| Cellule chiare < 25% | 1 | |
| Necrosi | 1 | |
| Citologia | Atipia nucleare | 1 |
| Mitosi > 5/50 campi ad alto ingrandimento (HPF) | 1 | |
| Mitosi atipiche | 1 | |
| Invasione | Venosa | 1 |
| Sinusale | 1 | |
| Capsulare | 1 | |
| Punteggio totale | 0-2: benigno 3: indeterminato > 3: maligno |
|
Pertanto, in alcuni casi dubbi, possono essere di ausilio altri sistemi di classificazione: indice di van Slooten, punteggio di Weiss modificato, punteggio di Helsinki e valutazione del reticolo. Quest’ultimo, detto anche “algoritmo del reticolo” si basa su due tappe: l’analisi della struttura del reticolo e successivamente, l’identificazione di almeno uno tra i tre criteri di malignità (necrosi, alto tasso mitotico e invasione venosa). L’accuratezza diagnostica è simile a quella del punteggio di Weiss, ma si è dimostrato più rapido, semplice da interpretare e riproducibile e la sua efficacia è stata confermata anche nelle varianti oncocitiche (33).
Riguardo al ruolo prognostico del punteggio di Weiss, alcuni studi hanno evidenziato come un valore > 6 sia associato a ridotta sopravvivenza libera da malattia (recurrence free survival – RFS) e sopravvivenza globale (overall survival – OS), altri invece hanno dato risultati contrastanti, pertanto non è raccomandato per la stratificazione prognostica (34). Tra i diversi criteri, solo l’attività mitotica si è dimostrata correlata alla prognosi ed è stato proposto un cut-off di 20 mitosi/50 HPF per differenziare basso e alto grado, anche se tale misurazione presenta limiti legati soprattutto alla scarsa riproducibilità (34).
Lo studio immuno-istochimico della proteina Ki-67 rappresenta una valida alternativa per la valutazione della proliferazione cellulare e del grading tumorale, ma la sua misurazione non è standardizzata e la sua distribuzione all’interno del tumore è eterogenea, per cui è necessaria la sua valutazione in diverse aree tumorali. È stato ampiamente dimostrato come l’espressione di Ki-67 svolga un importante ruolo prognostico nel predire il rischio di recidiva: Ki-67 > 20% si associa a prognosi peggiore, mentre Ki-67 < 10% si associa a basso rischio di recidiva (35). Una recente analisi multivariata conferma il suo ruolo prognostico patologico (36):
- grado I: Ki-67 < 10%;
- grado II: Ki-67 10-19%;
- grado III: Ki-67 ≥ 20%.
Oltre al punteggio di Weiss e all’espressione di Ki-67, le linee guida raccomandano che un referto anatomo-patologico includa anche lo stato dei margini di resezione, lo stadio tumorale e il convolgimento linfonodale, in quanto tutti questi parametri sono necessari per la stratificazione prognostica e quindi per la corretta gestione dei pazienti.
Lo stadio tumorale è il più importante fattore prognostico indipendente di sopravvivenza nei pazienti affetti da CCS. Nel 2008, l’European Network for the Study of Adrenal Tumors (ENSAT) ha proposto un sistema di classificazione riconosciuto oggi più accurato e più aderente alla prognosi del paziente, che ha sostituito i precedenti (37,38). La classificazione TNM proposta dall’ENSAT rispetto agli altri sistemi fornisce una più precisa differenziazione tra i diversi stadi; in particolare, ha meglio differenziato lo stadio III che comprende il tumore localmente avanzato (infiltrazione del tessuto circostante, trombosi cavale o renale, coinvolgimento linfonodale), dallo stadio IV che si ha solo in presenza di metastasi a distanza (tab 3). Quest’ultima revisione risulta attualmente la migliore nella predizione di mortalità cancro-specifica per CCS e ha consentito di definire una differente sopravvivenza stadio-dipendente a 5 anni (81%, 61%, 50%, 13%, rispettivamente, dallo stadio I allo stadio IV) (14,16,34,39).
| Tabella 3 Stadiazione ENSAT |
||
| Tumore | T1 | ≤ 5 cm |
| T2 | > 5 cm | |
| T3 | Infiltrazione neoplastica del tessuto circostante | |
| T4 | Invasione organi adiacenti o trombosi venosa cavale o renale | |
| Linfonodi | N0 | Nessun linfonodo coinvolto |
| N1 | Presenza di linfonodi positivi | |
| Metastasi | M0 | Nessuna localizzazione a distanza |
| M1 | Presenza di metastasi a distanza | |
| Stadio | I | T1, N0, M0 |
| II | T2, N0, M0 | |
| III | T1-T2, N1, M0 T3-T4, N0-N1, M0 |
|
| IV | T1-4, N0-N1, M1 | |
Importante è la definizione dei margini di resezione tumorale:
- R0: la resezione non coinvolge microscopicamente il tessuto neoplastico;
- R1: i margini sono coinvolti microscopicamente;
- R2: i margini sono coinvolti macroscopicamente;
- Rx: indica stato non noto dei margini di resezione.
Nel 20-30% dei casi R1/R2 sono associate a peggiore sopravvivenza, con progressiva riduzione di RFS, indipendentemente dagli altri fattori di rischio.
Oltre ai citati fattori, anche alcuni parametri clinici concorrono a determinare la prognosi:
- età: diversi studi hanno dimostrato che i pazienti molto giovani presentano prognosi più favorevole rispetto a quelli di età più avanzata, a suggerire un differente assetto molecolare (34);
- ipercortisolismo: l’esatto ruolo dell’ormono-sintesi non è chiaro; sebbene l’ipercortisolismo permetta in genere una diagnosi più precoce rispetto alle forme silenti, si associa a maggiori complicanze post-operatorie (infezioni, aumentato catabolismo proteico) e potrebbe essere responsabile di uno stato di immuno-soppressione in grado di favorire lo sviluppo del tumore. Comunque, le forme tumorali secernenti sono associate a comportamento tumorale più aggressivo anche dopo la risoluzione post-chirurgica dell’ipercortisolismo (40).
Infine, sono stati proposti diversi marcatori molecolari come possibili predittori di esito: metallo-proteasi tipo 2, GLUT1, SF1, BUB1B, micro-RNA (16). Ne è stato ipotizzato un potenziale come bersaglio terapeutico, ma al momento nessuno di essi ha ancora un ruolo preciso nella gestione clinica dei pazienti affetti da CCS.
Le ultime linee guida ESE/ENSAT sul CCS suggeriscono che i parametri sovra-descritti permettono di eseguire una stratificazione prognostica, che può essere d’ausilio nella gestione terapeutica dei pazienti affetti da CCS, basata sulla valutazione di:
- rischio di recidiva dei pazienti con CCS localizzato (in stadio I-III), per cui il panel distingue due classi di rischio:
- basso-moderato: stadio I-II, R0 e Ki-67 ≤ 10%;
- alto: stadio III, R1, o Ki-67 >10%;
- prognosi dei pazienti affetti da malattia avanzata (stadio IV o recidive ricorrenti non suscettibili di resezione completa o R2): peggiore con alto grado tumorale, elevata espressione di Ki-67 e sintomi non controllabili.
Nei pazienti sottoposti a intervento chirurgico radicale, in caso di recidiva di malattia, il più importante fattore prognostico è l’intervallo libero da malattia, insieme al quadro clinico, alla recidiva e alla resecabilità della stessa. Per guidare la strategia terapeutica, si raccomanda di ridefinire la prognosi a ogni evento significativo della storia del paziente (14).
Nei pazienti adulti con CCS si raccomanda di indagare la storia personale e familiare di neoplasie, al fine di individuare una eventuale predisposizione ereditaria.
Bibliografia
- Else T, Kim AC, Sabolch A, et al. Adrenocortical carcinoma. Endocr Rev 2014, 35: 282–326.
- Bielinska M, Parviainen H, Kiiveri S, et al. Origin and molecular pathology of adrenocortical neoplasms. Vet Pathol 2009, 46: 194-210.
- Patalano A, Brancato V, Mantero F. Adrenocortical cancer treatment. Hormone Res 2009, 71: 99-104.
- Berruti A, Baudin E, Gelderblom H, et al, on behalf of the ESMO Guideline Working Group. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012, 23 suppl 7: 131–8.
- Figueiredo BC, Stratakis CA, Sandrini R, et al. Comparative genomic hybridization analysis of adrenocortical tumors of childhood. J Clin Endocrinol Metab 1999, 84: 1116-21.
- Gicquel C, Bertagna X, Le Bouc Y. Recent advances in the pathogenesis of adrenocortical tumors. Eur J Endocrinol 1995, 133: 133–44.
- Yano T, Linehan M, Anglard P, et al. Genetic changes in human adrenocortical carcinomas. J Natl Cancer Inst 1989, 81: 518–23.
- Libè R, Fratticci A, Bertherat J. Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer 2007, 14: 13-28.
- Herrmann LJ, Heinze B, Fassnacht M, et al. TP53 germline mutations in adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab 2012, 97: E476–85.
- Fassnacht M, Allolio B. Clinical management of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab 2009, 23: 273-89.
- Allolio B, Fassnacht M. Clinical review: adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 2006, 91: 2027–37.
- Egoshi K, Masai M, Nagao K, Ito H. 11-Deoxycorticosterone-producing adrenocortical carcinoma. Urol Int 1998, 61: 251–3.
- Müssig K, Wehrmann M, Horger M, et al. Adrenocortical carcinoma producing 11-deoxycorticosterone: a rare cause of mineralocorticoid hypertension. J Endocrinol Invest 2005, 28: 61–5.
- Fassnacht M, Dekkers OM, Else T, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018, 179: G1-46.
- Terzolo M, Alì A, Osella G, et al. The value of dehydroepiandrosterone sulfate measurement in the differentiation between benign and malignant adrenal masses. Eur J Endocrinol 2000, 142: 611–7.
- Stigliano A, Chiodini I, Giordano R, et al. Management of adrenocortical carcinoma: a consensus statement of the Italian Society of Endocrinology (SIE). J Endocrinol Invest 2016, 39: 103-21.
- Terzolo M, Zaggia B, Daffara F, et al. Diagnosi e terapia del carcinoma corticosurrenalico. L'Endocrinologo 2014, 15: 22-7.
- Petersenn S, Richter PA, Broemel T, et al, for the German ACC Study Group. Computer tomography criteria for discrimination of adrenal adenomas and adrenocortical carcinomas: analysis of the German ACC registry. Eur J Endocrinol 2015, 172: 415–22.
- Lee MJ, Hahn PF, Papanicolau N, et al. Benign and malignant adrenal masses: CT distinction with attenuation coefficients, size, and observer analysis. Radiology 1991, 179: 415–8.
- Boland GW, Lee MJ, Gazelle GS, et al. Characterization of adrenal masses using unenhanced CT: an analysis of the CT literature. AJR 1998, 171: 201–4.
- Casola G, Nicolet V, van Sonnenberg E, et al. Unsuspected pheochromocytoma: risk of blood-pressure alterations during percutaneous adrenal biopsy. Radiology 1986, 159: 733-5.
- Park BK, Kim CK, Kwon GY, et al. Re-evaluation of pheochromocytomas on delayed contrast enhanced CT: washout enhancement and other imaging features. Eur Radiol 2007, 17: 2804–9.
- Terzolo M, Alì A, Osella G, et al. Prevalence of adrenal carcinoma among incidentally discovered adrenal masses. A retrospective study from 1989 to 1994. Arch Surg 1997, 132: 914-9.
- Zhang HM, Perrier ND, Grubbs EG, et al. CT features and quantification of the characteristics of adrenocortical carcinomas on unenhanced and contrast-enhanced studies. Clin Radiol 2012, 67: 38–46.
- Korobkin M, Brodeur FJ, Francis IR, et al. CT time-attenuation washout curves of adrenal adenomas and nonadenomas. AJR Am J Roentgenol 1998, 170: 747–52.
- Park BK, Kim CK, Kim B, Lee JH. Comparison of delayed enhanced CT and chemical shift MR for evaluating hyperattenuating incidental adrenal masses. Radiology 2007, 243: 760–5.
- Kandathil A, Wong K, Wale DJ, et al. Metabolic and anatomic characteristics of benign and malignant adrenal masses on positron emission tomography/computed tomography: a review of literature. Endocrine 2014, 49: 6-26.
- Ardito A, Massaglia C, Pelosi E, et al. 18F-FDG PET/CT in the post-operative monitoring of patients with adrenocortical carcinoma. Eur J Endocrinol 2015, 173: 749–56.
