Carcinoma a cellule di Merkel

Fernando Cirillo
Dipartimento di Chirurgia Generale, Unità di Chirurgia Generale, Gruppo Tumori Ormonali Rari, AO Istituti Ospitalieri, Cremona
DEFINIZIONE ED EPIDEMIOLOGIA
Il carcinoma a cellule di Merkel (MCC) è una neoplasia della cute rara e aggressiva, caratterizzata da frequenti recidive locali, rapida progressione della malattia, scarsa qualità di vita e breve sopravvivenza.
L’origine di questo tumore non è stata provata in modo definitivo e oggi si ritiene che la cellula di Merkel possa derivare da una cellula epiteliale totipotente, in grado di differenziarsi sia in senso neuroendocrino, sia come cheratinocita. A sostegno di questa teoria è la presenza di cellule transizionali simili sia ai cheratinociti che alle cellule di Merkel (1).
Recentemente è stato isolato un polyomavirus presente nell’80% dei MCC con una forte correlazione fra la presenza del virus e stati di alterata risposta immunitaria cellulo-mediata (2).
La reale prevalenza di MCC è sconosciuta. Esso colpisce più frequentemente (3/4 dei casi) pazienti anziani ultra60enni (range 7-95) soprattutto donne (F/M = 3/1). È più comune nelle popolazioni caucasiche, ma occasionalmente presente anche nei negri e nei polinesiani. È stata segnalata un’alta incidenza nei trapiantati, con caratteristiche di maggiore aggressività probabilmente secondaria ad alterazioni del sistema immunocompetente, in un paziente HIV positivo e in due casi di artrite reumatoide (4).
CLINICA, LOCALIZZAZIONE E DIAGNOSI
Le dimensioni della neoplasia possono essere del tutto varie, fino a 15 cm di diametro, con una media alla presentazione di circa 3 cm.
La sede più comune del tumore è la cute della regione testa-collo (50% dei casi), il 40% dei casi interessa le estremità e il rimanente 10% tronco e mucose. Sono stati anche riportati casi di sedi multiple della malattia.
La clinica per MCC permette solo una diagnosi presuntiva; per tale motivo la letteratura anglosassone ha coniato l’acronimo AEIOU (Asymptomatic/lack of tenderness, Expanding rapidly, Immune suppression, Older than age 50, and UV-exposed site on a person with fair skin)(3) per venire in aiuto alla diagnosi. Infatti, se la neoplasia si presenta tipicamente come una lesione solitaria, rilevata o a placca, talvolta peduncolata, di colore rosso-violaceo, a superficie lucida talvolta associata a vicine teleangectasie, nello stadio iniziale della malattia la diagnosi differenziale può risultare poco agevole: MCC può, infatti, essere confuso con basalioma, carcinoma spino-cellulare, granuloma piogenico, cherato-acantoma, melanoma, linfoma cutaneo, metastasi cutanee (da carcinoma a piccole cellule, carcinoma anaplastico, carcinoide, retinoblastoma, sarcoma di Ewing e neuroblastoma).
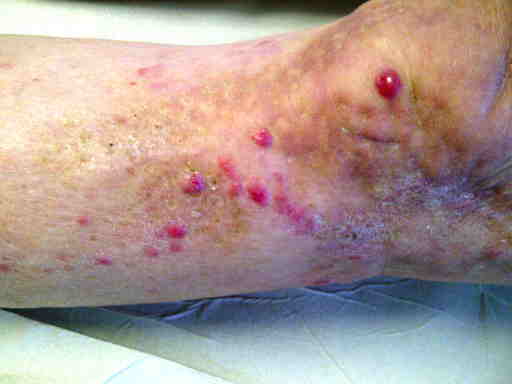
Merkel all'arto inferiore sinistro (lesione localmente avanzata dopo trattamento incongruo)
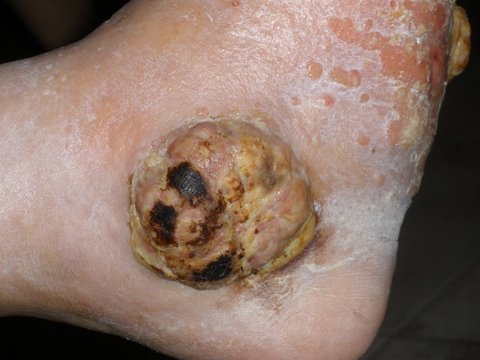
Merkel al piede destro
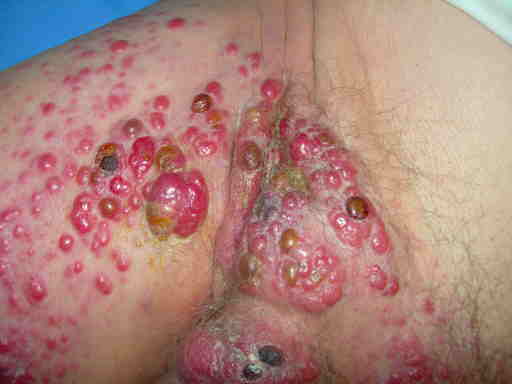
Merkel localmente avanzato da trattamento incongruo (non radicalizzazione, non RT)
I marcatori normalmente espressi da questo tumore sono NSE, cromogranine e sinaptofisina.
La stadiazione della malattia prevede la convenzionale tomografia multistrato (TC) e altre metodiche di immagine ancillari come 18F-FDG-PET-TC, utile come completamento nei pazienti con fattori di rischio più elevati e nel sospetto di recidiva, oppure 68Ga-DOTA-PET/TC per l’elevata espressione di recettori della somatostatina (2). Ciò vale anche per OctreoScan®, che rispetto alla PET con 68Ga presenta minore sensibilità diagnostica (1). Per la stadiazione di MCC viene utilizzata la classificazione AJCC del 2010 (5).
| Classificazione (5) | |||
| T | Tx | Il tumore primitivo non è valutabile | |
| T0 | Nessuna evidenza di tumore primitivo (per esempio il tumore è stato trovato nei linfonodi, ma il tumore principale non è stato trovato) | ||
| Tis | Carcinoma in situ: il tumore è confinato all’epidermide, lo strato più esterno della cute (questo è estremamente raro per il MCC) | ||
| T1 | Diametro tumorale < 2 cm | ||
| T2 | Diametro tumorale 2-5 cm | ||
| T3 | Diametro tumorale > 5 cm | ||
| T4 | Il tumore si è diffuso nei tessuti adiacenti (muscoli, osso, cartilagine) | ||
| N | Nx | I linfonodi regionali non sono valutabili | |
| N0 | I linfonodi regionali non sono interessati dal tumore | pN0: i linfonodi regionali non sono interessati dal tumore all’esame istologico | |
| cN0: i linfonodi regionali non sembrano interessati dal tumore (obiettivamente e agli esami strumentali), ma non sono stati sottoposti a biopsia | |||
| N1 | I linfonodi regionali sono interessati dal tumore all’esame istologico | N1a: ma non lo sembravano agli esami strumentali | |
| N1b: e lo sembravano agli esami strumentali | |||
| N2 | Il tumore si è diffuso verso i linfonodi regionali senza raggiungerli (metastasi in transito) | ||
| M | M0 | Nessuna diffusione metastatica agli organi a distanza | |
| M1a | Il tumore si è diffuso ad altre aree cutanee, ai tessuti sottocutanei, o ai linfonodi a distanza | ||
| M1b | Metastasi polmonari | ||
| M1c | Metastasi extra-polmonari | ||
| Stadiazione | |||
| Stadio | Caratteri | ||
| 0 | Tis, N0, M0 | ||
| I | IA | T1, pN0, M0 | |
| IB | T1, cN0, M0 | ||
| II | IIA | T2 o T3, pN0, M0 | |
| IIB | T2 o T3, cN0, M0 | ||
| IIC | T4, N0, M0 | ||
| III | IIIA | qualsiasi T, N1a, M0 | |
| IIIB | qualsiasi T, N1b o N2, M0 | ||
| IV | qualsiasi T, qualsiasi N, M1 (a, b, o c) | ||
TERAPIA
Trattamento chirurgico
Negli stadi iniziali della malattia (I e II) il trattamento di scelta è rappresentato dall’escissione radicale del tumore primitivo. Allo scopo di evitare la recidiva locale, è raccomandata un’adeguata rimozione della lesione, con margini di almeno 2 cm. La necessità di linfadenectomia elettiva è stata superata con la ricerca del linfonodo sentinella (LS) con tecnica radioisotopica, in grado di localizzare metastasi linfonodali occulte nel 29% dei casi (1). Anche se i pazienti con LS positivo presentano un maggior rischio di recidiva a distanza, sono possibili falsi negativi, con sviluppo di recidiva nel 9.8% dei casi (2).
Radioterapia
La maggior parte degli autori è a favore della radioterapia (RT) adiuvante. La scelta è motivata da una riduzione del rischio di recidiva locale nel I e II stadio della malattia, con presenza di metastasi linfonodali nel 40-73% dei casi e recidiva locale nel 23-60% (1). Questi dati sono suffragati dal programma governativo statunitense Surveillance, Epidemiology, and End Results Program e da quelli del gruppo australiano di Veness (1). Al contrario, la chemioterapia adiuvante non è in grado di ridurre la percentuale di recidiva locale né di migliorare la sopravvivenza. La RT è impiegata come monoterapia nei casi di malattia localmente avanzata in sedi anatomiche critiche con difficile resecabilità.
Chemioterapia
Tradizionalmente viene utilizzata nel setting avanzato, con intento palliativo. I chemioterapici più usati sono etoposide, cisplatino/carboplatino, doxorubicina, dacarbazina, vincristina, ciclofosfamide e metotrexate, sia in monoterapia che in combinazione. Non esistono studi randomizzati di confronto tra vari regimi chemioterapici. In un’analisi retrospettiva su 107 pazienti sono stati osservati tassi di attività con percentuali progressivamente in diminuzione in prima, seconda e terza linea, rispettivamente del 61%, 45%, e del 20%, con una durata di risposta da 3.5 a 15 mesi e sopravvivenza globale a 3 anni pari al 17% nei pazienti metastatici e al 35% in quelli con malattia localmente avanzata. In sostanza, MCC appare una neoplasia chemio-sensibile, ma non curabile con la chemioterapia (2). Lo studio prospettico di fase II TROG (Trans-Tasman Radiation Oncology Group) ha valutato il trattamento sincrono di cisplatino/etoposide associato a RT in pazienti con MCC localmente avanzato e ad alto rischio. Con un follow-up di 48 mesi, la sopravvivenza totale, il controllo loco-regionale e il controllo a distanza sono stati rispettivamente di 76%, 75% e 76%. Questo studio suggerisce che la chemio-radioterapia concomitante può influire positivamente sul controllo loco-regionale e sulla sopravvivenza globale nel MCC localmente avanzato (2).
Altre terapie
L’infiltrazione locale di IFNα-2b, l’utilizzo di Tumor Necrosis Factor (TNF), l’ipertermia associata a basse dosi di RT, la RT associata a TNF-α, IFN-γ e melphalan, l’elettro-chemioterapia e imiquimod associato a RT hanno mostrato aneddoticamente remissioni di malattia, con una sopravvivenza libera da progressione relativamente lunga (2).
I dati di letteratura sono scarsi ed eterogenei per gli analoghi della somatostatina (1,6). Non ci sono evidenze concrete che gli SSA siano attivi nel MCC. Tuttavia, in assenza di alternative terapeutiche, in presenza di gravi comorbilità, in MCC a decorso indolente con espressione di recettori per la somatostatina, gli SSA potrebbero essere presi in considerazione come trattamento palliativo (1).
La co-espressione di c-Kit in un’alta percentuale di MCC suggerisce un ruolo importante degli inibitori di tirosin-chinasi nella trasformazione neoplastica della cellula di Merkel. Su tale base sono state condotte esperienze cliniche con inibitori di c-Kit, che hanno dimostrato evidenze di risposta parziale a imatinib e pazopanib (2).
Terapia radiorecettoriale
Nei pazienti che presentano una positività alla PET/TC con SSA radiomarcati, può essere valutata la possibilità della PRRT con 90Y/177Lu DOTATOC/DOTATATE, di cui sono riportate al momento solo sporadiche esperienze, in base alle quali non è possibile esprimere un giudizio definitivo (2).
CONCLUSIONI
MCC è certamente una neoplasia molto aggressiva e altrettanto sconosciuta. Questi due fattori sono il risultato di numerose osservazioni di recidive locali o di malattia avanzata. Il sito italiano www.neuroendocrini.it dal 2002 ad oggi ha collezionato un considerevole numero di pazienti affetti da MCC con la possibilità di tracciare una mappa dei bisogni, delle differenti risorse culturali e tecnologiche da Regione a Regione (7). Il grande numero di richieste di informazione riguardo a MCC suggerisce che il tumore ha bisogno di attenzione e di expertise, così come oggi succede per il melanoma (8). In un programma di sorveglianza dovremmo di principio inserire il Medico di Medicina Generale, stimolandolo ad una maggiore attenzione alle lesioni della cute e per incoraggiare il paziente ad una precoce asportazione. La tempistica è parte integrante di un percorso appropriato per il management di MCC. Sia il Dermatologo che il Chirurgo e l’Oncologo dovrebbero collaborare per ridurre l’intervallo tra escissione e diagnosi patologica, con lo scopo di rendere il più possibile precoce l’inizio di altre terapie quando necessarie, favorendo una migliore qualità di vita del paziente. Le Linee Guida dovrebbero fornire quelle raccomandazioni chiare e indispensabili per la gestione della neoplasia, facendo il più possibile riferimento alle metodiche e alle terapie accessibili sul nostro territorio, negando i benefici di altre, fornendo anche informazioni sulla spesa sanitaria. Questi potrebbero essere alcuni dei motivi che fanno considerare utile il ruolo dell’informazione sanitaria in rete, soprattutto quando dedicata a patologie poco frequenti e scarsamente conosciute (8).
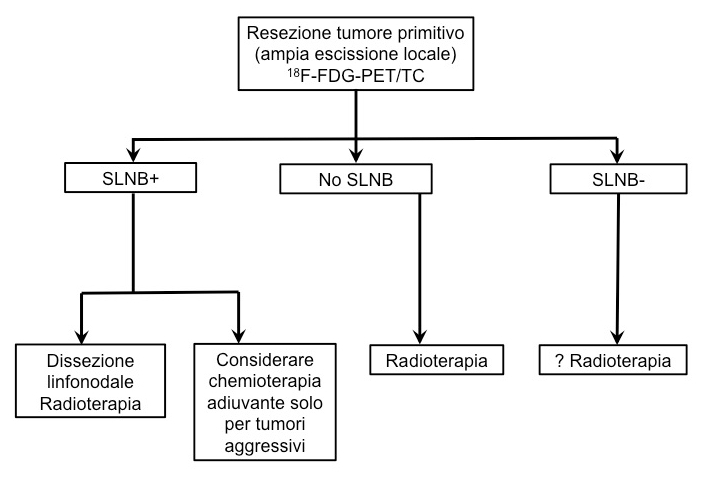
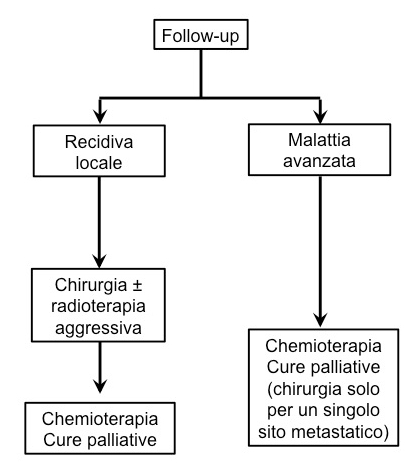
Strategia per MCC primitivo (sopra) e recidivo (sotto) (1).
BIBLIOGRAFIA
- Cirillo F, Vismarra M, Cafaro I, et al. Merkel cell carcinoma: a retrospective study on 48 cases and review of literature. J Oncol 2012, 2012: 749030.
- AIOM. Linee guida neoplasie neuroendocrine. AIOM/IT.A.NET 2014.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol 2008, 58: 375-81.
- Lima GF, Cirillo F, Buononato M, et al. Clinical experience on eight cases of Merkel cell carcinoma. Tumori 2003, 89: 146-51.
- American Joint Committee. Cancer Staging Manual and Handbook. Merkel Cell Carcinoma. Springer, New York, NY, USA, 2010.
- Cirillo F, Filippini L, Lima GF, et al. Tumore a cellule di Merkel: segnalazione di un caso e trattamento con octreotide. Minerva Chir 1997, 52: 1359-65.
- http://www.neuroendocrini.it/storia.php.
- Cirillo F. Merkel cell carcinoma: need for information and awareness. A case series of 47 patients from an Italian website. Tumori 2014, 100: 504-6.
Patologia neoplastica degli organi endocrini
Neoplasie tiroidee
Neoplasie ipofisarie
Neoplasie paratiroidee
Neoplasie surrenaliche
Neoplasie ovariche
Neoplasie testicolari
Tumori ormono-sensibili e ormono-dipendenti
Overview sui tumori ormono-sensibili e ormono-dipendenti
Valerio Adinolfi, Roberto Baldelli, Antonella Paoloni, Francesca Rota, Laura Rizza, Agnese Barnabei, Marialuisa Appetecchia
UOSD Endocrinologia, Istituto Nazionale Tumori Regina Elena – IRCCS, Roma
INTRODUZIONE
Per la prima volta nel 1963, uno studio pubblicato su Cancer Research (1) individuava tra le cause principali dello sviluppo dei tumori le radiazioni ionizzanti e le sostanze chimiche cancerogene. Tuttavia già in quello studio veniva attribuito un ruolo importante agli ormoni nella crescita di alcune neoplasie di mammella, prostata e utero. Nel 1983, un lavoro (2) concluse che i principali fattori che influenzavano positivamente o negativamente l’incidenza del cancro alla mammella erano le radiazioni ionizzanti, l’annessiectomia bilaterale, l’isterectomia e la terapia ormonale (contraccezione e/o terapia sostitutiva).
Gli estrogeni, attraverso i recettori ERα e ERβ, hanno un ruolo fondamentale nello sviluppo e nella morfogenesi della ghiandola mammaria; entrambi i recettori legano il 17β-estradiolo (E2) con la medesima affinità, ma sono in grado di attivare vie diverse con effetti diversi (3). Il signaling mediato dall’ERα promuove la proliferazione cellulare mentre quello di ERβ la inibisce (probabilmente per un effetto anche pro-apoptotico)(4).
Studi successivi hanno dimostrato un ruolo del complesso E2/ERα nella trascrizione di geni chiave per la proliferazione, differenziazione, sopravvivenza delle cellule sane, ma anche per l’invasività, la metastatizzazione e l’angiogenesi delle cellule neoplastiche (c-myc, ciclina D, A ed E, p21)(5), mentre ERβ inibisce la trascrizione ERα-mediata e la proliferazione E2-mediata nelle cellule di cancro mammario bloccando le cellule allo stadio G2 (6).
Circa il 70% dei cancri della mammella esprime gli ER, ma, a differenza delle cellule mammarie sane, esprimono più ERα che ERβ; inoltre possono esprimere il recettore del progesterone (PR) e l’erythroblastosis oncogene-B2 (ErbB-2, HER2/neu). Per tutti questi motivi, già da molti anni sono in uso e in studio nella terapia del carcinoma della mammella numerosi farmaci che agiscono sul sistema estrogenico.
Agonisti del GnRH
Nelle donne in pre- e peri-menopausa può esserne indicato l’utilizzo per sopprimere la produzione estrogenica ovarica. Questi farmaci si sono dimostrati efficaci al pari dell’annessiectomia bilaterale sulla sopravvivenza totale (OS) e libera da insuccesso (FFS)(7).
I farmaci che presentano come indicazione il carcinoma della mammella sono il triptorelin ed il leuprorelin. Nelle donne di età superiore ai 40 anni che vengono sottoposte a chemioterapia anti-neoplastica standard si verifica amenorrea permanente in quasi il 100% dei casi, per cui l’utilizzo degli analoghi del GnRH in queste pazienti va discusso, mentre l’associazione di chemioterapia tradizionale e castrazione farmacologica sembra essere l’approccio ottimale nelle donne in pre-menopausa più giovani (8,9).
Selective estrogen-receptor modulators (SERM)
Sono una classe di ligandi dell’ER che agiscono come anti-estrogeni o agonisti recettoriali, in base al tessuto che esprime il recettore; a livello mammario agiscono come anti-estrogeni.
Il capostipite è il tamoxifene, già in commercio da circa 30 anni, che viene generalmente somministrato per 5 anni dalla diagnosi e si è dimostrato in grado di ridurre il rischio di recidiva (RR 0.53 a 5 anni, 0.68 a 10 anni e 0.97 a 15 anni)(10), determinando un aumento di circa il 15% della sopravvivenza libera da recidiva (RFS)(11). Probabilmente la riduzione dell’effetto a lungo termine è associata a un meccanismo di resistenza, che può essere associato a isoforme dell’ER che non legano il farmaco o all’aumentata attività di co-attivatori oppure ad alterazioni di tappe della via di trasmissione del segnale intra-cellulare (12). È indicato in tutte le pazienti con carcinoma mammario ER+ e/o PR+. Il tamoxifene per agire deve essere metabolizzato dal citocromo P450-2D6: circa il 6-8% delle donne sono portatrici di forme meno attive di questo enzima e farmaci come gli SSRI lo inibiscono.
Altri membri di questa famiglia sono il raloxifene e il più nuovo toremifene, indicato nel carcinoma mammario metastatico nel post-menopausa.
Tra i possibili rischi di questi farmaci ci sono l’iperplasia endometriale fino al carcinoma dell’endometrio, per l’effetto agonista sull’utero, ed eventi trombo-embolici.
Inibitori dell'aromatasi (AI)
L’aromatasi è un’enzima che catalizza la reazione di trasformazione degli androgeni in estrogeni (estrone ed E2). È normalmente espresso in molti tessuti, compresa la mammella. Nella donna in post-menopausa la principale fonte di E2 non è più l’ovaio, ma sono i tessuti che contengono l’aromatasi, in particolare il tessuto adiposo.
Gli AI hanno come indicazione la terapia endocrina del carcinoma della mammella per 5 anni nelle donne in post-menopausa.
Membri di questa famiglia sono anastrozolo, letrozolo, exemestane. Vengono metabolizzati dal citocromo P450-3A4, per cui bisogna porre attenzione alle interazioni con farmaci induttori e inibitori di questo enzima (13).
Tra gli effetti indesiderati più importanti c’è la progressione dell’osteoporosi, per cui le pazienti devono effettuare una densitometria ossea prima e durante il trattamento e spesso è necessario associare una terapia con bisfosfonati.
Lo studio ATAC (Arimidex, Tamoxifen, Alone or in Combination) ha posto a confronto l’anastrozolo con il tamoxifene in donne in post-menopausa con carcinoma della mammella ER+: dopo un follow-up di 68 mesi, lo studio ha evidenziato che l’anastrozolo prolungava la sopravvivenza libera da malattia (DFS), il tempo alla recidiva, e riduceva le metastasi a distanza, con minori effetti secondari del tamoxifene, in particolare a livello ginecologico e cardiovascolare, suggerendo quindi di preferire gli AI ai SERM nelle donne in post-menopausa (14). Risultati simili sono stati ottenuti anche da uno studio successivo di confronto tra letrozolo e tamoxifene (15).
Selective estrogen-receptor down-regulators (SERD)
Sono una classe di steroidi puramente anti-estrogeni. L’unico farmaco di questa classe attualmente in uso è il fulvestrant, indicato nel trattamento in post-menopausa nelle donne in ricaduta di malattia durante o dopo terapia anti-estrogenica adiuvante. Presenta come potenziale rischio il tromboembolismo venoso. Il farmaco è in commercio in Italia, ma sottoposto a monitoraggio da parte dell’AIFA (13,16). Uno studio di confronto con il tamoxifene non ha evidenziato, nei pazienti ER+, differenze significative per i principali esiti (14,17).
Ligandi di ERβ
Non esistono ancora farmaci in commercio che agiscano da agonisti del recettore ERβ, ma, in considerazione dei suoi effetti anti-proliferativi e pro-apoptotici, potrebbe essere una classe di farmaci promettente per il trattamento del carcinoma della mammella.
Nelle pazienti che sovra-esprimono HER2 (che spesso sono anche resistenti alla terapia ormonale) è possibile associare all’eventuale terapia endocrina adiuvante il trastuzumab o il lapatinib, anticorpi monoclonali che legano il recettore HER2/neu, che normalmente lega il fattore di crescita dell’epidermide (EGF), determinando l’attivazione di diverse vie di segnale che promuovono la proliferazione cellulare e l’angiogenesi (18). Il lapatinib agisce inibendo l’attività tirosin-chinasica legata al recettore dell’EGF (EGF-R) e a HER2/neu.
Diversi studi hanno anche valutato l’utilizzo di questi approcci come terapia neo-adiuvante nel carcinoma della mammella: uno studio ha evidenziato la capacità del letrozolo di determinare un 55% di risposta tumorale oggettiva vs il 35% del tamoxifene e in circa il 50% dei casi è stato possibile evitare la mastectomia radicale, optando per una quadrantectomia (19).
L’IGF-1 e il suo recettore influenzano lo sviluppo e la crescita tumorale. L’IGF-1R appare sovra-espresso nella maggioranza dei carcinomi della mammella (90-95%) ed è spesso co-espresso con ER. Gli estrogeni inducono l’espressione di IGF-1R. L’obesità e dunque l’iperinsulinemia riducono la produzione di IGFBP-1 e IGFBP-2 (IGF-binding proteins) che normalmente legano e inibiscono l’azione dell’IGF-1; questo potrebbe essere uno dei motivi della maggior prevalenza di carcinoma mammario nelle donne obese (13,20). Per questo motivo sono state sviluppate molecole inibenti IGF-1R, come linsitinib, e sono al momento in fase di sperimentazione clinica per diversi tipi di neoplasie (21).
