Ipercalcemia acuta

Ernesto de Menis
Medicina Interna, Montebelluna
Classificazione ed eziopatogenesi
La classificazione dell’ipercalcemia può essere di tipo eziopatogenetico o basata sui livelli di calcio e la sintomatologia (tab 1). L’ipercalcemia severa può presentarsi talora come “crisi ipercalcemica”.
| Tabella 1 Classificazione dell’ipercalcemia in base ai valori di calcio |
|
| Grado | Calcemia (mg/dL) |
| Lieve | < 12 |
| Moderata | 12-14 |
| Severa | > 14 |
Le condizioni di ipercalcemia moderata e severa sono in genere osservate in ambito ospedaliero e due sono le cause principali (1): l’iperparatiroidismo primitivo e soprattutto l’ipercalcemia associata a neoplasie.
Le neoplasie determinano ipercalcemia con diversi meccanismi (2). La causa più frequente è la cosiddetta Ipercalcemia Umorale, cioè indotta da sostanze circolanti come il PTHrP (PTH-related peptide); in casi meno frequenti l’ipercalcemia è sostenuta da metastasi o da produzione anomala di metaboliti attivi della vitamina D.
Spesso l’aggravamento dell’ipercalcemia è causata da altri fattori, quali disidratazione (ad esempio iporessia, vomito e diarrea nel paziente oncologico), peggioramento della funzione renale o immobilizzazione. La disidratazione è legata al vomito, ma soprattutto alla perdita di sali e acqua a livello renale indotti dall’ipercalcemia: ciò determina un’insufficienza renale pre-renale, che a sua volta riduce l’escrezione renale di calcio (circolo vizioso).
Clinica e diagnostica
Il quadro clinico è condizionato dalla severità dell’ipercalcemia stessa e da altre condizioni, quali velocità di sviluppo dell’ipercalcemia, età del paziente, uso concomitante di farmaci come i sedativi. Il quadro è dominato da:
- sintomi neuro-muscolari, con astenia, rallentamento ideo-motorio e alterazioni progressive della vigilanza fino al coma;
- sintomi gastro-intestinali, con nausea, vomito, stipsi e talora quadri di pancreatite acuta;
- presenza di poliuria (con polidipsia) e frequente insufficienza renale almeno inizialmente di tipo pre-renale;
- interessamento cardio-vascolare, con ipertensione, accorciamento del QT, rischio di tossicità da digitale.
Nell’inquadramento iniziale dell’ipercalcemia acuta severa del paziente ospedalizzato la distinzione fondamentale è se l’ipercalcemia è o non è PTH-dipendente. Di fronte a valori aumentati di PTH, viene impostata la diagnostica dell’iperparatiroidismo, mentre in caso di valori soppressi o inappropriatamente normali di PTH il quadro è orientativo per una forma associata a neoplasia (senza dimenticare altre condizioni più rare come l’intossicazione da vitamina D attiva). In questa fase, in aggiunta al dosaggio del PTH, devono essere valutati fosforemia, equilibrio idro-elettrolitico, vitamina D (che è utile nell’interpretazione dei livelli di PTH) e soprattutto funzione renale. Il dosaggio del PTHrP viene eseguito solo in alcuni Centri e comunque ha valore primariamente di ricerca. Inoltre, deve essere valutato il grado di deplezione volemica.
Terapia
La terapia dell’ipercalcemia acuta richiede una valutazione della gravità generale del quadro. Nelle forme severe i punti cardine sono:
- correzione del deficit volemico;
- inibizione del riassorbimento osseo.
Misure generali, che hanno in genere un valore limitato ma sono consigliabili, sono:
- riduzione dell’apporto dietetico di calcio, soprattutto quando la nutrizione è enterale o parenterale;
- sospensione di farmaci che potenzialmente possono indurre ipercalcemia (supplementi di vitamina D, tiazidici, litio, ...);
- evitare il riassorbimento da immobilità e quindi favorire precocemente la mobilizzazione attiva;
- sospensione dei sedativi che aggravano il quadro neurologico;
- correzione dell’ipofosfatemia, ma non attraverso la somministrazione parenterale di fosforo.
La correzione dell’ipovolemia (anche subclinica) determina miglioramento della funzione renale e aumenta l’escrezione urinaria di calcio. Si utilizzano soluzioni isotoniche di cloruro di sodio, addizionate eventualmente di altri elettroliti (es. potassio e magnesio): inizialmente devono essere infuse quantità generose (anche 500 mL/h), poi la velocità di infusione viene regolata sulla base della diuresi e del rischio di sovraccarico idro-salino. I diuretici dell’ansa aumentano l’escrezione urinaria di calcio, per cui in passato sono stati utilizzati a dose elevate (concetto di diuresi salina forzata), tuttavia i risultati sono stati assolutamente sfavorevoli perché, aggravando l’ipovolemia, determinavano peggioramento dell’ipercalcemia. Pertanto, attualmente i diuretici dell’ansa vengono impiegati solo dopo ripristino della volemia, a dosi moderate e soprattutto nei pazienti a rischio di sovraccarico volemico (es. cardiopatici). Durante questa fase di ripristino della volemia ed eventuale utilizzo dei diuretici deve esservi un attento monitoraggio non solo della calcemia ma anche della funzione renale e degli altri elettroliti.
Il secondo punto cardine del trattamento dell’ipercalcemia severa prevede il blocco del riassorbimento osseo. Sono stati abbandonati mitramicina e gallio nitrato e i farmaci di riferimento sono oggi i bisfosfonati. In questo ambito sono maggiormente utilizzati pamidronato e zoledronato, in particolare quest’ultimo che presenta maggior velocità di azione e più comoda somministrazione. Un limite all’utilizzo dei bisfosfonati è rappresentato dalla presenza di insufficienza renale (controindicazione, limitazione della dose, aumento della durata di infusione) nei pazienti gravemente ipercalcemici (4). Si deve tuttavia rilevare che tale insufficienza renale presenta rapidamente un miglioramento dopo adeguata idratazione, permettendo l’uso successivo di tali farmaci.
Con la disponibilità dei bisfosfonati l’utilizzo della calcitonina nell’ipercalcemia severa si è molto ridotto, anche perché ha un effetto transitorio per lo sviluppo di tachifilassi.
L’utilizzo dei corticosteroidi è limitato a situazioni specifiche, come in caso di ipercalcemia sostenuta da 1,25-OH-vitamina D (intossicazione, malattie granulomatose, rare forme neoplastiche ematologiche) o nelle malattie ematologiche come il mieloma.
In situazioni in cui l’ipercalcemia severa non venga controllata dalle precedenti terapie o in casi in cui la compromissione renale sia tale da controindicare l’utilizzo dei bisfosfonati o ancora in casi con associato scompenso cardiaco, può venire utilizzata l’emodialisi.
Altri farmaci sono utilizzati nell’ipercalcemia in generale, sia da iperparatiroidismo sia associata a neoplasie. Riguardo il cinacalcet vi sono segnalazioni di utilizzo nelle forme severe di ipercalcemia; anche il denosumab appare efficace nel controllo degli eventi ipercalcemici nei pazienti neoplastici. Nell’ipercalcemia severa l’impiego di entrambi questi farmaci deve essere limitato a casi selezionati e per il cinacalcet come terapia aggiuntiva alle altre terapie.
Bibliografia
- Lindner G, et al. Hypercalcemia in the ED: prevalence, etiology, and outcome. Am J Emerg Med 2013, 31: 657–60.
- Stewart AF. Hypercalcemia associated with cancer. N Engl J Med 2005, 352: 373-9.
- Rosner MH. Onco-nephrology: the pathophysiology and treatment of malignancy-associated hypercalcemia. Clin J Am Soc Nephrol 2012, 7: 1722–9.
- Van Poznak CH, et al. American Society of Clinical Oncology clinical practice guideline update: recommendations on the role of bone-modifying agents in metastatic breast cancer. J Oncol Pract 2011, 7: 117-23.
Ipercalcemia cronica
Fabio Vescini
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria S. Maria della Misericordia, Udine
Definizione ed eziologia
Con il termine di ipercalcemia cronica si intende una condizione clinico-laboratoristica nella quale i valori plasmatici di calcio sono solo lievemente superiori alla norma. I pazienti con un aumento asintomatico della calcemia (calcio < 12 mg/dL) non necessitano di un trattamento immediato e possono essere seguiti in regime ambulatoriale, mentre la terapia delle forme acute richiede generalmente il ricovero ospedaliero (1-4).
Sia le forme acute che croniche riconoscono la stessa eziologia (tabella), ma, da un punto di vista clinico, si configurano come due entità nosologiche distinte.
| Cause di ipercalcemia | |
| Aumentata produzione di PTH | Iperparatiroidismo primitivo e terziario |
| Eccesso di 1,25(OH)2D3 | Intossicazione esogena Sarcoidosi e altre granulomatosi Ipofosfatemia severa Produzione neoplastica di 1,25(OH)2D3 (linfoma) |
| Aumentato riassorbimento osseo | Metastasi ossee (mammella, colon) PTHrP (carcinoma polmonare, renale) RANKL (mieloma) Prostaglandine Ipertiroidismo Immobilizzazione Malattia di Paget Intossicazione da vitamina A |
| Aumentato assorbimento intestinale di calcio | Intossicazione da vitamina D “Milk-alkali syndrome” |
| Diminuita escrezione renale di calcio | Ipercalcemia ipocalciurica familiare Tiazidici |
| Alterate osteoformazione e mineralizzazione | Intossicazione da alluminio “Adynamic bone disease” (basso turnover) Corticosteroidi Iposurrenalismo |
Le patologie che, più comunemente, possono indurre una condizione di ipercalcemia lieve/moderata sono le seguenti.
Iperparatiroidismo primitivo, forma “mild”: rappresenta la causa più frequente di ipercalcemia e il trattamento deve necessariamente essere rivolto sia al controllo della malattia, sia alla prevenzione e/o terapia delle complicanze (es. nefrolitiasi, osteoporosi) (1,5-6).
Linfoma, sarcoidosi o altre malattie granulomatose: i pazienti affetti da queste patologie presentano un aumentato assorbimento intestinale di calcio e, probabilmente, anche un esagerato riassorbimento osseo, conseguenti all’incremento della produzione endogena di calcitriolo. Normalmente l’attivazione del calcitriolo avviene a livello del tubulo renale prossimale attraverso l’azione della 1-alfa-idrossilasi; in questi pazienti, al contrario, è presente un’elevata attività biosintetica extra-renale all’interno delle cellule mononucleate attivate (soprattutto macrofagi), che nei polmoni e nei linfonodi sono in grado di convertire il calcifediolo in calcitriolo in maniera completamente indipendente dal PTH (5,7).
Intossicazione da calcitriolo: può avere una causa iatrogena (assunzione eccessiva) nei pazienti che assumono calcitriolo per la terapia dell’ipoparatiroidismo, ovvero in quelli in trattamento per iperparatiroidismo secondario a insufficienza renale cronica. Data la breve emivita del calcitriolo (circa 12 ore), queste forme tendono a risolversi spontaneamente in pochi giorni con la sospensione del farmaco (1,8).
Intossicazione da calcifediolo o colecalciferolo: è rarissima e generalmente accidentale. A causa della lunga emivita di questi composti, può avere una durata maggiore rispetto a quella da calcitriolo; in questi casi può essere necessaria una terapia di supporto (idratazione, furosemide, steroidi, bisfosfonati ev) (1,9).
Ipercalcemia ipocalciurica familiare: si tratta di un difetto genetico, autosomico dominante, del recettore sensibile al calcio (CaSR), in genere asintomatico, caratterizzato da un'ipercalcemia lieve associata a livelli normali o leggermente elevati di PTH, con relativa ipocalciuria. Lo screening sui familiari valuta le concentrazioni di calcio nel siero. La diagnosi è confermata dall'analisi delle mutazioni di CaSR. Generalmente non necessita di terapia specifica (10).
Clinica
I segni e i sintomi dell’ipercalcemia sono gli stessi, indipendentemente dalla causa. L’ipercalcemia lieve (< 12 mg/dL) non provoca, generalmente, sintomi e/o segni clinici rilevanti, anche se alcune complicanze della malattia (es. nefrolitiasi, nefrocalcinosi, osteoporosi) possono presentarsi comunque, specie nei casi con ipercalcemia cronica di lunga durata. Questi pazienti dovrebbero essere istruiti ad evitare le condizioni potenzialmente ipercalcemizzanti, quali l’uso di diuretici tiazidici, sali di litio, sali di calcio, diete ad elevato apporto di calcio (> 1000-1500 mg/die).
Anche l’ipercalcemia moderata (12-14 mg/dL) può decorrere asintomatica, ovvero presentare una sintomatologia di lieve entità; tuttavia in caso di incremento rapido, questi valori di calcemia sono in grado di produrre sintomi specifici, specie a livello neuro-sensoriale e gastro-intestinale, con necessità di terapia immediata (1).
Terapia
La terapia dell’ipercalcemia cronica, totalmente asintomatica, si fonda essenzialmente sull’idratazione del paziente, da attuarsi attraverso il consumo spontaneo di acqua (circa 2 litri/die). Non è consigliabile l’uso di diuretici dell’ansa che potrebbero, nel lungo termine, indurre altre disionie (ipokaliemia, ipomagnesiemia), ovvero deplezione del volume (1).
In caso di valori di calcemia compresi fra 12 e 14 mg/dL, andrà preso in considerazione un trattamento farmacologico. Sulla base dell’eziologia, potranno essere impiegati gli steroidi ovvero i calcio-mimetici.
Glucocorticoidi: trovano indicazione nell’ipercalcemia moderata e sintomatica dei pazienti affetti da linfomi, sarcoidosi o malattie granulomatose. Tali farmaci sono in grado, infatti, di diminuire l’assorbimento intestinale di calcio mediante un’inibizione della produzione macrofagica del calcitriolo. Una terapia con prednisone (20-40 mg/die) è in grado di ridurre significativamente la calcemia entro 2-5 giorni (1,11).
Calcio-mimetici (Cinacalcet): il suo uso è riservato ai pazienti con iperparatiroidismo nei quali la terapia chirurgica è giudicata inappropriata o controindicata, ovvero in quelli nei quali non sia possibile localizzare la/e paratiroide/i iperfunzionante/i; possono giovarsi di questo trattamento anche coloro che rifiutano l’intervento (12). Il trattamento con cinacalcet induce la normalizzazione della calcemia in circa il 70% dei pazienti trattati, che si accompagna ad una modesta, ma significativa, riduzione del PTH (12). Un recente studio ha dimostrato che la dose media efficace per riportare la calcemia nei limiti (< 10.3 mg/dL) è di 50.4 mg/die (12). Generalmente il trattamento viene iniziato con 30 mg x 2/die e, in caso di mancata risposta, la posologia del farmaco può essere incrementata. Gli studi condotti nell’iperparatiroidismo primitivo hanno raggiunto, in casi refrattari al trattamento, anche posologie di 90 mg x 4/die (12-16). La terapia con cinacalcet è stata protratta per cinque anni e si è dimostrata efficace e sicura (12-16).
Bibliografia
- Shane E, Berenson JR. Treatment of hypercalcemia. http://www.uptodate.com/contents/treatment-of-hypercalcemia
- Bilezikian JP. Clinical review 51: Management of hypercalcemia. J Clin Endocrinol Metab 1993, 77: 1445-9.
- Carroll MF, Schade DS. A practical approach to hypercalcemia. Am Fam Physician 2003, 67: 1959-66.
- Camozzi V, Luisetto G, Basso SM, et al. Treatment of chronic hypercalcemia. Med Chem 2012, 8: 556-63.
- Jacobs TP, Bilezikian JP. Clinical review: Rare causes of hypercalcemia. J Clin Endocrinol Metab 2005, 90: 6316-22.
