Deficit di 5-alfa-reduttasi

Antonio Balsamo1, Silvano Bertelloni2, Gianni Russo3
1Dipartimento di Scienze Mediche e Chirurgiche, UO Pediatria, Programma di Endocrinologia, AOU di Bologna, Bologna; 2Dipartimento Materno-infantile, UO Pediatria Universitaria, AOU Pisana, Pisa; 3UO Pediatria e Medicina dell’adolescenza, Università “Vita e Salute”, Ospedale San Raffaele, Milano
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Il deficit di 5α-reduttasi tipo 2 (OMIM n. 264600) è un raro 46,XY DSD, trasmesso con ereditarietà autosomico-recessiva e causato da mutazioni nel gene SRD5A2. L’anomalia genetica determina un deficit enzimatico, per cui il testosterone (T) non può essere convertito - in tutto o in parte - nel più potente diidro-testosterone (DHT). Quest’ultimo androgeno è responsabile della virilizzazione dei genitali esterni durante la vita intra-uterina, mentre i genitali interni sono virilizzati direttamente dal testosterone.
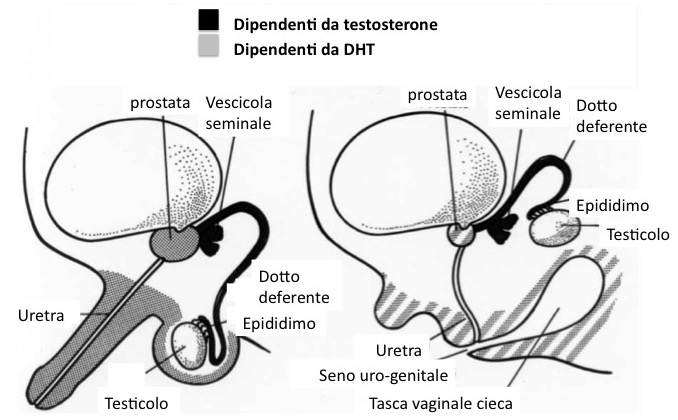
Ruolo di T e DHT nella differenziazione maschile in utero (sinistra); differenziazione dei genitali interni ed esterni nel deficit di 5α-RD2 (destra) (modificato da Shan Zhu YS & Imperato-McGinley J, Gynecology and Obstetrics, vol 5 cap 81).
Il deficit di 5α-reduttasi tipo 2 si manifesta prevalentemente in alcune aree geografiche dove è relativamente frequente il matrimonio tra consanguinei (Repubblica Dominicana, Nuova Guinea, alcune popolazioni arabe), ma – sebbene l’incidenza sia sconosciuta - non è raro neppure nei paesi occidentali, almeno in quelli dell’area del Mediterraneo.
Clinica
Alla nascita si ha generalmente un fenotipo esterno caratterizzato da genitali ambigui con grave ipospadia perineo-scrotale e con recesso vaginale a fondo cieco, ma l’aspetto dei genitali esterni può variare da un fenotipo quasi completamente femminile, con eventuale modesta clitorido-megalia, a bambini con ipospadia e/o micropene isolati (1). I genitali interni (epididimo, vasi deferenti, vescicole seminali) sono maschili normali. I testicoli sono endo-addominali o localizzati a vario livello lungo la loro normale via di discesa.
Alla pubertà, se i testicoli non sono stati rimossi, si ha uno sviluppo dell'apparato pilifero e delle masse muscolari di tipo maschile, assenza di ginecomastia e mascolinizzazione dei genitali di grado variabile in conseguenza di un’aumentata attività dell’enzima anomalo o per una maggiore attivazione dell’isoenzima 5α-reduttasi di tipo 1. Il fenotipo clinico è comunque non specifico, essendo ampiamente sovrapponibile a quello di altri 46,XY DSD da deficit di produzione o azione degli androgeni, tanto che non è possibile porre una diagnosi differenziale in base alla sola clinica (2).
La maggior parte dei soggetti affetti è oligo-azoospermica e quindi infertile, a causa del danno testicolare conseguente al criptorchidismo e alle complicazioni legate alla chirurgia genito-urinaria; l’analisi del seme evidenzia una riduzione del numero di spermatozoi e del volume seminale, con elevata viscosità, attribuibile allo sviluppo rudimentale della ghiandola prostatica e delle vescicole seminali (3,4).
Diagnosi
La diagnosi biochimica si basa su un elevato rapporto T/DHT secondario alla compromissione della conversione del T in DHT. Per una corretta valutazione di tale rapporto nei pazienti pre-puberi, è necessario stimolare la produzione di testosterone mediante somministrazione di hCG. Con una corretta metodica di dosaggio, che deve prevedere la separazione cromatografica degli steroidi (essendo la concentrazione del DHT circa dieci volte inferiore rispetto a quella del T), il rapporto T/DHT è patologico se > 15-18 (arriva a > 30 nei deficit più gravi). Tuttavia, un normale rapporto T/DHT non permette di escludere la diagnosi, poiché tale rapporto può variare con l’entità del deficit enzimatico (in caso di deficit parziale il rapporto può essere normale). Un elevato rapporto di metaboliti urinari 5β/5α (tetraidro-corticosterone/allotetraidro-corticosterone e etiocolanolone/androsterone) è suggestivo per deficit di 5α-RD2, anche in età pre-puberale e in pazienti orchiectomizzati; tuttavia tale modalità di analisi è disponibile solo in pochi centri.
Il deficit di 5 α-reduttasi può essere confermato dall’evidenza di una ridotta attività enzimatica su fibroblasti in coltura.
La diagnosi di certezza si ha con l’evidenza di mutazioni nel gene SRD5A2. Il gene codificante per la 5α-RD2 è costituito da cinque esoni e quattro introni. A oggi sono state descritte più di cinquanta mutazioni del gene. La mancanza di una completa correlazione genotipo/fenotipo nei pazienti portatori della stessa mutazione suggerisce il coinvolgimento di altri fattori, quali l’attività del recettore degli androgeni, i livelli di testosterone in utero o i fattori ambientali.
Trattamento
In questi pazienti deve essere eseguita un'attenta valutazione psicologica prima di qualsiasi trattamento chirurgico o ormonale. In conseguenza dell’esposizione in epoca pre-natale e post-natale del cervello agli androgeni, molti soggetti con deficit di 5α-RD2 (circa il 60%) cresciuti come femmine sviluppano poi un’identità di genere maschile e cambiano il sesso in epoca adolescenziale o adulta.
In caso di corretta e tempestiva diagnosi, è auspicabile l’attribuzione del sesso maschile e la crescita in tale sesso. In tal caso, è essenziale la correzione precoce del criptorchidismo per preservare la fertilità.
Prima dell’intervento chirurgico correttivo d’ipospadia è consigliata l’applicazione topica di crema al DHT, con lo scopo di migliorare le dimensioni peniene.
Non è generalmente necessaria la terapia sostitutiva con T in età puberale; può essere necessaria la somministrazione di T intramuscolare ad alte dosi o di gel DHT per migliorare le dimensioni peniene e lo sviluppo della peluria. L'ingrandimento massimo del pene si ottiene dopo circa sei mesi di trattamento. La maggior parte dei pazienti riferisce una performance sessuale soddisfacente in presenza di una dimensione peniena > 6 cm. Il trattamento con gel DHT presenta alcuni vantaggi: è circa cinquanta volte più attivo del T, promuovendo così un più rapido aumento della dimensione del pene; non è aromatizzato in periferia, per cui non influenza la maturazione ossea e non favorisce lo sviluppo della ghiandola mammaria.
Alcuni pazienti affetti hanno avuto figli con le attuali metodiche di fertilizzazione in vitro o d’inseminazione in utero.
In caso di attribuzione del sesso femminile, la gestione è forse ancora più complessa: la gonadectomia in epoca pre-puberale evita la virilizzazione, ma deve essere eseguita prima di poter avere il consenso del paziente; il mantenimento delle gonadi è probabilmente l’opzione preferibile, con l’utilizzo alla pubertà del GnRH analogo per bloccare transitoriamente la pubertà stessa e impedire la virilizzazione spontanea. In questo modo si può arrivare a un’età in cui il soggetto, correttamente informato e seguito dal punto di vista psicologico, può esprimere le proprie decisioni sul proprio presente e futuro (4).
Bibliografia
- Sinnecker GH, et al. Phenotypic classification of male pseudohermaphroditism due to steroid 5-Reductase 2 deficiency. Am J Med Genet 1996, 63: 223-30.
- Cheon CK. Practical approach to steroid 5alpha-reductase type 2 deficiency. Eur J Pediatr 2011, 170: 1-8.
- Imperato-McGinley J, Zhu YS. Androgens and male physiology: the syndrome of 5a-reductase-2 deficiency. Mol Cell Endocrinol 2002, 198: 51-9.
- Costa EMF, et al. DSD due to 5α-reductase 2 deficiency - from diagnosis to long term outcome. Semin Reprod Med 2012, 30: 427-31.
Insensibilità periferica agli androgeni
Silvano Bertelloni1, Lilia Baldazzi2, Antonio Balsamo3
1Dipartimento Materno-infantile, UO Pediatria, AOU Pisana, Pisa; 2Laboratorio di Genetica Molecolare, Dipartimento Salute della Donna, del Bambino e dell’Adolescente; 3Dipartimento di Scienze Mediche e Chirurgiche, UO Pediatria, Programma di Endocrinologia, AOU di Bologna, Bologna
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
La sindrome da insensibilità periferica agli androgeni (SIA), uno dei più frequenti disordini della differenziazione sessuale con cariotipo 46,XY, è una condizione genetica, X-linked (OMIM #300068), ad espressione nel solo sesso maschile, dovuta ad anomalie nel gene del recettore per gli androgeni (AR) (locus Xq11-12). A causa di tali anomalie si ha la sintesi di un recettore anomalo, per cui gli ormoni androgeni non possono esplicare la loro azione a livello dei tessuti bersaglio, sia durante la vita intra-uterina che in quella post-natale, derivandone un deficit totale o parziale dei caratteri sessuali maschili primitivi e secondari (1,2).
Aspetti clinici
La SIA si caratterizza per una notevole eterogeneità fenotipica. Nella forma completa (in passato indicata come sindrome di Morris, incidenza 1:20.000 - 1:99.000 nati/anno con cariotipo 46,XY), il fenotipo esterno è quello femminile normale (figura, fenotipo 5) sia prima che dopo la pubertà, ma con vagina a fondo cieco di lunghezza variabile. I genitali interni sono invece assenti per la normale azione dell’AMH durante la vita intra-uterina. I testicoli sono localizzati nell'addome, nel canale inguinale o nelle grandi labbra e possono determinare l'insorgenza di un'ernia inguinale, che rappresenta il segno clinico principale di esordio in una bambina pre-pubere. La vagina, sebbene di lunghezza ridotta, risulta solitamente adeguata per i rapporti sessuali senza la necessità di interventi chirurgici e/o dilatativi (3). Il menarca è assente ed è questo il motivo principale di consultazione medica in epoca puberale. Una bambina può inoltre giungere non infrequentemente all’osservazione per la presenza di una sorella affetta o di un altro soggetto nel ramo femminile della famiglia o per una discordanza tra cariotipo 46,XY all’amniocentesi e fenotipo femminile all’ultrasonografia prenatale o alla nascita (1,2).
Si ritiene oggi che la degenerazione maligna dei testicoli, causa in passato di gonadectomia precoce effettuata anche allo scopo di non dover rivelare il motivo dell’intervento in età adolescenziale/adulta, abbia un rischio relativamente molto basso, soprattutto prima del raggiungimento della maggiore età (3). Quindi, sebbene sia opportuno un accurato follow-up delle gonadi, può essere consigliato o il mantenimento delle stesse o la gonadectomia, se necessaria, solo dopo il completamento dello sviluppo puberale, tenendo conto dei vantaggi di ordine psicologico e pratico di una pubertà spontanea e del basso rischio neoplastico nelle prime decadi di vita (1,3).
Con il termine di SIA parziale si indicano invece tutte quelle forme di alterazione del gene AR che sono compatibili con una risposta parziale – seppure minima - agli androgeni, sia in epoca pre-natale che post-natale (1,2). I quadri clinici sono estremamente variabili in rapporto al diverso grado di resistenza (figura, fenotipi 2-4) (1). Usualmente, il sospetto diagnostico si pone fin dalla nascita per la presenza di genitali esterni incompletamente sviluppati in senso maschile o femminile (pliche labio-scrotali parzialmente fuse o scroto bifido, clitoride ipertrofico o micropene, ipospadia perineo-scrotale, talvolta seno uro-genitale con abbozzo vaginale a fondo cieco, testicoli palpabili – in un'ernia inguinale o nelle pliche labio-scrotali - o non palpabili). La forma più severa è caratterizzata da ipertrofia clitoridea isolata, mentre quella più lieve da micropene isolato. In alcune famiglie, lo stesso difetto genetico si può presentare con quadri fenotipici differenti (1,2).
Per SIA minima si intendono infine forme fenotipicamente poco rilevanti, come la sindrome del maschio infertile, cioè una condizione caratterizzata da un normale fenotipo maschile (figura, fenotipo 1) con isolata infertilità per azoospermia o grave oligospermia; può essere presente ginecomastia (1,2).