È inoltre interessante come diversi studi abbiano attribuito un effetto anti-neoplastico alla metformina: uno studio inglese ha evidenziato che l’uso continuo per almeno 5 anni della metformina ha determinato un odds ratio di 0.44 per lo sviluppo di carcinoma della mammella rispetto alle donne non in trattamento con metformina, probabilmente per un effetto indiretto di riduzione dei livelli circolanti di insulina (22).
Nuovi target terapeutici
Tra i nuovi target terapeutici, di particolare interesse sono i co-attivatori e co-repressori delle vie di trasduzione del segnale estrogenico: tra i co-attivatori, SRC-1 e SRC-3 sono frequentemente sovra-espressi nel carcinoma della mammella, soprattutto in quelli con peggiore prognosi. Il gossipolo, un prodotto naturale estratto dal seme del cotone, sembra essere in grado di down-regolare l’espressione di SRC-3 nelle cellule di carcinoma mammario; antagonizza mediatori anti-apoptotici e quindi rappresenta un possibile prototipo di molecole agenti sul sistema estrogenico (21,23).
L’espressione del recettore ERα può essere modulata in seguito all’esposizione a un inibitore dell’enzima istone-deacetilasi (HDAC) (Vorinostat, Belinostat, Panobinostat). Queste molecole sono in grado di bloccare il ciclo cellulare e di indurre apoptosi in molte cellule neoplastiche. Inoltre, questi farmaci sembrerebbero anche permettere la ricomparsa di ERα nelle cellule neoplastiche precedentemente ER- e determinano inoltre una riduzione dell’espressione dell’EGF-R, risensibilizzando le cellule alla terapia endocrina (24,25).
Sono in fase di studio per nuovi farmaci numerosi altri target molecolari delle complesse vie di trasduzione del segnale di ERα (PI3K/AKT, mTOR, Src, HSP90, …) (13).
Il cancro della prostata rappresenta la neoplasia più comune nell’uomo ed è dipendente dal sistema androgenico. Questa correlazione è nota a partire dagli anni ’70, da quando venne notata una regressione del carcinoma metastatico in uomini sottoposti a castrazione medica o chirurgica (26). Tuttavia, si notò subito come la deprivazione androgenica non era curativa, in quanto in molti uomini la neoplasia avanzava e portava a decesso nonostante livelli molto bassi di testosterone, con una sopravvivenza media di 2-3 anni (27).
I meccanismi di resistenza alla deprivazione androgenica possono essere molteplici: aumentata espressione di enzimi coinvolti nella steroidogenesi, aumentata espressione del recettore per gli androgeni (AR), mutazioni genetiche di AR o diversa specificità di ligando, attivazione di vie di segnale mediate da AR senza legame con il ligando, attivazione di vie indipendenti che possono superare le vie indotte dal blocco androgenico, proliferazione di cellule staminali prostatiche indipendenti dagli androgeni (28). È proprio la comprensione di questi meccanismi che ha portato allo sviluppo di farmaci sempre più nuovi e sempre più specifici per contrastare lo sviluppo di neoplasie cosiddette resistenti alla castrazione (CRPC). Inoltre, è interessante notare come in alcuni pazienti che sviluppano resistenza, in particolare nei pazienti in terapia con flutamide, la sospensione del farmaco determina una riduzione paradossa dei valori di PSA: questo potrebbe essere attribuito a una mutazione dell’AR, che farebbe comportare il farmaco come un agonista piuttosto che come antagonista. Pertanto, potrebbe essere utile in alcuni pazienti che presentano un rialzo del PSA in terapia ormonale adiuvante, tentare la sospensione del farmaco e ricontrollare a breve il PSA (29).
La terapia endocrina del carcinoma della prostata è indicata (29):
- come terapia adiuvante nei pazienti a rischio basso (T1-T2a, Gleason ≤ 6, PSA 10 anni in associazione o meno alla radioterapia;
- nei pazienti a rischio intermedio (T2b-T2c, G > 6 o PSA 10-20 ng/mL) con metastasi linfonodali in associazione o meno alla radioterapia;
- nei pazienti ad alto rischio (T3a o G 8-10 o PSA > 20 ng/mL), a rischio molto alto (T3b-T4) e metastatici (M1), in associazione alla radioterapia.
Agonisti e antagonisti del GnRH
Sono utilizzati in alternativa all’orchiectomia bilaterale per ottenere un’efficace deprivazione di androgeni determinando una castrazione farmacologica.
I farmaci che hanno indicazione per il carcinoma della prostata sono goserelin, triptorelin e leuprorelin e il più nuovo degarelix, che blocca invece l’interazione del GnRH con il suo recettore a livello ipofisario, inducendo una più rapida castrazione ed evitando l’iniziale aumento del testosterone associato all’inizio della terapia con gli analoghi (tuttavia necessita di somministrazioni mensili ed è associato a frequenti reazioni nel sito di inoculazione) (30). La castrazione ottenuta con questi farmaci è equivalente all’orchiectomia e non sembrerebbero esserci grandi differenze tra i vari farmaci (31). Inoltre, questi farmaci in associazione alla radioterapia aumentano significativamente la DFS rispetto alla sola radioterapia (32).
Tra gli effetti secondari più importanti sono la ginecomastia e i sintomi da ipogonadismo.
Ketoconazolo
Blocca la sintesi di androgeni, ma anche di corticosteroidi e mineralcorticoidi, per cui andrebbe somministrato in associazione a terapia corticosteroidea sostitutiva. Al momento non è utilizzato e la sua utilità è controversa.
Abiraterone acetato
È un potente inibitore della sintesi degli androgeni, agendo come potente e selettivo inibitore dell’enzima CYP17 (17α-idrossilasi/C17,20-liasi). Uno studio di fase III in uomini con CRPC avanzato, trattati in precedenza con chemioterapia tradizionale, ha evidenziato un aumento della sopravvivenza mediana rispetto al placebo. Il farmaco è commercializzato da aprile 2013 in Italia (con il nome di Zytiga)(33). È il primo farmaco endocrino a dimostrare un’efficacia nei CRPC (si parla infatti di terapia ormonale secondaria). Anche uno studio su pazienti CPRC naïve da chemioterapia tradizionale ha dimostrato una migliore PFS radiologica e un trend di aumento dell’OS rispetto al placebo (34).
Il farmaco determina un aumento di ACTH, che potrebbe determinare un eccesso di mineralcorticoidi, dal momento che quella è l’unica via della steroidogenesi non bloccata; questo potrebbe determinare un quadro di iperaldosteronismo primitivo, controllabile con l’aggiunta di prednisone 5 mg/die (30).
Orteronel (TAK-700)
È un inibitore selettivo di CYP17, con maggiore selettività per l’attività 17,20-liasica piuttosto che per la 17-idrossilasi. Evita la co-somministrazione del corticosteroide importante nella terapia con abiraterone (anche se a dosaggi elevati l’orteronel può a sua volta sopprimere la sintesi di corticosteroidi). I primi dati di fase I e II hanno evidenziato una buona efficacia del farmaco, sia in termini di riduzione dei livelli di testosterone e di DHEAS, sia di riduzione del PSA.
Galeterone (TOK-001)
Oltre a essere un inibitore di CYP17, blocca competitivamente il legame con gli androgeni e down-regola in vitro l’espressione dell’AR. Il farmaco è ancora in fase sperimentale.
Anti-androgeni non steroidei
Sono in uso da molti anni, i principali sono bicalutamide e flutamide. Si legano al dominio legante il ligando dell’AR (LBD). Sono efficaci nell’indurre il blocco androgenico, in associazione agli agonisti del GnRH. La loro somministrazione andrebbe iniziata almeno 3 giorni prima dell’inizio degli agonisti del GnRH, per attenuarne l’effetto di rialzo transitorio del testosterone.
Tra gli effetti secondari più importanti, la dolenzia mammaria e la ginecomastia, il calo della libido e la disfunzione erettile.
Diversi studi hanno valutato l’efficacia di questi farmaci in monoterapia o associati alla castrazione farmacologica: uno studio ha confrontato bicalutamide 150 mg vs la castrazione farmacologica o chirurgica ed ha evidenziato un’equivalenza dei due approcci nei pazienti con tumore localmente avanzato, ma non nei metastatici, in cui l’associazione determina risultati migliori (35). Una metanalisi ha riportato un maggior rischio di recidiva nei pazienti trattati con anti-androgeni in monoterapia piuttosto che in associazione con la castrazione farmacologica (36). Quindi le linee guida suggeriscono di effettuare il blocco androgenico combinato, mentre la monoterapia con gli anti-androgeni può essere discussa per la minore tossicità e per il minor effetto “ipogonadizzante” rispetto al blocco combinato con gli analoghi del GnRH.
Bloccanti le vie di trasduzione dell’AR
L’enzalutamide (MDV3100) è un anti-androgeno non steroideo con altissima affinità per AR, che, oltre ad inibire il recettore stesso, ne blocca la traslocazione nel nucleo. Negli studi di fase I e II nei pazienti con CRPC ha determinato una riduzione del PSA > 50% e una risposta radiologica nel 22% dei pazienti; gli studi di fase III vs placebo hanno evidenziato una maggiore sopravvivenza mediana nei pazienti trattati, con una riduzione del 37% del rischio decesso (37-39).
ARN-509 è un analogo dell’enzalutamide ma più potente: inibisce specificamente la crescita delle cellule che esprimono l’AR; impedisce anch’esso la traslocazione nucleare del recettore. È ancora in fase I e II di sperimentazione.
EPI-001
A differenza della maggior parte degli anti-androgeni che si legano al LBD, questo nuovo farmaco si lega al dominio N-terminale (NTD) e al dominio di legame del DNA (DBD), inibendo il legame del recettore attivato o transattivato al DNA, impedendo la trascrizione genica indipendentemente dal legame del ligando al recettore, by-passando quindi alcuni dei principali meccanismi di sviluppo di CRPC (30).
Il carcinoma dell’endometrio si suddivide in due tipi:
- endometrioide tipo 1 (90% dei casi, a basso grado di malignità) associato a fattori di rischio ormonali ben riconosciuti;
- endometrioide tipo 2 (ad alto grado di malignità).
Tutte le condizioni che causano un’eccessiva esposizione agli estrogeni endogeni o esogeni determinano uno stimolo proliferativo sull’endometrio, che a lungo termine può indurre iperplasia e quindi neoplasia endometriale. Il progesterone, invece, a differenza dell’effetto proliferativo sulla mammella, ha un effetto anti-mitogeno sull’endometrio e riduce la proliferazione cellulare.
Tutte le condizioni che causano iperestrogenismo in associazione ad anovularietà causano una stimolazione endometriale, in assenza di una controparte progestinica. Un esempio è l’obesità. Tumori ovarici e surrenalici che producono estrogeni rappresentano condizioni di rischio per lo sviluppo di carcinoma dell’endometrio. Altri fattori sono la nulliparità, la menopausa tardiva, la familiarità di primo grado, la terapia ormonale sostitutiva post-menopausa e la terapia con SERM nel carcinoma della mammella (40).
Una revisione Cochrane ha analizzato quale potesse essere il ruolo della terapia ormonale sostitutiva post-menopausa nell’insorgenza dell’iperplasia dell’endometrio: lo studio ha evidenziato un rischio aumentato nelle pazienti in terapia con soli estrogeni, mentre il rischio si azzerava nelle pazienti trattate anche con progestinici (41). Per l’uso di soli estrogeni il rischio di iperplasia e di carcinoma aumenta con il tempo: 1.30 a 2 anni, 4.50 da 5-10 anni. Per quanto riguarda l’uso di progestinici in schemi sequenziali, l’ideale è non somministrarli per meno di 10 giorni al mese, perché questo incrementa il rischio di cancro dell’endometrio (42). Per quanto riguarda il rischio di carcinoma endometriale nelle pazienti in terapia con SERM, alcuni studi non hanno evidenziato un rischio significativo con l’uso del raloxifene a differenza del tamoxifene (43).
Nell’ambito della terapia del carcinoma dell’endometrio, la terapia endocrina trova un posto limitato rispetto agli altri tumori ormono-sensibili già discussi: i progestinici sono in grado di ridurre l’espressione degli ER, riducono la trascrizione di geni coinvolti nella crescita cellulare ER-dipendente, attivano il gene onco-soppressore p21. Attualmente, sono disponibili progestinici di quarta generazione (dienogest), che non presentano attività androgenica, riducendo quindi tutti gli effetti secondari associati all’attività androgenica dei progestinici di prima, seconda e terza generazione. Inoltre, questi sembrano avere proprietà anti-angiogenetiche (44).
Anche per il carcinoma dell’endometrio, come per la mammella, sembrano promettenti farmaci di nuova generazione, come gli inibitori di mTOR (everolimus), gli inibitori dell’istone-deacetilasi (HDACi) e un ruolo interessante è svolto sempre dalla metformina, che sembra indurre l’espressione di PR sulle cellule endometriali, attraverso l’inibizione di IGF-1 e 2; promettente potrebbe quindi essere il suo uso in associazione a un progestinico.
BIBLIOGRAFIA
- Southam CM. The complex etiology of cancer. Cancer Res 1963, 23: 1105-15.
- Moore DH, et al. Breast carcinoma etiological factors. Adv Cancer Res 1983, 40: 189-253.
- Saville B, et al. Ligand-, cell-, and estrogen receptor subtype (alpha/beta)-dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem 2000, 275: 5379–87.
- Helguero LA. Estrogen receptors alfa (ERa) and beta (ERb) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 2005, 24: 6605–16.
- Musgrove EA, et al. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011, 11: 558–72.
- Ström A, et al. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA 2004, 101: 1566–71.
- Taylor CW, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998, 16: 994-9.
- Goodwin PJ, et al. Risk of menopause during the first year after breast cancer diagnosis. J Clin Oncol 1999, 17: 2365-70.
- Goel S, et al. LHRH agonists for adjuvant therapy of early breast cancer in premenopausal women. Cochrane Database Syst Rev 2009: CD004562.
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011, 378: 771-84.
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 1998, 351: 1451-67.
- Clarke R, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22: 7316–39.
- Renoir JM, et al. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol 2013, 85: 449–465.
- Howell A, et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years' adjuvant treatment for breast cancer. Lancet 2005, 365: 60-2.
- Coates AS, et al. Five years of letrozole compared with tamoxifen as initial adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer: update of study BIG 1-98. J Clin Oncol 2007, 25: 486-92.
- www.agenziafarmaco.gov.it
- Howell A, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol 2004, 22: 1605-13.
- Ménard S, et al. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22: 6570–8.
- Eiermann W, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: A randomized double-blind multicenter study. Ann Oncol 2001, 12: 1527-32.
- Patterson RE, et al. Metabolism and breast cancer risk: frontiers in research and practice. J Acad Nutr Diet 2013, 113: 288-96.
- www.clinicaltrials.gov
- Bodmer M, et al. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care 2010, 33: 1304-8.
- Wang Y, et al. Small molecule inhibition of the steroid receptor coactivators: SRC-3 and SRC-1. Mol Endocrinol 2011, 25: 2041–53.
- Urbinati G, et al. Liposomes loaded with histone deacetylase inhibitors for breast cancer therapy. Int J Pharm 2010, 397: 184–93.
- Zhou Q, et al. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther 2007, 6: 64–9.
- Huggins C, Hodges CV. Studies on prostatic cancer. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin 1972, 22: 232–40.
- Halabi S, et al. Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol 2003, 21: 1232–7.
- Amaral TM, et al. Castration-resistant prostate cancer: mechanisms, targets, and treatment. Prostate Cancer 2012, 2012: 327253.
- NCCN Clinical Practice Guidelines for Prostate Cancer 2013, www.nccn.org.
- Friedlander TW, Ryan CJ. Targeting the androgen receptor. Urol Clin North Am 2012, 39: 453-64.
- Seidenfeld J, et al. Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. Ann Intern Med 2000, 132: 566-77.
- Pilepich MV, et al. Androgen suppression adjuvant to definitive radiotherapy in prostate carcinoma--long-term results of phase III RTOG 85-31. Int J Radiat Oncol Biol Phys 2005, 61: 1285-90.
- De Bono JS, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 2008, 14: 6302–9.
- Ryan CJ. Interim analysis (IA) results of COU-AA-302, a randomized, Phase III study of abiraterone acetate (AA) in chemotherapy-naive patients (pts) with metastatic castration-resistant prostate cancer (mCRPC), American Society of Clinical Oncology Annual Meeting. Chicago, June 2, 2012.
- Iversen P, et al. Bicalutamide monotherapy compared with castration in patients with nonmetastatic locally advanced prostate cancer: 6.3 years of follow up. J Urol 2000, 164: 1579-82.
- Samson DJ, et al. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer 2002, 95: 361-76.
- Scher HI, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet 2010, 375: 1437-46.
- Scher HI, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012, 367: 1187-97.
- Tsao CK, et al. Targeting the androgen receptor signalling axis in castration-resistant prostate cancer (CRPC). BJU Int 2012, 110: 1580-8.
- Amant F, et al. Endometrial cancer. Lancet 2005, 366: 491-505.
- Furness S, et al. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia. Cochrane Database Syst Rev 2012: CD000402.
- Pike MC, et al. Estrogen-progestin replacement therapy and endometrial cancer. J Natl Cancer Inst 1997, 89: 1110-6.
- Martino S, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 2004, 96: 1751-61.
- Umene K, et al. New candidate therapeutic agents for endometrial cancer: potential for clinical practice (review). Oncol Rep 2013, 29: 855-60.
Il carcinoma della mammella ormono-sensibile
Gabriele Luppi & Elena Barbieri
Oncologia Medica, Azienda Ospedaliero-Universitaria di Modena
La neoplasia della mammella è quella con maggior frequenza e la seconda causa di morte per tumore nel sesso femminile [1].
Circa il 70% dei tumori della mammella esprime positività per il recettore per gli estrogeni e/o per il progesterone. Il recettore per gli estrogeni è il principale fattore di trascrizione che guida l’oncogenesi nelle neoplasie mammarie a recettori ormonali positive ed HER2 negative ed inoltre è un fattore predittivo di risposta alle terapie con anti-estrogeni.
Storicamente, il trattamento ormonale del carcinoma della mammella rappresenta uno dei primi esempi di terapia personalizzata in oncologia da quando, più di un secolo fa, Sir George Beatson osservò tre casi di carcinoma della mammella in fase avanzata che regredirono dopo ovariectomia [2], aprendo la strada all’utilizzo della manipolazione ormonale nel carcinoma della mammella.
Ad oggi la quantità di conoscenze acquisite ha permesso di identificare sottogruppi che differiscono dal punto di vista prognostico e predittivo di risposta alle terapie, rendendo il carcinoma della mammella l’esempio più tipico della sfida di personalizzare la terapia sulla base di caratteristiche peculiari della patologia e delle pazienti.
Perou et al. [3] individuarono profili molecolari che caratterizzano i carcinomi della mammella e ne permettono la classificazione in base a profili di espressione genica. In base a questa classificazione, i carcinomi mammari che esprimono i recettori ormonali si possono suddividere ulteriormente in due gruppi, Luminal A e Luminal B, che differiscono tra loro dal punto di vista genico e molecolare, con ricadute sul piano prognostico e predittivo [4]. Oltre a questi gruppi, ne sono stati identificati ulteriori due che tipicamente non esprimono i recettori ormonali e che corrispondono ai sottogruppi basal-like e HER2-enriched.
La tecnica dei micro-arrays, e di conseguenza la suddivisione in sottogruppi in base ai profili di espressione genica, non è ancora entrata nella pratica clinica di tutti i giorni e pertanto si è cercato di identificare tramite immunoistochimica caratteristiche proprie dei vari sottogruppi molecolari.
I tumori Luminal A sono caratterizzati dalla forte espressione dei recettori ormonali e da un basso indice di proliferazione (Ki-67 < 14%). Sono generalmente ad ottima prognosi, traggono scarso beneficio dall’aggiunta della chemioterapia alla manipolazione ormonale, che rimane il caposaldo del trattamento di queste neoplasie, e tipicamente hanno un andamento indolente, presentando recidive tardive (spesso ben oltre i 5-10 anni dalla diagnosi) e principalmente a livello osseo.
I tumori Luminal B presentano espressione meno marcata dei recettori ormonali e sovente mancano di espressione del recettore per il progesterone, hanno un’elevata velocità di proliferazione o la concomitante espressione della proteina HER2. Dal punto di vista clinico e sulla base delle sole caratteristiche biologiche, sono guidati nella crescita anche da altri fattori e non solo dalla via estrogenica, hanno un comportamento clinico più aggressivo, beneficiano della chemioterapia e tendono a recidivare più precocemente e soprattutto a livello viscerale [5].
I farmaci ad azione ormonale utilizzati nel trattamento del carcinoma della mammella possono essere classificati in base al meccanismo d’azione. I modulatori selettivi del recettore per gli estrogeni (SERMs), come il tamoxifene ed il raloxifene, sono agonisti/antagonisti del recettore per gli estrogeni. A livello del tessuto ghiandolare mammario, il tamoxifene agisce come antagonista, col risultato di interrompere la trascrizione dei geni la cui espressione è regolata dagli estrogeni e quindi il loro effetto di stimolo alla proliferazione. Il fulvestrant agisce anch’esso a livello del recettore per gli estrogeni, ma, a differenza del tamoxifene, è dotato solamente di attività antagonista, poiché porta alla degradazione del recettore estrogenico causandone la perdita.
Per indurre una deprivazione estrogenica le strategie in uso includono:
- nelle donne in pre-menopausa la soppressione della funzionalità ovarica (chirurgica, radioterapica o farmacologica);
- nelle donne in post-menopausa l’utilizzo di inibitori dell’aromatasi.
La scelta del tipo di terapia ormonale si basa su una serie di fattori, che includono lo stato menopausale ed il profilo di tossicità atteso.
Negli ultimi tempi sta emergendo l’utilizzo della terapia ormonale anche in fase pre-operatoria per le pazienti con malattia a recettori ormonali positivi/HER2 negativa. Questo tipo di approccio, finora riservato quasi esclusivamente alle pazienti anziane, sta assumendo sempre maggiore interesse nel campo della ricerca e parte dall’osservazione che in fase adiuvante, alle pazienti con neoplasia a recettori ormonali positivi ed HER2 negativa viene offerto generalmente il solo trattamento ormonale.
L’opportunità offerta dalla terapia sistemica primaria di valutare i biomarcatori prima e dopo il trattamento permette di identificare precocemente le pazienti che più probabilmente trarranno beneficio dal trattamento ormonale. Ad esempio è stato generato un punteggio prognostico (preoperative prognostic index, PEPI score) che si basa sulla dimensione tumorale, sul coinvolgimento linfonodale, sull’espressione del Ki-67 e del recettore per gli estrogeni secondo Allred dopo la terapia ormonale sistemica primaria [6].
Nonostante quanto detto in precedenza, alcune pazienti mostrano comunque una resistenza primaria o acquisita alla terapia ormonale, suggerendo di fatto il ruolo del cross-talk tra i recettori ormonali e altre vie molecolari intra-cellulari che portano a resistenza nei confronti della terapia endocrina. Bloccando entrambe le vie coinvolte, combinando la terapia ormonale con terapie a bersaglio molecolare, si può potenziare l’attività anti-tumorale e ristabilire la sensibilità all’ormono-terapia.
Ad esempio, in uno studio randomizzato di terapia pre-operatoria in donne con malattia a recettori ormonali positive e con espressione di EGFR, la monoterapia con l’inibitore tirosin-chinasico gefitinib ha comportato una riduzione delle dimensioni tumorali nel 54% delle pazienti, mentre la combinazione gefitinib + anastrozolo vs. gefitinib da solo ha permesso di inibire maggiormente la proliferazione tumorale [7].
Esperienze simili sono state condotte sia in fase metastatica, sia in fase neoadiuvante, aggiungendo alla terapia ormonale con letrozolo, lapatinib, un duplice inibitore di EGFR/HER2 ad attività tirosin-chinasica [8,9].
E’ stato dimostrato che la via di segnalazione intra-cellulare legata a PI3K/Akt/mTOR è legata alla resistenza alla terapia endocrina [10]. L’inibitore di mTOR everolimus ha aumentato in maniera significativa l’efficacia di letrozolo nel setting neoadiuvante: il tasso di risposta clinica nel braccio di combinazione letrozolo/everolimus è risultato maggiore rispetto al solo letrozolo (68.1% vs. 59.1%). Inoltre, dopo 15 giorni di trattamento si è osservata una riduzione dell’espressione di Ki-67 nel 57% di pazienti trattate con l’aggiunta di everolimus rispetto al 30% del braccio con placebo [11].
Nell’ambito della malattia metastatica recenti studi hanno dimostrato il beneficio dell’aggiunta di everolimus al trattamento ormonale in donne in post-menopausa progredite o recidivate dopo il trattamento con un’inibitore dell’aromatasi non steroideo (anastrozolo o letrozolo). Lo studio BOLERO-2 ha riportato un incremento di 2.4 volte della progression-free survival mediana con la combinazione di everolimus + exemestane vs. exemestane da solo [12]. Lo studio TAMRAD ha dimostrato un miglioramento in termini di beneficio clinico, tempo a progressione e sopravvivenza globale nelle pazienti trattate con everolimus + tamoxifene vs. tamoxifene da solo [13].
A più di un secolo dalla scoperta del suo ruolo nel trattamento del carcinoma mammario, la terapia endocrina ottimale per il carcinoma della mammella è in continua evoluzione. Le ricerche volte a caratterizzare in maniera sempre più precisa i carcinomi mammari ormono-sensibili perseguono l’obiettivo di personalizzare le terapie in base alle caratteristiche della malattia fino a livello molecolare, ed a quelle individuali di ogni paziente.
Bibliografia
- Siegel R, Naishadham D, Jemal A. Cancer Statistics 2012. CA Cancer J Clin 2012, 62: 10-29.
