Cateterismo selettivo delle vene surrenaliche

Soraya Puglisi & Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
(aggiornato al 28 gennaio 2020)
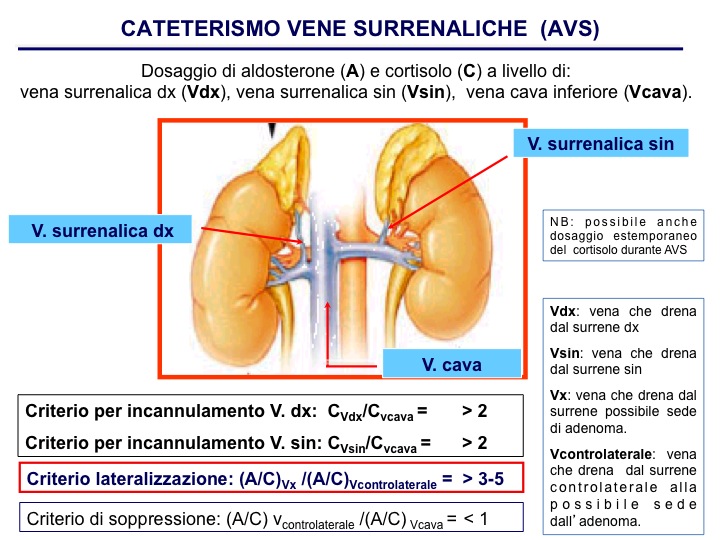
| Cateterismo venoso selettivo delle vene surrenaliche | |
| Indicazioni | Diagnosi differenziale dei sotto-tipi di iperaldosteronismo primario. Si effettua nei pazienti in cui gli esami biochimici di primo livello e di conferma sono diagnostici per iperaldosteronismo primario, suscettibili d’intervento chirurgico. Fanno eccezione pazienti giovani (< 35 anni), con fenotipo biochimico compatibile con iperaldosteronismo severo (ipopotassiemia spontanea, renina soppressa, aldosterone elevato > 20 ng/dL) e chiara evidenza radiologica di adenoma surrenalico unilaterale, che pertanto possono subito essere inviati a intervento di surrenectomia monolaterale, senza ulteriori indagini. Si presuppone, infatti, che una massa surrenalica in età giovanile abbia poche probabilità di essere un adenoma non secernente, soprattutto se il sospetto di iperaldosteronismo primitivo è sostenuto da chiari dati biochimici e di imaging. |
| Finalità | Evidenziare un gradiente secretorio di aldosterone tra i due surreni, dovuto alla presenza di un piccolo adenoma aldosterone-secernente o di una modesta iperplasia surrenalica monolaterale, non evidenziate con le tecniche di imaging. Confermare l’ipersecrezione di aldosterone da parte di una massa surrenalica individuata dagli accertamenti di imaging surrenalico. |
| Contro-indicazioni | Allergia al mezzo di contrasto iodato. Ipertensione non controllata. |
| Relazione con età, sesso, peso corporeo, gravidanza | Le popolazioni afro-americane hanno ridotti livelli di PRA e aldosterone, perciò dovrebbero essere utilizzati cut-off specifici. |
| Precauzioni | Anziani, pazienti con modesto aumento dei valori pressori. |
| Condizioni preliminari | Sospensione sempre degli anti-aldosteronici e, laddove possibile, dei farmaci interferenti con la secrezione di aldosterone (diuretici, ß-bloccanti, ACE-inibitori, sartani) (per i tempi, vedi diagnostica generale). Correzione dell’eventuale ipopotassiemia. Compenso pressorio (usando α-bloccanti e calcio-antagonisti). |
| Esecuzione |
|
| Possibili effetti collaterali | Il maggior rischio è l’emorragia surrenalica, che può essere minimizzato eseguendo preventivamente un'angiografia surrenalica e limitando l'utilizzo del mdc. Nei centri che dispongono di radiologi esperti, il tasso di rottura di una vena surrenalica è dello 0.61% ed è inversamente correlato con il numero di cateterismi eseguiti da ciascun radiologo. Sono stati riportati anche casi di dissezione, ematoma e trombosi venosa, durante il cateterismo simultaneo bilaterale, poiché è maggiore il tempo di ostruzione del vaso da parte del catetere, finché non viene cateterizzato con successo anche il contro-laterale. Queste possibili complicanze di solito vengono trattate in modo conservativo e non lasciano sequele a lungo termine. Reazione allergica al mezzo di contrasto iodato. Incremento dei livelli di creatininemia. Embolia colesterinica |
| Interpretazione | Condizione preliminare determinante per considerare il test idoneo (indicativa di un adeguato incannulamento dei vasi surrenalici) è il riscontro di un rapporto adeguato tra il valore di cortisolemia nelle vene surrenaliche e quello periferico (nella vena cava inferiore) - indice di selettività, SI. Per evitare errori determinati dalla diluizione del sangue refluo surrenalico nel caso in cui il catetere non sia posizionato perfettamente all’interno della vena surrenalica, i livelli di aldosterone vengono “corretti” per quelli di cortisolo (ossia espressi come rapporto aldosterone/cortisolo). Non c’è un consenso relativo al cut-off da utilizzare: per alcuni centri è sufficiente un SI > 1.1, altri usano criteri più restrittivi, quali SI > 2 (preferibilmente > 3) in condizioni basali e > 3 (preferibilmente > 5) durante l’infusione di ACTH. L’introduzione di metodiche di dosaggio rapido intra-procedurale del cortisolo permette al radiologo di ripetere subito il campionamento qualora l’incannulamento risulti scorretto, riducendo così la frequenza dei casi in cui è necessario ripetere l’intera procedura, i costi e il disagio per il paziente ed evitando inoltre il ritardo della diagnosi. L’indice di lateralizzazione (LI) è il rapporto tra la concentrazione di aldosterone/cortisolo in una vena surrenalica e la concentrazione di aldosterone/cortisolo nella vena surrenalica contro-laterale. Anche in questo caso non vi è omogeneità di interpretazione, dal momento che il rapporto considerato diagnostico varia tra 2 e 5. Monticone et al. suggeriscono di utilizzare un indice di lateralizzazione > 4 come diagnostico di patologia unilaterale e considerare i valori tra 3 e 4 come indeterminati. Alcuni centri al posto dell’indice di lateralizzazione, utilizzano un indice ipsi-laterale (rapporto > 2 tra aldosterone/cortisolo di una vena surrenalica e quello di una vena periferica) associato a un rapporto di soppressione contro-laterale (rapporto tra aldosterone/cortisolo della vena surrenalica contro-laterale e aldosterone/cortisolo della vena periferica, CLR) < 1. Per molti autori il CLR può essere utile nei pazienti in cui è stata incannulata una sola vena o che hanno un LI nella zona grigia. A oggi il ruolo del CLR è molto dibattuto, sia nell’indicazione alla surrenectomia che per la sua correlazione agli esiti clinici). |
| Attendibilità e ripetibilità dei risultati | La sensibilità diagnostica è intorno all’80% con una specificità del 100%. |
| Giudizio complessivo costo beneficio e costo-efficacia | L'AVS è una procedura tecnicamente difficile, che richiede l’intervento di radiologi esperti, associata a una scarsa standardizzazione dei protocolli utilizzati, sia nell’esecuzione della procedura che nell’interpretazione dei risultati. È un test costoso (necessita il regime ospedaliero di Day Hospital) e non privo di inconvenienti e rischi per il paziente. È inoltre necessario disporre di personale tecnico esperto, per evitare un’alta frequenza di inidoneità del test. È critica la manualità degli angio-radiologi, dalla quale dipende la percentuale di successo di cateterizzazione della vena surrenalica destra, che solitamente drena direttamente nella vena cava inferiore ed è perciò difficile da localizzare e da distinguere da altri vasi adiacenti. Secondo alcuni viene quindi ritenuto indispensabile quando, in presenza di un’alta probabilità di adenoma aldosterone-secernente, gli accertamenti di imaging non dimostrano una lesione monolaterale, in particolare nei pazienti oltre i 40 anni. Altri esperti giudicano sempre necessaria l’esecuzione del test, in quanto sensibilità e specificità della TC sono considerate non > 55% nella diagnosi di adenoma surrenalico aldosterono-secernente. |
| Bibliografia |
|
Classificazione degli iposurrenalismi primari
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
Gli iposurrenalismi primitivi sono classificabili dal punto di vista eziopatogenetico nelle seguenti forme (1,2):
- autoimmune (80-90% dei casi, forma più frequente nei paesi industrializzati): isolata (40%, più frequentemente nel sesso maschile) o parte di una sindrome poliendocrino-autoimmune (60%, più frequentemente nel sesso femminile, tipi 1 o 2);
- infettiva (TBC, HIV, CMV, miceti);
- infiltrativa (sarcoidosi, amiloidosi, emocromatosi, istiocitosi);
- emorragica (S. di Waterhouse-Friederichsen, terapia anti-coagulante, traumi);
- trombotica (LES, panarterite nodosa, sindrome da anticorpi anti-fosfolipidi, traumi);
- neoplastica (carcinoma surrenalico, metastasi);
- congenite (adrenoleucodistrofia, iperplasia surrenalica congenita, ipoplasia surrenalica congenita, sindromi familiari da resistenza all’ACTH);
- da farmaci (mitotane, aminoglutetimide, chetoconazolo, mifepristone);
- iatrogena (interventi di surrenectomia bilaterale).
Bibliografia
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003, 361: 1881-93.
- Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndrome: autoantibodies, autoantigens, and the applicability in diagnosis and disease prediction. Endocr Rev 2002, 23: 327-64.
Overview sugli iposurrenalismi primari
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
(aggiornato al 10 settembre 2015)
L’iposurrenalismo primario o malattia di Addison è una condizione clinica descritta per la prima volta nel 1855 da Thomas Addison, determinata dalla carente produzione surrenalica di glicocorticoidi, mineralcorticoidi ed androgeni, mentre il difetto della midollare è clinicamente irrilevante (1).
Si tratta di una patologia piuttosto rara, con prevalenza di 93-140 casi/milione, incidenza di 4.7-6.2 casi/milione/anno (1,2), quest’ultima stimata in ulteriore aumento nella popolazione occidentale (2), e che compare prevalentemente nell’età adulta, con una maggiore frequenza nella quarta decade di vita e nel sesso femminile (1).
L’iposurrenalismo primitivo è classificabile dal punto di vista eziopatogenetico in varie forme, tra cui quella autoimmune risulta essere più frequente nei paesi industrializzati e quella infettiva nel resto del mondo (1,3).
Il quadro clinico si differenzia in una forma acuta, che rappresenta una vera emergenza clinica con le manifestazioni cliniche dello shock ipovolemico, e una forma cronica, in cui frequentemente i segni e i sintomi presentano un esordio graduale e sono aspecifici (1,3). Nella storia naturale della forma autoimmune sono state peraltro descritte due fasi pre-cliniche, prive di segni o sintomi, e caratterizzate soltanto da alterazioni auto-anticorpali o biochimiche/ormonali (4).
Nell’insufficienza surrenalica acuta, le condizioni di urgenza del quadro clinico, che richiedono un trattamento tempestivo, non consentono una valutazione accurata della funzione surrenalica e pertanto ci si dovrà limitare al dosaggio di cortisolemia e di ACTH (1,3).
Nell’insufficienza surrenalica cronica, la diagnosi di iposurrenalismo primario è difficile nelle fasi pre-cliniche della forma autoimmune, ma la presenza di ipotensione arteriosa, collasso, iperpigmentazione cutanea, iposodiemia, iperpotassiemia, ipercalcemia, acidosi ed ipoglicemia non diversamente spiegabili sono suggestivi per la presenza di iposurrenalismo (1,3). In particolare, l’iposodiemia può essere presente in oltre il 90% dei casi (3).
In tutti i pazienti con sospetto clinico va eseguita una valutazione ormonale basale, con dosaggio al mattino (ore 7-9) di cortisolemia, ACTH, renina o attività reninica ed aldosterone; il dosaggio del DHEA-S non è attualmente riconosciuto come criterio diagnostico aggiuntivo (1,3-6).
In tutti i pazienti con forte sospetto clinico e valutazione ormonale basale di norma va comunque effettuata una valutazione ormonale dinamica, mediante test con ACTH sintetico alla dose di 250 µg con valutazione della cortisolemia (1,3-7). In pazienti con forme “mild” o subcliniche, è stato proposto da alcuni autori il test con ACTH sintetico alla dose di 1 µg, ma non è stato ancora universalmente accettato come test di provata superiorità diagnostica (8). Nel sospetto di una forma autoimmune vanno eseguiti specifici esami anticorpali (di non frequente disponibilità), quali anticorpi anti-corteccia surrenalica (ACA) e anticorpi anti-21-idrossilasi (3,4,9).
La terapia dell’insufficienza surrenalica acuta prevede l’impiego di idrocortisone per via e.v. e la correzione della disidratazione con un adeguato apporto idrico (3,10-13).
Nell’insufficienza surrenalica cronica il trattamento sostitutivo prevede l’impiego di glicocorticoidi, idrocortisone o cortisone acetato, quest'ultimo maggiormente impiegato nel nostro paese, e mineralcorticoidi, 9α-fluoro-idrocortisone. La terapia con DHEA è invece considerata opzionale (3,10-13).
Bibliografia
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003, 361: 1881-93.
- Løvås K, Husebye ES. High prevalence and increasing incidence of Addison's disease in western Norway. Clin Endocrinol (Oxf) 2002, 56: 787-91.
- Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014, 275: 104-15.
- Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndrome: autoantibodies, autoantigens, and the applicability in diagnosis and disease prediction. Endocr Rev 2002, 23: 327-64.
- Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab 1994, 79: 923-31.
- Dorin RI, Qualis CR, Crapo LM. Diagnosis of adrenal insufficiency. Ann Intern Med 2003, 139: 194-204.
- Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab 1992, 75: 259-64.
- Laureti S, Arvat E, Candeloro P, et al. Low dose (1 µg) ACTH test in the evaluation of adrenal dysfunction in pre-clinical Addison’s disease. Clin Endocrinol 2000, 53: 107-15.
- Falorni A, Nikoshkov A, Laureti S, et al. High diagnostic accuracy for idiopathic Addison's disease with a sensitive radiobinding assay for autoantibodies against recombinant human 21-hydroxylase. J Clin Endocrinol Metab 1995, 80: 2752-5.
- Arlt W. The approach to the adult with newly diagnosed adrenal insufficiency. J Clin Endocrinol Metab 2009, 94: 1059-67.
- Crown A, Lightman S. Why is the management of glucocorticoid deficiency still controversial: a review of the literature. Clin Endocrinol 2005, 63: 483-92.
- Hahner S, Allolio B. Therapeutic management of adrenal insufficiency. Best Pract Res Clin Endocrinol Metab 2009, 23: 167–79.
- Quinkler M, Hahner S. What is the best long-term management strategy for patients with primary adrenal insufficiency? Clin Endocrinol 2012, 76: 21–5.
Clinica e diagnostica iposurrenalismo primario
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
(aggiornato al 10 settembre 2015)
Clinica
Occorre distinguere il quadro clinico dell’insufficienza surrenalica acuta da quella cronica (1).
Nell’insufficienza surrenalica acuta, secondo uno studio tedesco (2) condizione più frequente di quanto si riteneva in passato (6.3 casi per 100 pazienti/anno), il quadro clinico è drammatico e, se non si realizza un intervento terapeutico immediato, porta rapidamente all’exitus. Spesso l’insufficienza surrenalica acuta si manifesta in conseguenza di stress intercorrenti (malattie febbrili, sepsi, traumi, interventi chirurgici) in un paziente affetto, talvolta inconsapevolmente, da insufficienza surrenalica cronica latente; più raramente è causata da un'emorragia surrenalica bilaterale in corso di sepsi, ritenuta oggi espressione di una variante particolarmente grave di coagulazione intravascolare disseminata (sindrome di Waterhouse-Friederichsen), oppure in seguito a vomito, diarrea o malattie febbrili intercorrenti in un paziente affetto da insufficienza surrenalica cronica che non ha modificato la propria terapia glucocorticoidea sostitutiva in maniera adeguata (1-3).
Il paziente presenta le manifestazioni cliniche dello shock ipovolemico, con profonda prostrazione, confusione, ipotensione arteriosa, tachicardia, nausea, vomito, disidratazione, talvolta dolori crampiformi all’addome (pseudo-addome acuto); la febbre può essere manifestazione di tale condizione o del processo morboso scatenante (1-3).
Nell’insufficienza surrenalica cronica frequentemente i segni e i sintomi presentano un esordio graduale, sono aspecifici, dovuti alla cronica carenza ormonale (1,3).
In particolare, tra i sintomi si annoverano astenia, anoressia, nausea, vomito, ricerca di cibi salati, epigastralgie e algie addominali, irritabilità e depressione, calo ponderale, polimialgie diffuse, vertigini, riduzione o perdita della libido (nel sesso femminile). Tali sintomi si aggravano col tempo. Inizialmente si ha infatti una fase di ridotta riserva surrenalica, con secrezione di glicocorticoidi conservata in condizioni basali, ma insufficiente in situazioni di stress; quando la perdita di tessuto corticale raggiunge circa il 90%, si ottiene il quadro completo di insufficienza surrenalica (1,3).