- Takeuchi S, Balachandran A, Habra MA, et al. Impact of 18F-FDG PET/CT on the management of adrenocortical carcinoma: analysis of 106 patients. Eur J Nucl Med Mol Imaging 2014, 41: 2066–73.
- Leboulleux S, Dromain C, Bonniaud G, et al. Diagnostic and prognostic value of 18-fluorodeoxyglucose positron emission tomography in adrenocortical carcinoma: a prospective comparison with computed tomography. J Clin Endocrinol Metab 2006, 91: 920–5.
- Hennings J, Lindhe O, Bergstrom M, et al. [11C]metomidate positron emission tomography of adrenocortical tumors in correlation with histopathological findings. J Clin Endocrinol Metab 2006, 91: 1410-4.
- Sbiera S, Schmull S, Assie G, et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J Clin Endocrinol Metab 2010, 95: 161–71.
- Duregon E, Fassina A, Volante M, et al. The reticulin algorithm for adrenocortical tumors diagnosis. A multicentric validation study on 245 unpublished cases. Am J Surg Pathol 2013, 37: 1433-40.
- Jouinot A, Bertherat J. Adrenocortical carcinoma: differentiating the good from the poor prognosis tumors. Eur J Endocrinol 2018, 178: R215-30.
- Beuschlein F, Weigel J, Saeger W, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab 2015, 100: 841–9.
- Morimoto R, Satoh F, Murakami O, et al. Immunohistochemistry of a proliferation marker Ki67/MIB1 in adrenocortical carcinomas: Ki67/MIB1 index is a predictor for recurrence of adrenocortical carcinomas. Endocr J 2008, 55: 49-55.
- Macfarlane DA. Cancer of the adrenal cortex. The natural history, prognosis and treatment in a study of fifty–five cases. Ann R Coll Surg Engl 1958, 23: 155–86.
- Sullivan M, Boileau M, Hodges CV. Adrenal cortical carcinoma. J Urol 1978, 120: 660-5.
- Lughezzani G, Sun M, Perrotte P, et al. The European Network for the Study of Adrenal Tumors staging system is prognostically superior to the international union against cancer-staging system: a North American validation. Eur J Cancer 2010, 46: 713-9.
- Berruti A, Fassnacht M, Haak H, et al. Prognostic role of overt hypercortisolism in completely operated patients with adrenocortical cancer. Eur Urol 2014, 65: 832–8.
Terapia carcinoma surrenalico
Pina Lardo & Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare, Ospedale Sant'Andrea, Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 18 gennaio 2021)
Obiettivi
Sono finalizzati a:
- rimozione della neoplasia;
- riduzione e/o remissione dei segni e sintomi clinici dell’ipercortisolismo;
- controllo della malattia a lungo termine.
Terapia chirurgica
L’intervento chirurgico radicale è l’unico potenziale approccio curativo con il maggiore impatto sulla prognosi (1). Pre-requisito fondamentale è l’esperienza chirurgica in ambito surrenalico e oncologico, a causa dell’anatomia specifica, dell’aggressività della neoplasia e della possibilità di dover eseguire una resezione in blocco multi-organo per garantire un risultato R0 (margini chirurgici indenni) con il minimo rischio di complicanze. Secondo le ultime linee guida, è richiesto un minimo di 6 surrenectomie/anno per garantire un’esperienza sufficiente nella chirurgia surrenalica, mentre sono richiesti > 20 interventi/anno per chi si occupa di chirurgia del CCS. Pertanto, è fondamentale che i pazienti con sospetto CCS siano gestiti presso centri di riferimento (2).
La probabilità di ottenere la guarigione è affidata alla radicalità dell’intervento chirurgico e agli stadi minori della neoplasia (I e II). Nonostante R0 sia considerato un parametro importante di sopravvivenza a lungo termine, recidiva loco-regionale o comparsa di metastasi a distanza rappresentano un'eventualità comune (85%), anche dopo resezione completa del tumore (3,4). Le probabilità di fallimento aumentano negli stadi avanzati della neoplasia: generalmente per masse > 12 cm di diametro, con elevato indice di proliferazione ed emorragie intra-lesionali (5,6).
Non è raccomandata l’enucleazione del nodo neoplastico o la resezione parziale del surrene, così come non è consigliata l’asportazione di routine del rene omolaterale in assenza di evidente infiltrazione. È di fondamentale importanza oncologica evitare la frammentazione tumorale, in quanto la disseminazione cellulare intra-operatoria ed R2 sono associati ad alto tasso di recidiva e ridotta sopravvivenza (7). Pertanto, è raccomandato l’approccio “open” nei casi di CCS confermato o fortemente sospetto e/o quando vi sia evidenza di invasione locale o sospette metastasi linfonodali (ENSAT stadio III) (8-10).
Considerando che la probabilità che una lesione sia benigna è maggiore per gli incidentalomi con diametro ≤ 6 cm (stadio ENSAT I e II), anche se tale cut-off è arbitrario e l’esperienza chirurgica è il fattore più importante, in questi casi è ragionevole un approccio laparoscopico, sempre dopo aver escluso un’invasione locale o linfonodale. La decisione a favore di questa scelta deriva dalla constatazione che per tumori > 6 cm è più alto il rischio di rottura capsulare durante l’intervento per via laparoscopica. In ogni caso, l’intervento deve essere immediatamente convertito in “open” se vi è rischio di rottura capsulare o disseminazione cellulare.
Nel 15-25% dei casi di CCS si verifica una trombosi venosa (surrenalica, renale o della cava inferiore). In queste situazioni l’approccio chirurgico deve essere individualizzato. Il tasso di sopravvivenza del 25% a 3 anni incoraggia la resezione venosa nei casi di interessamento della vena renale o della cava inferiore in assenza di metastasi a distanza.
Nei casi di resezione macroscopicamente incompleta (R2) è suggerito un nuovo ricorso alla chirurgia, rivolto a ottenere uno stadio R0. La presenza di recidive locali, in assenza di altre metastasi, indica una nuova chirurgia “open”. Nel caso di metastasi epatiche e/o polmonari deve essere presa in considerazione la resezione se è raggiungibile l’R0 e se l’asportazione si associa a basso tasso di mortalità e morbilità. Non è raccomandata, invece, la resezione di routine del tumore primitivo asintomatico in presenza di metastasi non resecabili, né il debulking o la resezione R2 del tumore primitivo, delle recidive e delle metastasi. Il debulking può essere considerato eccezionalmente per i tumori di grandi dimensioni, sintomatici e/o secernenti resistenti alla terapia medica, quando almeno l’80% della lesione è resecabile, valutando il rischio relativo di morbilità.
Qualunque sia l’approccio chirurgico, nei pazienti con ipercortisolismo è sempre indicata la terapia sostitutiva con glucocorticoidi, preferibilmente idrocortisone, in fase pre- e post-operatoria, per il rischio di insufficienza surrenalica conseguente alla resezione della lesione secernente (11).
Terapia adiuvante con mitotane
Cosa è. Il mitotane (MTT) appartiene alla categoria dei farmaci adrenolitici, capaci cioè di esercitare un effetto cito-tossico specifico diretto sulle cellule della corticale del surrene, provocando una degenerazione focale della zona fascicolata e maggiormente della zona reticolare della ghiandola (12,13), mentre gli effetti sulla zona glomerulare sono relativamente scarsi (14). È assunto per via orale, assorbito a livello del tratto gastro-intestinale per circa il 40% ed è immagazzinato soprattutto nel tessuto adiposo, a causa della lipofilicità.
Come agisce. Gli effetti sulla steroidogenesi sono conseguenti sia all’azione adrenolitica che alla capacità di inibire diversi enzimi steroidogenetici, principalmente il CYP11A1 e il CYP11B1 (15), effetti attraverso i quali è in grado di controllare l'ipersecrezione ormonale che di frequente si associa ai CCS funzionanti. Oltre gli effetti adrenolitici, il MTT possiede la capacità di potenziare l’azione di diversi farmaci chemioterapici (16).
A chi darlo. La terapia adiuvante con MTT è suggerita dopo l’intervento chirurgico nei pazienti senza residui macroscopici di malattia, ad alto rischio di recidiva (stadio III, o R1, o Ki-67 > 10%), mentre non ci sono evidenze sufficienti per potersi esprimere a favore o contro il suo utilizzo nei pazienti con rischio basso-moderato (stadio I-II, R0 e Ki-67 ≤ 10%) e le opzioni vanno discusse caso per caso (2). In tal senso, il primo studio prospettico randomizzato controllato (ADIUVO), recentemente terminato, ha valutato la sopravvivenza libera da malattia (RFS) della terapia adiuvante con MTT in pazienti con rischio medio-basso di recidiva (stadio I-III, R0 e Ki-67 ≤ 10%) (2). Tra gli studi attualmente in corso, ADIUVO-2 è un trial randomizzato che valuta l’efficacia della terapia adiuvante con MTT versus MTT + cisplatino-etoposide nei casi di CCS localizzato, ad alto rischio di recidiva (stadio I-III e Ki-67 > 10%).
Quando darlo. La terapia adiuvante con MTT deve essere iniziata precocemente dopo l’intervento chirurgico (idealmente entro 6 settimane, comunque non oltre 3 mesi) e proseguita per almeno 2 anni (a meno di effetti collaterali importanti), fino ad un massimo di 5 anni.
Come darlo. Si raccomanda di iniziare il trattamento con la dose massima tollerata, da incrementare progressivamente fino al raggiungimento del target terapeutico. In corso di terapia è necessario il monitoraggio dei livelli ematici del farmaco, con l’obiettivo di raggiungere concentrazioni ematiche comprese tra 14 e 20 mg/L, valore cui corrisponde la massima risposta anti-neoplastica e la migliore sopravvivenza. Sarebbe da evitare il superamento di questi livelli, che si associa a maggiore tossicità.
Attenzione. Tutti i pazienti in terapia con MTT devono ricevere una supplementazione con glucocorticoidi perché a rischio di insufficienza surrenalica: cortisone acetato o meglio idrocortisone devono essere somministrati a dosaggi maggiori rispetto ai pazienti affetti da insufficienza surrenalica di altra origine a causa dell’aumentata clearance metabolica dei glucocorticoidi indotta dal MTT. Diversi meccanismi portano a ridotta disponibilità di cortisolo:
- inibizione della steroidogenesi;
- induzione del CYP3A4, con conseguente rapida inattivazione di oltre il 50% della dose somministrata di idrocortisone;
- aumento dei livelli di CBG (cortisol-binding globulin);
- capacità di influenzare anche il metabolismo periferico del cortisolo attraverso l’induzione degli enzimi microsomiali epatici, determinando riduzione dei metaboliti urinari del cortisolo e aumento della produzione a livello epatico del 6β-idrossicortisolo, metabolita inattivo del cortisolo (17).
Visto il minore effetto del MTT sulla zona glomerulare, non è obbligatoria in tutti i pazienti la supplementazione con mineralcorticoidi, che dovrà essere guidata dal monitoraggio clinico e biochimico.
Anche la funzione gonadica è spesso compromessa, perché il MTT è in grado di inibire la produzione steroidea gonadica attraverso un effetto diretto sulle cellule ovariche della granulosa e della teca e sulle cellule di Leydig (18) e indirettamente mediante un aumento della SHBG, con riduzione dei livelli di testosterone libero, e inibendo la 5α-reduttasi, con conseguente ginecomastia. Inoltre, è possibile il riscontro di un quadro di ipotiroidismo centrale con ridotti livelli di FT4 senza aumento del TSH, dovuti all’azione inibente del MTT o all’aumento dei livelli di TBG. Tali condizioni devono essere valutate ogni 4-6 mesi ed eventualmente trattate (19).
La gestione della terapia con MTT è complessa e prevede una grande esperienza da parte dell’endocrinologo. È necessario che il paziente sia informato sugli effetti del farmaco, sulla corretta assunzione, generalmente dopo il pasto, e sulla necessità di monitorare periodicamente la concentrazione ematica del farmaco e alcuni parametri biochimici (emocromo, funzione epatica, lipidi, elettroliti). Il MTT è gravato da effetti collaterali più comuni ed altri che compaiono quando viene superata la concentrazione di 20 mg/L. Quelli più frequenti sono i disturbi gastro-intestinali indipendenti dai livelli di mitotanemia (nausea, vomito, anoressia, diarrea), che possono essere gestiti con terapia sintomatica o modulando la posologia del farmaco. La tossicità epatica è rara, mentre l’incremento dei valori di gamma-GT è sempre presente ma senza impatto clinico. La tossicità neurologica, con atassia, confusione, disgrafia, fatigue, era osservata più frequentemente in passato, per il superamento dei valori target del farmaco (12,20,21). Essendo il MTT un potente induttore di diversi enzimi epatici mitocondriali e del CYP3A4, è necessario tenere in considerazione questo aspetto nel valutare le terapie concomitanti somministrate (22-24).