- Beatson GW. On the treatment of inoperable cases of carcinoma of the mamma: suggestion of a new method of treatment with illustrative cases. The Lancet 1896, 2: 162-5.
- Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumor. Nature 2000, 406: 747-52.
- Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009, 101: 736-50.
- Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010, 28: 3271-7.
- Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008, 100: 1380-8.
- Polychronis A, Sinnett HD, Hadjiminas D, et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial. Lancet Oncol 2005, 383-91.
- Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with Letrozole and placebo as first-line therapy for postmenopausal hormone receptor positive metastatic breast cancer. J Clin Oncol 2009, 27: 5538-46.
- Conte PF, Guarneri V, Generali DG, et al. Double-blind, placebo-controlled, multicentric randomized phase IIb neoadjuvant study of letrozole-lapatinib in postmenopausal HER2-negative, hormone receptor-positive operable breast cancer. J Clin Oncol 2011, 29 suppl: abstr 550.
- Tokunaga E, Kimura Y, Mashino K, et al. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer 2006, 13: 137-44.
- Baselga J, Semiglazov V, van Dam P, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol 2009, 27: 2630-7.
- Baselga J, Campone M, Piccart M, et al. Everolimus for postmenopausal hormone receptor-positive advanced breast cancer. N Engl J Med 2012, 366: 520-9.
- Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol 2012, 30: 2718-24.
Carcinoma endometriale
Enrico Vizza, Benito Chiofalo, Valentina Bruno
UOC Ginecologia Oncologica, Istituto Nazionale Tumori "Regina Elena-IFO"
(aggiornato al 18 dicembre 2020)
EPIDEMIOLOGIA E FATTORI DI RISCHIO
Il carcinoma dell’endometrio è una neoplasia tipica dell’età post-menopausale: in oltre il 90% dei casi la diagnosi avviene dopo i 50 anni, con età media di 63 anni.
Nei Paesi economicamente più sviluppati rappresenta la neoplasia ginecologica più frequente, in Italia è il 5° tumore maligno più frequente nelle donne.
I principali fattori di rischio possono essere suddivisi in tre categorie.
Fattori ambientali: l’obesità e un regime alimentare ricco di grassi animali sono elementi comuni nell’anamnesi patologica delle pazienti affette da carcinoma dell’endometrio, essendo responsabili di un’aumentata produzione di estrogeni a seguito dell’aromatizzazione degli androgeni di provenienza surrenalica (1). In menopausa, quando la produzione estrogenica ovarica è praticamente nulla, il deidroepiandrosterone surrenalico viene convertito dagli adipociti in estrone, che ha un’azione stimolante sulle cellule endometriali. Altri fattori di rischio sono l’ipertensione arteriosa, il diabete mellito e le epatopatie croniche. Al contrario, attività fisica e diete ricche di fibre sembrano fattori protettivi (2).
Fattori ormonali: la presenza di un’attività estrogenica, sia endogena che esogena, non contro-bilanciata da una corretta quantità di progesterone sembra una delle principali cause di neoplasia endometriale. L’allungamento della finestra estrogenica (menarca precoce, menopausa tardiva e nulliparità) aumenta il rischio di sviluppare questa neoplasia. Una condizione clinica predisponente sembra la policistosi ovarica, soprattutto in un quadro di sindrome metabolica, a causa di cicli anovulatori, iperestrogenismo relativo e iperandrogenismo. Anche i tumori estrogeno-secernenti, come i tumori ovarici dei cordoni sessuali (cellule della granulosa, ecc), rappresentano fattori di rischio. Molti studi hanno messo in evidenza come il rischio di tumore dell’endometrio aumenti da 2 a 10 volte a seguito di terapia ormonale sostitutiva estrogenica in climaterio, senza l’associazione di progestinici (3-4). Le attuali linee guida per il trattamento dei disturbi associati alla menopausa raccomandano, quindi, la terapia estrogenica associata a progestinici nelle donne non sottoposte a pregressa isterectomia, mentre l’estrogeno può essere impiegato da solo nelle donne precedentemente isterectomizzate. Il rischio associato al trattamento con tamoxifene, agonista parziale degli estrogeni, è dibattuto in letteratura: secondo alcuni autori il pregresso carcinoma mammario è di per sé un fattore di rischio per la neoplasia endometriale, indipendentemente dal tipo di terapia ormonale utilizzata (5). Numerose evidenze scientifiche hanno infine mostrato che l’utilizzo di combinazioni estro-progestiniche in età fertile rappresenta un fattore protettivo, determinando una riduzione del rischio di tumore dell’endometrio di circa il 50%, con un effetto protettivo che si prolunga per più di 20 anni dopo la sospensione.
Fattori ereditari: esistono infine forme ereditarie di carcinoma endometriale. Nella sindrome di Lynch di tipo II, autosomica dominante, la predisposizione allo sviluppo di neoplasie è dovuta all’alterazione di geni che appartengono al sistema del mismatch repair (MMR). Oltre a una maggiore predisposizione genetica al solo carcinoma endometriale, è possibile una predisposizione familiare a sviluppare tumori maligni in diversi altri organi (6): il rischio è del 40-80% per tumore del colon, 40-60% per carcinoma dell’endometrio e 10-12% per carcinoma ovarico. È stato proposto che l’insorgenza in una donna giovane di una neoplasia endometriale possa essere considerato come evento sentinella della sindrome di Lynch.
ISTOPATOLOGIA
I tumori endometriali derivano dalle cellule ghiandolari di derivazione mulleriana. Nel 1983, Bokhman formulò l’ipotesi dell’esistenza di due varianti di carcinoma endometriale con diversa patogenesi:
- tipo I: comprende l’adenocarcinoma endometrioide, che rappresenta l’80% dei casi, è estrogeno-dipendente, insorge frequentemente su iperplasia endometriale e ha prognosi generalmente favorevole;
- tipo II: comprende l’adenocarcinoma sieroso-papillifero (< 10% dei casi) e l’adenocarcinoma a cellule chiare (2-4% dei casi). Non sono estrogeno-dipendenti, insorgono solitamente su endometrio atrofico, colpiscono soggetti più anziani e hanno elevata aggressività biologica, con prognosi sfavorevole (7).
Nel 2013 la caratterizzazione genomica di 373 campioni di carcinoma endometriale ha consentito una nuova classificazione biomolecolare più complessa, successivamente sviluppata nel tempo, con importanti risvolti diagnostici e terapeutici (8).
Dal punto di vista anatomo-patologico il carcinoma endometriale si presenta con differenti istotipi.
- Adenocarcinoma endometrioide: è l’istotipo più frequente (80%) ed è estrogeno-correlato. È generalmente puro, tuttavia può essere associato alla presenza di un carcinoma non endometrioide e la proporzione delle componenti influenza la diffusione della malattia e la prognosi.
- Adenocarcinoma sieroso-papillare: rappresenta circa il 5-10% dei carcinomi dell’endometrio e va sospettato nelle donne in una fascia di età di 10 anni superiore a quella dell’adenocarcinoma endometrioide. Spesso di alto grado, presenta solitamente un’infiltrazione miometriale profonda e particolare tropismo per i linfonodi. Diffonde rapidamente alla cavità peritoneale ed è pertanto a prognosi infausta (9).
- Adenocarcinoma a cellule chiare: è molto più raro dell’adenocarcinoma sieroso, rappresentando circa l’1% dei carcinomi dell’endometrio, è tipico dell’età avanzata e ha prognosi sfavorevole. Il carcinoma a cellule chiare tipico, per definizione, ha gli stessi caratteri istologici architetturali del carcinoma a cellule chiare di altre sedi genitali (10).
- Adenocarcinoma mucinoso: rappresenta circa l’1% dei tumori endometriali e va distinto dall’adenocarcinoma endo-cervicale primitivo, da cui si differenzia per l’abbondante presenza di mucina.
- Carcinoma squamoso: rappresenta meno dell’1% dei tumori endometriali e va distinto dall’adenocarcinoma a differenziazione squamosa (per l’assenza di differenziazione ghiandolare) e dai carcinomi a cellule squamose della cervice uterina diffusi a livello endometriale.
- Tumori misti: rappresentano meno dell’1% dei tumori endometriali e le diverse componenti superano ciascuna il 10% del totale.
- Carcinoma indifferenziato: sono tumori composti da masse solide di cellule indifferenziate. Possono essere associati ad adenocarcinoma endometrioide o rappresentare l’unica componente documentata nel tumore. Più rari sono i carcinomi indifferenziati a piccole cellule, simili a quelli di altri organi, che mostrano differenziazione neuroendocrina con positività per cromogranina, sinaptofisina e altri tipici marcatori.
- Carcinosarcoma: si compone di elementi sia sarcomatosi che carcinomatosi.
- Sarcoma endometriale stromale: quelli di alto grado sono molto aggressivi, con una sopravvivenza a 5 anni che va da 0 a 55%.
Oltre all’istotipo, i principali fattori di rischio per la comparsa di metastasi e quindi di un esito sfavorevole sono: il grado di differenziazione, il pattern molecolare, il grado di interessamento miometriale, l’invasione degli spazi linfatici e vascolari, l’interessamento cervicale e la diffusione extra-uterina della neoplasia.
Le sedi di più frequente localizzazione secondaria sono polmone, fegato, osso (prevalentemente vertebre) ed encefalo.
CLINICA E DIAGNOSI
L’esordio clinico del carcinoma endometriale è rappresentato, nella grande maggioranza dei casi, da un sanguinamento vaginale anomalo: perdita ematica vaginale in menopausa o sanguinamento inatteso, rispetto al flusso mestruale normale, in età fertile. Raramente invece la neoplasia decorre in maniera asintomatica e la diagnosi viene posta in modo accidentale.
In caso di sanguinamenti uterini anomali, il primo esame da eseguire è l’ecografia trans-vaginale per lo studio dell’utero e degli annessi, al fine di valutare lo spessore endometriale (sospetto se > 4 mm in menopausa) ed eventuali neoformazioni intra-uterine (11). L’aggiunta del Doppler (color e power) rende possibile studiare con accuratezza la perfusione ematica dell’endometrio e dei processi espansivi a suo carico (12). L’ecografia trans-vaginale eseguita da operatori esperti riesce a valutare con alta sensibilità l’infiltrazione miometriale, cervicale ed extra-uterina.
La diagnosi di carcinoma endometriale si basa essenzialmente sulla valutazione istologica del tessuto endometriale ottenuto attraverso prelievi bioptici.
Il gold standard per lo studio dell’endometrio è l’isteroscopia diagnostica, che consente la visualizzazione del canale cervicale e della cavità uterina, permettendo allo stesso tempo l’esecuzione di biopsie mirate (13-14).
Per una stadiazione clinica, la risonanza magnetica (RM) pelvica con mezzo di contrasto è in grado di valutare accuratamente il grado di infiltrazione miometriale (sensibilità 87%), l’infiltrazione dello stroma cervicale (sensibilità 80%) e delle pareti vaginali. Permette inoltre di valutare l’infiltrazione del tessuto parametriale e la presenza di linfadenomegalie pelviche o lombo-aortiche, con accuratezza del 76% (15).
La tomografia computerizzata (TC) presenta scarsa accuratezza diagnostica rispetto alla RM nella valutazione dell’infiltrazione miometriale, ma risulta utile nella valutazione delle eventuali sedi extra-uterine di malattia.
La tomografia ad emissione di positroni (PET) con 18F-fluorodeossiglucosio può essere impiegata sia per la rilevazione di metastasi linfonodali (N) durante la valutazione pre-chirurgica che nel follow-up post-operatorio (16).
La scintigrafia ossea è limitata invece ai casi clinicamente sospetti per localizzazione ossea.
Ulteriori indagini (cistoscopia, colonscopia, ecc) sono eseguite su indicazione clinica.
La stadiazione del carcinoma endometriale è anatomo-chirurgica. Quella attualmente utilizzata è la stadiazione FIGO 2009 (tabella), che è fondamentale per la decisione dell’iter terapeutico.
| Stadiazione del carcinoma endometriale | ||
| Stadio | Caratteristiche | |
| I | Tumore limitato al corpo del’utero | |
| Ia | Nessuna infiltrazione o < metà del miometrio | |
| Ib | Infiltrazione > metà del miometrio | |
| II | Tumore esteso allo stroma cervicale ma non fuori dell’utero | |
| III | Estensione locale o regionale | |
| IIIa | Alla sierosa uterina o alle ovaie | |
| IIIb | Alla vagina o alla regione parametriale | |
| IIIc | A linfonodi pelvici (IIIc1) o lombo-aortici (IIIc2, indipendentemente dai linfonodi pelvici) | |
| IV | Estensione alla mucosa vescicale o intestinale o metastasi a distanza | |
| IVa | Alla mucosa vescicale o intestinale | |
| IVb | Metastasi a distanza | |
TRATTAMENTO
Trattamento chirurgico
L’approccio chirurgico standard, nei primi stadi di malattia, è rappresentato dall’isterectomia totale extra-fasciale e dall’annessiectomia bilaterale, con o senza linfadenectomia pelvica e/o lombo-aortica.
Le possibili modalità di accesso chirurgico sono la via laparotomica, laparoscopica, robotica e vaginale. Numerosi studi hanno confrontato l’approccio laparotomico con quello laparoscopico, dimostrando che, a parità di sopravvivenza globale a 5 anni in entrambi i gruppi, la laparoscopia era associata a numerosi benefici in termini di durata dell’ospedalizzazione, complicanze, controllo del dolore e qualità di vita (17). Per quanto riguarda l’approccio mini-invasivo robotico, è riportato un numero di complicanze significativamente minore rispetto alla via laparotomica; inoltre, nelle pazienti obese, la chirurgia robotica sembra superiore anche all’approccio laparoscopico, associandosi a minore perdita ematica, minor tasso di conversione laparotomica, ridotti tempi operatori e durata della degenza (18-19).
Il ruolo della linfadenectomia in questo gruppo di pazienti è tuttora oggetto di discussione. Da uno studio italiano randomizzato (20), condotto su 514 pazienti con carcinoma dell’endometrio in stadio I, è emerso che la linfadenectomia non era associata a tassi maggiori di sopravvivenza libera da malattia (PFS). Un’ulteriore conferma di questi dati è derivata dallo studio ASTEC, in cui la linfadenectomia pelvica non ha dimostrato un beneficio in termini di sopravvivenza globale (OS) e intervallo di tempo libero da malattia (21). Il limite di questi studi è tuttavia rappresentato dall’inclusione di una popolazione in cui il rischio di presentare metastasi linfonodali era troppo basso per evidenziare un effetto positivo della linfadenectomia sulla sopravvivenza. Rimane pertanto irrisolto il ruolo della linfadenectomia nelle pazienti ad alto rischio. Alcuni autori suggeriscono di considerare una stadiazione chirurgica completa nelle pazienti con rischio intermedio-alto (stadio IA G3 e IB sec. FIGO 2009) per identificare quelle pazienti che richiedono un trattamento adiuvante post-operatorio. Lo studio SEPAL, pubblicato nel 2010, ha invece valutato l’estensione della linfadenectomia, confrontando la linfadenectomia pelvica vs linfadenectomia pelvica e lombo-aortica (22). Dai dati emersi, le pazienti ad alto rischio hanno beneficiato di una linfadenectomia pelvica e lombo-aortica in termini di OS. Altri studi randomizzati sono in corso per valutare il ruolo della linfadenectomia, con lo scopo di chiarire quale sia l’iter terapeutico più efficace per le pazienti ad alto rischio.
La biopsia del linfonodo sentinella ha mostrato una buona performance diagnostica e rappresenta un buon compromesso tra l’esecuzione di una stadiazione chirurgica completa e l’omissione di una linfadenectomia sistematica. In particolare, la biopsia del linfonodo sentinella è in grado di ridurre la morbilità potenzialmente derivabile dall’esecuzione di una linfadenectomia sistematica, mentre il suo “ultrastaging”, ovvero la sezione microscopica in più fette per una diagnosi più accurata, consente l’individuazione di micro-metastasi linfonodali spesso non diagnosticate con l’esame istologico convenzionale (anche in pazienti considerate a basso rischio). Uno studio osservazionale multicentrico sul linfonodo sentinella ha riportato una sensibilità dell’84% e un valore predittivo negativo del 97% in donne con carcinoma endometriale in stadio I-II sec. FIGO (23). La maggior parte degli studi pubblicati su questo argomento ha previsto l’identificazione del linfonodo sentinella mediante iniezione cervicale di un tracciante come il verde indocianina.
Trattamento conservativo
Nelle giovani donne desiderose di prole, affette da carcinoma endometriale con caratteristiche prognostiche favorevoli, come istotipo endometrioide, ben differenziato, invasione miometriale minima/assente, può essere ipotizzato un trattamento conservativo che preveda la somministrazione di un progestinico orale (medrossiprogesterone o megestrolo acetato) o endo-uterino (spirale levonorgestrel-medicata), associato o meno alla somministrazione di analoghi del GnRH. Il trattamento farmacologico può essere preceduto o meno da chirurgia resettoscopica. Il trattamento conservativo è da considerarsi tuttavia temporaneo e finalizzato all’ottenimento della gravidanza, che deve essere fortemente incoraggiata a partire dal riscontro di una risposta tumorale completa. Il trattamento chirurgico standard è comunque raccomandato in caso di progressione di malattia, mancata risposta tumorale completa a 12 mesi o al termine della gravidanza.
Trattamenti complementari e adiuvanti
Nella malattia diffusa (stadio III e IV), alcuni autori suggeriscono di considerare il massimo sforzo chirurgico nelle pazienti con un buon performance status, data la scarsa risposta ai regimi chemioterapici. Qualora la chirurgia non sia fattibile a causa di contro-indicazioni mediche (5-10% delle pazienti), oltre al trattamento medico con carboplatino e paclitaxel, può essere preso in considerazione un trattamento radioterapico, con o senza brachiterapia, per il controllo locale di malattia.
Dopo la chirurgia, in base alla presenza dei diversi fattori prognostici (età, stadio della malattia, istologia, grading, profondità di infiltrazione miometriale, interessamento degli spazi vascolari, coinvolgimento linfonodale), le pazienti vengono suddivise in classi di rischio per decidere se effettuare eventuali trattamenti adiuvanti (24).
La radioterapia ha un ruolo importante nel trattamento del carcinoma endometriale. Può essere utilizzata come trattamento adiuvante (radioterapia esterna con o senza brachiterapia) nelle pazienti ad alto rischio dopo la chirurgia, come terapia esclusiva nelle pazienti in cui la procedura chirurgica è improponibile, nelle pazienti con recidiva di malattia in associazione alla chemioterapia o come terapia palliativa negli stadi avanzati (25).
Anche per quanto riguarda il trattamento chemioterapico esistono diversi regimi attuabili a seconda dell’istotipo, stadio e classi di rischio.
Terapia ormonale
Il carcinoma endometriale può esprimere sia recettori per estrogeni (ER) che per progesterone (PR) e quindi può rispondere a una terapia sistemica ormonale.
La terapia ormonale è raccomandata solo per l’istologia endometrioide, in quanto neoplasia ormono-sensibile, e include, primariamente, l’uso di agenti progestinici. Vengono anche utilizzati il tamoxifene e gli inibitori dell'aromatasi sebbene vi siano meno evidenze (26-27).
Nel trattamento adiuvante non è stato dimostrato, all’interno di sette studi randomizzati, alcun vantaggio in termini di riduzioni del rischio di morte per malattia nelle donne trattate con ormonoterapia con progestinico verso la sola osservazione. Tuttavia, nelle pazienti sottoposte a trattamento ormonale vi è stata un’incidenza del 5% di eventi trombo-embolici (28).
Il trattamento ormonale viene quindi indicato nel trattamento della recidiva di malattia/malattia metastatica dei carcinomi ad istologia endometrioide, in particolare nelle pazienti non candidabili a chemioterapia sistemica. I fattori predittivi di risposta al trattamento nella malattia metastatica includono: tumori ben differenziati, lunga PFS, localizzazione metastatiche extra-pelviche (soprattutto polmonari) ed espressione di PR e/o ER (26,27,29-31). Quest’ultimo dato ha portato, nella pratica clinica, analogamente alle recidive di carcinoma mammario, a considerare una nuova procedura bioptica per determinare l’assetto recettoriale ormonale nelle pazienti con recidive di malattia, soprattutto se con lungo intervallo di PFS (32).
Più recentemente l’ormonoterapia nell’endometrio ha trovato un ulteriore spazio di utilizzo nel trattamento conservativo, in pazienti giovani e desiderose di prole, del carcinoma endometrioide stadio IA G1. Una recente revisione ha riportato un tasso di risposte patologiche complete durature variabile tra 57 e 76% con progesterone somministrato per via orale o uso topico con la spirale medicata, a fronte di una percentuale di non responders e di pazienti che vanno in progressione durante trattamento del 12-25% e di una quota di pazienti variabile tra il 25 e il 30% che dopo una iniziale risposta sviluppa una recidiva di malattia a un tempo mediano di 19 mesi (27,33).
I progestinici sono impiegati nel trattamento del carcinoma endometriale dagli anni ’50, con tassi di risposta obiettiva variabili tra l’11-56% e una PFS compresa tra i 2.5 e i 14 mesi (34). Una revisione più recente ha mostrato tassi di risposta variabili dal 15 al 30% e sopravvivenze globali di 7-11 mesi nel trattamento con progestinici nelle pazienti con recidiva di malattia o malattia metastatica (35).
Gli effetti collaterali legati al trattamento sono di solito minori: edemi, aumento ponderale. Da considerare l’aumentato rischio di TVP nelle pazienti con anamnesi positiva per tale patologia o con patologie della coagulazione.
Alte dosi di megestrolo o medrossiprogesterone acetato (MAP) non aumentano l’efficacia terapeutica, come dimostrato in uno studio randomizzato (36), che ha comparato MAP 200 mg/die verso 1000 mg/die. Rimane quindi raccomandata la dose di MAP 200 mg/die o megestrolo 160 mg/die (26).
Nei diversi studi clinici le risposte sono per la maggior parte parziali e di breve durata, nonostante alcune pazienti abbiano risposte di lunga durata (36-37).
Allo scopo di contrastare la resistenza ai trattamenti ormonali, è stata testata la combinazione di temsirolimus (inibitore di mTOR) con o senza megestrolo e tamoxifene nel trattamento dei carcinomi dell’endometrio avanzati o recidivati. Lo studio non ha dimostrato alcun vantaggio della combinazione ormonoterapia e temsirolimus, andando invece a incrementare gli eventi avversi, tra cui la TVP (38). L’attività di temsirolimus è, invece, preservata nelle pazienti sottoposte a precedente chemioterapia sistemica.
Altre terapie ormonali hanno dimostrato attività nei carcinomi endometriali in stadio avanzato o recidivato.
Per quanto concerne gli inibitori dell'aromatasi, i dati derivati da studi retrospettivi e studi di fase II si sono dimostrati molto modesti e contrastanti (39,40). Questi farmaci possono essere considerati in prima o seconda linea nelle pazienti non candidabili a trattamento chirurgico o con recidiva di malattia e che non possono ricevere progestinici (41). L’efficacia del trattamento non sembra correlata con l’espressione dei recettori ormonali o con mutazioni di determinate vie di segnale molecolare (42). Un recente studio di fase II (43) con la combinazione di letrozolo ed everolimus in pazienti pretrattate ha mostrato un alto tasso di beneficio clinico (40%) e di risposta (32%). In uno studio di fase II della Nordic Society of Gynecologic Oncology l’exemestane ha mostrato un tasso di risposta del 10%, mancata progressione di malattia dopo 6 mesi nel 35% delle pazienti trattate, con buon profilo di tossicità globale (44).
BIBLIOGRAFIA
- Crosbie EJ, Zwahlen M, Kitchener HC, et al. Body mass index, hormone replacement therapy, and endometrial cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2010, 19: 3119-30.
- World Cancer Research Fund/American Institute for Cancer Research. Diet, nutrition, physical activity, and prevention of cancer: a global perspective. Continuous Update Project Exper Report 2018.
- International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risks to hormones: vol. 72 Hormonal contraception and post-menopausal hormonal therapy. IARC Lyon, 1999.
- Bruno V, Corrado G, Baci D, et al. Endometrial cancer immune escape mechanisms: let us learn from the fetal-maternal interface. Front Oncol 2020, 10: 156.
- Chiofalo B, Mazzon I, Di Angelo Antonio S, et al. Hysteroscopic evaluation of endometrial changes in breast cancer women with or without hormone therapies: results from a large multicenter cohort study. J Minim Invasive Gynecol 2020, 27: 832‐9.
- Garg K, Shih K, Barakat R, et al. Endometrial carcinomas in women aged 40 years and younger: tumors associated with loss of DNA mismatch repair proteins comprise a distinct clinicopathologic subset. Am J Surg Pathol 2009, 33: 1869-77.
- Bockman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 1983, 15: 10-7.
- Kommoss S, McConechy MK, Kommoss F, et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann Oncol 2018, 29: 1180–8.
- Alkushi A, Kobel M, Kalloger SE, et al. High-grade endometrial carcinoma: serous and grade 3 endometrioid carcinomas have different immunophenotypes and outcomes. Int J Gynecol Pathol 2010, 29: 343-50.
- Cirisano FD, Robboy SJ, Dodge RK, Gilks CBl. The outcome of stage I-II clinically and surgically staged papillary serous and clear cell endometrial cancers when compared with endometrioid carcinoma. Gynecol Oncol 2000, 77: 55-65.
- Savelli L, Ceccarini M, Ludovisi M, et al. Preoperative local staging of endometrial cancer: transvaginal sonography vs. magnetic resonance imaging. Ultrasound Obstet Gynecol 2008, 31: 560-6.
- Epstein E, Van Holsbeke C, Mascilini F, et al. Gray-scale and color Doppler ultrasound characteristics of endometrial cancer in relation to stage, grade and tumor size. Ultrasound Obstet Gynecol 2011, 38: 586-93.