I segni clinici più tipici sono caratterizzati da iperpigmentazione cutanea o melanodermia (con localizzazione prevalentemente nelle zone esposte alla luce e allo sfregamento, quali volto, gomiti, ginocchia, pliche palmari, areole mammarie, cicatrici, e delle mucose della guancia, lingua, gengive, mucosa anale e vulvo-vaginale), ipotensione arteriosa, iposodiemia, iperpotassiemia, anemia, linfocitosi ed eosinofilia, TSH aumentato, ipercalcemia, ipoglicemia (più frequentemente si manifesta a digiuno, dopo attività fisica o dopo assunzione di alcolici), riduzione e successivamente scomparsa dei peli pubici e ascellari (nel sesso femminile), alterazioni mestruali di grado assai variabile (1,3).
Nella storia naturale della forma autoimmune sono state peraltro descritte due fasi pre-cliniche, prive di segni o sintomi, e caratterizzate da alterazioni auto-anticorpali (fase potenziale, stadio 0) o biochimiche/ormonali (fase subclinica, stadio 1, 2 e 3), come illustrato in figura 1 (4).
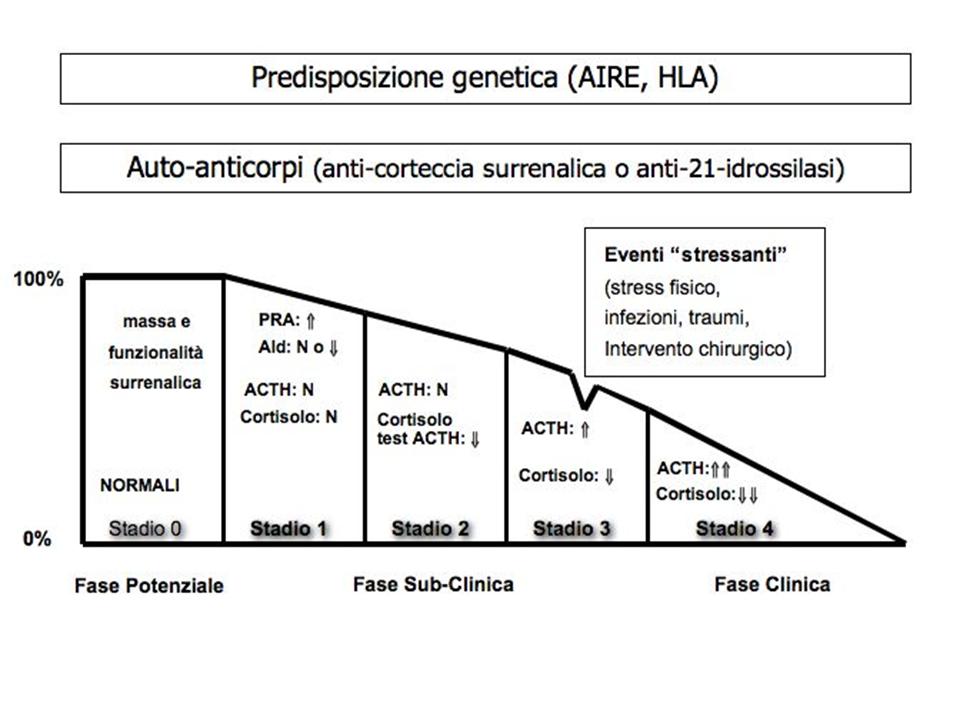
In alcune forme non autoimmuni (es. forma infettiva) può essere presente un deficit secretivo parziale della sola zona fascicolata, con preservata secrezione mineralcorticoide e mancanza di ipotensione arteriosa, iposodiemia ed iperpotassiemia (1,3).
Diagnostica
Nell’insufficienza surrenalica acuta, la condizioni di urgenza del quadro clinico, che richiede un trattamento tempestivo, non consente una valutazione accurata della funzione surrenalica e pertanto ci si dovrà limitare al dosaggio basale di cortisolemia e di ACTH (1-3,5).
Nell’insufficienza surrenalica cronica, la diagnosi di iposurrenalismo primario è difficile nelle fasi pre-cliniche della forma autoimmune, ma la melanodermia non diversamente spiegabile deve sempre suggerire la presenza di iposurrenalismo (1,3,5-7).
In tutti i pazienti con sospetto clinico va eseguita una valutazione ormonale basale, con dosaggio al mattino (ore 7-9) di cortisolemia, ACTH, renina o attività reninica ed aldosterone (1,3,5-7), anche se queste ultime valutazioni non sono universalmente eseguite per l’estrema variabilità dei risultati, sia legata al metodo di dosaggio che a fattori interferenti, e per una bassa predittività di malattia rispetto a cortisolemia e ACTH (3); il dosaggio del DHEA-S non è attualmente riconosciuto come criterio diagnostico aggiuntivo (1,3,5-7).
La diagnosi di iposurrenalismo primario viene posta in presenza di valori ridotti di cortisolemia ed aumentati di ACTH. In particolare secondo una recente Consensus Europea (3), valori di cortisolemia < 9 µg/dL (250 nmol/L) risultano diagnostici, mentre valori di cortisolemia < 14 µg/dL (400 nmol/L) risultano suggestivi per iposurrenalismo primario. Per quanto riguarda i valori di ACTH, si considerano aumentati se > 100 pg/mL (22 pmol/L) (1,5-7). Qualora si valutino anche renina o attività reninica ed aldosterone, sono da considerarsi diagnostici per iposurrenalismo primario valori aumentati di renina o attività reninica plasmatica (PRA > 3.0 ng/dL/h) e valori di aldosterone normali o ridotti (< 5 ng/dL) (1,5-7).
In tutti i pazienti con forte sospetto clinico, anche se la valutazione ormonale basale è normale, va comunque effettuata una valutazione ormonale dinamica mediante test con ACTH sintetico alla dose di 250 µg con valutazione della cortisolemia: un picco di cortisolemia < 500-550 nmol/L (18-22 µg/dL) va ritenuto indicativo di iposurrenalismo primitivo soltanto nell’ambito di un adeguato contesto clinico; è stato suggerito l’impiego di cut-off di picco di cortisolo più bassi (< 415 nmol/L, 15 µg/dL) per aumentare la sensibilità del test (1,3,5-7).
In pazienti con forme “mild” o subcliniche, è stato proposto da alcuni autori Il test con ACTH sintetico alla dose di 1 µg, ma non è stato ancora universalmente accettato come test di provata superiorità diagnostica (8).
Nel sospetto di una forma autoimmune vanno eseguiti specifici esami anticorpali (di non frequente disponibilità), quali anticorpi anti-corteccia surrenalica (ACA) e anticorpi anti-21-idrossilasi (3,4,9). Gli ACA e gli anti-21-idrossilasi sono immunoglobuline organo-specifiche, dimostrabili rispettivamente mediante tecniche di immunofluorescenza indiretta e di immunoprecipitazione o, più recentemente per gli anti-21-idrossilasi, mediante tecniche radioimmunologiche. Entrambi gli anticorpi riconoscono il loro auto-antigene nell’enzima 21-idrossilasi. Mentre la sensibilità dei due anticorpi è pressoché identica all’esordio della malattia, la sensibilità degli anti-21-idrossilasi è superiore in pazienti con lunga durata di malattia. La specificità diagnostica degli anti-21-idrossilasi è estremamente elevata e la loro presenza costituisce un fattore di rischio per lo sviluppo di insufficienza cortico-surrenalica clinica, in funzione del titolo anticorpale, dell’età del paziente (se presenti in età infantile il rischio a 15 anni è del 100%), del contesto clinico (presenza di altre patologie autoimmuni) (3,4,9).
Nella diagnostica dell’iposurrenalismo primario le indagini strumentali non sono generalmente necessarie nella forma ad eziologia autoimmune, possono essere invece indicate (TAC o RMN addome senza mdc) nel sospetto di forme infiltrative, emorragiche o neoplastiche (1,3).
Bibliografia
- Arlt W, Allolio B. Adrenal insufficiency. Lancet 2003, 361: 1881-93.
- Hahner S, Loeffler M, Bleicken B, et al. Epidemiology of adrenal crisis in chronic adrenal insufficiency: the need for new prevention strategies. Eur J Endocrinol 2010, 162: 597–602.
- Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014, 275: 104-15.
- Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndrome: autoantibodies, autoantigens, and the applicability in diagnosis and disease prediction. Endocr Rev 2002, 23: 327-64.
- Grinspoon SK, Biller BM. Clinical review 62: Laboratory assessment of adrenal insufficiency. J Clin Endocrinol Metab 1994, 79: 923-31.
- Dorin RI, Qualis CR, Crapo LM. Diagnosis of adrenal insufficiency. Ann Intern Med 2003, 139: 194-204.
- Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab 1992, 75: 259-64.
- Laureti S, Arvat E, Candeloro P, et al. Low dose (1 µg) ACTH test in the evaluation of adrenal dysfunction in pre-clinical Addison’s disease. Clin Endocrinol 2000, 53: 107-15.
- Falorni A, Nikoshkov A, Laureti S, et al. High diagnostic accuracy for idiopathic Addison's disease with a sensitive radiobinding assay for autoantibodies against recombinant human 21-hydroxylase. J Clin Endocrinol Metab 1995, 80: 2752-4.
Forme genetiche di iposurrenalismo primario
Chiara Sabbadin
Unità di Endocrinologia, Dipartimento di Medicina, Università di Padova
(aggiornato al 12 ottobre 2021)
L’iposurrenalismo primario (PAI) è una condizione clinica rara, potenzialmente fatale, che necessita di un tempestivo approccio diagnostico e terapeutico. Tra le forme congenite (1), la più nota è l’iperplasia surrenalica congenita (CAH), dovuta a difetti nella steroidogenesi, che, in base al tipo e alla gravità, alterano la sintesi di mineralcorticoidi, glucocorticoidi e ormoni sessuali, sia a livello surrenalico sia gonadico. La causa più frequente di CAH è il deficit di 21-idrossilasi, che può dare tre diversi fenotipi in base al grado di attività enzimatica residua:
- forma con perdita di sali (attività enzimatica < 1%), caratterizzata da PAI a insorgenza nelle prime settimane di vita e iperandrogenismo, responsabile in entrambi i sessi di pubertà precoce, nelle femmine di ambiguità dei genitali alla nascita e successivo quadro simile al fenotipo classico della sindrome dell’ovaio policistico (PCOS), mentre nei maschi comporta infertilità e disturbi metabolici in età adulta;
- forma virilizzante semplice (attività enzimatica residua 1-2%), con manifestazioni da iperandrogenismo analoghe alla forma con perdita di sali; manca il quadro di iposurrenalismo clinico, anche se questo potrebbe essere slatentizzato in situazioni di stress;
- forma non classica (attività enzimatica residua del 20-50%), responsabile di diversi quadri di iperandrogenismo clinico a esordio in età adolescenziale-adulta (late-onset), con fenotipo simile alla PCOS nella femmina e spesso del tutto asintomatica nel maschio.
Le prime due forme vengono definite classiche e interessano circa 1:16.000 nati; la forma non classica, invece, ha prevalenza maggiore, attorno a 1:1000 nella popolazione caucasica.
Altre forme congenite di iposurrenalismo meno frequenti sono altri difetti enzimatici responsabili di iperplasia surrenalica congenita (deficit di 17α-idrossilasi/17,20 liasi, deficit di 11-idrossilasi, deficit di 3-ß-idrossi-steroido-deidrogenasi, deficit di citocromo P450 ossido-reduttasi), la sindrome di Allgrove o della tripla A (acalasia, Addison e alacrimia) (2), l’adrenoleucodistrofia X-linked (malattia perossisomale, caratterizzata dall'accumulo plasmatico e tissutale di acidi grassi a catena lunga e molto lunga, che interessa principalmente i maschi e provoca diversi disturbi neurologici, spesso associati o preceduti dall’insorgenza di PAI e a volte di ipogonadismo primario in età adulta) (3) e il deficit familiare di glucocorticoidi (associato a mutazioni nel gene del recettore per l'ACTH) (4). Inoltre, negli ultimi anni sono state scoperte molte altre cause genetiche rare di PAI, la cui diagnosi ha importanti implicazioni non solo sui familiari, ma anche sulla gestione e il follow-up del paziente affetto.
Deficit della proteina StAR
StAR, codificata dall’omonimo gene sul cromosoma 8, è la proteina chiave per l’inizio della sintesi di tutti gli ormoni steroidei, in quanto media il trasporto del colesterolo dalla superficie esterna della membrana mitocondriale a quella interna, dove inizia la steroidogenesi per azione di P450side-chain (P450scc). Deficit severi di StAR sono associati ad iperplasia surrenalica lipoidea congenita (LCAH), caratterizzata da PAI ad esordio peri-natale, fenotipo femminile nei maschi e ipogonadismo ipergonadotropo nelle femmine (5). Deficit parziali di StAR, invece, sono associati a forme di LCAH non classica, caratterizzata solo da PAI, anche a esordio tardivo o con solo deficit dei glucocorticoidi, mimando un quadro simile al deficit familiare di glucocorticoidi (familial glucocorticoid deficiency), altra rara sindrome genetica, non del tutto chiarita, associata a mutazioni nel gene del recettore per l'ACTH (4). Nei soggetti con deficit parziali viene comunque raccomandato un controllo a lungo termine dell’asse gonadico, sia in età puberale sia in età adulta, proponendo cautelativamente anche metodiche di crio-conservazione dei gameti, in particolare nei maschi, per possibili alterazioni della fertilità nel tempo.
Deficit di P450scc
P450scc, codificato dal gene CYP11A1 sul cromosoma 15 e situato sulla parte interna della membrana mitocondriale, è l’altro fattore chiave assieme a StAR degli stadi iniziali della steroidogenesi, in quanto converte il colesterolo in pregnenolone. Mutazioni di P450scc, in base alla severità del deficit, provocano quadri simili a LCAH classica e non classica (6). Anche per i deficit parziali di P450scc, viene raccomandato il monitoraggio periodico della funzione sessuale soprattutto maschile, in quanto sono stati riportati casi di testicular adrenal rest tumors, soprattutto nei soggetti con scarso compenso surrenalico.
Mutazioni di recettori nucleari coinvolti nello sviluppo e nella funzione di surreni e gonadi
DAX-1: codificato dal gene NR0B1, localizzato sul braccio corto del cromosoma X, è stato descritto per la prima volta nel 1994 come responsabile di ipoplasia surrenalica congenita X-linked (7). Sono note numerose mutazioni genetiche, che, sempre sulla base del difetto di sintesi (completo o parziale), possono determinare la forma classica, caratterizzata da esordio precoce di PAI e ipogonadismo ipogonadotropo con infertilità, o non classica, caratterizzata da diversi fenotipi, a esordio tardivo, con ipogonadismo centrale parziale o anche con caratteristiche paradosse, come macropene e pubertà precoce. Il corretto inquadramento diagnostico di questi soggetti permette di ricercare anche eventuali forme di PAI subclinico nei familiari di sesso maschile della linea materna.
SF-1 (fattore steroidogenico 1): codificato dal gene NR5A1, localizzato sul cromosoma 9, è un altro fattore chiave che regola principalmente lo sviluppo testicolare e ovarico, mentre sembra meno determinante per lo sviluppo dei surreni. Infatti, alterazioni di SF-1 raramente provocano PAI, mentre risultano soprattutto associate a diversi spettri di disgenesia/disfunzione gonadica, descritti principalmente nel maschio: disturbi dello sviluppo sessuale, ipospadia, criptorchidismo, ipogonadismo e infertilità (8). Più recentemente sono state descritte mutazioni del gene NR5A1 anche in donne con insufficienza ovarica primaria sporadica o familiare. In tutti questi soggetti con disfunzioni gonadiche è raccomandato un follow-up a lungo-termine, perché resta ancora da chiarire se possano sviluppare nel tempo neoplasie gonadiche e PAI.
Disordini di crescita multi-sistemici
CDKN1C: inibitore della progressione del ciclo cellulare, viene espresso solamente dall’allele materno localizzato sul cromosoma 11, mimando una condizione X-linked. Mutazioni associate ad acquisizione di funzione di CDKN1C comportano una ridotta proliferazione cellulare associata alla sindrome IMAGe, caratterizzata da ritardo di crescita intra-uterino, displasia metafisaria (con arti corti), ipoplasia surrenalica congenita e anomalie genito-urinarie (9). Tale sindrome è stata descritta per la prima volta nel 1999 e risulta associata anche a tratti dismorfici (fronte prominente, setto nasale largo, orecchie basse) e diabete mellito. Di contro, mutazioni associate a perdita di funzione di CDKN1C sono state riscontrate nel 10% dei pazienti affetti da sindrome di Beckwith-Wiedemann, una malattia da iper-accrescimento, associata a malformazioni congenite e rischio di sviluppo di tumori, anche surrenalici.
SAMD9: è un altro inibitore della proliferazione cellulare, localizzato sul braccio lungo del cromosoma 7 ed espresso durante lo sviluppo fetale. Mutazioni associate ad acquisizione di funzione si associano alla sindrome MIRAGE (mielodisplasia, infezioni, ritardo di crescita intra-uterino, ipoplasia surrenalica congenita, disgenesia gonadica ed enteropatia), descritta per la prima volta nel 2016, caratterizzata da un ampio spettro di manifestazioni cliniche, con diversa gravità, e associata a elevata mortalità, prevalentemente per infezioni gravi entro il secondo anno di vita (10). Una caratteristica peculiare di alcuni bimbi affetti da mutazioni di SAMD9 è che spesso sviluppano nelle cellule ematopoietiche monosomia del cromosoma 7 o delezioni del suo braccio lungo o mutazioni associate a perdita di funzione di SAMD9: tali meccanismi compensatori, eliminando l’allele mutato, conferiscono un vantaggio di crescita clonale, che può comportare lo sviluppo di sindromi mielodisplastiche o forme leucemiche. Infine, sono stati riportati anche bimbi con fenotipi più lievi, dovute a forme di mosaicismo per l’allele con mutazione di SAMD9.
