Overview sulle ipoglicemie

Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
L’ipoglicemia è definita dalla triade di Whipple, ovvero il rilievo di livelli glicemici ridotti in presenza di sintomatologia suggestiva (autonomica e/o neuroglicopenica), che si risolve con l’assunzione di zuccheri e la normalizzazione della glicemia.
La classificazione tradizionale delle ipoglicemie distingue tra forme cosiddette spontanee, che si verificano in fase post-assorbitiva, e forme reattive o post-prandiali, sebbene la distinzione tra forme a digiuno e forme post-prandiali non sia mai netta (si veda, ad esempio, l’insulinoma). Una classificazione più recente preferisce, pertanto, distinguere tra ipoglicemie del paziente apparentemente sano e ipoglicemie del paziente con un importante carico di patologia e/o di terapia in atto.
L’eziologia dell’ipoglicemia è evidente in presenza di patologie concomitanti (malattia epatica, renale e/o cardiaca, iposurrenalismo, tumori non insulari), in corso di trattamento con alcuni farmaci o in soggetti affetti da diabete mellito in terapia insulinica e/o con farmaci secretagoghi. Nei soggetti non diabetici in apparente benessere, la diagnosi differenziale è tra ipoglicemia accidentale, factitia o maligna, e iperinsulinismo endogeno.
Nell’ipoglicemia lieve-moderata il trattamento terapeutico immediato è l’ingestione di cibi contenenti glucosio o carboidrati. Le ipoglicemie gravi richiedono l’assistenza di terzi per la terapia sistemica mediante soluzione glucosata. Nelle situazioni extra-ospedaliere di emergenza viene utilizzato prevalentemente il glucagone (per lo più im o sc).
Un accurato iter diagnostico consente di definire il trattamento terapeutico cronico o definitivo più adeguato.
Bibliografia
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009, 94: 709-28.
Classificazione delle ipoglicemie
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
Le principali sindromi ipoglicemiche sono classificate nella tabella, che propone per semplificazione una distinzione tra forme insulino-mediate (o del soggetto apparentemente sano) e forme non insulino-mediate (o del soggetto con terapie concomitanti e/o pluripatologie).
| Classificazione funzionale delle sindromi ipoglicemiche | |
| Insulino-mediate | Non insulino-mediate |
| A digiuno | |
| Insulinoma Autoimmune (Ab anti-insulina) Ipoglicemia factitia |
Autoimmune (mutazione del recettore per l’insulina, anticorpi anti-recettore per l’insulina) Insufficienza d’organo Deficit di substrati: chetosi dell’infanzia, uremia, denutrizione grave/sepsi, malattia delle urine a sciroppo d’acero Deficit ormonale: iposurrenalismo, deficit di GH, ipopituitarismo Tumori non insulari o secernenti IGF-2 |
| Reattive/post-prandiali | Indotte/iatrogene/factitia |
| Non-insulinoma pancreatogenous hypoglycemia (NIPHS) Nesidioblastosi primitiva o secondaria a by-pass gastrico Autoimmune (Ab anti-insulina) Alimentare Dumping syndrome By-pass gastrico Diabete mellito tipo 2 Idiopatica |
Alcool (inibisce la gluconeogenesi, si presenta dopo 12-72 ore) Farmaci anti-diabetici Altri farmaci |
Nel testo seguiremo la classificazione tradizionale tra forme:
- esogene
- endogene:
- sindromi ipoglicemiche cosiddette spontanee, che compaiono in fase post-assorbitiva, comprendono forme organiche o farmacologiche
- forme reattive, che compaiono nel post-prandiale, sono molto spesso, e soprattutto nell’adulto, disturbi funzionali.
I soggetti con ipoglicemia spontanea possono anche presentare un’ipoglicemia reattiva, come nel caso dell’insulinoma, che può produrre sintomi sia in fase post-assorbitiva sia, sebbene più raramente, nel periodo post-prandiale.
Ancora, le ipoglicemie factitiae si accompagnano all’insorgenza della sintomatologia in maniera erratica e casuale, indipendentemente dall’ingestione di cibo, mentre i disturbi autoimmuni generalmente danno luogo a sintomi nel passaggio dalla fase post-prandiale a quella post-assorbitiva.
Le forme esogene, legate per lo più all’assunzione di farmaci, sono più comuni negli anziani con comorbilità multiple e in chi assume terapie anti-diabetiche; causa frequente di ipoglicemia è inoltre rappresentata dall’alcol.
L’ipoglicemia a digiuno può verificarsi per ipersecrezione di insulina nel caso dell’insulinoma o in presenza di una normale secrezione insulinica per assenza/riduzione degli ormoni della contro-regolazione (iposurrenalismo, deficit di GH, ipopituitarismi multipli) o ancora per alterata biodisponibilità dell’insulina stessa (in presenza di auto-anticorpi contro l’insulina endogena o in pazienti esposti a farmaci contenenti gruppi sulfidrilici). L’ipoglicemia a digiuno può infine essere causata da tumori maligni o benigni, per consumo di glucosio e/o produzione di citochine specifiche (raro, ma paradigmatico, il caso della sindrome di Douge-Potter nel tumore fibroso solitario del polmone).
L’ipoglicemia post-prandiale può presentarsi per iperinsulinismo indotto da nesidioblastosi (ipertrofia e iperplasia con alterazioni nucleari delle ß-cellule), insulino-resistenza e, con frequenza ancora non nota, in pazienti sottoposti a chirurgia bariatrica, in particolare dopo by-pass gastrico.
Bibliografia
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009, 94: 709-28.
- Service FJ. Classification of hypoglycemic disorders. Endocrinol Metab Clin North Am 1999, 28: 501–17.
Clinica generale delle ipoglicemie
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
Tradizionalmente si parla di sindrome ipoglicemica in presenza della classica triade di Whipple: segni e/o sintomi di ipoglicemia con valori di glucosio plasmatico ridotti e scomparsa di tali sintomi dopo somministrazione di glucosio con correzione dei valori plasmatici.
I sintomi causati dall’ipoglicemia (tabella) si dividono in:
- neurogenici o autonomici, adrenergici e colinergici, indotti dall’attivazione del sistema nervoso da parte dell’ipoglicemia;
- neuroglicopenici, causati dalla deprivazione di glucosio a livello cerebrale, aspecifici in fase iniziale, arrivano fino alla perdita di coscienza con possibili danni cerebrali.
In relazione alla gravità, l’ipoglicemia si definisce:
- lieve, in presenza dei soli sintomi neurogenici;
- moderata, quando compaiono anche sintomi neuroglicopenici, quali debolezza e confusione mentale;
- severa, in presenza di alterazione dello stato di coscienza e/o quando è richiesto l’intervento di terzi.
Nell’ipoglicemia lieve o moderata il soggetto è in grado di rimediare autonomamente con l’assunzione di zuccheri semplici per bocca.
| Segni clinici di ipoglicemia | ||
| Autonomici | Adrenergici | Tachicardia Tremori Pallore Agitazione |
| Colinergici | Sudorazione Fame Parestesie |
|
| Neuroglicopenici | Difficoltà di concentrazione Alterazione del tono dell’umore Confusione Diplopia Visione offuscata Convulsioni Cefalea Coma |
|
Nella maggior parte dei casi sono i sintomi autonomici a rendere il paziente consapevole dell’ipoglicemia.