- Vitale SG, Bruni S, Chiofalo B, et al. Updates in office hysteroscopy: a practical decalogue to perform a correct procedure. Updates Surg 2020, 72: 967-76.
- Chiofalo B, Palmara V, Vilos GA, et al. Reproductive outcomes of infertile women undergoing "see and treat" office hysteroscopy: a retrospective observational study. Minim Invasive Ther Allied Technol 2019, Dec 19: 1‐7.
- Manfredi R, Mirk P, Maresca G, et al. Loco-regional staging of endometrial carcinoma: role of MR imaging in predicting surgical staging. Radiology 2004, 231: 372-8.
- Suga T, Nakamoto Y, Saga T, et al. Clinical value of FDG-PET for preoperative evaluation of endometrial cancer. Ann Nucl Med 2011, 25: 269-75.
- Walker JL, Piedmonte MR, Spirtos NM, et al. Recurrence and survival after random assignment to laparoscopy versus laparotomy for comprehensive surgical staging of uterine cancer: Gynecologic Oncology Group LAP2 Study. J Clin Oncol 2012, 30: 695-700.
- Burke WM, Gossner G, Goldman NA, et al. Robotic surgery in the obese gynecologic patient. Clin Obstet Gynecol 2011, 54: 420-30.
- Vizza E, Chiofalo B, Cutillo G, et al. Robotic single site radical hysterectomy plus pelvic lymphadenectomy in gynecological cancers. J Gynecol Oncol 2018, 29: e2.
- Benedetti Panici P, Basile S, Maneschi F, et al. Systematic pelvic lymphadenectomy vs no lymphadenectomy in early-stage endometrial carcinoma: randomized clinical trial. J Natl Cancer Inst 2008, 100: 1707-16.
- ASTEC study group, Kitchener H, Swart AM, Qian Q, et al. Efficacy of systematic pelvic lymphadenectomy in endometrial cancer (MRC ASTEC trial): a randomised study. Lancet 2009, 373: 125-36.
- Todo Y, Kato H, Kaneuchi M, et al. Survival effect of para-aortic lymphadenectomy in endometrial cancer (SEPAL study): a retrospective cohort analysis. Lancet 2010, 375: 1165-72.
- Ballester M, Dubernard G, Lécuru F, et al. Detection rate and diagnostic accuracy of sentinel-node biopsy in early stage endometrial cancer: a prospective multicentre study (SENTI-ENDO). Lancet Oncol 2011, 12: 469-76.
- Vizza E, Cutillo G, Bruno V, et al. Pattern of recurrence in patients with endometrial cancer: a retrospective study. Eur J Surg Oncol 2020, 46: 1697-702.
- Creutzberg CL, Nout RA, Lybeert MLM, et al. Fifteen-year radiotherapy outcomes of the randomized PORTEC-1 trial for endometrial carcinoma. Int J Radiat Oncol Biol Phys 2011, 81: 631–8.
- Colombo N, et al. Endometrial cancer: ESMO clinical practice guidelines. Ann Oncol 2013, 24 suppl 6: vi33-8.
- Linee guida AIOM. Neoplasie dell’utero, endometrio e cervice. Edizione 2019.
- Martin-Hirsch PP, Bryant A, Keep SL, et al. Adjuvant progestagens for endometrial cancer. Cochrane Database Syst Rev 2011, 6: CD001040.
- Singh M, et al. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol 2007, 106: 325-33.
- Chambers JT, et al. Estrogen and progestin receptor levels as prognosticators for survival in endometrial cancer. Gynecol Oncol 1988, 31: 65-81.
- Piver MS, et al. Medroxyprogesterone acetate (Depo-Provera) vs. hydroxyprogesterone caproate (Delalutin) in women with metastatic endometrial adenocarcinoma. Cancer 1980, 45: 268-72.
- Tangen IL, et al. Loss of progesterone receptor links to high proliferation and increases from primary to metastatic endometrial cancer lesions. Eur J Cancer 2014, 50: 3003–10.
- Bovicelli A, D'Andrilli G, Giordano A, De Iaco P. Conservative treatment of early endometrial cancer. J Cell Physiol 2013, 228: 1154-8.
- Decruze SB, Green JA. Hormone therapy in advanced and recurrent endometrial cancer: a systematic review. Int J Gynecol Cancer 2007, 17: 964-78.
- Kokka F, Brockbank E, Oram D, et al. Hormonal therapy in advanced or recurrent endometrial cancer. Cochrane Database Syst Rev 2010, 12: CD007926.
- Thingpen JT, et al. Oral medroxyprogesterone acetate in the treatment of advanced or recurrent endometrial carcinoma: a dose-response study by the Gynecologic Oncology Group. J Clin Oncol 1999, 17: 1736-44.
- Crespo C, et al. Metastatic endometrial cancer in lung and liver: complete and prolonged response to hormonal therapy with progestins. Gynecol Oncol 1999, 72: 250-5.
- Fleming GF, et al. Temsirolimus with or without megestrol acetate and tamoxifen for endometrial cancer: a Gynecologic Oncology Group Study. Gynecol Oncol 2014, 132: 585-92.
- Steed HL, Chu QS. Aromatase inhibition: a potential target for the management of recurrent or metastatic endometrial cancer by letrozole: more questions than answers? Expert Opin Investig Drugs 2011, 20: 681-90.
- Bogliolo S, Gardella B. Effectiveness of aromatase inhibitors in the treatment of advanced endometrial adenocarcinoma. Arch Gynecol Obstet 2016, 293: 701-8.
- Altman AD, Thompson J, Nelson G, et al. Use of aromatase inhibitors as first- and second-line medical therapy in patients with endometrial adenocarcinoma: a retrospective study. J Obstet Gynaecol Can 2012, 34: 664-72.
- Ma BB, Oza A, Eisenhauer E, et al. The activity of letrozole in patients with advanced or recurrent endometrial cancer and correlation with biological markers — a study of the National Cancer Institute of Canada Clinical Trials Group. Int J Gynecol Cancer 2004, 14: 650–8.
- Slomovitz BM, et al. Phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J Clin Oncol 2015, 33: 930-6.
- Lindemann K, Malander S, Christensen RD, et al. Examestane in advanced or recurrent endometrial carcinoma: a prospective phase II study by the Nordic Society of Gynecologic Oncology (NSGO). BMC Cancer 2014, 14: 68.
Sindromi paraneoplastiche endocrine
Ipercalcemia paraneoplastica
Fabio Vescini
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria S. Maria della Misericordia, Udine
L’ipercalcemia si può presentare in circa il 10% dei pazienti con carcinoma in stadio avanzato e, spesso, rappresenta un fattore prognostico sfavorevole, gravato da una mortalità di circa il 50% a 30 giorni dall’insorgenza. Questa forma di ipercalcemia è generalmente caratterizzata da una ridotta concentrazione di paratormone (PTH), al contrario di quella che si osserva nell’iperparatiroidismo primitivo, perché è conservato ed efficace il feed-back del calcio plasmatico sulle paratiroidi (1-2).
I meccanismi principali attraverso i quali un tumore può provocare ipercalcemia sono essenzialmente quattro (1-4).
- Produzione di sostanze in grado di mimare l’azione del PTH. Questa forma, che rappresenta circa l’80% delle ipercalcemie paraneoplastiche, è definita ipercalcemia neoplastica umorale (HHM) ed è generalmente sostenuta dalla produzione, da parte del tumore primitivo, o, più frequentemente, dalle metastasi tumorali, del peptide correlato al PTH (PTHrP). Le neoplasie in grado di produrre PTHrP sono quelle del polmone (squamocellulare), del rene, della mammella, del capo e collo (sempre forme a cellule squamose), della vescica, dell’utero e dell’ovaio. Occasionalmente anche i tumori neuroendocrini possono secernere PTHrP e, ancora più raramente, i sarcomi e alcune neoplasie ematologiche.
- Metastasi ossee osteolitiche, in grado di mobilizzare direttamente il calcio mediante un marcato incremento del riassorbimento osseo osteclasto-mediato. Esse rappresentano circa il 20% delle ipercalcemie paraneoplastiche e, in molti casi, sono sostenute da neoplasie della mammella, mieloma multiplo e linfomi.
- Produzione tumorale di vitamina D, ovvero sovra-espressione di 1-alfa-idrossilasi nel tessuto neoplastico, con conseguente aumento della produzione di calcitriolo; tale condizione è stata descritta in associazione a certi linfomi.
- Produzione metastatica di citochine con attività osteoclastogenica, ovvero del PTH vero e proprio (casi molto rari).
La HHM è sicuramente la forma più comune. Il PTHrP è un peptide monomerico, che esiste in diverse isoforme, di lunghezza variabile da 60 a 173 aminoacidi (5-6). Molti tessuti presentano una fisiologica, ancorché minima, produzione di PTHrP, ma il suo ruolo fisiologico non è stato ancora del tutto compreso. Dati sperimentali mostrano che il PTHrP regola l’ossificazione encondrale, controlla la proliferazione delle cartilagini articolari, promuove il differenziamento osteoblastico nel microambiente osseo e sicuramente partecipa al controllo del passaggio placentare del calcio (la delezione del gene è letale per i feti dei mammiferi)(5-6). In corso di HHM il PTHrP incrementa notevolmente e può agire in maniera sovrapponibile al PTH, legandosi ai recettori renali e ossei, dove è in grado di incrementare la calcemia e ridurre il riassorbimento tubulare di fosfati (1).
I pazienti affetti da HHM presentano la sintomatologia tipica dell’ipercalcemia, caratterizzata da nausea, vomito, letargia, insufficienza renale e, se non adeguatamente trattata, coma e morte. La gravità dei sintomi dipende sia dalla concentrazione plasmatica del calcio, sia dalla rapidità d’insorgenza dell’ipercalcemia, con le forme acute che possono rappresentare una vera e propria emergenza medica (1,2).
Il laboratorio mostra tutte le alterazioni tipiche dell’iperparatiroidismo primitivo (oltre all’ipercalcemia, ipofosforemia, iperfosfaturia, ipercalciuria ed elevazione della fosfatasi alcalina), ma con valori di PTH soppressi o inappropriatamente normali e, comunque, generalmente < 20-30 pg/mL. Anche in pazienti con PTH superiore a questa soglia, ma all’interno del range di normalità, in presenza di ipercalcemia deve essere sempre sospettata la presenza di una HHM, in modo particolare se si tratta di soggetti anziani, con fattori di rischio neoplastico o anamnesi positiva per pregresse neoplasie (1-3). In alcuni laboratori è possibile dosare il PTHrP, generalmente con metodiche immunometriche, ma va tenuto presente che, viste l’elevata concentrazione plasmatica di questo peptide nella HHM, è possibile avere dei falsi negativi a causa del cosiddetto “effetto gancio” (risultati artificialmente bassi ad alte concentrazioni di analita)(7).
Il trattamento ottimale dell’ipercalcemia paraneoplastica è, ovviamente, rappresentato dalla rimozione della neoplasia causale; tuttavia, non è infrequente l’impossibilità di procedere a questo tipo di approccio chirurgico e, in questo caso, devono essere adottate terapie mediche adeguate (tabella).
| Farmaci per il trattamento dell’ipercalcemia paraneoplastica | ||
| Via di somministrazione | Posologia | |
| Soluzione fisiologica | ev | 200-500 mL/ora |
| Furosemide | ev | 20-40 mg/die |
| Pamidronato | ev | 60-90 mg (ripetibile dopo 3-4 settimane) |
| Zoledronato | ev | 4 mg (ripetibile dopo 3-4 settimane) |
| Prednisone | po (nel mieloma e nei linfomi) | 40-100 mg/die |
| Calcitonina | sc/im | 4-8 UI/kg/12 ore |
Innanzi tutto è sempre raccomandabile rivalutare la terapia generale seguita dal paziente e, se possibile, sospendere i farmaci potenzialmente ipercalcemizzanti (supplementi di calcio, litio, diuretici tiazidici, anti-acidi a base di calcio, vitamina D)(1).
L’approccio di prima linea all’ipercalcemia neoplastica è rappresentato dalla terapia re-idratante con soluzione salina, al fine di aumentare la filtrazione glomerulare e, contemporaneamente, inibire il riassorbimento tubulare del calcio. Solo quando il volume plasmatico è stato ristabilito, potranno essere aggiunti alla terapia i diuretici dell’ansa, tenendo sempre sotto controllo la concentrazione plasmatica del potassio (1).
Il trattamento medico di scelta, tuttavia, è quello con bisfosfonati, sia per la loro efficacia nel ridurre la calcemia, sia per il loro profilo di sicurezza. La somministrazione endovenosa di pamidronato o zoledronato induce una diminuzione significativa della calcemia in 2-4 giorni, raggiungendo un nadir intorno ai 7 giorni dopo l’infusione. L’effetto ipocalcemizzante è sufficientemente duraturo, potendo persistere anche per 3-4 settimane (1,4).
Nel caso in cui non si possano utilizzare i bisfosfonati (es. insufficienza renale, pre-esistente osteonecrosi della mandibola), si potrà ricorrere alla calcitonina, ancorché il suo effetto ipocalcemizzante è minore e di durata inferiore rispetto ai bisfosfonati (8).
Solo pochi casi selezionati, trattati in centri ad elevatissima specializzazione, potranno giovarsi di una terapia con mitramicina ovvero con nitrato di gallio (8).
Infine nei pazienti con insufficienza renale e cardiaca, che non possono essere trattati con elevate quantità di liquidi e con bisfosfonati, andrà preso in considerazione il trattamento emodialitico (4).
Bibliografia
- Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc 2010, 85: 838-54.
- Guise TA, Mundy GR. Cancer and bone. Endocr Rev 1998, 19: 18-54.
- Jacobs TP, Bilezikian JP. Rare causes of hypercalcemia. J Clin Endocrinol Metab 2005, 90: 6316-22.
- Stewart AF. Hypercalcemia associated with cancer. N Engl J Med 2005, 352: 373-9.
- Mundy GR, Edwards JR. PTH-related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol 2008, 19: 672-5.
- Clemens TL, Cormier S, Eichinger A, et al. Parathyroid hormone-related protein and its receptors: nuclear functions and roles in the renal and cardiovascular systems, the placental trophoblasts and the pancreatic islets. Br J Pharmacol 2001, 134: 1113-36.
- Burtis WJ. Parathyroid hormone-related protein: structure, function, and measurement. Clin Chem 1992, 38: 2171-83.
- Lumachi F, Brunello A, Roma A, et al. Medical treatment of malignancy-associated hypercalcemia. Curr Med Chem 2008, 15: 415-21.
Ipoglicemia paraneoplastica
Ettore Seregni
SS Terapia Medico Nucleare ed Endocrinologia, Fondazione IRCCS Istituto Nazionale Tumori, Milano
In ambito oncologico il riscontro di ipoglicemia rappresenta un evento relativamente frequente. Una riduzione delle concentrazioni ematiche di glucosio può essere osservata, ad esempio, nei pazienti con neoplasie maligne che coinvolgono in maniera estesa e diffusa il parenchima epatico o surrenalico. In queste condizioni inadeguata gluconeogenesi e ridotta secrezione di ormoni glucocorticoidi sostengono il quadro metabolico. Inoltre, l’ipoglicemia è di frequente riscontro negli stadi terminali del malato oncologico e, ancora, severe crisi ipoglicemiche caratterizzano i tumori secernenti insulina (insulinomi pancreatici e tumori ectopici secernenti insulina).
Tuttavia, l’ipoglicemia paraneoplastica non è in senso stretto riconducibile alle cause sovramenzionate e, in particolare, alla presenza di un tumore insulare. La presenza di ipoglicemia in assenza di tumore insulare viene definita con l’acronimo di NICTH (da Non-Islet Cell Tumor Hypoglycemia)(1). Per definizione, quindi la NICTH non è sostenuta dalla produzione di insulina.
La NICHT è una condizione clinicamente molto rara (incidenza di circa 1 caso per milione/anno) e si stima che l’ipoglicemia da NICHT sia almeno quattro volte meno frequente di quella attribuibile alla presenza di un tumore insulare (insulinoma).
La sindrome di Douge-Potter (descritta in maniera indipendente dai due Autori nel 1930) rappresenta uno dei primi riscontri di ipoglicemia paraneoplastica e deriva dall’associazione di NICTH con un particolare tipo di neoplasia, il tumore fibroso solitario (SFT, acronimo di Solitary Fibrous Tumor)(2,3). Questo tipo di tumore mesenchimale insorge a livello intra-toracico dalla pleura polmonare e presenta un comportamento benigno nella maggior parte dei casi. In circa il 20% dei casi, invece, il tumore mostra un comportamento maligno, con recidive locali e metastasi a distanza, per lo più a livello epatico. Inoltre, sono descritti SFT a insorgenza extra-toracica, più frequentemente in sede retro-peritoneale peri-vescicale. Istologicamente, il tumore si caratterizza per la presenza di cellule fibroblastiche o miofibroblastiche fusiformi e per una tipica architettura vascolare di tipo emangiopericitoma, con positività per CD34. È da sottolineare, però, che il SFT si associa a NICTH solo nel 5-10% dei casi (4-7).
Oltre alla sindrome di Douge-Potter, la NICTH può riscontrarsi anche in altre neoplasie maligne: epatocarcinomi, tumori germinali, tumori stromali gastro-intestinali (GIST) e anche neoplasie di origine ematopoietica (8-14).
L’ipoglicemia riconosce come meccanismo fisio-patologico la produzione e il rilascio da parte delle cellule tumorali di un precursore a elevato peso molecolare dell’IGF-II (big IGF-II)(1,15,16). In condizioni normali la big IGF-II corrisponde a circa al 10-20% dell’IGF-II totale, mentre nelle situazioni di NICTH questa quota supera generalmente il 60%. L’eccesso di big IGF-II conduce a ipoglicemia attraverso esagerata stimolazione dei recettori bersaglio, quali i recettori per IGF-1 (IGF1-R) e per insulina (IR), che comporta aumentata captazione di glucosio da parte delle cellule muscolari e adipose, e riduzione della gluconeogenesi epatica. L’iperstimolazione di IGF1-R e IR è sostenuta dal fatto che la big IGF-II, a differenza dell’IGF-II, non forma complessi ternari sufficientemente stabili con le proteine leganti specifiche (IGFBP) e con la subunità acido-labile (ALS), dando luogo a forme binarie e libere che mostrano maggiore diffusione extra-vascolare e disponibilità per le cellule bersaglio. Inoltre, la soppressione ipofisaria della produzione di GH determina riduzione della sintesi di IGFBP-3 e di ALS, determinando ulteriore incremento delle forme libere o a più basso peso molecolare.
Poco è noto dei meccanismi molecolari che sostengono la iperproduzione di big IGF-2 da parte delle cellule tumorali. Alcuni studi sperimentali dimostrerebbero che le forme immature di big IGF-II sarebbero la conseguenza di un deficit cellulare dell’enzima pro-ormone convertasi 4 (17).
I criteri per la diagnosi di NICTH, proposti nel 1990 da Teale e Marks, includono valori inappropriatamente elevati di IGF-II associati a basse concentrazioni di IGF-I (rapporto concentrazioni IGF-II/IGF-I > 3)(18). Inoltre, si associano ridotte concentrazioni di insulina, peptide C e pro-insulina e soppressione di produzione di GH. La dimostrazione di elevati livelli di big IGF-II è patognomonica, ma non sono disponibili metodiche immunometriche di pronto utilizzo per tale determinazione, che necessita di più complesse tecniche di tipo cromatografico.
L’intervento terapeutico nei pazienti affetti da NICTH è complesso, multimodale e spesso frustrante (1,19,20). Nei pazienti portatori di SFT benigni l’asportazione della neoplasia determina la pronta risoluzione dell’ipoglicemia. Al contrario, la NICTH associata a tumori maligni in fase avanzata può essere affrontata attraverso l’adozione di:
- terapie anti-neoplastiche specificamente rivolte alla neoplasia responsabile del quadro sindromico;
- terapie di supporto finalizzate al mantenimento di adeguati valori glicemici (somministrazione ripetute di glucosio per os o per via infusionale);
- glucocorticoidi, che sembrano rappresentare l’approccio più efficace e in grado di garantire un miglior controllo nel tempo della glicemia. Viene fatto ricorso a dosi progressivamente crescenti fino ad arrivare a dosi equivalenti di prednisolone di 3 mg/kg/die;
- ormone della crescita (rhGH), in grado di stimolare la produzione di IGFB-3 e ALS e di aumentare la gluconeogenesi epatica. Può essere associato ai glucocorticoidi;
- approcci sperimentali. Considerando la patogenesi della NICHT, è prevedibile che l’utilizzo di inibitori molecolari della via di trasmissione del segnale innescata da IGF1-R possa portare a un miglioramento del quadro metabolico. Sono in fase di valutazione clinica diversi anticorpi in grado bloccare l’attività di IGF1-R. Tuttavia, nessuno di questi composti è stato utilizzato nel controllo della NICTH.
Bibliografia
- de Groot J, Rikhof B, van Doorn J, et al.Non-islet cell tumour-induced hypoglycaemia: a review of the literature including two new cases. Endocr Relat Cancer 2007, 14: 979–93.
- Doege KW. Fibrosarcoma of the mediastinum. Ann Surg 1930, 92: 955-60.
- Potter RE. Intrathoracic tumors. Radiology 1930, 14: 60-2.
- Kalebi A, Hale M, Wong M, et al. Surgically cured hypoglycemia secondary to pleural solitary fibrous tumour: case report and update review on the Doege-Potter syndrome. J Cardiothorac Surg 2009, 4: 45.
- Schutt R, Gordon T, Bhabhra R, et al. Doege-Potter syndrome presenting with hypoinsulinemic hypoglycemia in a patient with a malignant extrapleural solitary fibrous tumor: a case report. J Med Case Rep 2013, 7: 11.
- Tominaga N, Kawarasaki C, Kanemoto K et al. Recurrent solitary fibrous tumor of the pleura with malignant transformation and non-islet cell tumor-induced hypoglycemia due to paraneoplastic overexpression and secretion of high-molecular-weight Insulin-like Growth Factor II. Intern Med 2012, 51: 3267-72.
- Famà F, Le Bouc G, Barrande A, Villeneuve MGB. Solitary fibrous tumour of the liver with IGF-II-related hypoglycaemia. A case report. Langenbecks Arch Surg 2008, 393: 611-6.
- Nadler WA, Wolfer JH. Hepatogenic hypoglycemia associated with primary liver cell carcinoma. Arch Int Med 1929, 44: 700–5.
- Samlani-Sebbane Z, Azeddine Diffaa A, Khadija Krati K, et al. Fatal hypoglycaemia from IGF II hyperproduction as a complication of a mesenteric gastrointestinal stromal tumour. Arab J Gastroenterol 2011, 12: 171-2.
- Barra W, Castro G, Oliveira A, et al. Symptomatic hypoglycemia related to inappropriately high IGF-II serum levels in a patient with desmoplastic small round cell tumor. Case Rep Med 2010, 2010: 684045.
- Powter L, Phillips L, Husbands E. A case report of non-islet cell tumour hypoglycaemia associated with ovarian germ-cell tumour. Palliat Med 2013, 27: 281-3.
- Morioka T, Ohba K, Morita H, et al. Non–islet cell tumor-induced hypoglycemia associated with macronodular pulmonary metastases from poorly differentiated thyroid carcinoma. Thyroid 2013 Sep 11 (Epub ahead of print).
- Ida T, Morohashi T, Ohara H, et al. Gastric neuroendocrine carcinoma with non-islet cell tumor hypoglycemia associated with enhanced production of Insulin-like Growth Factor II. Intern Med 2013, 52: 757-60.
- Shibata Y, Ito Y, Fujita H, et al. A novel case of functional gastric neuroendocrine carcinoma occurred after endoscopic submucosal dissection. Case Rep Gastrointest Med 2013, 2013: 148761.
- Rikhof B, van Doorn J, Suurmeijer A, et al. Insulin-like growth factors and insulin-like growth factor-binding proteins in relation to disease status and incidence of hypoglycaemia in patients with a gastrointestinal stromal tumour. Ann Oncol 2009, 20: 1582-8.
- Qiu Q, Yan X, Bell M, et al. Mature IGF-II prevents the formation of ‘‘big” IGF-II/IGFBP-2 complex in the human circulation. Growth Horm IGF Res 2010, 20: 110-7.
- Tani Y, Tateno T, Izumiyama H, et al. Defective expression of prohormone convertase 4 and enhanced expression of insulin-like growth factor II by pleural solitary fibrous tumor causing hypoglycemia. Endocr J 2008, 55: 905-11.
- Teale JD, Marks V. Inappropriately elevated plasma insulin-like growth factor II in relation to suppressed insulin-like growth factor I in the diagnosis of non-islet cell tumour hypoglycaemia. Clin Endocrinol 1990, 33: 87–98.
- Davda R, Seddon BM. Mechanisms and management of non-islet cell tumour hypoglycaemia in gastrointestinal stromal tumour: case report and a review of published studies. Clin Oncol (R Coll Radiol) 2007, 19: 265-8.
- Okushina K, Asaokaa Y, Fukudab I, et al. IGF-II producing hepatocellular carcinoma treated with Sorafenib: metabolic complications and a foresight to molecular targeting therapy to the IGF signal. Case Rep Gastroenterol 2012, 6: 784-9.
Osteomalacia oncogenica
Alfredo Scillitani
Endocrinologia, Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG)
Fisiopatologia ed eziopatogenesi
L’osteomalacia tumorale (TIO, Tumor-Induced Osteomalacia) è una sindrome osteo-metabolica paraneoplastica rara, di cui sono stati descritti finora meno di 400 casi (1,4).