POLE1: fattore chiave nella replicazione del DNA, localizzato sul cromosoma 12. Mutazioni associate alla sua perdita di funzione sono state riscontrate in bimbi con sindrome IMAGe-like, caratterizzata principalmente da deficit di crescita, ipoplasia surrenalica congenita (con diversi gradi di PAI), deficit del sistema immunitario e tratti dismorfici (11).
Una nuova sfingolipidosi: deficit di sfingosina-1-fosfato liasi tipo 1 (SGPL1)
SGPL1 è un enzima coinvolto nella degradazione delle ceramidi, il cui gene è localizzato sul cromosoma 10 e il cui deficit comporta una forma di sfingolipidosi, in cui l’accumulo di sfingolipidi e ceramidi a livello intra-cellulare comporta anche PAI, oltre a manifestazioni multi-sistemiche simili a quelle della malattia di Fabry o di Gaucher (disfunzioni neurologiche, linfocitopenia, sindrome nefrosica) (12). Il corretto inquadramento diagnostico del deficit di SGPL1 risulta, pertanto, fondamentale anche per individuare precocemente un possibile concomitante iposurrenalismo, che potrebbe essere mascherato dai cicli di terapia steroidea per la sindrome nefrosica.
Conclusioni
Le forme genetiche di PAI sono rare e di alcune, come quelle di recente scoperta, sono stati riportati solo un centinaio di casi; tuttavia, il loro corretto inquadramento diagnostico è molto utile per le importanti implicazioni epidemiologiche, terapeutiche e di follow-up per il paziente e i suoi familiari. La diagnosi e successiva valutazione genetica, pertanto, devono essere eseguite in centri esperti e specializzati.
Cosa fare di fronte a un bimbo o adolescente con recente diagnosi di PAI:
- ricercare attraverso una dettagliata anamnesi familiare possibili parenti affetti, individuando trasmissioni X-linked o aree geografiche di provenienza note per l’elevata ricorrenza di alcune mutazioni, come ad esempio le zone centrali della Turchia, ad alta prevalenza di varianti di CYP11A1;
- individuare altre manifestazioni cliniche eventualmente associate a PAI, che potrebbero orientare verso la diagnosi, come segni di iperandrogenismo, anomalie genito-urinarie, ritardo di crescita intra-uterino, tratti dismorfici, patologie autoimmunitarie;
- sulla base del quadro anamnestico e clinico, indagare i livelli di 17OH-progesterone, acidi grassi a catena molto lunga, la funzione gonadica, la presenza di anticorpi anti-corticale del surrene e anti-21-idrossilasi, per richiedere successivamente un eventuale test genetico mirato a un singolo gene;
- nelle forme congenite che rimangono non chiarite, proseguire con le tecniche di next-generation sequencing, oramai sempre più diffuse in alcuni laboratori di genetica dei centri di III livello, che permetteranno di individuare non solo nuove cause, prevalenza e distribuzione di tali forme, ma anche pannelli di geni o esoni target, che potranno rendere ancora più rapida e meno dispendiosa tale ricerca in futuro.
Bibliografia
- Buonocore F, Achermann JC. Primary adrenal insufficiency: new genetic causes and their long-term consequences. Clin Endocrinol 2020, 92: 11-20.
- Alhassoun M, Almakadma AH, Almustanyir S, et al. Triple A multisystem disorder: Allgrove syndrome. Cureus 2021, 13: e17476.
- Zhu J, Eichler F, Biffi A, et al. The changing face of adrenoleukodystrophy. Endocr Rev 2020, 41: 577-93.
- Meimaridou E, Hughes CR, Kowalczyk J, et al. Familial glucocorticoid deficiency: new genes and mechanisms. Mol Cell Endocrinol 2013, 371: 195-200.
- Bose HS, Sugawara T, Strauss JF, Miller WL. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med 2002, 335: 1870-9.
- Baker BY, Lin L, Kim CJ, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab 2006, 91: 4781-45.
- Suntharalingham JP, Buonocore F, Duncan AJ, Achermann JC. DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab 2015, 29: 607-19.
- Orekhova AS, Kalinchenko N, Morozov IA, et al. A novel mutation in the critical P-box residue of steroidogenic factor-1 presenting with XY sex reversal and transient adrenal failure. Horm Res Paediatr 2018, 89: 450-4.
- Vilain E, Le Merrer M, Lecointre C, et al. IMAGe, a new clinical association of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies. J Clin Endocrinol Metab 1999, 84: 4335-40.
- Shima H, Hayashi M, Tachibana T, et al. MIRAGE syndrome is a rare cause of 46, XY DSD born SGA without adrenal insufficiency. PLoS One 2018, 13: e0206284.
- Logan CV, Murray JE, Parry DA, et al. DNA polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet 2018, 103: 1038-44.
- Prasad R, Hadjidemetriou I, Maharaj A, et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest 2017, 127: 942-53.
Terapia iposurrenalismo primario
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
(aggiornato al 10 settembre 2015)
INSUFFICIENZA SURRENALICA ACUTA
Vanno immediatamente iniettati per via ev 100 mg di idrocortisone e successivamente 100-300 mg dello steroide nelle 24 ore; questa dose totale può essere somministrata sia per infusione continua che divisa in boli ev o somministrazioni im ogni 6 ore (anche se i livelli di cortisolemia ottenuti sono superiori a quelli osservati nel soggetto normale in condizioni di stress) (1,2).
La crisi viene generalmente superata nelle prime 24 ore e pertanto nei giorni successivi le dosi di idrocortisone potranno essere progressivamente ridotte fino ad instaurare la terapia orale di mantenimento. Si dovranno comunque mantenere dosi elevate di steroide fino alla rimozione della causa scatenante la crisi (1,2).
Unitamente alla terapia steroidea, si dovrà correggere la disidratazione, con un adeguato apporto idrico (3-4 L di soluzione salina isotonica, 1 L nella prima ora e 500 mL/h nelle 2-3 ore successive) (1,2).
Non serve somministrare mineralcorticoidi, perchè le dosi utilizzate di glicocorticoidi sono sufficienti a saturare anche i recettori MR (1,2).
Ogni paziente portatore di insufficienza surrenalica, per evitare la possibilità che non venga riconosciuta ed adeguatamente trattata una crisi acuta, dovrebbe sempre portare con sè un alert (documento nel portafoglio, braccialetto, collana) che segnali la sua situazione e inviti l'eventuale soccorritore a prendere provvedimenti adeguati. Tra i vari documenti disponibili, quello dell'Associazione Pazienti con Addison (AIPAD) è particolarmente ben fatto: scarica il documento.
INSUFFICIENZA SURRENALICA CRONICA
Il trattamento sostitutivo prevede l’impiego di glicocorticoidi (idrocortisone o di cortisone acetato, quest'ultimo maggiormente impiegato nel nostro paese) e mineralcorticoidi (9α-fluoro-idrocortisone) (1-4).
Glucocorticoidi
Le dosi di impiegate oggi derivano dall’evidenza che la quantità di cortisolo fisiologicamente prodotta dai surreni varia tra i 5 e i 10 mg/m2 di superficie corporea/die, che corrispondono a una dose media di cortisolo variabile tra 8 e 17 mg/die (1-4).
Sebbene sia preferibile un dosaggio personalizzato per ogni singolo paziente, in base al peso oppure alla superficie corporea, è comune pratica clinica l’impiego di un dosaggio fisso di idrocortisone o cortisone acetato, variabile rispettivamente tra 15-25 mg/die e 18.75-31.25 mg/die (1,2). La dose totale viene frazionata:
- in 2 somministrazioni giornaliere: 2/3 del totale al mattino e 1/3 intorno alle h 15-16;
- in 3 somministrazioni giornaliere: 1/2 del totale al mattino, 1/4 a mezzogiorno e 1/4 a fine pomeriggio.
Non ci sono evidenze definitive a favore dell'uno o altro composto (1-4). Va peraltro ricordato che il cortisone acetato è un pro-ormone che richiede la conversione a idrocortisone durante il primo passaggio epatico da parte dell’enzima 11-ß-idrossi-steroido-deidrogenasi di tipo 1 e che quindi presenta un picco di azione un po’ più ritardato rispetto all’idrocortisone (2).
Numerosi farmaci possono interferire con l’efficacia di entrambi i composti per attivazione dell’enzima CYP3A4 e richiedono un aumento della dose: anti-epilettici e barbiturici, anti-TBC, anti-fungini, etomidate e topiramato. Viceversa, sostanze quali liquirizia e succo di ananas possono inibire l’attività dell’enzima CYP3A4 e richiedere una riduzione della dose (2).
L’efficacia del trattamento con entrambi gli steroidi è limitata dalla difficoltà di mantenere livelli fisiologici di cortisolemia nell’arco delle 24 ore, e di riprodurre il fisiologico profilo nictemerale del cortisolo, con picco al risveglio, secondo picco di minore intensità nelle prime ore del pomeriggio e secrezione quasi assente durante le ore notturne. Inoltre, a causa della farmacocinetica di entrambi gli steroidi, il paziente necessita inevitabilmente di dosi totali giornaliere che sono quasi costantemente sovra-fisiologiche. Nonostante gli sforzi di ottimizzazione della terapia sostitutiva, i pazienti con insufficienza cortico-surrenalica presentano inevitabilmente ampie oscillazioni della concentrazione sierica di cortisolo, con valori ampiamente sovra-fisiologici tra 90’ e 120’ dopo l’assunzione e concentrazioni inferiori alla norma prima dell’assunzione del farmaco (1-4). Negli ultimi anni sono stati sintetizzati composti a rilascio modificato dell’idrocortisone. In particolare sono stati studiati due tipi di composti impiegabili per via orale:
- un primo composto a rilascio modificato in due fasi, con un rivestimento esterno che garantisce la liberazione immediata del farmaco e un core interno che permette una liberazione lenta, disponibile in forma di compresse da 5 o 20 mg (Plenadren®) (5,6);
- un secondo composto a rilascio ritardato, che determina un incremento della cortisolemia a partire da 4 ore dopo l’assunzione orale e picco d’azione dopo 8 ore, disponibile in forma di compresse da 5 o 15 mg (Chronocort®) (7-11).
Mentre il primo farmaco è disponibile nel nostro paese ed è erogabile a totale carico del Servizio Sanitario Nazionale per il trattamento dell'insufficienza surrenalica negli adulti, previa compilazione di adeguato piano terapeutico, il secondo farmaco non è ancora disponibile nel nostro paese ed è stato studiato in soggetti normali e in pazienti con sindrome adreno-genitale.
Il composto a rilascio modificato in due fasi presenta il vantaggio di poter venir assunto una volta sola al giorno, al mattino, al risveglio, e presenta un profilo di rilascio simile a quello fisiologico del cortisolo, con bassi livelli sierici notturni. La disponibilità del preparato in due dosaggi da 5 e 20 mg permette, inoltre, la personalizzazione della dose ed incrementi della posologia in occasione di eventi stressanti concomitanti (5). In un recente studio clinico condotto in pazienti con ipocorticosurrenalismo primario, dopo 12 settimane di trattamento con tale farmaco sono stati osservati effetti cardiovascolari positivi, quali riduzione della pressione arteriosa e dell’HbA1c (6).
Il composto a rilascio ritardato richiede, invece, due somministrazioni giornaliere: la dose maggiore (circa 20 mg) deve essere assunta la sera (intorno alle ore 22-23), in modo da garantire un fisiologico picco di cortisolo plasmatico al mattino (tra le ore 6 e le ore 7), al risveglio, ed una seconda dose minore (circa 10 mg) va assunta al mattino al risveglio, per garantire il fabbisogno giornaliero e mimare il secondo picco fisiologico di cortisolo delle prime ore pomeridiane. Tale composto, rispetto al precedente, sembra presentare una maggiore esposizione sistemica e dei tessuti al cortisolo nelle prime ore notturne (7-9). Due studi clinici hanno dimostrato la superiorità di tale composto rispetto alla terapia tradizionale in pazienti con sindrome adreno-genitale (10,11).
Entrambi i composti non sono peraltro ancora in grado di garantire livelli costanti di cortisolemia nell’arco delle 24 ore, in particolare di impedire la caduta della cortisolemia durante la notte e i picchi nelle prime ore del mattino (5-11).
Mineralcorticoidi
La terapia con 9α-fluoro-idrocortisone viene somministrata in un’unica dose giornaliera di 0.05-0.2 mg al mattino. L’associazione del mineraloattivo evita la somministrazione di dosaggi sovra-fisiologici di glucocorticoidi e la comparsa di effetti secondari correlati all’eccesso di questi ultimi (1,2).
La nuova formulazione del farmaco prevede la conservazione in frigorifero, ma se conservato a temperatura ambiente è previsto un deterioramento dello 0.1% nei primi 6 mesi (2).
Alcuni farmaci, come diuretici, acetazolamide, carbenoxolone, contraccettivi contenenti drospirenone (progestinico con attività anti-aldosteronica), possono interferire con il 9α-fluoro-idrocortisone e andrebbero evitati (2). In presenza di ipertensione, occorre aggiungere un farmaco anti-ipertensivo e considerare una riduzione della terapia con 9α-fluoro-idrocortisone ma non la completa sospensione per il rischio di iposodiemia ed iperpotassiemia (2). È importante che l’alimentazione dei pazienti preveda l’introduzione libera di cibi salati ed eviti gli integratori a base di potassio (2).
Androgeni
La terapia con DHEA è invece considerata opzionale, da impiegare in casi selezionati solo nelle donne, per gli effetti positivi dimostrati sulla qualità di vita e sulla sfera sessuale, oltre che sul quadro lipidico, sulla sensibilità insulinica e sulla composizione corporea; gli studi esistenti condotti con dosi di 25-50 mg/die per periodi non superiori a 12 mesi non permettono al momento di indicare tale terapia come necessaria (12-16).
SITUAZIONI PARTICOLARI (1,2)
Stress fisico minore (febbre, malattia acuta infettiva intercorrente, intervento chirurgico con anestesia locale) o stress psichico maggiore e prolungato: la dose va duplicata o triplicata secondo alcuni autori (1), mentre la Consensus non suggerisce di modificare la terapia in corso se non in pazienti sintomatici, con l’aggiunta di una dose supplementare pari a 20 mg di idrocortisone (2).
Stress fisico di media o alta entità (trauma, interventi chirurgici con anestesia generale): se la via orale è impraticabile o sconsigliabile, come in presenza di vomito o diarrea, in previsione di manovre endoscopiche, si dovrà ricorrere alla somministrazione dello steroide per via parenterale, a dosaggi variabili in funzione della situazione clinica, evitando di impiegare in maniera indiscriminata dosi troppo elevate, simili a quelle impiegate nell’insufficienza surrenalica acuta e soprattutto cercando di ritornare alla terapia abituale nel più breve tempo possibile (1).
In corso di gravidanza è da preferire l’idrocortisone. Secondo una recente Consensus (2) nel 3° trimestre vanno aumentate la dose di idrocortisone (di 2.5-10 mg/die) e quella di fluoridrocortisone (per l’effetto anti-mineralcorticoideo del progesterone). Durante il travaglio va utilizzato idrocortisone a boli di 100 mg ev, ripetibili ogni 6 h se necessario e la dose orale va poi raddoppiata per 24-48 h dopo il parto.
| Tabella 1 Terapia "supplementare" con idrocortisone in corso di stress |
|
| Tipo di stress | Dose di idrocortisone |
| Minore (es. colonscopia, chirurgia addominale in Day-Surgery) | 25 mg in bolo ev (il giorno dell'evento) (1) 100 mg im prima della procedura o all’induzione dell’anestesia e raddoppio della dose orale per 24 h (2) |
| Moderato (es. intervento chirurgico in regime di ricovero ospedaliero ordinario) | 25-50 mg bolo ev (pre-anestesia) e poi 50-100 mg infusione continua ev in 24 h (1) 100 mg im (pre-anestesia) e poi 100 mg im ogni 6 h per 24-48 h e raddoppio della dose orale per 24-48 h (2) |
| Severo (pancreatite, politrauma, chirurgia maggiore inclusa cardiochirurgia e trapianti) | 50-100 mg bolo ev (pre-anestesia) e poi 100-150 mg infusione continua ev per 48-72 h (1) 100 mg im ogni 6 h fino alla ripresa dell’alimentazione orale e raddoppio della dose orale per almeno 48 h (2) |
| Critico (shock settico e ipovolemico) | 50-100 mg bolo ev ogni 6 h oppure 0.18 mg/kg/h infusione continua ev (per 48-72 h o fino a risoluzione) (1) |
MONITORAGGIO DELL'ADEGUATEZZA DELLA TERAPIA SOSTITUTIVA
Nessun dosaggio ormonale finora proposto (CLU, cortisolo random, curve giornaliere per cortisolemia sierica e salivare, ACTH, renina) permette di stabilire esattamente l’adeguatezza della terapia sostitutiva glico- e mineralcorticoide (1,2,17). Nel sospetto di sotto-dosaggio, possono essere utilizzate le curve giornaliere di cortisolo, sierico o salivare, con prelievi prima, 2, 4 e 6 h dopo la dose mattutina (1,2).