- Bilezikian JP. Primary hyperparathyroidism. When to observe and when to operate. Endocrinol Metab Clin North Am 2000, 29: 465-78.
- Inui N, Murayama A, Sasaki S, et al. Correlation between 25-hydroxyvitamin D3 1 alpha-hydroxylase gene expression in alveolar macrophages and the activity of sarcoidosis. Am J Med 2001, 110: 687-93.
- Adams JS. Vitamin D metabolite-mediated hypercalcemia. Endocrinol Metab Clin North Am 1989, 18: 765-78.
- Selby PL, Davies M, Marks JS, Mawer EB. Vitamin D intoxication causes hypercalcaemia by increased bone resorption which responds to pamidronate. Clin Endocrinol (Oxf) 1995, 43: 531-6.
- Christensen SE, Nissen PH, Vestergaard P, Mosekilde L. Familial hypocalciuric hypercalcaemia: a review. Curr Opin Endocrinol Diabetes Obes 2011, 18: 359-70.
- Sandler LM, Winearls CG, Fraher LJ, et al. Studies of the hypercalcaemia of sarcoidosis: effect of steroids and exogenous vitamin D3 on the circulating concentrations of 1,25-dihydroxy vitamin D3. Q J Med 1984, 53: 165-80.
- Schwarz P, Body JJ, Cap J, et al. The PRIMARA study: a prospective, descriptive, observational study to review cinacalcet use in patients with primary hyperparathyroidism in clinical practice. Eur J Endocrinol 2014, 171: 727-35.
- Peacock M, Bilezikian JP, Klassen PS, et al. Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab 2005, 90: 135-41.
- Peacock M, Bolognese MA, Borofsky M, et al. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab 2009, 94: 4860-7.
- Saponaro F, Faggiano A, Grimaldi F, et al. Cinacalcet in the management of primary hyperparathyroidism: post marketing experience of an Italian multicentre group. Clin Endocrinol 2013, 79: 20-6.
- Marcocci C, Bollerslev J, Khan AA, Shoback DM. Medical management of primary hyperparathyroidism: proceedings of the fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab 2014, 99: 3607-18.
Ipercalciurie
Gregorio Guabello
Ambulatorio di Patologia Osteo-Metabolica, UO Reumatologia, Istituto Ortopedico Galeazzi, Milano
Definizione e classificazione
Si definisce ipercalciuria un eccesso di escrezione di calcio nelle urine (> 4 mg/kg/die oppure > 300 mg/24 ore nel maschio e > 250 mg/24 ore nella donna).
L’interpretazione del dato della calciuria delle 24 ore deve tenere conto di alcune importanti considerazioni:
- la raccolta delle urine delle 24 ore deve essere eseguita correttamente (eliminando dalla raccolta la prima minzione del mattino del primo giorno);
- la determinazione della calciuria deve essere eseguita dopo normalizzazione della 25OH-D3, in quanto l’ipovitaminosi D può mascherare una ipercalciuria;
- il valore della calciuria andrebbe corretto per l’apporto giornaliero di calcio e per lo stato estrogenico (tabella 1) (1,2).
| Tabella 1 Dati normativi per calciuria (mg/24 h) in relazione ad apporto calcico e stato estrogenico |
||
| Apporto calcico (mg/die) | Pre-menopausa | Post-menopausa |
| < 500 | 39-194 | 32-252 |
| 500-1000 | 54-269 | 36-286 |
| > 1000 | 66-237 | 45-357 |
L’ipercalciuria riveste un importante ruolo come concausa, spesso sotto-diagnosticata, nell’ezio-patogenesi dell’osteoporosi primitiva, essendo presente nel 10-30% delle donne con osteoporosi post-menopausale (3).
Dal punto di vista ezio-patogenetico, l’ipercalciuria può essere idiopatica o secondaria.
L’ipercalciuria idiopatica, dal punto di vista fisiopatologico, può essere sostanzialmente di 3 tipi (tabella 2).
| Tabella 2 Classificazione patogenetica dell’ipercalciuria idiopatica |
|
| Iperassorbitiva | Fisiologica in corso di gravidanza Aumentato assorbimento intestinale di calcio per aumentata sensibilità intestinale alla vitamina D (la maggiore densità di VDR a livello intestinale condiziona aumento dei valori di calcemia e inibizione del PTH) |
| Renale (rara, 1-5% dei casi) | Tubulopatia calcio-disperdente (con perdita renale di calcio, iperparatiroidismo secondario e ipofosfatemia) Tubulopatia fosfaturica (la perdita renale di fosfato determina iperfosfaturia e ipofosfatemia; quest’ultima è un potente stimolo per la sintesi di 1,25OH-D3 e l’inibizione del PTH, con conseguente ipercalcemia e ipercalciuria; questa forma è da taluni considerata il corrispettivo nell’adulto del rachitismo ereditario ipofosfatemico con ipercalciuria-HHRH) |
| Idiopatica (scheletrica) | Aumento primitivo del riassorbimento osseo con inibizione del PTH (maggiore densità di VDR a livello osseo, dieta acidogena, aumento di citochine pro-osteoclastiche con aumento di RANKL) |
La classificazione patogenetica dell’ipercalciuria idiopatica non sempre trova corrispettivo sul piano clinico, potendoci essere un “overlap” tra le diverse forme (1,2).
Per quanto riguarda la dieta acidogena, ricordiamo che l’alimentazione tipica dei Paesi occidentali ricca in proteine animali, unitamente alla carenza estrogenica tipica della menopausa e all’invecchiamento, apporta un eccessivo carico acido, che condiziona nel tempo uno stato di lieve acidosi metabolica cronica protratta, con importanti effetti a livello osseo: passaggio di calcio/tamponi (carbonato/P) nel liquido extra-cellulare, attivazione del sistema RANKL-RANK, inibizione degli osteoblasti, alterazione degli osteociti, aumentata solubilità dell’idrossi-apatite; l’effetto finale può esitare in ipercalciuria e depauperamento osseo (osteopenia/osteoporosi) (4).
L’ipercalciuria secondaria è facilmente inquadrabile nella storia clinica del malato; le principali cause di ipercalciuria secondaria sono elencate nella tabella 3.
| Tabella 3 Cause di ipercalciuria secondaria |
|
| Ipercalcemia |
Iperparatiroidismo primario |
| Aumento dell’assorbimento intestinale di calcio | Dietetica:
Sarcoidosi Sindrome da latte e alcali |
| Aumento dell’escrezione renale di calcio | Ipocalcemia ipercalciurica autosomica dominante (ADHH) da mutazione attivante del CaSR (calcium sensing receptor) Sindrome di Cushing Farmaci: steroidi, sali di calcio, diuretici (furosemide, spironolattone, acido etacrinico, acetazolamide), teriparatide Acidosi tubulare renale distale (tipo I), da ridotta rigenerazione distale di bicarbonato Iperaldosteronismo primario |
| Aumento del riassorbimento osseo | Morbo di Paget osseo Ipertiroidismo Immobilizzazione prolungata |
| Deplezione di fosfato | Tubulopatia fosfaturica Enteropatie con malassorbimento intestinale Anti-acidi chelanti del fosforo |
Clinica
Il quadro clinico del paziente con ipercalciuria idiopatica comprende la possibile e variabile presenza di nefrolitiasi (la calcolosi delle vie urinarie è costituita da calcoli di ossalato o fosfato di calcio nell’80% dei casi e nel 40-50% dei casi è presente una condizione di ipercalciuria primitiva o secondaria) e/o malattia ossea (generalmente rappresentata da una condizione di osteomalacia per ridotta mineralizzazione della matrice osteoide) (1,2). Nelle forme secondarie prevale il quadro clinico della malattia sottostante.
Diagnosi
Di fronte a un paziente con riscontro di ipercalciuria nelle urine delle 24 ore, il primo step diagnostico consiste nell’esclusione delle forme secondarie, in particolare di iperparatiroidismo primario (ipercalciuria associata a valori elevati di PTH e di calcemia corretta per albumina). Esclusa la forma secondaria, si fa diagnosi di ipercalciuria idiopatica.
La diagnosi differenziale delle diverse forme di ipercalciuria idiopatica è poco sfruttabile nella pratica clinica, oltre a non avere grosse implicazioni sul piano clinico e terapeutico. In linea teorica essa prevede:
- una prima raccolta di urine delle 24 ore da eseguire dopo 7 giorni di dieta ipocalcica (400 mg/die), normoproteica (0.8 g/kg/die) e normosodica (100 mEq/die);
- il mattino seguente una seconda raccolta di urina di 2 ore (dalle 7 alle 9) per il calcolo del rapporto calciuria (mg/dL)/creatininuria (mg/dL).
La combinazione della calciuria delle 24 ore con il rapporto calciuria/creatininuria permetterebbe di distinguere le diverse forme di ipercalciuria sul piano fisio-patologico (1,2):
- ipercalciuria iperassorbitiva: normalizzazione della calciuria 24 ore e rapporto nella norma (< 0.11);
- ipercalciuria renale/idiopatica: persistenza dell’ipercalciuria 24 ore e rapporto aumentato (> 0.11), con livelli di PTH:
- normali/alti nella forma renale da tubulopatia calcio-disperdente;
- normali/bassi nella forma idiopatica e nella forma renale da tubulopatia fosfato-disperdente.
Terapia
Quella dell'ipercalciuria idiopatica poggia su 2 cardini.
Dieta: in particolare normocalcica (1000-1200 mg/die), idropinica (acqua a contenuto calcico normale/elevato, diuresi > 2 L/die), povera di sale (< 6 g/die) e proteine animali (0.8-1 g/kg/die), povera di zuccheri semplici e alcool, povera di alimenti contenenti ossalato (noci, cioccolato, legumi, soia, vegetali a foglia verde, uova, rabarbaro, tè, caffè), ricca di alimenti vegetali.
Diuretico calcio-ritentore: idroclorotiazide 25 mg/die in mono-somministrazione, fino a un massimo di 50-100 mg/die in due somministrazioni; clortalidone 25 mg/die fino ad un massimo di 50-100 mg/die in mono-somministrazione.
È possibile associare, al fine di ridurre il rischio di ipopotassiemia, un diuretico risparmiatore di potassio (amiloride 5-10 mg/die, controindicato il triamterene).
Utile il periodico monitoraggio di calciuria e sodiuria 24 ore (obiettivo < 100 mEq/24 ore).
In caso di persistenza di ipercalciuria o di intolleranza al tiazidico, valutare la terapia con alcali del potassio e del magnesio (citrato di potassio e magnesio alla dose di 2-4 g in 2 somministrazioni/die); i sali di fosfato sono utili nelle forme associate a ipofosfatemia (1,2).
In caso di trattamento con farmaci anti-osteoporosi nel paziente con ipercalciuria idiopatica, non esiste una controindicazione assoluta alla somministrazione di vitamina D e calcio. Per quanto concerne la vitamina D, deve essere garantito, in genere con la supplementazione, un valore di 25OH-D3 > 30 ng/mL (necessario a volte per slatentizzare un’ipercalciuria altrimenti misconosciuta). Per quanto riguarda il calcio, vale la regola generale di ottimizzare un adeguato apporto di calcio con la dieta e di ricorrere ai supplementi (calcio citrato anziché carbonato nel paziente con ipercalciuria idiopatica) qualora la correzione dietetica non sia sufficiente.
La terapia dell’ipercalciuria secondaria risiede nel trattamento della patologia sottostante.
Acidosi tubulare renale distale di tipo I
Merita un approfondimento a parte nell’ambito delle cause renali di ipercalciuria secondaria.Tale condizione si definisce come l’incapacità del tubulo renale di acidificare le urine per difetto della rigenerazione distale di HCO3- a livello del tubulo contorto distale.
La tabella 4 elenca le cause.
| Tabella 4 Cause di acidosi tubulare renale distale di tipo I |
||
| Genetica | Autosomica dominante Autosomica recessiva |
|
| Secondaria | Malattie autoimmuni |
Cirrosi biliare primitiva |
| Malattie granulomatose | Sarcoidosi | |
| Malattie endocrine | Ipertiroidismo Iperparatiroidismo primario |
|
| Nefro-uropatie | Nefrocalcinosi Infezioni croniche delle vie urinarie Ostruzione cronica delle vie urinarie |
|
| Malattie epatiche | Epatite cronica attiva | |
| Emopatie | Anemia a cellule falciformi | |
| Farmaci | Amfotericina B FANS Toluene Litio |
|
| Trapianto renale e rigetto di trapianto di rene | ||
Dal punto di vista clinico, si presenta nel bambino con ritardo di crescita, rachitismo, vomito, ipotonia, sordità e nell’adulto con osteomalacia e nefrolitiasi/nefrocalcinosi.
Il sospetto diagnostico si pone in presenza di acidosi metabolica (bicarbonati ematici ≤ 20 mEq/L) non altrimenti spiegabile, con gap anionico normale, in associazione con ipercloremia, ipopotassiemia, ipercalciuria, ipofosfatemia, ipocitraturia; la diagnosi viene confermata dalla valutazione del pH urinario (> 5.3-5.5) e dell’escrezione urinaria di ammonio delle 24 ore (ridotta).
La terapia consiste nella correzione dell’acidosi con sodio bicarbonato o sodio citrato per os e nella supplementazione di potassio con potassio citrato, che possiede anche effetto ipocalciurico (5).
Bibliografia
- Liebman SE, Taylor JG, Bushinsky DA. Idiopathic hypercalciuria. Curr Rheumatol Rep 2006, 8: 70-5.
- Ryan LE, Ing SW. Idiopathic hypercalciuria and bone health. Curr Osteoporos Rep 2012, 10: 286-95.
- Giannini S, Nobile M, Dalle Carbonare L, et al. Hypercalciuria is a common and important finding in postmenopausal women with osteoporosis. Eur J Endocrinol 2003, 149: 209-13.
- Jajoo R, Song L, Rasmussen H, et al. Dietary acid-base balance, bone resorption, and calcium excretion. J Am Coll Nutr 2006, 25: 224-30.
- Seidowsky A, Moulonguet-Doleris L, Hanslik T, et al. Tubular renal acidosis. Rev Med Interne 2014, 35: 45-55.
Ipopotassiemia
Paola Sartorato
Dipartimento di Medicina Interna, Ospedale di Montebelluna (TV)
FISIOLOGIA DEL POTASSIO
Il potassio è il catione più abbondante nell’organismo: il 98% circa è intra-cellulare, mentre solo il 2% è nel compartimento extra-cellulare.
L’elevato gradiente di concentrazione di potassio tra i compartimenti intra- ed extra-cellulari contribuisce al potenziale di membrana.
Il potassio è un fattore importante nella regolazione del volume cellulare e dell’osmolalità dei liquidi dell’organismo; svolge inoltre un ruolo fondamentale come cofattore nelle reazioni enzimatiche e nell’omeostasi acido-base.
L’omeostasi del potassio si basa su tre elementi chiave: passaggio cellulare, escrezione renale e in misura minore perdita gastro-intestinale. L’escrezione renale risulta dalla somma di carico di potassio filtrato, riassorbito e secreto: il potassio viene liberamente filtrato nel glomerulo, quasi completamente riassorbito nel tubulo prossimale e secreto secondo le esigenze nel tubulo distale e collettore; l’escrezione distale, a sua volta, è regolata da aldosterone, equilibrio acido-base ed entità del flusso urinario nel nefrone distale.
La concentrazione sierica di potassio è strettamente regolata e risulta compresa fra 3.5 e 5 mEq/L.
DEFINIZIONE ED EZIOLOGIA
L’ipopotassiemia è definita da valori di potassio < 3.5 mEq/L (1):
- lieve = 3-3.5 mEq/L;
- moderata = 2.5-3 mEq/L;
- grave: < 2.5 mEq/L.