È dovuta alla secrezione da parte di piccoli tumori mesenchimali di FGF-23. Questo è un ormone che legandosi fisiologicamente al proprio recettore e interagendo con altri cofattori (Klotho), riduce trascrizione ed espressione dei cotrasportatori sodio-fosfato 2a e 2c. L’effetto finale è quello di diminuire il riassorbimento tubulare del fosforo, con conseguente iperfosfaturia e ipofosforemia. Inoltre, FGF-23 riduce l’espressione della 1α-idrossilasi renale e aumenta quella della 24-idrossilasi renale, favorendo così l’idrossilazione in posizione 24 della 25OH-vitaminaD, determinando alla fine una riduzione dei livelli di 1-25(OH)2vitaminaD. Tale riduzione è di fatto più importante, se si considera che l’ipofosforemia normalmente aumenta espressione e attività dell’1α-idrossilasi.
TIO è in genere dovuto a piccoli tumori mesenchimali a lenta crescita, con caratteristiche istologiche di benignità (solo nel 5% dei casi sono maligni con metastasi generalmente polmonari), che possono infiltrare il tessuto connettivo circostante. Originano dai tessuti molli (55% casi) o dall’osso (40% casi), sono localizzati alle estremità, al distretto testa/collo o alla mandibola (strange tumors in strange places). Dal punto di vista istologico, i più frequenti sono displasia fibrosa, emangiopericitoma, osteosarcoma, condroblastoma, fibroma condro-mixoide, istiocitoma fibroso maligno, tumore a cellule giganti ed emangioma (all’esame microscopico sono presenti cellule stellate o fusiformi con basso indice mitotico in matrice mixoide che può contenere osso lamellare/cartilagine/osteoide/cellule giganti). La possibile infiltrazione del tessuto circostante si traduce da un punto di vista pratico nella necessità di eseguire al momento dell’intervento un’ampia resezione dei margini chirurgici, per evitare la persistenza o la recidiva della malattia (3). La diagnosi istologica di malignità è difficile e infatti nelle rare metastasi le caratteristiche istologiche delle lesioni metastatiche sembrano benigne (3). Oossono recidivare anche dopo anni dalla chirurgia.
Il tumore produce fosfatonine (FGF-23 ma anche MEPE, SFRP-4, FGF-7), che causano ipofosfatemia attraverso 3 meccanismi:
- inibizione del riassorbimento tubulare del fosforo, con aumento di fosfaturia e ipofosfatemia;
- inibizione della sintesi di 1,25OHD3 con ridotto assorbimento intestinale di fosforo e calcio e conseguente iperparatiroidismo secondario;
- inibizione della mineralizzazione della matrice osteoide.
Da un punto di vista isto-morfometrico, tutto questo si traduce a livello osseo in una grave osteomalacia, con importante deficit di mineralizzazione e numerose fratture.
Clinica
La sintomatologia è aspecifica e peggiora progressivamente negli anni. Sono presenti dolori ossei, debolezza muscolare progressiva fino all’allettamento, fratture multiple; i pazienti pediatrici hanno un quadro di rachitismo con ritardo di crescita.
È stato riportato un tempo di 2.5–28 anni tra l’inizio dei sintomi e la diagnosi (2), mentre sono in media necessari 5 anni per l’identificazione della neoplasia dopo la diagnosi (1).
La malattia deve essere sospettata per la presenza della sintomatologia associata a ipofosforemia.
Diagnosi
Da un punto di vista biochimico si osservano ipofosforemia, iperfosfaturia (ridotto TmPO4/GFR) e valori bassi o inappropriatamente normali in rapporto all’ipofosforemia di 1-25(OH)2vitaminaD, aumentati livelli di fosfatasi alcalina (indice di aumentato rimodellamento scheletrico), mentre PTH e calcemia sono nella norma.
Dopo aver confermato l’ipofosforemia, deve essere calcolato il TmPO4/GFR, cioè la capacità di riassorbimento massimale tubulare del fosfato, che, se risulta basso, indica che la perdita renale di fosfati è la causa dell’ipofosforemia. Il TmPO4/GFR, si determina misurando la fosforemia e creatininemia sierica e la fosfaturia e creatininuria a digiuno sulla 2° minzione del mattino; per il calcolo possono essere utilizzati vari algoritmi presenti sul web (3).
A questo punto un ulteriore supporto alla diagnosi può essere la misurazione dei livelli serici di 1-25(OH)2vitaminaD, che risulteranno bassi o inappropriatamente normali (in riferimento all’ipofosforemia).
Infine, può essere dosato il livello plasmatico di FGF23, che risulterà molto elevato e confermerà la diagnosi di malattia di perdita di fosfato FGF23-dipendente.
La diagnosi differenziale deve considerare essenzialmente alcune malattie genetiche, che sono delle fenocopie, e per potersi orientare può essere importante considerare l’età di inizio della sintomatologia, la storia familiare, possibili patologie dentarie. Generalmente più giovane è il paziente, più probabile è la causa genetica. Per un’accurata diagnosi differenziale si rimanda alla letteratura (3). Ci sono peraltro cause acquisite che possono determinare un quadro clinico e biochimico sostanzialmente sovrapponibile a quello del TIO. Tra esse ricordiamo le tubulopatie da metalli pesanti e da farmaci (i.e. da tenofovir, aminoglicosidici). In queste ultime forme acquisite i livelli di FGF23 sono bassi.
Localizzazione del tumore
Il tumore può essere presente in qualsiasi parte del corpo, dalla testa ai piedi, ed è spesso localizzato nelle estremità. Questa informazione è importante, perché testa, gambe e piedi, braccia e mani devono essere sempre valutate nei vari esami eseguiti per la localizzazione della neoplasia. Pertanto, un’accuratissima visita del paziente, con particolare osservazione di qualsiasi minima lesione presente nel sottocute dalla testa ai piedi, potrebbe già essere di aiuto per la localizzazione. Metodiche funzionali utili sono:
- 18F-FDG-PET/TC che è una metodica sensibile;
- scintigrafia con Octreoscan (recettori della somatostatina sono stati identificati su molti TIO) eventualmente combinata con metodica TC;
- 68Ga-DOTANOC PET/TC, che combinerebbe la specificità dell’octreotide con la sensibilità della PET/TC.
In queste metodiche “funzionali” alcune aree del corpo non sono ben visualizzate: l’encefalo da 18F-FDG-PET; fegato e milza da octreotide. Se anche dopo aver sottoposto il paziente a tali indagini la neoplasia non è stata identificata, è indicata l’esecuzione di altri esami come TC o RM (3,4).
Talora è stato eseguito un campionamento venoso con dosaggio di FGF23, per confermare che una lesione evidenziata con altri esami è responsabile della sindrome (1).
Nonostante tutte le indagini eseguite, il tumore potrebbe non essere localizzato. In tale evenienza è opportuno ripetere gli esami di localizzazione a distanza (dopo un anno).
Terapia e follow-up
Il trattamento risolutivo è l’asportazione chirurgica della lesione con ampio margine di resezione, che determina una rapida normalizzazione del quadro biochimico e quindi una rimineralizzazione dell’osso.
Comunque, se dopo aver posto la diagnosi la neoplasia non è stata individuata o, se individuata, non è stata asportata del tutto, è indicata la terapia medica con fosfato (1-3 g/die, suddivisi in 3-4 somministrazioni) e calcitriolo (1-3 µg/die). La terapia medica deve essere individualizzata, per migliorare i sintomi e cercare di normalizzare la fosfatasi alcalina. Tale terapia riduce il dolore osseo e muscolare e favorisce la mineralizzazione.
È in corso di studio l’anticorpo monoclonale anti-FGF23.
Bisogna monitorare il quadro biochimico per prevenire gli effetti negativi della terapia (ipercalcemia, calcolosi renale e nefrolitiasi). Pertanto, è necessario misurare calcio serico ed urinario, funzione renale e PTH, inizialmente ogni mese, quindi ogni 3 mesi. È riportata un’aumentata incidenza di iperparatiroidismo dopo prolungato trattamento con fosforo. In pazienti intolleranti a tale terapia, sono stati descritti casi singoli trattati con cinacalcet e octreotide (2).
Bibliografia
- Ruppe MD, Jane de Beur SM. Disorders of Phosphate Homeostasis. In: Rosen CJ, et al, Editors; Primer on the metabolic bone diseases and disorders of mineral metabolism; 8th Edition, Wiley-Blackwell 2013: 601-12.
- Chiam P, Tan HC, Bee YM, Chandran M. Oncogenic osteomalacia – Hypophosphatemic spectrum from “benignancy” to “malignancy”. Bone 2013, 53: 182-7.
- Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer 2011, 18: R53-77.
- Jiang Y, Xia WB, Xing XP, et al. Tumor-induced osteomalacia: an important cause of adult-onset hypophosphatemic osteomalacia in China: report of 39 cases and review of the literature. J Bone Miner Res 2012, 27: 1967-75.
Complicanze endocrine delle patologie oncologiche, della target therapy e nei cancer survivors
I primi studi sui pazienti affetti da malattia neoplastica il cui trattamento aveva dato una lunga sopravvivenza (per questo chiamati "cancer survivors") sono iniziati nel 1986 a seguito del primo Congresso della National Coalition for Cancer Survivorship che si è svolto a Albuquerque nel New Mexico. Risalgono però a numerosi anni dopo, dal 1997 in poi, i primi studi sistematici longitudinali sugli effetti collaterali delle terapie anti-neoplastiche nei bambini sopravvissuti. Le possibili sequele dei pregressi trattamenti anti-neoplastici, si presentavano a carico di numerosi organi e apparati: in particolare le prime descrizioni a carico del sistema endocrino sono state a carico dell'asse ipotalamo-ipofisario e delle ghiandole corticosurrenaliche.
Complicanze tiroidee delle patologie o terapie oncologiche
Daniele Barbaro
Sezione Endocrinologia, ASL 6, Livorno
INTRODUZIONE
La tiroide può essere interessata in corso di patologie oncologiche sia per coinvolgimento diretto, con metastasi, che nell’ambito di effetti collaterali di terapie oncologiche.
Nell’ambito delle terapie mediche, negli ultimi anni sono state sviluppate molecole in grado di agire su specifici bersagli molecolari implicati nella progressione neoplastica, la cosiddetta target therapy. In questo ambito, in particolare, gli inibitori multi-kinasici hanno mostrato di poter indurre con vari meccanismi sia ipotiroidismo che ipertiroidismo. La radioterapia esterna nella regione del collo può indurre ipotiroidismo con latenza di insorgenza, in alcuni studi, anche di pochi mesi. Essa, inoltre, può essere responsabile di sequele tardive, ipotiroidismo e seconda neoplasia tiroidea, nei long-survivors oncologici.
EFFETTI TIROIDEI DEGLI INIBITORI MULTI-KINASICI
Premesse
La target therapy comprende trattamenti in campo oncologico basati su molecole in grado di andare ad interagire con specifici bersagli molecolari implicati nella progressione neoplastica. Rientrano nella target therapy i trattamenti con inbitori multi-kinasici, i quali vanno ad interagire primariamente ma non esclusivamente con recettori di membrana che innescano varie kinasi implicate nella regolazione della proliferazione cellulare. Questi farmaci sono impiegati nel trattamento di neoplasie solide e in alcune forme ematologiche. Essi inibiscono il segnale in una varietà di recettori, tra cui vascular endothelial growth factor receptor (VEGF-R) 1, 2 e 3, platelet–derived growth factor receptor (PDGF-R), c-RET, macrophage colony-stimulating factor 1 (CSF1), FMS-like tyrosine kinase 3 receptor (FLT3), e c-KIT. Per alcune molecole è stata inoltre evidenziata la capacità di bloccare kinasi intra-cellulari, quali RAF (1).
Nel 2004 fu osservato che sunitinib poteva indurre ipotiroidismo e nel 2005 fu osservato che imatinib induceva ipotiroidismo in un paziente già tiroidectomizzato che assumeva levo-tiroxina (2). Successivamente, in seguito all’uso sempre più diffuso di questi farmaci, gli effetti avversi sulla funzione tiroidea sono stati meglio caratterizzati e dimostrati praticamente per tutti (6-12). Questi farmaci possono indurre ipotiroidismo, tireotossicosi con meccanismo distruttivo e per alcuni è stato anche ipotizzato un effetto sul metabolismo della T4. Il meccanismo che sta alla base non è del tutto chiarito, anche se l’inibizione dei VEGF-R sembra costituire una delle ipotesi principali. La tiroide è infatti l’organo con il più alto flusso ematico per unità di peso e il segnale mediato da VEGF-R (in particolare da VEGF-R1 e VGF-R2), espresso sulle cellule endoteliali, è ritenuto fondamentale per il mantenimento della vascolarizzazione. La riduzione del flusso potrebbe rappresentare un fattore determinante (1,3). In un caso di ipotiroidismo è stato chiaramente documentata una riduzione del flusso e in molti casi di ipotiroidismo è stata documentata una chiara diminuzione di dimensioni. Le fasi tireotossiche possono essere seguite da ipotiroidismo e anche in questi casi è stata documentata marcata riduzione volumetrica (1). Dopo l’interruzione eventuale del farmaco, i parametri della vascolarizzazione migliorano e la funzione tiroidea può tornare nella norma. Analogamente la plausibile spiegazione per la tireotossicosi è un effetto distruttivo legato a ischemia più severa ed acuta. Peraltro questo non appare l’unico meccanismo, in quanto anche inibitori multi-kinasici che non interagiscono con i VEGF-R possono, seppur raramente, indurre ipotiroidismo e il bevacizumab, anticorpo monoclonale contro il VEGF-A, pur prevenendo il legame di questo al suo recettore, può indurre ipotiroidismo con una prevalenza decisamente più bassa del sunitinib. Appare dunque verosimile che per indurre l’ischemia tiroidea sia indispensabile l’inibizione di almeno due VEGF-R e probabilmente anche il blocco di ulteriori fattori di crescita (1-3). Alcuni studi suggeriscono inoltre che un meccanismo aggiuntivo nell’indurre ipotiroidismo possa essere l’inibizione della tireo-perossidasi (4) e il blocco della captazione dello iodio (5).
Questi farmaci sono inoltre in grado di produrre anche alterazioni dei test funzionali tiroidei per effetti periferici. Sono, infatti, state descritte diminuzione del rapporto T3/T4 e T3/rT3 e modificazioni dei valori di TSH. I livelli di TSH sono spesso inappropriatamente alti rispetto ai valori di T4. Nel ratto è stata dimostrata inibizione della 5-desiodasi tipo 1 e tipo 3 e in un case report l’axitinib produceva ipotiroidismo in un paziente tiroidectomizzato in terapia con L-tiroxina.
Con quali farmaci è più probabile che si verifichi un’alterazione della funzione tiroidea e di quale tipo?
Se si presuppone che alla base dello sviluppo di ipo e ipertiroidismo ci sia principalmente l’inibizione dei VEFG-R, è ragionevole pensare che siano maggiormente associati a tali problemi i farmaci meno selettivi e con più alto potere inibente dei VEFG-R. In effetti, il sunitinib è quello per il quale vi sono state più segnalazioni. Il primo studio prospettico eseguito su 42 pazienti per un periodo mediano di 37 mesi mostrò che il 36% sviluppava ipotiroidismo, il 10% aveva TSH soppresso e il 17% innalzamento del TSH (6). Ulteriori studi prospettici e retrospettivi hanno riconfermato tali dati, con prevalenza di ipotiroidismo fino all’85% e alcuni casi di tireotossicosi franca (7,8,10,11,13). I dati della letteratura sembrano mostrare che gli altri due farmaci per i quali vi può essere associata frequentemente disfunzione tiroidea sono il sorafenib e l’axinitib (7,9,11,12). Per il sorafenib sono descritti casi di tireotossicosi, mentre l’axitinib sembra avere la maggior percentuale di effetti tiroidei (addirittura ipotiroidismo nel 100% dei casi in uno studio, nella popolazione giapponese, dove vi sono evidenze di una maggiore sensibilità a questi farmaci). Pazopanib appare avere la minore prevalenza, mentre per alcuni farmaci quali motesanib e vandetanib vi è evidenza di innalzamento del TSH, durante terapia sostitutiva con L-T4, nei pazienti tiroidectomizzati e trattati per carcinoma midollare in progressione (1,3,9). In particolare uno studio ha mostrato l’innalzamento del TSH nel 100% dei casi con vandetanib. Comunque, seppur con alcune differenze sopra descritte, praticamente per ogni farmaco di questa classe sono descritti effetti sulla funzione tiroidea. I dati derivati dai vari studi sono riportati nella tabella.
| Alterazioni della funzione tiroidea in corso di target-therapy | |||
| Farmaci | Numero di studi | Ipotiroidismo C/S | Tireotossicosi |
| Axitinib | 3 | 31-89% | NR |
| Cediranib | 2 | 13-56% | NR |
| Dasatinib | 1 | 50% | NR |
| Imatinib* | 2 | 90-100% | - |
| Linifanib | 3 | 5-44% | NR |
| Motesanib* | 3 | 8-41% | - |
| Nilotinib | 1 | 12% | NR |
| Pazopanib | 1 | 10% | NR |
| Sorafenib | 5 | 8-68% | 24% |
| Sunitinib | 10 | 18-85% | 6-40% |
| Vandetanib* | 1 | 100% | - |
| Vatalanib | 1 | 2% | NR |
* Tutti i pazienti erano tiroidectomizzati
C/S: clinico/subclinico
NR: non riportato
A tutt’oggi non si individuano fattori predittivi certi per lo sviluppo di tali problematiche tranne l’etnia giapponese. In uno studio la tireotossicosi è risultata più frequente in pazienti che avevano ipotiroidismo prima del trattamento e in cui vi era pre-esistenza di autoimmunità.
Dopo quanto tempo dobbiamo aspettarci un’eventuale alterazione della funzione tiroidea?
La probabilità che si sviluppi ipotiroidismo dipende dal tempo di trattamento e il tempo per lo sviluppo delle anomalie di funzione sembra anche diverso per i vari farmaci, in particolare sarebbe più breve per axitinib (mediana 3 settimane) vs sunitinib e sorafenib (16 settimane). Il monitoraggio della funzione tiroidea con TSH e FT4 prima del trattamento e il successivo monitoraggio del TSH come primo esame sembra dunque corretto, soprattutto in caso di uso di farmaci per i quali vi è maggiore evidenza di indurre alterazioni della funzione tiroidea. Non esiste nessuna raccomandazione formale circa la tempistica del monitoraggio del TSH, ma sembra ragionevole eseguire l’esame ogni 3 settimane nei primi quattro mesi ed eventualmente in caso di sospetto clinico. Nei casi in cui il farmaco è prescritto a cicli con periodo di wash-out, è probabilmente preferibile eseguire il dosaggio prima dello stop. Da sottolineare inoltre che i sintomi generali degli inibitori multi-kinasici, quali astenia e fatica, possono assomigliare ai sintomi dell’ipotiroidismo e dunque la clinica non ci viene in grande aiuto.
Quale è il comportamento di fronte a tale problematiche?
In caso di sviluppo di ipotiroidismo franco, il trattamento con L-tiroxina sarà senz’altro opportuno ed è verosimilmente preferibile iniziare con un dosaggio un po’ più basso per il possibile sviluppo di tireotossicosi. Di più difficile interpretazione potrebbe essere il caso di innalzamento del TSH con FT4 normale, in quanto più che un reale ipotiroidismo subclinico potrebbe trattarsi di un’anomalia correlata all’alterato metabolismo periferico e all’alterata desiodazione ipofisaria. Comunque, l’ipotiroidismo franco e a maggior ragione il solo innalzamento del TSH non dovrebbe costituire motivo di interruzione della terapia. La tireotossicosi rappresenta come sempre una preoccupazione maggiore: può essere autolimitante e sfociare nell’ipotiroidismo; in almeno un caso descritto vi è stata risposta al cortisone (22).
Lo sviluppo di un’alterazione della funzione tiroidea è predittivo dell’effetto del farmaco sulla neoplasia?
Non vi è accordo su questo punto oppure sull’ipotesi che l’ipotiroidismo possa migliorare l’efficacia terapeutica (13-15). Certamente la comparsa di alterazione della funzione tiroidea, soprattutto di ipotiroidismo, non dovrebbe costituire motivo per interrompere una terapia che si dimostri efficace.
EFFETTI TIROIDEI DELLA RADIOTERAPIA
Gli effetti tiroidei dell’esposizione a radiazioni ionizzanti sul collo sono noti e descritti già in una storica review da Greenspan nel 1977 (17). L’esposizione a basse dosi in età infantile aumenta il rischio di carcinoma tiroideo in modo lineare da 15 a 1500 Rad, per poi tendere a diminuire, in quanto dosi maggiori producono effetto distruttivo e ipotiroidismo. Da allora molte cose sono cambiate, in quanto è scomparso l’uso di radiazioni ionizzanti per la cura di patologie benigne ed inoltre il miglioramento dei trattamenti oncologici ha migliorato drasticamente la prognosi di molti pazienti, sia in età pediatrica che adulti.
Per quanto riguarda l’ipotiroidismo, l’incidenza nelle varie casistiche oscilla fra il 3 e il 40% (19,21) in base alla dose e al tempo del follow-up. Il picco di incidenza è fra i 2 e i 3 anni ed è stato osservato anche dopo soli tre mesi dalla radioterapia (RT). Non esiste un minimo soglia di dose, anche se la maggior parte dei lavori sono relativi a dosi > 30 Gy (3000 Rad) erogate sulla tiroide (17,18). Il concomitante uso di chemioterapia non ha mostrato un incremento di ipotiroidismo, tranne che in uno studio. Il problema ipotiroidismo post-RT è piuttosto semplice: basta porsi il dubbio e il trattamento con L-T4 non differisce dall’ipotiroidismo di altra origine.
Il problema dello sviluppo di carcinoma tiroideo nei pazienti sottoposti a RT riguarda sostanzialmente i “long-term survivors” e principalmente i pazienti sottoposti a trattamento in età pediatrica. Inoltre il miglioramento del trattamento dei tumori in età pediatrica coincide con un significativo decremento di mortalità e dunque è maggiore la probabilità di seconda neoplasia. Circa il 10% dei secondi tumori in questi pazienti è tiroideo e il rischio è più alto per esposizioni comprese fra 15 e 30 Gy alla tiroide. Esistono alcuni modelli che permettono una predizione di rischio assoluto per una seconda neoplasia e un recente lavoro descrive un analogo modello per la valutazione del rischio di secondo cancro primario tiroideo (17). A parte i vari modelli, spesso di difficile applicazione, il concetto fondamentale è la possibilità che pazienti esposti a RT in età infantile possano sviluppare una seconda neoplasia in sede tiroidea. Ipotiroidismo e carcinoma tiroideo sono inoltre stati segnalati in pazienti in età infantile trattati con 131I-MIBG per neuroblastoma (20). I pazienti esposti a radiazioni ionizzanti in età infantile necessitano dunque di controlli periodici della funzione tiroidea ed ecografici. I noduli in questi pazienti con “high-risk history” necessitano di FNA, indipendentemente da criteri dimensionali e ultrasonografici.
BIBLIOGRAFIA
- Makita R, Liri T. Tyrosine Kinase Inhibitor – induced thyroid disorders: a review and hypothesis. Thyroid 2013, 23: 151-9.
- De Groot JW, Links TP, Vandergraaf WT. Thyrosine kinase inhibitors causing hypothyroidism in a patients on levothyroxine. Ann Oncol 2006, 17: 1719-24.
- Brown RL. Tyrosine kinase inhibitor-induced hypothyroidism: incidence, etiology, and management. Target Oncol 2011, 6: 217-26.
- Wong E, Rosen LS, Mulay M, et al. Sunitinib induces hypothyroidism in advanced cancer patients and may inhibit thyroid peroxidase activity. Thyroid 2007, 17: 351-5.
- Mannavola D, Coco P, Vannucchi G, et al. A novel tyrosine kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J Clin Endocrinol Metab 2007, 92: 3531-4.
- Desai J, Yassa L, Marqusee E, et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med 2006, 145: 660-4.
- Clemons J, Gao D, Naam M, et al. Thyroid dysfunction in patients treated with sunitinib or sorafenib. Clin Genitourin Cancer 2012, 10: 225-31.
- Funakoshi T, Shimada YJ. Risk of hypothyroidism in patients with cancer treated with sunitinib: a systematic review and meta-analysis. Acta Oncol 2013, 52: 691-702.
- Torino F, Corsello SM, Longo R, et al. Hypothyroidism related to thyrosine kinase inhibitors: an emerging toxic effect of targeted therapy. Nat Rev Clin Oncol 2009, 6: 219-28.
- Grossmann M, Premaratne E, Desai J, Davis ID. Thyrotoxicosis during sunitinib treatment for renal cell carcinoma. Clin Endocrinol 2008, 69: 669-72.
- Daimon M, Kato T, Kaino W, et al. Thyroid dysfunction in patients treated with thyrosine kinase inhibitors, sunitinib, sorafenib and axitinib, for metastatic renal cell carcinoma. Jpn J Clin Oncol 2012, 42: 742-7.
- Tamaskar I, Bokowski R, Elson P, et al. Thyroid function test abnormalities in patients with metastatic renal cell carcinoma treated with sorafenib. Ann Oncol 2008, 19: 265-8.
- Baldazzi V, Tassi R, Lapini A, et al. The impact of sunitinib induced hypothyroidism in progressive-free survival of metastatic renal cancer patients: a prospective single-center study. Urol Oncol 2012, 29: 807-13.
- Schmidinger M, Vogl UM, Bojic M, et al. Hypothyroidism in patients with renal cell carcinoma: blessing or curse? Cancer 2011, 117: 534-44.
- Sabatier R, Eymard JC, Walz J, et al. Could thyroid dysfunction influence outcome in sunitinib-treated metastatic renal cell carcinoma? Ann Oncol 2012, 23: 714-21.
- Greenspan FS. Radiation exposure and thyroid cancer. JAMA 1977, 237: 2089-91.
- Donovan EM, James H, Bonora M, et al. Second cancer incidence risk estimates using BEIR VII models for standard and complex external beam radiotherapy for early breast cancer. Med Phys 2012, 39: 5814-24.