I parametri indiretti relativi all’azione degli steroidi (elettroliti, conta leucocitaria, glicemia, colesterolemia, marcatori di citolisi e colestasi epatica) sono utili soprattutto nel prevenire gli effetti collaterali di un sovradosaggio terapeutico, ma non sempre permettono di esprimere un giudizio corretto sulla terapia sostitutiva in corso (1).
A tutt’oggi il mezzo migliore per esprimere un giudizio di adeguatezza della terapia sostitutiva è la valutazione clinica del benessere del paziente e della qualità di vita attraverso specifici questionari (18), associata alla ricerca di sintomi e segni di sotto-dosaggio (astenia, algie addominali o muscolari, calo ponderale, melanodermia, ipotensione arteriosa, edemi declivi) o sovra-dosaggio (obesità, osteoporosi, iperglicemia, dislipidemia, ipertensione arteriosa) (1,2,19).
BIBLIOGRAFIA
- Hahner S, Allolio B. Therapeutic management of adrenal insufficiency. Best Pract Res Clin Endocrinol Metab 2009, 23: 167–79.
- Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014, 275: 104-15.
- Crown A, Lightman S. Why is the management of glucocorticoid deficiency still controversial: a review of the literature. Clin Endocrinol 2005, 63: 483-92.
- Quinkler M, Hahner S. What is the best long-term management strategy for patients with primary adrenal insufficiency? Clin Endocrinol 2012, 76: 21–5.
- Johannsson G, Bergthorsdottir R, Nilsson AG, et al. Improving glucocorticoid replacement therapy using a novel modified-release hydrocortisone tablet: a pharmacokinetic study. Eur J Endocrinol 2009, 161: 119-30.
- Johannsson G, Nilsson AG, Bergthorsdottir R, et al. Improved cortisol exposure-time profile and outcome in patients with adrenal insufficiency: a prospective randomized trial of a novel hydrocortisone dual-release formulation. J Clin Endocrinol Metab 2012, 97: 473-81.
- Debono M, Ghobadi C, Rostami-Hodjegan A, et al. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 2009, 94: 1548-54.
- Debono M, Ross RJ, Newell-Price J. Inadequacies of glucocorticoid replacement and improvements by physiological circadian therapy. Eur J Endocrinol 2009, 160: 719-29.
- Newell-Price J, Whiteman M, Rostami-Hodjegan A, et al. Modified-release hydrocortisone for circadian therapy: a proof-of-principle study in dexamethasone-suppressed normal volunteers. Clin Endocrinol (Oxf) 2008, 68: 130-5.
- Verma S, Vanryzin C, Sinaii N, et al. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2010, 72: 441-7.
- Mallappa A, Sinaii N, Kumar P, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2015, 100: 1137-45.
- Arlt W, Callies F, van Vlijmen JC, et al. Dehydroepiandrosterone replacement in women with adrenal insufficiency. N Engl J Med 1999, 341: 1013-20.
- Gebre-Medhin G, Husebye ES, Mallmin H, et al. Oral dehydroepiandrosterone (DHEA) replacement therapy in women with Addison's disease. Clin Endocrinol 2000, 52: 775-80.
- Tchernof A, Labrie F. Dehydroepiandrosterone, obesity and cardiovascular disease risk: a review of human studies. Eur J Endocrinol 2004, 151: 1–14.
- Gurnell EM, Hunt PJ, Curran SE, et al. Long-term DHEA replacement in primary adrenal insufficiency: a randomized, controlled trial. J Clin Endocrinol Metab 2008, 93: 400-9.
- Rice SP, Agarwal N, Bolusani H, et al. Effects of dehydroepiandrosterone replacement on vascular function in primary and secondary adrenal insufficiency: a randomized crossover trial. J Clin Endocrinol Metab 2009, 94: 1966-72.
- Oelkers W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison’s disease: rapid adrenocorticotropin (ACTH) test and measurement of plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab 1992, 75: 259-64.
- Oksnes M, Bensing S, Hulting AL, et al. Quality of life in European patients with Addison's disease: validity of the disease-specific questionnaire AddiQoL. J Clin Endocrinol Metab 2012, 97: 568-76.
- Leelarathna L, Breen L, Powrie JK, et al. Co-morbidities, management and clinical outcome of auto-immune Addison's disease. Endocrine 2010, 38: 113-7.
Iposurrenalismo in gravidanza
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
Diagnosi
La diagnosi di iposurrenalismo primario è molto difficile in gravidanza.
Molti sintomi gravidici possono simulare quelli dell’insufficienza surrenalica e non sono così specifici (stanchezza, nausea, vomito).
La valutazione ormonale basale con dosaggio al mattino (h 7-9) di cortisolemia può risultare spesso ancora nella norma, dal momento che lo stato gravidico si caratterizza per un fisiologico progressivo incremento dei livelli circolanti di cortisolo a partire dal terzo trimestre di gravidanza, a causa di un incremento della proteina legante (CBG).
Secondo alcuni Autori, il sospetto di iposurrenalismo primario può essere posto in presenza di livelli di cortisolemia al mattino:
- I trimestre: < 11 µg/dL;
- II trimestre: < 16.3 µg/dL;
- III trimestre: < 22 µg/dL.
In tutte le pazienti gravide con sospetto clinico di iposurrenalismo primario, il miglior test diagnostico è rappresentato dal test con ACTH alla dose di 250 µg, con valutazione della risposta di cortisolo secondo cut-off più alti rispetto a quelli impiegati nelle pazienti non gravide e variabili in base al trimestre di gravidanza:
- I trimestre: < 25 µg/dL;
- II trimestre: < 29 µg/dL;
- III trimestre: < 32 µg/dL.
La valutazione di renina o attività reninica ed aldosterone non presenta invece alcuna affidabilità diagnostica, in quanto tali ormoni possono subire modificazioni indotte dallo stato gravidico stesso.
Terapia
In gravidanza è preferibile impiegare l’idrocortisone rispetto al cortisone acetato, prednisolone, o prednisone, in quanto è degradato dll’11ß-HSD tipo 2 placentale e non agisce sul feto, ed è da evitare il desametasone, in quanto invece non è inattivato dalla placenta.
È importante monitorare le pazienti gravide dal punto di vista clinico, ricercando sintomi e segni di sotto- e sovra-dosaggio (es. normale incremento corporeo, “fatigue”, ipotensione posturale o ipertensione, iperglicemia), con almeno una valutazione ogni trimestre.
In corso di gravidanza, nel terzo trimestre, è preferibile aumentare la dose di terapia steroidea che era in corso prima della gravidanza, con aggiustamenti non standardizzati e secondo le recenti LG dell’Endocrine Society variabili da caso a caso sulla base dell’andamento clinico della gravidanza stessa. Altri autori suggeriscono invece aumenti precisi della dose di glucocorticoide, da 2.5 a 10 mg/die di idrocortisone.
Inoltre è preferibile aumentare anche la dose di fluoridrocortisone, per l’effetto anti-mineralcorticoideo del progesterone, in maniera variabile a seconda della valutazione della pressione arteriosa e degli elettroliti.
Durante il travaglio va utilizzato idrocortisone: un bolo di 100 mg ev, ripetibile ogni 6 h o seguito da infusione continua ev di 200 mg/24 h se necessario. Secondo alcuni autori la dose orale va poi raddoppiata per 24-48 h dopo il parto.
L’allattamento non è sconsigliato, ma non esiste alcuna indicazione sui tempi consigliati rispetto a donne non affette da insufficienza surrenalica. E’ importante ricordare che i glucocorticoidi assunti dalla madre passano nel latte materno e raggiungono il neonato.
Bibliografia
- Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016, 101: 364-89.
- Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med 2014, 275: 104-15.
- Yuen KCJ, Chong LE, Koch CA. Adrenal insufficiency in pregnancy: challenging issues in diagnosis and management. Endocrine 2013, 44: 283–92.
- Lutwyche V. Successful lactation in Addison's disease. Br Med J 1953, 1: 313-4.
Insufficienza surrenalica acuta
Giuseppe Reimondo & Isabella Tabaro
Dipartimento di Scienze Cliniche e Biologiche, Università di Torino; Medicina Interna I ad Indirizzo Endocrinologico, AOU San Luigi Gonzaga, Orbassano (TO)
Epidemiologia
La crisi iposurrenalica acuta (AC) è una complicanza grave, determinata dal non adeguato incremento della produzione endogena di cortisolo in risposta a un aumentato fabbisogno. Si tratta di un’emergenza clinica, che contribuisce all’aumento di mortalità nei pazienti con iposurrenalismo cronico (tasso di mortalità per AC è 0.5/100 pazienti/anno), pertanto richiede un trattamento tempestivo (1).
Si è osservata un’incidenza di circa 5-10 crisi iposurrenaliche/100 pazienti/anno con insufficienza surrenalica in terapia sostitutiva (1). In un recente studio prospettico, in cui sono stati arruolati 423 pazienti, di cui 222 con iposurrenalismo primario e 201 con forma secondaria, si sono verificate 64 crisi iposurrenaliche (8.3/100 pazienti/anno), risultate fatali nel 6.3% dei casi (2).
Definizione
È stata recentemente proposta una variazione della definizione diagnostica (1,2) di AC, che associa i criteri A e B:
- criterio A: peggioramento delle condizioni generali in presenza di almeno 2 dei seguenti segni/sintomi:
- ipotensione (pressione sistolica < 100 mmHg);
- nausea o vomito;
- astenia severa;
- febbre;
- sonnolenza;
- iposodiemia o iperkaliemia;
- ipoglicemia;
- criterio B: somministrazione parenterale di glucocorticoidi con successivo miglioramento clinico.
Sono stati definiti 4 livelli di gravità:
- trattamento ambulatoriale
- ricovero ordinario
- ricovero in terapia intensiva
- decesso.
Eziologia
Le cause più frequente di AC sono le infezioni, in particolare del tratto gastroenterico (23-29%) negli adulti (3) e respiratorie nei bambini (4), seguite da interventi chirurgici, esercizio fisico intenso, stress emotivi, incidenti, interruzione o inadeguata assunzione della terapia steroidea.
Bisogna tener presente che molti farmaci possono alterare il metabolismo del cortisolo, come per esempio la levo-tiroxina, gli induttori del CYP3A4 (carbamazepina, barbiturici, mitotane, anti-fungini) e gli induttori del CytP450 (rifampicina, fenitoina) che ne aumentano tutti la clearance.
I pazienti con iposurrenalismo primario sono più a rischio di AC, poiché il deficit di mineralcorticoidi aumenta il rischio di disidratazione e ipovolemia; anche nei pazienti con concomitante diabete insipido il rischio di AC è aumentato. Altri fattori di rischio sono il diabete mellito, l’ipogonadismo, la tireotossicosi, la gravidanza, la terapia anti-coagulante (3). In circa il 10% dei casi l’eziologia rimane sconosciuta (1).
Anche l’utilizzo di desametasone come terapia sostitutiva, se non somministrata insieme al fludrocortisone, può essere un fattore scatenante per AC, poiché non ha un’attività mineralcorticoide (4).
Presentazione clinica
I pazienti si presentano tipicamente ipovolemici e ipotesi, talvolta anche in shock ipovolemico.
I sintomi più comuni sono una severa astenia e sintomi gastrointestinali, come nausea, vomito, anoressia, diarrea e dolore addominale. Spesso è presente febbre. Possono manifestarsi anche sintomi depressivi e pazienti con Addison non diagnosticato possono presentare iperpigmentazione cutanea. Nei casi più gravi può verificarsi un’alterazione del sensorio fino alla perdita di coscienza.
Agli ematochimici si possono osservare iposodiemia, iperpotassiemia (nella forma primaria), ipoglicemia (più comune nei bambini), ipercalcemia.
Terapia
Nei pazienti in cui la diagnosi di iposurrenalismo non è nota, in caso di sospetto clinico la terapia va comunque iniziata prima di ottenere i risultati degli esami ormonali. Una cortisolemia > 20 µg/dL può escludere la diagnosi, mentre una cortisolemia < 5 µg/dL al mattino o in uno stato di stress corrobora il sospetto di insufficienza surrenalica (3).
La terapia consigliata consiste nell’immediata somministrazione parenterale di 100 mg di idrocortisone in bolo (50 mg/m2 nei bambini), seguiti da adeguata idratazione e 200 mg di idrocortisone nelle successive 24 ore (50-100 mg/m2 nei bambini) in infusione continua endovena oppure suddivisa in boli ogni 6 ore (1,4).
Idratazione: si consiglia l’infusione rapida di Soluzione Fisiologica (SF) 0.9% 1000 cc nella prima ora, a seguire infusione continua di SF 0.9% in base alla clinica; nei bambini è suggerito un bolo di 20 mL/kg di SF 0.9%, che può essere ripetuto in caso di shock, fino ad un totale di 60 mL/kg nella prima ora (4).
Una dose di idrocortisone > 50 mg/die ha anche un effetto mineralcorticoide, per cui non è necessario il trattamento con mineralcorticoidi. Se l’idrocortisone non fosse disponibile, è suggerito in alternativa l’utilizzo di prednisolone alla dose equivalente, mentre il desametasone è considerato come ultima scelta e va utilizzato solo nel caso in cui nessun altro glucocorticoide fosse disponibile.
Generalmente il quadro acuto si risolve entro 24 ore dall’inizio della terapia e nei giorni successivi le dosi di steroidi per via parenterale possono essere scalate (il secondo giorno idrocortisone 100 mg/die), fino alla ripresa della terapia orale di mantenimento. In caso di diagnosi di iposurrenalismo di primo riscontro, una volta superata la fase acuta, iniziare anche la terapia orale con fludrocortisone 50-100 µg/die. È ovviamente fondamentale mantenere il monitoraggio emodinamico, elettrolitico e della glicemia. A seconda dell’eziologia, andrà valutata inoltre l’eventuale terapia antibiotica e profilassi eparinica.
Prevenzione
Si raccomanda di aggiustare le dosi della terapia sostitutiva cronica steroidea in base agli eventi intercorrenti:
- in caso di febbre, raddoppiare o triplicare (se T > 39 C°) la dose fino ad avvenuta guarigione e ridurla entro 2 giorni fino al dosaggio abituale;
- in caso di gastroenterite-vomito-diarrea, infezione severa, o impossibilità di assumere la terapia orale è indicata la somministrazione di idrocortisone 100 mg intramuscolo, endovena o sottocute, che può essere ripetuta ogni 6-12 ore fino a guarigione;
- la gravidanza richiede generalmente un aumentato dosaggio di idrocortisone, in particolare durante il terzo trimestre (4);
- alcuni autori consigliano di aumentare il dosaggio dell’idrocortisone di 10-20 mg in caso di stress emotivo e di assumere 10 mg di idrocortisone 30-60 minuti prima di effettuare un esercizio fisico intenso (1);
- la posologia va adeguata anche in base al tipo di intervento chirurgico/procedura invasiva che il paziente deve eseguire e in alcuni casi è necessaria l’infusione pre- e post-procedurale di idrocortisone endovena, come è indicato per esempio durante il travaglio e il parto.
Tutti i pazienti affetti da iposurrenalismo dovrebbero possedere sia una carta di emergenza che li identifichi, sia un kit di emergenza per l’iniezione di idrocortisone.
Bibliografia
- Allolio B. Extensive expertise in endocrinology: adrenal crisis. Eur J Endocrinol 2015, 172: R115-24.
- Haner S, et al. High incidence of adrenal crisis in educated patients with chronic adrenal insufficiency - a prospective study. J Clin Endocrinol Metab 2015, 100: 407-16.
- Puar TH, et al. Adrenal crisis: still a deadly event in the 21st. Am J Med 2016, 129: 339.e1-9.
- Stefan R, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016, 101: 364-89.
Scheda glucocorticoidi
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
Meccanismo d’azione
Interazione con i recettori per i glucocorticoidi.
Preparazioni, via di somministrazione, posologia
Disponibili vari composti (vedi tabella).
- Cortisone acetato: cp 25 mg (Cortone)
- Idrocortisone:
- cp 10 mg (Idrocortisone);
- cp solubili da 10 e 20 mg (Idrocortisone Bruno)
- cp a rilascio modificato da 5 e da 20 mg (Efmody, Plenadren);
- fl 100 mg/2 mL (Flebocortid), fl 500 mg/5 mL (Flebocortid), fl 1 g/10 mL (Flebocortid)
- Metilprednisolone:
- cp 4 mg (Medrol Farma, Medrol Farmaroc, Medrol Farmed, Medrol Gekofar, Medrol GMM, Medrol New Pharmashop, Medrol Pfizer, Medrol Price Tag, Medrol Programmi Sanitari Integrati, Metildrol, metilprednisolone DOC, metilprednisolone EG, Urbason), cp 16 mg (Medrol Pfizer, Metildrol, metilprednisolone DOC, metilprednisolone EG, )
- fl 20 mg/1 mL (Urbason solubile), fl 40 mg/mL (Depo-Medrol, Solu-Medrol, Urbason solubile), fl 80 mg/1.5 ml (Metilbetasone), fl 125 mg/2 mL (Solu-Medrol), fl 250 mg/5 mL (Urbason solubile), fl 500 mg/8 mL (Solu-Medrol), fl 1 g/16 mL (Solu-Medrol), fl 2 g/32 mL (Solu-Medrol).
- polvere per sol. iniettabile 20 mg (Cortrium, Lisamethyle, Urbason), 40 mg (Cortrium, Lisamethyle, Metilprednisolone Hikma, Urbason), 125 mg (Cortrium, Lisamethyle, Metilprednisolone Hikma), 250 mg (Urbason), 500 mg (Cortrium, Lisamethyle, Metilprednisolone Hikma), 1000 mg (Metilprednisolone Hikma).