Le principali cause di ipokaliemia sono: ridotto apporto, shift trans-cellulare, eccessive perdite corporee e deplezione di magnesio (tabella).
| Cause di ipopotassiemia (modif da 1) | ||
| Ridotto apporto | Ridotto apporto di K con la dieta (< 1 g/die) | |
| Shift trans-cellulare | Terapia insulina/glucosio Stimolazione ß2-adrenergica (es salbutamolo) Alcalosi Paralisi periodica ipokaliemica |
|
| Aumentate perdite | Farmaci | Diuretici Abuso di lassativi Liquirizia Steroidi |
| Gastro-intestinali | Diarrea Vomito Ileostomia Fistola intestinale Adenomi villosi |
|
| Renali | Disordini tubulari renali Sindrome di Bartter Sindrome di Liddle Sindrome di Gitelman Diabete insipido nefrogenico |
|
| Endocrinopatie | Iperaldosteronismo primitivo Sindrome di Cushing Iperplasia surrenalica congenita |
|
| Dialisi | ||
| Deplezione di magnesio (aumentata perdita renale di potassio) | Ridotto apporto di Mg con la dieta Aumentate perdite di magnesio |
|
La quota quantitativamente più significativa è rappresentata dalle ipokaliemie iatrogene e da patologie gastro-intestinali.
Ipopotassiemia da ridotto apporto
La restrizione di potassio nella dieta determina raramente ipokaliemia, essendo controbilanciata da ridotta escrezione renale. Anche se l’assunzione di potassio fosse pari a 0 (osservabile in situazioni di grave e persistente disturbo alimentare, es anoressia nervosa), sarebbero necessarie 2-3 settimane per raggiungere valori di potassio < 3 mEq/L; anche in queste situazioni il deficit deriva spesso da concomitanti perdite renali, gastro-intestinali o cutanee.
Ipopotassiemia da shift trans-cellulare
L’ingresso di potassio nelle cellule è promosso dall’alcalosi, dall’insulina, dalla stimolazione ß-adrenergica e dalle xantine (es. caffeina).
L’insulina stimola la captazione di potassio, in particolare a livello del muscolo scheletrico, agendo sulla pompa Na+/K+-ATPasi, stimolandone la rapida traslocazione dai depositi intra-cellulari sulla superficie cellulare.
Le catecolamine attivano i recettori alfa1-adrenergici e ß2-adrenergici, riducendo o aumentando rispettivamente la captazione di potassio, agendo direttamente sull’attività Na+/K+-ATPasi a livello muscolare ed epatico.
Diversi case report hanno descritto ipokaliemie severe dopo somministrazione di clorochina, anti-psicotici quali risperidone o quetiapina, avvelenamenti da cesio e bario; la pericolosità di queste situazioni deriva dall’induzione di aritmie gravi, potenzialmente fatali (2).
Altre rare cause di ipopotassiemia sono la paralisi periodica ipokaliemica autosomica dominante (HPP) e l’ipokaliemia nella tireotossicosi. I ridotti livelli di potassio nella tireotossicosi sembrano legati in parte ad un’aumentata sensibilità alle catecolamine, ma anche a un effetto diretto della tiroxina sulla pompa Na+/K+-ATPasi (3).
Ipopotassiemia da aumentate perdite da cause extra-renali
In questo ambito la causa più frequente è la diarrea cronica o severa (incluse quella da abuso di lassativi). Le perdite gastro-intestinali di potassio si possono osservare anche nella celiachia e negli adenomi villosi secretivi. Nell'ipotassiemia da vomito prolungato contribuiscono le perdite renali di potassio secondarie alla combinazione di un quadro di iperaldosteronismo secondario a deplezione di volume e alcalosi.
Le ipopotassiemie possono derivare anche da perdite cutanee in seguito ad eccessiva sudorazione dopo esercizio fisico intenso.
Ipopotassiemia da aumentate perdite renali
In questo ambito tre elementi rivestono un ruolo centrale per una corretta diagnosi differenziale: la potassiuria (intesa come valori inappropriatamente elevati per i valori sierici di potassio), l’equilibrio acido-base, la valutazione dei livelli pressori e del volume circolante efficace (figura).
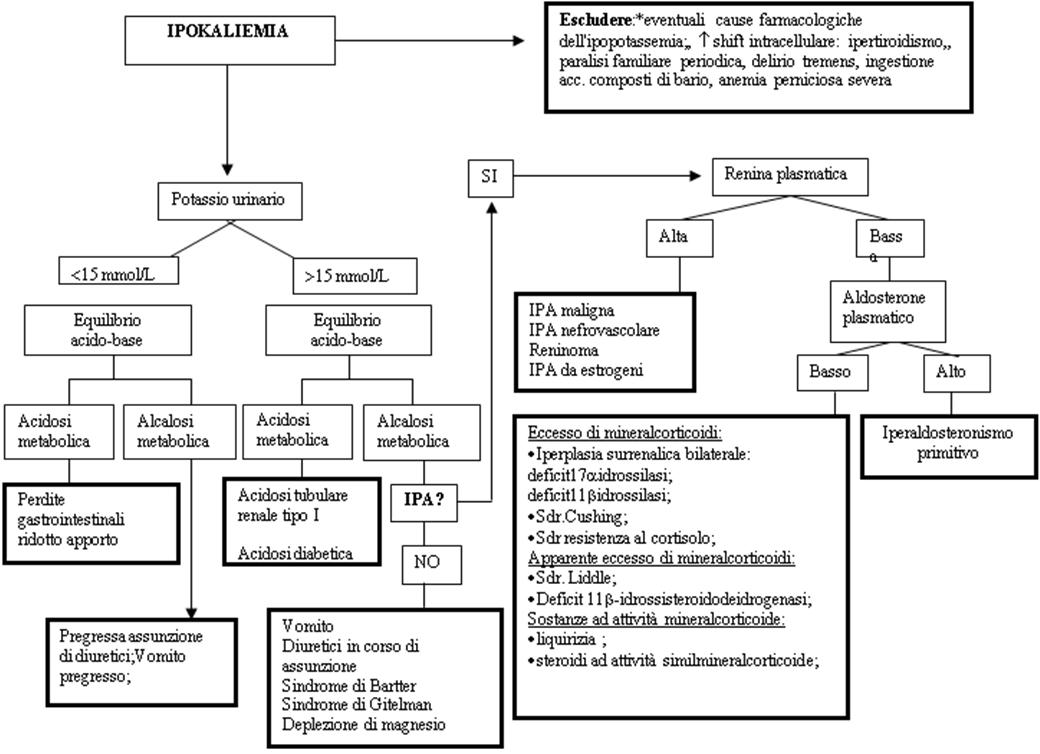
PRESENTAZIONE CLINICA
Molti pazienti ipopotassiemici sono asintomatici, soprattutto se l’ipokaliemia è lieve.
I sintomi muscolari includono malessere generale, stanchezza, letargia e debolezza; alcune volte si possono presentare anche tetanie/fascicolazioni e rabdomiolisi. Nelle ipopotassiemie severe vi può essere anche una paralisi ascendente dei muscoli respiratori.
I pazienti possono presentare anche poliuria e polidipsia, per incapacità di concentrare le urine.
A livello gastro-enterico i pazienti possono presentare nausea, vomito, stipsi ed ileo dinamico (4).
L’ipopotassiemia predispone a diverse aritmie: prolungamento dell’intervallo PR fino a blocco A-V di I e II grado, fibrillazione atriale e i quadri più severi di tachicardia ventricolare, torsione di punta e arresto cardiaco. I pazienti che assumono digitale sono più suscettibili alle aritmie.
Le manifestazioni cliniche dell'ipopotassiemia sono correlate alla gravità del deficit, alla rapidità di insorgenza e alla presenza di una concomitante patologia cardiaca (5).
DIAGNOSI
In presenza di ipopotassiemia l’esecuzione di un ECG rappresenta il test iniziale che può guidare le scelte terapeutiche più appropriate. Le alterazioni tipiche all’ECG sono: riduzione di voltaggio dell’onda T, sotto-slivellamento del tratto ST, comparsa dell’onda U, allungamento dell’intervallo QT e le anomalie aritmiche descritte sopra.
È cruciale riconoscere se l’ipokaliemia è secondaria a perdita renale di potassio. È necessario valutare se vi sono valori di potassiuria inappropriatamente elevati per i valori sierici di potassio: in presenza di potassiuria > 20 mEq/24h, si sospetta una perdita renale. Utilizzando un campione di urine spot (circa 10 mL), è possibile calcolare il gradiente di concentrazione trans-tubulare di potassio (TTGK) attraverso questa formula:
TTGK = ([K+urine]/[K+plasma])/(OsmU/OsmP)
Si tratta di un indice semi-quantitativo della potassiuria aggiustata per la potassiemia e per l’acqua riassorbita dai dotti collettori nella midollare. TTGK > 2 è indicativo di inappropriate perdite renali di potassio.
Tutti i pazienti ipokaliemici devono eseguire un’emogasanalisi arteriosa: nell’ipokaliemia il rene non può eliminare bicarbonato, mentre elimina protoni e cloro, determinando alcalosi metabolica.
Esiste una stretta correlazione tra ipomagnesiemia e ipopotassiemia, pertanto in tutti i pazienti con potassio ridotto deve essere dosata anche la magnesiemia (6).
TRATTAMENTO
Il trattamento dell’ipokaliemia prevede la correzione della causa responsabile e la terapia “sintomatica”. L’approccio terapeutico dipende dalla severità dell’ipokaliemia e dalla presenza di sintomi o anomalie all’ECG.
La somministrazione di potassio, in particolare per via endovenosa, non è esente da rischi; la correzione deve avvenire gradualmente evitando l’iperkaliemia.
Molti pazienti carenti di potassio sono anche deficitari di magnesio, che è importante per l’assorbimento di potassio e per il mantenimento dei livelli intra-cellulari di potassio, in particolare nel miocardio. Il deficit combinato dei due ioni potenzia il rischio di insorgenza di aritmie, la correzione del deficit di magnesio facilita la correzione dei livelli di potassio ed è raccomandata in particolare nelle ipopotassiemie severe (7).
Nei pazienti stabili con livelli di potassio tra 3 e 3.5 mEql/L, è sufficiente la supplementazione di potassio per os, al dosaggio di 20-80 mEq/die (il potassio è lesivo della mucosa gastrica e va assunto durante i pasti).
In caso di somministrazione di potassio per via ev, è necessario il monitoraggio ECG e la potassiemia deve essere monitorata strettamente (ogni 2 h). Il potassio deve essere diluito in soluzione fisiologica oppure in soluzione ripolarizzante (500 mL di soluzione glucosata al 10%, 1 unità di insulina ogni 3 g di glucosio e 40 mEq di cloruro di potassio), che favorisce l’ingresso di potassio nella cellula. In assenza di tossicità da digossina o patologie cardiache severe, l’ipopotassiemia deve essere corretta gradualmente, al dosaggio di 10 mEq/h nei pazienti asintomatici. La supplementazione con magnesio in questi casi è indicata solo se il deficit è documentato.
In condizioni di emergenza con aritmie documentate, la dose massima raccomandata di potassio per via ev è di 20 mEq/h, anche se nelle aritmie instabili a rischio di arresto può essere indicata una correzione più rapida (infusione iniziale di 2 mEq/min per 10 minuti seguita da 10 mEq in 5-10 minuti). Dovrebbero essere evitati i boli rapidi, perché gravati da elevato rischio di arresto cardiaco. In questi pazienti il magnesio dovrebbe essere somministrato precocemente dopo l’inizio dell’infusione del potassio, indipendentemente dai livelli di magnesiemia (1).
BIBLIOGRAFIA
- Alfonzo AVM, Isles C, Geddes C, et al. Potassium disorders-clinical spectrum and emergency management. Resuscitation 2006, 70: 10-25.
- Clausen T. Hormonal and pharmacological modification of plasma potassium homeostasis. Fundam Clin Pharmacol 2010, 24: 595-605.
- Lin S-H, Lin Y-F, Halperin ML. Hypokalaemia and paralysis. QJM 2001,94, 133–9.
- Medford-Davis L, Rafique Z. Derangements of potassium. Emerg Med Clin North Am 2014, 32: 329-47.
- Gennari FJ. Hypokalaemia. New Eng J Med 1998, 339: 451-8.
- Unwin RJ, Luft FC, Shirley DG. Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011, 7: 75-84.
- Cohn JN, Kowey PR, Whelton PK, et al. New guidelines for potassium replacement in clinical practice. Arch Intern Med 2000,160: 2429-36.
Iperpotassiemia
Andrea Guarnieri
SC Nefrologia e Dialisi - AO S. Croce e Carle – Cuneo
L’iperpotassiemia è definita da una concentrazione sierica > 5.5 mEq/L negli adulti. Livelli > 7 mEq/L possono dare significativi sintomi neurologici ed emodinamici, mentre una potassiemia > 8.5 mEq/L può essere rapidamente fatale, causando paralisi respiratoria o arresto cardiaco.
Spesso l’iperkaliemia è asintomatica. I sintomi, qualora presenti, sono aspecifici: debolezza e affaticamento, occasionalmente parestesie, paralisi muscolare, dispnea, palpitazioni, dolore toracico, nausea o vomito.
L’esame obiettivo non consente generalmente la diagnosi.
L’iperpotassiemia è un problema clinico comune; la prevalenza nella popolazione generale non è nota; si ritiene che si verifichi in percentuale dell’1-10% nei pazienti ospedalizzati (1). È relativamente rara nelle persone sane, poiché la risposta cellulare (passaggio trans-membrana) e l’adattamento dell’eliminazione urinaria prevengono l’accumulo del K+ nel comparto extra-cellulare, inoltre la capacità di eliminazione del K+ aumenta con la quantità di ione introdotta (2); pertanto, l’iperpotassiemia è raramente causata dal solo aumento dell’introito alimentare o farmacologico.
Le principali cause di iperpotassiemia sono riconducibili a due meccanismi: l’aumento del rilascio di K+ dalle cellule e la ridotta eliminazione urinaria. Il rilascio dello ione dal comparto intra-cellulare determina in genere solo un incremento transitorio della potassiemia, mentre un’iperkaliemia persistente richiede un’alterata escrezione urinaria.
AUMENTATO RILASCIO DALLE CELLULE
Pseudo-iperpotassiemia
L’iperpotassiemia “spuria” si presenta quando i valori di K+ riportati dal laboratorio non riflettono la concentrazione in vivo, generalmente perché piastrine, leucociti o eritrociti hanno rilasciato K+ in vitro. Può essere esclusa ripetendo il prelievo o misurando simultaneamente la potassiemia su siero e su plasma: la concentrazione sierica è generalmente maggiore di 0.2–0.4 mEq/L a causa del rilascio di ione durante la coagulazione del prelievo. Questa condizione andrebbe esclusa in pazienti asintomatici, senza alterazioni ECG o in apparente assenza di altre cause. La pseudo-iperpotassiemia può essere espressione di emolisi causata dall’uso di un ago di calibro troppo piccolo o di un laccio emostatico eccessivamente stretto, da venipuntura traumatica, meno frequentemente da raffreddamento o ritardata elaborazione del campione. Anche in presenza di severa trombocitosi o leucocitosi (es. nella leucemia linfocitica cronica) è possibile iperpotassiemia in vitro da lisi cellulare. Nella pseudo-iperkaliemia familiare il K+ esce dagli eritrociti per incremento della permeabilità passiva.