In letteratura vengono riportati valori soglia per l’ipoglicemia compresi tra 45 e 75 mg/dL. Durante l’ipoglicemia indotta sperimentalmente in soggetti non diabetici, i sintomi autonomici vengono avvertiti a una soglia plasmatica della glicemia pari a 60 mg/dL, mentre l’alterazione delle funzioni cerebrali associata a sintomi neuroglicopenici si evidenzia a glicemie pari a 54 mg/dL; tale soglia si abbassa in pazienti con ipoglicemie ricorrenti.
La comparsa del quadro clinico di ipoglicemia è condizionata da tre variabili:
- la risposta dell’organismo al digiuno;
- la soglia di comparsa dei sintomi neuroglicopenici;
- la soglia di comparsa della risposta contro-regolatoria.
L’assenza di sintomi di ipoglicemia può verificarsi in presenza di digiuno prolungato, specialmente in donne e bambini, per passaggio all’utilizzo dei chetoni come fonte alternativa di nutrimento a livello cerebrale, che ritarda la comparsa dei sintomi neuroglicopenici.
La risposta all’ipoglicemia è condizionata dall’età del paziente e dal sesso:
- dopo digiuno di 24 h, la glicemia è in genere > 55 mg/dL negli uomini e > 35 mg/dL nelle donne;
- dopo digiuno di 48 e 72 h, la glicemia è > 50 mg/dL negli uomini e > 35 mg/dL nelle donne.
A glicemia normalizzata, si assiste alla scomparsa dei sintomi, anche se in rare occasioni il recupero neurologico è ritardato. Un’ipoglicemia severa e prolungata può causare la morte cerebrale.
La risposta contro-regolatoria inizia già a livelli di glicemia non particolarmente ridotti: la soppressione della secrezione insulinica ha luogo per glicemia pari a circa 70 mg/dL e il rilascio di ormoni contro-regolatori (catecolamine, glucagone, cortisolo e GH) si determina quando la glicemia scende al di sotto di 65 mg/dL.
Nella pratica clinica possono presentarsi con il sospetto di ipoglicemia sia pazienti con storia di multipli episodi compatibili con tale diagnosi, sia pazienti con un singolo riscontro occasionale di valori di glicemia a digiuno ridotti. Poiché i sintomi possono essere aspecifici, è essenziale che in qualsiasi soggetto sintomatico sia misurata la glicemia plasmatica durante un episodio di sospetta ipoglicemia. Si suggerisce di proseguire nell’accertamento diagnostico solo nei soggetti in cui è stata confermata la triade di Whipple, pertanto è importante escludere una pseudo-ipoglicemia, legata per esempio a un’errata raccolta del campione di sangue venoso (in provette non contenenti l’inibitore della glicolisi).
L’anamnesi e la raccolta dettagliata delle informazioni relative all’evento ipoglicemico sono essenziali per interpretare l’ipoglicemia e definirne l’eziologia.
L’esame obiettivo consente di valutare il grado di alterazione cognitiva del soggetto e limita gli errori diagnostici e terapeutici anche in corso di valutazione iniziale. La presenza di sudorazione fredda con cute pallida e vaso-costretta si associa all’ipoglicemia, mentre negli stati di chetosi la sudorazione è associata a vaso-dilatazione.
La relazione tra un evento ipoglicemico e la recente assunzione di un pasto è di notevole importanza:
- un’ipoglicemia che insorge 2 o 3 ore dopo un pasto è suggestiva di una condizione di iperinsulinismo o di una malattia come la glicogenosi;
- un episodio ipoglicemico che si manifesta dopo un digiuno prolungato suggerisce un alterato meccanismo legato alla gluconeogenesi;
- un’ipoglicemia post-prandiale può essere indicativa di galattosemia, intolleranza ereditaria al fruttosio, insulinoma, dumping syndrome, sindrome autoimmune.
Bibliografia
- Towler DA, Havlin CE, Craft S, Cryer P. Mechanism of awareness of hypoglycemia. Perception of neurogenic (predominantly cholinergic) rather than neuroglycopenic symptoms. Diabetes 1993, 42: 1791–8.
- Guettier JM, Gorden P. Hypoglycemia. Endocrinol Metab Clin North Am 2006, 35: 753–66.
Approccio diagnostico ragionato alle ipoglicemie
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
L’approccio al paziente è differente se questo si presenti in apparente buona salute o in corso di malattia intercorrente o con pluri-patologie croniche e in base all’assunzione concomitante di farmaci.
Nella malattia cronica, infatti, non è rara l’insorgenza di ipoglicemie, in particolare in presenza di sepsi o insufficienza cardiaca, epatica o renale, ustioni o in casi di malnutrizione; inoltre, va esclusa la presenza di deficit ormonali e considerata l’ipoglicemia secondaria a tumori non insulari.
È fondamentale, inoltre, indagare i farmaci assunti dal paziente, in quanto questi rappresentano una causa frequente di ipoglicemia: oltre alla terapia con insulina o secretagoghi nei pazienti diabetici, anche altri farmaci possono indurre tale condizione, quali ad esempio indometacina, litio e anti-malarici.
Nel paziente in buona salute il sospetto va indirizzato, invece, verso cause di iperinsulinismo endogene o autoimmuni; inoltre, con l’incremento degli interventi di chirurgia bariatrica, sono in aumento le ipoglicemie da tale causa.
Va escluso che il dato di laboratorio sia artefattuale (per problemi legati al prelievo o ritardo nell’esaminazione del campione, o per interferenza biochimica, come in caso di leucemie acute) come pure i casi di ipoglicemia auto-provocata.
In presenza della classica triade di Whipple, le ipoglicemie vanno considerate rispetto al momento di insorgenza, previa esclusione di una pseudo-ipoglicemia.
È fondamentale verificare la storia clinica degli episodi: il momento di insorgenza e la loro durata, la presenza di fattori aggravanti o comunque clinicamente rilevanti. In presenza di soli sintomi neuroglicopenici, è improbabile una reale ipoglicemia, ma è necessario comunque un corretto inquadramento diagnostico. Si parte quindi da un’accurata anamnesi, anche farmacologica, dall’esame obiettivo e da un’attenta valutazione dei dati di laboratorio disponibili: frequentemente questi dati sono sufficienti per orientare verso una causa specifica di ipoglicemia, o quantomeno permettono di escludere ipoglicemie legate all’utilizzo di farmaci, malattie croniche, difetti ormonali o neoplasie non insulari.
Per quanto l’insufficienza corticosurrenalica sia una causa non comune di ipoglicemia, la valutazione della funzione surrenalica può comunque avere un suo ruolo, anche in assenza di altri segni e sintomi specifici per ipocorticosurrenalismo. Va detto che il riscontro di una cortisolemia basale ridotta in corso di un episodio ipoglicemico non pone diagnosi di insufficienza corticosurrenalica, in quanto può dipendere dall’abbassamento della soglia della risposta all’ipoglicemia causata da episodi ipoglicemici subentranti. Sono inoltre da tenere in considerazione le sindromi ipoglicemiche del paziente pancreasectomizzato (deficit di glucagone) e quelle del paziente surrenectomizzato (deficit di adrenalina).