- Prednisone:
- cp 1 mg rilascio modificato (Lodotra), cp 2 mg rilascio modificato (Lodotra), cp 5 mg rilascio modificato (Lodotra), cp 2.5 mg (Idelt), cp 5 mg (Cison, Cortirex, Deltacortene, Idelt, Prednisone DOC, Prednisone EG, Prednisone Mylan, Prednisone TEVA, Prednisone Zentiva, Soflon), cp 10 mg (Idelt), cp 20 mg (Cison, Cortirex, Idelt), cp 25 mg (Cison, Cortirex, Deltacortene, Prednisone DOC, Prednisone EG, Prednisone Mylan, Prednisone TEVA, Prednisone Zentiva, Soflon).
- Prednisolone:
- cp 2.5 mg (Idelt), 5 mg (Idelt), 10 mg (Idelt), 20 mg (Idelt)
- soluzione orale 1 mg/mL (Sintredius)
- Triamcinolone:
- fl 40 mg/mL (Kenacort, Triacort, Triamvirgi, Triesence), fl 80 mg/2 mL (Triacort, Triamvirgi)
- Betametasone:
- cp efferv 0.5 mg (Bentelan, Betametasone DOC, Betametasone EG Stada, Betametasone Zentiva, Etason), cp efferv 1 mg (Bentelan, Betametasone DOC, Betametasone EG Stada, Betametasone Zentiva, Etason)
- fl 1.5 mg/2 mL (Albaflo, Bentelan, Betametasone DOC, Betametasone EG, Ibet), fl 4 mg/1 mL (Betametasone LFM), fl 4 mg/2 mL (Albaflo, Bentelan, Betametasone DOC, Betametasone EG, Betametasone LFM, Ibet)
- Desametasone:
- cp 0.5 mg (Decadron), cp 0.75 mg (Decadron), cp 40 mg (Neofordex)
- cp effervescenti 2 mg (Varcodes), 4 mg (Varcodes), 8 mg (Varcodes),
- gocce orali 2 mg/mL (Decadron), 0.2% (Soldesam)
- fl 4 mg/mL (Decadron, desametasone Hameln, desametasone Kalceks, desametasone Pfizer, Soldesam), fl 8 mg/2 mL (Decadron, desametasone Pfizer, Soldesam)
| Tabella comparativa glucocorticoidi | |||||
| Steroide | Dose equivalente (mg) | potenza relativa | Emivita (h) | ||
| anti-infiammatoria | mineralcorticoide | plasmatica | biologica | ||
| Cortisone | 25 | 0.8 | 2 | 0.5 | 8-12 |
| Idrocortisone | 20 | 1 | 2 | 1.5-2 | 8-12 |
| Metilprednisolone | 4 | 5 | 0 | 1.5-3 | 18-36 |
| Prednisone | 5 | 4 | 1 | 1 | 18-36 |
| Prednisolone | 5 | 4 | 1 | 2-3.5 | 18-36 |
| Triamcinolone | 4 | 5 | 0 | 3.5-4 | 18-36 |
| Betametasone | 0.6-0.75 | 20-30 | 0 | 5.5 | 36-54 |
| Desametasone | 0.75 | 20-30 | 0 | 2-3.5 | 36-54 |
Indicazioni
Terapia sostitutiva nell'insufficienza surrenalica primitiva e secondaria, azione anti-infiammatoria ed anti-allergica.
Contro-indicazioni
Ulcera peptica, diabete mellito, scompenso cardiaco, ipertensione arteriosa, obesità grave.
Effetti collaterali
Danni alla mucosa gastrica, sovrappeso-obesità, osteoporosi, iperglicemia, diminuzione della resistenza alle infezioni, ritenzione di sodio ed acqua, rischio trombotico, aumento del tempo di cicatrizzazione delle ferite, acne, irsutismo, disturbi mestruali.
Limitazioni prescrittive
Idrocortisone cp non è disponibile in Italia, ma è ottenibile con richiesta diretta alla ditta Sanofi (modulo richiesta, aggiornato 2021).
Scheda mineralcorticoidi
Roberta Giordano
Dipartimento di Scienze Cliniche e Biologiche, Università degli Studi di Torino
Meccanismo d’azione
Interazione con i recettori per i mineralcorticoidi.
Preparazioni, via di somministrazione, posologia
Fludrocortisone acetato (Florinef), compresse da 0.1 mg, confezione da 56 compresse, somministrabile per via orale.
Adulti compresi gli anziani: la dose giornaliera varia da 0.05 a 0.3 mg una volta al giorno al mattino. Pazienti con una terapia a lungo termine possono, in periodi di stress o malattia, richiedere l’aggiunta di altri tipi di farmaci steroidei.
Bambini: la dose dovrà esser aggiustata secondo l’altezza ed il peso, ma dovrà essere sempre tenuta il più bassa possibile.
Indicazioni
Insufficienza surrenalica primitiva.
Contro-indicazioni
Pazienti con precedenti reazioni allergiche a farmaci simili o ad uno qualsiasi dei componenti di Florinef compresse.
Precauzioni d'uso
Da assumere con un bicchiere di acqua.
Effetti collaterali
Aumento di appetito, aumento di peso, indigestione, nausea, debolezza, stanchezza, aumentato rischio di infezioni, osteoporosi, ritenzione idrica, battito cardiaco irregolare, ipertensione arteriosa, rischio trombotico, problemi dermatologici o oculari, irregolarità mestruali, alterazioni del tono dell’umore, insonnia ed emicrania.
Limitazioni prescrittive
Non disponibile in Italia, ma è ottenibile con richiesta diretta alla ditta Mylan (modulo richiesta-2023).
Classificazione delle iperplasie surrenaliche congenite
Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
Queste patologie genetiche vengono classificate sulla base del relativo difetto genetico a cui devono la loro patogenesi e alla diversa penetranza con cui il genotipo si esprime clinicamente. Pertanto la loro classificazione, che segue la frequenza dei difetti riscontrati nella popolazione di origine caucasica, riporta i nomi degli enzimi della steroidogenesi coinvolti:
- Deficit di 21-idrossilasi (gene CYP21A2)
-
forma classica:
- forma grave con perdita salina
- forma meno grave senza perdita salina
- forma non classica (deficit parziale di 21-idrossilasi)
- Deficit di 11-idrossilasi (gene CYP11B1)
- Deficit di 3-ß-idrossi-steroido-deidrogenasi (gene 3-ß-HSD BII)
- Deficit di 17-alfa-idrossilasi/17,20 liasi (gene CYPc17)
- Sindrome adrenogenitale lipoidea congenita (gene StAR)
Overview iperplasie surrenaliche congenite
Pina Lardo & Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 29/3/2020)
Le iperplasie surrenaliche congenite (ISC) sono un gruppo di patologie genetiche, autosomiche recessive, dipendenti dal difetto di una delle tappe biosintetiche della steroidogenesi (tabella).
L’incidenza mondiale della ISC varia da 1:14.000 a 1:18.000 nuovi nati.
Si distinguono forme classiche o severe, che si manifestano in età neonatale o nelle prime fasi dell’infanzia, con virilizzazione e insufficienza surrenalica con o senza perdita salina, e forme non classiche o “mild”, che si manifestano tardivamente, nella fase finale dell’infanzia o nell’età adulta, con un quadro variabile di iperandrogenismo o a decorso asintomatico.
Le manifestazioni cliniche e la severità della patologia correlano con il tipo di mutazione del gene affetto, quindi con l’attività enzimatica residua, assente nelle forme classiche con perdita salina, pari all’1% nelle forme classiche virilizzanti e al 30-50% nelle forme non classiche (1).
| Caratteristiche delle differenti forme di iperplasia surrenalica congenita | |||||
| Deficit 21-idrossilasi | Deficit 11-idrossilasi | Deficit 17α-idrossilasi | Deficit 3-ß-idrossi-steroido-deidrogenasi | Iperplasia lipoidea | |
| Gene mutato | CYP21A2 | CYP11B1 | CYP17 | HSD3B2 | StAR |
| Cromosoma | 6p21.3 | 8q24.3 | 10q24.3 | 1p13.1 | 8p11.2 |
| Ambiguità genitali | + nelle F | + nelle F | + nei M Non pubertà nelle F |
+ nei M Moderata nelle F |
+ nei M Non pubertà nelle F |
| Crisi addisoniane | + | Rare | No | + | ++ |
| Incidenza | 1:10-18.000 | 1:100.000 | Rara | Rara | Rara |
| Glucocorticoidi | ↓ | ↓ | Normale corticosterone | ↓ | ↓ |
| Mineralcorticoidi | ↓ | ↑ | ↑ | ↓ | ↓ |
| Androgeni | ↑ | ↑ | ↓ | ↓ nei M ↑ nelle F |
↓ |
| Pressione arteriosa | ↓ | ↑ | ↑ | ↓ | ↓ |
| Na | ↓ | ↑ | ↑ | ↓ | ↓ |
| K | ↑ | ↓ | ↓ | ↑ | ↑ |
| Metaboliti elevati | 17OHP | DOC, 11-deossi-cortisolo | DOC, corticosterone | DHEA, 17∆5Preg | Nessuno |
| Legenda: 17OHP = 17-idrossiprogesterone; DOC = desossicorticosterone; DHEA = deidroepiandrosterone; 17∆5Preg = 17∆5-idrossipregnenolone. | |||||
Caratteristiche comuni alle diverse forme sono:
- ipocortisolismo relativo con aumento di CRH e ACTH (per la mancanza del feed-back negativo) con gradi diversi di iperplasia surrenalica; biochimicamente si realizza un’esaltata sintesi dei precursori a monte della tappa biosintetica bloccata, i quali vengono deviati verso la sintesi degli ormoni non bloccati (soprattutto testosterone e diidrotestosterone, e in misura minore estrogeni, estrone ed estradiolo);
- iperandrogenismo, responsabile nelle forme classiche di:
- ambiguità dei genitali femminili, mentre quelli maschili possono essere interessati in vario modo, con sola accentuazione dei caratteri o ambiguità dei genitali, a seconda del deficit enzimatico;
- virilizzazione in età infantile;
- livelli bassi di aldosterone, mentre quelli di renina sono differenti a seconda delle forme.
Bibliografia
- Speiser PW, Arlt W, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018, 103: 4043-88.
Deficit di 21-idrossilasi
Pina Lardo & Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 29/3/2020)
FORMA CLASSICA
Epidemiologia e patogenesi
Questa è la forma più frequente tra le ICS (95% dei casi), con incidenza altamente variabile in rapporto all'etnia e all'area geografica (1:7.000 in Italia, 1:10-15.000 in Europa e negli Stati Uniti, 1:20.000 in Giappone). Si associa ad un particolare aplotipo del sistema HLA (A3, Bw47, DR7 e Bw60).
Il difetto enzimatico responsabile di questa sindrome deriva da delezioni o mutazioni in omozigosi o in eterozigosi composta (mutazioni uguali o diverse in loci differenti) del gene CYP21A2 (1).
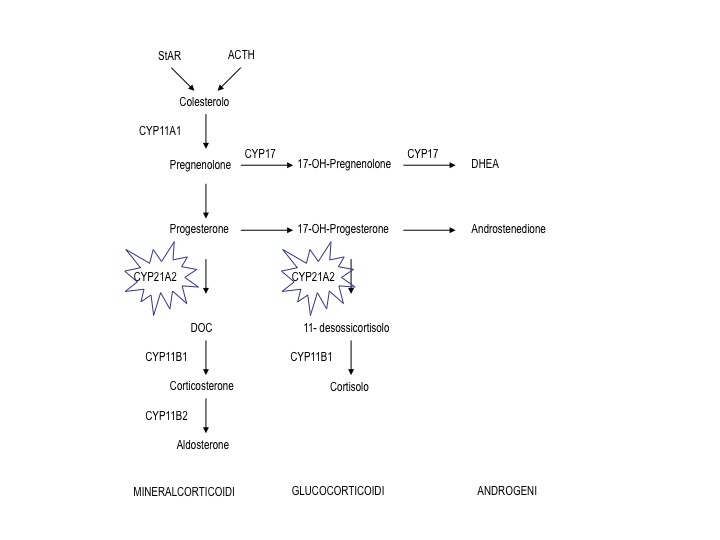
Figura 1. Localizzazione del blocco enzimatico nella cascata steroidogenetica
| Tabella 1 Caratteristiche delle diverse forme del deficit di 21-idrossilasi |
||||||
| Classica | Non classica | |||||
| Fenotipo | con perdita salina | virilizzante semplice | ||||
| M | F | M | F | M | F | |
| Genitali | Normali | Ambigui | Normali | Ambigui | Normali | +/- ipertrofia clitoridea |
| Aldosterone | ↓ | N | N | |||
| Renina | ↑ | talvolta ↑ | N | |||
| Cortisolo | ↓ | ↓ | N | |||
| 17-OH-Progesterone | > 20.000 ng/dL | 10.000-20.000 ng/dL | 1500-10.000 ng/dL (ACTH-stimolato) | |||
| Testosterone | ↑ solo pre- pubere | ↑ | ↑ solo pre-pubere | ↑ | ↑ variabile solo pre-pubere | ↑ variabile |
| Terapia | GC + MC (+ Na) | GC + MC | GC, se sintomi | |||
| Crescita staturale | - 2-3 DS | - 1-2 DS | Variabile -1 DS | |||
| Incidenza | 1/20.000 | 1/60.000 | 1/1000 | |||
| Mutazione | Delezione conversione nt 656g G110∆8nt I236N/V237E/M239K Q318X, R356W |
I172N nt656g |
V281L P30L |
|||
| Attività enzimatica residua | 0 | 1% | 20-50% | |||
| Legenda. GC = glucocorticoidi; MC = mineralcorticoidi | ||||||
Clinica
Si identificano gradi diversi della malattia, in rapporto alla severità del difetto genetico e sono circa 300 le mutazioni del CYP21A2 conosciute, associate a un ampio spettro di fenotipi (tabella 1).
La forma più grave, definita con perdita salina, si contraddistingue per un fenotipo caratterizzato in entrambi i sessi da disidratazione iponatremica e iperandrogenismo.
Disidratazione iponatremica con ipotensione e possibile shock ipovolemico da deficit di ormoni mineraloattivi: la carenza di cortisolo peggiora tale quadro, con riduzione della contrattilità cardiaca, della sensibilità cardiaca e vascolare alle catecolamine. Inoltre, l’accumulo dei precursori steroidei può direttamente antagonizzare l’effetto mineraloattivo, contribuendo al peggioramento del quadro clinico. Le manifestazioni cliniche si hanno verso la fine della 1° o dalla 2-4° settimana, oppure durante i primi anni di vita in coincidenza di condizioni stressanti. Sintomi e segni sono caratterizzati da scarso appetito, vomito, letargia, perdita di peso, iponatremia, iperkaliemia, iper-reninemia fino allo shock ipovolemico, soprattutto nei nati maschi, nei quali la diagnosi può essere più tardiva a causa della mancanza di ambiguità dei genitali.
L’iperandrogenismo è responsabile di:
- ambiguità dei genitali con macrogenitosomia:
- nel sesso femminile chiusura delle grandi labbra (aspetto scrotale), ipertrofia clitoridea, seno urogenitale comune, fino ad un quadro più grave di pseudoermafroditismo femminile, con aspetto di maschio criptorchide con ipospadia. Tali manifestazioni consentono generalmente una diagnosi precoce alla nascita e quindi un trattamento immediato. L’entità della virilizzazione è valutata secondo la scala di Prader (figura 2) e la variabilità delle manifestazioni cliniche è correlata non solo al livello di androgeni, ma anche alla conversione dei precursori in androgeni più potenti, all’espressione e all’attività trascrizionale dei recettori androgenici;
- nel sesso maschile l’eccesso di precursori androgenici non influisce significativamente sulla differenziazione dei genitali, e si manifesta con aumento di dimensioni del pene, iperpigmentazione dello scroto, per cui, la diagnosi spesso è misconosciuta alla nascita;
- virilizzazione in età infantile: il primo segno è la rapida crescita staturale con aumento dell’età ossea e precoce chiusura delle cartilagini epifisarie e bassa statura finale;
- nel maschio si osserva: pubarca prematuro, aumento delle dimensioni del pene in assenza di aumento del volume testicolare, modificazioni del tono di voce;
- nella femmina si manifesta con: progressiva ipertrofia clitoridea, orientamento del comportamento sessuale di tipo maschile (1-3) e durante l’adolescenza predisposizione alla sindrome dell’ovaio policistico (PCOS) con irsutismo, acne, disturbi del ciclo mestruale, infertilità, spesso ridotta sensibilità insulinica (4) e possibile ipotrofia mammaria da eccesso di androgeni e deficit di cortisolo.
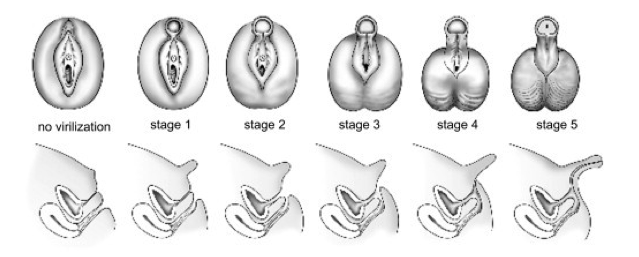
Figura 2. Scala di Prader per la valutazione della virilizzazione
In entrambi i sessi, la pubertà compare in forte anticipo, a causa di una precoce sensibilità ipotalamica indotta dagli aumentati livelli circolanti di androgeni (1-3). È importante nel sesso maschile la distinzione tra pseudo-pubertà precoce e pubertà precoce vera. Infatti, nelle ICS si verifica pubarca prematuro con sviluppo del pene, in contrasto con il volume testicolare che rimane di tipo pre-puberale (pseudo-pubertà), seppur una lunga esposizione a elevati livelli di androgeni può determinare l’attivazione dell’asse ipotalamo-ipofisi-gonade, con induzione della pubertà precoce vera (5).