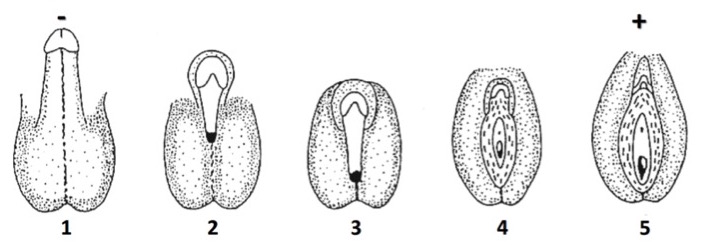
Fenotipi clinici della sindrome da insensibilità agli androgeni, con il progressivo spostamento dall’estrema sinistra (fenotipo 1, normale mascolinizzazione) all’estrema destra (fenotipo 5, con insensibilità completa e aspetto femminile normale). Fenotipo 2: prevalentemente maschile (ipospadia isolata/ipospadia + micropene). Fenotipo 3: ambiguo (ipospadia perineo-scrotale, scroto bifido simil-labia, micropene simil-clitoride). Fenotipo 4: prevalentemente femminile (clitoridomegalia + fusione labia, seno uro-genitale, vagina di lunghezza ridotta a fondo cieco)
Caratteristiche endocrine
I pochi dati a disposizione suggeriscono che nei primi mesi di vita l’assenza del picco post-natale di LH e testosterone nelle bambine sia completa, mentre nei lattanti con SIA parziale i livelli basali di LH e di testosterone sono normali o aumentati (1). Il DHT può essere normale o ridotto (1).
Nel periodo pre-puberale, i dati endocrinologici sono usualmente non informativi.
Dopo la pubertà, nei soggetti con gonadi in sede si hanno valori normali o aumentati di testosterone. I livelli di FSH sono nella norma o di poco aumentati. Si ritrovano inoltre maggiori livelli circolanti di estrogeni, in parte dovuti a una maggiore secrezione testicolare e in parte causati da un'aumentata aromatizzazione a livello periferico del testosterone (1,4).
La femminilizzazione del fenotipo dipende sia dagli aumentati livelli estrogenici, sia dal fatto che la loro azione periferica non viene contrastata, in tutto o in parte, dagli steroidi sessuali maschili (1).
Difetti recettoriali e genetica molecolare
La valutazione della capacità di legame degli androgeni su colture primarie di fibroblasti ottenute da biopsie di cute genitale può mettere in evidenza una capacità di legame assente o molto ridotta tipica delle forme complete, o una ridotta affinità di legame, usualmente presente nelle forme parziali (1).
Ai fini diagnostici più frequentemente si effettua oggi l’analisi del gene AR. Sono state identificate più di 400 mutazioni differenti: nella maggioranza si tratta di mutazioni puntiformi, ma possono essere presenti inserzioni nucleotidiche, duplicazioni di esoni e mutazioni introniche con conseguente alterazione dei siti di splicing. Solo poche donne con SIA completa presentano delezioni estese, che condizionano una mancata sintesi del recettore o di ampie porzioni dello stesso (www.androgendb.mcgill.ca).
In circa il 70% delle persone con SIA si ha trasmissione da madre a figlio (5); nel restante 30% dei casi si tratta invece di mutazioni ex-novo, che possono originare nella linea germinale materna o direttamente nello zigote durante le prime fasi di divisione (5). Nel secondo caso non vi è rischio di ricorrenza in altre gravidanze.
Qualche indicazione di trattamento medico
Nella SIA completa non vi sono dubbi sull'identità somatica e psicologica femminile. La rimozione delle gonadi, se necessaria, dovrebbe essere effettuata dopo la pubertà per garantire uno sviluppo puberale spontaneo e assicurare maggiore capacità decisionali alla donna sulle proprie scelte.
Dopo l’intervento deve essere assicurata un’adeguata terapia ormonale sostitutiva (6). Si è dimostrata utile una terapia psicologica di supporto, che deve essere eseguita da professionisti esperti nel settore. È importante un sostegno psicologico anche nella fase di diagnosi, soprattutto se pre- o neonatale e nella primissima infanzia.
Nella SIA parziale le decisioni terapeutiche presentano maggiori difficoltà e non vi sono al momento indicazioni “evidence based” che possano guidare le scelte terapeutiche.
Bibliografia
- Hughes IA, Deeb A. Androgen resistance. Best Pract Res Clin Endocrinol Metab 2006, 20: 577-98.
- Hughes IA, Werner R, Bunch T, Hiort O. Androgen insensitivity syndrome. Semin Reprod Med 2012, 30: 432-42.
- Cools M, Drop SL, Wolffenbuttel KP, et al. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev 2006, 27: 468-84.
- Doehnert U, Bertelloni S, Werner R, et al. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex Dev 2015, 9: 69-74.
- Hiort O, Sinnecker GH, Holterhus PM, et al. Inherited and de novo androgen receptor gene mutations: investigation of single-case families. J Pediatr 1998, 132: 939-43.
- Bertelloni S, Dati E, Baroncelli GI, Hiort O. Hormonal management of complete androgen insensitivity syndrome from adolescence onward. Horm Res Paediatr 2011, 76: 428-33.
Deficit recettore LH
Gianni Russo, Silvia Meroni
UO Pediatria e Medicina dell’adolescenza, Università “Vita e Salute”, Ospedale San Raffaele, Milano
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
LH e hCG si legano allo stesso recettore (LH-R) situato sulla membrana cellulare delle cellule bersaglio. Il gene codificante per il recettore è situato sul braccio corto del cromosoma 2 (2p21) ed è formato da undici esoni. Il recettore è costituito da un’ampia porzione extra-cellulare, responsabile del legame con l’ormone, sette domini trans-membrana e una coda C-terminale intra-cellulare. La sua azione è mediata da una proteina G e, quindi, dalle variazioni dei livelli intra-cellulari di cAMP.
Tale recettore è essenziale per una corretta crescita e differenziazione delle cellule di Leydig fetali e per la produzione di androgeni. Le mutazioni inattivanti del LH-R (tipicamente situate nel dominio trans-membrana) determinano un’assente responsività delle cellule di Leydig a hCG e LH. I soggetti 46,XY con omozigosi o doppia eterozigosi per mutazioni del LH-R (autosomica recessiva) presentano ipoplasia/agenesia delle cellule di Leydig (evidenziabile all’esame istologico) e difetto di virilizzazione in utero e alla pubertà (1).
La forma completa presenta le seguenti caratteristiche:
- fenotipo femminile con assegnazione del sesso femminile;
- assente sviluppo dei caratteri sessuali secondari alla pubertà;
- testicoli non discesi di dimensioni leggermente inferiori rispetto alla norma, con conservazione dei tubuli seminiferi, ma assenza di cellule di Leydig mature;
- presenza di strutture Wolffiane ipoplasiche e assenza di strutture mülleriane;
- ridotta/assente concentrazione di T, nonostante elevati livelli di gonadotropine (LH > FSH);
- assente risposta al test da stimolo con hCG, in assenza di deficit nella sintesi del testosterone (non accumulo di precursori).
Se presente una parziale responsività del LH-R, con conseguenti livelli sub-ottimali di testosterone, il fenotipo è prevalentemente maschile con deficit di virilizzazione, quali micropene e/o ipospadia; i testicoli possono essere criptorchidi o localizzati nello scroto. Alla pubertà si assiste a una parziale virilizzazione con dimensioni testicolari normali o solo lievemente ridotte, ma con evidente compromissione della crescita peniena (1).
Bibliografia
- Segaloff DL. Diseases associated with mutations of the human lutropin receptor. Prog Mol Biol Transl Sci 2009, 89: 97-114.
Disordini da alterata secrezione o azione dell’AMH
Soara Menabò1, Antonio Balsamo2
1Laboratorio di Genetica Molecolare, Dipartimento Salute della Donna, del Bambino e dell’Adolescente, Programma Endocrinologia Pediatrica, AOU S. Orsola-Malpighi, Bologna; 2Dipartimento di Scienze Mediche e Chirurgiche, UO Pediatria, Programma di Endocrinologia, AOU di Bologna, Bologna
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Fisiopatologia e genetica
L’ormone Anti-Mülleriano (AMH) è una proteina secreta dalle cellule del Sertoli, che nel feto maschio è responsabile della regressione dei dotti di Müller (che vanno a costituire utero, tube e parte alta della vagina). L’azione più specifica, mediata dal recettore di tipo II, è l’induzione dell’apoptosi nei dotti di Müller fetali omolaterali: ogni testicolo sopprime lo sviluppo delle strutture Mülleriane solo dalla sua parte (1).
Il gene AMH, localizzato in 19p13.3, è formato da 5 esoni e codifica per una proteina di 535 aminoacidi, membro della famiglia TGF-β. Le mutazioni sono per lo più missenso, disperse in tutto il gene (non ci sono hotspot mutazionali). I pazienti sono in prevalenza omozigoti e la mutazione è privata.
Il gene AMHR2, localizzato in 12q13, è formato da 11 esoni e codifica per una proteina recettoriale trans-membrana. Circa la metà dei pazienti presenta la stessa mutazione, una delezione di 27-bp nell’esone 10 (2).
La regolazione dell’AMH è finemente controllata da diversi fattori, poiché l’espressione deve avvenire in uno specifico lasso temporale, tra l’8° e la 10° settimana di gestazione. L’SRY, per esempio, è importante per l’attivazione di SOX9, il quale assieme a SF1 e DAX1 stimolano in modo diretto l’espressione di AMH nei testicoli fetali. Intervengono anche altri fattori, quali GATA e FSH. Poiché il recettore è espresso anche nei neuroni degli embrioni di topo, probabilmente gioca un ruolo anche nello sviluppo sessuale dimorfico del cervello.
L’espressione di AMH avviene anche nelle femmine, ma con un timing completamente diverso: viene infatti secreto dalle cellule della granulosa dei follicoli ovarici e può essere usato come marcatore del numero di cellule follicolari di riserva per predire l’età della menopausa (3).
L’AMH è probabilmente coinvolto anche nell’inizio della pubertà in entrambi i sessi (2).
I livelli di AMH nel sangue variano in base a età e sesso:
- nei maschi restano alti fino a prima della pubertà, poi scendono e sono bassi nel maschio pubere e adulto;
- nelle femmine avviene il contrario, infatti restano indosabili fino alla pubertà e poi aumentano.
Clinica
La sindrome da persistenza dei dotti di Müller (PMDS, OMIM 261550) include due forme diverse:
- la PMDS di tipo I è causata da difetti di secrezione dell’AMH, riconducibili a mutazioni nel gene AMH (OMIM 600957);
- la PMDS di tipo II è causata da difetti di azione per mancata responsività del recettore, attribuibili a mutazioni nel gene AMHR2 (OMIM 600956), che codifica per il recettore di tipo II dell’AMH (4).
Si tratta di una sindrome rara a trasmissione autosomica recessiva con prevalenza ignota.
Circa un 13-15% di pazienti con PMDS rimane senza diagnosi genetica, non avendo mutazioni nè nel gene AMH nè nel gene AMHR2. Le femmine con entrambi gli alleli mutati per uno di questi geni non presentano alterazioni dei genitali.
La manifestazione fenotipica è identica per entrambi i tipi: maschi XY con normale virilizzazione, sviluppo dei genitali esterni e caratteri sessuali secondari, con presenza di un utero rudimentale. Alcuni pazienti hanno criptorchidismo bilaterale, altri hanno un testicolo disceso, che ha trascinato la tuba omolaterale nel canale inguinale generando un’ernia, e il testicolo contro-laterale localizzato a livello addominale. È stata riportata infertilità (azoospermia) in diversi pazienti.
Diagnosi
Generalmente, in assenza di criptorchidismo, la diagnosi di PMDS avviene solo in modo accidentale (ecografia addominale, interventi chirurgici, ecc.). Il criptorchidismo, invece, può far sì che la diagnosi avvenga fin dalla nascita.
Il dosaggio di AMH rispecchia il tipo di difetto: se è basso o indosabile è indicativo di difetti sul gene AMH, se è normale o alto è indicativo di difetti sul gene del recettore AMHR2. Sono riportati tuttavia casi eccezionali di livelli normali in pazienti con mutazione nel gene AMH; si tratta di mutazioni che affliggono la bioattività ma non la secrezione (5).
Terapia
La rimozione chirurgica dei residui mülleriani, un tempo evitata per non danneggiare i vasi deferenti che sono spesso intimamente aderenti a queste strutture, è invece ora raccomandata, dal momento che si è osservata degenerazione neoplastica (carcinoma) in alcuni pazienti (6).
Bibliografia
- Josso N, Lamarre I, Picard JY, et al. Antimüllerian hormone in early human development. Early Hum Dev 1993, 33: 91–9.
- Josso N, Belville C, di Clemente N, Picard JY. AMH and AMH receptor defects in Persistent Müllerian Duct Syndrome. Hum Reprod Update 2005, 11: 351–6.
- Belville C, Josso N, Picard JY. Pesistence of müllerian derivatives in males. Am J Med Genet 1999, 89: 218–23.
- Imbeaud S, Belville C, Messica-Zeitoun L, et al. A 27 base-pair deletion of the anti-müllerian type II receptor gene is the most common cause of the Persistent Müllerian Duct Syndrome. Hum Mol Genet 1996, 5: 1269–77.
- Menabò S, Balsamo A, Nicoletti A, et al.Three novel AMH gene mutations in a patient with persistent mullerian duct syndrome and normal AMH serum dosage. Horm Res 2008, 70: 124-8.
- Lindhardt Johansen M, Hagen CP, Johannsen TH, et al. Anti-Müllerian hormone and its clinical use in pediatrics with special emphasis on disorders of sex development. Int J Endocrinol 2013, 2013: 198698.
Da eccesso di androgeni fetali

Da eccesso di androgeni feto-placentari
Da eccesso di androgeni materni
DSD con ovotestis
Forme cliniche di DSD di difficile classificazione
Protocollo diagnostico e management in età neonatale
Paolo Ghirri1, Rosa T Scaramuzzo2, Nella Augusta Greggio3, Maria Carolina Salerno4
1UO Neonatologia, AOU Pisana, Pisa; 2Istituto di Scienze della Vita, Scuola Superiore S. Anna, Pisa; 3UOS Endocrinologia Pediatrica e Adolescentologia, DAIS per la Salute della Donna e del Bambino, AOU Padova; 4Sezione Pediatrica, Dipartimento di Scienze Mediche Traslazionali, Università Federico II, Napoli
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
La nascita di un neonato con difetto della differenziazione sessuale (DSD) associato ad anomalie dei genitali esterni rappresenta un evento emotivamente molto stressante per i genitori. Inoltre, alcuni DSD, anche non necessariamente associati a malformazioni clinicamente evidenti alla nascita, possono porre a rischio la vita stessa del neonato, qualora non tempestivamente diagnosticate e trattate (si pensi all’iperplasia surrenalica congenita con perdita di sali).