Acidosi metabolica
In presenza di acidosi metabolica gli ioni H+ entrano nelle cellule, mentre Na+ e K+ passano dallo spazio intra- a quello extra-cellulare, con conseguente incremento di potassiemia; se il paziente ha acidosi metabolica con incremento dell’eliminazione di K+ (per sindrome diarroica o acidosi tubulare renale), la potassiemia può essere normale, anche se risulta elevata se rapportata alle perdite. L’aumentato passaggio trans-membrana di K+ non avviene nell’acidosi lattica e nella cheto-acidosi, probabilmente per la capacità dello ione organico e dello ione idrogeno di entrare nelle cellule mediante un meccanismo di co-trasporto sodio-anione. Per motivi non ben definiti, le condizioni di acidosi respiratoria non hanno significativo effetto sulla potassiemia.
Deficit d’insulina, iperglicemia, iperosmolarità
L’insulina promuove l’entrata di K+ nelle cellule. Nel diabete non controllato la combinazione di deficit d’insulina (sia alterata secrezione sia resistenza periferica) e di iperosmolarità secondaria all’iperglicemia causa frequentemente aumento di potassiemia. Altre condizioni sono state associate a iperpotassiemia secondaria a deficit d’insulina e/o iperosmolarità: uso di somatostatina in pazienti con marcata riduzione della funzione renale, terapia con immunoglobuline, somministrazione di mezzo di contrasto, mannitolo.
Paralisi periodica iperkaliemica
È un disturbo muscolare caratterizzato da attacchi episodici di debolezza muscolare, associati ad aumento della concentrazione di potassio nel siero, usualmente scatenati dal riposo dopo l’esercizio, dal digiuno e dall’esposizione al freddo. Altri fattori scatenanti sono l’ingestione di cibi ricchi di potassio, lo stress, le infezioni, la somministrazione di glucocorticoidi, l’anestesia e la gravidanza. È dovuta a mutazioni puntiformi del gene SCN4A, che codifica per la subunità alfa del canale del sodio voltaggio-dipendente del muscolo scheletrico.
Altre cause
In numerose altre condizioni è possibile rilevare un incremento della potassiemia, anche transitorio: esercizio fisico, necrosi tissutale (rabdomiolisi, sindrome da lisi tumorale, ustioni estese, manovre rianimatorie, ipotermia severa), terapia con ß-bloccanti, sovradosaggio di digitale, trasfusioni di emazie, somministrazione di succinil-colina (in pazienti con ustioni, traumi estesi, immobilizzazione prolungata, infezioni croniche, malattie degenerative neuro-muscolari), arginina o acido amino-caproico, farmaci che attivano il canale di membrana per il potassio ATP-dipendente (ciclosporina, tacrolimus, minoxidil, isoflurano).
RIDOTTA ESCREZIONE URINARIA
Ridotta secrezione di aldosterone
Ogni causa in grado di ridurre la secrezione di aldosterone, come l'ipoaldosteronismo iporeninemico o farmaci quali ACE-inibitori, sartani, inibitori della renina, anti-infiammatori non steroidei, inibitori della calcineurina, eparina e anti-fungini (ketoconazolo, fluconazolo, itraconazolo), può ridurre l’efficacia della secrezione di K+, portando ad acidosi metabolica e iperkaliemia (acidosi tubulare renale tipo 4). Quest’ultima stimola direttamente la secrezione di K+, controbilanciando almeno in parte l’effetto del relativo deficit di aldosterone; tale effetto può tuttavia mancare in presenza di ridotta funzione renale. L’ipoaldosteronismo può anche essere causato da disordini primitivi del surrene (come nella malattia di Addison o nei deficit congeniti di enzimi steroidogenetici, più frequentemente il deficit di 21-idrossilasi). Tuttavia, in genere un livello patologico di ormoni mineralcorticoidi non produce iperpotassiemia se al nefrone distale arriva una quantità sufficiente di sodio, pertanto i pazienti affetti da Addison non manifestano iperpotassiemia se hanno un sufficiente apporto alimentare di sale.
Ridotta risposta all’aldosterone
Le cause più comuni di resistenza all’aldosterone sono l’uso di diuretici risparmiatori di potassio (antagonisti dell’aldosterone: spironolattone ed eplerenone; inibitori dei canali del sodio del dotto collettore: amiloride e triamterene) e l’insufficienza renale sia acuta sia cronica.
Insufficienza renale acuta e cronica
L’iperpotassiema è una complicanza comune in presenza di ridotta funzione renale. Nei pazienti con danno renale acuto è più frequente in condizioni di oliguria. Nell’insufficienza renale cronica la capacità di eliminare il K+ a livelli pressoché normali persiste sino a quando è preservata la responsività all’aldosterone ed è mantenuto l’apporto distale di acqua e sodio. È più frequente nell’IRC di grado avanzato (Classe K-DOQI IV-V: e-GFR < 30 mL/min/1.73 m2).
Riduzione del volume circolante
In condizioni di ipovolemia vera (perdite gastro-intestinali o renali) o relativa (scompenso cardiaco, cirrosi), la quantità di acqua e sodio che raggiunge le parti del nefrone distale deputate alla secrezione del K+ può ridursi sensibilmente, alterandone la secrezione e, conseguentemente, l'escrezione (3).
Alterazione selettiva della secrezione di potassio
Alcuni pazienti possono manifestare iperpotassiemia malgrado una normale secrezione di aldosterone e un normale apporto distale di acqua e sodio. Questa alterazione (apparentemente) selettiva della secrezione di K+ è stata descritta nella nefrite lupoide, nel rigetto acuto, nell’anemia falciforme. Anche nei pazienti in trattamento con inibitori della calcineurina si può avere una riduzione selettiva della capacità di secrezione tubulare del K+.
Acidosi tubulare renale distale voltaggio-dipendente
Descritta in pazienti affetti da uropatia ostruttiva, nefrite lupoide, amiloidosi renale, anemia falciforme, è caratterizzata da acidosi metabolica ipercloremica, aumento delle perdite urinarie di sale e aldosteronemia normale o aumentata. Il meccanismo alla base è un’alterazione della pompa Na-K ATP-dipendente.
Pseudo-ipoaldosteronismo tipo 1
Raro disordine ereditario caratterizzato da resistenza all’aldosterone. Nella forma autosomica recessiva interessa il canale del sodio del dotto collettore, in quella autosomica dominante il recettore dei mineralcorticoidi (nella maggioranza dei pazienti).
Pseudoipoaldosteronismo tipo 2 (sindrome di Gordon), chiamata anche ipertensione familiare iperkaliemica
Rara forma ereditaria di ipertensione, caratterizzata da iperkaliemia, acidosi metabolica ipercloremica, espansione del volume extra-cellulare e funzione renale conservata. L’ipoaldosteronismo rappresenta una riduzione appropriata della secrezione di aldosterone in risposta all’espansione volemica.
Uretero-ileostomia
Un aumento della potassiemia può manifestarsi in pazienti portatori di diversioni urinarie per assorbimento di potassio dall’ansa intestinale.
TRATTAMENTO
Il trattamento dell’iperkaliemia deve essere adattato alla potenziale pericolosità della disionia che, a sua volta, può dipendere dalla velocità di incremento del K+, dal valore assoluto raggiunto e dall’evidenza di tossicità cardiaca. La terapia può essere schematizzata in alcune tappe, anche se spesso i vari interventi sono applicati contemporaneamente.
- Somministrazione parenterale di calcio per ridurre la tossicità cardiaca: 10 mL di calcio gluconato 10% endovena in 2 minuti. Se non ci sono miglioramenti, può essere somministrata un'ulteriore dose ogni 10 minuti. N.B.: se il paziente è in terapia con digossina, la dose va somministrata lentamente (in almeno 20 minuti) in 10 mL di soluzione glucosata al 10%.
- Identificazione e rimozione delle fonti di K+: supplementi orali o parenterali, sostituti del sale.
- Incremento del passaggio del K+ nel compartimento intra-cellulare:
- infusione di glucosio e insulina: 25 mL di soluzione glucosata 50% + 10 UI in 10 minuti. Efficace entro 20–30 minuti, durata variabile, in genere tra le 2 e le 6 ore. È possibile mantenere l’infusione per prolungare l’effetto. L’insulina parenterale può causare ipoglicemia, particolarmente nei pazienti con danno renale acuto o cronico; è pertanto necessario uno stretto controllo (ogni due ore) dei livelli di glucosio e K+;
- bicarbonato di sodio per via parenterale: NaHCO3- 5% 15-20 mL/h;
- ß-agonisti per aerosol o per via parenterale (probabilmente da preferire alla somministrazione di bicarbonato nei pazienti con insufficienza renale): salbutamolo 10-20 mg in 4 mL di NaCl 0.9% in aerosol oppure 3-20 µg/minuto per via parenterale;
- Incremento dell’escrezione:
- somministrazione di soluzione fisiologica e diuretici dell’ansa: NaCl 0.9% 1 mL/kg/h + furosemide ev 10 mg x 3/die nei soggetti con funzione renale conservata (è necessario sospendere i diuretici risparmiatori di K+ e i farmaci interferenti con il Sistema Renina Angiotensina);
- l’escrezione renale può essere aumentata anche dalla somministrazione di un analogo dell’aldosterone come il fluoro-idrocortisone acetato (0.05-0.1 mg/die); tale farmaco è utile in particolare in pazienti con iporeninemia o ipoaldosteronismo. In genere il K+ si normalizza dopo circa 48 ore;
- uso di resine a scambio cationico somministrate per via orale o rettale per favorire l’eliminazione intestinale (calcio polistiren-sulfonato, sodio polistiren-sulfonato): 15-30 grammi x 1-2 volte/die. L’utilizzo non è indicato per il controllo acuto dell’iperpotassiemia, poiché l’azione inizia dopo circa due ore e raggiunge il massimo a 4-6 ore;
- emodialisi: terapia di elezione nei pazienti con iperpotassiemia potenzialmente fatale, che non rispondono rapidamente al trattamento farmacologico o in presenza di severa compromissione della funzione renale.
La terapia dell’iperpotassiemia deve essere comunque individualizzata.
Se il paziente ha solo un moderato incremento dei livelli di K+ e nessuna alterazione elettrocardiografica, è possibile intervenire sospendendo fonti di K+ e farmaci interferenti e aumentando l'escrezione dello ione con una resina a scambio cationico o un diuretico.
In pazienti con severa iperpotassiemia e alterazioni ECG, il trattamento comprenderà più interventi focalizzati su:
- immediata stabilizzazione della membrana cellulare;
- passaggio dello ione nello spazio intra-cellulare;
- eliminazione del K+ totale corporeo;
- sospensione immediata di risorse esogene di K+ e di tutti i farmaci in grado di provocare iperpotassiemia.
La terapia definitiva è l’emodialisi.
BIBLIOGRAFIA
- Acker CG, et al. Hyperkalemia in hospitalized patients: causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Arch Intern Med 1998, 158: 917-24.
- Rabelink TJ, Koomans HA, Hené RJ, et al. Early and late adjustment to potassium loading in humans. Kidney Int 1990, 38: 942-7.
- Stokes JB. Potassium secretion by cortical collecting tubule: relation to sodium absorpion, luminal sodium concentration and transepithelial voltage. Am J Physiol 1981, 241: F395-402.
Ipomagnesiemia
Alessandro Piovesan
Endocrinologia Oncologica, AO Città della Salute e della Scienza, Torino
EPIDEMIOLOGIA
Anche se l’introito alimentare di Mg è insufficiente nel 75% della popolazione americana, l‘incidenza di ipomagnesiemia clinicamente rilevante è circa del 2%. Può raggiungere il 10-20% nei pazienti ospedalizzati e il 50-60% nei ricoverati in reparti di terapia intensiva. È comune negli alcolisti (30-80%) ed è presente fino al 25% dei pazienti diabetici. È più frequente nei neonati.
EZIOPATOGENESI
Le cause più frequenti sono riconducibili a patologie del tratto gastro-enterico o dell’emuntorio renale (tabella).
| Cause di ipomagnesiemia | ||
| Perdite gastro-intestinali | Diarrea, steatorrea Malassorbimento, chirurgia bariatrica Pancreatite acuta Malattie ereditarie (ipomagnesiemia intestinale con ipocalcemia secondaria) |
|
| Iatrogene | Inibitori di pompa protonica | |
| Perdite renali | Iatrogene | Diuretici dell’ansa e tiazidici Antibiotici (amfotericina, aminoglicosidi) Cisplatino Anticorpi monoclonali anti-EGF (cetuximab, panitumumab, matuzumab) |
| Espansione del volume plasmatico Diabete scompensato Alcolismo Ipercalcemia Diabete a esordio precoce Malformazioni renali |
||
| Disfunzione tubulare acquisita | Post necrosi tubulare acuta Diuresi post-ostruttiva Post trapianto renale |
|
| Malattie genetiche | Sindromi di Bartter o Gitelman Ipomagnesiemia familiare con ipercalciuria e nefrocalcinosi Ipomagnesiemia isolata autosomica dominante Ipomagnesiemia isolata autosomica recessiva |
|
Perdite e ridotto assorbimento intestinale
Diarrea cronica, steatorrea e malassorbimento, specie quello indotto dalla chirurgia bariatrica sono le cause più frequenti di ipomagnesiemia. Una piccola quota di Mg viene persa attraverso i secreti intestinali in maniera continua e non regolata, quota che può diventare rilevante in caso di ridotto introito alimentare. Anche la pancreatite può indurre ipomagnesiemia, attraverso la saponificazione di Mg e calcio.
L’ipomagnesiemia con ipocalcemia secondaria è una malattia genetica da difetto selettivo dell’assorbimento intestinale di Mg: la forma recessiva è dovuta ad una mutazione del gene TRPM6, che riduce l’assorbimento intestinale e il riassorbimento renale di Mg.
L’uso di inibitori di pompa protonica (PPI) è associato all’ipomagnesiemia specie in corso di terapia diuretica. Si è ipotizzato che i PPI possano inibire i canali TMRP6- e TMP7-dipendenti. L’FDA nel 2011 ha emesso un “safety warning” con il suggerimento di verificare i livelli di Mg in tutti i pazienti in terapia cronica con PPI, suggerendo un'integrazione di Mg in caso di livelli carenziali. L’ipomagnesiemia è reversibile con la sospensione dei PPI.
Perdite renali
I diuretici tiazidici e dell’ansa inibiscono il riassorbimento del Mg, mentre i risparmiatori di potassio lo incrementano. L’ipomagnesiemia indotta dai diuretici è solitamente di grado moderato.
L’ipomagnesiemia grave può invece essere indotta da farmaci che causino necrosi tubulare acuta, quali derivati del platino, aminoglicosidi, ciclosporina, anfotericina B.
L’espansione di volume, riducendo il riassorbimento di sodio, riduce anche il riassorbimento di Mg, provocando ipomagnesiemia.
Anche l’alcolismo può indurre ipomagnesiemia, presumibilmente attraverso tossicità acuta a livello tubulare.
Nel diabete scompensato, la diuresi osmotica aumenta l’escrezione urinaria di Mg. L’ADA suggerisce la supplementazione di Mg nei pazienti diabetici con ipomagnesiemia.
L’ipercalcemia favorisce lo sviluppo di ipomagnesiemia presumibilmente attraverso l’inibizione dei canali ROMK e delle claudine 16 e 19, mediata dall’azione sul CaSR che favorisce l’escrezione urinaria di Mg.
Le forme familiari di ipomagnesiemia da perdita urinaria di Mg hanno meccanismi d’azione diversi:
- nelle sindromi di Bartter e di Gitelman l’ipomagnesiemia è dovuta a mutazione del co-trasportatore Na-HCl SLC12A3 sensibile ai tiazidici;
- nell’ipomagnesiemia familiare con ipercalciuria e nefrocalcinosi – FHHNC - autosomica recessiva è dovuta alla mutazione nel gene di claudina 16 e talora si associa a diabete insipido;
- una mutazione del gene di claudina 19 può indurre ipomagnesiemia associata a miopia grave;
- le mutazioni di EGF hanno meccanismi d’azione simili a quelli responsabili dell’ipomagnesiemia da anticorpi anti-EGF (cetuximab, panutinimab, matuzumab).