La fertilità femminile è compromessa in questa forma a causa degli elevati livelli di androgeni e 17OHP, responsabili di cicli anovulatori e degli elevati livelli di progesterone che hanno effetto contraccettivo. Un’appropriata terapia con glucocorticoidi, migliorando il quadro ormonale, rende possibile il concepimento.
Nel sesso maschile, la fertilità è severamente compromessa, principalmente per la presenza di ipogonadismo iper - o ipogonadotropo.
La forma meno grave, definita senza perdita salina, è caratterizzata da un fenotipo con semplice virilizzazione, spesso non precocemente individuabile per l'esordio più tardivo.
È importante considerare la possibile associazione con alcuni tipi di tumori. ll 60% dei pazienti affetti da incidentaloma surrenalico presenta un’aumentata risposta del 17OHP allo stimolo con ACTH. La frequenza di mutazioni germinali del CYP21 è bassa, ma l’incidenza di masse surrenaliche sembra essere maggiore nei pazienti affetti da ISC ed in eterozigosi rispetto alla popolazione generale. La prevalenza aumenta con l’età, e si possono riscontrare adenomi, mielolipomi, emangiomi, i quali, non necessariamente richiedono l’asportazione chirurgica, in quanto possono regredire con la terapia glucocorticoide.
Diagnosi
Nella storia anamnestica possono risultare familiari affetti da irsutismo, disturbi del ciclo mestruale, pubertà anticipata e bassa statura finale.
Nella forma con perdita salina, un importante criterio diagnostico è la comparsa di sintomi secondari a deficit di aldosterone nel periodo neonatale (verso la fine della 1° settimana di vita o nella 2-3° settimana) e durante i primi anni di vita in coincidenza di situazioni stressanti.
La forma con virilizzazione semplice, senza perdita salina, è generalmente sospettata più tardivamente, con caratteristiche cliniche differenziali diverse tra femmina e maschio (5).
La diagnosi differenziale di queste patologie va posta con le altre forme di ICS e con il pubarca prematuro idiopatico (6). Elemento differenziante è caratterizzato dall'incremento annuale dell'età ossea (DBA), che nelle iperplasie da deficit di 21-idrossilasi è maggiore rispetto all'età cronologica (DCA): DBA/DCA = 3.2 ± 0.6; al contrario, nel pubarca prematuro si aggira intorno all'unità o poco superiore (DBA/DCA = 1.03 ± 1.1).
La diagnostica ormonale basale, nella forma virilizzante semplice, da eseguire nelle donne in età fertile in fase follicolare precoce mostra:
- aumento degli androgeni (testosterone, DHEA-S, androstenedione), aumento di 17OHP, aumento dei livelli di renina e potassiemia;
- riduzione della sodiemia soltanto nella forma con perdita salina.
Il test con ACTH non è necessario se i valori di 17OHP basali sono già molto elevati (circa 1000 ng/dL). Viceversa, ha indicazione per valori di 17OHP solo relativamente elevati (≤ 1000 ng/dL) (7) (fig. 3).
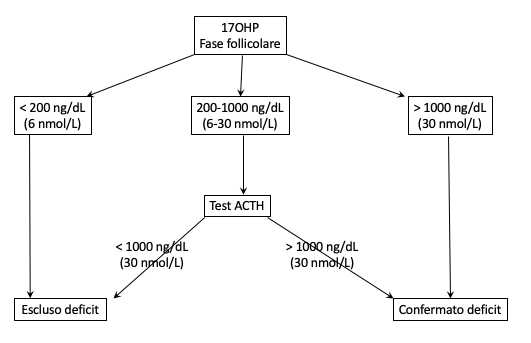
Figura 3. Diagnosi deficit 21-idrossilasi
Tuttavia, per questa forma, può essere eseguita l'analisi molecolare del gene CYP21A2, che dimostra delezioni o mutazioni puntiformi in omozigosi o in eterozigosi composta. L’esame genetico è indicato solo quando il risultato del test di stimolo con ACTH non è dirimente.
Allo stesso modo, in caso di gravidanza a rischio è possibile effettuare la diagnosi pre-natale sul feto mediante analisi del DNA ottenuto con la villocentesi (8).
Fisiopatologia
Inizialmente le gonadi maschili e femminili sono indifferenziate e bipotenti.
Durante la 7° settimana di gestazione le gonadi maschili iniziano a differenziarsi. ll gene SRY (sex-determining region), localizzato sul cromosoma Y, determina il fenotipo maschile con la formazione dei testicoli. Lo sviluppo dei genitali interni maschili, derivanti dai dotti di Wolff (epididimo, dotti deferenti, dotti eiaculatori e tubuli seminiferi), richiede alte concentrazioni di testosterone, secreto dalle cellule di Leydig a partire dalla 7° settimana. Lo sviluppo dei genitali esterni maschili dipende dal testosterone e dal suo metabolita, diidrostestosterone (DHT).
Le ovaie sono riconoscibili verso la 10° settimana. In assenza della secrezione dell’ormone anti-Mulleriano (AMH), procede lo sviluppo delle strutture Mulleriane: tube di Falloppio, utero, cervice e III superiore della vagina.
Nelle ISC, l’eccesso dei precursori androgenici non influenza significativamente la differenziazione sessuale maschile, mentre, nelle femmine affette dalle forme classiche del deficit di 21-idrossilasi e 11-idrossilasi, i livelli di androgeni circolanti sono in grado di determinare virilizzazione, impedendo la formazione di un canale uretrale e vaginale separato, inducendo clitoridomegalia e fusione labiale. Diversamente dalla virilizzazione dei genitali esterni, i genitali interni (utero, tube di Falloppio e ovaio) si sviluppano normalmente, essendo di derivazione Mulleriana e non responsivi agli androgeni. Tuttavia, alcune femmine affette in modo severo, possono sviluppare strutture genitali interne tipicamente maschili, e presentarsi quindi con genitali esterni apparentemente maschili, ipospadia, testicoli ritenuti (7).
DEFICIT PARZIALE DI 21-IDROSSILASI (FORMA NON CLASSICA)
Epidemiologia e patogenesi
La prevalenza di questa variante del deficit di 21-idrossilasi varia a seconda del gruppo etnico: 0.3% nei bianchi americani, 1.6% negli slavi, 1.9% negli spagnoli, 3.7% negli ebrei dell'Est europeo. Si associa a un particolare aplotipo del sistema HLA (B14 e DR1).
Il difetto enzimatico deriva da mutazioni puntiformi in eterozigosi del gene CYP21A2 (1).
Clinica
I segni clinici che caratterizzano questa variante sono molto sfumati rispetto alla forma classica, variano con il sesso e l’età e sono generalmente sostenuti nella femmina dagli effetti dell'iperandrogenismo.
Nei bambini prima dei 10 anni è comune l’adrenarca prematuro (87%), dovuto agli elevati livelli di DHEA-S, tipica manifestazione delle ISC non classiche non trattate, mentre non si osserva nelle forme classiche per l’effetto soppressivo del trattamento glucocorticoideo.
In entrambi i sessi l'età del pubarca è prematura, con incremento annuale dell'età ossea maggiore rispetto all'età cronologica, per la conversione periferica degli androgeni in estrogeni (1-3,7), con possibile compromissione dell’altezza definitiva. È sempre assente la perdita salina.
In età adolescenziale-adulta le manifestazioni possono essere:
- nella donna acne pre- e post-puberale, irsutismo, alopecia androgenetica, disturbi del ciclo mestruale con oligomenorrea, amenorrea primitiva o secondaria e modesta ipertrofia clitoridea senza ambiguità dei genitali;
- nel maschio, invece, sono generalmente assenti e la diagnosi è posta dopo la pubertà per la presenza di acne, talvolta ginecomastia, infertilità. È possibile lo sviluppo di isole di tessuto surrenalico nel testicolo (adrenal rest), potenzialmente responsabili di oligospermia, azoospermia e infertilità.
I soggetti portatori della mutazione in eterozigosi sono clinicamente asintomatici. Rispetto ai soggetti non affetti, si caratterizzano solo per valori di 17OHP lievemente aumentati dopo test con ACTH (200-1000 ng/dL a 60’) e in genere l’esame genetico è dirimente per la diagnosi.
Diagnosi
Anche la storia anamnestica di questa forma può risultare positiva per irsutismo, disturbi mestruali, pubertà anticipata e bassa statura finale. L'iperandrogenismo di origine surrenalica va monitorato e trattato, poichè molte pazienti sviluppano secondariamente un iperandrogenismo di origine ovarica, fino al quadro clinico completo di PCOS (1-3).
La diagnosi differenziale di questa forma comporta difficoltà maggiori rispetto alla variante classica, poichè vanno prese in considerazione:
- nella femmina tutte le forme di irsutismo/virilismo;
- nel maschio tutte le forme di pseudo-pubertà precoce di origine surrenalica.
La diagnostica ormonale mostra nella maggior parte dei casi:
- livelli di ACTH normali;
- livelli basali di cortisolo normali e dopo stimolo normali o lievemente alterati;
- livelli basali aumentati di androgeni (testosterone, DHEA-S, androstenedione) e dei loro precursori della via ∆4 e ∆5 (valori di 17OHP < 200 ng/dL escludono la diagnosi);
- i valori basali di 17OHP possono essere usati per lo screening, ma la misurazione a 60’ dopo stimolo con ACTH (da eseguire per valori di17OHP compresi tra 200·e 1000 ng/dL) rappresenta il gold standard (fig. 3).
L'analisi molecolare del gene CYP21A2 è raccomandata per la definizione diagnostica e per guidare il trattamento e il follow-up.
Bibliografia
- Miller WI. Genetic, diagnosis and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab 1994, 78: 241-6.
- Merke DP, Bornstein SR. Congenital Adrenal Hyperplasia. Lancet 2005, 365: 2125-36.
- Nimkarn S, Lin-Su K, New MI. Steroid 21 hydroxylase deficiency congenital adrenal hyperplasia. Pediatr Clin North Am 2011, 58: 1281-300.
- Premawardhana LDKE, Hunhest IA, Read GF, Scanlon F. Longer term outcome in females with congenital adrenal hyperplasia (CAH): the Cardiff experience. Clin Endocrinol 1997, 46: 327-32.
- Speiser PW, Arlt W, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018, 103: 4043-88.
- Dacau-Voutetakis C, Dracopoulou M. High incidence of molecolar defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab 1999, 84: 1570-5.
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000, 21: 245-91.
- Olney RC, Mougey EB, Wang J, et al. Using real-time, quantitative PCR for rapid genotyping of the steroid 21-hydroxylase gene in a north Florida population. J Clin Endocrinol Metab 2002, 87: 735-41.
Terapia deficit 21-idrossilasi (e iperplasie surrenaliche congenite)
Pina Lardo & Antonio Stigliano
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 14 aprile 2020)
DEFICIT 21-IDROSSILASI FORMA CLASSICA
Tutti i pazienti affetti dalla forma classica del deficit di 21-idrossilasi devono essere trattati con glucocorticoidi (GC).
La terapia con mineralcorticoidi (MC) è indicata nella forma classica con perdita salina: anche se il dosaggio ottimale è ancora oggetto di studio, è generalmente utilizzato il fludrocortisone al dosaggio di 0.1-0.2 mg/die. Con l’età la necessità di MC si riduce, in quanto alla nascita i recettori per MC a livello renale sono scarsamente espressi e il contenuto di sodio nella dieta è ridotto; la necessità di terapia sostitutiva con MC deve essere quindi rivalutata nella transizione dall’età pediatrica a quella adulta.
Nei neonati affetti, appena diagnosticato il deficit, è necessaria la somministrazione di GC, in genere a un dosaggio maggiore rispetto a quello fisiologicamente prodotto, allo scopo di:
- evitare la comparsa di crisi addisoniane;
- mantenere un normale livello di ACTH, evitando lo sviluppo di tumori surrenalici;
- evitare la crescita di adrenal rest presenti nel testicolo (1);
- sopprimere la produzione di androgeni surrenalici, riducendo l'iperandrogenismo nel sesso femminile ed evitando che, in età fertile, questo comprometta l'attività ovulatoria e sia la causa di irsutismo e virilizzazione.
La terapia steroidea da preferire in età pediatrica è l'idrocortisone (IC), alla posologia di 15-20 mg/m2 die (solitamente non < 10 mg/m2), suddiviso in 2 o 3 somministrazioni. La breve emivita dell’IC minimizza l’impatto sulla crescita e gli altri possibili effetti avversi. Il Cortone acetato invece, non è il farmaco di scelta per le ISC, avendo solo l’80% di biodisponibilità rispetto all’IC e circa 2/3 della sua potenza. Il dosaggio deve essere personalizzato, considerando appropriato il più basso compatibile con una velocità di crescita normale con il raggiungimento del target genetico, l’avanzamento dell'età ossea parallelo all'età anagrafica e un’androgenizzazione fisiologica. Dunque, l’obiettivo è il raggiungimento di un equilibrio tra iperandrogenismo e ipercortisolismo.
L’aggiunta, in età pediatrica, del GH, da solo o in combinazione con agonisti del GnRH, allo scopo di raggiungere la massima altezza finale, sembra essere promettente, ma non può ancora essere considerato uno schema terapeutico standard (7).
Nei casi di pubertà precoce vera, la cui diagnosi viene effettuata mediante l’esecuzione del test di stimolo con GnRH, è necessario anche il trattamento con GnRH agonisti, volto a sopprimere la produzione di gonadotropine ipofisarie e quindi la produzione di steroidi gonadici sessuali, prevenendo una precoce saldatura delle cartilagini epifisarie.
Il trattamento dei sintomi dell’iperandrogenismo nelle giovani donne può spesso richiedere l’associazione di farmaci con attività anti-androgena. Sono efficaci contraccettivi orali contenenti drospirenone, mentre lo spironolattone è relativamente controindicato in caso di perdita salina, essendo anche un antagonista dei recettori dei MC.
Riguardo il trattamento chirurgico delle bambine affette da ambiguità genitale, è utile la correzione precoce, mentre nelle forme con lieve virilizzazione il trattamento può essere effettuato in età pediatrica o adulta.
Il monitoraggio ormonale della terapia in età pediatrica prevede la normalizzazione dell'androstenedione per età e sesso, della renina plasmatica e la riduzione del livello del 17OHP, il cui ritorno ai parametri di normalità è invece espressione di un trattamento eccessivo, mentre il dosaggio di ACTH non è considerato un parametro utile (2).
L’attento monitoraggio della terapia sostitutiva è importante per evitare gli effetti legati ad un possibile sovra-dosaggio dei GC e dei MC, mediante il controllo di velocità di crescita, body mass index (BMI), pressione arteriosa, esame obiettivo completo, unitamente al dosaggio di sodio, potassio e renina. In particolare:
- in età pediatrica è consigliata una valutazione annuale dell’età ossea fino al raggiungimento della statura finale;
- in età adulta un controllo biochimico annuale volto a evitare la soppressione degli steroidi surrenalici. Viene suggerita inoltre la valutazione della densitometria ossea nei soggetti sottoposti a trattamento con alte dosi di steroidi e nei casi di fratture non traumatiche.
L’imaging surrenalico non è raccomandato di routine, ma va riservato ai casi che sviluppano masse surrenaliche. Nei maschi affetti dalla forma classica è raccomandata l’esecuzione periodica dell’ecografia testicolare per monitorare gli adrenal rest, a partire dall’adolescenza con cadenza ogni 1-2 anni nei pazienti asintomatici o più spesso in presenza di sintomi (8).
Terapia pre-natale
Nelle gravidanze a rischio deve essere valutato il trattamento pre-natale con desametasone. In particolare, se in una coppia, un partner presenta una mutazione severa del gene CYP21 (forma classica, forma non classica o eterozigosi), è necessario eseguire lo studio genetico del partner, in quanto se entrambi sono affetti da mutazione severa, vi è rischio fetale e necessità di terapia pre-natale.
L’obiettivo è la soppressione dell’iperproduzione di androgeni nel feto, la prevenzione della virilizzazione dei genitali esterni nelle femmine e delle ripercussioni psicologiche che in età adulta questa condizione può comportare (9).
La terapia con desametasone, non inattivato dall’enzima 11β-idrossisteroido-deidrogenasi placentare di tipo 2, deve essere iniziata tra la 6° e la 7° settimana di gestazione, perchè l’attività surrenalica secretiva fetale inizia dalla 7° settimana. Pertanto, una femmina affetta dalla forma classica (deficit 21-idrossilasi o 11-idrossilasi), è esposta agli androgeni nella fase critica della differenziazione sessuale (tra l’8° e la 12° settimana).
Il trattamento, che prevede un dosaggio di desametasone pari a 20 µg/kg peso materno pre-gravidico, fino ad un massimo di 1.5 mg/d, va poi proseguito fino alla 10-12° settimana, quando si rende possibile la diagnosi pre-natale: se il feto è maschio o non affetto la terapia deve essere interrotta.