Terapia radiante
Il ruolo della radioterapia (RT) nell'ambito del CCS non è stato ancora definito e i dati a riguardo sono contrastanti: alcuni autori la considerano inefficace (25,26), altri ne hanno documentato un'azione terapeutica, sostenendo la radio-sensibiltà del tumore (11,27).
Le linee guida suggeriscono la RT adiuvante in associazione al MTT in pazienti con resezione R1 o Rx o in stadio III, mentre non è consigliata di routine in pazienti in stadio I-II e resezione R0 (2).
Il trattamento combinato radioterapia + MTT è biologicamente sinergico, perchè il farmaco sembra avere un effetto radio-sensibilizzante sulle cellule neoplastiche, potenziandone l’effetto anti-proliferativo (28). Un recente studio pre-clinico nell’animale ha dimostrato che il trattamento combinato inibisce la crescita del CCS, la steroidogenesi e interferisce in vitro con il ciclo cellulare (29).
I dati della letteratura mostrano come la RT sia in grado di prevenire le recidive locali, senza però un effetto sulle metastasi a distanza né sulla sopravvivenza globale (30).
Questo trattamento, quando indicato, dovrebbe essere iniziato il prima possibile dopo la chirurgia, secondo gli schemi della terapia adiuvante, alla dose di 50-60 Gy in frazioni da circa 2 Gy (31-33).
La RT trova attualmente la sua indicazione clinica anche nel trattamento delle recidive loco-regionali sintomatiche e nel trattamento delle metastasi ossee, cerebrali e in altre sedi (34,35).
Per l’alta affinità di legame all’11ß-idrossilasi e all’aldosterone-sintetasi surrenalici, recentemente è stato sperimentato, su piccole casistiche di pazienti affetti da malattia avanzata, l’impiego del 131I-iodometomidate come terapia radiometabolica. I risultati ottenuti sono promettenti ma necessitano di studi clinici di conferma (36).
Terapia della malattia avanzata o recidivante
L’indicazione principale per il CCS metastatico è rappresentata da MTT.
Nei pazienti con malattia oligo-metastatica si suggerisce, se possibile, di procedere alla resezione chirurgica delle lesioni. Non è raccomandato, invece, l’intervento chirurgico nei casi di malattia diffusa con numerose localizzazioni secondarie.
In caso di mancata risposta alla terapia con MTT, associata o meno a terapie loco-regionali, è consigliato aggiungere la combinazione etoposide-doxorubicina-cisplatino (EDP-M), che ha mostrato i migliori risultati nello studio prospettico randomizzato di fase III, FIRM-ACT (37). Considerata l’importante tossicità del regime EDP-M, possono essere considerate opzioni alternative che prevedono l’impiego di uno o due di questi farmaci da soli o in combinazione con MTT.
Nei pazienti con recidiva e un intervallo libero da malattia di almeno 12 mesi, è raccomandata, se possibile, la resezione chirurgica e/o altre terapie loco-regionali, in quanto tale condizione si associa a una migliore prognosi e risposta alle terapie locali; se l’intervallo libero da malattia è < 6 mesi, come prima scelta è raccomandato lo schema EDP+MTT.
Per il controllo della malattia avanzata trovano impiego in associazione alla chirurgia altre terapie loco-regionali (es. radioterapia, radiofrequenza, crio-ablazione, ablazione con micro-onde, chemio-embolizzazione). Non esistono studi di confronto delle diverse tecniche, pertanto la scelta della metodica va personalizzata con un team multi-disciplinare esperto, in base a caratteristiche della malattia, esperienza dei centri e preferenze del paziente.
Nei pazienti in progressione in corso di EDP-M, si suggerisce di considerare l’inserimento in studi clinici. In caso di mancata risposta al suddetto regime chemioterapico, sono da considerare due regimi citotossici di seconda linea: gemcitabina + capecitabina (± MTT) e streptozocina + MTT (37). Il razionale alla base dell’associazione con MTT deriva dalla dimostrazione in vitro della capacità di potenziare l’efficacia dei farmaci chemioterapici bloccando la proteina di resistenza ai farmaci 1/P-glicoproteina attiva nel trasporto dei farmaci cito-tossici fuori dalla cellula (16). La prosecuzione del trattamento con MTT è controversa: alcuni autori suggeriscono una continuazione per 2-3 anni dall’inizio del trattamento, altri preferiscono la prosecuzione della terapia per almeno 5 anni (37).
Follow-up
Dopo l’intervento chirurgico si suggerisce l’esecuzione di un imaging radiologico ogni tre mesi per due anni, poi ogni 3-6 mesi per ulteriori 3 anni; la maggior parte degli autori suggerisce comunque una prosecuzione del follow-up radiologico per i 5 anni successivi mediante un imaging annuale. Anche se non vi sono dati pubblicati, ciò deriva dalla considerazione che solo pochi CCS recidivano dopo i 5 anni e quindi una sorveglianza a 5 anni permetterebbe di includere più del 90% delle recidive. In ogni caso, la tempistica dovrà essere individualizzata sulla base del trattamento in atto e sulla risposta ad esso. È infatti importante una ri-classificazione del rischio ad ogni follow-up e la presenza di recidiva è il maggior fattore prognostico sfavorevole, in particolare il minor tempo intercorso tra chirurgia iniziale e comparsa di recidiva. Anche la valutazione ormonale è importante, in quanto può permettere una diagnosi precoce delle recidive, oltre che richiedere terapie farmacologiche specifiche (38).
Terapie targeted
I recenti progressi nella conoscenza delle alterazioni genetiche coinvolte nel CCS hanno permesso l’identificazione di bersagli molecolari per lo sviluppo di una terapia selettiva. Le strategie molecolari hanno preso in considerazione alcune delle vie più frequentemente coinvolte nell’oncogenesi cortico-surrenalica: le più promettenti sono quelle dell’IGF-2 (39,40), del VEGF e del suo recettore VEGF-R (41), di Wnt/ß-catenina (42).
Studi recenti di fase I e II hanno valutato l’efficacia di figitumumab, anticorpo monoclonale anti-IGF-1R, e di OSI-906, inibitore tirosin-chinasico anti-IGF-1R, mostrandone una parziale efficacia (43). Discordanti sono i risultati ottenuti da studi che hanno valutato l’efficacia di sunitinib e sorafenib, inibitori tirosin-chinasici anti-VEGF-R (41).
L’immuno-terapia, ovvero la stimolazione della risposta immune verso antigeni cellulari neoplastici specifici, potrebbe rappresentare una nuova opzione terapeutica, ma uno studio in tal senso non ha mostrato benefici (44). Sono attualmente in corso studi per la valutazione dell’effetto combinato immuno-terapia e inibitori tirosin-chinasici (studio clinico E02401: NCT04187404).
Quindi, pur rappresentando il futuro nella ricerca farmacologica del CCS, al momento l’impiego di queste molecole è riservato solo ai trial clinici dedicati.
Terapia per l'eccesso ormonale
Nei CCS secernenti è raccomandata la terapia farmacologica per ridurre il rischio di complicanze legate all’eccesso ormonale.
Tenendo conto dei suoi tempi di azione, il MTT può non essere sufficiente, in particolare nelle forme di ipercortisolismo severo. Gli inibitori della steroidogenesi e gli antagonisti dei recettori steroidei, in associazione o meno al MTT, sono in grado di controllare i livelli ormonali. Nonostante l’assenza di studi clinici comparativi, la maggior parte degli autori delle ultime linee guida considera il metirapone il farmaco di scelta (2), ben tollerato e somministrabile in associazione a MTT e chemioterapia (26). Il chetoconazolo, inibitore della steroidogenesi a diversi livelli, è una possibile alternativa in grado di inibire anche la quota androgenica, ma richiede un attento monitoraggio della funzionalità epatica, e dovrebbe essere evitato nella fase iniziale del trattamento con MTT, essendo entrambi epato-tossici.
Il mifepristone, antagonista del recettore dei glucocorticodi, è anch’esso impiegato nella terapia ormonale del CCS. Questo farmaco, non disponibile in Italia, è comunque scarsamente impiegato per la difficoltà di gestione clinica.
Nei casi di eccesso androgenico, è possibile valutare il ricorso a farmaci ad attività anti-androgenica (bicalutamide, flutamide o spironolattone), avendo cura di selezionare il paziente sulla base del quadro clinico e delle interazioni farmacologiche.
Altri trattamenti
Si raccomanda di intraprendere terapia anti-riassorbitiva ossea nei pazienti con ipercortisolismo endogeno e nelle metastasi ossee.
Si raccomandano cure palliative (fisiche, psichiche) volte a migliorare la qualità di vita dei pazienti e delle famiglie.
Si suggerisce un servizio di counseling per i soggetti in età fertile che debbano essere sottoposti a trattamenti sistemici (chemioterapia e mono-terapia con MTT).
Gravidanza
In caso di sospetto CCS rilevato in corso di gravidanza, si raccomanda di procedere con l’intervento appena possibile, indipendentemente dall’epoca della gravidanza.
Le pazienti devono essere informate della possibilità che una gravidanza possa favorire la recidiva della patologia o accelerarne la progressione.
Si raccomanda di evitare la gravidanza in corso di terapia con MTT. Sono da preferire misure contraccettive non contenenti estrogeni. Infatti, il CCS può esprimere recettori per estrogeni e vi sono dati pre-clinici (45) che mostrano la possibilità che gli estrogeni possano favorire lo sviluppo e la progressione tumorale.
Bibliografia
- Terzolo M, Daffara F, Ardito A, et al. Management of adrenal cancer: a 2013 update. J Endocrinol Invest 2014, 37: 207–17.
- Fassnacht M, Dekkers OM, Else T, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018, 179: G1-46.
- Fassnacht M, Johanssen S, Quinkler M, et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a revised TNM classification. Cancer 2009, 115: 243-50.
- Schulick RD, Brennan MF. Long–term survival after complete resection and repeat resection in patients with adrenocortical carcinoma. Ann Surg Oncol 1999, 6: 719–26.
- Stojadinovic A, Ghossein RA, Hoos A, et al. Adrenocortical carcinoma: clinical, morphologic, and molecular characterization. J Clin Oncol 2002, 20: 941-50.
- Harrison LE, Gaudin PB, Brennan MF. Pathologic features of prognostic significance for adrenocortical carcinoma after curative resection. Arch Surg 1999, 134: 181–5.
- Crucitti F, Bellantone R, Ferrante A, et al. The ACC Italian Registry Study Group. The Italian registry for adrenal cortical carcinoma: analysis of a multiinstitutional series of 129 patients. Surgery 1996, 119: 161–70.
- Fassnacht M, Arlt W, Bancos I, et al. Management of adrenal incidentalomas: European Society of Endocrinology clinical practice guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016, 175: G1–34.
- Zheng G, Li H, Deng J, et al. Open adrenalectomy versus laparoscopic adrenalectomy for adrenocortical carcinoma: a retrospective comparative study on short-term oncologic prognosis. Onco Targets Ther 2018, 11: 1625–32.
- Panjwani S, Moore MD, Gray KD, et al. The impact of nodal dissection on staging in adrenocortical carcinoma. Ann Surg Oncol 2017, 24: 3617-23.
- Fassnacht M, Allolio B. Clinical management of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab 2009, 23: 273-89.
- Hahner S, Fassnacht M. Mitotane for adrenocortical carcinoma treatment. Curr Opin Invest Drugs 2005, 6: 386–94.
- Touitou Y, Bogdan A, Luton JP. Changes in corticosteroid synthesis of the human adrenal cortex in vitro, induced by treatment with o,p'-DDD for Cushing's syndrome: evidence for the sites of action of the drug. J Ster Biochem 1978, 9: 1217-24.
- Bergenstal DM, Hertz R, Lipsett MB, Moy RH. Chemotherapy of adrenocortical cancer with o,p’-DDD. Ann Int Med 1960, 53: 672-82.
- Berruti A, Fassnacht M, Baudin E, et al. Adjuvant therapy in patients with adrenocortical carcinoma: a position of an international panel. J Clin Oncol 2010, 28: e401-2.
- Bates SE, Shieh CY, Mickley LA, et al. Mitotane enhances cytotoxicity of chemotherapy in cell lines expressing a multidrug resistance gene (mdr–1/P–glycoprotein) which is also expressed by adrenocortical carcinomas. J Clin Endocrinol Metab 1991, 73: 18–29.