In assenza di una causa evidente di ipoglicemia, l’indicazione è quindi di effettuare una determinazione di glicemia, insulina, C-peptide, proinsulina e idrossi-butirrato (quando disponibile) ed effettuare uno screening per l’assunzione di ipoglicemizzanti orali (teoricamente sia sulfaniluree sia glinidi) in corso di un episodio ipoglicemico spontaneo. Questi dati consentono di discriminare tra iperinsulinismo (sia esso endogeno o esogeno) e altre cause di ipoglicemia. Gli anticorpi anti-insulina, che possono essere misurati indipendentemente dall’episodio ipoglicemico, sono diagnostici per le forme su base autoimmune.
Livelli di idrossibutirrato < 2.7 mmol/L e incremento della glicemia di almeno 25 mg/dL dopo somministrazione di glucagone 1 mg ev dimostrano la presenza di un eccesso di insulina (o di IGF-1); in assenza di tali dati, ci si orienta quindi verso un’ipoglicemia non mediata da eccesso di insulina (quindi ad esempio da deficit ormonale).
Valori di insulina, C-peptide e proinsulina inappropriatamente elevati per i contestuali livelli di glicemia (e non necessariamente francamente elevati) sono diagnostici di iperinsulinismo endogeno: in presenza di glicemia < 55 mg/dL e in assenza di patologie interferenti quali l’insufficienza renale, si considerano significativi valori plasmatici di insulina di almeno 3 mUI/L (18 pmol/L), C-peptide di almeno 0.6 ng/mL (0.22 nmol/L) e proinsulina di almeno 5 pmol/L.
Se invece questo quadro si presenta in concomitanza con il riscontro di livelli di sulfaniluree o glinidi dosabili, la diagnosi è di una ipoglicemia factitia da ipoglicemizzanti orali; la presenza dei soli valori di insulinemia aumentati, con C-peptide e proinsulina normalmente inibiti depone invece per un’ipoglicemia da insulina esogena. Fondamentali per questo tipo di diagnosi rimangono comunque gli elementi di sospetto clinico, legati sia alle caratteristiche degli episodi ipoglicemici sia all’accessibilità ad agenti ipoglicemizzanti per il paziente e/o i suoi familiari.
Nel caso in cui non sia stato possibile effettuare tali esami in corso di un episodio spontaneo di ipoglicemia, è indicato ricreare le circostanze in cui l’ipoglicemia si era presentata, andando quindi a misurare i medesimi analiti ed effettuando, a ipoglicemia documentata, la somministrazione di glucagone 1 mg ev.
In pazienti in cui l’ipoglicemia si presenta tipicamente a digiuno, l’indicazione è di effettuare il test del digiuno; viceversa, in presenza di episodi ipoglicemici post-prandiali è utile effettuare il test del pasto misto. Il pasto deve includere eventuali elementi individuati dal paziente come verosimilmente responsabili dell’ipoglicemia, ma talora vengono utilizzati pasti misti con integratori nutrizionali; il test dovrebbe essere prolungato per 5 ore. Il test prolungato dopo carico orale di glucosio (OGTT) non dispone di standard specifici per l’effettuazione e l’interpretazione dei dati: il 10% dei soggetti normali presenta infatti nadir glicemico < 50 mg/dL durante il test. Comunemente per la conferma diagnostica si utilizzano i medesimi criteri proposti in presenza di ipoglicemia spontanea.
Infine, nei casi in cui il paziente richieda un’infusione di glucosio ev per prevenire ulteriori ipoglicemie, la diagnosi verrà effettuata da prelievi seriati effettuati in ambiente protetto in corso di temporanea sospensione dell’infusione.
Nel complesso, il riscontro di livelli inappropriatamente elevati di insulina, C-peptide e proinsulina plasmatica in pazienti con triade di Whipple documentata, in assenza di ipoglicemizzanti orali e anticorpi anti-insulina dosabili nel plasma, depone verosimilmente per una diagnosi di insulinoma. Esistono, tuttavia, anche altre possibilità di iperinsulinismo endogeno: si parla, infatti, di nesidioblastosi nei casi di incremento della componente ß-cellulare, sebbene non sempre all’esame istologico sia confermato il riscontro di insule ipersviluppate. È inoltre segnalata una forma di nesidioblastosi legata al prolungato utilizzo factitio di sulfaniluree. La secrezione ectopica di insulina, di cui sono stati descritti alcuni casi in letteratura, è invece da considerarsi una forma estremamente rara. Rare sono anche le forme di ipoglicemia iperinsulinemica legate a mutazioni del recettore dell’insulina e di iperinsulinemia indotta dall’esercizio fisico.
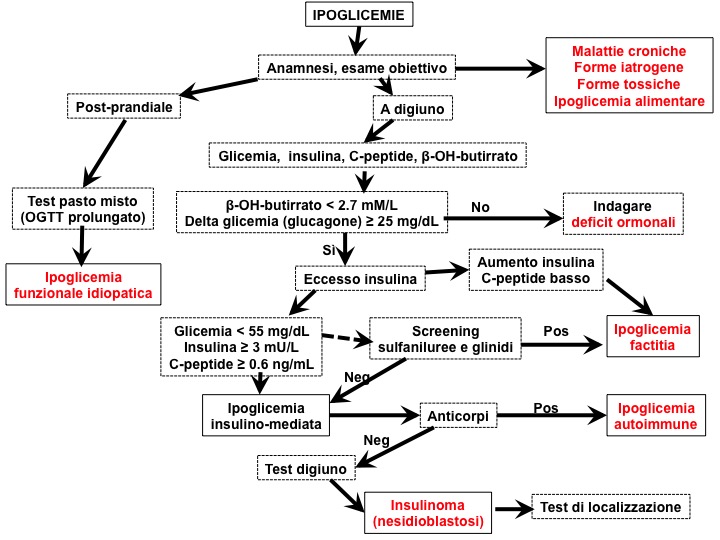
In presenza di evidenze cliniche e laboratoristiche suggestive per ipersecrezione insulinica, bisogna, quindi, proseguire con una sua localizzazione mediante tecniche di imaging. Non esiste al momento una tecnica radiologica di elezione nell’individuazione dell’insulinoma e le sensibilità e specificità delle metodiche variano notevolmente da centro a centro; in ogni caso le tecniche di radiologia tradizionale (ecografia addominale, TC e RM) sono in grado di identificare la maggior parte degli insulinomi, specialmente se di dimensioni > 1-2 cm. Con queste tecniche vengono, inoltre, individuate frequentemente le metastasi che possono essere presenti nei casi di insulinomi maligni (circa il 10% del totale). Tuttavia, poichè sono frequenti insulinomi di dimensioni < 1 cm, la negatività dell’imaging non esclude tale diagnosi (la sensibilità della TC arriva all’85% se spirale e della RM all’85-95%). Il ruolo della scintigrafia con analoghi della somatostatina rimane marginale (sensibilità 80% se SPECT, 30-50% se planare). La PET con Gallio (o DOPA) raggiunge una sensibilità del 97%, LA PET-FDG è utile nelle forme maligne.
L’ecoendoscopia pancreatica, che dà la possibilità di ago-biopsia con ago sottile, presenta maggiore invasività, ma in alcuni centri ha sensibilità > 90% per le lesioni testa-corpo. La combinazione di tali tecniche consente quindi di identificare pre-operatoriamente la gran parte degli insulinomi.