Bibliografia
- Cakir ED, Mutlu FS, Eren E, et al. Testicular adrenal rest tumors in patients with congenital adrenal hyperplasia. J Clin Res Pediatr Endocrinol 2012, 4: 94-100.
- Falhammar H, Thoren M. Clinical outcome in the management of congenital adrenal hyperplasia. Endocrine 2012, 4: 355-73.
- Witchel SF, Miller WL. Prenatal treatment of congenital adrenal hyperplasia: not standard of care. J Genet Couns 2012, 21: 615-24.
- Bonfig W, Schmidt H, Schwarz HP. Growth patterns in the first three years of life in children with classical congenital adrenal hyperplasia diagnosed by newborn screening and treated with low doses of hydrocortisone. Horm Res Paediatr 2011, 75: 32-7.
- Bonfig W, Schwarz HP. Growth pattern of untreated boys with simple virilizing congenital adrenal hyperplasia indicates relative androgen insensitivity during the first six months of life. Horm Res Paediatr 2011, 75: 264-8.
- Claahsen-van der Grinten HL, Noordam K, Born GF, Otten BJ. Absence of increased height velocity in the first year of life in untreated children with simple virilizing congenital adrenal hyperplasia. J Clin Endocrinol Metab 2006, 91: 1205-9.
- Lin-Su K, Harbison MD, Lekarev O, et al. Final adult height in children with congenital adrenal hyperplasia treated with growth hormone. J Clin Endocrinol Metab 2011, 96: 1710-7.
- Speiser PW, Arlt W, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018, 103: 4043-88.
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000, 21: 245-91.
DEFICIT 21-IDROSSILASI FORMA NON CLASSICA
A chi riservare il trattamento
Il trattamento è rivolto alla risoluzione dei segni clinici che il paziente presenta nel corso della vita. Poiché il paziente affetto da questa forma presenta un'adeguata risposta allo stress, la terapia con GC non ha indicazione solo per la correzione del difetto biochimico e nei bambini in assenza di segni clinici (1). Al contrario, deve essere presa in considerazione nel caso di bambini o adolescenti che presentino pubarca precoce e un significativo aumento della maturazione scheletrica, infertilità e storia di pregressa abortività (2). Nei maschi affetti da questa forma, raramente si riscontra la presenza di adrenal rest (lesioni bilaterali benigne che possono causare azoospermia ostruttiva ed infertilità). Sono spesso osservati nei pazienti non trattati adeguatamente, ma possono anche persistere nonostante il trattamento, particolarmente nei maschi affetti dalla forma classica con perdita salina. Queste forme possono beneficiare del trattamento soppressivo con glucocorticoidi, essendo generalmente ACTH-responsive (2).
Indicazioni comuni ad un trattamento sintomatico sono oligomenorrea e acne nelle giovani donne. Infatti, l’irsutismo, risponde difficilmente al trattamento con i soli GC e, come per altre forme di iperandrogenismo, il miglior approccio terapeutico è l’utilizzo di contraccettivi orali con o senza anti-androgeno. Tale trattamento è in grado di ridurre i livelli di testosterone mediante l’effetto inibitorio sulla produzione androgenica ovarica e l’aumentata sintesi di SHBG a livello epatico. In tutti questi casi è suggerita la rivalutazione della terapia con GC, appena risolti i segni clinici.
Obiettivi del trattamento con GC
Il successo della terapia prevede la normalizzazione della velocità di crescita lineare, l’appropriata maturazione scheletrica, il regolare sviluppo puberale e il raggiungimento del normale peso corporeo. Nel sesso femminile, in età fertile, la terapia ha l’obiettivo di regolarizzare la ciclicità mestruale e di promuovere la fertilità.
Scopo, non secondario, è la regressione dei segni clinici riconducibili all’eccesso androgenico, quali l’irsutismo e l’acne (3).
Quali presidi terapeutici impiegare
- Donne con cicli anovulatori che desiderino una gravidanza o maschi con adrenal rest: glucocorticoidi (4).
- Forme importanti di iperandrogenismo (irsutismo grave e alopecia androgenica): anti-androgeni quali spironolattone, flutamide, ciproterone acetato, finasteride, ecc.
- Condizioni di lieve iperandrogenismo e donne adulte che non desiderino una gravidanza: soli contraccettivi orali (5).
- Limitazione dell’irsutismo: rimedi estetici, quali laser, creme depilatorie o elettrolisi.
Come trattare
Il GC di scelta è l’IC, alla posologia di 6-15 mg/m2, suddiviso in tre dosi giornaliere (6).
Durante l’infanzia è da preferire l’IC, in quanto ha minore effetto sulla crescita rispetto alle preparazioni long-acting.
Negli adulti possono essere invece impiegati anche prednisone, prednisolone e desametasone (7).
Nelle donne in età fertile, allo scopo di evitare gli effetti indesiderati degli steroidi sul feto, è consigliabile l’impiego di una formulazione di GC inattivata dalla 11ß-idrossisteroido-deidrogenasi di tipo 2 placentare, quale IC, prednisone e prednisolone (2,6,8).
La consulenza genetica è ovviamente indicata per tutte le pazienti prima del concepimento. L’impiego di GC durante il concepimento sembra ridurre il rischio di aborti spontanei (9).
In età matura il dosaggio della terapia steroidea può essere ridotto per la ridotta secrezione androgenica surrenalica che si verifica in rapporto all’età (10).
Gestione terapeutica in caso di stress
In tutti i pazienti affetti da ISC che richiedono trattamento con GC, in caso di situazioni stressanti (febbre > 38°C, gastroenterite con disidratazione, interventi chirurgici maggiori e traumi maggiori) è indicato l’incremento del dosaggio. Fondamentale è l’educazione del paziente nella gestione di eventuali crisi surrenaliche e la distribuzione di card identificative relativamente alla diagnosi e alla terapia da somministrare in caso di emergenza.
Nei soggetti affetti dalla forma non classica è suggerito l’incremento del dosaggio solo in caso di risposta sub-ottimale del cortisolo al test di stimolo con ACTH (< 14 o < 18 µg/dL) (2).
Bibliografia
- Trapp CM, Oberfield SE. Recommendations for treatment of nonclassic congenital adrenal hyperplasia (NCCAH): an update. Steroids 2012, 77: 342-6.
- Speiser PW, Arlt W, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018, 103: 4043-88.
- Auchus RJ. Congenital adrenal hyperplasia in adults. Curr Opin Endocrinol Diabetes Obes 2010, 17: 210-6.
- White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000, 21: 245-91.
- Wiebe RH, Morris CV. Effect of an oral contraceptive on adrenal and ovarian androgenic steroids. Obstet Gynecol 1984, 63: 12-4.
- Merke DP. Approach to the adult with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2008, 93: 653-60.
- Claahsen-van der Grinten HL, Stikkelbroeck NM, Otten BJ, Hermus AR. Congenital adrenal hyperplasia: pharmacologic interventions from the prenatal phase to adulthood. Pharmacol Ther 2011, 132: 1-14.
- Clayton PE, Miller WL, Oberfield SE. ESPE/LWPES CAH Working Group. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrine Society. Horm Res 2002, 58: 188-95.
- Bidet M, Bellannè-Chantelot C, Galand-Portier M-B, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2010, 95: 1182-90.
- Azziz R, Koulianos G. Adrenal androgens and reproductive aging in females. Semin Reprod Endocrinol 1991, 9: 249-60.
NUOVE STRATEGIE TERAPEUTICHE
GC a rilascio modificato
Recentemente, è stata impiegata una formulazione di idrocortisone orale multi-particolato a rilascio modificato (Chronocort, Diurnal, UK), da somministrare la sera prima di coricarsi per ridurre l’aumento dell’ACTH notturno e con esso l’aumento degli androgeni (1). Tuttavia, questo schema si è dimostrato insufficiente nella soppressione di 17OHP e androstenedione (2).
Una migliore efficacia nella riduzione del livello androgenico è stata osservata in un successivo studio di fase 2, in cui la stessa formulazione di idrocortisone a rilascio modificato è stata somministrata in due periodi diversi della giornata con diversa posologia (10 mg alle ore 7:00 e 20 mg alle ore 23:00) (3).
Terapia GC infusionale
È ancora controversa la somministrazione di idrocortisone in modalità iniettiva attraverso sistemi di pompa infusionale. La maggior parte degli studi è stata condotta con riguardo al ripristino della ritmicità circadiana del cortisolo nei pazienti con insufficienza surrenalica. Quindi attualmente sono pochi i dati per poter proporre queste metodiche per la riduzione del livello androgenico in pazienti affetti da ISC (4).
Inibitori della sintesi androgenica
La strategia di impiego di questi farmaci è l'inibizione degli enzimi chiave della biosintesi degli androgeni, come la 17a-idrossilasi/17,20-lasi (CYP17A1, P450c17).
L’abiraterone acetato è un potente inibitore del CYP17A1 e quindi della sintesi di testosterone, impiegato nella terapia del carcinoma della prostata resistente alla castrazione (5). L’esperienza con questo farmaco in donne adulte si è rivelata positiva nella riduzione delle concentrazioni sieriche di androstenedione e testosterone (6). Tuttavia, l’impiego di questo inibitore ha dei limiti rappresentati dall’età delle pazienti: l’inibizione del CYP17A1 inibisce la produzione di steroidi gonadici, rendendone sconsigliato l’uso in età pre-puberale. La terapia trova un maggior razionale nella forma non classica di deficit di 2i-idrossilasi, ma in questo caso l’inibizione della sintesi del cortisolo complica la terapia.
Antagonisti recettoriali del CRH
Questo approccio è stato recentemente indagato mediante l’impiego di un antagonista selettivo del recettore CRH di tipo 1, NBI-77860. Un piccolo studio in pazienti di sesso femminile, in singolo cieco, controllato con placebo, ha mostrato promettenti risultati sulla capacità dose-dipendente del farmaco di ridurre le concentrazioni di ACTH e 17OHP, con riduzioni variabili nella concentrazione di androstenedione e testosterone (7). Attualmente sono in corso studi clinici di maggiori dimensioni, per studiare sicurezza ed efficacia di questo tipo di inibitore.
Bibliografia
- Debono M, Ghobadi C, Rostami-Hodjegan A, et al. Modified-release hydro-cortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 2009, 94: 1548–54.
- Verma S, Vanryzin C, Sinaii N, et al. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2010, 72: 441–7.
- Mallappa A, Sinaii N, Kumar P, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2015, 100: 1137–45.
- Merza Z, Rostami-Hodjegan A, Memmott A, et al. Circadian hydrocortisone infusions in patients with adrenal insufficiency and congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2006, 65: 45–50.
- Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017a (17a-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem 1995, 38: 2463–71.
- Auchus RJ, Buschur EO, Chang AY, et al. Abiraterone acetate to lower androgens in women with classic 21-hydroxylase deficiency. J Clin Endocrinol Metab 2014, 99: 2763–70.
- Turcu AF, Spencer-Segal JL, Farber RH, et al. Single-dose study of a corticotropin-releasing factor receptor-1 antagonist in women with 21-hydroxylase deficiency. J Clin Endocrinol Metab 2016, 101: 1174–80.
Deficit di 11-idrossilasi
Antonio Stigliano & Pina Lardo
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 14 aprile 2020)
Patogenesi
Rappresenta la seconda causa di sindrome adrenogenitale, legata eziologicamente a difetto del gene CYP11B1 (1,2).
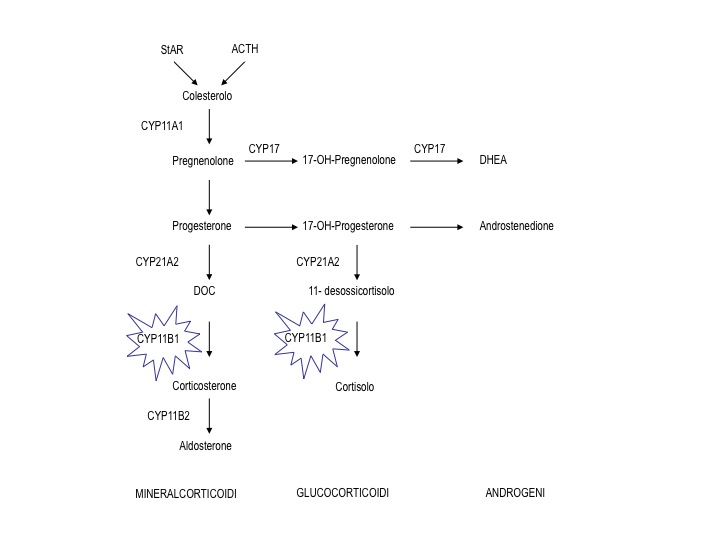
Clinica
Clinicamente questa forma di sindrome adrenogenitale si caratterizza per ipertensione arteriosa in entrambi i sessi (3) e per la presenza di:
- nel sesso femminile: genitali ambigui con clitoridomegalia, chiusura delle grandi labbra, disturbi del ciclo mestruale con oligomenorrea, irsutismo;
- nel sesso maschile: ambiguità dei genitali con aumento di sviluppo del pene, pubertà precoce, acne (4-6).
Le manifestazioni cliniche più evidenti che distinguono questa forma dal difetto di 21-idrossilasi sono l’ipertensione arteriosa e l’ipopotassiemia, indotte dall’aumento dei precursori degli ormoni glucoattivi e dei mineraloattivi dovuto al blocco enzimatico (3,4,7).
Diagnosi
La diagnostica ormonale mostra aumento degli androgeni e dei loro precursori della via Δ4 e Δ5 (testosterone, DHEA-S, androstenedione, 17-OH-progesterone), di 11-desossicorticosterone e 11-desossicortisolo. I livelli di renina e di potassio sono diminuiti, perché, pur in assenza di aldosterone, il DOC ha azione mineraloattiva, quindi aumenta l’escrezione di potassio, sopprime la renina e causa ipertensione (4,8).
Può essere eseguita l’analisi molecolare che dimostra delezioni o mutazioni puntiformi del gene CYP11B1.
Terapia
La terapia con glucocorticoidi ha l’obiettivo di sostituire il deficit di cortisolo, ridurre i livelli di androgeni e dei precursori mineralcorticoidi in eccesso. Dosi e formulazioni sono sovrapponibili a quelli descritti per il deficit di 21-idrossilasi.
L’obiettivo del trattamento è quello di prevenire la virilizzazione, migliorare il controllo pressorio, ottimizzare la crescita, preservare la fertilità e normalizzare i livelli di DOC e renina. Utile pertanto il monitoraggio clinico e biochimico mediante il dosaggio di 11-desossicortisolo, DOC e attività reninica plasmatica.
In presenza di valori pressori elevati nonostante un adeguato trattamento sostitutivo, deve essere aggiunta una terapia anti-ipertensiva: spironolattone o amiloride possono essere utilizzati da soli o combinati con calcio-antagonisti, mentre sono da evitare farmaci che agiscono sul sistema renina-angiotensina, essendo questo soppresso dagli elevati livelli dei precursori mineralcorticoidi. Inoltre, in presenza di ipopotassiemia, i diuretici tiazidici devono essere evitati o combinati con diuretici risparmiatori di potassio (9).
Bibliografia
- Curnow KM, Slutsker L, Vitek J, et al. Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc Natl Acad Sci USA 1993, 90: 4552-6.
- Krone N, Riepe FG, Gotze D, et al. Congenital adrenal hyperplasia due to 11-hydroxylase deficiency: functional characterization of two novel point mutations and a three-base pair deletion in the CYP11B1 gene. J Clin Endocrinol Metab 2005, 90: 3724-30.
- White PC, Curnow KM, Pascoe L. Disorders of steroid 11β-hydroxylase isozymes. Endocr Rev 1994, 15: 421-38.
- Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab 1983, 56: 222-9.
- Cathelineau G, Brerault JL, Fiet J, et al. Adrenocortical 11β-hydroxylation defect in adult women with postmenarcheal onset of symptoms. J Clin Endocrinol Metab 1980, 51: 287-91.
- Lucky AW, Rosenfield RL, McGuire J, et al. Adrenal androgen hyperresponsiveness to adrenocorticotropin in women with acne and/or hirsutism: adrenal enzyme defects and exaggerated adrenarche. J Clin Endocrinol Metab 1986, 62: 840-8.
- De Simone G, Tommaselli AP, Rossi R, et al. Partial deficiency of adrenal 11-hydroxylase. A possible cause of primary hypertension. Hypertension 1985, 7: 204-10.
- White PC, New MI, Dupont B. Congenital adrenal hyperplasia (2). N Engl J Med 1987, 316: 1580-6.
- Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Endocrine 2017, 55: 19-36.
Deficit di 3-ß-idrossi-steroido-deidrogenasi
Antonio Stigliano & Pina Lardo
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 14 aprile 2020)
Epidemiologia e patogenesi
L’incidenza di questa forma è piuttosto rara nella popolazione caucasica. In rapporto all’entità del difetto, rappresentato da delezioni, mutazioni puntiformi o inserzioni del gene 3-ß-HSD BII espresso nelle gonadi e nel surrene (1-4), si distinguono due forme:
- la forma classica, riconducibile ad un difetto completo;
- la forma non classica, dovuta ad un difetto parziale dell’enzima.