Pertanto, i DSD con esordio in età neonatale devono essere considerati emergenze mediche e necessitano di una gestione attenta e ragionata. Il coinvolgimento di un team multi-disciplinare ed esperto (neonatologo, endocrinologo pediatra, urologo pediatra, psicologo, psichiatra, genetista clinico) è presupposto imprescindibile per un inquadramento corretto e completo (1,2).
L’inquadramento diagnostico è ragionato sulla base della classificazione della Consensus Conference di Chicago del 2006, che ha revisionato completamente l’argomento, proponendo una classificazione sulla base dell’assetto cariotipico, delle più recenti conoscenze di biologia molecolare e una nuova nomenclatura, che ha eliminato definizioni gravate da pesanti stigmate socio-culturali (3). Resta comunque complesso, sia perché varie forme possono avere il medesimo fenotipo clinico, sia perché sussistono limiti negli esami di laboratorio e strumentali. Pertanto, un congruo numero di casi di DSD, in particolar modo tra quelli con cariotipo 46,XY, resta privo di diagnosi eziologica definitiva, il che pone dubbi gravosi sull’adeguatezza della stessa gestione clinica e sull’assegnazione del sesso (4,5).
Gestione clinica del neonato
La necessità di un algoritmo appropriato, da applicare sin dai primi giorni di vita allo scopo di formulare una diagnosi precoce, deriva dalla considerazione che l’eziologia del DSD ha conseguenze variabili per diversi aspetti di notevole rilevanza clinica: assegnazione del sesso, terapia ormonale sostitutiva, stima del rischio di neoplasie maligne delle gonadi, indicazione a intervento chirurgico (6). È opportuno ritardare la registrazione della nascita, informando l’ufficio anagrafe, perché per la legge italiana al momento della registrazione è necessario indicare il sesso maschile o femminile; così facendo, sarà possibile guadagnare qualche settimana per poter arrivare a una diagnosi e un’assegnazione appropriata del sesso.
La valutazione biologica dei vari quadri clinici si basa su una sequenza logica e ordinata di esami ormonali e genetici, dettata da anamnesi approfondita (familiare, gravidica anche con focus su eventuali endocrine disruptors, neonatale) ed esame obiettivo dettagliato, comprensivo di ricerca di eventuali gonadi palpabili, misurazione delle dimensioni del micropene/clitoride, osservazione della posizione del meato uretrale e del grado di eventuale ipospadia, dell’aspetto dello scroto e di sua eventuale iperpigmentazione, nonché della ricerca di eventuali altre malformazioni, di restrizione di crescita intra-uterina (IUGR) e di ipotonia. La caratterizzazione completa del fenotipo può richiedere anche l’esecuzione di un’ecografia per l’individuazione dell’utero (o suoi residui) ed eventualmente delle gonadi, nonché di una genitografia per la descrizione di un seno uro-genitale (1,7).
Esami ormonali. L’attivazione fisiologica dell’asse ipofisi-gonadi nei primi giorni di vita consente di misurare le gonadotropine e gli steroidi sessuali, con risultati molto attendibili (4). Nel maschio i livelli di androstenedione, testosterone e DHT sono elevati nei primi 2 giorni di vita, si riducono successivamente con un nadir alla fine della 1° settimana, e si elevano nuovamente dalla fine della 2° settimana di vita (“mini-pubertà”, fino al 3°-4° mese) (8). In presenza di ridotti livelli di testosterone, nel sospetto di un disordine dello sviluppo testicolare, è necessario considerare che possono essere associate altre anomalie congenite a seconda del gene alterato: ipoplasia surrenalica e ipogonadismo ipogonadotropo (geni SF1, DAX1), tumore di Wilms (gene WT1, sindrome di Dennis Drash), anomalie congenite multiple (gene DMRT1), atassia, teleangectasie, ritardo mentale (gene ATRX), displasia campomelica (gene SOX9).
Gli ormoni surrenalici sono generalmente elevati nei primi 2-3 giorni di vita, anche nei neonati senza difetti enzimatici della sintesi degli steroidi. in neonati pretermine sono stati rilevati alla nascita aumentati livelli di 17-idrossi-progesterone, con successiva diminuzione e normalizzazione a un’età post-concezionale pari al termine (9). Il dosaggio dell’AMH, secreto dalle cellule del Sertoli, completa lo studio della funzione testicolare (4). In presenza di IUGR e/o dismorfismi associati, è indicato il dosaggio del 7-deidro-colesterolo plasmatico, per riconoscere la sindrome di Smith-Lemli-Opitz, caratterizzata da ridotti livelli di colesterolo e di tutti gli steroidi. La figura 1 schematizza l’algoritmo proposto. Si noti che nei primi giorni di vita è necessario il controllo pressoché quotidiano di glicemia ed elettroliti plasmatici (sodio, potassio), nel sospetto di iperplasia surrenale congenita (7).
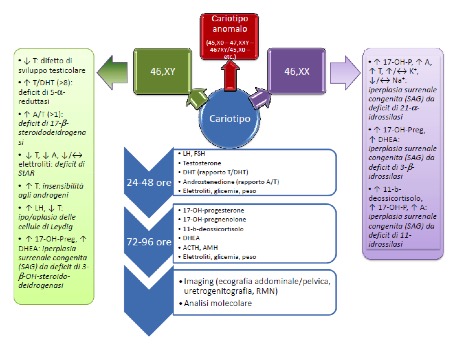
Figura 1. Algoritmo diagnostico dei DSD in età neonatale (1)
Per quanto attiene specificamente alla diagnostica genetica, il cariotipo è da considerarsi indubbiamente esame di prima linea. In modo mirato, in seguito all’approfondimento ormonale, si procederà poi alla valutazione di singoli geni candidati: i DSD, infatti, possono derivare dalla mutazione di uno qualunque dei geni che presiedono allo sviluppo e alla differenziazione sessuale. Inoltre, un fattore critico può essere il timing di espressione dei trascritti codificanti (si pensi ad esempio alla persistenza di strutture Mulleriane in caso di normale ma ritardata secrezione di androgeni e AMH nello sviluppo fetale) (2).
Un nuovo approccio genetico-molecolare, la Next-Generation Sequencing (NGS), permette la sequenza simultanea di numerosi geni all’interno di piattaforme che si possono configurare a seconda dell’applicazione clinica (fig. 2), e appare uno strumento importante per una diagnosi precoce, soprattutto nei neonati con 46 XY DSD. Una volta individuata l’alterata sequenza di un gene con la NGS, si procede alla conferma diagnostica con un sequenziamento classico tipo Sanger.
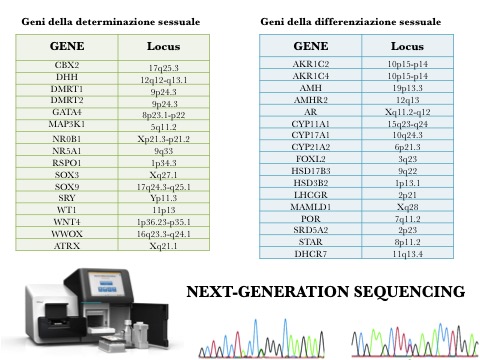
Figura 2. Piattaforma dei geni per sequenza con Next-Generation Sequencing – UO Genetica Molecolare, AOU Pisana, Pisa
Una nuova frontiera della diagnostica, attualmente solo a livello di ricerca, è costituita dallo studio in vitro della sensibilità dei tessuti all'azione degli ormoni, in particolare degli androgeni (4).
L’assegnazione del sesso
È conclusione diretta della diagnosi eziopatogenetica del DSD: per questo, rimane controversa in alcuni quadri 46,XY di definizione incerta (tabella). Nella decisione, condivisa tra genitori e staff sanitario, è necessario considerare le conseguenze mediche e i dati disponibili in letteratura e quindi le possibili previsioni a lungo termine riguardo al potenziale di fertilità, alla possibilità di una stabile identità di genere, alle possibili opzioni chirurgiche, alla necessità di terapie sostitutive, alla possibilità di una normale funzione e soddisfazione sessuale, al rischio oncogenico.
| Indicazioni per l'assegnazione del sesso in 46,XY DSD (modificato da 5) | ||
| Forma clinica | Sesso da assegnare | Note |
| Deficit di sintesi del testosterone | Preferibilmente maschile (soprattutto se diagnosi precoce) | Considerare la possibilità di riassegnazione del sesso maschile in soggetti allevati come femmine, in rapporto a diagnosi molecolare, valutazione psico-comportamentale e storia di altre persone con stesso difetto genetico (se presenti in letteratura) |
| Deficit di 5-alfa-reduttasi | ||
| SIA completa | Femminile | Sesso femminile mai in dubbio, orientamento sessuale e attitudini femminili |
| SIA parziale | Femminile o maschile | Da valutare caso per caso anche in rapporto alla teorica possibilità di androgenizzazione alla pubertà e alla storia di altre persone con stesso difetto genetico (se presenti in letteratura) |
| Deficit di virilizzazione grave “senza diagnosi" | Da valutare caso per caso anche in rapporto al livello fenotipico di androgenizzazione | |
Gestione della famiglia
La comunicazione della diagnosi ai genitori del neonato con DSD è questione estremamente delicata, sia nella fase iniziale di sospetto clinico che nelle successive di indagine diagnostica e definizione della prognosi. L’informazione deve essere necessariamente completa, per diritto umano universale prima ancora che per vincoli strettamente legati al consenso per le procedure diagnostiche: questo tipo di approccio comunicativo dovrebbe essere la costante metodologica del team multi-disciplinare, in grado di fornire conoscenze, competenze ed esperienze, fornire qualità e cura, e affrontare le barriere emotive e culturali/religiose (10). Solo in virtù di una comunicazione simile, la famiglia prima e il paziente stesso nelle età successive, saranno in grado di compiere scelte libere e consapevoli su eventuali interventi terapeutici e, in generale, sulla gestione dell’identità di genere e del ruolo di genere.
Sfortunatamente, non esistono regole prestabilite: ogni situazione richiede un’approfondita valutazione degli aspetti biologici, della storia naturale della specifica condizione, quando conosciuta, e anche del contesto socio-culturale ed etnico in cui il bambino e la famiglia vivono. Ogni informazione può, infatti, sollevare problematiche rilevanti e psicologicamente impegnative, per cui ogni messaggio deve essere autorevole, comprensibile, ripetuto/integrato negli anni e adeguato alla capacità di comprensione dei genitori e del bambino stesso, man mano che questi cresce. La “care” deve essere individualizzata, sia per il supporto psicologico alle famiglie sia per l’educazione appropriata all’età per i piccoli pazienti. Le associazioni dei genitori rappresentano, in questo contesto complesso, un valore aggiunto per gli stimoli che possono dare (5).
Consorzi per la clinica e la ricerca
I DSD sono, considerati singolarmente, patologie rare e storicamente ciò ha ostacolato sia la ricerca clinica che l’assistenza clinica basata sull’evidenza. Riconoscendo i limiti connessi a questa epidemiologia, le società scientifiche europee e statunitensi negli ultimi anni hanno prodotto dichiarazioni politiche (ad esempio: Consensus Statement on Management of Intersex Disorders) e documenti scientifici, che sanciscono la necessità di team multidisciplinari e reti di collaborazione per l’assistenza sanitaria ottimale dei pazienti con DSD. Inoltre, viene sempre più promossa la costituzione di consorzi tra centri specializzati (11,12), la creazione di registri internazionali (Euro-DSD, I-DSD) e le iniziative di e-learning, allo scopo di tesaurizzare le energie per la ricerca, oltre che standardizzare la gestione clinica (13-15). Nella medesima ottica, risorsa preziosa costituiscono le associazioni di pazienti che si pongono anche l’obiettivo della divulgazione scientifica (ad esempio: AISIA) (16).
Bibliografia
- Ghirri P, Scaramuzzo RT. Approccio clinico-diagnostico al neonato con genitali ambigui. Area Pediatrica 2011, 12: 208-12.
- Wisniewski AB, Krishnan S. Ambiguous genitalia in the newborn. In: De Groot LJ, Beck-Peccoz P, Chrousos G, et al (Eds). Endotext (aggiornamento 2015).
- Hughes IA, Houk C, Ahmed SF, Lee PA; LWPES Consensus Group; ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child 2006, 91: 554-63.
- Hughes IA, Morel Y, McElreavey K, Rogol A. Biological assessment of abnormal genitalia. J Pediatr Urol 2012, 8: 592-6.
- Bertelloni S, Dati E, Ghirri P, et al. Gestione clinica dei disturbi della differenziazione sessuale con cariotipo 46,XY: aspetti emergenti. Prospettive in Pediatria 2013, 41: 110-20.
- Cools M, Wolffenbuttel KP, Drop SLS, et al. Gonadal development and tumor formation at the crossroads of male and female sex determination. Sex Dev 2011, 5: 167-80.
- Balsamo A, Cicognani A, Ghirri P, et al. Disorders of sexual development. In: Buonocore G, et al (eds). Neonatology. A practical approach to neonatal diseases. Springer-Verlag Italia, 2011: cap. 123: 1004.
- Migeon CJ, Berkovitz GD, Brown TR. Sexual differentiation and ambiguity. In: Wilkins L (eds). The diagnosis and treatment of endocrine disorders in childhood and adolescence, 4th edition. Charles C Thomas Publisher, Springfield, 1994.
- Nykänen P, Heinonen K, Riepe FG, et al. Serum concentrations of adrenal steroids and their precursors as a measure of maturity of adrenocortical function in very premature newborns. Horm Res Paediatr 2010, 74: 358-64.