- Kovalchik SA, Ronckers CM, Veiga et al. Absolute risk prediction of second primary thyroid cancer among 5-year survivors of childhood cancer. J Clin Oncol 2013, 31: 119-27.
- Srikantia N, Rishi KS, Janaki MG, et al. How common is hypothyroidism after external radiotherapy to neck in head and neck cancer patients? Indian J Med Paediatr Oncol 2011, 32: 143-8.
- Van Santen HM, Tytgat G, Wetering MV, et al. Differentiated thyroid carcinoma after 131I-MIBG treatment for neuroblastoma during childhood: description of the first 2 cases. Thyroid 2012, 22: 643-6.
- Cella L, Liuzzi R, Conson M, et al. Development of multivariate NTCP models for radiation-induced hypothyroidism: a comparative analysis. Radiat Oncol 2012, 7: 224.
- Barbaro D. Sorafenib and thyrotoxicosis. J Endocrinol Invest 2010, 33: 436.
Complicanze ipofisarie delle patologie o terapie oncologiche
Alessandro Scoppola
Endocrinologia IDI - IRCCS, Roma
(aggiornato al 4 aprile 2016)
Trattamento radioterapico
Mentre le prime osservazioni avevano documentato che il trattamento chirurgico a livello encefalico determinava immediate alterazioni dell'integrità ipotalamo-ipofisaria, il trattamento radioterapico induce un danno più tardivo sulla secrezione delle tropine ipofisarie, anche a 10 anni dal primo trattamento, con sintomatologie aspecifiche e talvolta di difficile inquadramento, quali fatigue, aumento di peso e deficit di crescita (1,2).
Esiste una forte correlazione tra dose totale di radiazione somministrata e sviluppo del deficit ipofisario, come documentato durante il trattamento della leucemia linfoblastica acuta o dei tumori cerebrali: per dosi di 18-50 Gy si sviluppa deficit isolato di GH, per dosaggi superiori (> 60 Gy) sono stati descritti casi di panipopituitarismo. Sono stati anche descritti casi di diabete insipido (3). La somministrazione di una dose totale, ma frazionata in un breve periodo, determina lo stesso danno ipofisario rispetto alla somministrazione della stessa dose per un lungo periodo con numerosi intervalli.
L'ipotalamo è maggiormente sensibile alle radiazioni rispetto all'ipofisi e questo va considerato in particolare per dosaggi < 50 Gy (4). Pertanto, nei bambini trattati affetti da neoplasia cerebrale è prevalente il deficit di secrezione di GH, mentre il deficit degli altri ormoni ipofisari si presenta nel 2-6% dei casi dopo circa 10 anni dalla somministrazione di 40-50 Gy (5).
La secrezione di GH è la più radiosensibile, seguita dalle gonadotropine, dall'ACTH e molto raramente dal TSH. La maggior parte degli studi è concorde nell'affermare che, usando il test di tolleranza con l'insulina (ITT), il 100% dei bambini presenta una ridotta risposta del GH per dosaggi > 30 Gy, mentre la risposta è normale nel 35% dei bambini trattati con dosaggi < 30 Gy (6). Tali risposte possono comparire dopo 3 mesi come dopo 12 mesi dal trattamento.
Per quanto riguarda la secrezione gonadotropinica, una dose di radiazioni > 50 Gy può rendere i bambini ipogonadotropi, mentre dosi inferiori, paradossalmente, possono determinare pubertà precoce.
Possono esserci importanti ripercussioni anche sulla secrezione di ACTH, con un deficit nel 19% dei casi a distanza di 15 anni. Questa comparsa tardiva è una caratteristica peculiare, descritta per la prima volta nel 2003 (7). La dose di radiazioni che riduce questa prevalenza è compresa tra 18-24 Gy nel caso di leucemia linfoblastica acuta, come dimostrato dopo un follow-up di 3.6-10 anni (8).
Target therapy
La più recente scoperta di specifici anticorpi monoclonali diretti verso il CTLA-4, che è un recettore presente sui linfociti T attivati e che regola normalmente la risposta immunitaria, ha aperto nuove importanti possibilità terapeutiche per i pazienti affetti da melanoma metastatico (IV stadio) (9-10). Il legame del CTLA-4 con il suo ligando B7 (CD86) genera un segnale negativo, che viene utilizzato dalle cellule tumorali per indurre uno stato di anergia nei linfociti, disattivando l'attivazione immunitaria. Il legame dell'anticorpo anti-CTLA-4 alla suddetta molecola, impedisce l'innesco del segnale negativo che si traduce in un potenziamento della difesa immunitaria. Per questa sua caratteristica, l'Ipilimumab, eliminando un freno inibitore alla risposta immunitaria, che peraltro contribuisce allo stabilizzarsi della risposta immunogenica, è associato al rischio di effetti collaterali immuno-correlati. Attualmente in Italia l'Ipilimumab è indicato solo per i pazienti affetti da melanoma metastatico avanzato pretrattati (seconda linea), con uno schema di trattamento per via endovenosa di 3 mg/kg ogni tre settimane per 4 cicli. Tra i più importanti e frequenti effetti collaterali immuno-mediati, ci sono diarrea, epatotossicità ed eruzione cutanea. Più rare, ma talvolta insidiose per le modalità di presentazione, sono le tossicità neurologiche, oculari, articolari, polmonari ed endocrinologiche. Queste ultime si presentano a carico della tiroide nel 2.4% dei casi e a carico dell'ipofisi e dei surreni in < 1% dei casi.
Più in particolare a carico dell'ipofisi, la manifestazione più frequente a seguito della somministrazione di Ipilimumab, è l'ipofisite, con una frequenza, a seconda delle casistiche, che varia dallo 0% al 17% nei pazienti trattati per melanoma metastatico, per neoplasia renale e neoplasia prostatica (11-12). In un importante studio di Fase III su 676 pazienti affetti da melanoma metastatico al II e IV stadio, l'incidenza di ipofisite era dell’1.5% in combinazione con altri farmaci immuno-modulatori e dello 0.5% utilizzando solo Ipilimumab (13). Il rischio di ipofisite immuno-correlata a seguito di terapia con Ipilimumab al dosaggio maggiormente impiegato (3 o 10 mg/kg) può variare dall'1% al 6%. Simili percentuali sono state documentate con un altro farmaco analogo, il Tremelimumab.
Rispetto alle altre forme di ipofisite linfocitaria o autoimmune, quella da Ipilimumab è più frequente nei maschi (83-87%) e presenta ipogonadismo ipogonadotropo (14-17). La sintomatologia è caratterizzata da segni e sintomi non specifici, quali cefalea, disturbi del visus, fatigue, debolezza, confusione, perdita della memoria, disfunzione erettile e perdita della libido, anoressia, insonnia, intolleranza alla temperatura, sensazione soggettiva di febbre e brividi. È importante considerare ed escludere, in presenza di questa sintomatologia, la nuova comparsa di metastasi cerebrali. Sono stati descritti un caso di diabete insipido ed un caso di iponatremia secondaria a SIADH (18) e recentemente un caso di ipofisite associato ad insufficienza renale (19). La tossicità è stata suddivisa in 5 gradi secondo il National Cancer Institute (2012) per una migliore uniformità di valutazione e di intervento: Grado 1) assenza o lieve presenza di sintomi; diagnosi prevalentemente clinica per cui è consigliata l'osservazione; non sono necessari interventi farmacologici. Grado 2) sintomatologia moderata; limitazione iniziale delle abitudini di vita quotidiane, più significative in funzione dell'età; possibile documentazione di alterazione ipofisaria senza segni di sovvertimento ghiandolare; sono necessari interventi farmacologici. Grado 3) sintomatologia severa pur senza importanti rischi per la vita; necessario il ricovero ospedaliero e terapia appropriata; disabilità e parziale limitazione nelle abitudini di vita quotidiane. Grado 4) grave rischio di vita; possibili conseguenze irreversibili; necessità di intervento urgente. Grado 5) morte.
I sintomi generalmente compaiono dopo 2-6 mesi dall'inizio del trattamento. Al dosaggio di Ipilimumab più usuale nella pratica clinica (3 mg/kg) i sintomi più rilevanti compaiono dopo una media di 11 settimane, corrispondente a poco prima della IV somministrazione e questo è interpretabile in funzione dell'accumulo di dose.
Attraverso la valutazione radiologica con RMN con contrasto è possibile documentare un marcato aumento del volume della ghiandola ipofisaria, spesso con assottigliamento del peduncolo. In particolare sono stati riportati incrementi dell'altezza ipofisaria sul piano sagittale da valori basali di 3.4-6 mm a 7.7-11.8 mm (16,20). L'aumento del volume ipofisario è talvolta omogeneo, altre volte irregolare e si modifica durante la terapia corticosteroidea. Recentemente è stato descritto un caso di ipofisite, dall'esordio alla completa risoluzione, attraverso l'impiego della 18F-FDG PET/CT (21)
I livelli circolanti di ACTH, TSH, GH, prolattina, FSH e LH sono variabili in funzione dei vari gradi di ipopituitarismo e del tempo di comparsa dei sintomi. Per questo motivo è sempre opportuna la determinazione delle tropine ipofisarie, come di altri ormoni circolanti (FT4, cortisolemia, elettroliti plasmatici, funzione epatica, ecc.) prima dell'inizio della terapia. In alcuni casi ove si sospetti un iposurrenalismo subclinico associato al frequente sintomo che riferiscono questi pazienti, la fatigue, è opportuno eseguire un test di stimolo con ACTH anche prima dell'inizio della terapia. È necessario prestare attenzione alla possibilità di rilevare ridotti livelli di ACTH e di cortisolo plasmatico pur in presenza di sintomatologia addisoniana o di normali livelli di ACTH e bassi livelli circolanti di cortisolo. Similarmente è possibile documentare ridotti livelli circolanti di FT4 e TSH con sintomatologia ipotiroidea. La prolattina può essere elevata, normale o bassa (17).
Il trattamento si fonda sull'impiego di corticosteroidi, la cui posologia va individuata in funzione del grado di ipofisite: per il grado 3-4 è opportuno sospendere temporaneamente l'Ipilimumab ed iniziare il desametasone 4 mg ev ogni 6 ore per circa 7 giorni, scalando poi gradualmente fino a 0.5 mg/die e quindi sostituire con prednisone (1-2 mg/kg/die per os) o dosi analoghe di idrocortisone, con graduale riduzione dopo la 4° settimana ed eventuale successiva sostituzione degli ormoni deficitari.
In corso di terapia con Ipilimumab è necessario sempre sorvegliare la possibile insorgenza di una crisi iposurrenalica acuta. Generalmente tutti i pazienti che sviluppano un quadro di ipofisite presentano una graduale risoluzione della sintomatologia dopo pochi giorni dall'inizio della terapia corticosteroidea. Nel corso della storia clinica è stato anche possibile documentare l'evoluzione con valutazioni RMN seriate. Talvolta la funzione ipofisaria può rimanere deficitaria per lunghi periodi (in media 20 settimane, ma anche per tutta la vita), tanto da dover richiedere un trattamento prolungato con corticosteroidi (22). Più selettivamente è stata riportata la ripresa funzionale dell'asse ipofisi-tiroide nel 37-50% dei casi (23), e dell'asse ipofisi-gonadi nel 52% dei casi (24).
Episodicamente è possibile documentare un isolato deficit tiroideo e/o corticosurrenalico senza un completo ipopituitarismo. Il rischio del panipopituitarismo spesso non rappresenta un problema a causa della scarsa sopravvivenza dei pazienti affetti da melanoma metastatico. Non è mai stato possibile, prima del trattamento con farmaci anti CTLA-4, predire quali pazienti potessero sviluppare un ipopituitarismo persistente. Non è stato ancora documentato il possibile ruolo protettivo dei corticosteroidi per ridurre incidenza e severità di tali effetti collaterali.
Bibliografia
- Shalet SM, Beardwell CG, Pearson D, Jones PH. The effect of varying doses of cerebral irradiation on growth hormone production in childhood. Clin Endocrinol 1976, 5: 287-90.
- Constine LS, Woolf PD, Cann D, et al. Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med 1993, 328: 87-94.
- Jyotsna VP, Singh SK, Chaturvedi R, et al. Cranial irradiation- an unusual case for diabetes insipidus. J Assoc Physicians India 2000, 48: 1107-8.
- Livesey EA, Hindmarsh PC, Brook CG, et al. Endocrine disorders following treatment of childhood brain tumours. British J Cancer 1990, 61: 622-5.
- Shalet SM, Beardwell CG, MacFarlane IA, et al. Endocrine morbidity in adults treated with cerebral irradiation for brain tumours during childhood. Acta Endocrinol 1977, 84: 673-80.
- Clayton PE, Shalet SM. Dose dependency of time of onset of radiation-induced growth hormone deficiency. J Ped 1991, 118: 226-8.
- Schmiegelow M, Feldt-Rasmussen U, Rasmussen AK, et al. Assessment of the hypothalamo-pituitary-adrenal axis in patients treated with radiotherapy and chemotherapy for childhood brain tumor. J Clin Endocrinol Metab 2003, 88: 3149-54.
- Crowne EC, Wallace WH, Gibson S, et al. Adrenocorticotrophin and cortisol secretion in children after low dose cranial irradiation. Clin Endocrinol 1993, 39: 297-305.
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol 2005, 23: 515-48.
- Peggs KS, Quezada SA, Korman AJ. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol 2006, 18: 206-13.
- Maker AV, Yang JC, Sherry RM, et al. Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother 2006, 29: 455-63.
- Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol 2010, 37: 499-507.
- Hodi FS, O'Day SJ, McDermott DF. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010, 363: 711-23.
- Ribas A, Hauschild A, Kefford R. Phase III, open-label, randomized, comparative study of tremelimumab (CP-675,206) and chemotherapy (temozolomide [TMZ] or dacarbazine [DTIC]) in patients with advanced melanoma. J Clin Oncol 2008, 26 (15 suppl).
- Ribas A, Camacho LH, Lopez-Berestein G. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol 2005, 23: 8968-77.
- Wallis N, Bulanhagui CA, Dorazio PC. Safety of tremelimumab (CP-675,206) in patients with advanced cancer. J Clin Oncol 2008, 26 (15 suppl): 3040.
- Kirkwood JM, Lorigan P, Hersey P. Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin Cancer Res 2010, 16: 1042-8.
- Barnard ZR, Walcott BP, Kahle KT, et al. Hyponatremia associated with ipilimumab-induced hypophysitis. Med Oncol 2012, 29: 374-7.
- Forde PM, Rock K, Wilson G, O'Byrne KJ. Ipilimumab induced immune-related renal failure: a case report. Anticancer Res 2012, 32: 4607-8.
- Carpenter KJ, Murtagh RD, Lilienfeld H, et al. Ipilimumab-induced hypophysitis: MR imaging findings. AJNR 2009, 30: 1751-3.
- van der Hiel B, Blank CU, Haanen JB, Stokkel MP. Detection of early onset of hypophysitis by 18F-FDG PET/CT in a patient with advanced stage melanoma treated with Ipilimumab. Clin Nucl Med 2013, 38: e182-4.
- Blansfield JA, Beck KE, Tran K, et al. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 2005, 28: 593-8.
- Juszczak A, Gupta A, Karavitaki N, et al. Ipilimumab: a novel immunomodulating therapy causing autoimmune hypophysitis: a case report and review. Eur J Endocrinol 2012, 167: 1-5.
- Blansfield JA, Beck KE, TranKet. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 2005, 28: 593-8.
Aggiornamento 2016
Sull’argomento relativo alle complicanze della terapia con ipilimumab per il melanoma metastatico sull’ipofisi, sono stati presentati in letteratura due casi di tiroidite di Hashimoto con elevati titoli anticorpali (1) e vari casi di malattia di Graves con elevati anticorpi anti-TSH-R (2-3), associati a gravi disturbi centrali, compatibili con un quadro clinico di encefalite in assenza di alterazioni infettive, infiltrative, neoplastiche o metaboliche del liquor. Talvolta, infatti, specialmente nelle prime fasi del manifestarsi dei disturbi caratterizzati da confusione e letargia, senza anomalie morfologiche del sistema nervoso centrale e del liquido cefalo-rachidiano, il quadro clinico dell’ipofisite può essere confuso con una più complessa e grave sindrome encefalitica. La terapia steroidea è stata impiegata con successo per la risoluzione del quadro encefalitico, come già descritto in passato in merito alla encefalopatia e tiroidite di Hashimoto (encefalite di Hashimoto).
Sono stati anche descritti casi di meningite immuno-correlata e sindrome di Guillain-Barrè in pazienti con melanoma in trattamento con Ipilimumab (4). Questo è importante, poiché, al fine di evitare lo slatentizzarsi di altre importanti patologie, prima di intraprendere una terapia con i nuovi farmaci interferenti con il sistema immunitario, si rende sempre più necessaria un’attenta analisi preventiva delle possibili malattie autoimmuni, tra cui quelle di interesse del sistema endocrino.
Bibliografia
- Carl D, Grüllich C, Hering S, Schabet M. Steroid responsive encephalopathy associated with autoimmune thyroiditis following ipilimumab therapy: a case report. BMC Res Notes 2015, 8: 316.
- Min L, Vaidya A, Becker C. Thyroid autoimmunity and ophthalmopathy related to melanoma biological therapy. Eur J Endocrinol 2011, 164: 303–7.
- Borodic G, Hinkle DM, Cia Y. Drug-induced Graves’ disease from CTLA-4 receptor suppression. Ophthal Plast Reconstr Surg 2011, 27: 87–8.
- Bot I, Blank CU, Boogerd W, Brandsma D. Neurological immune-related adverse events of ipilimumab. Pract Neurol 2013, 13: 278–80.
Complicanze surrenaliche delle patologie o terapie oncologiche
Alessandro Scoppola
Endocrinologia IDI - IRCCS, Roma
La valutazione della funzione corticosurrenalica viene talvolta sottostimata nei pazienti affetti da neoplasia trattati sia con radioterapia che chemioterapia, poichè molti dei segni e sintomi caratteristici sono di frequente riscontro in questa categoria di pazienti. È stato documentato che il trattamento radiante cerebrale per neoplasie non di origine ipofisaria o ipotalamica, determina un iposurrenalismo associato anche a ipotiroidismo in meno del 5% dei casi (1).
La maggior parte degli inibitori della steroidogenesi impiegati da anni nel carcinoma surrenalico, quali Mitotane, Ketoconazolo, Metopirone ed Etomidate, determina un blocco della secrezione ormonale che necessita un’immediata sostituzione.
Diverso è il caso invece di un’altra categoria di farmaci, gli Inibitori della Tirosin-Kinasi (TKI) largamente impiegati per la terapia delle neoplasie stromali gastrointestinali (GIST), del cancro del rene avanzato e più recentemente di numerose altre neoplasie solide e della serie ematica, che proprio per il selettivo meccanismo di azione, interferiscono con la vascolarizzazione capillare e conseguente secrezione ormonale. Studi di fase III sull’impiego dei TKI nei pazienti con neoplasia renale metastatica nell’uomo non hanno documentato quadri di insufficienza surrenalica conclamata, sebbene questa sia stata osservata negli animali da esperimento per esposizioni farmacologiche modestamente superiori a quelle impiegate negli studi clinici (2). Parallelamente nelle sezioni istologiche dei surreni di questi animali sono state documentate aree di necrosi, emorragie, congestione, ipertrofia e infiammazione, ma ripetute valutazioni con RMN e CT in 336 pazienti trattati con Sunitinib non hanno dimostrato alcuna alterazione. La valutazione funzionale con il test di stimolo con ACTH in 400 pazienti trattati con Sunitinib ha documentato un deficit funzionale importante in un solo soggetto, mentre 11 pazienti avevano un ritardo di risposta con un picco del cortisolo di 12-16.4 µg/dL (normale > 18 µg/dL). Tuttavia, nessun paziente ha riportato evidenza clinica di iposurrenalismo (2). Recentemente è stato documentato per la prima volta durante trattamento con Sunitinib per neoplasia metastatica del rene un caso di insufficienza surrenalica acuta associato alla transitoria fase di tireotossicosi che può precedere l’ipotiroidismo nel corso di trattamento in questi pazienti (3). È noto infatti da tempo che l’insufficienza surrenalica subclinica può essere smascherata dall’ipertiroidismo, come avviene durante un eccesso di tiroxina o nella tireotossicosi da carbonato di litio (4,5).
L’asse ipotalamo-ipofisi-surrene è stato valutato anche nei pazienti con leucemia mieloide cronica in trattamento con Imatinib (un altro TKI), documentando in 12/25 casi la presenza di ipocortisolismo sub-clinico (cortisolo dopo stimolazione < 18 µg/dL) attraverso l’impiego dei test con glucagone e con ACTH. Solo due pazienti presentavano un quadro di ipocortisolismo conclamato. Tuttavia, nel medesimo studio non è stato possibile documentare un’associazione tra deficit di glucocorticoidi e durata o dose del trattamento.
L’effetto anti-secretivo (inibizione della sintesi di cortisolo) degli inibitori di mTOR è stato confermato in vitro in cellule di carcinoma corticosurrenalico umane.
Per tutti questi motivi la FDA nell’approvare il trattamento di queste neoplasie con i TKI ha indicato di porre la massima attenzione alle possibili manifestazioni sub-cliniche e pertanto è raccomandato il monitoraggio della funzione surrenalica in quei pazienti affetti da malattie importanti e/o da sottoporre ad interventi chirurgici.
Bibliografia
- Constine LS, Woolf PD, Cann D, et al. Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med 1993, 328: 87-94.
- Lodish MB, Stratakis CA. Endocrine side effects of broad-acting kinase inhibitors. Endocr Relat Cancer 2010, 17: R233-44.
- Yoshino T, Kawai K, Miyazaki J, et al. A case of acute adrenal insufficiency unmasked during sunitinib treatment for metastatic renal cell carcinoma. Jpn J Clin Oncol 2012, 42: 764-6.
- Shaikh MG, Lewis P, Kirk JM. Thyroxine unmasks Addison's disease. Acta Paediatr 2004, 93: 1663-5.
- Nagai Y, Ohsawa K, Hayakawa T. Acute adrenal insufficiency after unilateral adrenalectomy in Cushing's syndrome: precipitation by lithium-induced thyrotoxicosis during cortisol replacement. Endocr J 1994, 41: 177-82.
Complicanze ossee delle patologie o terapie oncologiche
Metastasi ossee da carcinoma della mammella
Stefania Bonadonna e Sonia Baruffi
UOS Dipartimentale di Riabilitazione Neuromotoria a indirizzo Oncologico, Pio Albergo Trivulzio, Milano
Negli Stati Uniti il tumore della mammella colpisce 1 donna su 8 ed è la seconda causa di morte per tumore, anche se la sopravvivenza delle pazienti affette da tale neoplasia è aumentata negli anni. Il Women’s Health Initiative Observational Study (WHI) (1) ha evidenziato che le donne che sopravvivono al tumore mammario hanno un rischio più elevato di fratture cliniche vertebrali, fratture di polso e in generale di tutte le fratture (esclusa l’anca). Tutto ciò verosimilmente è dovuto al fatto che la maggior parte dei tumori sono ormono-dipendenti, cioè hanno recettori per estrogeni o progesterone che rendono la neoplasia responsiva ad un trattamento mirato alla riduzione del tasso estrogenico (modulatori selettivi per i recettori estrogenici –SERM–, inibitori dell’aromatasi, ovariectomia o analoghi del GnRH).
Nonostante i progressi, soprattutto nella diagnosi precoce del tumore mammario e nelle terapie adiuvanti, la recidiva da neoplasia mammaria rimane una sfida aperta, con percentuali di recidiva di malattia che arrivano al 4.3% (durante i primi 2 anni di trattamento adiuvante con tamoxifene dopo resezione chirurgica del tumore); di questo 4.3% circa il 75% è rappresentato da metastasi a distanza. Questo dato appare molto significativo, in quanto la presenza di metastasi a distanza rende la malattia suscettibile solo di terapie palliative. La sopravvivenza per tumore alla mammella correla direttamente con lo stadio di malattia, poiché a a 5 anni è del:
- 98% per gli stadi I e II
- 84% per lo stadio III
- 28% per lo stadio IV (metastasi a distanza).
Per tale motivo l’identificazione di ulteriori sedi di malattia ed il trattamento mirato di tali foci viene ad assumere una notevole importanza in termini di sopravvivenza e qualità di vita.
Naume e coll. (2) hanno evidenziato che in alcune pazienti con tumore mammario in fase precoce il numero di cellule tumorali circolanti può essere calcolato da campioni di sangue periferico e la biopsia osteo-midollare rivela la presenza di cellule tumorali disseminate nel midollo. Queste sono le pazienti a più alto rischio di recidiva a distanza e con prognosi peggiore. Infatti, una volta che la cellula tumorale ha metastatizzato l’osso, interrompe il normale processo di rimodellamento osseo, causando lesioni osteolitiche che sono il risultato dell’attività osteoclastica incrementata grazie al rilascio, da parte della cellula tumorale stessa, di numerosi fattori di crescita, come PTHrP, VEGF, IGF e citochine.
Nelle pazienti affette da tumore mammario sono stati condotti numerosi studi sugli effetti di diversi bisfosfonati (clodronato, pamidronato, acido zoledronico). I bisfosfonati, da soli o in associazione con altri agenti anti-tumorali, hanno dimostrato in studi preclinici di essere in grado di bloccare diverse tappe nel processo di metastatizzazione. Nell’uomo, la capacità dei bisfosfonati di modificare il microambiente osseo può essere di supporto alla terapia adiuvante o neo-adiuvante del tumore della mammella, migliorando l’esito clinico.
Tre sono i principali studi che prenderemo in considerazione.