Va inoltre ricordato che l’ipomagnesiemia può verificarsi durante nutrizione parenterale, dopo trapianto epatico e in corso di “hungry bone syndrome”.
MANIFESTAZIONI CLINICHE
L’ipomagnesiemia si può manifestare con segni e sintomi che interessano diversi organi e apparati: neuro-muscolari, cardio-vascolari, ipokaliemia, alterazioni nel metabolismo del calcio; è stata inoltre associata a sindrome metabolica, ipertensione arteriosa e cefalea.
Sintomi neuro-muscolari
Il Mg è essenziale per la stabilizzazione assonale: l’ipomagnesiemia aumenta la soglia di eccitabilità e la velocità di conduzione nervosa, favorisce l’eccitabilità di nervi e muscoli inducendo aumento del calcio intra-cellulare. I sintomi insorgono precocemente e possono andare da ipereccitabilità, irritabilità neuro-muscolare, tremori, fascicolazioni (di particolare rilievo può essere la ridotta efficienza dei muscoli respiratori nei pazienti in terapia intensiva) e giungere fino alla tetania o a vere e proprie crisi convulsive. Si possono d’altra parte osservare apatia, astenia, depressione, potendo arrivare, talora, al coma.
Sintomi cardio-vascolari
Il Mg agisce sui flussi ionici delle cellule miocardiche con meccanismi complessi. L’ipomagnesiemia può inibire la funzione del co-trasportatore NA-K ATPasi-dipendente, inducendo così modificazioni dell’ECG da ripolarizzazione anomala, che possono andare dall’allargamento del QRS o alle T appuntite, in caso di modesta ipomagnesiemia, fino al prolungamento del PR, all’ulteriore allargamento del QRS e all’appiattimento delle onde T in caso di ipomagnesiemia severa. In particolari condizioni cliniche (by-pass aorto-coronarico, esiti di IMA), l’ipomagnesiemia predispone alla fibrillazione atriale e a pericolose aritmie ventricolari (fibrillazione ventricolare e torsione di punta). L’ipomagnesiemia può, inoltre, potenziare gli effetti tossici della digitale, agendo sinergicamente sul co-trasportatore Na-K ATPasi-dipendente, favorendo quindi la deplezione di potassio.
Il Mg sembra avere un ruolo nel regolare i livelli pressori: nell’ipertensione si osserva una riduzione nel Mg intra-cellulare, che potrebbe indurre un incremento delle resistenze periferiche.
Nei pazienti con coronaropatia, l’ipomagnesiemia è più frequente che nella popolazione generale e si è ipotizzato che possa contribuire alla patogenesi dell’IMA, tuttavia, il trattamento con Mg non ne ha modificato la mortalità. È stata comunque suggerita la valutazione dei livelli di Mg nei pazienti con malattia coronarica e, in caso di conferma dell’ipomagnesiemia, auspicata una sua correzione farmacologica.
Alterazioni elettrolitiche
L’ipokaliemia è frequentemente associata all’ipomagnesiemia (dal 40 al 60% dei casi) a causa di condizioni cliniche (diarrea, impiego di diuretici) che provocano la perdita di entrambi gli elettroliti. In alcuni pazienti ipomagnesiemici, tuttavia, la secrezione di potassio nel dotto collettore tubulare e corticale è aumentata grazie all’inibizione dei canali ROMK da parte del Mg intra-cellulare. In questi casi, l’ipokaliemia è poco responsiva alla supplementazione di potassio, ma viene corretta dalla supplementazione di Mg.
L’ipomagnesiemia può indurre ipocalcemia attraverso molteplici meccanismi:
- l’ipoparatiroidismo, dovuto all’inibizione dell’adenosin-ciclasi AMP-dipendente che media il rilascio di PTH;
- la resistenza scheletrica all’azione del PTH: l’ipomagnesiemia induce la rimozione del Mg dai depositi scheletrici; il Mg è scambiato con il Ca circolante, che viene depositato nello scheletro;
- l’ipovitaminosi D: l’ipomagnesiemia riduce la conversione di 25-OH-vitamina D in 1,25-OH2-vitamina D, oltre che attraverso l’ipoparatiroidismo, anche per azione diretta a livello renale.
La supplementazione di Mg è in grado di risolvere l’ipocalcemia nei pazienti con ipomagnesiemia e, talora la calcemia si normalizza in corso di supplementazione di Mg, anche in pazienti con normali valori di Mg.
L’ipomagnesiemia è considerata un fattore di rischio per osteoporosi e favorente altre condizioni patologiche, quali il diabete di tipo 2, l’emicrania, la sindrome da fatica cronica.
DIAGNOSI
La determinazione della magnesiemia non è un esame routinario: viene solitamente richiesta in condizioni a rischio di ipomagnesiemia (diarrea cronica, terapia con prazolici, alcoolismo, ipocalcemia, ipopotassiemia, aritmie ricorrenti). Se l’ipomagnesiemia è confermata, è indispensabile discriminare tra perdite gastro-intestinali e renali di Mg, valutando l’escrezione urinaria di Mg. Bassi livelli di magnesiuria 24 ore (< 30 mg) sono diagnostici per perdita renale. La frazione di escrezione del Mg (FEmg) in una raccolta spot urinaria può essere ricavata dalla seguente formula:
FEMg = {(magnesiuria x creatininemia)/[(0.7* x magnesiemia) x creatininuria]}x100
(il Mg plasmatico è moltiplicato per 0.7 poiché solo il 70% del Mg è libero e filtrabile a livello glomerulare).
I pazienti con ipomagnesiemia da perdita renale di Mg hanno FEMg > 4%, mentre i pazienti con perdite extra-renali hanno una frazione di escrezione < 2%.
TERAPIA
Dosi e modalità di trattamento (parenterale o enterale) dipendono dalla gravità dell’ipomagnesiemia e delle sue manifestazioni cliniche.
In presenza di gravi sintomi è indispensabile la somministrazione endovenosa che, nel caso di aritmie gravi deve essere rapida: 1-2 g di MgSO4 (8-16 mEq) in 2-15 minuti, mentre nei pazienti emodinamicamente stabili può essere prolungata fino a 60 min. I livelli di Mg vanno controllati 6-12 ore dopo l’infusione di Mg: oltre il 50% del Mg infuso viene escreto nelle urine per l’inibizione del riassorbimento nell’ansa di Henle indotto dal carico tubulare elevato. La dose può essere ripetuta per 3-5 giorni, fino a ottenere una magnesiemia stabilmente > 1 mg/dL.
Nei pazienti con insufficienza renale (clearance creatinina < 30 mL/min) a rischio di ipermagnesiemia, la dose va ridotta del 50% e la magnesiemia controllata più spesso.
Il ripristino dei depositi intra-cellulari richiede che il trattamento sia protratto per almeno 2 giorni dopo aver raggiunto normali valori di magnesiemia.
In caso di trattamento pediatrico, viene suggerito di infondere a velocità ridotta da 25 a 50 mg/kg, fino ad un massimo di 2 g.
La supplementazione per os è consigliata nei pazienti asintomatici: gli effetti collaterali (dispepsia, diarrea) indotti dalle dosi elevate di Mg, fanno comunque preferire la somministrazione parenterale nei pazienti ospedalizzati. Nei pazienti ambulatoriali vengono somministrati sali di Mg che sono però scarsamente biodisponibili per os. La dose suggerita nei soggetti asintomatici va da 240 a 1000 mg/die. I preparati “slow release” riducono l’escrezione renale e limitano gli effetti collaterali.
In caso di deplezione severa (Mg < 1 mg/dL), bisogna risolvere, se possibile, la causa di ipomagnesiemia (patologiche, farmacologiche). Nei pazienti con ipomagnesiemia secondaria all’impiego di tiazidici o diuretici dell’ansa, possono essere utilizzati risparmiatori di potassio (amiloride, spironolattone o kanreonato), che favoriscono il riassorbimento distale di Mg. Sono infatti efficaci nel controllare l’ipomagnesiemia anche nelle sindromi di Bartter, Gitelman e nella tossicità da cisplatino.
In caso di insufficienza renale grave o di pazienti dializzati, vanno monitorati i livelli sierici di Mg e i segni clinici di ipermagnesiemia.
BIBLIOGRAFIA
- Agus ZS. Hypomagnesiemia. J Am Soc Nephrol 1999, 10: 1616-22.
- Tong GM, Rude RK. Magnesium deficiency in critical illness. J Intensive Care Med 2005, 20: 3-17.
- Kass L, Weekes J, Carpenter L. Effect of magnesium supplementation on blood pressure: a metaanalysis. Eur J Clin Nutr 2012, 66: 411-8.
- Vallee BL, Wacker WE, Ulmer DD. The magnesium-deficiency tetany syndrome in man. N Engl J Med 1960, 262: 155-61.
- Yu ALS, Goldfarb S, Forman JP. Evaluation and treatment of hypomagnesiemia. Uptodate 2015.
- Fulop T, Batuman V. Hypomagnesiemia. Medscape overview.
- Del Gobbo L, Imamura F, Wu JHY, et al. Circulating and dietary magnesium and risk of cardiovascular disease: a systematic review and meta-analysis of prospective studies. Am J Clin Nutr 2013, 98: 160–73.
Ipermagnesiemia
Alessandro Piovesan
Endocrinologia Oncologica, AO Città della Salute e della Scienza, Torino
L’ipermagnesiemia è una condizione rara e ancor più rara è quella sintomatica.
EZIOPATOGENESI
Poiché il rene è il principale regolatore del metabolismo del Mg, l’ipermagnesiemia può verificarsi nell’insufficienza renale o in caso di sovradosaggio di Mg (per via parenterale, per os o attraverso clisteri).
Insufficienza renale cronica
L’ipermagnesiemia può essere rilevata fino al 15% dei pazienti ospedalizzati, solitamente in quelli con insufficienza renale. La magnesiemia aumenta progressivamente con la riduzione della funzione renale (nei pazienti con insufficienza renale grave la magnesiemia è tra 2.4 e 3.6 mg/dL). L’assunzione orale di Mg è il principale determinante della magnesiemia nei pazienti dializzati; gli anti-acidi e i diuretici (ricchi in Mg) sono pertanto controindicati in questi pazienti. Nei pazienti con insufficienza renale acuta, l’ipermagnesiemia compare durante la fase oligurica e si riduce nella fase poliurica. L’assunzione di solfato di Mg può portare a ipermagnesiemia severa in caso di acidosi.
Cause iatrogene
Infusione di Mg: è frequentemente impiegata nell’eclampsia o pre-eclampsia: tra le conseguenze di tale trattamento sono state riportate ipocalcemia e iperkaliemia materna, così come l’ipocalcemia neonatale
Ingestione orale: sebbene raramente, il carico orale di Mg può superare le capacità escretorie dell’emuntorio renale, specie in caso di modesta insufficienza renale cronica. L’ipermagnesiemia severa con sintomi gravi può essere indotta da abuso di lassativi e dal trattamento con Mg in caso di intossicazione da farmaci. Si calcola che l’ingestione di circa 9 g di citrato di magnesio possa incrementare i livelli plasmatici di Mg attorno ai 3 mg/dL. L’assunzione di anti-acidi a base di Mg in corso di ulcera o gastrite può provocare ipermagnesiemia.
Clisteri: l’ipermagnesiemia può essere provocata dall’assorbimento intestinale di clisteri contenenti grandi quantità di Mg solfato, quindi tale pratica va evitata nei pazienti in insufficienza renale severa.
Altre cause
L’iperparatiroidismo primitivo può indurre ipermagnesiemia in un numero limitato di casi. L’ipermagnesiemia può essere presente nell’ipercalcemia ipocalciurica familiare, nella chetoacidosi diabetica, in stati ipercatabolici (lisi tumorale, ustioni estese, shock settico), nell’intossicazione da litio, nella milk-alkali syndrome, nell’insufficienza cortico-surrenalica, nell’ipotiroidismo e nella depressione.
CLINICA
Sono evidenti sintomi per valori di magnesiemia > 4.8 mg/dL. L’insorgenza dei sintomi può essere facilitata da ipocalcemia, iperkaliemia e uremia.
Neuro-muscolari
Sono i più frequenti: l’ipermagnesiemia può indurre blocco della trasmissione neuro-muscolare, attraverso l’inibizione del rilascio dell’acetil-colina pre-sinaptica e selettivamente del flusso intra-cellulare di calcio nei canali pre-sinaptici. Le conseguenze possono andare dall’attenuazione del riflesso tendineo, alle parestesie. Per livelli di Mg più elevati (> 5 mg/dL) si manifesta debolezza muscolare, che può arrivare alla paralisi flaccida e alla depressione respiratoria, fino all’apnea.
Cardiaci
L’ipermagnesiemia rallenta il sistema di conduzione cardiaco e il tono simpatico, incrementa i livelli pressori, mentre un ulteriore incremento della magnesiemia si associa a ipotensione. A concentrazioni più elevate può accompagnarsi a bradicardia, fino al blocco AV completo e all’arresto cardiaco in caso di livelli > 15 mg/dL.
Altre
L’ipermagnesiemia può indurre ileo paralitico (particolarmente rischioso in corso di terapia della pre-eclampsia), indurre uno stato pro-coagulativo attraverso l’interferenza con l’adesività piastrinica e la generazione di trombina; può inoltre associarsi a ipoparatiroidismo o a resistenza degli organi bersaglio all’azione del PTH.
TERAPIA
La prevenzione è la terapia più efficace: evitare l’assunzione di composti contenti Mg in caso di insufficienza renale e se, non evitabili, monitorare frequentemente la magnesiemia.
Nei casi più gravi, resistenti e sintomatici, può essere necessaria l’emodialisi (più rapida) o la dialisi peritoneale. In questi casi, nell’attesa che la dialisi manifesti la sua efficacia, può essere indicato infondere calcio gluconato per antagonizzare gli effetti del Mg (solitamente da 100 a 200 mg in 5-10 min).
BIBLIOGRAFIA
- Fulop T, Batuman V. Hypermagnesiemia update. Medscape reference 28-05-2014.
- Yu ASL, Goldfarb S, Forman JP. Causes and treatment of hypermagnesiemia. Uptodate 2015.
- Chang WT, Radin B, McCurdy MT. Calcium, magnesium, and phosphate abnormalities in the emergency department. Emerg Med Clin North Am 2014, 32: 349-66.
Ipofosfatemia
Paola Sartorato1 & Gregorio Guabello2
1Dipartimento di Medicina Interna, Ospedale di Montebelluna (TV)
2Ambulatorio di Patologia Osteo-Metabolica, UO Reumatologia, IRCCS Istituto Ortopedico Galeazzi; Ambulatorio di Endocrinologia Oncologica, IRCCS Ospedale San Raffaele, Milano
(aggiornato al 20 marzo 2020)
FISIOLOGIA DEL FOSFORO
Lo ione fosfato svolge un ruolo critico in molti processi biologici, tra cui il metabolismo energetico, la fosforilazione di proteine necessarie alla trasmissione cellulare, il metabolismo degli acidi nucleici e il mantenimento dell’integrità della membrana cellulare e della mineralizzazione ossea.
L’omeostasi del fosforo è sostenuta da una complessa rete di interazioni tra intestino, rene e osso. L’85% del totale è contenuto nello scheletro come idrossi-apatite (il fosforo deriva dalla defosforilazione del pirofosfato inorganico per opera della fosfatasi alcalina localizzata nella membrana plasmatica degli osteoblasti), mentre il 15% è nel comparto extra-scheletrico (fosfoproteine, ATP, fosfolipidi, acidi nucleici).
Gli ormoni coinvolti nell’omeostasi del fosforo sono essenzialmente PTH, calcitriolo e FGF-23.