- D’Alberton F. Disclosing disorders of sex development and opening the doors. Sex Devel 2010, 4: 304-9.
- Consortium on the management of disorders of sex development. Clinical guidelines for the management of disorders of sex development in childhood. 2006.
- COST BM1303 DSDnet.
- Ahmed SF, Rodie M, Jiang J, Sinnott RO. The European disorder of sex development registry: a virtual research environment. Sex Dev 2010, 4: 192-8.
- Ahmed SF, Bryce J, Hiort O. International networks for supporting research and clinical care in the field of disorders of sex development. Endocr Dev 2014, 27: 284-92.
- Sandberg DE, Callens N, Wisniewski AB. Disorders of sex development (DSD): networking and standardization considerations. Horm Metab Res 2015, 47: 387-93.
- AISIA, Associazione Italiana Sindrome Insensibilità Androgeni. Linee guida cliniche per il trattamento dei DSD in età infantile. 2012.
- Società Italiana di Neonatologia. Genitali ambigui in età neonatale: gestione clinica.
Attribuzione del genere e correzione delle ambiguità genitali
Inquadramento diagnostico della pubertà precoce
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
INTRODUZIONE
Viene definita come pubertà precoce una condizione caratterizzata dalla comparsa di segni di sviluppo puberale prima di quanto atteso rispetto agli standard di normalità: tradizionalmente il limite di età preso in considerazione è rappresentato dagli 8 anni nelle bambine e 9 anni nei maschi, che corrispondono a -2.5/-3.0 DS al di sotto dell’età media di insorgenza della pubertà (1-3).
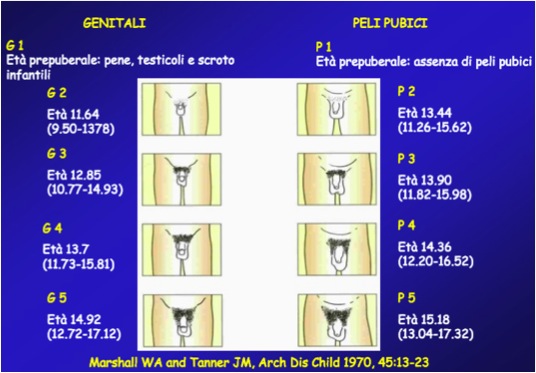
La valutazione del grado di sviluppo puberale viene generalmente effettuata utilizzando la scala di Tanner (figura) e l’inizio della pubertà è indicato dall’incremento del volume mammario (stadio B2) o del volume testicolare (stadio G2: volume testicolare ≥ 4 mL). Viene valutata anche la presenza di peluria pubica, il cosiddetto pubarca, che, pur rispondendo a un diverso meccanismo rispetto a quello che regola l’asse ipotalamo-ipofisi-gonadi, accompagna e partecipa al cambiamento corporeo tipico della pubertà.
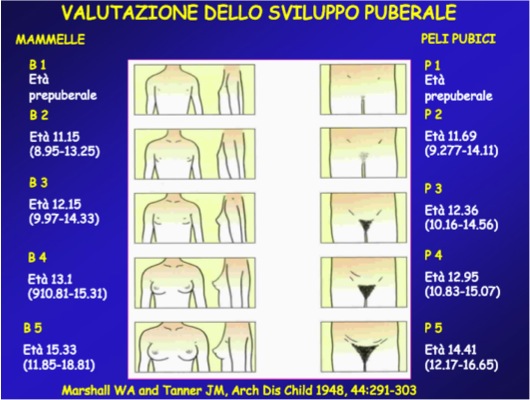
La definizione e i limiti temporali della pubertà precoce si basano sugli studi di Tanner (1,2), tuttavia pubblicazioni più recenti hanno dimostrato (tabella 1) come un’elevata percentuale di bambine (10-40%) presenta un iniziale sviluppo mammario (B2) e peluria pubica prima degli 8 anni di età (4), mentre altri studi, pur confermando questo dato, non hanno trovato una reale anticipazione dell’età del menarca (5).
| Tabella 1 Proporzione di bambine con sviluppo mammario e peluria pubica in 3 studi US (PROS, NHANES III e Biro)(modif da 4) |
|||
| Sviluppo mammario | Peli pubici | ||
| 7 anni | Caucasiche | 5-10.4% | 2.8-6.4% |
| Nere | 15.4-23.4% | 17.6-20% | |
| Ispaniche | 14.9% | 6.7% | |
| 8 anni | Caucasiche | 10.5-18.3% | 6.5-10.1% |
| Nere | 27.8-42.9% | 30.4-31.6% | |
| Ispaniche | 25.4-30.9% | 6.7-18.9% | |
Queste evidenze suggeriscono che esiste una classe di bambine che, seppure con precoci segni di sviluppo mammario, ha una lenta progressione puberale e non necessita di interventi terapeutici, ma solo di osservazione (4).
CLASSIFICAZIONE ED EZIOLOGIA
La pubertà precoce può essere classificata in due diverse forme, all’interno delle quali troviamo diverse possibili cause (tabella 2):
- GnRH-dipendente (Pubertà Precoce Vera o Centrale, PPC);
- GnRH-indipendente (Pseudo-pubertà o Pubertà Precoce Periferica, PPP).
Accanto a queste troviamo i casi di cosiddetta “pubertà precoce incompleta”, rappresentati da adrenarca e telarca prematuri, condizioni benigne che devono comunque essere monitorate nel tempo.
| Tabella 2 Classificazione eziologica delle pubertà precoci |
|||
| GnRH-dipendenti (o vere o centrali) | Idiopatica o familiare | ||
| Adozioni internazionali | |||
| Danni acquisiti SNC | Neoplasie cerebrali: astrocitoma, pinealoma, glioma vie ottiche, craniofaringioma Cisti Idrocefalo Post-infettivo Post-traumatico Post-radioterapia Malattie granulomatose Paralisi cerebrale |
||
| Amartoma ipotalamico | |||
| Neurofibromatosi tipo 1 | |||
| Sclerosi tuberosa | |||
| Displasia setto-ottica | |||
| Sindrome di Sturge-Weber | |||
| Mutazioni attivanti nei geni che codificano kisspeptina e il suo recettore | |||
| Mutazioni inattivanti di MKRN3 | |||
| GnRH-indipendenti (o pseudo o periferiche) | Testotossicosi | ||
| Sindrome di McCune-Albright | |||
| Cisti o tumori ovarici | |||
| Tumori surrenalici estrogeno-secernenti | |||
| Sindrome di Peutz-Jegher | |||
| Ipotiroidismo primario | |||
| Sindrome da eccesso di aromatasi | |||
| Neoplasie secernenti hCG: coriocarcinoma, epatoblastoma, tumori a cellule germinali | |||
Forma GnRH-dipendente (PPC)
È caratterizzata da precoce attivazione dell’asse ipotalamo-ipofisi-gonadi, con incremento dei livelli plasmatici basali e GnRH-stimolati di LH, incremento degli ormoni sessuali, progressivo e armonico sviluppo puberale, accelerazione della velocità di crescita e avanzamento dell’età ossea.
Può essere espressione di una patologia organica del SNC, riscontrabile in circa l’8% delle ragazze e nel 40-90% dei ragazzi con PPC (6). Con l’avanzare dell’età, si riduce la possibilità della presenza di una patologia intra-cranica (7-9).
La forma idiopatica è la più frequente (> 80% dei casi). I pazienti affetti presentano appunto un prematuro sviluppo puberale, senza evidenza clinica e/o radiologica di lesioni del SNC e/o neoplasie cerebrali. La forma idiopatica è molto più comune nelle ragazze, in cui rappresenta circa il 90% dei casi (10).
Una certa proporzione dei pazienti con pubertà precoce idiopatica presenta anamnesi familiare positiva per pubertà precoce, suggerendo la presenza di un meccanismo genetico all’origine dell’alterato timing puberale. Al momento sono state riportate in letteratura due diverse alterazioni genetiche responsabili di pubertà precoce (11):
- mutazioni attivanti di Kiss1 o Kiss1-R (molto rare), con inizio dello sviluppo puberale intorno al primo anno di età;
- mutazioni inattivanti di MKRN3; il gene MKRN3, localizzato sul braccio lungo del cromosoma 15, nella stessa regione implicata nella genesi della sindrome di Prader Willi, codifica per la proteina Makorina 3. Il deficit di questo gene rappresenta la più frequente causa genetica di pubertà precoce familiare. L’allele materno di questo gene è silenziato, mentre quello paterno è espresso. Non è chiaro il preciso meccanismo che porta alla precoce attivazione del processo puberale che comincia intorno ai 6 anni nelle ragazze e 8.8 anni nei ragazzi.
Anche vari tumori cerebrali sono descritti come responsabili di PPC (12), ma l’amartoma del tuber cinereum risulta senza dubbio quello più frequentemente associato (13).
Altre possibili cause sono diverse lesioni del SNC (14) e l’esposizione alla radioterapia per neoplasie cerebrali in età pre-puberale (15).
Da non dimenticare la PPC descritta nei sempre più frequenti casi di adozione internazionale.
Forma GnRH-indipendente (PPP)
La PPP è caratterizzata da un autonomo incremento degli ormoni sessuali, non associato ad attivazione dell’asse ipotalamo-ipofisi-gonadi, quindi con valori di LH bassi/soppressi, progressione puberale talvolta non armonica, aumento di velocità di crescita ed età ossea.
La PPP riconosce cause diverse nel maschio e nella femmina.
Nelle bambine ricordiamo:
- sindrome di McCune-Albright, definita da tre segni clinici: displasia fibrosa delle ossa, macchie “caffè-latte”, pubertà precoce periferica. La mutazione a livello ovarico determina l’attivazione costituzionale del recettore per LH, con precoce sintesi estrogenica e comparsa di segni puberali (17);
- tumori dello stroma ovarico, compresi i tumori delle cellule della granulosa, rari, specialmente nelle bambine (solo il 4-5% si presenta in età infantile). La manifestazione clinica più comune, determinata dalla sintesi di estradiolo da parte del tumore, è appunto la pubertà precoce (18);
- ipotiroidismo: l’ipotiroidismo grave può raramente essere associato a un precoce sviluppo dei caratteri sessuali secondari (cosiddetta sindromre di Van Wyk-Grumbach) con galattorrea e/o sanguinamento vaginale. I dosaggi ormonali mostrano TSH elevato, con estradiolo e FSH nel range o al di sopra dello stesso e LH soppresso. Nei maschi si osserva aumento del volume testicolare senza virilizzazione (19).
Nei maschi troviamo tra le diverse possibili cause di PPP:
- testotossicosi (familial male limited precocious puberty): raro disordine causato dalla presenza di una mutazione attivante nel gene che codifica per il recettore di LH. La conseguenza di questa mutazione, che si manifesta clinicamente solo nei maschi, è una prematura maturazione delle cellule di Leydig e secrezione di testosterone. La pubertà può insorgere tra uno e quattro anni di età (20);
- neoplasie secernenti hCG: possono insorgere nei testicoli, cervello, retro-peritoneo, mediastino posteriore, fegato (21);
- tumori a cellule di Leydig: devono essere sospettati nel caso di un bambino con ingrandimento asimmetrico dei testicoli (22).
Adrenarca prematuro
Con questo temine viene indicata una condizione benigna caratterizzata dalla comparsa precoce (prima di 8 o 9 anni) di peluria pubica, il cosidetto “pubarca”, associata generalmente a modificazione dell’odore del sudore e cute “grassa”, in assenza di sviluppo mammario o incremento del volume testicolare. Talvolta può essere osservata una transitoria accelerazione della velocità di crescita (4), con incremento dell’età ossea (23).
L’adrenarca, che procede indipendentemente dalla maturazione di ovaio e testicolo (24), è sostenuto dalla secrezione di androgeni deboli (DHEA) da parte del surrene, che vengono poi convertiti in androgeni attivi nei tessuti periferici e nelle gonadi (25). Tale secrezione è già osservabile nei bambini tra 6 e 8 anni, ma non in tutti si associa a pubarca (26). Un possibile fattore di rischio per adrenarca prematuro sembra essere il BMI (27).
La diagnosi differenziale deve essere effettuata con:
- neoplasie androgeno-secernenti, per altro caratterizzate da elevati livelli di DHEAS e testosterone, segni clinici di rapida virilizzazione in entrambi i sessi, nei maschi testicoli pre-puberi e nelle femmine assenza di sviluppo mammario e/o ipertrofia clitoridea, incremento della velocità di crescita e avanzamento dell’età ossea (28);
- forma non classica di iperplasia surrenalica congenita, che sembra essere diagnosticabile in una percentuale del 4-8% nei bambini con adrenarca prematuro (4). Anche in questo caso possiamo osservare aumento della peluria pubica e ascellare, cute seborroica, incremento della velocità di crescita (28);
- pubertà precoce: la peluria pubica sarà accompagnata da sviluppo mammario o testicolare, aumento della velocità di crescita e avanzamento dell'età ossea.
Il bambino con adrenarca prematuro, nel quale sia stata esclusa clinicamente e/o biochimicamente una delle condizioni suddette, non necessita di alcun intervento terapeutico, poiché si tratta di una condizione benigna che non interferisce con il raggiungimento del target staturale (29). Alcuni studi hanno invece osservato nelle bambine con adrenarca prematuro, soprattutto se sovrappeso o obese, un incremento del rischio di sindrome metabolica e sindrome dell’ovaio policistico (30-31).
Telarca prematuro
Un’altra possibile fonte di preoccupazione per i genitori e i medici è rappresentata dal telarca prematuro, isolato e transitorio sviluppo della ghiandola mammaria che può essere osservato molto più frequentemente nelle bambine nel primo anno di vita e poi dopo i 5-6 anni (31). L’incremento ghiandolare è quasi sempre bilaterale, raramente raggiunge lo stadio B3, in molti casi non c’è modificazione della velocità di crescita e la peluria pubica è per lo più assente (32). In circa il 30% delle bambine, soprattutto nelle più grandi, non si osserva una regressione del volume mammario allo stadio pre-pubere e in alcuni casi il telarca si presenta ciclicamente (32).