- Terzolo M, Zaggia B, Allasino B, De Francia S. Practical treatment using mitotane for adrenocortical carcinoma. Curr Opin Endocrinol Diabetes Obes 2014, 21: 159-65.
- Sparagana M. Primary hypogonadism associated with o,p-DDD (mitotane) therapy. J Toxicol Clin Toxicol 1987, 25: 463–72.
- Zatelli MC, Gentilin E, Daffara F, et al. Therapeutic concentrations of mitotane (o,p'-DDD) inhibit thyrotroph cell viability and TSH expression and secretion in a mouse cell line model in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocrinology 2010, 151: 2453-61.
- Sbiera S, Leich E, Liebisch G, et al. Mitotane inhibits sterol-O-acyl transferase 1 triggering lipid-mediated endoplasmic reticulum stress and apoptosis in adrenocortical carcinoma cells. Endocrinology 2015, 156: 3895–908.
- Hermsen IG, Fassnachr M, Terzolo M, et al. Plasma concentrations of o,p’DDD, o,p’DDA and o,p’DDE as predictors of tumor response to mitotane in adrenocortical carcinoma: results of a retrospective ENS@T multicenter study. J Clin Endocrinol Metab 2011, 96: 1844-51.
- Kroiss M, Quinkler M, Lutz WK, et al. Drug interactions with mitotane by induction of CYP3A4 metabolism in the clinical management of adrenocortical carcinoma. Clin Endocrinol 2011, 75: 585–91.
- Volante M, Terzolo M, Fassnacht M, et al. Ribonucleotide reductase large subunit (RRM1) gene expression may predict efficacy of adjuvant mitotane in adrenocortical cancer. Clin Cancer Res 2012, 18: 3452–61.
- D'Avolio A, De Francia S, Basile V, et al. Influence of the CYP2B6 polymorphism on the pharmacokinetics of mitotane. Pharmacogenet Genomics 2013, 23: 293-300.
- Libè R, Fratticci A, Bertherat J. Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer 2007, 14: 13-28.
- Claps M, Cerri S, Grisanti S, et al. Adding metyrapone to chemotherapy plus mitotane for Cushing’s syndrome due to advanced adrenocortical carcinoma. Endocrine 2017, 61: 169-72.
- Sabolch A, Feng M, Griffith K, et al. Adjuvant and definitive radiotherapy for adrenocortical carcinoma. Int J Radiat Oncol Biol Phys 2010, 80: 1477-84.
- Cerquetti C, Sampaoli D, Amendola B, et al. Mitotane sensitizes adrenocortical cancer cells to ionizing radiations by involvement of the cyclin B1/CDK complex in G2 arrest and mismatch repair enzymes modulation. Int J Oncol 2010, 37: 493-501.
- Cerquetti L, Bucci B, Carpinelli G, et al. Antineoplastic effect of mitotane treatment/ionizing radiation in adrenocortical carcinoma: a preclinical study. Cancers (Basel) 2019, 11: 1768.
- Sabolch A, Else T, Griffith KA, et al. Adjuvant radiation therapy improves local control after surgical resection in patients with localized adrenocortical carcinoma. Int J Radiat Oncol Biol Phys 2015, 92: 252-9.
- Habra MA, Ejaz S, Feng L, et al. A retrospective cohort analysis of the efficacy of adjuvant radiotherapy after primary surgical resection in patients with adrenocortical carcinoma. J Clin Endocrinol Metab 2013, 98: 192–7.
- Luo Y, Chen SS, Zheng XG, et al. The efficacy of radiation therapy in adrenocortical carcinoma. A propensity score analysis of a population-based study. Medicine 2017, 96: e6741.
- Fassnacht M, Hahner S, Polat B, et al. Efficacy of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. J Clin Endocrinol Metab 2006, 91: 4501-4.
- Schteingart DE, Doherty GM, Gauger PG, et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer 2005, 12: 667–80.
- Polat B, Fassnacht M, Pfreundner L, et al. Radiotherapy in adrenocortical carcinoma. Cancer 2009, 115: 2816-23.
- Hahner S, Kreissl MC, Fassnacht M, et al. (131I)Iodometomidate for targeted radionuclide therapy of advanced adrenocortical carcinoma. J Clin Endocrinol Metab 2012, 97: 914-22.
- Fassnacht M, Terzolo M, Allolio B, et al, for the FIRM-ACT Study Group. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 2012, 366: 2189-97.
- Berruti A, Baudin E, Gelderblom H, et al, on behalf of the ESMO Guideline Working Group. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2012, 23 suppl 7: 131–8.
- Barlaskar FM, Spalding AC, Heaton JH, et al. Preclinical targeting of the type I Insulin-Like Growth Factor Receptor in adrenocortical carcinoma. J Clin Endocrinol Metab 2009, 94: 204-12.
- Ragazzon B, Assié G, Bertherat J. Transcriptome analysis of adrenocortical cancers: from molecular classification to the identification of new treatments. Endocr Related Cancer 2011, 18: R15-27.
- Tacon LJ, Prichard RS, Soon PSH, et al. Current and emerging therapies for advanced adrenocortical carcinoma. Oncologist 2011, 16: 36-48.
- Garber K. Drugging the Wnt pathway: problems and progress. J Natl Cancer Inst 2009, 101: 548-50.
- Haluska P, Worden F, Olmos D, et al. Safety, tolerability and pharmacokinetics of the anti-IGF-1E monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chem Pharm 2010, 65: 765-73.
- Papewalis C, Fassnacht M, Willenberg HS, et al. Dentritic cells as potential adjuvant for immunotherapy in adrenocortical carcinoma. Clin Endocrinol 2006, 65: 215-22.
- Sirianni R, Zolea F, Chimento A, et al. Targeting estrogen receptor reduces adrenocortical cancer (ACC) cell growth in vitro and in vivo: potential therapeutic role of selective estrogen receptor modulators (SERMs) for ACC treatment. J Clin Endocrinol Metab 2012, 97: 2238-50.
Scheda mitotane
Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
Meccanismo d'azione
Il mitotane, 1,1-dichloro-2-(o-chlorophenyl)-2-(p-chloro-phenyl) ethane (o,p’-DDD) appartiene alla categoria dei farmaci adrenolitici, capaci cioè di esercitare un effetto citotossico specifico diretto sulle cellule della corticale del surrene, provocando una degenerazione focale della zona fascicolata e maggiormente della zona reticolare della ghiandola (1,2); minori sono invece gli effetti sulla zona glomerulare (3). Il farmaco è quindi in grado di controllare l'eccesso ormonale che frequentemente accompagna il quadro clinico dei carcinomi funzionanti.
L'effetto adrenolitico si realizza attraverso attivazione metabolica mediante una reazione di idrossilazione seguita da una di deidroclorinazione con produzione di un acil-cloride. Questo composto reattivo libero lega covalentemente macromolecole intracellulari e proteine mitocondriali, esercitando il suo effetto biologico. Quindi l'attivazione del farmaco avviene principalmente all'interno dei mitocondri delle cellule della corticale del surrene, mentre una piccola quota subisce l'azione degli enzimi microsomiali epatici. L’attivazione enzimatica del mitotano nel fegato induce la formazione di due metaboliti, di cui uno attivo l’o,p’-DDA ed un altro inattivo l’o,p’-DDE (4,5).
La somministrazione del farmaco altera il metabolismo extra-surrenalico del cortisolo, con conseguente riduzione dei 17-idrossi-corticosteroidi (1). Il mitotano induce un aumento delle proteine di trasporto degli ormoni. Esso, inoltre, esercita effetti inibitori anche a livello delle altre ghiandole endocrine steroido–secernenti, con relativa inibizione della produzione testicolare degli androgeni, comportandosi da antagonista sul recettore per il progesterone e per gli androgeni e da agonista verso il recettore per gli estrogeni (1).
L’interferenza del mitotano sulla farmacocinetica di altri farmaci non è completamente nota. Recentemente è stato osservato che il mitotano induce l’attività del CYP3A4 epatico interferendo potenzialmente con l’efficacia terapeutica di altre molecole, comprese quelle ad azione anti-neoplastica metabolizzate da questo enzima (6).
Oltre gli effetti adrenolitici, il mitotano possiede la capacità di inibire la glicoproteina P, una multidrug resistance, prodotta dal gene MDR-1, potenziando l’azione di diversi farmaci chemioterapici (7).
Indicazioni
Attualmente il mitotano trova la sua indicazione terapeutica nel trattamento del carcinoma corticosurrenalico non suscettibile di terapia chirurgica, metastatico o recidivante. Tuttavia, l'elevata percentuale di recidiva del carcinoma corticosurrenalico dopo resezione chirurgica totale ne giustifica l’impiego anche in terapia adiuvante post-chirurgica (8,9).
Effetti collaterali
I più frequenti sono quelli a carico dell’apparato gastrointestinale, con la comparsa di anoressia, nausea, vomito e diarrea, ipertransaminasemia. Importanti quelli a livello del SNC, con letargia, sonnolenza, depressione, vertigini e atassia. I disturbi metabolici comprendono un aumento del colesterolo plasmatico o dei trigliceridi. Meno frequenti sono: comparsa di rash cutanei, leucopenia, maculopatia, diplopia (10-12).
Preparazioni, via di somministrazione, posologia
Il farmaco, disponibile in compresse da 500 mg (Lysodren), viene somministrato per os ad una posologia generalmente superiore ai 4 g/die e comunque rispettando una finestra terapeutica che prevede un livello di mitotanemia compreso tra 14 e 20 mg/L. Al di sopra di 20 mg è stata documentata tossicità neurologica. E' necessario eseguire una determinazione plasmatica del farmaco ad intervalli frequenti (ogni 15 giorni) fino al raggiungimento della dose ottimale. Nella terapia di mantenimento è opportuno dosare la mitotanemia mensilmente, poichè le variazioni di dosaggio del farmaco non riflettono immediate variazioni nei livelli plasmatici dello stesso (13-16).
L’emivita plasmatica è di circa 2-3 ore, aumentando progressivamente dall'inizio della terapia in relazione alle sempre maggiori quantità che si accumulano nel tessuto adiposo, sino a raggiungere un plateau che ne condiziona la farmacocinetica (10).
La quota di farmaco assorbita giornalmente viene escreta a livello nelle urine sotto forma di metaboliti inattivi; una percentuale più bassa, invece, compare nelle feci attraverso la via di eliminazione del circolo epato–biliare. Poiché il farmaco si accumula nel tessuto adiposo, l’eliminazione plasmatica è molto lunga (18-159 giorni) (11).
Precauzioni d'uso
Per l’effetto sulla soppressione della sintesi di cortisolo e sull’aumento del suo catabolismo periferico è fondamentale istituire, nel paziente in trattamento con mitotano, una terapia sostitutiva con glucocorticoidi (preferibilmente idrocortisone 50 mg /die) (1).
Per gli effetti neurotossici deve essere rispettata la finestra terapeutica di 14-20 mg/L.
Poichè la sede metabolica del mitotano è il fegato, se ne raccomanda l'impiego con cautela nei pazienti con alterazione lieve della funzionalità epatica. Il farmaco non è raccomandato in pazienti affetti da insufficienza epatica.
Nel trattamento del paziente obeso il livello del farmaco può aumentare, nonostante la somministrazione di una dose costante, per via dell'accumulo del mitotano nel tessuto adiposo.
Nei pazienti in trattamento, destinati ad intervento chirurgico, va considerato un prolungamento dei tempi di sanguinamento indotti dal mitotano.
Il farmaco induce un'alterazione della funzione tiroidea, con un quadro di ipotiroidismo centrale nell'adulto e un ritardo neuro-psicologico in età pediatrica. Pertanto è necessaria una valutazione periodica dell'asse ipofisi-tiroide.
Bibliografia
- Hahner S, Fassnacht M. Mitotane for adrenocortical carcinoma treatement. Cur Opin Invest Drugs 2005, 6: 386–94.
- Touitou Y, Bogdan A, Luton JP. Changes in corticosteroid synthesis of the human adrenal cortex in vitro, induced by treatment with o,p'-DDD for Cushing's syndrome: evidence for the sites of action of the drug. J Ster Biochem 1978, 9: 1217-24.
- Bergenstal DM, Hertz R, Lipsett MB, Moy RH. Chemotherapy of adrenocortical cancer with o,p’-DDD”. Ann Int Med 1960, 53: 672-82.