In presenza di imaging non univoco o anche negativo, può esserci indicazione all’arteriografia selettiva pancreatica con test da stimolo con calcio, che consente una localizzazione precisa dell’insulinoma in un’alta percentuale di casi. È inoltre utilizzata per diagnosticare le forme di ipoglicemia a genesi pancreatica non dipendenti da insulinoma e quelle successive a by-pass gastrico Roux-en-Y.
Infine, in corso di intervento chirurgico, l’effettuazione di ecografia pancreatica intra-operatoria identifica in mani esperte la quasi totalità degli insulinomi.
Bibliografia
- Guettier JM, Gorden P. Hypoglycemia. Endocrinol Metab Clin North Am 2006, 35: 753–66.
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009, 94: 709-28.
Angiografia selettiva e stimolazione intra-arteriosa con calcio gluconato
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
Razionale
Gli insulinomi si presentano nella maggioranza dei casi come neoformazioni solitarie, intra-pancreatiche e di modeste dimensioni, per cui la chirurgia ottiene la guarigione nella grande maggioranza dei casi. La difficoltà clinica consiste nell’individuazione pre-chirurgica della corretta localizzazione. Infatti, una pancreasectomia distale “alla cieca” è controindicata per la ridotta probabilità di guarigione e l’elevato tasso di complicanze. La presenza di una corretta localizzazione incrementa le probabilità di successo, riduce la durata dell’atto operatorio e riduce il rischio di re-intervento con il correlato incremento della morbilità.
Nonostante il miglioramento delle tecniche non invasive di imaging, i tumori di dimensioni < 2 cm rimangono di difficile localizzazione. Per questo motivo è stata sviluppata nel 1989 da Doppman una tecnica di iniezione selettiva intra-arteriosa di calcio con successivi prelievi venosi, per individuare isole di cellule tumorali secernenti insulina. Questa tecnica si basa sulla differente risposta allo stimolo con iniezione intra-arteriosa di calcio tra le cellule tumorali e le ß-cellule normali. Nel tempo sono state sviluppate alcune modifiche alla tecnica iniziale, mantenendo nei diversi studi una sensibilità intorno all’80-85%. Questa tecnica può anche trovare applicazione nella localizzazione di lesioni secernenti insulina associate con forme congenite di iperinsulinismo.
Procedura
Si effettua un’arteriografia con mezzo di contrasto non ionico a livello delle arterie mesenterica superiore, prossimale e splenica, gastro-duodenale ed epatica propria; normalmente a seguito di tali incannulamenti risultano perfuse anche la pancreatica dorsale e la pancreatica magna. Dopo l’acquisizione delle immagini, viene effettuata un’infusione di calcio gluconato (tradizionalmente al 10%), con dosaggio adattato al peso del paziente (con correttivi per i pazienti obesi). Vengono prelevati campioni di sangue dalle vene epatiche destra e sinistra prima e 20-40-60 (e talora 90) secondi dopo ogni infusione, per la successiva determinazione dei livelli di insulina.
Interpretazione
Vengono considerate come risposte positive gli incrementi di almeno 2 volte (secondo alcuni di 5 volte) delle concentrazioni venose di insulina a qualsiasi tempo successivo:
- una risposta positiva dall’infusione a livello dell’arteria gastro-duodenale o mesenterica superiore suggerisce la localizzazione a livello della testa/collo del pancreas;
- una risposta positiva dall’infusione nella splenica prossimale o meso-splenica predice una localizzazione a livello di corpo/coda;
- una risposta positiva dopo iniezione nell’arteria epatica propria depone per la presenza di metastasi epatiche.
In presenza di più di una risposta positiva, si considera quella con l’incremento maggiore, dal momento che non è raro un parziale overlap della vascolarizzazione a livello del tumore e questo è quasi sempre di modeste dimensioni.
Giudizio
Complessivamente si tratta di una tecnica invasiva, con necessità di sedazione, da riservarsi ai casi in cui non sia stato possibile ottenere una localizzazione mediante tecniche non invasive; l’utilizzo di questa tecnica piuttosto che dell’ecoendoscopia dipende dall’esperienza e dalla disponibilità presso i vari centri, con complicanze di modesta entità in centri dedicati. L’angiografia sembrerebbe in qualche studio avere una sensibilità complessivamente simile, ma superiore per le lesioni a livello della coda del pancreas. Ancora più importante risulta l’utilizzo di queste tecniche nell’ottica del passaggio a una chirurgia di tipo laparoscopico.
Un caso a parte è rappresentato dagli insulinomi nel contesto della MEN-1, che si presentano più frequentemente come multipli e per i quali è normalmente raccomandata una pancreasectomia distale; in questi casi l’arteriografia con stimolo di calcio, se positiva per lesioni a livello della testa/collo, deve indurre a effettuare anche un’accurata esplorazione intra-operatoria di tali regioni.
Bibliografia
- Guettier JM, Kam A, Chang R, Casey RT, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab 2009, 94: 1074-80.
Terapia generale delle ipoglicemie
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
TERAPIA ACUTA
Ipoglicemia iatrogena lieve-moderata
L’ipoglicemia lieve-moderata con soggetto cosciente va trattata assumendo per bocca 15 g di carboidrati (0.3 g/kg nei bambini) o saccarosio (3-4 cucchiaini di zucchero), preferibilmente glucosio in tavolette o saccarosio in grani o sciolto in acqua o 2 bustine di zucchero da bar o 125 mL di una bibita zuccherata o di un succo di frutta o un cucchiaio da tavola di miele. Si rimisura la glicemia dopo 15 minuti e il trattamento deve essere ripetuto con altri 15 g di carboidrati sino a che la glicemia non risulti > 100 mg/dL. Questo comportamento terapeutico è noto come “la regola del 15”.
La somministrazione di questa quantità di zucchero determina un aumento glicemico medio di 60-80 mg/dL a 15 minuti. L’effetto della soluzione ha un picco a 15-20 minuti, ma la glicemia ricomincia a scendere già dopo 1 ora e l’effetto si esaurisce dopo 2 ore. I carboidrati contenuti in altri alimenti (latte, pane, ecc) non sono sufficienti a dare una risposta glicemica analoga. Il glucosio, infatti, agisce più precocemente rispetto al saccarosio o ad altri tipi di zuccheri semplici.
L’effetto del trattamento sull’ipoglicemia può essere solo temporaneo. Per questo è opportuno continuare a rilevare la glicemia capillare ogni 15 minuti, fino al riscontro di almeno due valori normali in assenza di ulteriore trattamento tra le due misurazioni.
In caso di ipoglicemia in soggetti trattati con acarbosio, la somministrazione orale di disaccaridi o altri carboidrati complessi non permette di risolvere l’episodio ipoglicemico. È raccomandabile, pertanto, l’utilizzo di monosaccaridi (destrosio) o meglio la somministrazione di glucagone o glucosio ev.
In caso di ipoglicemia ripetuta entro 2 ore dall’ultima somministrazione di insulina o se conseguente ad altre ipoglicemie (Hypo begets Hypo) o se in uso schema diverso da basal-bolus con analoghi rapidi e lenti, è utile assumere anche carboidrati a lento assorbimento. In caso di terapia con microinfusore, va preso in considerazione l’utilizzo delle basali temporanee alla ripresa dell’erogazione insulinica. Sono sconsigliati zuccheri semplici associati a grassi.