Generalmente il telarca prematuro richiede solo un’osservazione periodica per identificare quella piccola percentuale di ragazze in cui può essere progressivo (4).
INQUADRAMENTO DIAGNOSTICO
La diagnosi di pubertà precoce prevede una serie di tappe successive, che saranno tanto più articolate quanto più piccolo è il bambino che giunge alla nostra osservazione.
Dal colloquio con i genitori è importante capire da quanto tempo sono comparsi i segni di attivazione puberale (bottone mammario, aumento del volume testicolare, peluria pubica) e quanto velocemente questi sono progrediti, poiché una estrema rapidità può già da sola essere indicativa di una sottostante patologia organica. Al contrario, l’assenza di modificazioni nello stadio puberale negli ultimi 6 mesi può essere considerato un segno prognostico favorevole e, soprattutto nelle ragazzine più grandi, può indurci semplicemente ad effettuare controlli seriati nel tempo senza altri interventi (4). Altre informazioni utili ci vengono dalla conoscenza dell’età di sviluppo puberale dei genitori, dalla visione, quando disponibile, della curva di crescita compilata dal pediatra di famiglia nelle visite precedenti alla nostra, poiché l’accelerazione della velocità di crescita può suggerire una possibile pubertà precoce (33). Anche la presenza di cefalea, convulsioni, dolori addominali o pregressi traumi, radioterapie o infezioni che abbiano interessato il SNC, può orientarci nelle successive tappe diagnostiche.
Durante la visita, oltre a un accurato esame obiettivo, durante il quale verificare l'eventuale presenza di segni particolari (es. dismorfie, chiazze caffè-latte, irsutismo, ipertrofia clitoridea), dobbiamo effettuare la valutazione dello stadio puberale, utilizzando il sistema di Tanner, che prende in considerazione lo sviluppo della ghiandola mammaria (non solo osservazione ma anche palpazione!), il volume testicolare e la presenza di peluria pubica. È opportuno ricordare che nei maschi con PPP, il volume testicolare può essere di tipo pre-pubere (< 4 mL), quindi fortemente in contrasto con i segni generali di virilizzazione.
Un’attenta anamnesi, un accurato esame fisico e la valutazione della curva di crescita potrebbero essere elementi sufficienti per poter distinguere una forma di pubertà precoce da un forma puberale incompleta (pubarca o telarca prematuro).
Nel caso di pubarca prematuro, il dosaggio di 17OH-progesterone (> 200 ng/dL), DEAS, testosterone e delta4-androstenedione sono generalmente sufficienti per una diagnosi differenziale tra le diverse condizioni sopracitate (28).
La radiografia della mano e polso per la valutazione dell’età ossea rappresenta un ulteriore importante elemento di valutazione in caso di sospetto oppure di dubbio diagnostico. Infatti, nei casi di pubertà precoce l’età ossea sarà generalmente superiore (+2 DS) a quella anagrafica (34). Da ricordare, tuttavia, che nei casi in cui l’indagine radiologica sia stata effettuata molto precocemente, l’età ossea sarà solo lievemente aumentata.
Nelle bambine l’ecografia pelvica (nelle pre-puberi il volume ovarico è < 2 mL e la lunghezza dell’utero < 4 cm con endometrio sottile) (35) può essere di ausilio nel processo diagnostico, anche se non è dotata di sufficiente sensibilità per distinguere tra le diverse forme di pubertà precoce (36).
La tappa successiva consiste nel richiedere il dosaggio di LH basale e dopo stimolo con GnRH, al fine di effettuare una diagnosi differenziale tra le due diverse forme di pubertà precoce, PPC vs PPP. Sebbene siano ormai disponibili metodiche ultrasensibili per il dosaggio dell’LH, vi sono in letteratura indicazioni non univoche rispetto al valore basale da considerare diagnostico; tuttavia, il valore di 0.3 UI/L è l’unico ad avere una sensibilità del 100% (10,37,38). Il dosaggio basale dell’LH permette quindi di confermare ma non di escludere la diagnosi di pubertà precoce.
Il gold standard rimane il GnRH test, sebbene anche con questo test siano state osservate delle risposte falsamente negative (39). Viene considerato diagnostico un picco di LH > 5 U/L (38). Un elevato rapporto LH/FSH sotto stimolo può rappresentare un ulteriore elemento di conferma (34). Oltre che con il GnRH, il test di stimolo può essere effettuato con un analogo del GnRH stesso (leuprolide). Anche in questo caso è considerato diagnostico un valore di LH > 5 UI/L a 3 ore dall’iniezione del farmaco (40).
Le metodiche di dosaggio usate routinariamente per la valutazione degli ormoni sessuali non hanno una sufficiente sensibilità per i livelli di estradiolo e testosterone tipici della fase pre-puberale o di iniziale pubertà (41,42). Un valore di estradiolo > 20 pg/mL e un testosterone > 50-100 ng/dL possono solo confermare una diagnosi già formulata su altri criteri (4).
Quando sia stato dimostrato un livello di LH di tipo puberale, è necessaria l’esecuzione di una RMN encefalo, al fine di escludere possibili lesioni di varia origine del SNC (7,8,43). Questa indagine va certamente prescritta a tutti i maschi con segni di PPC e a tutte le bambine con esordio della pubertà prima dei 6 anni. Per le bambine di età compresa tra i 6 e gli 8 anni, secondo alcuni la richiesta di RMN può seguire un criterio clinico (solo in caso di veloce progressione dello sviluppo puberale, cefalea), visto che questa fascia di età presenta un rischio molto basso di patologia organica (44).
Nel caso di una diagnosi di PPP e in funzione della clinica dovremo richiedere indagini ormonali diverse, come TSH e ß-hCG, ed esami radiologici, quali TC surreni, ecografia pelvica o testicolare, TC addome o torace.
Alla luce di quanto riportato, appare chiaro come la diagnosi di pubertà precoce sia complessa, con la necessità di una stretta integrazione tra valutazione clinica accurata, e spesso ripetuta nel tempo, e valutazione biochimico/strumentale.
BIBLIOGRAFIA
- Marshall WA, Tanner JM. Variation in pattern of pubertal changes in girls. Arch Dis Child 1969, 44: 291-303.
- Marshall WA, Tanner JM. Variation in pattern of pubertal changes in boys. Arch Dis Child 1970, 45: 13-23.
- Precocious Puberty. In: Reproductive Endocrinology, Surgery and Technology. Lippincott-Raven, Philadelphia 1996, Vol 1: p 989.
- Kaplowitz P. Update on precocious puberty: girls are showing sign of puberty earlier but most do not require treatment. Adv Pediatr 2011, 58: 243-58.
- Aksglaede L, et al. Recent decline in age at breast development: the Copenhagen Puberty Study. Pediatrics 2009, 123: e932–9.
- Carel JC, Léger J. Precocious puberty. N Engl J Med 2008, 358: 2366-77.
- Chalumeau M, et al. Selecting girls with precocious puberty for brain imaging: validation of European evidence-based diagnosis rule. J Pediatr 2003, 143: 445–50.
- Cisternino M, et al. Etiology and age incidence of precocious puberty in girls: a multicentric study. J Pediatr Endocrinol Metab 2000, 13 suppl 1: 695–701.
- De Sanctis V, et al. Etiology of central precocious puberty in males: the results of the Italian Study Group for Physiopathology of Puberty. J Pediatr Endocrinol Metab 2000, 13 suppl 1: 687–93.
- Fuqua JS. Treatment and outcomes of precocious puberty: an update. J Clin Endocrinol Metab 2013, 98: 2198–207.
- Macedo DB, et al. New causes of central precocious puberty: the role of the genetic factors. Neuroendocrinology 2014, 100: 1-8.
- Listernick R, et al. Intracranial gliomas in neurofibromatosis type. Am J Med Genet 1999, 89: 38-44.
- Mahachoklertwattana P, et al. The luteinizing hormone-releasing hormone-secreting hypotalamic hamartoma is a congenital malformation: natural history. J Clin Endocrinol Metab 1993, 77: 118-24.
- Trivin C, et al. Presentation and evolution of organic central precocious puberty according to the type of CNS lesion. Clin Endocrinol 2006, 65: 239–45.
- Armstrong GT, et al. Alterations in pubertal timing following therapy for childhood malignancies. Endocr Dev 2009, 15: 25-39.
- Diaz A, et al. McCune-Albright syndrome and disorders due to activating mutations of GNAS1. J Pediatr Endocrinol Metab 2007, 20: 853-80.
- Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008, 3: 12.
- Hashemipour M, et al. Granulosa cell tumor in a six-year-old girl presented as precocious puberty. J Res Med Sci 2010, 15: 240–2.
- Van Wyk JJ, et al. Syndrome of precocious menstruation and galactorrhea in juvenile hypothyroidism: an example of hormonal overlap in pituitary feedback. J Pediatr 1960, 57: 416–35.
- Shenker A, et al. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature 1993, 365: 652-4.
- Englund AT, et al. Pediatric germ cell and human chorionic gonadotropin-producing tumors. Clinical and laboratory features. Am J Dis Child 1991, 145: 1294-7.
- Urban MD, et al. The diagnosis of Leydig cell tumor in childhood. Am J Dis Child 1978, 132: 494-7.
- DeSalvo D, et al. In patients with premature adrenarche, bone age advancement by 2 or more years is relatively common and generally benign. Annual Meeting of the Pediatric Academic Societies. Denver (CO), April 30–May 3, 2011.
- Sklar CA, et al. Evidence for dissociation between adrenarche and gonadarche: studies in patients with idiopathic precocious puberty, gonadal dysgenesis, isolated gonadotropin deficiency and constitutional delayed growth and adolescence. J Clin Endocrinol Metab 1980, 51: 548-66.
- Berberoglu M. Precocious puberty and normal variant puberty: definition, etiology, diagnosis and current management. J Clin Res Pediatr Endocrinol 2009, 1: 164-74.
- Idkowiak J, et al. Premature adrenarche: novel lesson from early onset androgen excess. Eur J Endocrinol 2011, 65: 189-207.
- Rosenfield RL, et al. Thelarche, pubarche, and menarche attainment in children with normal and elevated body mass index. Pediatrics 2009, 123: 84–8.
- Williams RM, et al. Premature adrenarche. Arch Dis Child 2012, 97: 250–4.
- Pere A, et al. Follow up of growth and steroids in premature adrenarche. Eur J Pediatr 1995, 154: 346-52.
- van Weissenbruch MM. Premature adrenarche, polycystic ovary syndrome and intrauterine growth retardation: does a relationship exist? Curr Opin Endocrinol Diabetes Obes 2007, 14: 35–40.
- Ibáñez L, et al. Clinical spectrum of premature pubarche: links to metabolic syndrome and ovarian hyperandrogenism. Rev Endocr Metab Dis 2009, 10: 63-76.
- De Vries L, et al. Ultrasonografic and clinical parameters for early differentiation between precocious puberty and premature thelarche. Eur J Endocrinol 2006, 154: 891-8.
- De Vries L, et al. Premature thelarche: age at presentation affects clinical course but not clinical characteristics or risk to progress to precocious puberty. J Pediatr 2010, 156: 466–71.
- Nam HK, et al. Factors to predict positive results of gonadotropin releasing hormone stimulation test in girls with suspected precocious puberty. J Korean Med Sci 2012, 27: 194-9.
- Martin DD, et al. The use of bone age in clinical practice. Horm Res Paediatr 2011, 76: 10–6.
- Ziereisen F, et al. Sonographic imaging of the paediatric female pelvis. Eur Radiol 2005, 15: 1296-309.
- Sathasivam A, et al. Pelvic ultrasonography in the evaluation of central precocious puberty: comparison with leuprolide stimulation test. J Pediatr 2011, 159: 490–5.
- Pasternak Y, et al. The utility of basal serum LH in prediction of central precocious puberty in girls. Eur J Endocrinol 2012, 166: 295-9.
- Houk CP, et al, Adequacy of a single unstimulated luteinizing hormone level to diagnose central precocious puberty in girls. Pediatrics 2009, 123: e1059–63.
- Pescovitz OH, et al. Premature thelarche and central precocious puberty: the relationship between clinical presentation and the gonadotropin response to luteinizing hormone-releasing hormone. J Clin Endocrinol Metab 1988, 67: 474–9.
- Yazdani P, et al. A single sample GnRHa stimulation test in the diagnosis of precocious puberty. Int J Pediatr Endocrinol 2012, 2012: 23.
- Bay K, et al. Estradiol levels in prepubertal boys and girls: analytical challenges. Int J Androl 2004, 27: 266–73.
- Wang C, et al. Measurement of total serum testosterone in adult men: comparison of current laboratory methods versus liquid chromatography-tandem mass spectrometry. J Clin Endocrinol Metab 2004, 89: 534–43.
- Choi KH, et al. Boys with precocious or early puberty: incidence of pathological brain magnetic resonance imaging findings and factors related to newly developed brain lesions. Ann Pediatr Endocrinol Metab 2013, 18: 183-90.
Approccio terapeutico alla pubertà precoce
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
PUBERTÀ PRECOCE CENTRALE
La terapia della PPC si basa sull’uso degli analoghi del GnRH, che hanno lo scopo di inibire, dopo un’iniziale stimolazione, la secrezione del GnRH e delle gonadotropine, con conseguente riduzione dei livelli di ormoni sessuali e rallentamento/arresto della progressione puberale.
Nel 2009 è stata pubblicata una Consensus (1) che chiarisce indicazioni, finalità e limiti della terapia con analoghi. Questa deve essere utilizzata:
- quando venga documentata una progressione puberale e della velocità di crescita tali da poter compromettere il raggiungimento dell’altezza target;
- in tutti i maschi con comparsa di segni puberali prima dei 9 anni;
- in tutte le bambine con comparsa dei segni puberali prima dei 6 anni;
Nelle bambine di età compresa tra i 6 e gli 8 anni, poiché i vantaggi in termini staturali non sono provati, la decisione di iniziare la terapia deve essere individualizzata.