Il trasporto intra-cellulare e la concentrazione sierica del fosfato sono mediati da due principali famiglie di proteine: SCL34 (NPT2a, NPT2b, NPT2c) o co-trasportatori di tipo II e SCL20 (Pit-1 e Pit-2) o co-trasportatori di tipo III (1).
A livello osseo il PTH ha azione diretta, stimolando la degradazione della matrice, con passaggio di fosforo dall’osso al sangue.
A livello intestinale il calcitriolo attiva l’assorbimento intestinale di fosforo attraverso il co-trasportatore sodio-fosforo IIb e il PTH ha azione indiretta, attraverso lo stimolo della sintesi renale di calcitriolo.
A livello renale il fosforo è filtrato dal glomerulo e riassorbito per circa l’85% nel tubulo prossimale, dove sulla membrana apicale delle cellule sono espressi i due co-trasportatori sodio-fosforo NPT2a e NPT2c. L’azione dei co-trasportatori di tipo II è regolata da PTH e FGF-23. Il PTH agisce internalizzando NPT2a dalla membrana apicale delle cellule del tubulo prossimale e quindi aumentando l’escrezione urinaria di fosfato. L’FGF-23 o fosfatonina è un peptide prodotto principalmente dall’osso e dal tessuto connettivo, che agisce riducendo l’espressione di mRNA di NPT2a e NPT2c e quindi ha anch’esso azione fosfaturica. FGF-23, inoltre, ha azione opposta al PTH sull’attivazione della vitamina D: inibisce l’espressione della 1-alfa-idrossilasi a livello del tubulo renale prossimale e stimola la 24-idrossilasi, convertendo il calcitriolo e la 25-OH vitamina D in metaboliti inattivi (2).
La tabella 1 riporta i fattori che influenzano il riassorbimento di fosforo a livello del tubulo contorto prossimale (3).
| Tabella 1 Fattori che influenzano il riassorbimento di fosforo a livello del tubulo contorto prossimale |
|
| Riducono il riassorbimento | PTH FGF-23 Acidosi Elevato apporto dietetico di fosforo Peptide natriuretico atriale Cortisolo Dopamina |
| Aumentano il riassorbimento | Alcalosi Basso apporto dietetico di fosforo Ipoparatiroidismo Ormone tiroideo Calcitriolo GH-IGF-I |
CLINICA DELL’IPOFOSFATEMIA
L’ipofosfatemia è definita da livelli di fosfato sierico inferiori a 2.5 mg/dL (0.8 mmol/L) e può essere acuta o cronica. L’ipofosfatemia acuta, frequente in ambito ospedaliero, in particolare nelle unità di terapia intensiva, può essere di diversa gravità (tab. 2) (4).
| Tabella 2 Classificazione ipofosfatemia in relazione alla gravità |
|
| Lieve | 2-2.5 mg/dL |
| Moderata | 1-1.9 mg/dL |
| Severa | < 1 mg/dL |
I sintomi dell’ipofosfatemia sono aspecifici e legati alla causa, durata e severità del deficit di fosfati; molti pazienti sono asintomatici. L’ipofosfatemia può determinare debolezza muscolare, in particolare ai muscoli respiratori, e se severa può determinare disfunzione miocardica, aritmie ventricolari, rabdomiolisi e alterazione dello stato mentale. L’ipofosfatemia cronica a qualsiasi età altera la mineralizzazione ossea, causando rachitismo/osteomalacia.
EZIOPATOGENESI DELL’IPOFOSFATEMIA
L’ipofosfatemia può avere molteplici cause (5), che possono essere classificate in tre categorie (tab. 3).
| Tabella 3 Cause di ipofosfatemia |
||
| Meccanismo patogenetico | Patologie | Caratteristiche |
| Aumentata escrezione renale mediata da FGF-23 (FGF-23 elevato, o inappropriatamente normale per i livelli di ipofosfatemia, iperfosfaturia isolata) | Rachitismo ipofosfatemico autosomico dominante (ADHR) | Mutazione attivante di FGF-23, che diventa resistente a clivaggio/degradazione, con conseguente eccesso di FGF-23. Livelli di calcitriolo normali/bassi. |
| Rachitismo ipofosfatemico autosomico recessivo (ARHR) | Mutazione inattivante di DMP1* (alterata differenziazione degli osteociti con aumentata produzione di FGF-23) o di ENPP1* (aumentata produzione di FGF-23). Livelli di calcitriolo normali/bassi. |
|
| Rachitismo ipofosfatemico dominante legato all’X (XLH) | Mutazione inattivante di PHEX*-endopeptidasi, con aumento di trascrizione di FGF-23. È il più frequente (prevalenza di 1:20.000). Livelli di calcitriolo normali/bassi. |
|
| Osteomalacia oncogenica (TIO, tumor-induced osteomalacia) | Sindrome paraneoplastica acquisita. | |
| Displasia fibrosa/ sindrome di McCune-Albright | Mutazione della sub-unità alfa della proteina G stimolatoria, con produzione di FGF-23 da tessuto fibroso. | |
| Ipofosfatemia post-trapianto renale | Produzione terziaria di FGF-23. | |
| Forme rare | Sindrome dei nevi sebacei lineari: produzione di FGF-23 da lesioni cutanee. | |
| Neurofibromatosi. | ||
| Displasia osteoglofonica (macroglossia e disfonia). | ||
| Rachitismo ipofosfatemico con iperparatiroidismo. | ||
| Aumentata escrezione renale non mediata da FGF-23 (FGF-23 normale/basso, iperfosfaturia non isolata) | Iperparatiroidismo primario e secondario. | |
| Secrezione paraneoplastica di PTH-RP. | ||
| Ipercortisolismo endogeno/terapia steroidea cronica. | ||
| Sindrome di Fanconi | Su base genetica: mutazione inattivante del co-trasportatore Na/P IIa del tubulo contorto prossimale, malattia di Dent, cistinosi, altre. | |
| Iatrogena (vedi oltre). | ||
| Iperfosfaturia associata a ipercalciuria, glicosuria, aminoaciduria, acidosi, disionie. | ||
| Rachitismo ereditario ipofosfatemico con ipercalciuria (HHRH) | Mutazione inattivante autosomica recessiva del co-trasportatore Na/P IIc del tubulo contorto prossimale. Livelli di calcitriolo elevati con PTH e FGF-23 bassi. Iperfosfaturia associata a ipercalciuria. |
|
| Acidosi tubulare distale | Difetto genetico o acquisito della rigenerazione distale del bicarbonato. L’acidosi metabolica determina perdita renale di fosforo. Iperfosfaturia associata a ipercalciuria e acidosi |
|
| Ridotto assorbimento intestinale (NB: l’ipofosfatemia può determinare up-regulation di calcitriolo, con potenziale ipercalcemia e ipercalciuria) | Ridotto apporto. | |
| Prematurità. | ||
| Etilismo. | ||
| Malassorbimento. | ||
| Malnutrizione. | ||
| Diarrea, vomito. | ||
| Sondino naso-gastrico. | ||
| Carenza di vitamina D | By-pass digiuno-ileali. | |
| Steatorrea, diarrea, enteropatia da glutine. | ||
| Resistenza alla vitamina D | Deficit 1alfa-idrossilasi. | |
| Deficit recettore VDR. | ||
| Ingresso di fosforo nelle cellule (malattie acute) | Sindrome da refeeding (infusione di fluidi/glucosio) + ipopotassiemia e ipomagnesiemia. | |
| Terapia della cheto-acidosi diabetica (infusione di insulina). | ||
| Alcalosi respiratoria acuta. | Avvelenamento da salicilati. | |
| Ventilazione meccanica. | ||
| Rapida captazione cellulare (sindrome dell’osso affamato). | ||
| Rapida proliferazione cellulare (leucemia acuta). | ||
| Sepsi. | ||
| Fase post-operatoria chirurgie maggiori (epatica e cardiaca). | ||
| Traumi. | ||
| Ustioni. | ||
| Elevati livelli di catecolamine. | ||
| Iatrogeno | Vedi oltre. | |
| *DMP1 (dentin matrix protein 1), ENPP1 (ectonucleotide pyrophosphatase/phosphodiesterase 1), PHEX (PHosphate regulating gene with homology to Endopeptidases on the X chromosome) | ||
Ipofosfatemia secondaria a inadeguato apporto o ridotto assorbimento intestinale
Raramente l’ipofosfatemia è secondaria a ridotto introito, mentre il malassorbimento intestinale è causa frequente di ipofosforemia. Pazienti in trattamento per lungo tempo con anti-acidi chelanti il fosforo possono presentare ipofosforemia, ipofosfaturia con ipercalciuria e nefrolitiasi. La carenza di vitamina D è associata a ipofosfatemia, essendo il metabolita attivo della vitamina D il maggior regolatore dell’assorbimento intestinale di fosfati. Nei pazienti sottoposti a by-pass digiuno-ileali o con enteropatia da glutine una compromissione dei recettori intestinali per la vitamina D può ridurre l’assorbimento di fosfato (6). Non solo la carenza, ma anche la resistenza alla vitamina D, secondaria ad alterazioni genetiche della 1alfa-idrossilasi o del recettore della vitamina D possono determinare un quadro di rachitismo/osteomalacia con ipofosforemia (7).
Ipofosfatemia secondaria a redistribuzione del fosfato dai compartimenti extra-cellulari ai compartimenti intra-cellulari
La redistribuzione del fosfato attraverso la membrana cellulare è la causa più comune di ipofosfatemia nei pazienti in terapia intensiva e può essere secondaria a molteplici condizioni, quali alcalosi respiratoria acuta, cheto-acidosi diabetica, elevati livelli sierici di catecolamine, sia endogene che esogene. L’ipofosforemia si osserva in corso di sepsi, nel post-operatorio di chirurgie maggiori (epatica e cardiaca), in pazienti traumatizzati (traumi cerebrali, ustioni). Una rapida redistribuzione del fosfato tra i compartimenti, con ipofosforemia anche severa, può avvenire nei pazienti malnutriti dopo alimentazione forzata parenterale (sindrome da refeeding) e nella sindrome dell’osso affamato post-paratiroidectomia (8).
Ipofosfatemia da eccessiva perdita urinaria di fosfati mediata da elevati livelli di FGF-23
Le forme genetiche (5) di malattie renali ipofosfatemiche accomunate da un eccesso di FGF-23 includono l’ipofosfatemia X-linked (XLH), il rachitismo ipofosfatemico autosomico dominante (ADHR) e il rachitismo ipofosfatemico autosomico recessivo (ARHR).
L’ipofosfatemia mediata da FGF-23 può presentarsi anche in condizioni cliniche in cui la fosfatonina viene prodotta localmente in siti anomali, che includono: displasia fibrosa, sindrome del nevo sebaceo lineare e osteomalacia oncogenica.
La displasia fibrosa dipende da una mutazione acquisita somatica attivante della subunità a della proteina Gs nelle cellule staminali mesenchimali, con formazione di osteoblasti aberranti (cellule osteogeniche), che depongono matrice osteoide anomala (tessuto osseo fibroso con trabecole anormali in numero, distribuzione e forma), producono (50% dei casi) FGF-23, con conseguente osteomalacia da ipofosfatemia, e infine producono interleukina-6 e up-regolano RANKL, con conseguente osteoclastogenesi inappropriata. Si parla di sindrome di McCune-Albright quando alla displasia fibrosa si accompagnano manifestazioni cutanee (macchie cutanee caffè-latte) ed endocrinopatie (pubertà precoce, ipertiroidismo, acromegalia, Cushing) (9).
L’ipofosfatemia post-trapianto renale dipende in primis da una produzione terziaria di FGF-23; fattori concomitanti sono rappresentati dall’iperparatiroidismo secondario e dall’azione di tacrolimus e steroide, che riducono l’espressione del co-trasportatore sodio/fosforo IIa (NPT IIa) a livello del tubulo contorto prossimale (10).
Ipofosfatemia da eccessiva perdita urinaria di fosfati indipendente dai livelli di FGF-23
Gli steroidi (endogeni o esogeni) possono determinare perdita renale primaria di fosfato, in quanto riducono l’espressione di NPT IIa a livello del tubulo contorto prossimale (10).
La perdita renale di fosfati può essere secondaria ad anomalie tubulari. La forma ereditaria di rachitismo ipofosfatemico con ipercalciuria (HHRH) è causata da mutazioni del gene codificante per il co-trasportatore NPTC2 e si caratterizza, oltre che per l’ipercalciuria con normocalcemia, anche per il riscontro di elevati livelli di calcitriolo e soppressione della secrezione di PTH.
La sindrome di Fanconi è una disfunzione complessa della capacità di riassorbimento del tubulo contorto prossimale: oltre al fosfato, può riguardare glucosio, aminoacidi, proteine, bicarbonati (acidosi tubulare prossimale tipo II), acido urico ed elettroliti (calcio, sodio, potassio). Ne esistono due forme distinte:
- genetica, da mutazione inattivante del co-trasportatore Na/P NPT IIa del tubulo contorto prossimale, malattia di Dent, cistinosi;
- iatrogena, che causa una disfunzione mitocondriale.
Le manifestazioni cliniche sono principalmente rappresentate da osteomalacia ipofosfatemica, acidosi metabolica e sintomi da deplezione proteica (11).
Dal punto di vista laboratoristico, all’ipofosfatemia si possono associare ipercalciuria, glicosuria, aminoaciduria, proteinuria tubulare, acidosi metabolica (bicarbonati venosi < 20 mEq/L), disionie (iposodiemia, ipopotassiemia, ipocalcemia).
La terapia si basa sulla supplementazione delle sostanze perse a livello renale (fosforo, aminoacidi/proteine, bicarbonato sodio/potassio citrato) (11).
L’acidosi tubulare distale (tipo I) dipende da un difetto della rigenerazione distale dei bicarbonati, con acidosi metabolica che causa perdita renale di fosforo. Può essere genetica (autosomica dominante o recessiva) o secondaria (malattie autoimmuni, granulomatosi, farmaci, trapianto di rene, ipertiroidismo, iperparatiroidismo, infezioni croniche delle vie urinarie, epatite cronica attiva, nefrocalcinosi, anemia a cellule falciformi).
Le principali manifestazioni cliniche dell’adulto sono osteomalacia e nefrolitiasi/nefrocalcinosi (12).
Le principali alterazioni laboratoristiche sono: bicarbonati venosi < 20 mEq/L, ipofosfatemia, ipopotassiemia, pH urinario > 5.5-6 e ipercalciuria.
La terapia si basa sulla correzione dell’acidosi metabolica (bicarbonato sodio o citrato sodio per os) e sulla supplementazione di potassio (potassio citrato che ha anche effetto ipocalciurico) (12).