Lo studio ZO-FAST (Zometa-Femara Adjuvant Synergy Trial) (3) ha reclutato 1066 donne post-menopausa, con età mediana di 58 anni, affette da tumore mammario ER-positivo in stadio da I a IIIa, con l’obiettivo di dimostrare che le donne che ricevono acido zoledronico all’inizio del trattamento con impiego di un inibitore dell’aromatasi come il letrozolo hanno meno perdita ossea rispetto alle donne in cui il trattamento è stato posticipato. Il T-score basale era < -1 in 718 pazienti e tra -1 e -2 in 368 pazienti. Le pazienti sono state randomizzate a ricevere acido zoledronico (4 mg ogni 6 mesi) da subito o dopo un periodo di latenza dall’inizio del trattamento con letrozolo, quando la BMD scendeva al di sotto di -2 DS di T-score, oppure se avevano una frattura non traumatica o asintomatica. L’end-point primario era rappresentato da cambiamenti della BMD a livello lombare. A 12 mesi, le donne nel gruppo di trattamento con periodo di latenza presentavano una perdita di BMD, in media del 3.5% a livello della colonna lombare e del 2% all’anca, rispetto ad un lieve incremento di BMD nel gruppo che, invece, aveva ricevuto un trattamento immediato (p < 0.0001). Nel corso di tutto lo studio, le donne trattate dopo latenza hanno avuto una perdita di BMD del 6% vs nessuna perdita nelle donne trattate da subito con acido zoledronico. Riguardo all’incidenza di fratture, a 12 mesi non è stata osservata alcuna differenza tra i due gruppi di trattamento, immediato o ritardato.
Lo studio AZURE (Adjuvant Zoledronic acid to redUce Recurrence) è uno studio randomizzato, in aperto, multicentrico, a gruppi paralleli, che ha arruolato 3.360 donne provenienti da 174 centri in 7 Paesi, per determinare se l’acido zoledronico in aggiunta alla terapia ormonale dopo l'intervento chirurgico offrisse un beneficio nella prevenzione delle recidive in donne in pre-menopausa e post-menopausa con carcinoma mammario precoce. Alcune pazienti (n = 1681) hanno preso parte a una fase di trattamento di 5 anni con acido zoledronico con successivo periodo osservazionale di 5 anni, altre no (gruppo controllo, n = 1678). Un piccolo sottogruppo di pazienti ha anche ricevuto una terapia neo-adiuvante. L'end-point primario di sopravvivenza libera da malattia è stato fissato dopo 940 eventi di malattia. Gli end-point secondari comprendevano la sopravvivenza libera da malattia invasiva, la sopravvivenza globale, la sopravvivenza libera da metastasi ossee, la sicurezza. I risultati della seconda analisi ad interim dello studio (4), effettuata dopo un follow-up mediano di 59 mesi, quando si era presentato almeno il 75% (n = 752) degli eventi finali, hanno mostrato che l'acido zoledronico non migliora la sopravvivenza libera da malattia quando aggiunto alla chemioterapia adiuvante standard e/o alla terapia ormonale: hazard ratio (HR) = 0.98 (P = 0.79). Non è risultata statisticamente significativa la tendenza verso un miglioramento della sopravvivenza globale nelle pazienti del braccio trattato (HR = 0.85, P = 0.0726). In un'analisi pre-pianificata in base allo stato menopausale, nel braccio di trattamento è stato osservato un beneficio nella sopravvivenza libera da malattia e nella sopravvivenza globale nelle donne in menopausa ben consolidata: miglioramento del 32% della sopravvivenza libera da malattia (HR = 0.68, P = 0.009) e del 29% della sopravvivenza globale (HR = 0.71, P = 0.017).
Lo studio ABCSG-12 (Austrian Breast and Colorectal Cancer Study Group trial-12) è uno studio randomizzato, controllato, in aperto e multicentrico, condotto per 3 anni su 1803 donne in pre-menopausa con carcinoma mammario ER-positivo in stadio I-II, in trattamento con goserelin (3.6 mg ogni 28 giorni), rispetto ad efficacia e sicurezza di anastrozolo (1 mg/die) o tamoxifene (20 mg/die) con o senza acido zoledronico (4 mg ogni 6 mesi). La randomizzazione in un rapporto 1:1:1:1 ha bilanciato i 4 bracci di trattamento su 8 variabili prognostiche (età, chemioterapia neoadiuvante, stadio patologico del tumore, coinvolgimento linfonodale, tipo di chirurgia o terapia loco-regionale, dissezione ascellare completa, radioterapia intra-operatoria e regione geografica). L’end-point primario era la sopravvivenza libera da malattia (definita come recidiva di malattia o decesso). L’analisi a 48 mesi di follow-up (5) ha mostrato che l’aggiunta di acido zoledronico migliora significativamente la sopravvivenza libera da malattia (HR = 0.68, p = 0.009), ma non il rischio di decesso (HR = 0.67, p = 0.09).
In conclusione, i dati clinici da 3 principali studi di fase 3 (ABCSG-12, ZO-FAST e AZURE) dimostrano un importante miglioramento nella sopravvivenza libera da malattia con acido zoledronico aggiunto a terapia endocrina adiuvante in una popolazione di pazienti con tumore mammario in fase iniziale. Anche se i risultati ad interim provenienti dallo studio AZURE non mostravano una sopravvivenza libera da malattia nella popolazione generale, un'analisi per sottogruppi ha dimostrato che l’acido zoledronico migliora significativamente la sopravvivenza libera da malattia in donne già in stato post-menopausale ad inizio trattamento. Allo stesso modo, una sottoanalisi dello studio ABCSG-12 dimostra un importante beneficio dal trattamento con zoledronato in donne > 40 anni, che abbiano raggiunto una completa soppressione dell’attività ovarica. Questi dati insieme supporterebbero un ruolo potenziale dell’acido zoledronico che va oltre il benessere dell’osso e suggerirebbero che il substrato endocrino potrebbe influenzare l’effetto anti-tumorale dello zoledronato (6)
Uno studio randomizzato (7) ha confrontato denosumab, un anticorpo monoclonale umano contro il RANK-ligando, con acido zoledronico nel ritardare o prevenire gli eventi scheletrici in pazienti con carcinoma mammario con metastasi ossee. Le pazienti sono state randomizzate a denosumab (120 mg sc) e placebo ev (n = 1026) o acido zoledronico (4 mg ev, con aggiustamento basato sulla clearance della creatinina) e placebo sc (n = 1020) ogni 4 settimane. A tutte le pazienti è stato raccomandato di assumere supplementazione giornaliera di calcio e vitamina D. L'end-point primario era il tempo al primo evento scheletrico nel corso dello studio, definito come frattura patologica, terapia radiante o chirurgica all'osso o compressione della colonna spinale. Denosumab è risultato superiore ad acido zoledronico nel ritardare il tempo al primo evento scheletrico (HR = 0.82, P = 0.01) e il tempo al primo e ai successivi (multipli) eventi scheletrici (rate ratio, RR = 0.77, P = 0.001). La riduzione nei marcatori del turn-over osseo è risultata maggiore con denosumab. La sopravvivenza generale, la progressione della malattia e i tassi di eventi avversi ed eventi avversi gravi sono risultati simili tra i gruppi.
Alla luce di questi dati, alleghiamo le recenti linee guida prodotte dall’American Society of Clinical Oncology (ASCO) sul ruolo dei farmaci attivi sull’osso nel tumore della mammella metastatico (8).
| Linee guida della Società Americana di Oncologia Clinica (ASCO) sul ruolo dei farmaci attivi sull’osso nel carcinoma mammario metastatico |
|
| Rivolte a | Oncologi, Radioterapisti, Oncochirurghi, Palliativisti |
| Raccomandazioni chiave |
|
| Metodo | revisione sistematica della letteratura e analisi da parte del comitato ad hoc del pannello di esperti |
| Ulteriori informazioni | sono disponibili il riassunto esecutivo, le linee-guida complete e i supplementi (www.asco.org/guidelines/bisphosbreast) |
Sono in corso due importanti studi di fase 3 con denosumab nella neoplasia mammaria:
- come terapia adiuvante in donne con alto rischio di recidiva che ricevono terapia adiuvante o neo-adiuvante (end-point primario sopravvivenza libera da metastasi ossee, end-point secondari sopravvivenza assoluta, sopravvivenza libera da recidiva), termine dello studio previsto per ottobre 2016
- in pazienti in terapia con inibitori dell’aromatasi (end-point primario fratture cliniche, end-point secondario nuove fratture vertebrali, variazione della BMD a livello vertebrale e femorale), termine del reclutamento previsto per novembre 2013.
Bibliografia
- Chen Z, Maricic M, Bassford TL, et al. Fracture risk among breast cancer survivors: results from the Women's Health Initiative Observational Study. Arch Intern Med 2005, 165: 552-8.
- Naume B, Zhao X, Synnestvedt M, et al. Presence of bone marrow micrometastasis is associated with different recurrence risk within molecular subtypes of breast cancer. Mol Oncol 2007, 1: 160-71.
- Aapro M. Improving bone health in patients with early breast cancer by adding bisphosphonates to letrozole: the Z-ZO-E-ZO-FAST program. Breast 2006, 15 Suppl 1: S30-40.
- Coleman RE, Marshall H, Cameron D, et al; AZURE Investigators. Breast-cancer adjuvant therapy with zoledronic acid. N Engl J Med 2011, 365: 1396-405.
- Gnant M, Mlineritsch B, Stoeger H, et al; Austrian Breast and Colorectal Cancer Study Group, Vienna, Austria. Adjuvant endocrine therapy plus zoledronic acid in premenopausal women with early-stage breast cancer: 62-month follow-up from the ABCSG-12 randomised trial. Lancet Oncol 2011, 12: 631-41.
- Gnant M. Zoledronic acid in the treatment of early-stage breast cancer: is there a final verdict? Curr Oncol Rep 2012, 14: 35-43.
- Stopeck AT, Lipton A, Body JJ, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol 2010, 28: 5132-9.
- Van Poznak CH, Von Roenn JH, Temin S. American society of clinical oncology clinical practice guideline update: recommendations on the role of bone-modifying agents in metastatic breast cancer. J Oncol Pract 2011, 7: 117-21.
Metastasi ossee da carcinoma prostatico
Stefania Bonadonna e Sonia Baruffi
UOS Dipartimentale di Riabilitazione Neuromotoria a indirizzo Oncologico, Pio Albergo Trivulzio, Milano
I tumori del tratto genito-urinario rappresentano una delle patologie più rilevanti, soprattutto nei soggetti di sesso maschile, con la neoplasia della prostata al primo posto (192.000 nuovi casi diagnosticati all’anno negli Stati Uniti, che rappresentano il 25% dei tumori diagnosticati nell’uomo e causano oltre 27.000 morti per malattia metastatica). Queste neoplasie tendono a metastatizzare prevalentemente nello scheletro: la percentuale di metastatizzazione all’osso arriva al 65-75% nei pazienti con tumore della prostata avanzato, all’80-90% nei pazienti con neoplasia resistente alla terapia da deprivazione androgenica.
I siti più comunemente coinvolti sono colonna vertebrale, pelvi, coste e ossa lunghe, cioè le aree di ematopoiesi nell’adulto, che si ritiene siano le aree che forniscono un miglior microambiente di crescita alle cellule tumorali. Le lesioni ossee di origine maligna distruggono la normale omeostasi ossea data dall’interazione tra osteoclasti (riassorbimento osseo) e osteoblasti (neoformazione ossea), responsabile di mantenere l’osso sano, portando quindi a una maggior fragilità ossea.
All’imaging, le lesioni provenienti da tumori prostatici possono essere osteolitiche, osteoblastiche (in prevalenza) o di entrambi i tipi. Indipendentemente dal tipo di lesione, si osserva sempre un incremento dei marcatori di turn-over osseo, soprattutto quelli indicativi di aumentata attività osteoclastica. L’incremento di attività osteoclastica è associato in maniera indipendente all’aumentato rischio di complicanze scheletriche, incluse fratture patologiche, compressione midollare, necessità di chirurgia sull’osso o di radioterapia palliativa o dell’ipercalcemia neoplastica. In assenza di terapia diretta sull’osso, la maggior parte dei pazienti affetti da tumore della prostata va incontro ad un evento avverso scheletrico nel corso della malattia (1).
La terapia del tumore prostatico, anche allo stadio iniziale, prevede la deprivazione androgenica, che può essere accompagnata da orchiectomia bilaterale o da terapia cronica con GnRH-agonista o antagonista. In entrambi i casi si ottiene un ipogonadismo severo, con valori di testosterone < 0.2 ng/mL. La castrazione si accompagna a riduzione della BMD e quindi a una maggior incidenza di eventi ossei. Le terapie studiate ad oggi sono quello dirette sugli osteoclasti: bisfosfonati e denosumab.
L’acido zoledronico (4 mg ogni 3-4 settimane) è al momento il farmaco approvato dall’FDA come terapia standard preventiva degli eventi avversi scheletrici in pazienti affetti da tumore prostatico, indipendentemente dalla risposta tumorale alla terapia ormonale di prima linea (castrazione).
Un gruppo di ricerca giapponese ha valutato in 40 uomini con carcinoma prostatico naive per terapia ormonale l’efficacia di una singola infusione di acido zoledronico (4 mg ev al giorno 1) dopo la terapia di deprivazione androgenica vs nessuna infusione, misurando a 6 e 12 mesi la BMD del femore prossimale e della colonna lombare con DEXA e l’N-telopeptide urinario (uNTx) (2). A 6 mesi, la BMD media (senza differenze significative al basale) della colonna spinale antero-posteriore è diminuita del 4.6% nei controlli ed è aumentata del 5.1% nei pazienti trattati (P = 0.0002). Anche a 12 mesi, la differenza di BMD era significativa nella regione lombare (P = 0.0004), dimostrando che l’acido zoledronico mantiene la densità minerale ossea. Anche uNTx era significativamente diminuito a 6 mesi nei trattati, dimostrando una diminuzione del turn-over osseo.
Le fratture scheletriche nei pazienti con tumore della prostata sono associate ad una ridotta sopravvivenza. In 643 pazienti con carcinoma della prostata avanzato ed almeno un sito di metastasi ossea, è stata valutata l’efficacia dell’acido zoledronico nel prevenire fratture ossee: 208 pazienti hanno completato lo studio della durata di 15 mesi e 186 di questi hanno preso parte ad una fase di estensione. Dopo 24 mesi, il 38% dei pazienti trattati con acido zoledronico ha presentato complicanze ossee, contro il 49% dei pazienti nel gruppo placebo (p = 0.028). Inoltre l’acido zoledronico ha ritardato il tempo alla prima complicanza ossea (488 vs 321 giorni, p = 0.009), il tempo alla prima frattura patologica (0.77 vs 1.47 eventi/anno, P = 0.005) e ha ridotto il dolore, valutato mediante la scala Brief Pain Inventory, per tutto il periodo dello studio (3).
Il Denosumab HALT Prostate Cancer Study (4) è uno studio multicentrico, in doppio cieco, in cui uomini sottoposti a terapia di deprivazione androgenica per tumore della prostata non-metastatico sono stati randomizzati a denosumab, un anticorpo monoclonale umano contro RANK-L (60 mg sc ogni 6 mesi), oppure placebo (734 pazienti in ciascun braccio). L'end-point primario era la percentuale di cambiamento della BMD lombare a 24 mesi. Gli end-point secondari comprendevano i cambiamenti percentuali di BMD al collo del femore e all'anca a 24 mesi, e a tutti i siti a 36 mesi, così come l'incidenza di nuove fratture vertebrali. A 24 mesi, la BMD lombare è aumentata del 5.6% nel gruppo denosumab rispetto a una perdita dell'1% nel gruppo placebo (P = 0.001); le differenze tra i 2 gruppi si sono mantenute significative fino a 36 mesi. La terapia con denosumab è risultata anche associata a un aumento significativo di BMD durante tutte le valutazioni all'anca, al collo del femore e al terzo distale del radio. I pazienti trattati con denosumab hanno mostrato una diminuzione nell'incidenza di nuove fratture vertebrali a 36 mesi (1.5% vs 3.9% con placebo, RR = 0.38, P = 0.006).
Il Denosumab 147 è uno studio in doppio cieco, su 1.432 uomini con tumore alla prostata resistente alla castrazione, localmente progressivo, o con metastasi a qualsiasi linfonodo, randomizzati a denosumab (120 mg sc) o placebo sc ogni 4 settimane. Il 55% dei pazienti non aveva ricevuto alcun precedente trattamento locale (5). Sono stati inclusi pazienti considerati ad alto rischio di metastasi ossee, secondo almeno uno di due criteri: livello di PSA ≥ 8 ng/mL o tempo di raddoppiamento del PSA ≤ 10 mesi. L'end-point primario era la sopravvivenza libera da metastasi a livello osseo; gli end-point secondari erano il tempo alla prima metastasi ossea e la sopravvivenza globale. L'analisi finale dello studio ha dimostrato una mediana di sopravvivenza libera da metastasi ossee di 29.5 mesi per il braccio denosumab e 25.2 mesi per il braccio placebo (HR = 0.85, P = 0.028). Il tempo medio alla prima metastasi ossea è stato 33.2 mesi per il braccio denosumab e 29.5 mesi per il braccio placebo (HR = 0.84, P = 0.032). La sopravvivenza mediana è stata di 43.9 mesi per il braccio Denosumab e 44.8 mesi per il placebo (HR = 1.01, P = 0.91). Il trattamento con denosumab non ha portato a miglioramento nella sopravvivenza complessiva o nella sopravvivenza libera da progressione. Sebbene lo studio abbia incontrato l’end-point primario pre-specificato, con un prolungamento statisticamente significativo nella sopravvivenza libera da metastasi ossee, non è chiaro se un miglioramento del valore mediano della sopravvivenza di 4.2 mesi nei pazienti con tumore prostatico resistente alla castrazione, ad alto rischio di metastasi ossee, sia una misura adeguata di beneficio clinico. È anche poco chiaro se la prevenzione delle metastasi (pazienti senza evidenza di metastasi) aggiunga beneficio ai pazienti trattati con denosumab nel contesto metastatico (pazienti con evidenza di metastasi).
Un editoriale su Lancet (6), a commento dello studio denosumab 147, ha sollevato perplessità sull'opportunità di somministrare denosumab come farmaco preventivo per le metastasi ossee. Sulla base dei risultati dello studio, l'editoriale afferma che i risultati riportati sostengono l’uso di denosumab come alternativa all’acido zoledronico, ma non sostengono il suo più ampio utilizzo come agente preventivo per le metastasi ossee nel cancro alla prostata.
Alleghiamo le linee guida dell’Associazione Europea di Oncologia (EAU) (7) sulla terapia del carcinoma della prostata avanzato.
| Linee guida della Società Europea di Urologia per la terapia palliativa del carcinoma prostatico resistente alla castrazione |
|
| Raccomandazione | Grado |
| Il trattamento farmacologico non prolunga la sopravvivenza nei pazienti con metastasi ossee estese e sintomatiche | A |
| Il trattamento di questi pazienti deve mirare a migliorare la qualità della vita e a ridurre il dolore | A |
| L’obiettivo terapeutico principale è una terapia con la massima efficacia e la minima comparsa di effetti collaterali | A |
| Nei pazienti con metastasi scheletriche dovrebbero essere usati i bisfosfonati (p.e. acido zoledronico) per prevenire le complicanze ossee. Però bisogna valutare il rapporto con i possibili rischi di tossicità, in particolare la necrosi mascellare | A |
| Nel trattamento delle metastasi ossee dolorose devono essere presi precocemente in considerazione trattamenti palliativi, quali i radioisotopi, la radioterapia esterna e un adeguato uso di antalgici | B |
| Nei pazienti con sintomi neurologici critici, bisogna prendere in considerazione provvedimenti d’emergenza quali chirurgia spinale o radioterapia decompressiva | A |
Bibliografia
- Lee RJ, Saylor PJ, Smith MR. Treatment and prevention of bone complications from prostate cancer. Bone 2011, 48: 88-95.
- Satoh T, Kimura M, Matsumoto K, et al. Single infusion of zoledronic acid to prevent androgen deprivation therapy-induced bone loss in men with hormone-naive prostate carcinoma. Cancer 2009, 115: 3468-74.
- Saad F, Lipton A. Zoledronic acid is effective in preventing and delaying skeletal events in patients with bone metastases secondary to genitourinary cancers. BJU Int 2006, 97: 1351-2.
- Smith MR, Egerdie B, Hernández Toriz N, et al; Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 2009, 361: 745-55.
- Saylor PJ, Michaelson MD. Should the denosumab metastasis prevention trial change practice for men with nonmetastatic prostate cancer? Oncologist 2012, 17: 288-90.
- Logothetis CJ. Treatment of prostate cancer metastases: more than semantics. Lancet 2012, 379: 4-6.
- Mottet N, Bellmunt J, Bolla M, et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol 2011, 59: 572-83.
Gestione del diabete nel paziente in terapia oncologica
Edoardo Guastamacchia
Endocrinologia, Università di Bari
I dati epidemiologici rilevano che la coesistenza di diabete e cancro oscilla tra l'8 e il 18% (1); le due patologie si influenzano negativamente e ciò pone enormi difficoltà ai clinici che si occupano della cura di questa complessa popolazione di pazienti (2,3).
I dati sulla gestione dei diabetici in terapia oncologica sono scarsi ed insufficienti le evidenze di come meglio trattare la malattia diabetica, mentre simultaneamente è necessario curare la neoplasia.
Il cancro può complicare un diabete pre-esistente o favorire l'iperglicemia e l'insorgenza di nuovi casi di diabete attraverso vari meccanismi: rilascio di citochine o di altre sostanze, ridotta attività fisica, cachessia e quindi accentuazione dell'insulino-resistenza (4, 5).
Inoltre, è noto come la terapia oncologica (chemioterapia, radioterapia, glucocorticoidi) possa, con gli effetti collaterali che la caratterizzano (nausea, vomito, perdita di peso, esacerbazione dell'insulino-resistenza), favorire condizioni di scompenso glico-metabolico (2,6,7).
D'altra parte, le complicanze acute e croniche del diabete possono a loro volta ritardare il trattamento neoplastico ed indurne modificazioni; infatti, la terapia oncologica, con i possibili effetti nefrotossici, cardiotossici, neurotossici ed epatotossici, incontrerà notevoli difficoltà ad essere praticata al meglio in pazienti diabetici con insufficienza renale, cardiaca ed epatica, quali sono i diabetici di lunga durata più esposti all'insorgenza di cancro (6,8-10).
Il successo di una terapia oncologica completa richiede l'uso dell'85% della chemioterapia, percentuale che nel diabetico, per i motivi sopra menzionati, potrebbe risultare particolarmente tossica; d'altra parte è evidente che ogni modificazione posologica, della modalità di somministrazione o la sostituzione del chemioterapico si tradurrà in una ridotta risposta al trattamento e in una riduzione della sopravvivenza (11).
Non sorprende pertanto il fatto che i pazienti con cancro e diabete abbiano una prognosi peggiore se confrontati con pazienti senza diabete: remissioni più brevi, maggiori episodi infettivi, sopravvivenza media più breve e più alti tassi di mortalità; quanto detto vale anche per i pazienti con iperglicemia evidenziata per la prima volta durante la terapia oncologica (2,12-16).
Il trattamento della malattia diabetica e gli obiettivi da raggiungere nei soggetti con cancro saranno differenti e dovranno tenere presente la storia clinica del paziente e la sua aspettativa di vita.
Il controllo rigoroso della glicemia, che è solitamente richiesto per evitare le complicanze a lungo termine del diabete, non è più da ritenersi appropriato in soggetti con cancro sottoposti a terapia oncologica, per i quali lo scopo della cura dovrà essere un miglioramento della qualità della vita.
Ovviamente, devono essere prevenute iperglicemie severe, che compromettono il benessere dei pazienti e gli stessi esiti delle 2 patologie: pertanto si potrebbe, in fase non terminale, perseguire un range glicemico compreso tra 140 e 200 mg/dL (2).
Se lo stato nutrizionale del paziente è stabile, si potrà mantenere la terapia in atto qualunque essa sia. Se in seguito alla patologia neoplastica e alla terapia oncologica si manifesterà una perdita di peso o anoressia (nausea, vomito), sarà necessario nel diabete tipo 1 ridurre la posologia dell'insulina in atto e nel diabete tipo 2 ridurre la posologia degli antidiabetici orali o sostituirli con gli analoghi insulinici short-acting (2,17).
La metformina, com'è noto, è il farmaco di prima scelta, se non controindicata, nella cura del diabete mellito tipo 2, inoltre numerose e recenti evidenze mostrano che l'uso di essa è associato con una più bassa incidenza di cancro e mortalità per cancro rispetto ad altri ipoglicemizzanti (18-24); nonostante ciò, l'uso di essa dovrebbe essere evitato in questa popolazione di pazienti durante i cicli di terapia oncologica, perchè potrebbe esacerbarne gli effetti collaterali (nausea, vomito, enterite) (4,25,26).
Gli ipoglicemizzanti orali da preferire, per il controllo della glicemia post-prandiale, sono i secretagoghi di breve durata quali nateglinide e repaglinide, mentre sono possibilmente da evitare le sulfoniluree (glimepiride, glipizide, gliburide); ciò è importante perchè si può adattare la terapia ai pazienti, considerato che essi sono spesso affetti da nausea, vomito e quindi riluttanti a mangiare (2). Ovviamente, sono utilizzabili anche gli analoghi short-acting dell'insulina, in alcuni casi preferibili agli ipoglicemizzanti orali, per la loro rapidità d'azione, per la capacità di compensare l'iperglicemia post-prandiale e per la flessibilità nella soluzione della qualità e quantità dei cibi.
Durante l'utilizzo di glucocorticoidi (previsti in molti protocolli per il trattamento anti-neoplastico ed anti-emetico) (25,26) i pazienti con pre-esistente diabete potrebbero continuare ad assumere la terapia orale, confortati da un più accurato monitoraggio, ma solitamente agli ipoglicemizzanti si preferisce la terapia insulinica con schema basal-bolus, che è la sola in grado di compensare precocemente l'esacerbazione dell'insulinoresistenza determinata dai cortisonici (27). La posologia insulinica dovrà tener conto del peso del paziente e della capacità di alimentarsi. Potrà essere prevalentemente praticata sottocute, ma in alcuni casi potrebbe essere infusa ev, con la cautela di prevenire glicemie > 180 mg/dL ed evitare glicemie < 110 mg/dL (27). La posologia insulinica sarà poi modificata parallelemente alla necessità e alla diminuzione dei glucocorticoidi per evitare l'ipoglicemia (27).