I farmaci disponibili sono diversi (tabella) e si differenziano per dosaggio e modalità di somministrazione, ma sembrano tutti efficaci nel controllare la progressione puberale (1). In Italia sono approvati per il trattamento della pubertà precoce centrale (nota AIFA 51) gli analoghi Leuprolide e Triptorelina, delle quali esistono formulazioni a somministrazione intra-muscolo o sottocute mensile o trimestrale.
| Formulazioni long-acting di analoghi del GnRH | |
| Preparato | Dosaggio |
| Goserelin | 3.6 mg/mese o 10.8 mg/3 mesi |
| Buserelin | 6.3 mg/2 mesi |
| Leuprolide | 3.75 mg/mese o 11.25 mg/3 mesi |
| Triptorelina | 3 o 3.75 mg/mese o 11.25 mg/3 mesi |
Generalmente la terapia con analoghi è ben tollerata (1). Gli effetti collaterali più comunemente riportati sono flushing e cefalea, comunque solitamente di breve durata. In più del 10% dei pazienti viene riferito dolore nella sede di iniezione, dove raramente sono stati descritti ascessi sterili. In questo caso è necessario cambiare farmaco (1).
Nelle bambine la stimolazione ovarica iniziale all’avvio del trattamento, seguita dalla sospensione estrogenica indotta dal trattamento, può causare, durante il primo mese, un sanguinamento vaginale di intensità lieve o moderata (3).
Monitoraggio della terapia
L’efficacia della terapia può essere valutata con due diversi criteri:
- criterio clinico (2): rallentamento della velocità di crescita e della progressione puberale, che devono essere valutati ogni 3-6 mesi; la progressione della sola peluria pubica è indicativa di normale pubarca;
- criterio biochimico (2,4,5):
- dosaggio random dell’LH con metodica ultrasensibile: quando elevato esprime una mancata soppressione;
- dosaggio LH sotto stimolo con GnRH: un valore ≤ 3UI/L è indicativo di efficace soppressione della secrezione gonadotropinica.
Il trattamento con analoghi del GnRH è efficace nel migliorare la statura definitiva dei bambini affetti da PPC, tuttavia il risultato è fortemente influenzato da diverse variabili, quali altezza, età ossea, età anagrafica e stadio puberale alla diagnosi (5).
Sospensione della terapia
Poiché appare complesso determinare l’influenza delle singole variabili che possono concorrere al risultato finale in termini di altezza adulta, al momento non vi sono chiare indicazioni su quale sia il momento migliore per interrompere la terapia con analoghi. Generalmente si attende il raggiungimento di un’età ossea di circa 12-12.5 anni, che per alcuni autori permette di ottenere il massimo effetto sulla statura definitiva (6). Comunque, la decisione deve essere condivisa con i familiari e il paziente, al quale dobbiamo cercare di assicurare un percorso di maturazione puberale più sincrono possibile ai suoi coetanei (1). Il menarca compare mediamente dopo 16 mesi dalla sospensione della terapia, che non sembra interferire in alcun modo con la futura capacità riproduttiva del paziente (1).
PUBERTÀ PRECOCE PERIFERICA O PSEUDO-PUBERTÀ
La terapia è chiaramente diversa in funzione dell’eziologia.
Nella testotossicosi, la terapia mira a ridurre/annullare gli effetti negativi della precoce secrezione di testosterone, soprattutto sull’altezza definitiva, che risulta invariabilmente compromessa se il paziente non è trattato. Attualmente la terapia che sembra più efficace in termini di modalità di somministrazione, risultati e costi, è data dalla combinazione di un potente anti-androgeno, la bicalutamide, con l’anastrozolo, un inibitore dell’aromatasi di terza generazione (7).
Nel caso della sindrome di McCune-Albright, sono stati riportati risultati positivi con il letrozolo, un inibitore dell’aromatasi di terza generazione, il tamoxifene e il fulvestrano, un inibitore puro del recettore degli estrogeni (8).
BIBLIOGRAFIA
- Carel JC, et al. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics 2009, 123: e752-62.
- Roger M, et al. Long term treatment of male and female precocious puberty by periodic administration of a long-acting preparation of D-Trp-luteinizing hormone-releasing hormone microcapsules. J Clin Endocrinol Metab 1986, 62: 670–7.
- Carel JC, et al. Treatment of central precocious puberty with depot leuprorelin. French Leuprorelin Trial Group. Eur J Endocrinol 1995, 132: 699-704.
- Lee PA, et al. Efficacy of leuprolide acetate 1-month depot for central precocious puberty (CPP): growth outcomes during a prospective, longitudinal study. Int J Pediatr Endocrinol 2011, 2011: 7.
- Willemsen RH, et al. Pros and cons of GnRHa treatment for early puberty in girls. Nature Rev Endocrinol 2014, 10: 352–63.
- Arrigo T, et al. Analysis of the factors affecting auxological response to GnRH agonist treatment and final height outcome in girls with idiopathic central precocious puberty. Eur J Endocrinol 1999, 141: 140-4.
- Lenz AM, et al. Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics 2010, 126: 728-33.
- Collins MT, et al. McCune Albright syndrome and the extraskeletal manifestation of fibrous dysplasia. Orphanet J Rare Dis 2012, 7: S4.
Scheda GnRH agonisti
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
Meccanismo d’azione
Il GnRH è un decapeptide sintetizzato da un gruppo specializzato di circa 1000 neuroni localizzati nell’ipotalamo. Viene rilasciato in maniera ritmica, con dei pulse che si succedono ogni 30-120 minuti e ai quali corrisponde un’altrettanto ritmica secrezione di LH.
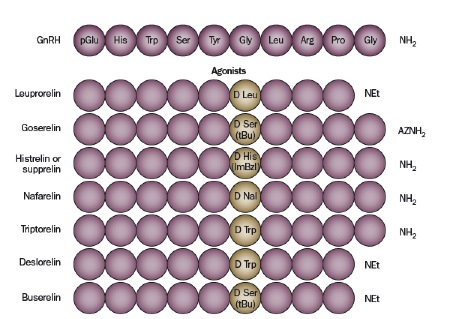
La particolare struttura spaziale ha lo scopo di preservare la posizione di alcuni aminoacidi localizzati in particolari punti chiave, fondamentali per la modulazione dell’affinità recettoriale. La sostituzione della glicina in posizione 6 con diversi altri aminoacidi ha dato vita ad altri peptidi dotati di maggiore affinità recettoriale e maggiore resistenza agli enzimi proteolitici rispetto al GnRH nativo (figura 1) (1).
Dopo un’iniziale e transitoria iperstimolazione, nota come “flare up”, la prolungata esposizione agli analoghi induce una progressiva riduzione della secrezione delle gonadotropine, indotta da diversi meccanismi verosimilmente rappresentati da un’alterazione della trasmissione del messaggio intra-cellulare e/o della sintesi ed esocitosi delle gonadotropine stesse. L’effetto finale è l’inibizione dell’attività gonadica.
Indicazioni
Trattamento della pubertà precoce centrale (2,3), in cui rappresentano il trattamento di scelta dall’inizio degli anni ’80.
Trattamento adiuvante nel cancro della mammella. Nelle donne in pre-menopausa affette da cancro della mammella, l’uso degli analoghi associato ad altri farmaci (anti-estrogeni) sembra ridurre il rischio di recidiva di malattia (4,5).
Trattamento del carcinoma prostatico. Il cancro della prostata, che rappresenta nel mondo la causa principale di morte per la popolazione maschile, ha patogenesi complessa, ma appare evidente una relazione con gli ormoni sessuali (6). Per tale ragione la castrazione o la privazione farmacologica degli androgeni appaiono momenti fondamentali della terapia (7). Gli analoghi più comunemente utilizzati sono leuprolide e goserelin.
Trattamento del dolore da endometriosi. È una patologia estrogeno-dipendente e per questa ragione il trattamento con analoghi è in grado di migliorare il dolore associato a questa condizione (8,9).
Trattamento leiomiomatosi uterina. I leiomiomi rappresentano il tumore benigno più comune nelle donne in età fertile. I sintomi più frequenti sono sanguinamento mestruale eccessivo con relativa anemia, dolore e sensazione di peso nella pelvi e in addome, dolore o fastidio durante i rapporti sessuali, infertilità e aborti ricorrenti (10,11). Gli analoghi del GnRH si sono dimostrati efficaci nel ridurre il volume dei leiomiomi e dell’utero, ma l’effetto è limitato al periodo di trattamento. Sembrano inoltre migliorare l’outcome chirurgico delle donne sottoposte ad isterectomia (12).
Protocolli di procreazione medicalmente assistita, in cui vengono utilizzati da molti anni per prevenire il picco endogeno di LH e scegliere il momento migliore per il prelievo dell’ovocita (13,14).
Prevenzione della menopausa precoce (POF) nelle pazienti oncologiche. La tossicità ovarica è un possibile effetto della chemioterapia e si può manifestare con infertilità, amenorrea transitoria o menopausa precoce. La soppressione dell’attività ovarica indotta dagli analoghi del GnRH potrebbe, attraverso diversi meccanismi, preservare la fertilità della paziente. In letteratura sono presenti diversi lavori che sembrano suggerire con l’uso degli analoghi un minore rischio di POF nelle donne in pre-menopausa sottoposte a chemioterapia, tuttavia al momento il loro utilizzo è controverso, non codificato e quindi off-label (15-17).
Preparazioni, via di somministrazione, posologia
Esistono diverse formulazioni (18):
- forme somministrabili per via nasale o sottocute (tabella 1), che necessitano più somministrazioni giornaliere;
- forme a lento rilascio intra-muscolo o sotto-cute (tabella 2), che assicurano una liberazione costante per 28 giorni o tre mesi. Tra queste certamente quella più utilizzata è la formulazione a somministrazione mensile della leuprolide acetato (19-21);
- impianto sottocute di istrelina in grado di rilasciare il farmaco per un anno (22). L’efficacia sembra analoga a quella ottenuta con la somministrazione di leuprolide, con il grande vantaggio di un lungo intervallo di somministrazione (23,24).
| Tabella 1 Formulazioni short-acting di analoghi del GnRH |
|
| Preparato | Dosaggio/die |
| Nafarelin | spray 800 µg x 2 |
| Buserelin | spray 20-40 µg/kg 1200-1800 µg sc |
| Leuprolide | 50 µg/kg sc |
| Triptorelina | 20-40 µg/kg sc |
| Deslorelina | 4-8 µg/kg sc |
| Istrelina | 8-10 µg/kg sc |
| Tabella 2 Formulazioni long-acting di analoghi del GnRH |
|
| Preparato | Dosaggio |
| Goserelin | 3.6 mg/mese o 10.8 mg/3 mesi |
| Buserelin | 6.3 mg/2 mesi |
| Leuprolide | 3.75 mg/mese o 11.25 mg/3 mesi |
| Triptorelina | 3 o 3.75 mg/mese o 11.25 mg/3 mesi |
| Istrelina | 50 mg impianto annuale |
Tutti i diversi analoghi disponibili sembrano avere analoga efficacia nel trattamento della pubertà precoce centrale (18).
- Leuprolide:
- soluzione 1 mg/0.2 mL (Enantone)
- siringa preriempita 3.6 mg (Leptoprol), 3.75 mg (Enantone, Politrate), 5 mg (Leptoprol), 7.5 mg (Eligard), 11.25 mg (Enantone), 22.5 mg (Eligard), 45 mg (Eligard). Nel bambino si usano le fl (intera se > 20 kg, ½ se < 20 kg) da 3.75 mg mensile o da 11.25 mg trimestrale.
- Triptorelina: soluzione 0.1 mg/mL (Decapeptyl, Fertipeptil), 3.75 mg/2 mL (Decapeptyl, Gonapeptyl depot), 11.25 mg/2 mL (Decapeptyl), 22.5 mg/2 mL (Decapeptyl). Nel bambino si usano le fl (½ se < 20 kg, 2/3 tra 20 e 30 kg, intera se > 30 kg,) da 3.75 mg mensile o da 11.25 mg trimestrale.
- Goserelin: siringa preriempita da 3.6 mg (Novimp, Zoladex), 10.8 mg (Zoladex)
- Buserelin (Suprefact): spray nasale 0.1 mg/puff, soluzione 1 mg/mL, siringa preriempita 6.3 mg, siringa preriempita 9.45 mg
Contro-indicazioni
Gravidanza e allattamento.
Effetti collaterali
Sono collegati alla riduzione della sintesi degli ormoni sessuali, caratteristica propria di questa classe di farmaci.
Più comunemente riportati flushing e cefalea, comunque solitamente di breve durata.
In più del 10% dei pazienti viene riferito dolore nella sede di iniezione, dove raramente sono stati descritti ascessi sterili.
Nei pazienti con carcinoma della prostata, il trattamento induce impotenza, riduzione della libido, osteopenia e disturbi delle vie urinarie (25).
Vampate (più dell’80% delle pazienti), riduzione della libido, secchezza e sanguinamenti vaginali, irritabilità, depressione, riduzione della densità minerale ossea (26).
Limitazioni prescrittive
Nota AIFA 51 per Leuprolide e Triptorelina nella pubertà precoce
Bibliografia
- Millar RP, et al. Current and future applications of GnRH, kisspeptin and neurokinin B analogues. Nat Rev Endocrinol 2013, 9: 451–66.
- Berberoglu M. Precocious puberty and normal variant puberty: definition, etiology, diagnosis and current management. J Clin Res Pediatr Endocrinol 2009, 1: 164–74.
- Lahlou N, et al. Pharmacokinetics and pharmacodynamics of GnRH agonists: clinical implications in pediatrics. J Pediatr Endocrinol Metab 2000, 13 suppl 1: 723–37.
