Terapia delle amenorree e dei disturbi del ciclo

Costanzo Moretti & Fedra Mori
*Cattedra di Endocrinologia, Università di Torvergata, Roma
**UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
(aggiornato al 10 novembre 2015)
Il trattamento dell'amenorrea deve essere finalizzato, ove possibile, alla correzione del meccanismo che la determina e alla prevenzione delle sue complicanze.
Certamente nei casi di disgenesia gonadica con cariotipo XY è necessaria innanzitutto la rimozione chirurgica delle gonadi ritenute, mentre la creazione di una neovagina per pazienti che presentano difetti della differenziazione mulleriana viene in genere rimandata a un’età più matura, in relazione alla condizione psicologica ed alla necessità della fisioterapia post-operatoria (1).
In caso di sindrome di Asherman, è indicata la lisi delle aderenze presenti nell’utero attraverso isteroscopia operativa, seguita da un lungo periodo di terapia estrogenica.
Per quanto riguarda invece l'amenorrea che accompagna l'anoressia, ancora non esistono in letteratura chiare indicazioni ad un trattamento, rappresentato unicamente dal recupero di peso e regime alimentare adeguato. I contraccettivi orali possono indurre in queste pazienti la falsa convinzione di benessere per la ripresa del flusso mestruale, ma non si sono dimostrati utili nel prevenire la perdita di massa ossea (2), risultato che sembra invece raggiungibile con la somministrazione di estradiolo transdermico più progestinico per os (3), un protocollo che pare inoltre modificare positivamente la percezione dell'ansia in queste pazienti (4). Da ricordare, inoltre, che con il DSM V l'amenorrea finalmente non rappresenta più un criterio diagnostico per l'anoressia, facilitando quindi un approccio diagnostico e terapeutico più precoce al problema.
Nelle pazienti con disturbo alimentare, dove l’amenorrea ancora rappresenta uno dei criteri diagnostici ed è il prodotto di una serie di complesse alterazioni endocrine secondarie alla malnutrizione, non esistono al momento indicazioni terapeutiche precise. La terapia estroprogestinica non sembra in grado di prevenire/trattare la consensuale osteoporosi da cui sono affette queste pazienti e solo il recupero di una adeguata alimentazione e del peso può (ma non necessariamente) essere accompagnato dal ripristino della ciclicità mestruale (5). La somministrazione di leptina sottocute (a livello sperimentale, non esistendo un prodotto commerciale) sembra migliorare la secrezione e pulsatilità delle gonadotropine, determinando in qualche caso la ripresa della funzionalità ovarica, accompagnata purtroppo da un'ulteriore riduzione di tessuto adiposo e peso (6).
L’iperprolattinemia, in funzione della patogenesi, dovrà essere trattata (7) con farmaci dopamino-agonisti, che rappresentano il principale presidio terapeutico per l'iperprolattinemia da adenoma ipofisario, riuscendo a normalizzare i livelli ormonali nell'80-90% dei casi (8) o con la sospensione (ove possibile) delle terapie farmacologiche responsabili dell’aumentata secrezione dell’ormone. Questa stessa classe di farmaci può essere utilizzata nell'iperprolattinemia che si associa alla terapia con anti-psicotici (9), nel caso in cui non sia possibile un’interruzione o una modificazione della terapia psichiatrica.
La terapia estroprogestinica è riservata a due distinti gruppi di pazienti e con finalità completamente diverse:
- da un lato avremo pazienti con amenorrea primaria da ipogonadismo (iper o ipogonadotropo), nelle quali è necessario indurre le caratteristiche sessuali secondarie, favorire un corretto spurt puberale, supportare e mantenere una normale mineralizzazione ossea e promuovere un adeguato sviluppo psico-fisico (10);
- dall’altro, invece, avremo donne adulte con insufficienza ovarica prematura, nelle quali può essere utile prevenire gli effetti del deficit estrogenico (11).
La terapia nelle bambine con ipogonadismo deve essere iniziata non prima dei 12 anni d’età e prevede l’iniziale somministrazione di estrogeni in diverse formulazioni e vie di somministrazione (10,12). Lo schema generalmente utilizzato prevede alternativamente:
- etinilestradiolo per os 2 µg/die, da incrementare a 5 µg/die dopo 6-12 mesi;
- 17β-estradiolo per os 5 µg/kg/die, da incrementare a 10 µg/kg/die in 6-12 mesi;
- 17β-estradiolo transdermico 5 µg nelle 24 ore, da aumentare di 5 µg ogni 6 mesi;
- estrogeni coniugati 0.1625 mg/die per os per 6-12 mesi, con progressivi aggiustamenti fino a 0.325 mg/die.