Ipoglicemia iatrogena grave
Se ci si trova in una situazione di emergenza fuori da un ospedale e lontano da un pronto soccorso o quando non sia possibile fare un’iniezione endovenosa, negli adulti e nei bambini di età > 12 anni si possono utilizzare siringhe pre-riempite di glucagone; per i bambini < 12 anni la dose è 0.5 mg (nei bambini < 20 kg è 15 µg/kg). Le persone a stretto contatto con i diabetici e/o i care-giver devono essere istruiti alla somministrazione del farmaco per via intramuscolare o sottocutanea.
Il glucagone determina un rapido incremento glicemico (medio di 150 mg in 15’), con picco a 1 ora; poiché la glicemia ricomincia a scendere dopo 90 minuti, al risveglio del paziente devono essere assunti carboidrati per bocca.
Nel diabetico tipo 2 l’efficacia di questa terapia è minore, poiché il glucagone stimola la secrezione di insulina endogena.
Nel caso sia possibile eseguire rapidamente un’iniezione per via endovenosa, è indicata l’infusione di 7-10 grammi di glucosio in bolo ev in 1-3 minuti (2-3 fl da 10 mL di soluzione glucosata 33% oppure 80 mL di soluzione glucosata al 20%), eventualmente ripetibile, seguita dall’infusione di soluzione glucosata al 5-10%. Nei bambini è consigliata una dose di 200-500 mg/kg.
L'incremento glicemico dopo bolo ev è transitorio, soprattutto se l’ipoglicemia è causata da sulfaniluree. In tal caso alcuni autori suggeriscono la somministrazione di soluzione glucosata al 20%, in associazione con octreotide 0.1 mg sc/12 ore. L’octreotide sopprime l’increzione endogena di insulina, prevenendo quindi successive crisi ipoglicemiche.
TERAPIA CRONICA O DEFINITIVA
Iperinsulinismo endogeno
Intervento dietetico (pasti piccoli e frequenti), inibitori dell’alfa-glicosidasi (acarbosio), diazossido (3-8 mg/kg/die nel paziente adulto, frazionato in due-tre somministrazioni fino a 1000 mg/die), analoghi della somatostatina (octreotide o lanreotide), pancreasectomia totale o subtotale.
Nelle sindromi ipoglicemiche refrattarie (insulinoma maligno), accanto alle terapie convenzionali (diazossido, octreotide/lanreotide, steroidi, ß-bloccanti e diuretici) e alla terapia in continuo con glucosata infusiva e/o glucagone sottocute, si comincia ad avere esperienza dell’utilizzo di pasireotide (analogo della somatostatina che riduce l'insulina e non il glucagone) ed everolimus, inibitore di mTOR che agisce a più livelli (aumento dell’insulino-resistenza attraverso inibizione di GLUT1 e riduzione del trasporto di glucosio dentro la cellula e riduzione della pro-insulina) determinando iperglicemia.
La terapia radiorecettoriale (Y90, Lu177) rappresenta un nuovo approccio terapeutico.
Ipoglicemie autoimmuni
Il trattamento è dietetico (dieta equilibrata, normocalorica, priva di zuccheri semplici, ricca in fibre, frazionata in piccoli pasti, amido di mais prima di andare a dormire). Quando necessario, si possono associare farmaci immuno-soppressori (prednisone), acarbosio, diazossido e, in casi isolati, si ricorre alla plasmaferesi.
Il monitoraggio glicemico continuo costituisce senz’altro un valido supporto terapeutico.
Ipoglicemia reattiva
Pasti piccoli e frequenti (non più di tre ore tra un pasto e l’altro), con preferenza per gli zuccheri complessi rispetto a quelli semplici, a basso indice glicemico (yogurt, piselli, mele, pesche, riso parboiled, latte, fagioli, noci), alimenti integrali e ricchi di fibre che rallentano l’assorbimento degli zuccheri a livello intestinale, frutta e verdura.
Non assumere alcolici e alimenti ricchi di zuccheri a stomaco vuoto.
Non svolgere attività fisica a digiuno, ma praticare regolarmente attività fisica moderata, mai eccessiva.
Paziente ospedalizzato
Supporto nutrizionale parenterale o enterale, monitoraggio continuo della glicemia, glucosio ev.
BIBLIOGRAFIA
- Villani M, de Courten B, Zoungas S. Emergency treatment of hypoglycaemia: a guideline and evidence review. Diabet Med 2017, 34: 1205-11.
- Challis BG, Poulson AS, et al. Adult-onset hyperinsulinaemic hypoglycaemia in clinical practice: diagnosis, aetiology and management. Endocr Connect 2017, 6: 540-8.
Scheda diazossido
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
Meccanismo d’azione
Inibisce la secrezione insulinica.
Indicazioni
Trattamento dell’ipoglicemia sintomatica da iperinsulinismo di diversa eziologia (ipoglicemia idiopatica dell’infanzia, tumori insulari funzionanti, neoplasie extra-pancreatiche determinanti ipoglicemia, ipoglicemia neonatale, tesaurismosi glicogenica, ipoglicemia ad eziologia sconosciuta). Può essere usato in periodo pre-operatorio come trattamento temporaneo e dopo l’intervento se l’ipoglicemia persiste.
Contro-indicazioni
Ipoglicemia funzionale, ipersensibilità ad altri tiazidici. Gravidanza e allattamento. Rari problemi ereditari di intolleranza al galattosio, deficit di lattasi congenito o acquisito o malassorbimento di glucosio-galattosio.
Preparazioni, via di somministrazione, posologia
cp 25 e 100 mg (Proglicem), 100-200 mg/die in 3 somministrazioni (3-8 mg/kg da somministrare frazionati in 2-3 dosi uguali).
Precauzioni
Eseguire regolarmente esami ematologici per escludere alterazioni del quadro leucocitario e delle piastrine, oltre al controllo periodico di glicemia, uricemia, glicosuria e chetonuria.
La proprietà anti-diuretica del diazossido può comportare significativa ritenzione di fluidi, che nei pazienti con compromessa riserva cardiaca può sfociare in insufficienza cardiaca congestizia.
Sono stati riferiti chetoacidosi e coma iperosmolare non chetosico in corso di patologie concomitanti.
Raramente l’ipopotassiemia rappresenta un problema, anche se il diazossido viene associato a un diuretico tiazidico.
Nei pazienti con insufficienza renale, si ha allungamento dell’emivita plasmatica del medicinale, con la necessità di ridurre opportunamente il dosaggio e controllare gli elettroliti sierici.
Non sono disponibili specifiche informazioni sull’uso negli anziani. Nei bambini si deve valutare la crescita e lo sviluppo osseo e psichico.
Effetti collaterali
Comuni: ritenzione idrosodica, tachicardia, palpitazioni, ipertricosi, anoressia, nausea, vomito, dolori addominali, ipertensione, trombocitopenia, iperglicemia e glicosuria, sintomi extra-piramidali.
Non comuni: pancreatite acuta, dolore toracico, diarrea.
Limitazioni prescrittive
Rimborsabile dal SSN con Ricetta Ripetibile Limitativa.