- van Slooten H, Moolenaar AJ, van Seters AP, Smeenk D. The treatment of adrenocortical carcinoma with o,p’-DDD: prognostic implications of serum level monitoring. Eur J Cancer Clin Oncol 1984, 20: 47-53.
- Hermsen IG, Fassnachr M, Terzolo M, et al. Plasma concentrations of o,p’DDD, o,p’DDA and o,p’DDE as predictors of tumor response to mitotane in adrenocortical carcinoma: results of a retrospective ENS@T multicenter study. J Clin Endocrinol Metab 2011, 96: 1844-51.
- van Herp NP, Guchelaar HJ, Ploeger BA, et al. Mitotane has a strong and a durable inducing effect on CYP34A activity. Eur J Endocrinol 2011, 164: 621-6.
- Bates SE, Shieh CY, Mickley LA, et al. Mitotane enhances cytotoxicity of chemotherapy in cell lines expressing a multidrug resistance gene (mdr–1/P–glycoprotein) which is also expressed by adrenocortical carcinomas. J Clin Endocrinol Metab 1991, 73: 18–29.
- Terzolo M, Angeli A, Fassnacht M, et al. Adjuvant Mitotane Treatment for Adrenocortical Carcinoma. N Engl J Med 2007, 356: 2372–80.
- Lacroix A. Approach to the patient with adrenocortical carcinoma. J Clin Endocrinol Metab 2010, 95: 4812-22.
- van Slooten H, van Seters AP, Smeenk D, Moolenaar AJ. O,p’-DDD (mitotane) levels in plasma and tissues during chemotherapy and at autopsy. Cancer Chemother Pharmacol 1982, 9: 85-8.
- Baudin E, Pellegriti G, Bonnay M, et al. Impact of monitoring plasma 1,1 dichlorodiphenildichloroethane (o,p’DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer 2001, 92: 1385-92.
- Libè R, Fratticci A, Bertherat J. Adrenocortical cancer: pathophysiology and clinical management. Endocr Relat Cancer 2007, 14: 13-28.
- Luton JP, Cerdas S, Billaud L, et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med 1990, 322: 1195–201.
- Haak HR, Hermans J, Van de Velde CJH, et al. Optimal treatment of adrenal cortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer 1994, 69: 947–51.
- Allolio B, Fassnacht M. Clinical review: adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab 2006, 91: 2027–37.
- Fassnacht M, Allolio B. Clinical management of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab 2009, 23: 273-89.
Masse surrenaliche: cosa considerare in aggiunta ad adenomi, carcinomi e feocromocitomi?
Alessandro Prete1,2
1 Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, UK
2 Department of Endocrinology, Queen Elizabeth Hospital Birmingham, University Hospitals Birmingham NHS Foundation Trust, Birmingham, UK
(aggiornato a 9 settembre 2021)
Introduzione
La maggior parte delle masse surrenaliche viene riscontrata in maniera incidentale (incidentalomi surrenalici, fino al 5-7% della popolazione adulta) (1,2). L’incidenza delle masse surrenaliche è aumentata di dieci volte nel corso delle ultime due decadi, in maniera parallela alla diffusione di TC e RM nella pratica clinica (3). Questo aumento dell’incidenza è da mettere in relazione, in primo luogo, al riscontro incidentale di adenomi cortico-surrenalici di piccole dimensioni in soggetti di età > 65 anni, mentre il numero di tumori maligni e di grandi dimensioni è rimasto pressoché invariato (3). Le masse surrenaliche sono diagnosticate quasi alla pari in uomini (45%) e donne (55%), con un’età mediana alla diagnosi di 62 anni. Al contrario, le masse surrenaliche sono estremamente rare in età pediatrica: solo l’1% dei casi sono riscontrati in soggetti < 18 anni (3).
Le masse surrenaliche possono essere suddivise in cinque categorie (tabella 1 e figura 1) (2,4,5):
- adenomi cortico-surrenalici (inclusa l’iperplasia nodulare);
- altre lesioni surrenaliche benigne (come mielolipomi, cisti ed ematomi);
- carcinomi cortico-surrenalici;
- altri tumori surrenalici maligni (come metastasi, sarcomi e linfoma);
- tumori della midollare surrenalica (feocromocitomi, ganglioneuromi).
| Tabella 1 Classificazione delle masse surrenaliche (adattata dalla classificazione 2017 dell’OMS) (4,5) |
||
| Adenomi cortico-surrenalici (inclusa iperplasia nodulare) | ||
| Altre lesioni surrenaliche benigne (escluse lesioni della midollare surrenalica) |
Mielolipoma
|
|
| Carcinoma cortico-surrenalico | ||
| Altri tumori surrenalici maligni (escluse lesioni della midollare surrenalica) | Primitivi | Linfoma Sarcoma (liposarcoma, leiomiosarcoma, angiosarcoma) Tumori stromali dei cordoni sessuali Tumori neuroblastici (neuroblastoma, ganglio-neuroblastoma) Teratoma |
| Secondari (metastasi) | ||
| Tumori della midollare |
Feocromocitoma
Neuroblastoma |
|
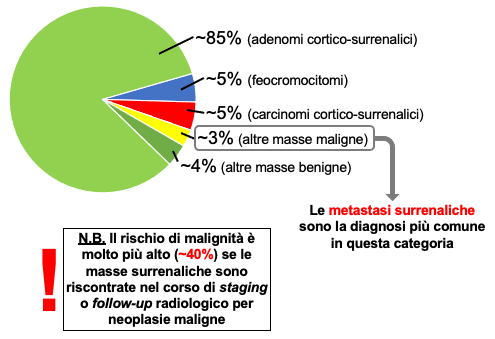
Figura 1. Eziologia di masse surrenaliche di nuovo riscontro.
Le percentuali riportate sono un’approssimazione e sono tipiche di centri di riferimento per la gestione di patologie surrenaliche (e pertanto potrebbero non essere rappresentative della popolazione generale).
Rischio di malignità. L’anamnesi positiva per neoplasie maligne aumenta sostanzialmente il rischio che masse surrenaliche di nuovo riscontro siano metastasi (soprattutto in caso di masse bilaterali). Ulteriori fattori associati col rischio di malignità includono:
- età (pazienti più giovani, soprattutto se < 40 anni, sono più a rischio);
- dimensioni della massa (un diametro > 4 cm aumenta considerevolmente il rischio);
- unità Hounsfield in corso di TC senza mezzo di contrasto (se < 10 il rischio di malignità è pressoché zero);
- sindromi genetiche che predispongono allo sviluppo di carcinoma cortico-surrenalico e feocromocitoma maligno.
Metastasi surrenaliche
Le metastasi sono la massa surrenalica più frequente dopo gli adenomi cortico-surrenalici e – di conseguenza – costituiscono l’eziologia più comune in caso di lesioni surrenaliche maligne (6). Un recente studio di popolazione ha dimostrato come i tumori maligni costituiscono l’8.6% delle masse surrenaliche di nuovo riscontro, di cui la maggior parte (86%) è costituita da metastasi e solo lo 0.3% da carcinomi cortico-surrenalici (3). Questo dato è in contrasto con l’esperienza di endocrinologi che operano in centri di II e III livello, dove la maggior parte dei pazienti con tumori maligni del surrene viene diagnosticata con carcinoma cortico-surrenalico e solo una minoranza con altri tumori maligni (figura 1). Questo è probabilmente legato al fatto che i pazienti con sospette metastasi o altri tumori maligni (linfoma, sarcoma, ecc) vengono valutati direttamente da altri specialisti, quali oncologo, pneumologo, chirurgo e urologo. Infatti, un recente lavoro in pazienti con metastasi surrenaliche ha rilevato che una valutazione endocrinologica veniva effettuata soltanto nel 30% dei casi (7). Inoltre, soltanto il 10% dei pazienti che eseguivano una biopsia surrenalica venivano gestiti in ambiente endocrinologico (8).
Le neoplasie che più frequentemente metastatizzano ai surreni sono i carcinomi (in particolar modo adeno-carcinomi) e l’eziologia più comune è il tumore polmonare (~35% dei casi; tabella 2) (9).
| Tabella 2 Origini principali delle metastasi surrenaliche (9) |
|
| Tipo di tumore (% approssimativa dei casi) | |
|
Carcinomi (~90%):
|
|
|
Altre neoplasie maligne (~10%):
|
|
| a In ordine decrescente di frequenza: adenocarcinoma (~70% dei casi di metastasi surrenaliche da carcinoma del polmone), carcinoma squamoso, carcinoma a grandi cellule, microcitoma. b In ordine decrescente di frequenza: linfoma non-Hodgkin (~85% dei casi di metastasi surrenaliche da neoplasie ematologiche), linfomi di Hodgkin, leucemie. c Espressa come percentuale dei pazienti con carcinomi. d Espressa come percentuale dei pazienti con altre neoplasie maligne. |
Anche se la maggior parte delle metastasi surrenaliche viene diagnosticata nel corso dello staging e follow-up di neoplasie maligne, il 36% è di riscontro incidentale (7). Solitamente, il periodo di latenza tra diagnosi del tumore e il riscontro di metastasi surrenaliche è breve (7 mesi in media) (9). Tuttavia, non è infrequente diagnosticare metastasi surrenaliche oltre 2 anni dopo il riscontro del tumore primitivo (6) e sono stati descritti rari casi di diagnosi dopo i 5 anni (fino a 22 anni) (6,7).
Vi sono alcune caratteristiche delle metastasi surrenaliche che possono essere di aiuto nella diagnosi differenziale (figura 2):
- solitamente le dimensioni sono modeste al momento della diagnosi (diametro mediano 3 cm), anche se variano considerevolmente (da 0.5 a 20 cm) (7);
- tendono a rapido accrescimento nel corso del follow-up (> 1 cm ogni 3-6 mesi), contrariamente ad adenomi cortico-surrenalici, feocromocitomi e altre masse benigne (2);
- la bilateralità è di riscontro comune (24% al momento della diagnosi e 43% nel corso del follow-up) (7);
- unità Hounsfield (HU) alla TC pre-contrasto > 10 nel 100% dei casi (e > 20 nel 96-98%) (2).
Figura 2. Metastasi surrenaliche bilaterali con ampie aree di necrosi (paziente con diagnosi di adeno-carcinoma polmonare)
L’utilizzo della PET-TC (in particolar modo con 18F-FDG) è spesso utile nel confermare la diagnosi di metastasi surrenaliche, soprattutto nel corso di stadiazione di neoplasie maligne (10,11). Carcinoma cortico-surrenalico, feocromocitoma maligno e altre neoplasie maligne (linfoma surrenalico, sarcomi, ...) sono ovviamente importanti diagnosi differenziali in caso di masse surrenaliche di grandi dimensioni con ipercaptazione di 18F-FDG. Tuttavia, possono esserci falsi positivi per ipercaptazione in corso di 18F-FDG-PET-TC di lesioni benigne: adenomi cortico-surrenalici (soprattutto se funzionanti e/o di grandi dimensioni), iperplasia surrenalica (es. sindrome di Cushing ACTH-dipendente), feocromocitomi, emorragia surrenalica acuta o cronica (probabilmente per reazione infiammatoria locale), mielolipomi, ganglioneuromi, emangiomi e infezione tubercolare (12-14). Inoltre, alcune neoplasie maligne possono non captare 18F-FDG dando origine a falsi negativi; esempi includono carcinoma a cellule renali, carcinoma bronchiolo-alveolare, carcinoidi e carcinoma epato-cellulare.
Infine, occorre ricordare che la presenza di metastasi bilaterali è un fattore di rischio per lo sviluppo di iposurrenalismo, a causa della distruzione della corticale surrenalica, soprattutto in caso di lesioni di grandi dimensioni. Di fatti, il 12% dei pazienti con metastasi surrenaliche bilaterali sviluppa iposurrenalismo (7). Pertanto, è essenziale seguire i pazienti nel tempo e investigare la comparsa di segni e sintomi di iposurrenalismo.
Mielolipoma surrenalico
È un tumore benigno, contenente elementi di tessuto adiposo e midollo osseo. Mielolipomi surrenalici sono diagnosticati nel 3-7% dei pazienti con masse surrenaliche (5,15,16). Nella maggior parte dei casi si tratta di lesioni unilaterali, con diametro mediano di 2-2.5 cm (alcuni casi raggiungono dimensioni > 15 cm) (15). Mielolipomi bilaterali si riscontrano soltanto nel 5% dei casi, ma questa percentuale sale al 20% in caso di tumori > 6 cm (15).