La terapia ciclica (10 giorni/mese) con un progestinico va aggiunta dopo due anni dall’inizio della terapia estrogenica, per permettere un adeguato sviluppo della ghiandola mammaria e comunque appena compare il sanguinamento uterino.
Nelle donne adulte con insufficienza ovarica prematura deve essere presa in considerazione la terapia ormonale sostitutiva, poiché appare ormai evidente, in questa condizione, un aumentato rischio di osteoporosi, malattia cardiovascolare e di più rapido declino cognitivo (13).
Bibliografia
- Deans R, Creighton SM, Liao LM, et al. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): patient preferences and clinical evidence. Clin Endocrinol (Oxf) 2012, 76: 894-8.
- Bergstrom I, Crisby M, Engstrom AM, et al. Women with anorexia nervosa should not be treated with estrogen or birth control pills in a bone-sparing effect. Acta Obstetr Gynecol Scand 2013, 92: 877–80.
- Misra M, Katzman D, Miller KK, et al. Physiologic estrogen replacement increases bone density in adolescent girls with anorexia nervosa. J Bone Miner Res 2011, 26: 2430–8.
- Misra M, Katzman DK, Estella NM, et al. Impact of physiologic estrogen replacement on anxiety symptoms, body shape perception and eating attitudes in adolescent girls with anorexia nervosa: data from a randomized controlled trial. J Clin Psychiatry 2013, 74: e765-71.
- Muñoz MT, Argente J. Anorexia nervosa: hypogonadotrophic hypogonadism and bone mineral density. Horm Res 2002, 57 suppl 2: 57-62.
- Chou SH, Chamberland JP, Liu X, et al. Leptin is an effective treatment for hypothalamic amenorrhea. Proc Natl Acad Sci USA 2011, 108: 6585-90.
- Melmed S, Casanueva FF, Hoffman AR, et al; Endocrine Society. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 273-88.
- Glezer A, Bronstein MD. Prolactinomas. Endocrinol Metab Clin North Amer 2015, 44: 71–8.
- Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs 2014, 28: 421–53.
- Heinz M. Hormonal development therapy (HDT) in hypogonadism in long-term view. Best Pract Res Clin Obstet Gynaecol 2010, 24: 149-55.
- Viswanathan V, Eugster EA. Etiology and treatment of hypogonadism in adolescents. Pediatr Clin North Am 2011, 58: 1181-200.
- Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med 2012, 366: 443-53.
- Vujovic S, Brincat M, Erel T, et al; European Menopause and Andropause Society. EMAS position statement: Managing women with premature ovarian failure. Maturitas 2010, 67: 91-3. Erratum in: Maturitas 2011, 69: e4.
Classificazione ed eziopatogenesi degli ipogonadismi femminili
Roberto Mioni1 e Vincenzo Toscano2
1 Clinica Medica 3, Dipartimento di Medicina, Università di Padova, Azienda Ospedaliera di Padova
2DAI Scienze Mediche, Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea
L’ipogonadismo femminile può essere definito come un quadro clinico caratterizzato da un’alterazione, o disfunzione, dell’asse ipotalamo–ipofiso-ovarico con conseguente riduzione, talvolta assenza, dei livelli circolanti di estrogeni e una riduzione della frequenza e, nei casi più conclamati, assenza o scomparsa dei cicli mestruali.
Il sistema ipotalamo-ipofiso-ovarico, dopo un breve e limitato periodo di attivazione post-natale, rimane quiescente fino al periodo puberale. Il meccanismo con cui tale asse viene attivato in prossimità della pubertà rimane ancora in discussione e nell’ultimo decennio sono stati dimostrati essere coinvolti più sistemi, sia centrali che periferici.