La tabella 4 riporta la diagnosi differenziale biochimica dei disturbi da perdita renale di fosforo.
| Tabella 4 Diagnosi differenziale biochimica dei disturbi da perdita renale di fosfato |
||||||
| Patologia | PTH | Calcio | Fosfato TmP/GFR |
25-OH-D3 1,25-OH-D3 |
Calciuria 24 ore | Altre |
| Con FGF-23 N/alto | ||||||
|
XLH/ ADHR/ ARHR/ TIO/ displasia fibrosa |
N/alto | N | Bassi | N N/bassa |
N/bassa | - |
| Post-trapianto renale | N/alto | N | Bassi | N Bassa |
N/bassa | - |
| Ferro EV | N/alto | N | Bassi | N N/bassa |
N/bassa | - |
| Con FGF-23 N/basso | ||||||
| N/alto | Alto | N/bassi | N N/alta |
N/alta | - | |
| Diuretici | N/basso/alto | N/basso/alto | Bassi | N N/alta |
N/alta/bassa | - |
| Steroidi | N/alto | N/basso | Bassi | N N/bassa |
N/alta | - |
| HHRH | N/basso | N | Bassi | N Alta |
Alta | - |
| Fanconi | N/alto | N/basso | Bassi | N N |
Alta | Glicosuria, aminoaciduria, proteinuria tubulare, acidosi metabolica (bicarbonati venosi < 20 mEq/L), disionie (ipoNa, ipoK, ipoCa) |
| Acidosi tubulare distale | N/basso | N | Bassi | N N |
Alta | Acidosi metabolica (bicarbonati venosi < 20 mEq/L), ipoK, pH urinario > 5.5-6 |
APPROCCIO DIAGNOSTICO AL PAZIENTE CON IPOFOSFATEMIA
Di fronte al riscontro biochimico di ipofosfatemia (valore < 2.7-2.5 mg/dl, se la fosfatemia è espressa in mmol/L è necessario dividere per 0.323 per ottenere il valore in mg/dL), la prima cosa da valutare è la presenza o assenza di sintomi:
- un lieve abbassamento dei valori di fosfatemia è in genere asintomatico, talvolta possono essere presenti astenia o debolezza muscolare;
- il primo segno clinico di ipofosfatemia cronica può essere un evento fratturativo nell’ambito di un quadro di osteomalacia misconosciuta;
- in caso di ipofosfatemia severa e/o acuta (fosfatemia < 1.5 mg/dL), il quadro clinico invece può essere rilevante e drammatico, con grave miopatia, fratture multiple, encefalopatia fino al coma e cardiomiopatia con scompenso di circolo.
Dopo la valutazione della presenza o meno di sintomatologia clinica, il passo successivo è quello di comprendere la causa.
Il primo passo in assoluto è rappresentato da un’attenta anamnesi farmacologica, in quanto numerosi farmaci possono causare, con meccanismi complessi e spesso non completamente noti, un abbassamento più o meno severo dei livelli plasmatici di fosfato (13). La tabella elenca tutti i farmaci per i quali esistono attuali evidenze di possibile nesso causale con ipofosfatemia, evidenziando per ciascuno il meccanismo fisiopatologico e l’effetto finale (riportato alle tre categorie sovra-descritte).
| Tabella 5 Farmaci che possono causare ipofosfatemia |
|||
| Farmaco | Meccanismo fisiopatologico | Effetto finale | |
| Infusione di fluidi e glucosio/fruttosio, nutrizione parenterale (refeeding syndrome) Infusione di insulina (terapia della chetoacidosi diabetica) |
Stimolazione intra-cellulare della glicolisi, con incremento della formazione di composti intermedi fosforilati nel fegato e nel muscolo | Shift intra-cellulare | |
| Eritropoietina e GM-CSF | Proliferazione cellulare acuta | Shift intra-cellulare | |
| Simpatico-mimetici | Adrenalina | Attivazione della glicolisi | Shift intra-cellulare |
| Dopamina, teofillina | Attivazione della glicolisi + effetto sul tubulo contorto prossimale (TCP) | Shift intra-cellulare + perdita renale | |
| Steroidi | Effetto sull’intestino e sul TCP | Ridotto assorbimento + perdita renale | |
| Diuretici | Acetazolamide | Inibizione dell’anidrasi carbonica (AC) nel TCP e conseguente acidosi metabolica | Perdita renale |
| Tiazidici | Inibizione dell'AC, riduzione del riassorbimento tubulare distale, induzione di ipo-K+ e ipo-Mg++ | ||
| Furosemide | Inibizione lieve dell’AC | ||
| Ferro endovena (soprattutto carbossi-maltosio, ma anche isomaltoside, oxitolo, polimaltosio) | Aumento di FGF-23 per inibizione della degradazione Effetto tossico diretto del ferro sul TCP |
Perdita renale | |
| Inibitori delle tirosin-chinasi (sorafenib, ibrutinib, imatinib, sunitinib, dasatanib, dabrafenib, ceritinib, nilotinib) | Disfunzione tubulare (s Fanconi iatrogena) Ipocalcemia con iperparatiroidismo secondario |
Perdita renale | |
| Chemioterapici (ifosfamide, cisplatino, ciclofosfamide, streptozocina, azacitidina, 6-mercaptopurina, suramina) | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale | |
| Inibitori di mTOR (everolimus, temsirolimus, sirolimus) | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale | |
| Anti-epilettici | Carbamazepina, fenitoina, fenobarbitale | Induzione del citocromo P450 con aumentato catabolismo del calcifediolo (ridotto assorbimento intestinale del fosforo e ipocalcemia con iperparatiroidismo secondario) | Ridotto assorbimento intestinale + perdita renale |
| Valproato | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale | |
| Antibiotici | Linezolid (alla sospensione) | Riattivazione mitocondriale | Shift intra-cellulare |
| Aminoglicosidi, rifampicina, tetracicline | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale | |
| Isoniazide | Induzione del citocromo P450 con aumentato catabolismo del calcifediolo (ridotto assorbimento intestinale del fosforo e ipocalcemia con iperparatiroidismo secondario) | Ridotto assorbimento intestinale + perdita renale | |
| Inibitori trascrittasi inversa | Adefovir, tenofovir, cidofovir, abacavir | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale |
| Efavirenz | Induzione del citocromo P450 con aumentato catabolismo del calcifediolo (ridotto assorbimento intestinale del fosforo e ipocalcemia con iperparatiroidismo secondario) | Ridotto assorbimento intestinale + perdita renale | |
| Inibitori proteasi (darunavir, lopinavir, atazanavir) | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale | |
| Anti-virali (HCV) (ribavirina, interferone) | |||
| Anti-virali (HBV) (lamivudina, entecavir) | |||
| Anti-virali (Herpes Simplex) (acyclovir) | |||
| Anti-riassorbitivi (amino-bisfosfonati, denosumab) | Ipocalcemia con iperparatiroidismo secondario | Perdita renale | |
| Anabolizzanti dell’osso (teriparatide) | Azione sul tubulo renale | Perdita renale | |
| Estrogeni (TOS, pillola EP) | Down-regulation del co-trasportatore Na/P IIa e IIc del TCP | Perdita renale | |
| FANS (ibuprofene ad alte dosi) | Inibizione dell’AC (acidosi tubulare distale) | Perdita renale | |
| Cannabis | Iperemesi, etilismo, denutrizione | Ridotto assorbimento intestinale | |
| Etanolo (etilismo) | Malnutrizione | Ridotto apporto | |
| Azione tossica diretta sul TCP | Perdita renale | ||
| Refeeding syndrome | Shift intra-cellulare | ||
| Gadolinio (dosi ripetute) | Fibrosi sistemica | Alterazione dell’omeostasi del fosforo | |
| Anti-acidi contenenti Ca/Mg/Al | Chelanti del fosforo | Ridotto assorbimento intestinale | |
| Anti-psoriasici (acido fumarico, apremilast) | Disfunzione tubulare (s Fanconi iatrogena) | Perdita renale | |
Quando la causa dell’ipofosfatemia non emerge dalla storia clinica e dall’esame obiettivo, può essere utile valutare se l’escrezione urinaria di fosfati è appropriata. Il tasso di riassorbimento massimo tubulare per i fosfati (TmP), espresso come funzione del tasso di filtrazione glomerulare (TmP/GFR) è un indice che esprime il riassorbimento di fosfato come funzione sia della concentrazione sierica di fosfato sia del tasso di filtrazione glomerulare. La formula per calcolare l’escrezione frazionata su una raccolta urinaria mattutina di 2 h è:
CPO4/Ccreat = (creatininemia x fosfaturia)/(fosfatemia x creatininuria).
In condizioni fisiologiche questo rapporto è compreso tra 0.05 e 0.15. Tale valore può essere espresso in forma percentuale, quindi come riassorbimento tubulare del fosfato (RTP):
RTP = (1- CPO4/Ccreat) x 100.
Tale indice si può calcolare attraverso l’utilizzo di un algoritmo on-line, inserendo i 4 parametri biochimici (fosfatemia, fosfaturia spot, creatininemia, creatininuria spot): vengono calcolati il riassorbimento tubulare massimo del fosforo per unità di filtrato glomerulare (TmP/GFR: valore normale 2.5-4.5 mg/dL, ma nei bambini in fase di crescita il rapporto varia tra 4.5 e 5 mg/dL, riflettendo gli effetti del GH sull’assorbimento tubulare di fosfato (2)) e la percentuale del riassorbimento tubulare del fosforo (RTP: valore normale 82-90%) (14):
- se RTP è ≤ 86%, la relazione fra fosfatemia e fosfaturia è lineare e l’algoritmo fornisce già il valore reale del TmP/GFR (TmP/GFR = RTP * fosfatemia);
- se invece RTP è > 86%, la relazione fra fosfatemia e fosfaturia non è lineare e il TmP/GFR fornito dall’algoritmo deve essere ricalcolato, secondo la formula:
TmP/GFR = (0.3 * RTP) / [1 – (0.8 * RTP)] * fosfatemia.
I dati di RTP e/o di TmP/GFR vengono interpretati orientativamente in questo modo:
- basso valore orienta per perdita renale;
- valore normale/alto orienta per malassorbimento intestinale.
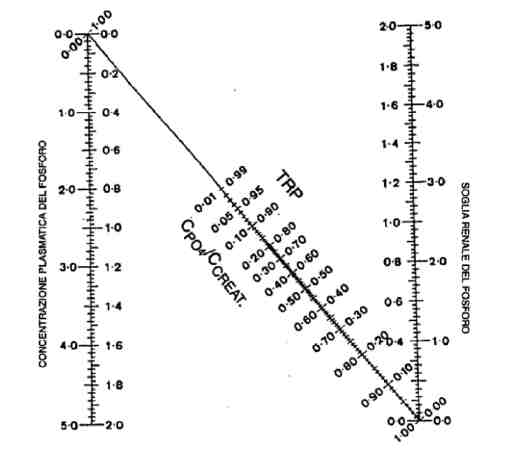
Nomogramma di Walton e Bijovoet
In caso di ipofosfatemia da perdita renale, il dosaggio di FGF-23 permette di differenziare tra le forme mediate e non mediate da FGF-23. È oggi disponibile in alcuni laboratori un dosaggio di FGF-23 intatto, valutato in un campione di 908 soggetti francesi sani, di 18-89 anni, che ha dimostrato eccellenti caratteristiche analitiche. Il range normale di riferimento è risultato 22.7-93.1 pg/mL. In 11 pazienti con tumor-induced osteomalacia (TIO) e in 8 pazienti con rachitismo ipofosfatemico X-linked (XLH) i valori di FGF-23 sono risultati, rispettivamente, 73.2-361.2 pg/mL e 122.3-179.5 pg/mL (15).
Quando si verifica una perdita renale primaria di fosfato, per un meccanismo fisiologico di feed-back, la sintesi di FGF-23 viene down-regolata fino a sopprimersi; la presenza quindi di un basso livello di fosfato associato a FGF-23 alto o inappropriatamente normale permette di fare diagnosi di forma mediata da FGF-23: dopo avere escluso il post-trapianto renale e la terapia marziale ev, l’anamnesi e l’esame obiettivo sono fondamentali per distinguere una forma geneticamente determinata (XLH, ADHR, ARHR), che in virtù dello spettro clinico molto variabile può essere diagnosticata anche in età adulta, da una forma acquisita (displasia fibrosa, TIO).
In presenza invece di bassi valori di fosfato con FGF-23 basso o nel range basso della norma, la diagnosi orienta verso una forma non mediata da FGF-23: in questo caso, dopo avere escluso iperparatiroidismo e terapia diuretica e steroidea, l’ipercalciuria (sempre presente) associata o meno ad acidosi metabolica e ad altre alterazioni urinarie (aminoaciduria, proteinuria, …) permette di differenziare l’HHRH dalla sindrome di Fanconi e dall’acidosi tubulare distale.
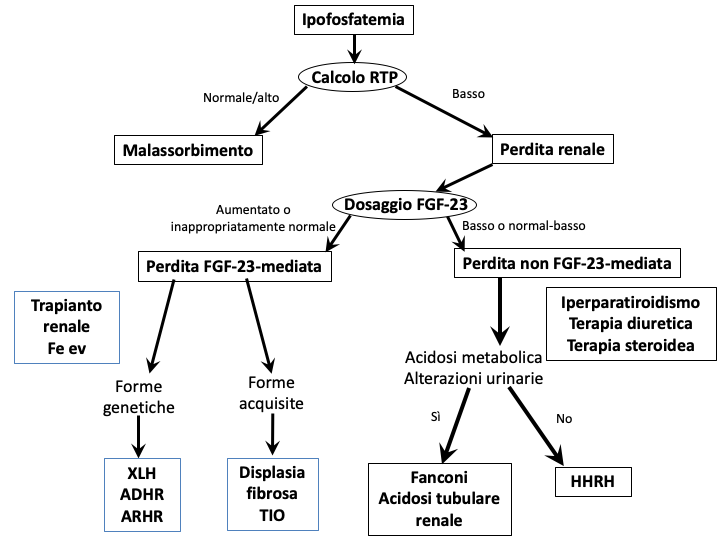
TRATTAMENTO
Il trattamento dell’ipofosfatemia è legato alla causa che la determina e alla gravità del deficit. Il trattamento deve rimuovere, quando possibile, la causa di fondo, gli eventuali farmaci responsabili e correggere le eventuali carenze.
La terapia consiste nella somministrazione orale o endovena di fosforo elementare, con alcune importanti raccomandazioni:
- il fosforo ev dovrebbe essere usato con cautela, per il rischio di ipocalcemia acuta e iperfosfatemia da ipercorrezione, ed evitato nei pazienti iper/ipocalcemici;
- il fosforo per os è la più sicura forma di trattamento e deve essere la prima scelta;
- la supplementazione aggressiva di fosforo (per os/ev) può determinare grave ipocalcemia, il cui rischio può essere aumentato nell’insufficienza renale cronica;
- nella pratica clinica come sali di fosfato si usano potassio fosfato e sodio fosfato;
- il fosfato di potassio non dovrebbe essere usato in caso di iperpotassiemia e/o insufficienza renale cronica;
- la terapia dell’ipofosfatemia da perdita renale mediata da FGF-23 richiede sia il fosforo sia il calcitriolo, in quanto l’FGF-23 determina da un lato perdita renale di fosforo e dall’altro inibizione della sintesi renale di calcitriolo.