Ipoglicemie a digiuno insulino-mediate
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
Non-insulinoma pancreatogenous hypoglycemia syndrome (NIPHS - nesidioblastosi)
Clinicamente è caratterizzata da episodi di neuroglicopenia, generalmente ma non esclusivamente post-prandiali, secondari a iperinsulinismo. È più frequente nel sesso maschile. Istologicamente è caratterizzata da diffuso interessamento delle ß-cellule con nesidioblastosi (ipertrofia della ß-cellula, iperplasia dei nuclei della ß-cellula che risultano ipercromici e allargati). Il test del digiuno protratto è spesso negativo, così come le procedure diagnostiche radiologiche per l’identificazione di un insulinoma. La conferma dell’iperfunzione delle ß-cellule viene dal test di stimolazione intra-arteriosa selettiva con calcio. La base genetica sottostante la NIPHS risulta ancora sconosciuta. La terapia è chirurgica: pancreasectomia parziale guidata da test di stimolo con calcio.
La forma secondaria a by-pass gastrico tipo Roux-en-Y si manifesta anche a distanza di alcuni mesi dall’intervento e si caratterizza per neuroglicopenia prevalentemente nel periodo post-prandiale. La terapia è medica (acarbosio, diazossido, octreotide) e in seconda battuta chirurgica (pancreasectomia parziale).
La forma transitoria, dovuta a deficit di substrati, si riscontra in alcune condizioni patologiche del neonato: neonato di madre diabetica, asfissia neonatale, malattia emolitica del neonato, ritardo di crescita intra-uterino, sindrome di Beckwith-Wiedemann.
L'iperinsulinismo familiare (FHI, da mutazione di ABCC8, KCNJ11, GCK, GDH, HADH, HNF4A, SLC16A1) si manifesta in genere in epoca neonatale, da poche ore a due giorni dalla nascita, con sintomatologia di grado variabile. La malattia dell'infanzia si manifesta durante i primi mesi o anni di vita. In casi gravi, le concentrazioni di glucosio nel sangue sono tipicamente estremamente basse e quindi facilmente riconoscibili, mentre in casi più lievi l'ipoglicemia variabile e lieve può rendere più difficile la diagnosi. Anche all'interno della stessa famiglia, le manifestazioni della malattia possono variare da lievi a gravi. Le forme lievi rispondono alla terapia con diazossido.
Bibliografia
- Kaczirek K, Soleiman A, Schindl M, et al. Nesidioblastosis in adults: a challenging cause of organic hyperinsulinism. Eur J Clin Invest 2003, 33: 488–92.
- Yorifuji T, Horikawa R, Hasegawa T, et al. Clinical practice guidelines for congenital hyperinsulinism. Clin Pediatr Endocrinol 2017, 26: 127-52.
Ipoglicemie a digiuno non insulino-mediate
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
Da deficit ormonale
Iposurrenalismi primari
Deficit di GH
Ipopituitarismo
Ipoglicemie dovute a malattie acquisite
Numerosi stati di malattia acuta (fegato, cuore, reni), la sepsi, la malnutrizione grave e tutte le condizioni associate a marcata atrofia delle masse muscolari, si associano allo sviluppo di ipoglicemie.
Un’epatopatia diffusa può causare ipoglicemia a digiuno quando sono compromesse la glicogenolisi e la gluconeogenesi. Questa situazione si verifica nel caso di necrosi epatica acuta (da epatite virale, tossine, sindrome di Reye), glicogenosi, deficit di enzimi gluconeogenetici e in altre cause di insufficienza epatica grave.
L'ipoglicemia a digiuno si manifesta inoltre occasionalmente nei pazienti con insufficienza renale grave e negli stati uremici. La nefropatia nei diabetici in trattamento insulinico può provocare ipoglicemia, diminuendo la degradazione renale dell'insulina.
La cachessia e lo shock endotossinico possono provocare ipoglicemia a digiuno in qualunque fascia d'età.
Ipoglicemia dovuta a difetti congeniti del metabolismo
Un’ipoglicemia può riscontrarsi nel deficit dei substrati della gluconeogenesi. Il basso turnover di alanina nell’infanzia o ipoglicemia idiopatica chetotica, si caratterizza per episodi ricorrenti di ipoglicemia a digiuno, con elevati livelli plasmatici di acidi grassi liberi e corpi chetonici, in presenza di lattacidemia solitamente nella norma. L’ipoglicemia idiopatica chetotica esordisce di solito tra i 18 mesi e i 5 anni e si risolve spontaneamente intorno agli 8-9 anni. Le crisi compaiono soprattutto dopo il digiuno notturno o in concomitanza di episodi febbrili. Colpiscono più spesso bambini di basso peso e con storia di vomito frequente.
I pazienti con difetti congeniti degli enzimi coinvolti nella gluconeogenesi (piruvato carbossilasi, fosfoenolpiruvato-carbossilasi, fruttosio 1,6-difosfatasi e glucosio 6-fosfatasi) manifestano un quadro clinico caratterizzato da ipoglicemia e acidosi lattica. Il più frequente è la glicogenosi di tipo 1 (malattia di Von Gierke), legata al deficit dell’enzima glucosio 6-fosfatasi. I pazienti affetti da tale patologia presentano episodi ricorrenti di ipoglicemia associata ad acidosi lattica, iperuricemia e iperlipidemia. Nel deficit di piruvato-carbossilasi il quadro si presenta più variegato e comprende, oltre all’acidosi lattica, anche ritardo mentale e acidosi tubulare renale prossimale. Infine, il deficit primitivo di carnitina è un disordine autosomico recessivo dell’ossidazione degli acidi grassi, che può manifestarsi in diverse fasi dell’età evolutiva, associato a episodi di ipoglicemia non chetosica, miopatia e cardiomiopatia.
Le ipoglicemie in età infantile possono essere causate da alterazioni metaboliche ereditarie, quali galattosemia, fruttosemia, tirosinemia, acidemie organiche (acido propionico, metilmalonico), la malattia a sciroppo d’acero e i difetti della catena respiratoria mitocondriale. L’intolleranza al fruttosio è una condizione ereditaria recessiva conseguente a deficit dell’enzima aldolasi B (fruttosio 1,6-fosfato aldolasi), nella quale i soggetti omozigoti sviluppano ipoglicemia e importanti disturbi addominali dopo l’assunzione di cibi contenenti fruttosio. Le sindromi CDG (Congeniti Disturbi della Glicosilazione) sono un gruppo di malattie autosomiche recessive, che interessano la sintesi glicoproteica.
Bibliografia
- Eckert-Norton M, Kirk S. Non-diabetic hypoglycemia. J Clin Endocrinol Metab 2013, 98: 39A-40A.
Ipoglicemie autoimmuni
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
La sindrome insulinica autoimmune (IAS o sindrome di Hirata) è una rara condizione clinica caratterizzata dallo sviluppo di un elevato titolo di anticorpi anti-insulina (AIAA) in assenza di anomalie della ß-cellula e di precedente esposizione all’insulina esogena. Risulta estremamente rara nella popolazione caucasica, mentre sono stati riportati diversi casi nella popolazione giapponese e coreana.
I pazienti affetti dalla sindrome, per lo più di sesso femminile e di ogni età, presentano una storia clinica di patologie autoimmuni (Graves, artrite reumatoide, LES, polimiosite) o esposizione a farmaci contenenti gruppi sulfidrilici (metimazolo, glutatione, captopril, acido α-lipoico - ampiamente impiegato nella neuropatia diabetica), che interagiscono con i ponti disolfuro all’interno della molecola dell’insulina, rendendola molto più immunogenica. La IAS è tipicamente descritta in pazienti giapponesi con m. di Graves trattati con metimazolo, per la presenza di suscettibilità genetica.