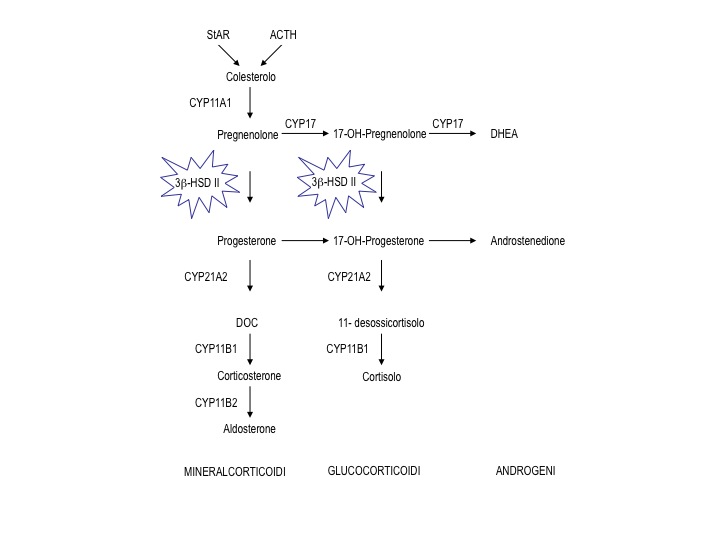
Clinica
Nella femmina è presente un modesto grado di virilizzazione alla nascita, pubarca prematuro, irsutismo, oligomenorrea.
Nel maschio vi sono ambiguità dei genitali con ipospadia, femminilizzazione, criptorchidismo in epoca neonatale.
Questo aspetto permette la diagnosi differenziale con il deficit di 21-idrossilasi, in presenza di elevati valori plasmatici di 17OHP. In entrambi i sessi, nella forma classica, si verifica disidratazione per perdita salina alla fine della prima settimana di vita neonatale (5-7). Questo aspetto pone dei dubbi interpretativi rispetto al difetto completo di 21-idrossilasi, soprattutto nel sesso femminile. Più agevole risulta la diagnosi differenziale della SAG da deficit di 21-idrossilasi dopo il periodo neonatale:
- generalmente livelli di 17OHP > 5000 ng/dL in 4°-5° giornata di vita indicano un deficit di 21-idrossilasi;
- al contrario, valori inizialmente elevati che tendono a ridursi durante le prime settimane di vita depongono per un deficit di 3-β-idrossi-steroido-deidrogenasi.
Diagnosi
Il difetto parziale dell’enzima va considerato nella diagnostica differenziale del pubarca prematuro, specie in quei soggetti in cui durante un anno di osservazione la maturazione ossea procede più velocemente della crescita staturale, con riduzione progressiva della prognosi della statura definitiva.
La diagnostica ormonale dimostra:
- aumento degli androgeni e dei loro precursori della via Δ5;
- ridotta produzione di glucocorticoidi, mineralcorticoidi e androgeni Δ4.
La formazione periferica degli steroidi della via Δ4 (androgeni in particolare, come androstenedione) è possibile e questo può essere responsabile della virilizzazione della femmina, ma non è sufficiente a mascolinizzare il maschio.
La diagnostica prevede l’esecuzione di esami a diversi livelli (8,9):
- 1° livello: dosaggio di 17-OH-progesterone (generalmente normale o modicamente elevato), 17-OH-pregnenolone (> 300 ng/dL, con rapporto 17-PGN/17OHP >3), DHEA, DHEA-S (> 700-800 ng/dL, con rapporto DHEA/DHEA-S > 5-6);
- 2° livello: test con ACTH, dopo il cui stimolo:
- 17-OH-pregnenolone > 1500 ng/dL (vn < 966 ng/dL);
- rapporto 17-PGN/17OHP > 9.5 (vn 4.2 ± 2);
- rapporto 17-PGN/cortisolo 68 ± 24 (vn 29 ± 11);
- 3° livello: analisi molecolare del gene 3-ß-HSD BII, che dimostra delezioni, mutazioni puntiformi o inserzioni.
Bibliografia
- Rhéaume E, Simard J, Morel Y, et al. Congenital adrenal hyperplasia due to point mutations in the type II 3 beta-hydroxysteroid dehydrogenase gene. Nat Genet 1992, 1: 239-45.
- Mason JI. The 3ß-hydrossisteroid dehydrogenase gene family of enzymes. Trend Endocrinol Metabol 1993, 6: 199-203.
- Simard J, Rheaume E, Sanchez R, et al. Molecular basis of congenital adrenal hyperplasia due to 3 beta-hydroxysteroid dehydrogenase deficiency. Mol Endocrinol 1993, 7: 716-28.
- Zhang L, Mason Ji, Naiki Y, et al. Characterization of two novel homozigous missense mutations involving codon 6 and 259 of type II 3ß-hydroxysteroid dehydrogenase (3ßHSD) gene causing, respectively, nonsalt wasting and salt-wasting 3ßHSD deficiency disorder. J Clin Endocr Metab 2000, 85: 1678-85.
- Pang S, LS Levine, E Stoner, et al. Non salt-losing congenital adrenal hyperplasia due to 3ß-hydroxysteroid dehydrogenase deficiency with normal glomerulosa function. J Clin Endocrinol Metab 1983, 56: 808-18.
- Pang S, Lerner AJ, Stoner E. Late-onset adrenal steroid 3ß-hydroxysteroid dehydrogenase deficiency. A cause of hirsutism in pubertal and postpubertal women. J Clin Endocrinol Metab 1985, 60: 428-39.
- Codner E, Okuma C, Iniguez G, et al. Molecular study of the 3ß-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. J Clin Endocrinol Metab 2004, 89: 957-64.
- Lutfallah C, Wang W, Mason JI, et al. Newly proposed hormonal criteria via genotypic proof for type II 3ß-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab 2002, 87: 2611-22.
- Mermejo LM, Elias LL, Marui S, et al. Refining hormonal diagnosis of type II 3ß-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. J Clin Endocr Metab 2005, 90: 1287-93.
Deficit di 17α-idrossilasi/17,20 liasi
Antonio Stigliano & Pina Lardo
Dipartimento di Medicina Clinica e Molecolare - Ospedale Sant'Andrea - Facoltà di Medicina e Psicologia, "Sapienza" Università di Roma
(aggiornato al 14 aprile 2020)
Epidemiologia e patogenesi
La prevalenza di questa forma è molto rara.
È dovuta ad un difetto isolato a carico dell'enzima 17-idrossilasi o dell'enzima 17,20 liasi. Più comunemente il difetto, ereditato come tratto autosomico recessivo, coinvolge ambedue le attività enzimatiche, per mutazioni del gene CYPc17 che codifica per il citocromo microsomiale P450c17 (1).
Il difetto di attività della 17-idrossilasi riduce la quota di ormoni glucoattivi e degli ormoni sessuali, privilegiando la quota degli ormoni mineraloattivi e dei loro precursori (2-4). La ridotta o mancante attività della 17,20 liasi comporta una combinata riduzione della sintesi di androgeni ed estrogeni. L'espressione del citocromo P450c17 anche a livello gonadico rende ragione di una ridotta steroidogenesi a livello ovarico e testicolare nei pazienti affetti (4).
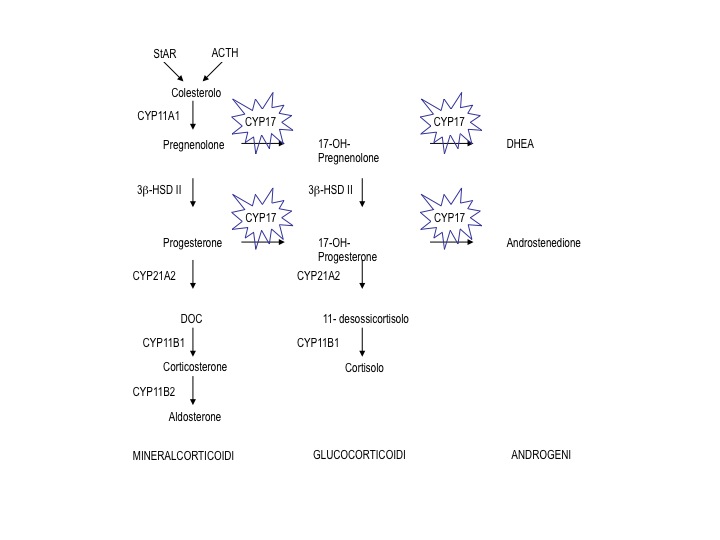
Clinica
La clinica, nella femmina, è sostenuta dal mancato pubarca o dall'amenorrea, mentre il maschio presenta genitali ambigui con gradi diversi di pseudoermafroditismo, criptorchidismo, mancato pubarca o ritardo puberale con comparsa di ginecomastia. Nel sesso maschile queste caratteristiche possono indurre un riconoscimento fenotipico femminile, ritardando la diagnosi, che avviene tardivamente per la mancanza dello sviluppo puberale. In entrambi i sessi è frequente il riscontro di ipertensione e di ipopotassiemia, mentre solo il 15% circa di questi pazienti è normoteso al momento della diagnosi. L'insufficienza surrenalica dipende dal grado del difetto nella sintesi di cortisolo (4-6).
Diagnosi
Dal punto di vista biochimico è frequente il riscontro di ipopotassiemia e la diagnostica ormonale segnala:
- a livello surrenalico:
- un aumento di desossicorticosterone e corticosterone, progesterone, pregnenolone;
- ridotta produzione di testosterone (non stimolabile dopo hCG), renina, DHEA, DHEA-S, 17-OH-progesterone, androstenedione;
- a livello gonadico: ridotta sintesi di androgeni ed estrogeni.
È possibile eseguire l'analisi molecolare del gene CYPc17, che dimostra inserzioni, delezioni o mutazioni puntiformi.
Bibliografia
- Winter JSD, Couch RM, Muller J, et al. Combined 17-hydroxylase and 17,20-desmolase deficiencies: evidence for synthesis of a defective cytochrome P450c17. J Clin Endocrinol Metab 1989, 68: 309-16.
- Griffing GT, Wilson TE, Hoolbrook MM, et al. Plasma and urinary 19-nor-deoxycorticosterone in 17 alpha-hydroxylase deficiency syndrome. J Clin Endocrinol Metab 1984, 59: 1011-5.
- Fardella CE, Hum DW, Homokij J, Miller WL. Point mutation of Arg 440 to His in cytocrome P450 c17 causes severe 17 alfa hydroxylase deficiency. J Clin Endocrinol Metab 1994, 79: 160-4.
- Kater C A, Biglieri EG. Disorders of steroid 17α-hydroxylase deficiency. Endocr Metab N Amer 1994, 23: 341-57.
- Geller DH, Auchus RJ, Mendonca BB, Miller WL. The genetic and functional basis of isolated 17,20 lyase deficiency. Nat Genet 1997, 17: 201-5.
- Arlt W, Walker EA, Draper N, et al. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 2004, 363: 2128-35.
Deficit di StAR e di P450scc
Stefano Tumini
Dipartimento di Pediatria, Università di Chieti.
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
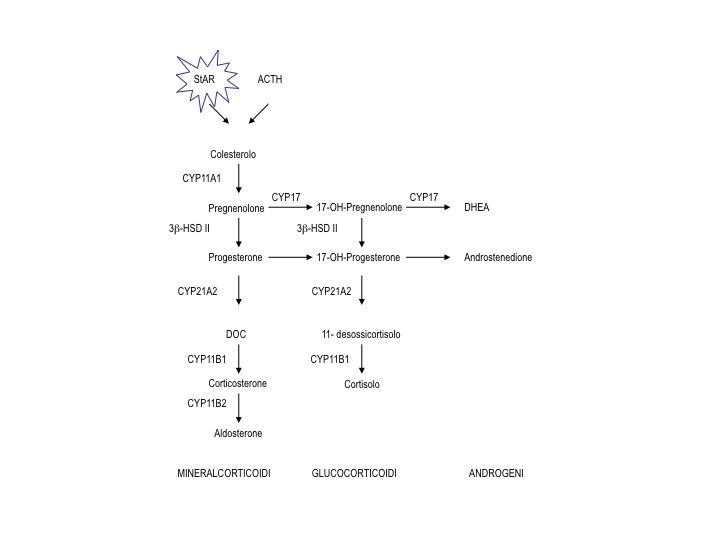
Deficit di StAR (Steroidogenic Acute Regulatory protein)
L’iperplasia congenita lipoidea (Congenital Lipoid Adrenal Hyperplasia – LCAH) (OMIM: 201701) è una malattia autosomica recessiva, molto rara in Europa e Nord America, mentre nei Paesi Asiatici la sua prevalenza è di circa 1:300.000 nati. Determina blocco completo della steroidogenesi. Il difetto fu identificato inizialmente come deficit di conversione del colesterolo in pregnenolone e definito deficit di 20-22 desmolasi. L’osservazione che P450scc è normale nei soggetti LCAH portò all’individuazione nel gene della Steroidogenic Acute Regulatory protein (StAR) di mutazioni responsabili di LCAH (1). StAR regola il passaggio del colesterolo dalla superficie esterna della membrana mitocondriale a quella interna, dove si avvia la steroidogenesi per azione di P450scc. Il corrispondente gene è situato sul locus 8p11.2, è costituito da sette esoni e sei introni ed è espresso nel surrene, nel testicolo, nel cervello ma non nella placenta (2). Sono state descritte oltre 40 mutazioni.
Il modello “two-hits” spiega la sequenza delle manifestazioni cliniche (3); la deposizione di colesterolo nelle cellule steroidogenetiche distrugge (4):
- nel maschio le cellule di Leydig a causa della stimolazione fetale da parte di hCG;
- in entrambi i sessi precocemente la zona fetale del surrene e alla nascita la zona glomerulare e fascicolata;
- nella femmina alla pubertà l’ovaio in seguito alla stimolazione di LH e FSH.
LCAH si manifesta clinicamente nei primi mesi di vita con insufficienza surrenalica severa (vomito, diarrea, disidratazione, iponatremia, ipopotassiemia) (5). Ci sono tuttavia casi in cui sono presenti genitali maschili normali e l’insufficienza surrenalica si manifesta tardivamente (non-classical LCAH) (6).
Nelle forme classiche l’insufficienza surrenalica con perdita di sali presenta manifestazioni cliniche sovrapponibili nei soggetti 46,XX e 46,XY e l’insufficienza testicolare si manifesta nei soggetti 46,XY con “sex reversal” (7). Nei soggetti 46,XX la funzione ovarica può permettere la comparsa di pubertà e menarca con cicli anovulatori e si associa ad ipogonadismo ipergonadotropo (8). Dopo l’inizio della pubertà è stata descritta la comparsa di cisti follicolari con possibilità di torsione ovarica (9) e l’associazione con quadri malformativi del SNC e la sindrome di Chiari tipo 1 (7).
Deficit di P450scc (Cholesterol Side-Chain Cleavage Enzyme)
L’enzima P450scc è codificato dal gene CYP11A1, situato sulla parte interna della membrana mitocondriale e converte il colesterolo in pregnenolone, dando inizio alla steroidogenesi (10). Il deficit di P450scc (OMIM: 118485) compromette la steroidogenesi surrenalica e gonadica. Sono stati riportati solo 19 casi in letteratura (11). Le cellule surrenaliche e gonadiche che esprimono StAR esprimono anche P450scc. Il quadro clinico è indistinguibile dai pazienti con LCAH classica e non classica. Le forme con parziale compromissione di p450scc possono presentarsi tardivamente senza DSD.
Nei pazienti LCAH i surreni appaiono massivamente aumentati di volume, mentre nei pazienti con deficit di P450scc sono di volume diminuito. La diagnosi definitiva viene posta dopo conferma genetica.
Bibliografia
- Lin D, Sugawara T, Strauss J, et al. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 1995, 267: 1828–31.
- Sugawara T, Holt JA, Driscoll D, et al. Human steroidogenic acute regulatory protein: functional activity in COS-1 cells, tissue-specific expression, and mapping of the structural gene to 8p11.2 and a pseudogene to chromosome 13. Proc Natl Acad Sci USA 1995, 92: 4778–82.
- Miller WL. Mitochondrial specificity of the early steps in steroidogenesis. J Steroid Biochem Mol Biol 1995, 55: 607–16.
- Metherell LA, Naville D, Halaby G, et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J Clin Endocrinol Metab 2009, 94: 3865–71.
- King SR, Bhangoo A, Stocco DM. Functional and physiological consequences of StAR deficiency: role in lipoid congenital adrenal hyperplasia. Endocr Dev 2011, 20: 47–53.
- Baker BY, Lin L, Kim CJ, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab 2006, 91: 4781–5.
- Sertedaki A, Dracopoulou M, Voutetakis A, et al. Long-term clinical data and molecular defects in the STAR gene in five Greek patients. Eur J Endocrinol 2013, 168: 351–9.
- Bhangoo A, Buyuk E, Oktay K, Ten S. Phenotypic features of 46, XX females with StAR protein mutations. Pediatr Endocrinol Rev 2007, 5: 633–41.
- Kaku U, Kameyama K, Izawa M, et al. Ovarian histological findings in an adult patient with the steroidogenic acute regulatory protein (StAR) deficiency reveal the impairment of steroidogenesis by lipoid deposition. Endocr J 2008, 55: 1043–9.
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011, 32: 81–151.
- Tee MK, Abramsohn M, Loewenthal N, et al. Varied clinical presentations of seven patients with mutations in CYP11A1 encoding the cholesterol side-chain cleavage enzyme, P450scc. J Clin Endocrinol Metab 2013, 98: 713–20.
- Bose HS, Sugawara T, Strauss III JF, Miller WL. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med 1996, 335: 1870-9.
- Fujieda K, Okuhara K, Abe S, et al. Molecular pathogenesis of lipoid adrenal hyperplasia and adrenal hypoplasia congenita. J Steroid Biochem Mol Biol 2003, 85: 483-9.