L'iperglicemia è una frequente complicanza della nutrizione enterale e parenterale totale, solitamente usate in oncologia per supplementare e rimpiazzare la dieta in pazienti che non si alimentano normalmente. In tal caso l'aggiunta di insulina regolare alla soluzione della parenterale totale può favorire il controllo dei livelli glicemici. Anche l'insulina long-acting può essere usata in questi pazienti.
Nel caso della nutrizione enterale si potrà utilizzare una long-acting (glargine, detemir, lispro-protamina) la cui scelta dipenderà dalla durata della nutrizione enterale stessa; il monitoraggio glicemico si potrà effettuare ogni 4-6 ore e il controllo glicemico potrà beneficiare della somministrazione estemporanea di analoghi rapidi.
Importante è evitare la brusca interruzione della nutrizione parenterale totale, perchè essa potrebbe favorire l'ipoglicemia; si suggerisce una graduale riduzione dell'infusione almeno 1 ora prima della definitiva interruzione (2).
I pazienti diabetici con cancro in fase terminale sono a più alto rischio di iperglicemia (28,29), ma non vi è purtroppo accordo, nè vi sono chiare linee guida, relativamente ai range glicemici auspicabli in questi pazienti (30): gli oncologi tollerano valori tra 270 e 360 mg/dL, i diabetologi valori più bassi fra 180 e 270 mg/dL (30,31), altri ancora valori compresi tra 180 e 360 mg/dL (3).
L'iperglicemia in questa fase potrebbe non essere trattata fino a livelli di 250 mg/dL, ma al di sopra di questi valori la terapia insulinica diviene necessaria per evitare poliuria, polidipsia e disidratazione, che peggiorano ulteriormente la qualità della vita dei pazienti.
Assolutamente da evitare è l'ipoglicemia, che si manifesta per molteplici cause (anoressia, alterato assorbimento, vomito). Per minimizzare tale evento, i livelli glicemici in questi pazienti dovranno essere elevati e sicuri, compresi cioè tra 150 e 250 mg/dL (29) o addirittura più alti (tra 180-360 mg/dL) (3).
In questa fase l'uso di una long-acting sembra essere la terapia insulinica più appropriata per diversi vantaggi, quali un profilo glicemico piatto, minimo rischio ipoglicemico ed una sola iniezione al giorno (30).
Anche la dieta dovrà essere più elastica e si potrà permettere ai pazienti di assumere i cibi che più gradiscono; infine, il monitoraggio glicemico effettuato dal paziente o più frequentemente dai familiari sarà meno rigoroso, fino ad essere del tutto interrotto (17,32) o essere sostituito dalla determinazione della HbA1c molto meno invasiva (33).
Bisognerà parlare con i pazienti e i familiari per far comprendere loro che ciò che era valido in passato da un punto di vista terapeutico e di monitoraggio, potrebbe in questa fase rappresentare solo un ulteriore fardello per il paziente, al quale bisogna solo assicurare una dignitosa qualità della vita (3,17,28,29).
Bibliografia
- Ko C, Chaudhry S. The need for a multidisciplinary approach to cancer care. J Surg Res 2002, 105: 53-7.
- Psarakis HM. Clinical challenges in caring for patients with diabetes and cancer. Diabetes Spectrum 2006, 19: 157–62.
- McCoubrie R, Jeffrey D, Paton C, Dawes L. Managing diabetes mellitus in patients with advanced cancer: a case note audit and guidelines. Eur J Cancer Care (Engl) 2005, 14: 244-8.
- Diabetes and cancer.
- Argilés JM, Busquets S, Toledo M, López-Soriano FJ. The role of cytokines in cancer cachexia. Curr Opin Support Palliat Care 2009, 3: 263-8.
- Renehan AG, Yeh HC, Johnson JA, et al; Diabetes and Cancer Research Consortium. Diabetes and cancer (2): evaluating the impact of diabetes on mortality in patients with cancer. Diabetologia 2012, 55: 1619-32.
- Nguyen NP, Vos P, Vinh-Hung V, et al. Altered glucose metabolism during chemoradiation for head and neck cancer. Anticancer Res 2009, 29: 4683-7.
- van de Poll-Franse LV, Houterman S, Janssen-Heijnen ML, et al. Less aggressive treatment and worse overall survival in cancer patients with diabetes: a large population based analysis. Int J Cancer 2007, 120: 1986-92.
- Feng YH, Lin CY, Huang WT, et al. Diabetes mellitus impairs the response to intra-arterial chemotherapy in hepatocellular carcinoma. Med Oncol 2011, 28: 1080-8.
- Caudle AS, Kim HJ, Tepper JE, et al. Diabetes mellitus affects response to neoadjuvant chemoradiotherapy in the management of rectal cancer. Ann Surg Oncol 2008, 15: 1931-6.
- Richardson LC, Pollack LA. Therapy insight: Influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat Clin Pract Oncol 2005, 2: 48-53.
- Huang YC, Lin JK, Chen WS, et al. Diabetes mellitus negatively impacts survival of patients with colon cancer, particularly in stage II disease. J Cancer Res Clin Oncol 2011, 137: 211-20.
- Dehal AN, Newton CC, Jacobs EJ, et al. Impact of diabetes mellitus and insulin use on survival after colorectal cancer diagnosis: the Cancer Prevention Study-II Nutrition Cohort. J Clin Oncol 2012, 30: 53-9.
- Peairs KS, Barone BB, Snyder CF, et al. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol 2011, 29: 40-6.
- van de Poll-Franse LV, Haak HR, Coebergh JW, et al. Disease-specific mortality among stage I-III colorectal cancer patients with diabetes: a large population-based analysis. Diabetologia 2012, 55: 2163-72.
- Thong MS, van de Poll-Franse L, Hoffman RM, et al. Diabetes mellitus and health-related quality of life in prostate cancer: 5-year results from the Prostate Cancer Outcomes Study. BJU Int 2011, 107: 1223-31.
- Tice MA. Diabetes management at the end of life: transitioning from tight glycemic control to comfort. Home Healthc Nurse 2006, 24: 290-3.
- Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res 2010, 16: 1695-700.
- Del Barco S, Vazquez-Martin A, Cufí S, et al. Metformin: multi-faceted protection against cancer. Oncotarget 2011, 2: 896-917.
- Bayraktar S, Hernadez-Aya LF, Lei X, et al. Effect of metformin on survival outcomes in diabetic patients with triple receptor-negative breast cancer. Cancer 2012, 118: 1202-11.
- Berstein LM, Boyarkina MP, Teslenko SY. Familial diabetes is associated with reduced risk of cancer in diabetic patients: a possible role for metformin. Med Oncol 2012, 29: 1308-13.
- Viollet B, Guigas B, Sanz Garcia N, et al. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012, 122: 253-70.
- Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. J Clin Endocrinol Metab 2012, 97: E510-20.
- Lee DJ, Kim B, Lee JH, et al. The effect of metformin on responses to chemotherapy and survival in stage IV colorectal cancer with diabetes. Korean J Gastroenterol 2012, 60: 355-61.
- NCCN Clinical Practice Guidelines in Oncology (NCCN GuidelinesTM)- Antiemesis - Version 1.2012.
- Navari RM. Management of chemotherapy-induced nausea and vomiting: focus on newer agents and new uses for older agents. Drugs 2013, 73: 249-62.
- Oyer DS, Shah A, Bettenhausen S. How to manage steroid diabetes in the patient with cancer. J Support Oncol 2006, 4: 479-83.
- Boyd K. Diabetes mellitus in hospice patients: some guidelines. Palliat Med 1993, 7: 163-4.
- Poulson J. The management of diabetes in patients with advanced cancer. J Pain Symptom Manage 1997, 13: 339-46.
- Ford-Dunn S, Smith A, Quin J. Management of diabetes during the last days of life: attitudes of consultant diabetologists and consultant palliative care physicians in the UK. Palliat Med 2006, 20: 197-203.
- King EJ, Haboubi H, Evans D, et al. The management of diabetes in terminal illness related to cancer. QJM 2012, 105: 3-9.
- Quinn K, Hudson P, Dunning T. Diabetes management in patients receiving palliative care. J Pain Symptom Manage 2006, 32: 275-86.
- Kondo S, Kondo M, Kondo A. Glycemia control using A1C level in terminal cancer patients with preexisting type 2 diabetes. J Palliat Med 2013, 16: 790-3.
Problematiche relative alla fertilità e alla procreazione nel paziente oncologico
Maria Anna Sarno e Fedro Alessandro Peccatori
Unità di Fertilità e Procreazione in Oncologia, Istituto Europeo di Oncologia, Milano
Introduzione
Sono in aumento le neoplasie diagnosticate sotto i quarant’anni, che comprendono tumori mammari, tumori ginecologici (neoplasie della cervice uterina e tumori ovarici, prevalentemente germinali), tumori del testicolo e neoplasie ematologiche (linfomi, leucemie). Nonostante l’aumento d’incidenza, la sopravvivenza globale delle neoplasie dei giovani adulti è migliorata, grazie a diagnosi più tempestive, all’utilizzo di protocolli di cura standardizzati e all’avvento di nuovi farmaci.
In questo complesso scenario diventa prioritario considerare aspetti legati alla qualità di vita dei pazienti, che comprendono sempre i temi della fertilità e della procreazione. Il posticipo della maternità e della paternità, che si è affermato negli ultimi 2 decenni, rende ancor più urgente affrontare queste problematiche, offrendo risposte adeguate alle richieste dei pazienti.
Patogenesi dell’infertilità in ambito oncologico
I trattamenti chemio- e radioterapici riducono la fertilità con differenti meccanismi.
Sia radio che chemioterapia determinano un danno sulla gonade maschile, che si manifesta essenzialmente in una riduzione del numero, della motilità, della morfologia e dell’integrità del DNA degli spermatozoi. Ciò si traduce in una riduzione del potenziale fertile e un aumento del rischio malformativo fetale nel periodo immediatamente successivo al trattamento stesso.
Per quanto riguarda la donna, il danno gonadico da chemioterapia si esplica sia a livello dei follicoli primordiali che dei follicoli in diversa fase di maturazione, determinando, in acuto, un’amenorrea temporanea chemio-indotta e, a lungo termine, un più precoce esaurimento follicolare, quindi una ridotta fertilità. Il rischio di infertilità legato ai trattamenti chemioterapici dipende essenzialmente da 3 fattori:
- la classe di farmaci utilizzati, con potenziale gonado-tossico basso, medio ed alto. Gli agenti alchilanti, tra tutti la ciclofosfamide, sono i chemioterapici a più alto potere gonado-tossico, perchè non essendo ciclo-specifici possono colpire anche cellule che non sono in attiva proliferazione, come gli ovociti e i follicoli primordiali;
- la dose totale somministrata: maggiore è la dose somministrata, maggiore è il danno a livello gonadico, quindi dose e numero di cicli sono direttamente correlati alla tossicità ovarica;
- l’età della paziente svolge un ruolo preminente. Ogni donna alla nascita possiede un numero di follicoli primordiali (circa 300.000), che costituiscono il proprio patrimonio gametico. Nel corso della vita, questo patrimonio gametico viene per così dire “depauperato”, fino ad arrivare alla menopausa con circa 1000 follicoli residui. Per il solo effetto del passare degli anni, la riserva ovarica diminuisce progressivamente, con un’accelerazione piuttosto marcata dopo i 35 anni.
È difficile fare una stima precisa dell’entità del danno ovarico da chemioterapia, ma certamente la riserva gametica al termine del trattamento sarà ridotta di una quota percentuale che è funzione dell’età al momento del trattamento, della dose e del tipo di trattamento. Una donna in età più avanzata ha una ridotta riserva ovarica, pertanto il danno ovarico dei farmaci anti-blastici sarà proporzionalmente maggiore rispetto ad una donna più giovane, che possiede già in partenza un maggiore numero di follicoli residui (1,2).
In che modo la chemioterapia vada a depauperare il pool di follicoli primordiali è ancora oggetto di studio. Sono state proposte diverse teorie a riguardo, ma le più accreditate sono 3:
- quella dell’apoptosi follicolare ipotizza che i farmaci anti-blastici inducano una riduzione del numero di follicoli mediante l’induzione dell’apoptosi follicolare;
- quella del danno vascolare ipotizza un danno dei chemioterapici a carico dell’endotelio dei vasi della corticale ovarica, con iniziale ispessimento e ialinizzazione dei vasi, successiva proliferazione corticale di piccoli vasi (neo-vascolarizzazione) e poi formazione di focali zone di fibrosi della corticale con deposizione di collagene;
- quella del “burn-out” (esaurimento) follicolare attualmente gode di maggiore credito. Quando sono esposti alla chemioterapia, i follicoli in fase di crescita vengono distrutti, determinando una riduzione dei fattori di crescita, in primis l’AMH, implicati nel reclutamento follicolare. Ciò si traduce in un maggiore reclutamento follicolare con più rapido e precoce esaurimento della riserva di follicoli primordiali (3,4).
Come già accennato, la chemioterapia induce anche un danno ovarico a breve termine, per l’effetto sulle cellule gonadiche in attiva fase di proliferazione (follicoli antrali, pre-antrali e pre-ovulatori), causando amenorrea temporanea, che solitamente si manifesta già con i primi cicli di trattamento. Questo effetto è nella maggior parte dei casi transitorio, con un successivo recupero nella funzionalità gonadica al termine dei trattamenti chemioterapici, in un intervallo di tempo variabile dai 6 ai 12 mesi. Talvolta l’amenorrea temporanea può esitare in amenorrea permanente e quindi menopausa precoce. Ancora una volta, al di là della classe di farmaci somministrata, il principale determinante della amenorrea chemio-indotta è l’età della paziente. Donne di età più giovane hanno, infatti, un più rapido recupero dei cicli mestruali e un minor rischio di amenorrea permanente, con una percentuale di ripresa dei cicli mestruali pari all’85% per le donne di età < 35 anni, circa 60% nella fascia di età compresa tra i 35 e 40 anni, 40 anni, che sono quindi più vicine alla menopausa fisiologica.
Il tipo di chemioterapia somministrata influenza più che altro i tempi e l’andamento dei cicli mestruali post-trattamento. Ad esempio, in corso di terapia con antracicline si ha un precoce arresto dei cicli mestruali, con progressivo recupero di cicli regolari entro i 6-12 mesi dal termine del trattamento. L’aggiunta di taxani alla chemioterapia con antracicline determina una maggiore percentuale di amenorrea, con curve di ripresa dei cicli che seguono andamento analogo. Situazione differente si osserva invece dopo terapia con agenti alchilanti. L’amenorrea si instaura più lentamente ed in una minore percentuale di donne, ma, allo stesso tempo, si assiste ad un recupero più lento dei cicli mestruali, per poi seguire una più rapida evoluzione verso la menopausa (5).
Anche la radioterapia, quando il campo di irradiazione include le gonadi, determina una riduzione della riserva ovarica e quindi riduzione del potenziale fertile della donna. Il danno da radioterapia è proporzionale all’età della paziente e alla dose totale erogata. La dose sterilizzante nella maggior parte delle donne è pari a 5-15 Gy, con una tossicità maggiore per gli schemi di trattamento frazionato rispetto alla singola dose. Altro ostacolo della radioterapia pelvica al concepimento è l’irradiazione del viscere uterino, che porta solitamente a un utero ipoplasico, fibrotico, con pareti rigide che hanno perso elasticità e distensibilità, requisito fondamentale per accogliere il prodotto del concepimento. Oltre ai trattamenti diretti sulla pelvi, anche i trattamenti radianti sull’ipofisi possono indurre sterilità con un meccanismo differente che porta allo sviluppo di ipogonadismo ipogonadotropo. Per ridurre il danno ovarico da radioterapia è indicata, quando tecnicamente possibile e oncologicamente sicura, la trasposizione chirurgica delle ovaie al di fuori del campo di irradiazione, prima dell’avvio del trattamento (3).
Se la gonado-tossicità di chemioterapia e radioterapia è nota, non è dimostrato, invece, un rischio di infertilità dopo sola terapia endocrina. Il rischio globale di menopausa correlato alla sola assunzione di tamoxifene, farmaco cardine per la terapia ormonale del carcinoma mammario in pre-menopausa, è basso e sembrerebbe essere legato più all’età della paziente che non al profilo di tossicità ovarica del farmaco. Il rischio di ridotta fertilità dopo terapia endocrina è infatti da attribuirsi all’età materna avanzata al termine del trattamento, che solitamente prevede 5 anni di cura, durante i quali è fortemente sconsigliata la ricerca di una gravidanza per la nota teratogenicità del tamoxifene. Allo stesso modo non è nota una tossicità ovarica da parte dei farmaci biologici, che oramai sono diventati parte integrante dei trattamenti oncologici (1,2).
Valutazione e preservazione della fertilità
Gli approcci e le modalità sono differenti per uomini e donne.
Se le gonadi maschili sono più suscettibili al danno gonado-tossico di quelle femminili, allo stesso tempo le metodiche di preservazione della fertilità sono di più facile esecuzione e maggiore semplicità, avvalendosi essenzialmente della crio-preservazione dello sperma. La raccolta e il congelamento di liquido seminale devono essere proposte, indipendentemente dall’età, a tutti gli uomini che esprimano un desiderio di paternità, prima dell’avvio delle cure oncologiche. Per garantire il futuro risultato con maggiore sicurezza, devono essere raccolti da 1 a 3 campioni di sperma, che poi verranno conservati in apposite banche del seme (6).
La questione della preservazione della fertilità per le donne è invece più complessa e attiene a molteplici considerazioni etiche e tecniche, che sono meno standardizzate e logisticamente più articolate. Nell’ambito dei trattamenti oncologici bisogna dunque tener conto del rischio di infertilità dovuto alle terapie, dell’età della paziente, della possibilità di riutilizzare successivamente i gameti in relazione alla durata delle terapie. Sarebbe quindi opportuno che ogni donna, prima dell’avvio di un trattamento oncologico, ricevesse una valutazione da parte di uno specialista in onco-fertilità per poter vagliare le diverse opzioni di preservazione della fertilità adatte alla specifica situazione. Ogni procedura deve essere messa in atto prima dell’avvio delle cure, pertanto è di fondamentale importanza il tempestivo invio della paziente allo specialista, per evitare inutili e talvolta anche dannosi ritardi nell’inizio dei trattamenti oncologici (7,8).
Una stima indiretta della riserva ovarica può essere fatta mediante il dosaggio dei livelli circolanti di ormone anti-mulleriano (AMH) e la conta ecografica dei follicoli antrali. Il dosaggio dell’AMH, supportato dalla conta dei follicoli antrali, può essere utile sia prima dell’avvio del trattamento chemioterapico per una stima della riserva ovarica basale, sia al termine del trattamento stesso, per una stima della fertilità residua. L’AMH (glicoproteina dimerica prodotta dai follicoli antrali) svolge un ruolo inibitorio sulla maturazione dei follicoli primordiali, riducendone pertanto il reclutamento. I livelli di AMH subiscono un incremento progressivo dopo il menarca, con un picco nella terza decade, per poi ridursi successivamente fino a raggiungere livelli indosabili in menopausa. In corso di chemioterapia i livelli di AMH si riducono progressivamente e sembrerebbero rimanere soppressi anche dopo il termine della chemioterapia. Spesso la ripresa di cicli mestruali dopo chemioterapia viene erroneamente ritenuta un indice di fertilità. Molte donne che pur mestruano regolarmente e hanno livelli non menopausali di estradiolo, FSH e LH non riescono tuttavia a concepire, probabilmente per una misconosciuta ridotta riserva ovarica o per fattori endometriali (9).
Le tecniche di preservazione della fertilità in pazienti candidate a un trattamento chemioterapico sono: la crio-preservazione di ovociti o di embrioni, la crio-preservazione di tessuto ovarico o l’utilizzo di analoghi dell’LHRH in concomitanza alla chemioterapia.
La crio-preservazione degli ovociti viene proposta a donne di età ≤ 40 anni, con buona riserva ovarica sia in termini di numero che qualità ovocitaria. Mentre il numero degli ovociti può essere stimato, come suddetto, col dosaggio dell’AMH e la conta dei follicoli antrali, la qualità è in relazione all’età della paziente (donne più giovani hanno una migliore qualità ovocitaria). La raccolta di ovociti può avvenire su ciclo spontaneo, ma solitamente, si preferisce procedere dopo una stimolazione ormonale, per ottenere un numero maggiore di uova da congelare. È quindi necessario attendere il primo giorno del ciclo mestruale, iniziare la stimolazione ormonale monitorando il momento dell’ovulazione e quindi procedere alla raccolta (pick-up) degli ovociti.
Recenti dati suggeriscono che si possa iniziare la stimolazione anche in diverse fasi del ciclo mestruale, inducendo la luteolisi con antagonisti di LHRH.
La stimolazione ormonale può essere effettuata anche in donne affette da neoplasie endocrino-responsive, purchè la neoplasia sia già stata asportata chirurgicamente, e si utilizzino protocolli di stimolazione adattati con tamoxifene o letrozolo.
La raccolta degli ovociti viene effettuata per via vaginale con una procedura mini-invasiva. Una volta prelevati, gli ovociti vengono congelati mediante congelamento lento (slow-freezing), che ha tassi di sopravvivenza dopo scongelamento pari al 60-80%, o la più recente vitrificazione, che raggiunge una sopravvivenza ovocitaria fino al 90%. Per ottenere un risultato finale soddisfacente è necessario congelare tra gli 8-15 ovociti totali, considerando che parte del materiale viene perso nelle manovre di congelamento e scongelamento e quindi non è più fecondabile.
Dopo lo scongelamento, è indispensabile una fecondazione in vitro con ICSI, con percentuali di successo finali stimate intorno al 30%.
Il congelamento di ovociti immaturi che vengono poi maturati in vitro, prima o dopo il congelamento, è un’altra possibilità che è stata valutata di recente e per la quale si attendono i risultati degli studi in corso.
Il congelamento di embrioni, per quanto abbia tassi di successo maggiori del congelamento di ovociti, non è permesso in Italia e prevede comunque la presenza di un partner al momento della raccolta. Dopo la raccolta degli ovociti, infatti, si procede direttamente alla creazione di embrioni in vitro, che verranno poi congelati.
Il congelamento di tessuto ovarico è l’altra metodica utilizzata per preservare la fertilità femminile. Il vantaggio è che la procedura può essere fatta in qualsiasi momento del ciclo mestruale, evitando così ritardi nell’avvio della terapia, e che la successiva procreazione può avvenire spontaneamente. Lo svantaggio è la necessità di un intervento chirurgico, per altro eseguibile anche per via laparoscopica, sia per la rimozione del tessuto ovarico che per il successivo reimpianto.
La procedura di espianto è tecnicamente semplice e la quantità di tessuto ovarico da asportare dipende dalla riserva ovarica. È importante effettuare un esame istologico sul tessuto ovarico prima del congelamento, per escludere la presenza di metastasi che potrebbero poi essere reintrodotte con il successivo reimpianto. Il reimpianto può avvenire nella stessa sede anatomica (impianto ortotopico) o in una sede differente (impianto eterotopico), solitamente all’interno dell’addome o sotto la pelle dell’avambraccio, in ogni caso in zone particolarmente vascolarizzate. Con l’impianto ortotopico sono state ottenute 25 gravidanze.
Quando il tempo a disposizione prima dell’avvio delle cure non è abbastanza o le pazienti non desiderino sottoporsi a manovre invasive, può essere proposta la somministrazione di analoghi dell’LHRH concomitanti alla chemioterapia. Questa terapia sembra ridurre l’effetto gonadotossico dei chemioterapici ed è associata a minor probabilità di menopausa dopo il trattamento. Non è ancora chiaro se aumenti il tasso di gravidanze post-chemioterapia (10,11).
Bibliografia
- Patridge A, et al. Fertility and adjuvant treatment in young women with breast cancer. Breast 2007, 16: 175-81.
- Litton JK. Breast cancer and fertility. Curr Treat Options Oncol 2012, 13: 137-45.
- Meirow D, et al. Toxicity of chemotherapy and radiation on female reproduction. Clin Obstet Gynecol 2010, 53: 727-39.
- Meirow D, et al. Cortical fibrosis and blood-vessels damage in human ovaries exposed to chemotherapy. Potential mechanisms of ovarian injury. Hum Reprod 2007, 22: 1626-33.
- Petrek J, et al. Incidence, time course, and determinants of menstrual bleeding after breast cancer treatment: a prospective study. J Clin Oncol 2006, 24: 1045-51.
- Del Mastro L, Anserini P, Tomirotti M, Lambertini M, Peccatori F. Preservazione della fertilità in oncologia. Linee guida AIOM 2012.
- Divya S, et al. Medical, ethical, and legal considerations in fertility preservations. Int J Gynaecol Obstet 2011, 115: 11-5.
- Wallace W, et al. Fertility preservation for young patients with cancer: who is at risk and what can be offered? Lancet Oncol 2005, 6: 209-18.
- Anderson RA, et al. Measuring anti-Mullerian Hormone for the assessment of ovarian reserve: when and for whom is indicated? Maturitas 2012, 71: 28-33.
- Del Mastro L, Boni L, et al. Effect of the gonadotropin-releasing hormone analogue triptorelin on the occurrence of chemotherapy-induced early menopause in premenopausal women with breast cancer: a randomized trial. JAMA 2011, 306: 269-76.
- Demeestere I, et al. Gonadotropin-releasing hormone agonist for the prevention of chemotherapy-induced ovarian failure in patients with lymphoma: 1 year follow-up of a prospective randomized trial. J Clin Oncol 2013, 31: 903-9.