La sintomatologia, prevalentemente neuroglicopenica, si manifesta nel periodo post-prandiale tardivo e a digiuno. Gli episodi di ipoglicemia sono causati dal legame e poi dal rilascio dell’insulina da parte degli anticorpi, asincrono rispetto ai livelli di glucosio. È frequente in questi pazienti il riscontro di iperglicemie post-prandiali correlate al legame tra anticorpi e insulina secreta dopo un pasto. Questo legame riduce la biodisponibilità dell’insulina a livello epatico e nei tessuti periferici, determinando iperglicemia e ulteriore secrezione insulinica (diagnosi differenziale con esordio di diabete mellito tipo 2 per possibile riscontro di HbA1c elevata) e successiva comparsa di ipoglicemia.
Le concentrazioni di insulina circolanti durante l’ipoglicemia sono in genere estremamente elevate (> 100 mU/L) per un artefatto nel dosaggio causato dagli autoanticorpi, l’insulina libera è normale o elevata, la proinsulina e il C-peptide sono elevati (quest’ultimo in misura minore). Gli anticorpi anti-insulina mostrano un’elevata percentuale di legame con l’insulina in soggetti non precedentemente trattati con insulina (diagnosi differenziale con insulinoma) e sono più alti rispetto all’ipoglicemia da insulina esogena (diagnosi differenziale con ipoglicemia factitia, in cui sono spesso riscontrabili anticorpi anti-insulina). Gli anticorpi anti-insulina possono essere evidenziati mediante precipitazione con PEG (la stessa metodica che si usa per la valutare la presenza di macroprolattinemia) dei complessi antigene-anticorpo marcati con insulina radioattiva. Un’accurata anamnesi clinico-farmacologica e la presenza di HLADRB1*0403 (nella popolazione caucasica) costituiscono un valido supporto alla diagnosi.
La sindrome è caratterizzata da risoluzione spontanea nell’arco di 1-3 mesi dall’esordio/sospensione del farmaco causale (vedi acido α-lipoico).
Il trattamento è dietetico: dieta equilibrata, normocalorica, priva di zuccheri semplici, ricca in fibre, frazionata in piccoli pasti; quando necessario, si possono associare farmaci immuno-soppressori (prednisone), acarbosio e, in casi isolati, si ricorre alla plasmaferesi. Il monitoraggio glicemico continuo costituisce senz’altro un valido supporto terapeutico.
La comparsa di autoanticorpi anti-insulina è descritta anche a seguito della cronica esposizione all’insulina esogena nei pazienti diabetici. In tale caso comporta un elevato fabbisogno insulinico con importante variabilità glicemica.
Gli anticorpi anti-recettore dell’insulina (AIRA) caratterizzano una rara forma di diabete insulino-resistente, che colpisce gli adulti, prevalentemente di sesso femminile, con presenza di acanthosis nigricans e irsutismo, in un quadro di ovaio policistico. Gli AIRA mimano gli effetti dell'insulina, provocando ipoglicemia a digiuno, che può essere estremamente severa. L’insulina, dopo la sua secrezione pancreatica, è rapidamente rimossa dalla circolazione attraverso un meccanismo in parte mediato dal suo recettore specifico: una rapida clearance dell’insulina rappresenta il punto critico per la normale omeostasi glicidica. Gli AIRA hanno la capacità di attivare il recettore in modo inappropriato, indipendentemente dai livelli di insulina circolante. Ciò può causare gravi ipoglicemie, oppure una condizione di insulino-resistenza, che si evidenzia quando la cronica presenza di questi anticorpi è in grado di desensibilizzare il recettore dell’insulina (sindrome da insulino-resistenza di tipo B). Anche questa sindrome si associa a altre patologie autoimmuni o a malattie linfo-proliferative (Hodgkin).
La diagnosi si basa sul quadro clinico, sui risultati delle analisi di laboratorio e sull'individuazione di autoanticorpi diretti contro il recettore dell'insulina nel siero.
Il trattamento si basa sull'uso di immuno-soppressori associati a insulina in caso di iperglicemia. La prognosi è sfavorevole nei casi con ipoglicemia (porta alla morte nel 50% dei casi).
Il leprecaunismo è una malattia congenita molto rara (meno di un caso ogni milione di nati) da insulino-resistenza estrema, trasmessa come carattere autosomico recessivo. È dovuta alle mutazioni omozigoti o eterozigoti composte del gene del recettore dell'insulina (INSR; 19p13.3-p13.2).
La malattia è caratterizzata da grave ritardo della crescita già in epoca pre-natale e si associa a facies dismorfica caratteristica (che ricorda gli gnomi del folclore irlandese, i leprecani), lipo-atrofia del tessuto adiposo sottocutaneo e ipotrofia muscolare. Nelle giovani donne sono spesso presenti segni di virilizzazione. Si osservano episodi di ipo- e iper-glicemia, con costante iperinsulinemia.
La diagnosi differenziale si pone con le altre forme di insulino-resistenza estrema (la sindrome di Rabson-Mendenhall, le lipodistrofie e le sindromi dette di tipo A e B).
Può essere considerato il trattamento con IGF-I ricombinante. La prognosi non è certa, la crescita è gravemente compromessa e l’attesa di vita raramente supera i due mesi.
Bibliografia
- Gullo D. La sindrome ipoglicemica da anticorpi anti-insulina (sindrome di Hirata). AME News 46/2013.
Ipoglicemie post-prandiali o reattive
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
Ipoglicemia alimentare
È caratterizzata dalla comparsa di ipoglicemia post-prandiale precoce in soggetti sottoposti a intervento di gastro-resezione, gastro-digiunostomia, piloro-plastica con vagotomia. Riguarda il 10% dei pazienti operati per ulcera gastro-duodenale. La forma funzionale idiopatica si può verificare anche nel paziente non gastrectomizzato, per iperattività vagale dopo riempimento gastrico.
Compare da ½ ora a 3 ore dopo il pasto e si caratterizza per senso di ripienezza, nausea, vomito, tachicardia.
È dovuta all’accelerato svuotamento gastrico, con conseguente ipersecrezione insulinica da rapido assorbimento intestinale di glucosio.
La terapia consiste nell’assumere pasti piccoli e frequenti, prevalentemente solidi e poveri di carboidrati, possibilmente a temperatura corporea; bere lontano dai pasti; anti-colinergici per rallentare la motilità intestinale nella forma funzionale.
Dumping syndrome tardiva
È una sindrome clinica caratterizzata dalla diminuzione dei livelli plasmatici di glucosio, che si verifica entro 2-3 ore dall’assunzione di un pasto, per lo più ad alto tenore di carboidrati. Non è infrequente nei soggetti grandi obesi che si sottopongono a by-pass gastrico: il rapido passaggio dei nutrienti nel piccolo intestino comporta un aumento di GLP-1, con ipertrofia e iperfunzione delle ß-cellule.
La diagnosi è clinica e confermata dalla comparsa di ipoglicemia durante test OGTT prolungato (5 ore), con dosaggio di glicemia e insulina.