- Cheer SM, et al. Goserelin. A review of its use in the treatment of early breast cancer in premenopausal and perimenopausal women. Drugs 2005, 65: 2639-55.
- Goel S. et al. LHRH agonists for adjuvant therapy of early breast cancer in premenopausal women. Cochrane Database Syst Rev 2009: CD004562.
- Gann PH, et al. Prospective study of sex hormone levels and risk of prostate cancer. J Natl Cancer Inst 1996, 88: 1118–26.
- Shen MM, et al. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010, 24: 1967–2000.
- Kennedy S, et al. ESHRE guideline for the diagnosis and treatment of endometriosis. Hum Reprod 2005, 20: 2698–704.
- Brown J, et al. Endometriosis: an overview of Cochrane Reviews. Cochrane Database Syst Rev 2014: CD009590.
- Evans P, et al. Uterine fibroid tumors: diagnosis and treatment. Am Fam Physician 2007, 75: 1503–8.
- Al-Nafussi A. Uterine smooth-muscle tumours: practical approach to diagnosis. Curr Diagn Pathol 2004, 10: 140–56.
- Stovall TG, et al. GnRH agonist and iron versus placebo and iron in the anemic patient before surgery for leiomyomas: a randomized controlled trial. Leuprolide Acetate Study Group. Obstet Gynecol 1995, 86: 65-71.
- Cetel NS, et al. The dynamics of gonadotropin inhibition in women induced by an antagonistic analog of gonadotropin-releasing hormone. J Clin Endocrinol Metab 1983, 57: 62–5.
- Albano C, et al. Ovarian stimulation with HMG: results of a prospective randomized phase III European study comparing the luteinizing hormone-releasing hormone (LHRH)-antagonist cetrorelix and the LHRH-agonist buserelin. European Cetrorelix Study Group. Hum Reprod 2000, 15: 526–31.
- Chen H, et al. Adjuvant gonadotropin-releasing hormone analogues for the prevention of chemotherapy induced premature ovarian failure in premenopausal women. Cochrane Database Syst Rev 2011: CD008018.
- Partridge A, et al. Age at menopause among women who remain premenopausal following treatment for early breast cancer. Long-term results from International Breast Cancer Study Group Trials V and VI. Eur J Cancer 2007, 43: 1646–53.
- Del Mastro L, et al. Gonadotropin-releasing hormone analogues for the prevention of chemotherapy-induced premature ovarian failure in cancer women: systematic review and meta-analysis of randomized trials. Cancer Treat Rev 2014, 40: 675–83.
- Carel JC, et al. Consensus Statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics 2009, 123: e752-62.
- Lee PA, et al. Efficacy and safety of leuprolide acetate 3-month depot 11.25 milligrams or 30 milligrams for the treatment of central precocious puberty. J Clin Endocrinol Metab 2012, 97: 1572–80.
- Lee PA, et al. Efficacy of leuprolide acetate 1-month depot for central precocious puberty (CPP): growth outcomes during a prospective, longitudinal study. Int J Pediatr Endocrinol 2011, 2011: 7.
- Neely EK, et al. Leuprolide acetate 1-month depot for central precocious puberty: hormonal suppression and recovery. Int J Pediatr Endocrinol 2010, 2010: 398639.
- Eugster EA, et al. Efficacy and safety of histrelin subdermal implant in children with central precocious puberty: a multicenter trial. J Clin Endocrinol Metab 2007, 92: 1697–704.
- Rahhal S, et al. Results of a second year of therapy with the 12-month histrelin implant for the treatment of central precocious puberty. Int J Pediatr Endocrinol 2009, 2009: 812517.
- Gillis D, et al. Time to menarche and final height after histrelin implant treatment for central precocious puberty. J Pediatr 2013, 163: 532-6.
- Choi S, Lee AK. Efficacy and safety of gonadotropin-releasing hormone agonists used in the treatment of prostate cancer. Drug Healthc Patient Saf 2011, 3: 107–19.
- Magon N. Gonadotropin releasing hormone agonists: Expanding vistas. Indian J Endocrinol Metab 2011, 15: 261-7.
Scheda inibitori aromatasi
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
Meccanismo d’azione
L’aromatasi è un enzima che appartiene alla famiglia del citocromo P450 ed è espresso ubiquitariamente nelle cellule umane (es. cellule della granulosa, cellule di Leydig, tessuto adiposo, cervello, fibroblasti, sincizio-trofoblasto) (1,2).
La sua espressione nelle cellule della granulosa è fondamentale per il processo di sintesi dell’estradiolo, poichè permette la conversione dell’androstenedione in estrone. In menopausa l’espressione principale dell’aromatasi si trova invece a livello del tessuto adiposo.
Gli inibitori dell’aromatasi quindi riducono la sintesi degli estrogeni in tutti i tessuti e trovano applicazione nel trattamento di tutte quelle condizioni che sono estrogeno-dipendenti (3).
Negli anni si è passati da una prima generazione di inibitori dell’aromatasi (aminoglutetimide) gravati da un importante effetto collaterale rappresentato dall’insufficienza surrenalica, a una seconda generazione costituita da farmaci che devono essere somministrati im, a quelli più recenti, di cosiddetta terza generazione (letrozolo, anastrozolo, exemestano), che sono somministrati per via orale e appaiono selettivi, potenti e reversibili (3).
Indicazioni
Cancro della mammella. Questa rappresenta al momento l’unica indicazione approvata per l’uso di questi farmaci. La terapia adiuvante con inibitori dell’aromatasi sembra ridurre l’incidenza di cancro controlaterale, di invasività e carcinoma duttale in situ nelle donne in menopausa e positività del recettore per estrogeni (4-7). Questa rappresenta al momento l’unica indicazione approvata per questa classe di farmaci che possono essere prescritti ormai (det. AIFA 30.10.12; GU n. 267 del 15-11-2012) senza piano terapeutico e senza alcuna nota. Gli inibitori dell’aromatasi vengono utilizzati anche nel trattamento del carcinoma mammario del maschio (8).
Endometriosi. Il trattamento con analoghi di terza generazione si è dimostrato efficace nel ridurre il dolore pelvico. Possono essere utilizzati inoltre in associazione con altri farmaci quali gli analoghi del GnRH, con una migliore soppressione dei livelli estrogenici (9).
Induzione dell’ovulazione. Sebbene al momento non siano raccomandati routinariamente, vi sono in letteratura numerose evidenze che identificano gli inibitori dell’aromatasi come farmaci utili nell’induzione dell’ovulazione in donne affette da PCO o infertilità inspiegata, con effetti comparabili al clomifene citrato in termini di safety e rischio di gravidanze multiple (3,10).
In pediatria gli inibitori dell’aromatasi hanno trovato diverse applicazioni off-label (11):
- Sindrome di Mc Cune Albright: gli studi attualmente a disposizione hanno dimostrato solo una parziale e transitoria efficacia del letrozolo nel controllare la progressione puberale delle bambine affette da questa sindrome (12,13);
- Testotossicosi: migliori risultati in termini di riduzione della velocità di crescità e maturazione ossea sembrano derivare dall’uso contemporaneo di anti-androgeni (bicalutamide, ciproterone) associati a letrozolo o anastrozolo (14,15);
- Bassa statura idiopatica: poichè gli estrogeni hanno un ruolo importante sia nell’induzione dello spurt puberale che nella maturazione e chiusura delle epifisi, gli inibitori dell’aromatasi sono stati utilizzati, da soli o in associazione con altri farmaci, nel trattamento di bambini con bassa statura idiopatica o ritardo puberale costituzionale. Sebbene alcuni studi dimostrino un vantaggio in termini di statura finale nei bambini trattati rispetto ai controlli, ci sono anche evidenze di un incremento delle deformità vertebrali nei trattati e mancano studi dei possibili effetti collaterali a lungo termine (11,16,17).
Contro-indicazioni
Pazienti in pre-menopausa, poiché a causa del ridotto feed-back degli estrogeni, inducono un aumento della secrezione delle gonadotropine e in alcuni esperimenti su animali determinano un aumento delle dimensioni e del peso delle ovaie. Le pazienti con carcinomi mammari che risultano privi di recettori per gli estrogeni sono usualmente non responsive ai trattamenti ormonali.
Preparazioni, via di somministrazione, posologia
- Anastrozolo: cp 1 mg (Anastrozolo Accord Healthcare, Anastrozolo Actavis, Anastrozolo Alter, Anastrozolo Bluefish, Anastrozolo Crinos, Anastrozolo DOC generici, Anastrozolo Hicma, Anastrozolo Kabi, Anastrozolo Mylan generics, Anastrozolo Professionalcare, Anastrozolo Sandoz, Anastrozolo Sun, Anastrozolo TEVA, Anastrozolo Zentiva, Arimidex, Eristrol, Extroplex, Griset, Iniben, Keyfen, Raoloz, Renazole)
- Letrozolo: cp 2.5 mg (Brestoral, Calantha, Femara, Letrix, Letrocet, Letroveres, Letrozolo Actavis, Letrozolo AHCL, Letrozolo Crinos, Letrozolo DOC generici, Letrozolo Fidia, Letrozolo Germed, Letrozolo Kabi, Letrozolo Mylan Generics, Letrozolo Mylan Pharma, Letrozolo Sandoz, Letrozolo Technigen, Letrozolo TEVA, Letrozolo Zentiva, Zoltron)
- Exemestane: cp 25 mg (Aromasin, Axelta, Exegen, Exemestane Accord Healthcare, Exemestane Actavis, Exemestane DOC Generici, Exemestane EG, Exemestane Hikma, Exemestane Mylan Generics, Exemestane Pfizer, Exemestane Pharos Generics, Exemestane Sandoz, Exemestane TEVA, Exemestane Zentiva, Mestane).
Effetti collaterali
Effetti collaterali a medio e lungo temine: raramente vampate, secchezza vaginale, modesta cefalea, dolore e tumefazione articolare, compresa la sindrome del tunnel carpale. I disturbi articolari sembrano responsabili di circa il 10% dei casi di interruzione precoce della terapia (18).
L’uso prolungato può indurre, in entrambi i sessi, osteopenia e osteoporosi, con incremento del rischio di frattura (19,20). L'effetto negativo osseo cessa immediatamente alla sospensione della terapia (21). L’uso di bisfosfonati è in grado di prevenire completamente la perdita minerale ossea indotta dagli inibitori (22).
Possibile peggioramento del profilo lipidico, sebbene non sia stata dimostrato un incremento del rischio cardiovascolare in confronto al tamoxifene (23).
Limitazioni prescrittive
Senza piano terapeutico e senza nota per il carcinoma mammario.
Bibliografia
- Simpson ER, et al. Aromatase gene expression in adipose tissue: relationship to breast cancer. Int J Fertil Menopausal Stud 1994, 39 suppl 2: 75–83.
- Grodin JM, et al. Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab 1973, 36: 207–14.
- Pavone ME, Bulun SE. The use of aromatase inhibitors for ovulation induction and superovulation. J Clin Endocrinol Metab 2013, 98: 1838–44.
- Baum M, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet 2002, 359: 2131–9.
- Goss PE, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med 2003, 349: 1793–802.
- Goss PE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med 2011, 364: 2381–91.
- Advani P, et al. Current strategies for the prevention of breast cancer. Breast Cancer 2014, 6: 59–71.
- Bradley KL, et al. Contemporary systemic therapy for male breast cancer. Clin Breast Cancer 2014, 14: 31-9.
- Pavone ME, Bulun SE. Aromatase inhibitor for the treatment of endometriosis: a review. Fertil Steril 2012, 98: 1370–9.
- Eckmann KR, Kockler DR. Aromatase inhibitors for ovulation and pregnancy in polycystic ovary syndrome. Ann Pharmacother 2009, 43: 1338-4.
- Wit JM, et al. Aromatase inhibitors in pediatrics. Nat Rev Endocrinol 2012, 8: 135–47.
- Feuillan P, et al. Letrozole treatment of precocious puberty in girls with the McCune–Albright syndrome: a pilot study. J Clin Endocrinol Metab 2007, 92: 2100–6.
- Mieszczak J, et al. The aromatase inhibitor anastrozole is ineffective in the treatment of precocious puberty in girls with McCune–Albright Syndrome. J Clin Endocrinol Metab 2008, 93: 2751–4.
- Lenz AM, et al. Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics 2010, 126: e728–33.
- Eyssette-Guerreau S, et al. Effectiveness of anastrozole and cyproterone acetate in two brothers with familial male precocious puberty. J Pediatr Endocrinol Metab 2008, 21: 995–1002.
- Diaz-Thomas A, et al. Use of aromatase inhibitors in children and adolescents: what’s new? Curr Opin Pediatr 2010, 22: 501–7.
- Ranke MB. Treatment of children and adolescents with idiopathic short stature. Nat Rev Endocrinol 2013, 9: 325-34.
- Adjuvant aromatase inhibitors in early breast cancer – toxicity and adherence. Important observations in clinical practice. Breast Cancer Res Treatm 2007, 106 (S1): S111.
- Buzdar AU. The ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial: an update. Clin Breast Cancer 2004, 5 suppl 1: S6–12.
- Jakesz R, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years' adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet 2005, 366: 455–62.
- Eastell R, et al. Effect of anastrozole on bone mineral density: 5-year results from the anastrozole, tamoxifen, alone or in combination trial 18233230. J Clin Oncol 2008, 26: 1051–8.
- Gnant M, et al. Adjuvant endocrine therapy plus zoledronic acid in premenopausal women with early-stage breast cancer: 5-year follow-up of the ABCSG-12 bone-mineral density substudy. Lancet Oncol 2008, 9: 840–9.
- Geisler J, et al. Changes in bone and lipid metabolism in postmenopausal women with early breast cancer after terminating 2-year treatment with exemestane: a randomised, placebo-controlled study. Eur J Cancer 2006, 42: 2968–75.