Pazienti con iperplasia surrenalica congenita possono sviluppare mielolipomi bilaterali spesso di grandi dimensioni; pertanto, in caso di riscontro di mielolipomi bilaterali è consigliato misurare il 17-OH-progesterone, per escludere difetti congeniti della steroidogenesi surrenalica (2).
Dal punto di vista radiologico, il mielolipoma si presenta come massa ben circoscritta, con aree ipodense interne compatibili con tessuto adiposo (HU < -20) (figura 3). Calcificazioni sono descritte in ~25% dei casi (17).
Figura 3. Mielolipoma surrenalico destro di 9 cm con macro-aree di tessuto adiposo
Nonostante siano tumori benigni e non funzionanti, i mielolipomi di maggiori dimensioni (> 3.5 cm) tendono a crescere nel tempo e vi è un rischio (modesto) di sanguinamento e di sviluppare sintomi compressivi se il diametro supera i 6 cm. Pertanto, nei mielolipomi di maggiori dimensioni è ragionevole considerare la ripetizione di TC o RM nel corso del follow-up, soprattutto in caso di comparsa di sintomi locali.
Ganglioneuroma surrenalico
È un raro tumore benigno della midollare (0.2-0.4% dei tumori surrenalici), costituito da cellule gangliari, cellule di Schwann e fibre nervose (18). Alcuni ganglioneuromi compositi (10-45% dei casi, a seconda delle casistiche) includono elementi istologici tipici sia di ganglioneuroma che di feocromocitoma (19). Sono descritti rari casi associati a MEN-2 e neurofibromatosi di tipo 1 (19).
È spesso difficile distinguere i ganglioneuromi da altre masse surrenaliche, in quanto mancano di caratteristiche radiologiche specifiche (13) e vengono spesso diagnosticati a seguito di rimozione chirurgica di masse con caratteristiche sospette.
Di seguito alcune considerazioni di rilievo (figura 4) (18-20):
- distribuzione simile tra i sessi;
- più frequenti in adolescenti e giovani adulti (età media alla diagnosi 39 anni), anche se i tumori con caratteristiche istologiche composite sono tipici dell’età adulta (età mediana 63 anni);
- i ganglioneuromi con caratteristiche istologiche composite tendono ad essere più piccoli di quelli classici (diametro mediano 3.9 cm vs 7 cm) e meno frequentemente di riscontro incidentale (65% vs 84%), probabilmente perchè tali tumori possono secernere catecolamine ed essere sintomatici;
- quasi invariabilmente unilaterali (99%);
- HU solitamente > 20, ma il range riportato in letteratura è estremamente variabile (da -118 a +49);
- omogenei all’imaging nel ~70% dei casi e tipicamente presentano intensificazione di segnale dopo mezzo di contrasto;
- calcificazioni nel ~40% dei casi;
- lobulati nel ~40% dei casi.
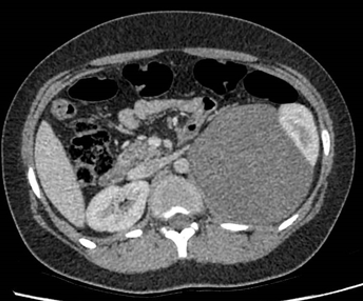
Figura 4. Ganglioneuroma surrenalico sinistro di 16 cm
Oncocitoma surrenalico
Tumore estremamente raro (poco più di 100 casi descritti in letteratura) (21).
L’età mediana alla diagnosi è 46 anni, con lieve predilezione per il sesso femminile (21).
Nella maggior parte dei casi si tratta di tumori unilaterali di grandi dimensioni (diametro mediano 8.5 cm), anche se sono stati descritti casi < 3 cm (21,22). In circa il 30% dei casi l’oncocitoma è stato associato a ipersecrezione ormonale (eccesso di cortisolo, virilizzazione e femminilizzazione) (21).
L’oncocitoma surrenalico ha potenziale malignità. I criteri isto-patologici di Lin-Weiss-Bisceglia (tab 3), proposti per definire il rischio di malignità (23), permettono di distinguere oncocitomi (21):
- benigni (~30% dei casi);
- a potenziale incerto di malignità (~45% dei casi), con recidiva/metastasi nell’8% dei casi;
- maligni (~20% dei casi), con recidiva/metastasi nel 75% dei casi.
| Tabella 3 Criteri istopatologici di Lin-Weiss-Bisceglia |
|
Maggiori:
|
Oncocitomi benigni: nessun criterio presente. Oncocitomi a potenziale incerto di malignità: ≥ 1 criteri minori presenti. Oncocitomi maligni: ≥ 1 criteri maggiori presenti. |
Minori:
|
|
Linfoma primitivo surrenalico
Il coinvolgimento dei surreni è piuttosto comune in pazienti con linfoma non-Hodgkin con malattia disseminata (fino al 25% dei casi) (24). Al contrario, il linfoma primitivo surrenalico è un’entità estremamente rara (< 1% di tutti i casi di linfoma non-Hodgkin) (25). L’istotipo più frequente è il linfoma non-Hodgkin a cellule B (~90% dei casi), in particolare il sottotipo diffuso a grandi cellule B (~75% dei casi) (26).
Il sesso maschile è più frequentemente interessato (70% dei casi) e l’età mediana alla diagnosi è 66 anni (range 25-79).
Una casistica retrospettiva di 81 pazienti ha mostrato che solo in una minoranza di casi il linfoma primitivo surrenalico interessava esclusivamente il surrene, mentre nel 77% dei pazienti era dimostrabile un coinvolgimento extra-surrenalico al momento della diagnosi (26).
Dal punto di vista clinico erano comuni sintomi generali (55%), astenia (45%) e dolore addominale (35%) (26).
Nonostante la rarità del linfoma surrenalico primitivo, alcune caratteristiche clinico-radiologiche possono aiutare nella diagnosi differenziale e gestione clinica:
- interessamento surrenalico bilaterale in circa metà dei pazienti (26);
- circa il 70% dei casi con interessamento surrenalico bilaterale sviluppa iposurrenalismo (26);
- massa/e solitamente di grandi dimensioni (diametro mediano 8 cm, range 7-18), omogenea o eterogenea in corso di TC, HU in media > 30, spesso invade le strutture circostanti (25,26);
- la 18F-FDG-PET-TC è utile nell’individuare il coinvolgimento extra-surrenalico in una percentuale sostanziale di pazienti, mentre la TC da sola non è in grado di identificare localizzazioni a distanza nel 50% dei pazienti (figura 5) (26);
- livelli ematici di LDH elevati in circa l’80% dei pazienti, che possono essere d’aiuto nel sospettare la diagnosi (26,27); altri parametri di laboratorio spesso alterati includono β2-microglobulina, proteina C reattiva e ferritina (27).
Figura 5. Paziente con linfoma surrenalico diffuso a grandi cellule B bilaterale: la 18F-FDG-PET-TC dimostra intensa ipercaptazione surrenalica bilaterale (massa destra 7 cm, massa sinistra 4 cm), nonché milza ingrandita con aree focali di ipercaptazione
Emorragie surrenaliche
È un evento relativamente raro, le cui possibili cause includono (28):
- traumi addominali (la causa più comune);
- uso di anti-coagulanti;
- coagulopatie (es. sindrome da anticorpi anti-fosfolipidi);
- infezioni e stress maggiori (es. sepsi da meningococco, COVID-19, ustioni estese);
- emorragia nel contesto di un tumore surrenalico (soprattutto maligno);
- emorragia nel contesto di una cisti surrenalica;
- emorragia idiopatica.
L’emorragia unilaterale è solitamente post-traumatica o associata con lesione surrenalica pre-esistente (es. tumore maligno, cisti), mentre emorragie bilaterali sono solitamente associate a eventi non traumatici (farmaci, coagulopatie, infezioni, stress, ustioni estese) (figura 6) (13).
Sia TC che RM permettono in genere di confermare agevolmente la diagnosi nell’adulto, mentre l’esame di scelta nel neonato è l’ecografia (28). La RM è inoltre in grado di differenziare sanguinamenti più o meno recenti (28). In casi dubbi, è spesso utile ripetere l’esame radiologico a distanza: l’evoluzione temporale (con il riassorbimento dell’emorragia e la conseguente riduzione volumetrica) rafforza infatti il sospetto diagnostico. È importante notare che masse surrenaliche (soprattutto maligne) possono sanguinare; pertanto, è essenziale escludere una componente solida nel contesto dell’emorragia (13).
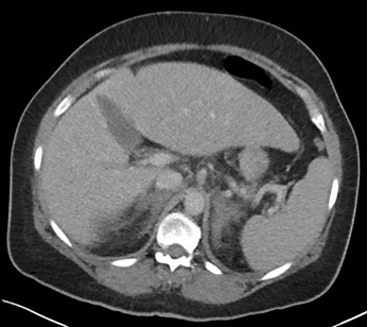
Figura 6. Ingrandimento surrenalico bilaterale secondario a emorragia spontanea in paziente con sindrome da anticorpi anti-fosfolipidi
Tubercolosi surrenalica
È una malattia infettiva tutt’oggi di frequente riscontro, con 10 milioni di nuovi casi riportati a livello mondiale nel 2019 (29). Al di là di ovvie differenze geografiche nella prevalenza di TBC, fattori di rischio includono immuno-soppressione (es. infezione da HIV), età avanzata, professioni con aumentato rischio di esposizione (es. operatori sanitari) e condizioni di vita (es. sovraffollamento abitativo, carcerazione).
Il BK ha un particolare tropismo per il surrene ed è una delle principali cause di iposurrenalismo primario nei paesi in via di sviluppo. L’infezione tubercolare dei surreni è solitamente bilaterale (~90%), caratterizzata da ingrandimento che rassomiglia a masse, piuttosto che a iperplasia (30). La TC frequentemente documenta lesioni con centro ipodenso (necrosi caseosa) e margini che captano il mezzo di contrasto.
Nel lungo termine l’ingrandimento dei surreni tende a ridursi e compaiono calcificazioni, tipiche di una pregressa infezione tubercolare (31).
Bibliografia
- Reimondo G, Castellano E, Grosso M, et al. Adrenal incidentalomas are tied to increased risk of diabetes: findings from a prospective study. J Clin Endocrinol Metab 2020, 105: e973-81.
- Bancos I, Prete A. Approach to the patient with adrenal incidentaloma. J Clin Endocrinol Metab 2021, DOI: 10.1210/clinem/dgab512.
- Ebbehoj A, Li D, Kaur RJ, et al. Epidemiology of adrenal tumours in Olmsted County, Minnesota, USA: a population-based cohort study. Lancet Diabetes Endocrinol 2020, 8: 894-902.
- Bychkov A. Adrenal ganglia and paraganglia. General WHO classification. Aggiornamento 2021. (Accessed September 7th, 2021).
- Lam AK. Lipomatous tumours in adrenal gland: WHO updates and clinical implications. Endocr Relat Cancer 2017, 24: R65-79.
- Cingam SR, Mukkamalla SKR, Karanchi H. Adrenal metastasis. Treasure Island (FL) 2021.
- Mao JJ, Dages KN, Suresh M, Bancos I. Presentation, disease progression and outcomes of adrenal gland metastases. Clin Endocrinol (Oxf) 2020, 93: 546-54.
- Delivanis DA, Erickson D, Atwell TD, et al. Procedural and clinical outcomes of percutaneous adrenal biopsy in a high-risk population for adrenal malignancy. Clin Endocrinol (Oxf) 2016, 85: 710-6.
- Lam KY, Lo CY. Metastatic tumours of the adrenal glands: a 30-year experience in a teaching hospital. Clin Endocrinol (Oxf) 2002, 56: 95-101.
- Park SY, Park BK, Kim CK. The value of adding 18F-FDG PET/CT to adrenal protocol CT for characterizing adrenal metastasis (≥ 10 mm) in oncologic patients. AJR Am J Roentgenol 2014, 202: W153-60.
- Wu Q, Luo W, Zhao Y, et al. The utility of 18F-FDG PET/CT for the diagnosis of adrenal metastasis in lung cancer: a PRISMA-compliant meta-analysis. Nucl Med Commun 2017, 38: 1117-24.
- Akkus G, Guney IB, Ok F, et al. Diagnostic efficacy of 18F-FDG PET/CT in patients with adrenal incidentaloma. Endocr Connect 2019, 8: 838-45.
- Dong A, Cui Y, Wang Y, et al. 18F-FDG PET/CT of adrenal lesions. AJR Am J Roentgenol 2014, 203: 245-52.
- Ciftci E, Turgut B, Cakmakcilar A, Erturk SA. Diagnostic importance of 18F-FDG PET/CT parameters and total lesion glycolysis in differentiating between benign and malignant adrenal lesions. Nucl Med Commun 2017, 38: 788-94.