La terapia consiste nell’assunzione di pasti piccoli e frequenti, poveri di carboidrati ad alto indice glicemico, eliminazione dell’alcool e introduzione di fibre di emicellulosa e guar o acarbosio per ridurre l’assorbimento del glucosio.
Ipoglicemia funzionale idiopatica
Si definisce tale un’ipoglicemia precoce post-prandiale senza altre cause. Compare da ½ ora a 3 ore dopo il pasto e si caratterizza per fatica, ansietà, irritabilità, debolezza, difficoltà alla concentrazione, senso di fame dopo il pasto e tremore generalizzato.
La causa è un’aumentata sensibilità individuale all’insulina.
La diagnosi è clinica e per comparsa di ipoglicemia precoce (ma a volte compaiono i sintomi e non l’ipoglicemia!) durante test con carico orale di glucosio (OGTT) prolungato. È spesso una diagnosi di esclusione.
Un'ipoglicemia di questo tipo può rappresentare una fase precoce del diabete tipo 2, come conseguenza della secrezione aumentata e/o ritardata di insulina.
La terapia consiste nel ridurre o eliminare gli zuccheri raffinati dalla dieta, ridurre il volume dei pasti e aumentarne la frequenza; può essere utile un appoggio psicologico.
Bibliografia
- AMD-SID. Standard italiani per la cura del diabete mellito. 2016.
Ipoglicemie esogene
Micaela Pellegrino & Francesca Garino
Endocrinologia Diabete e Metabolismo, Ospedale S Croce e Carle, Cuneo
(aggiornato al 19 settembre 2017)
Alcool
Le ipoglicemie indotte dall’assunzione di alcool sono frequenti e legate a stati di deplezione di glicogeno, indipendentemente dal fatto che il digiuno sia precedente o successivo all’assunzione. L’alcool inibisce la gluconeogenesi, riducendo la risposta degli ormoni contro-regolatori, la captazione dei precursori della gluconeogenesi e inibendo l’ossidazione di lattato e glutammato a livello di fegato e rene per carenza di NAD (l’etanolo è ossidato ad acetaldeide, con la conversione di NAD in NADH; glicerolo, aminoacidi come l’alanina, e acido lattico necessitano di NAD per entrare nella via gluconeogenetica).
Il paziente presenta sintomi neuroglicopenici (compromissione dello stato di coscienza, stupor, fino al coma), che possono essere confusi con gli effetti neurotossici dell’alcol.
Si associa frequentemente all'aumento dei livelli plasmatici di lattato e chetoni e ad acidosi metabolica. Non esiste correlazione tra livelli plasmatici di etanolo e grado di ipoglicemia, che può insorgere anche quando i livelli ematici di etanolo sono in diminuzione. I livelli plasmatici di insulina sono inappropriatamente bassi, ma i meccanismi contro-regolatori per ripristinare l’euglicemia sono inadeguati, per cui è necessaria l’ingestione o l’infusione di glucosio.
Tossine
Da segnalare l’ipoglicemia da intossicazione acuta da frutto ackee acerbo (detta anche malattia del vomito giamaicana), frequente nei paesi caraibici e dell’Africa occidentale; è causata dall’effetto dell’ipoglicina A contenuta nei semi (che si riduce progressivamente con la maturazione del frutto), che ha azione cito-tossica sulle ß-cellule e inibisce la gluconeogenesi. La stessa tossina è contenuta nel litchi.
L’ipoglicemia è una tra le cause di morte da Amanita Phalloide.
Farmaci
I farmaci di uso più comune che causano ipoglicemia sono riportati in tabella.
L’insulina e gli altri farmaci che ne stimolano la secrezione sono quelli più frequentemente responsabili di ipoglicemie, intenzionalmente (nel caso delle ipoglicemie factitie) o in caso di ridotta assunzione di cibo o di errore nella somministrazione/assunzione del farmaco.
Nel soggetto non diabetico, i farmaci più frequentemente imputati per l’insorgenza di ipoglicemie sono propranololo, sulfonamidi e salicilati.
I ß-bloccanti non selettivi, tra i quali il propranololo, inibiscono il rilascio di glucosio a livello epatico e renale, aumentano la sensibilità dei tessuti periferici all’insulina, mascherando i sintomi di insorgenza dell’ipoglicemia, primo tra tutti la tachicardia.
Le sulfonamidi possono incrementare il rilascio di insulina, con un meccanismo simile a quello delle sulfaniluree.
Il meccanismo con cui i salicilati sono in grado di provocare ipoglicemie non è chiaro, ma potrebbe coinvolgere sia l’inibizione del rilascio di glucosio a livello epatico, sia lo stimolo della secrezione insulinica; le ipoglicemie da salicilati sono descritte più frequentemente nei bambini.
La pentamidina, utilizzata nel trattamento della polmonite da Pneumocystis in pazienti con AIDS, può indurre ipoglicemia con un meccanismo particolare: è infatti citotossica sulle ß-cellule e determina il rilascio dei depositi di insulina e conseguente ipoglicemia, che è successivamente seguita da un condizione di iperglicemia fino a un franco diabete.
Il chinino causa ipoglicemia con un meccanismo insulino-secretagogo.
Un abbassamento dei livelli di glucosio può verificarsi anche in seguito all’assunzione di cimetidina e ranitidina (che interferiscono con il metabolismo delle sulfaniluree), indometacina, difenidramina e altri anti-istaminici, lidocaina, litio, aloperidolo e tramadolo, e, in casi rari, durante assunzione di ACE-inibitori e sartani (per incremento della sensibilità insulinica). Questi farmaci sono stati segnalati come possibili cause di ipoglicemia, prevalentemente se assunti in concomitanza con farmaci ipoglicemizzanti o in presenza di comorbilità importanti, quali l’insufficienza epatica o renale severa.
L’ipoglicemia è stata recentemente descritta come effetto collaterale degli inibitori della tirosin-chinasi, in uso nel trattamento di alcune neoplasie maligne. Il meccanismo non è noto, ma potrebbero interferire con il recettore dell’insulina, recettore trans-membrana appartenente alla famiglia dei recettori tirosin-chinasici. L’uso di questi farmaci nei pazienti diabetici può implicare la necessità di ridurre le terapie anti-diabetiche in atto.
| Farmaci che possono indurre ipoglicemia | ||
| Meccanismo | Farmaco | |
| Insulino-mediato | Aumento dell’insulinemia | Insulina Sulfaniluree Disopiramide Chinino Pentamidina Ritodrine Isoniazide Sulfonamidi |
| Aumento della sensibilità insulinica | ß-bloccanti ACE-inibitori Biguanidi Agonisti PPAR-γ |
|
| Autoimmune | Idralazina Isoniazide Procainamide α-interferone Farmaci contenenti gruppi sulfidrilici: metimazolo, captopril, penicillamina, acido α-lipoico |
|
| Non definito | Salicilati Anti-coagulanti dicumarolici Anti-infiammatori: indometacina, colchicina, paracetamolo, fenilbutazone Lidocaina Tramadolo Anti-istaminici: cimetidina, ranitidina, difenidramina Anti-psicotici: aloperidolo, clorpromazina Litio Fenitoina Chetoconazolo Octreotide Inibitori tirosin-chinasi |
|
Bibliografia
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009, 94: 709-28.