Inquadramento diagnostico della pubertà ritardata
Rossella Gaudino, con la collaborazione di Sarah Dal Ben
Dipartimento di Scienze della Vita e Riproduzione, UO Complessa Pediatria, Policlinico Verona, AOUI di Verona
Endocrinologia e Auxologia Pediatrica e dell'Adolescenza
DEFINIZIONE ED EPIDEMIOLOGIA
Si definisce pubertà ritardata o interrotta nella donna:
- l'assenza di sviluppo del bottone mammario all'età di 13 anni;
- la mancanza dello sviluppo del seno in età maggiore di 2-2.5 DS rispetto a quella della popolazione generale (13 anni nelle ragazze italiane);
- l'assenza di menarca all'età di 16 anni o entro 5 anni dall'inizio della pubertà;
- una progressione dello sviluppo troppo lenta secondo la classificazione di Tanner, che superi i limiti fisiologici e con necessità di più di 12 mesi per il passaggio da uno stadio all'altro.
Si definisce pubertà ritardata nell'uomo:
- la mancanza di ingrandimento testicolare (testicolo < 4 mL) in età maggiore di 2-2.5 DS rispetto a quella della popolazione generale (circa 14 anni nei maschi italiani).
Lo sviluppo della peluria pubica non è generalmente considerato nella definizione di pubertà ritardata, in quando dipende dalla maturazione del surrene (adrenarca) ed è indipendente dall'asse ipotalamo-ipofisi-gonadi (1).
La pubertà ritardata è una condizione clinica rara nel sesso femminile e colpisce circa il 2.5% delle ragazze (2); di queste circa il 30% ha un ritardo costituzionale di crescita e pubertà (CDGP). Nel maschio, invece, la pubertà ritardata colpisce circa il 3% dei ragazzi (3) e, molto frequentemente, rappresenta una semplice variabilità nel normale accrescimento puberale, con un pattern di accrescimento riferibile a CDGP nel 65% dei casi.
EZIOLOGIA
La maggior parte dei ritardi puberali è secondaria a patologie croniche, mentre più raramente il ritardo puberale è primitivo (ipogonadismo ipergonadotropo, tabella 1).
Ritardo costituzionale (CDGP, constitutional delay of growth and puberty)
Si tratta di una variante del normale processo di crescita, più comune nel sesso maschile rispetto a quello femminile. Il pattern di sviluppo puberale nei bambini affetti è normale, ma in ritardo rispetto ai coetanei. Questi bambini, inoltre, hanno anche bassa statura (di circa 2 DS inferiore rispetto a quella attesa per età), che riflette una maturazione ossea in ritardo di 2-4 anni rispetto all'età cronologica (4). Molti di questi pazienti continuano la crescita lineare fino ai 20 anni, raggiungendo un'altezza appropriata rispetto al target genetico; in questi bambini la maturazione sessuale è più correlata all'età ossea che a quella anagrafica (2).
Le cause del CDGP non sono note, ma sembrano esserci basi genetiche: si stima che il 50-80% della variazione nello sviluppo puberale sia dovuta a fattori genetici e che circa l'80% dei pazienti con CDGP abbia una storia familiare di pubertà ritardata (5,6). Per quanto riguarda le modalità di trasmissione del CDGP, è stata vista una variabilità, probabilmente dovuta alla trasmissione poligenica, ma in molti casi è emersa una trasmissione autosomica dominante (6).
Ipogonadismo ipogonadotropo (HH)
Secondo in frequenza, è una condizione clinica caratterizzata dal difetto nella secrezione pulsatile di gonadotropine, per un problema di origine centrale coinvolgente ipofisi e/o ipotalamo. Si può distinguere in ipogonadismo ipogonadotropo permanente e funzionale (tabella 1).
L'ipogonadismo ipogonadotropo ha una frequenza del 30% nei maschi con pubertà ritardata e del 40% delle femmine (1).
Ipogonadismo ipergonadotropo
Condizione caratterizzata da insufficienza gonadica associata ad alti livelli di gonadotropine per mancanza del feed-back ormonale. L'ipogonadismo ipergonadotropo si può distinguere in:
- congenito: comprende la disgenesia gonadica familiare o sporadica, la sindrome di Turner, la s. di Klinefelter oltre ad altre anomalie genetiche rare;
- acquisito: iatrogeno, traumatico, infettivo o autoimmune.
L'ipogonadismo ipergonadotropo ha una frequenza del 5-10% nei maschi con pubertà ritardata e del 25% nelle femmine (1) (tabella 1).
Le cause più frequenti di pubertà ritardata, quindi, sono il CDGP e l'Ipogonadismo Ipogonadotropo. La diagnosi differenziale tra queste due condizioni non è sempre facile inizialmente; infatti, il gold standard per la diagnosi sarebbe la valutazione di un progressivo sviluppo sessuale fino ai 18 anni che si ha nel CDGP. Per cercare di ottenere una diagnosi quanto più accurata e precoce sono stati messi a punti vari test diagnostici (7).
| Tabella 1 Cause di ritardo puberale |
||
| Ipogonadismo ipergonadotropo | S. Turner S. Noonan S. X fragile Criptorchidismo Disgenesia gonadica Trauma/Torsione testicolare Chemio/Radioterapia Infezioni delle gonadi Galattosemia Difetti della steroidogenesi |
|
| Ipogonadismo ipogonadotropo | Permanente | Tumori SNC/ patologie infiltrative Cisti della tasca di Rathke S. Kallmann S. Prader-Willi S. di Bardet-Biedl Malattia di Gaucher Difetti della linea mediana Chemio/Radioterapia Trauma |
| Funzionale | Malnutrizione Anoressia nervosa/Bulimia Fibrosi Cistica Asma Patologie intestinali infiammatorie croniche Celiachia Artrite reumatoide giovanile Emosiderosi Anemia falciforme Patologie renali croniche AIDS Diabete Mellito Ipotiroidismo Iperprolattinemia Deficit di GH S. Cushing |
|
VALUTAZIONE DEL RITARDO PUBERALE
Nonostante il CDGP sia la causa più frequente di pubertà ritardata in entrambi i sessi, la diagnosi può essere fatta solo dopo aver escluso tutte le altre cause di pubertà ritardata (1). Lo scopo della valutazione iniziale è quindi quello di escludere disordini sottostanti che possano causare il ritardo puberale, come ipogonadismi ipogonadotropi o ipergonadotropi. Per questo sono molto importanti una corretta anamnesi ed un corretto esame obiettivo.
Anamnesi
È molto importante eseguire un'anamnesi accurata che vada ad indagare il periodo perinatale e l'infanzia del paziente; fondamentale è anche una corretta anamnesi per familiarità nel ritardo puberale (età di sviluppo sessuale dei genitori).
Vanno valutate anche malattie croniche con particolare attenzione per celiachia e patologie tiroidee ma non solo; si devono indagare anche patologie renali, cardiache, genitali, intestinali, respiratorie e diabete, oltre a terapie farmacologiche in atto o pregresse e stato nutrizionale.
La presenza di ritardo mentale e dismorfismi corporei deve far pensare a condizioni genetiche.
Esame obiettivo
La valutazione iniziale deve comprendere la misurazione del peso e dell'altezza del paziente, in modo da poter delineare la curva di crescita. La pubertà ritardata, infatti, è spesso associata con bassa altezza e crescita inferiore rispetto alla media per età. Durante l'adolescenza in entrambi i sessi una velocità di crescita < 3 cm/anno è suggestiva di disturbi dello sviluppo (1). Per escludere altre condizioni di bassa statura, l'altezza del paziente va confrontata anche con il target genetico (tutte le misure sono in cm):
- maschio = [(altezza paterna + altezza materna + 12.5)/2] ± 10;
- femmina = [(altezza paterna + altezza materna - 12.5)/2] ± 9.
Vanno valutati eventuali dismorfismi corporei nell'ipotesi che il ritardo di sviluppo sia associato a quadri sindromici.
Infine va valutata la scala di Tanner:
- nelle ragazze la comparsa del bottone mammario è generalmente il primo segno di pubertà;
- nel maschio segno caratteristico dello sviluppo puberale è un aumento delle dimensioni dello scroto e dei testicoli; molto importante è la misurazione del volume testicolare: un volume testicolare > 3 mL indica un inizio di pubertà centrale.
Indagini di primo livello
Permettono un inquadramento generale del paziente e consentono un'iniziale diagnosi differenziale tra le possibili cause di pubertà ritardata; permettono una valutazione dello stato nutrizionale del paziente e di eventuali patologie croniche associate alla pubertà ritardata (tabella 2).
| Tabella 2 Indagini di primo livello |
|
| Emocromo con formula leucocitaria, sideremia, ferritina, LDH, creatininemia, azotemia, VES, PCR, EMA, Ab anti-TTG, TSH, FT3, FT4 | Analisi preliminari per indagare lo stato di nutrizione del paziente e valutare la presenza di eventuali malattie croniche come celiachia e ipotiroidismo |
| Rx età ossea (Rx mano e polso di sinistra) | Ha lo scopo di determinare la maturazione dello scheletro andando a vedere le modificazioni delle cartilagini "d'accrescimento". L'Rx va poi confrontata con un atlante di riferimento come quello di Greulich e Pyle per stabilire l'età ossea del bambino. Un'età ossea in ritardo ≥ 2 anni è caratteristica ma non diagnostica di CDGP; può essere presente anche in patologie croniche, ipogonadismi ipogonadotropi o ipergonadotropi |
| Dosaggio LH | Nel CDGP e nell'ipogonadismo ipogonadotropo si riscontrano valori di LH bassi, mentre nel caso di ipogonadismo ipergonadotropo si rilevano valori elevati associati a pubertà ritardata. In generale LH è un marcatore migliore dell'inizio dello sviluppo puberale rispetto a FSH. In ogni caso la valutazione dei valori basali di gonadotropine non è utile per la diagnosi differenziale tra CDGP e ipogonadismo ipogonadotropo: infatti, nonostante siano stata riscontrati livelli di gonadotropine più bassi in pazienti con ipogodadismo ipogonadotropo rispetto a quelli di pazienti con CDGP, la differenza di valori tra i due gruppi non è molto significativa |
|
Dosaggio FSH |
Valori di FSH < 0.2 IU/L con ICMA sono suggestivi ma non diagnostici per ipogonadismo ipogonadotropo, mentre valori superiori al limite sono indice molto sensibile e specifico di insufficienza gonadica primitiva e di deficit di Inibina B |
| Dosaggio IGF-1 | Anche i livelli sierici di IGF-1 potrebbero essere utili nella valutazione di un ritardo di crescita, ma la loro interpretazione è molto più complessa, in quanto si è visto che possono essere bassi anche in condizioni di normale accrescimento |
|
Dosaggio testosterone (maschi) |
Livelli di testosterone > 20 ng/dL indicano la possibilità di un inizio di sviluppo puberale in circa 12-15 mesi |
| Ecografia pelvi (femmine) | Permette di precisare il grado di maturazione estrogenica dell'utero e valutare eventuali anomalie ovariche. I parametri ritenuti segno di estrogenizzazione sono l'aumento dei diametri longitudinale e traverso (diametro longitudinale > 3.5 cm, volume > 2 mL), la prevalenza del corpo sul collo e la presenza di una linea di vacuità endometriale. Per quanto riguarda le ovaie, sono molto importanti le dimensioni (un ovaio > 1 cm³ è molto probabilmente un ovaio stimolato), mentre non sembra rilevante la presenza di follicoli, dal momento che si possono ritrovare a tutte le età |
Indagini di secondo livello
Sono molto utili in caso di diagnosi differenziale tra ipogonadismo ipogonadotropo e CDGP e nel ricercare altre cause di pubertà ritardata (tabella 3).
Nonostante tutte queste indagini, in molti casi la diagnosi differenziale tra CDGP e ipogonadismo ipogonadotropo rimane complessa (1).
| Tabella 3 Indagini di secondo livello |
|
| GnRH test | Una risposta maggiore dell'LH rispetto all'FSH o un picco di LH tra 5-8 IU/L suggeriscono l'attivazione del sistema di controllo centrale della pubertà. In ogni caso il test presenta dei limiti dovuti al fatto che la risposta, anche in pazienti con CDGP, potrebbe essere molto bassa e paragonabile a quella del soggetto prepubere |
| Test con hCG | Il picco di testosterone è minore nei pazienti con ipogonadismo ipogonadotropo rispetto a quelli con CDGP |
| Dosaggio PRL | Per escludere adenomi ipofisari o altri tumori centrali |
| Dosaggio inibina B sierica | Maschi prepuberi con livelli di inibina B > 35 pg/mL hanno maggiori probabilità di essere affetti da CDGP. Nei maschi livelli di inibina B non misurabili sono indice di insufficienza gonadica primitiva |
| Cariotipo e altre indagini genetiche | Sia in presenza di note dismorfiche e/o di ritardo mentale associati a bassa statura o alta statura o in caso di importante obesità, nel sospetto di sindrome di Turner, di Klinefelter o di Prader-Willi |
| RMN | Per valutare la presenza di eventuali lesioni centrali |
BIBLIOGRAFIA
- Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med 2012, 366: 443-53.
- Hoffman B, Bradshaw KD. Delayed puberty and amenorrhea. Semin Reprod Med 2003, 21: 353-62.
- Busiah K, Belien V, Dallot N, et al. Diagnosis of delayed puberty. Arch Pediatr 2007, 14: 1101-10.
- Carr BR. Disorders of the ovary and female reproductive tract. Harrison's Principles of Internal Medicine 15th ed New York: McGraw-Hill 2001: 2154.
- Gajdos ZK, Henderson KD, Hirschhorn JN, Palmert MR. Genetic determinants of pubertal timing in the general population. Mol Cell Endocrinol 2010, 324: 21-9.
- Wehkalampi K, Widen E, Laine T, et al. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J Clin Endocrinol Metab 2008, 93: 723-8.
- Harrington J, Palmert MR. Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab 2012, 97: 3056-67.