- Hamidi O, Raman R, Lazik N, et al. Clinical course of adrenal myelolipoma: A long-term longitudinal follow-up study. Clin Endocrinol (Oxf) 2020, 93: 11-8.
- Calissendorff J, Juhlin CC, Sundin A, et al. Adrenal myelolipomas. Lancet Diabetes Endocrinol 2021, DOI: 10.1016/S2213-8587(21)00178-9.
- Lattin GE Jr, Sturgill ED, Tujo CA, et al. From the radiologic pathology archives. Adrenal tumors and tumor-like conditions in the adult: radiologic-pathologic correlation. Radiographics 2014, 34: 805-29.
- Rha SE, Byun JY, Jung SE, et al. Neurogenic tumors in the abdomen: tumor types and imaging characteristics. Radiographics 2003, 23: 29-43.
- Dages KN, Kohlenberg JD, Young WF Jr, et al. Presentation and outcomes of adrenal ganglioneuromas: A cohort study and a systematic review of literature. Clin Endocrinol (Oxf) 2021, 95: 47-57.
- Radin R, David CL, Goldfarb H, Francis IR. Adrenal and extra-adrenal retroperitoneal ganglioneuroma: imaging findings in 13 adults. Radiology 1997, 202: 703-7.
- Wong DD, Spagnolo DV, Bisceglia M, et al. Oncocytic adrenocortical neoplasms--a clinicopathologic study of 13 new cases emphasizing the importance of their recognition. Hum Pathol 2011, 42: 489-99.
- Hong Y, Hao Y, Hu J, et al. Adrenocortical oncocytoma. 11 Case reports and review of the literature. Medicine (Baltimore) 2017, 96: e8750.
- Bisceglia M, Ludovico O, Di Mattia A, et al. Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol 2004, 12: 231-43.
- Singh D, Kumar L, Sharma A, et al. Adrenal involvement in non-Hodgkin's lymphoma: four cases and review of literature. Leuk Lymphoma 2004, 45: 789-94.
- Zhou L, Peng W, Wang C, et al. Primary adrenal lymphoma: radiological; pathological, clinical correlation. Eur J Radiol 2012, 81: 401-5.
- Majidi F, Martino S, Kondakci M, et al. Clinical spectrum of primary adrenal lymphoma: results of a multicenter cohort study. Eur J Endocrinol 2020, 183: 453-62.
- Laurent C, Casasnovas O, Martin L, et al. Adrenal lymphoma: presentation, management and prognosis. QJM 2017, 110: 103-9.
- Mehmood KT, Sharman T. Adrenal Hemorrhage. StatPearls. Treasure Island (FL) 2021.
- Chakaya J, Khan M, Ntoumi F, et al. Global Tuberculosis Report 2020 - Reflections on the Global TB burden, treatment and prevention efforts. Int J Infect Dis 2021, DOI: 10.1016/j.ijid.2021.02.107.
- Guo YK, Yang ZG, Li Y, et al. Addison's disease due to adrenal tuberculosis: contrast-enhanced CT features and clinical duration correlation. Eur J Radiol 2007, 62: 126-31.
- Ma ES, Yang ZG, Li Y, et al. Tuberculous Addison's disease: morphological and quantitative evaluation with multidetector-row CT. Eur J Radiol 2007, 62: 352-8.
Biosintesi, trasporto e metabolismo delle catecolamine
Massimo Mannelli
Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
Le catecolamine sono molecole costituite da un anello benzenico con due gruppi ossidrilici adiacenti (anello catecolico), legato ad un gruppo aminico.
La loro sintesi avviene a partire dalla tirosina, aminoacido assunto con la dieta o derivato dall’idrossilazione della fenilalanina. Nel tessuto nervoso la tirosina viene idrossilata a diidrossifenilalanina (DOPA) per azione di una tirosina-idrossilasi, enzima presente soltanto nei tessuti che sintetizzano catecolamine.
La conversione della DOPA a dopamina, primo composto catecolaminico, avviene per azione di una decarbossilasi, enzima che non è specifico del tessuto nervoso, essendo presente anche in altri tessuti come fegato e rene. Nei neuroni dopaminergici e molto parzialmente nella midollare surrenale, la sintesi si arresta a dopamina. Nei terminali nervosi simpatici e parzialmente nella midollare surrenale, la dopamina viene invece idrossilata a noradrenalina, altro composto catecolaminico, da parte di un enzima, la DA-ß-idrossilasi che è specifico del tessuto nervoso.
La noradrenalina è il neurotrasmettitore liberato dai terminali simpatici periferici che, attraverso l’attivazione dei recettori nei tessuti bersaglio innervati dal simpatico, determina gli effetti biologici tipici dell’attivazione simpatica.
A livello della midollare del surrene la sintesi delle catecolamine prosegue per azione di un enzima, la fenil-etanol-amina-N-metil-transferasi (PMNT), che trasforma la noradrenalina in adrenalina, ultimo composto catecolaminico. L’adrenalina è da considerarsi un vero e proprio ormone, poiché viene rilasciato nel torrente circolatorio e veicolato attraverso il sangue ai tessuti bersaglio, non necessariamente innervati, dove esercita le proprie azioni biologiche.
È interessante notare come la ghiandola surrenale costituisca di fatto un esempio di stretta interrelazione fra sistema nervoso e sistema endocrino; infatti la PMNT è un enzima che per la sua attivazione necessita di alte dosi di corticosteroidi e quest’ultimi si trovano in alta concentrazione nel sangue venoso refluo che, proveniente dal cortico-surrene, irrora, assieme a quello arterioso, le cellule della midollare del surrene.
Le catecolamine, dopo la sintesi sono immagazzinate in granuli citoplasmatici dove permangono, complessate con molecole di ATP tramite una proteina denominata cromogranina A. Al momento del rilascio, che avviene sotto l’impulso dello stimolo nervoso, le catecolamine vengono liberate nel vallo inter-sinaptico o nei capillari venosi surrenalici.
La noradrenalina, quale neurotrasmettitore del secondo neurone simpatico, una volta liberata nel vallo inter-sinaptico, subisce un destino diverso; una piccola quota sfugge nel circolo dove può venire misurata, una parte si lega ai recettori post-sinaptici attraverso i quali esercita le sue azioni biologiche, la maggior parte viene ricaptata dai terminali nervosi all’interno dei quali o viene metabolizzata dalle monoaminoossidasi (MAO), o viene re-immagazzinata nei granuli. Esistono farmaci, come gli anfetaminici e gli anti-depressivi triciclici, che inibendo la ricaptazione determinano un aumento della noradrenalina nel vallo intersinaptico.
A livello dei tessuti periferici, inoltre, la noradrenalina viene metabolizzata dalle catecol-orto-metil-transferasi (COMT). Questi enzimi, presenti soprattutto nel fegato e nel rene, sono la principale via di metabolizzazione delle catecolamine circolanti e pertanto dell’adrenalina. Una piccolissima quota di catecolamine viene escreta come tale nelle urine.
I principali prodotti del metabolismo delle catecolamine sono l’acido vanilmandelico (VMA), derivante da noradrenalina e adrenalina per azione combinata di MAO e COMT, e le metanefrine derivanti da adrenalina (metanefrina) e noradrenalina (normetanefrina) per azione delle COMT.
La dopamina subisce lo stesso processo di metabolizzazione ed i suoi metaboliti principali nel sangue e nelle urine sono l’acido omovanillico e la metoxitiramina. E’ bene ricordare che la dopamina urinaria non rispecchia i livelli di dopamina plasmatica, perché i tubuli renali sono ricchi di DOPA-decarbossilasi e pertanto capaci di sintetizzare dopamina e liberarla nelle urine in risposta a stimoli quali, per esempio il carico salino. Pertanto, la dopamina urinaria è indice dell'attività sintetica del rene.
Una volta immesse nel circolo, sia per secrezione diretta dal surrene che per sfuggita dai terminali nervosi, le catecolamine vengono per una elevata percentuale coniugate come solfati (99% la dopamina, 85% la adrenalina, 70% la noradrenalina). La forma coniugata non è biologicamente attiva e non viene misurata dai comuni metodi di dosaggio.
Bibliografia
- Catecholamines. Bridging basic science with clinical medicine. Goldstein D, Eisenhofer G, McCarty R Eds, Academic Press, London, 1998.
Azioni e regolazione delle catecolamine
Massimo Mannelli
Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
Il sistema
Il sistema simpatoadrenergico (SSA) costituisce una parte del sistema nervoso autonomo (1) e concorre, con l’altra parte del sistema nervoso autonomo, il sistema parasimpatico, al mantenimento dell’omeostasi di molte funzioni vitali, prima fra tutte quella cardiocircolatoria. Il SSA è responsabile del mantenimento dell’omeostasi di molte funzioni vitali, quale quella cardiocircolatoria.
Le caratteristiche anatomo-chimiche del SSA permettono risposte istantanee agli stimoli ambientali e rendono pertanto tale sistema il mezzo più rapido attraverso cui l’organismo mantiene le proprie condizioni di equilibrio.
Il SSA è funzionalmente integrato con altri sistemi omeostatici, come quello endocrino, le cui risposte, più lente e più durature nel tempo, possono a loro volta modulare la funzione del SSA. D’altra parte, il SSA comprende in se stesso sia elementi funzionali tipici della funzione neuronale (terminali nervosi delle fibre simpatiche) che elementi tipici della funzione endocrina (cellule cromaffini della midollare surrenale).
I mediatori chimici delle risposte simpatiche sono le catecolamine sostanze sintetizzate e rilasciate esclusivamente dal tessuto neuroendocrino (2,3).
In modo schematico possiamo affermare che il SSA esercita le proprie azioni attraverso due vie effettrici, le terminazioni nervose e la midollare surrenale.
Le terminazioni nervose, ultime diramazioni del neurone simpatico post-gangliare, sono le sedi di giunzione sinaptica (neuro-tissutale) dove viene liberato il neurotrasmettitore noradrenalina (NA). Questa, riversata nel vallo inter-sinaptico, viene poi a contatto con i recettori presenti sulle cellule del tessuto innervato. I neuroni post-gangliari hanno i loro nuclei nei gangli simpatici periferici, disposti per lo più in catene parallele al decorso dei grossi vasi nel torace e nell’addome. Questi neuroni sono, a livello gangliare, a loro volta innervati e pertanto modulati nella loro funzione, da altri neuroni, detti pre-gangliari, che liberano come neurotrasmettitore l’acetilcolina (Ach) e che hanno il loro nucleo nella regione intermedio-laterale del midollo spinale, in una zona compresa fra T1 ed L2. L’attività di questi neuroni è regolata da vie nervose bulbo-spinali discendenti da centri superiori del sistema nervoso centrale siti nel bulbo. Tali centri costituiscono la sede d’integrazione di moltissime afferenze provenienti sia dalla periferia (barocettori, chemocettori, termocettori, algocettori) che dai centri corticali superiori (ipotalamo e corteccia). Si viene così a costituire un sistema integrato, capace di registrare le più diverse alterazioni ambientali e di organizzare una conseguente risposta adeguata al mantenimento dell’equilibrio delle funzioni organiche.
La midollare surrenale si inserisce in questo sistema come un ganglio simpatico modificato. Essa riceve, come il neurone post-gangliare, un’innervazione acetilcolinica da fibre pre-gangliari raccolte nel nervo splancnico, ma le sue cellule non posseggono diramazioni assoniche e non liberano i prodotti di sintesi in un vallo sinaptico, ma nei capillari venosi.
Risulta pertanto evidente che il SSA mantiene costantemente un livello di attività che è determinato e regolato dagli stimoli afferenti continuamente dalla periferia e dai centri superiori ai suoi centri di integrazione. Ne consegue che innumerevoli sono gli stimoli capaci di modificare l’attività simpatica e pertanto la secrezione di catecolamine.Pertanto una valutazione corretta dell’attività simpatica risulta spesso difficoltosa e deve necessariamente tenere conto delle condizioni ambientali evitando, per quanto possibile, i principali fattori di stimolazione (4).
Il più basso livello di attivazione simpatica si ha fisiologicamente durante il sonno, quando la posizione è clinostatica e le stimolazioni esterne, comprese quelle visive, sono minime. È infatti durante il sonno che la pressione arteriosa diminuisce e la frequenza cardiaca rallenta.
Innumerevoli sono i fattori capaci di modificare l’attività del SSA. Fra questi vi sono stimoli fisici (postura, freddo, esercizio fisico, traumi, ecc), chimici (caffeina, nicotina, la maggior parte dei farmaci), metabolici (ipoglicemia), emotivi, ecc.