La somministrazione parenterale di fosfati è riservata ai pazienti con ipofosfatemia acuta severa (< 1.5 mg/dL), contribuendo questa all’instabilità emodinamica e gravando sul rischio di mortalità (8). Il fosfato per via endovenosa deve essere utilizzato con cautela. Esistono pochi studi non controllati sul trattamento dell’ipofosforemia severa con vari regimi proposti e non validati nell’infanzia (tab 6) (5). La risposta alla terapia con fosfato ev è molto variabile e non prevedibile: nel paziente trattato sono raccomandati controlli almeno ogni 6 ore di fosfatemia, calcemia, potassiemia, magnesiemia e monitoraggio con telemetria. I rischi maggiori legati alla terapia con fosfato per via ev sono ipocalcemia severa con tetania e convulsioni, modifiche ECG e shock. Il trattamento non deve essere somministrato a pazienti ipocalcemici ed è gravato da ulteriori rischi nei pazienti con alterazioni della funzionalità renale. Quando l’ipofosfatemia è attesa, secondaria ad altri trattamenti (per esempio nella sindrome da refeeding, nell’alcolismo e nella cheto-acidosi diabetica), il fosfato è infuso come fosfato di potassio per prevenire o trattare la carenza.
| Tabella 6 Trattamento ipofosfatemia acuta |
||
| Dose di fosfato da somministrare (vari regimi pubblicati) | Controlli suggeriti | |
| Terapia ev (se P < 1.5 mg/dL) | 15 mM (464 mg) in 100 mL di fisiologica in 2 ore, da ripetere dopo 6 h se necessario (max 45 mmol/24h) 0.32 mmol/kg (9.9 mg/kg) infusi in 12 h e da ripetere ogni 12 h fino a P > 2 mg/dL 0.64 mmol/kg (19.8 mg/kg) in 8-12 h Mantenimento: 70-160 mg/kg/die di principio attivo, pari a 1-2 flaconi/die di Esafosfina 5 g/50 mL (velocità di infusione 10 mL/min) |
Ogni 6 h: Ca, P, Mg, K, creatinina, ECG |
| Terapia orale | 30-40 mg/kg/die in 4-5 dosi | Ogni 12-24 h: Ca, P, Mg, K, creatinina |
| Sindrome da refeeding | In genere potassio fosfato: 0.5-0.8 mmol/kg/die (15-25 mg/kg/die) nell’idratazione ev | Ogni 12-24 h: Ca, P, Mg, K, creatinina |
| Tabella 7 Terapia dell’ipofosfatemia cronica (soprattutto da perdita renale) |
|
| Somministrare | Monitorare |
| Fosforo elementare per os: 20-40 mg/kg/die in 4 somministrazioni (effetto collaterale: diarrea). Calcitriolo (se perdita renale mediata da FGF-23): 20-30 ng/kg/die in 2 somministrazioni. |
Ca, P, K, creatinina una volta al mese fino a stabilità, quindi ogni 3 mesi + fosfatasi alcalina. PTH ogni 6-12 mesi. Ecografia renale ogni 1-2 anni. |
| Tabella 8 Formulazioni commerciali di fosfati |
||
| Via di somministrazione | Sale | Contenuto di fosforo |
| Orale | Sodio fosfato dibasico/potassio (Reducto-spezial)* | In ogni cp 612 mg (+ 360 mg di K) |
| Miscela di acido fosforico e bifosfato sodico (soluzione di Joulie) | 50 mg/mL | |
| Phosphate Sandoz* | In ogni cp effervescente 500 mg + potassio 3.1 mEq | |
| Infusionale | Fruttosio difosfato sodico (Esafosfina) | 0.5 g/10 mL pari a 0.235 mEq/mL 5 g/50 mL pari a 0.47 mEq/mL 10 g/100 mL pari a 0.47 mEq/mL |
| * disponibili in Svizzera | ||
In generale, la somministrazione di fosfato per os è sicura e raccomandata nei pazienti stabili con ipofosfatemia cronica ed acuta. I dosaggi suggeriti di sali di fosfato variano tra 30-40 mg/kg/die suddivisi in 4-5 somministrazioni, con titolazione della dose (16).
Il trattamento dei rachitismi ipofosfatemici si basa sulla somministrazione di fosfato per os e calcitriolo. Nell’infanzia il trattamento inizia dal momento della diagnosi e prosegue fino al raggiungimento della statura definitiva. La terapia riesce parzialmente a correggere le deformità degli arti, riduce il numero di interventi chirurgici e migliora la prognosi staturale. Nelle forme familiari note è suggerito l’inizio precoce del trattamento, prima dell’anno di vita. I dosaggi di calcitriolo raccomandati variano tra 20 e 30 ng/kg/die, suddivisi in 2-3 dosi/die e quelli di fosforo tra 20-40 mg/kg/die (in 3-5 dosi). La somministrazione di fosfato viene titolata per minimizzare gli effetti collaterali gastro-intestinali. Gli obiettivi della terapia nell’adulto sono ridurre la sintomatologia dolorosa, l’estensione dell’osteomalacia e favorire la guarigione delle fratture, ma l’efficacia della terapia convenzionale per queste indicazioni è supportata da dati clinici limitati. Il trattamento deve essere individualizzato in relazione a età, peso corporeo, funzionalità renale e paratiroidea. Un adulto normocalcemico con PTH normale o di poco aumentato, inizia la terapia con 0.5-0.75 µg/die di calcitriolo in 2 somministrazioni; dopo una settimana si introduce il trattamento con fosforo, generalmente 250 mg/die, che viene aumentato dopo 4 giorni fino a 750-1000 mg/die da distribuire in 3-4 somministrazioni/die. I ridotti livelli endogeni di calcitriolo in questi pazienti possono spiegare il rischio di sviluppare iperparatiroidismo secondario e addirittura terziario se il trattamento con fosfato non è bilanciato.
In questi pazienti adulti con iperparatiroidismo secondario o terziario accompagnato da elevati livelli di ALP è stato utilizzato anche il cinacalcet, con risposte al trattamento variabili. Il trattamento del rachitismo HHRH prevede solo l’utilizzo di fosfato (17).
Il monitoraggio della terapia richiede controlli periodici di calcemia, fosforemia, creatininemia, calciuria, fosfaturia, creatininuria, fosfatasi alcalina e PTH.
BIBLIOGRAFIA
- Penido MG, Alon US. Phosphate homeostasis and its role in bone health. Pediatr Nephrol 2012, 27: 2039-48.
- Lee JY, Imel EA. The changing face of hypophosphatemic disorders in the FGF-23 era. Pediatr Endocrinol Rev 2013, 10 suppl 2: 367-79.
- Wagner CA, Rubio-Aliaga I, Biber J, Hernando N. Genetic diseases of renal phosphate handling. Nephrol Dial Transplant 2014, 29 suppl 4: 45-54.
- Felsenfeld AJ, Levine BS. Approach to treatment of hypophosphatemia. Am J Kidney Dis 2012, 60: 655-61.
- Imel EA, Econs MJ. Approach to the hypophosphatemic patient. J Clin Endocrinol Metab 2012, 97: 696-706.
- Assadi F. Hypophosphatemia: an evidence-based problem-solving approach to clinical cases. Iran J Kidney Dis 2010, 4: 195-201.
- Elder CJ, Bishop NJ. Rickets. Lancet 2014, 383: 1665-76.
- Geerse DA, Bindels AJ, Kuiper MA, et al. Treatment of hypophosphatemia in the intensive care unit: a review. Crit Care 2010, 14: R147.
- Boyce AM, Florenzano P, de Castro LF, Collins MT. In: Adam MP, Ardinger HH, Pagon RA, et al (Eds). Fibrous dysplasia/McCune-Albright syndrome. GeneReviews® [Internet]. University of Washington, Seattle; 1993-2018.
- Manghat P, Sodi R, Swaminathan R. Phosphate homeostasis and disorders. Ann Clin Biochem 2014, 51: 631–56.
- Karatzas A, Paridis D, Kozyrakis D, et al. Fanconi syndrome in the adulthood. The role of early diagnosis and treatment. J Musculoskelet Neuronal Interact 2017, 17: 303-6.
- Seidowsky A, Moulonguet-Doleris L, Hanslik T, et al. Tubular renal acidosis. Rev Med Interne 2014, 35: 45-55.
- Megapanou E, Florentin M, Milionis H, et al. Drug-induced hypophosphatemia: current insights. Drug Saf 2020, 43: 197-210.
- Barth JH, Jones RG, Payne RB. Calculation of renal tubular reabsorption of phosphate: the algorithm performs better than the nomogram. Ann Clin Biochem 2000, 37 (Pt 1): 79-81.
- Souberbielle JC, Prié D, Piketty ML, et al. Evaluation of a new fully automated assay for plasma intact FGF23. Calcif Tissue Int 2017, 101: 510–8.
- Miller DW, Slovis CM. Hypophosphatemia in the emergency department therapeutics. Am J Emerg Med 2000, 18: 457–61.
- Carpenter TO, Imel EA, Holm IA, et al. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res 2011, 26: 1381-8.
Iperfosfatemia
Andrea Guarnieri
SC Nefrologia e Dialisi - AO S. Croce e Carle – Cuneo
CLINICA
In letteratura mancano dati che individuano la concentrazione sierica di fosfato che si accompagna alla comparsa di sintomi. Nei pazienti con insufficienza renale cronica è infrequente la presenza di disturbi per valori < 7 mg/dL.
L’iperfosfatemia è generalmente asintomatica; occasionalmente i pazienti possono manifestare sintomi da ipocalcemia, quali crampi, tetania, intorpidimento o formicolio peri-orale, più raramente dolori osteo-articolari, prurito ed eruzioni cutanee. I sintomi più frequentemente accusati sono quelli relativi alla condizione causa di iperfosfatemia, il più delle volte l’uremia: affaticamento, dispnea, anoressia, nausea e vomito, disturbi del sonno.
Nell’iperfosfatemia acuta, causata in particolare dalla somministrazione parenterale di fosfato, il paziente può essere ipoteso o manifestare segni di ipocalcemia grave, quali segno di Trousseau o di Chvostek, iper-reflessia, spasmo carpo-pedalico o convulsioni.
EZIOPATOGENESI
L’iperfosfatemia può manifestarsi in due condizioni:
- entrata nei liquidi extra-cellulari di fosfato in eccesso rispetto alla quantità che può essere eliminata;
- aumento della soglia renale per l’escrezione del fosfato.
Le cause sono riassumibili in quattro circostanze: carico acuto di fosfato, passaggio acuto di fosfato nello spazio extra-cellulare, danno renale acuto o cronico, incremento primario del riassorbimento tubulare del fosfato.
Carico acuto di fosfato
Un carico di fosfato sufficiente a superare la capacità renale di escrezione può derivare sia da risorse endogene sia esogene. Poiché il fosfato è il principale anione intra-cellulare, ogni causa di marcata lisi cellulare può portare a un rilascio di fosfato nei liquidi extra-cellulari. L’iperfosfatemia che ne consegue, può provocare la precipitazione di fosfato di calcio nei tessuti, con conseguente ipocalcemia.
Sindrome da lisi tumorale. Normalmente causata da terapie cito-tossiche, ma occasionalmente spontanea, in pazienti con una grossa massa tumorale caratterizzata da alto turn-over cellulare (linfoma di Burkitt, linfoma non-Hodgkin, alcune leucemie; più raramente descritta in pazienti con tumori solidi, come epatoblastoma e neuroblastoma in fase avanzata). La lisi cellulare comporta, oltre all’iperfosfatemia, un rapido sviluppo di iperuricemia, iperkaliemia, ipocalcemia e, frequentemente, danno renale acuto (1).
Rabdomiolisi. Anche il danno muscolare porta a un rilascio di fosfato nel comparto extra-cellulare. Poiché una delle conseguenze più frequenti della rabdomiolisi è il danno renale, la gravità dell’iperfosforemia può essere ancora maggiore qualora si abbia una riduzione acuta del filtrato glomerulare.
Fosfato esogeno. L’iperfosfatemia esogena è più frequentemente indotta dall’ingestione di grosse quantità di lassativi contenenti fosfato (anche come preparazione per la colonscopia). In questi pazienti si può anche manifestare una nefropatia acuta con insufficienza renale (2). Un’iperfosfatemia esogena è stata raramente descritta anche in pazienti con convulsioni trattati con alte dosi di fosfenitoina.
Passaggio acuto di fosfato nell’extra-cellulare
Un passaggio massivo di fosfato dalle cellule al comparto extra-cellulare è una causa rara di iperfosfatemia, ma è stata descritta in presenza di acidosi lattica e cheto-acidosi diabetica. L’acidosi metabolica, oltre a promuovere il passaggio del fosfato nell’extra-cellulare, può diminuire la glicolisi e quindi l’utilizzo cellulare del fosfato, con conseguente ulteriore aumento della concentrazione sierica. Per ragioni non chiare, altre forme di acidosi sono meno comunemente associate a iperfosfatemia.
Malattia renale acuta o cronica
Solo una percentuale del 5–20% del fosfato filtrato dai reni è escreto con le urine, essendo la maggior parte riassorbita a livello del tubulo prossimale. Una riduzione sia acuta sia cronica del filtrato glomerulare diminuirà la filtrazione e l’escrezione del fosfato. Inizialmente il bilancio può essere mantenuto in equilibrio riducendo il riassorbimento prossimale, grazie all’aumento dei livelli di PTH e FGF-23 con azione fosfaturica. Per valori di GFR < 20-25 mL/min il riassorbimento del fosfato è massimamente soppresso e l’escrezione urinaria non è più in grado di mantenere livelli sierici normali; a questo punto la fosfatemia aumenta riequilibrando il bilancio.
Aumento del riassorbimento tubulare
L’escrezione del fosfato può essere ridotta anche per un incremento del riassorbimento tubulare prossimale.
Ipoparatiroidismo. Sia il deficit di PTH sia la resistenza renale all’azione dell’ormone (pseudo-ipoparatiroidismo) comportano un incremento del riassorbimento del fosfato, con conseguente iperfosfatemia.
Acromegalia. In alcuni pazienti con acromegalia può manifestarsi iperfosfatemia, probabilmente per uno stimolo diretto all’assorbimento del fosfato da parte di GH o IGF-1.
Bisfosfonati. Principalmente l’etidronato può causare modesta iperfosfatemia, per stimolo diretto dell’assorbimento renale.
Tossicità da vitamina D. La vitamina D aumenta l’assorbimento di calcio e fosfato e l’ipercalcemia diminuisce l’escrezione urinaria di fosfato.
Calcinosi Tumorale Familiare (CT). La CT è una malattia congenita, ereditata con modalità autosomica recessiva, caratterizzata dalla formazione, nei primi 20 anni di vita, di depositi di calcio a livello della cute e delle strutture muscolo-tendinee che circondano le articolazioni, in particolare quelle di bacino, spalle, piedi e mani (3). La CT è caratterizzata da normocalcemia, iperfosfatemia, livelli di vitamina D nella norma ma inadeguati per i valori di fosfato. Nei pazienti con CT sono state riscontrate mutazioni di vario tipo e gravità in tre diversi geni deputati al controllo dei livelli circolanti di fosfato e vitamina D: GALNT3 (il cui prodotto è la glicosil-trasferasi, un enzima che previene la degradazione di FGF-23), FGF-23 e Klotho. Complessivamente, le mutazioni descritte determinerebbero un deficit di FGF-23 o una resistenza periferica alla sua azione.
Pseudo-iperfosfatemia. Un’iperfosfatemia “spuria”, dovuta a interferenza con i metodi analitici, può manifestarsi in pazienti con iperglobulinemia (mieloma multiplo, macroglobulinemia di Waldenstrom, gammopatie monoclonali), iperlipidemia, emolisi e iperbilirubinemia. È stata anche descritta una pseudo-iperfosfatemia secondaria a terapia con alte dosi di amfotericina B liposomiale e a contaminazione del campione con attivatore del plasminogeno tissutale ricombinante ed eparina.
TRATTAMENTO
Iperfosfatemia acuta
Se la funzione renale è preservata, si risolve spontaneamente in 6–12 ore. L’escrezione urinaria può essere aumentata con infusione di soluzione fisiologica. In pazienti con ipocalcemia sintomatica è spesso indicata l’emodialisi.
Iperfosfatemia cronica
Si manifesta in pazienti affetti da insufficienza renale cronica o calcinosi tumorale familiare. La terapia consiste nel ridurre l’apporto alimentare di fosfato e nell’utilizzo di chelanti.
BIBLIOGRAFIA
- Arrambide K, Toto RD. Tumor lysis syndrome. Semin Nephrol 1993, 13: 273-80.
- Gonlusen G, Akgun H, Ertan A, et al. Renal failure and nephrocalcinosis associated with oral sodium phosphate bowel cleansing: clinical patterns and renal biopsy findings. Arch Pathol Lab Med 2006, 130: 101-6.
- Slavin RE, Wen J, Kumar D, et al. Familiar tumoral calcinosis. A clinical, histophatologic, and ultrastructural study with an analysis of its calcifying process and pathogenesis. Am J Surg Pathol 1993, 17: 788-802.