I diversi tipi di stimolo non determinano tutti quanti una stessa attivazione quantitativa e qualitativa del sistema. Esistono cioè stimoli preferenziali per l’attivazione neuronale (es. postura eretta) o per l’attivazione surrenalica (ipoglicemia insulinica). Inoltre l’attivazione neuronale non è generalizzata, ma, sempre in rapporto allo stimolo, prevalentemente confinata in alcuni distretti. Tutto ciò permette una risposta differenziata e pertanto più finemente adeguata al controllo omeostatico.
Per convenzione, riguardo l’uomo, si considera attività simpatica basale quella valutata in soggetto sano, esaminato in posizione supina da almeno 30 minuti, in ambiente silenzioso e in penombra, con temperatura ambientale fra i 20 ed i 25°C, in assenza di stimoli emotivi, digiuno da circa 12 ore e senza che siano state assunte nelle 12 ore precedenti sostanze stimolanti quali teina, caffeina o nicotina. Qualora la valutazione venga poi effettuata tramite dosaggio delle catecolamine ematiche, un vaso periferico dovrà essere incannulato almeno 30 minuti prima del prelievo.
Recettori
Gli effetti dell’attivazione simpatica si esplicano attraverso l’attivazione dei recettori da parte delle catecolamine. La risposta globale dipende pertanto sia dall’entità del rilascio catecolaminico, sia dalla responsività dei tessuti periferici. Questa non è sempre costante, ma può variare, poiché i recettori subiscono una modulazione sia per azione delle stesse catecolamine (autologa), sia da parte di altre sostanze (eterologa). In particolare, la cronica esposizione del tessuto ad alte concentrazioni di catecolamine determina una diminuzione dei recettori adrenergici (down regulation), mentre la denervazione causa un aumento dei recettori (ipersensibilità alle amine esogene).
Gli effetti dell’attivazione simpatica si esplicano attraverso l’attivazione dei recettori da parte delle catecolamine. La risposta globale dipende pertanto sia dall’entità del rilascio catecolaminico, sia dalla responsività dei tessuti periferici. Questa non è sempre costante, ma può variare poiché i recettori subiscono una modulazione sia per azione delle stesse catecolamine (autologa), sia da parte di altre sostanze (eterologa). In particolare la cronica esposizione del tessuto ad alte concentrazioni di catecolamine determina una diminuzione dei recettori adrenergici (down regulation), mentre la denervazione causa un aumento dei recettori (ipersensibilità alle amine esogene).
I recettori adrenergici (5) si distinguono in base all’affinità relativa per i composti amminici (isoproterenolo, adrenalina, noradrenalina), in alfa (poco sensibili all’isoproterenolo) e beta (molto sensibili)(6-9), a loro volta suddivisi in alfa-1 e 2 e beta-1 e 2 in base all’affinità relativa alle amine endogene (10).
I recettori adrenergici possono anche essere suddivisi, in relazione alla loro distribuzione in giunzionali ed extra-giunzionali. I recettori giunzionali possono a loro volta distinguersi in pre e post-sinaptici.
È stato anche notato che la distribuzione dei recettori di tipo 1 è particolarmente ricca nei tessuti innervati, in zona post-sinaptica dove cioè viene liberata la noradrenalina che ha alta affinità per tali recettori, mentre i recettori di tipo 2, particolarmente sensibili all'adrenalina, sono presenti specialmente nei tessuti non innervati e nei tessuti innervati, ma in sede extra-giunzionale. I recettori di tipo 2 sono inoltre presenti in sede giunzionale pre-sinaptica (auto-recettori) dove modulano, insieme ad altri recettori, il rilascio di catecolamine.
I recettori adrenergici appartengono tutti alla famiglia caratterizzata da 7 domini transmembrana uniti da tre ponti extra-cellulari e tre ponti intra-cellulari e legati a strutture intra-citoplasmatiche denominate proteine G, la cui attivazione trasmette il messaggio all’interno della cellula in vario modo: stimolando la produzione di cAMP (recettori beta), inibendo la produzione di cAMP (recettori alfa-2), facilitando l’ingresso del calcio dentro la cellula (recettori alfa-1).
Esistono anche recettori specifici per la dopamina, suddivisi in D1 (la stimolazione determina aumento del cAMP intra-cellulare) e D2 (la stimolazione determina riduzione del cAMP e del calcio intra-cellulare).
La stimolazione dei recettori adrenergici non determina effetti univoci; ad esempio i recettori vasali beta sono vasodilatanti e gli alfa vasocostrittori. La risposta biologica globale dipende pertanto anche dal tipo di recettore prevalentemente presente in quel tessuto, oltre che dalla quantità e dal tipo di amina che si lega al recettore.
L’entità della liberazione di catecolamine dipende a sua volta sia dal tipo e dall’intensità dello stimolo neuronale, che dagli effetti modulanti esercitati da recettori, adrenergici e non, presenti sulle terminazioni nervose in sede pre-sinaptica (cioè sul terminale stesso) e sulle cellule cromaffini della midollare surrenale. Questi recettori, se attivati, sono capaci, a parità di stimolazione neuronale, di facilitare il rilascio di catecolamine (beta 2, angiotensinici) o di limitarlo (alfa 2, DA2, oppioidergici). I bloccanti di tali recettori mediano azioni opposte (11).
Azioni
Gli effetti clinici di una rapida ed intensa attivazione simpatica sono rappresentati dal quadro clinico denominato con il termine anglosassone del “fight or flight”. Esso è caratterizzato da tachicardia, tachipnea, aumento del metabolismo, ecc.
A livello cardiocircolatorio i principali effetti della noradrenalina sono dati da aumento della gittata cardiaca (per aumento della frequenza e della contrattilità miocardia), aumento della resistenze vascolari periferiche, con aumento della pressione arteriosa ed aumento del flusso coronarico. L'adrenalina causa anch’essa aumento della gittata cardiaca, aumento della pressione arteriosa sistolica, diminuzione della diastolica, aumento del flusso muscolare e riduzione di quello renale e cutaneo (12-14). Va ricordato che il SSA non è l’unico meccanismo che regola la ridistribuzione dei flussi distrettuali, ma che a livello periferico assumono grande importanza le modificazioni tissutali indotte dalla contrazione muscolare, come l’ipossia, l’acidosi, l’aumento della pCO2, dell’ADP, del magnesio, del calcio e della temperatura. Sono questi stimoli locali che, nonostante l’aumento della scarica simpatica, inducono una vasodilatazione che pertanto risulta preferenziale nei gruppi muscolari attivi, a scapito di quelli inattivi e degli organi meno coinvolti nell’esercizio fisico.
A livello renale le catecolamine determinano ritenzione di sodio ed acqua, sia per stimolo diretto sui tubuli, sia per via indiretta attraverso la stimolazione del sistema renina-angiotensina-aldosterone (15).
A livello respiratorio le catecolamine determinano broncodilatazione e stimolazione del centro del respiro.
Le catecolamine, specie l'adrenalina, hanno profondi effetti sul metabolismo, determinando quale risultato finale una rapida disponibilità e utilizzazione delle fonti energetiche. Infatti, aumentano la produzione e l’utilizzazione di glucosio, facilitando la glicogenolisi e la gluconeogenesi sia a livello epatico che muscolare, facilitano la lipolisi a livello epatico, muscolare e adiposo, inibiscono la glicogenosintesi a tutti i livelli. Tutte queste azioni sono sia dirette che indirette attraverso l’inibizione della secrezione insulinica e la liberazione di glucagone.
A livello muscolare, specialmente l'adrenalina facilita la degradazione del glicogeno ad acido lattico con formazione di ATP, sorgente di energia indispensabile per la contrazione muscolare.
Infine, le catecolamine facilitano l’ossidazione del glucosio e la chetogenesi a livello epatico.
Metodi di valutazione dell’attività simpatica
Il metodo più disponibile per la valutazione dell’attività simpatica è quello indiretto, attraverso la misurazione dei parametri clinici prevalentemente controllati dal sistema simpatico, come la frequenza cardiaca, la pressione arteriosa, ecc. Tale metodo, di facile realizzazione, è però impreciso e poco sensibile, poiché numerosi altri sono i fattori che controllano tali variabili.
Ulteriore metodo indiretto di misurazione del tono neurovegetativo a livello miocardico è l’analisi della variabilità dell’intervallo R-R attraverso l’elettrocardiografia dinamica secondo Holter; una riduzione della variabilità dell’intervallo R-R è indice di aumentato tono simpatico, mentre un aumento della variabilità è conseguente ad aumento del tono vagale.
In tempi recenti si sono resi disponibili metodi più diretti.
Uno di questi è la misura delle catecolamine nei liquidi biologici, che può essere effettuata con metodiche (radioenzimatiche, HPLC) sufficientemente sensibili per determinarne correttamente i livelli campionando per piccoli volumi. La misura delle catecolamine ematiche fornisce indicazioni sull’attività immediata del SSA, mentre la misura nelle urine fornisce una valutazione più complessiva riguardo al periodo di raccolta del campione. La NA plasmatica riflette per lo più la quota liberata dai terminali simpatici dei muscoli del distretto dove si campiona. Durante esercizio fisico con impegno delle principale masse muscolari, la misura della NA nel sangue venoso periferico costituisce un buon indice dell’attivazione del SSA. La misura della A plasmatica fornisce un indice accurato di attivazione della midollare surrenalica. Poiché la A viene estratta dai tessuti periferici, la campionatura del sangue arterioso è da considerarsi preferibile a quella nel sangue venoso, dove i livelli sono perciò inferiori, anche se per lo più i valori sono fra loro correlati.
Altro metodo diretto per la misura dell’attività simpatica è la misurazione della scarica neuronale attraverso microelettrodi di tungsteno impiantati in nervi periferici, sia nella cute che nel distretto muscolare. È stata dimostrata una stretta correlazione fra i livelli di NA plasmatica e la scarica neuronale muscolare, sia a riposo che durante esercizio fisico dinamico.
Infine, è possibile valutare la sensibilità dei tessuti periferici alle catecolamine attraverso lo stimolo o il blocco farmacologico dei recettori adrenergici e la misura delle variazioni dei parametri biologici corrispondenti.
Bibliografia
- Gilbey MP, Spyer KM. Essential organization of the sympathetic nervous system. In: Catecholamines, Bailliere’s Clin Endocrinol Metab, Bailliere Tindall, London PMG Bouloux Ed, 1993: pag 259-78.
- Esler M, Haskinh GJ, Willett IR, et al. Noradrenaline release and sympathetic nervous system activity. J Hypertension 1985, 3: 117-29.
- Esler M, Jennings G, Lambert G, et al. Overflow of the catecholamine neurotransmitters to the circulation: source, fate and functions. Physiol Rev 1990, 70: 963-85.
- Goldstein D. Stress, catecholamines and cardiovascular disease. Oxford University Press, 1995.
- Raymond JR, Hnatowich M, Lefkowitz RJ, et al. Adrenergic receptors. Models for regulation of signal transduction processes. Hypertension 1990, 15: 119-31.
- Ahlquist RP. Study of adrenotropic receptors. Am J Physiol 1948, 153: 586-600.
- Lands AM, Arnold A, McAuliff JP, et al. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967, 214: 597-8.
- Berthelson S, Pettinger WA. A functional basis for classification of alpha-adrenergic receptors. Life Sci 1977, 1: 171-83.
- Starke K. Alfa-adrenoceptor subclassification. Rev Physiol Biochem Pharmacol 1981, 88: 199-236.
- Bylund DB. Subtypes of alpha2-adrenoceptors. Pharmacological and molecular biological evidence converge. Trends Pharmacol Sci 1988 9: 356-61.
- Cortez V, Santana M, Marques AP, et al. Regulation of catecholamine release in human adrenal chromaffin cells by β-adrenoceptors. Neurochem Int 2012, 60: 387-93.
- Reis DJ, Morrison S, Ruggiero DA. The C1 area of the brainstem in tonic and reflex control of blood pressure. Hypertension 1988, ll (suppl I): I8-13.
- van Zwieten PA, Jie K, van Brummelen P. Postsynaptic a1 and a2 adrenoceptor changes in hypertension. Cardiovasc Pharmacol 1987,10 (suppl 4): 568-75.
- Brodde O-E. Cardiac beta-adrenergic receptors. ISI Atlas of Scv Pharmacol 1987, l: 107-12.
- Keeton TK, Campbell WB. The pharmacologic alteration of renin release. Pharmacol Rev 1981, 32: 81-227.