Il patologico coinvolgimento di uno dei sistemi di regolazione HPG (ipotalamo-ipofisi-gonadi) è caratterizzato da un quadro ormonale e clinico solitamente ben definito, sebbene non specifico. È intuitivo che nella maggior parte dei casi il sintomo più facilmente riconoscibile di ipogonadismo nel sesso femminile rimane l’amenorrea, ad eccezione di tipiche stigmate in peculiari malattie a carattere genetico. Questa si definisce come assenza di ciclo mestruale, o di un’adeguata attività endometriale, in un soggetto di sesso femminile oltre l’età media di 9.8 anni (1). Viene distinta in:
- amenorrea primaria se viene diagnosticata in una paziente che non ha mai presentato flusso mestruale fino all’età di 16 anni, se sono presenti i caratteri sessuali secondari, e di 14 anni, se questi ultimi non sono rilevabili. In considerazione dei suggerimenti dell’American Society for Reproductive Medicine (ASRM) (1) e delle epoche relative di menarca e sviluppo mammario (solitamente il telarca, precede il menarca di circa 2.5 anni e lo stato di maturazione della ghiandola mammaria è solitamente allo stadio IV di Tanner in coincidenza del menarca), le caratteristiche cliniche del soggetto in esame modificano l’età oltre la quale è consigliato eseguire accertamenti:
- 13 anni (2 SD dall’età media di 11 anni) se il soggetto non presenta comparsa di caratteri sessuali secondari (adrenarca, telarca e pubarca), oppure non ha menarca dopo almeno 5 anni dal solo telarca (avvenuto prima dei 10 anni);
- 15 anni (2 SD dall’età di 13 anni) in presenza di caratteri sessuali secondari;
- amenorrea secondaria se l’arresto del flusso mestruale, o dell’attività endometriale, si registra dopo la comparsa del normale menarca. Tuttavia, anche in questo caso devono essere considerati criteri temporali differenti a seconda delle caratteristiche del flusso mestruale quando era presente:
- 6 mesi nei soggetti in cui non sono ancora trascorsi 2 anni dal menarca o che abbiano presentato da sempre un flusso mestruale caratterizzato da estrema irregolarità (45-60 giorni o oltre);
- 3 mesi in quei soggetti che avevano presentato flussi mestruali con intervalli regolari (21-35 giorni).
Sebbene le alterazioni del ciclo mestruale e quindi l’oligo- e/o amenorrea si possano considerare il sintomo più frequente del quadro di ipogonadismo femminile, tale da suggerirne l’utilizzo per una classificazione funzionale di tale patologia, rimane importante ricordare che la classificazione degli ipogonadismi avviene tuttora in:
- ipogonadismo primario da coinvolgimento, o prevalente coinvolgimento, gonadico (caratterizzato verosimilmente da un ipogonadismo ipergonadotropo);
- ipogonadismo secondario da coinvolgimento, o prevalente coinvolgimento, extra-gonadico (caratterizzato da un quadro di ipogonadismo ipogonadotropo).
Sebbene l’utilizzo di recenti metodiche di sequenziamento genico stia permettendo il riconoscimento di patologie precedentemente definite idiopatiche, suggerendone l’inclusione in un gruppo ad eziologia genetica, le forme primitive e secondarie vengono ulteriormente suddivise in idiopatiche, acquisite (secondarie a processi e/o patologie infiammatorie locali e/o sistemiche, autoimmuni, proliferative, iatrogene, post-attiniche, traumatiche etc.), funzionali e multifattoriali. Gli autori nel presente capitolo suddivideranno le cause di ipogonadismo femminile seguendo un ordine a “cascata” secondo l’asse ipotalamo-ipofiso-gonadico.
| Tabella 1 Classificazione degli ipogonadismi femminili |
|||
| Primario | Ovarico | Genetico | Sindrome di Turner Difetto di 17alfa-idrossilasi |
| Acquisito | Ovarite:
Menopausa precoce |
||
| Secondario | Ipogonadismo ipogonadotropo da cause ipotalamiche | Genetiche | S. di Kalmann S. di Laurence–Moon-Biedl S. di Prader-Willi |
| Acquisite |
|
||
| Ipogonadismo ipogonadotropo da cause ipofisarie | Genetiche | Prop-1 HESX1/Rpx LHX3 SOX3 FSH-ß |
|
| Tumori o masse | Adenomi Craniofaringiomi Meningiomi Cordomi Gliomi Germinomi Metastasi |
||
| Cause infiammatorie-infiltrative | Sarcoidosi Malattia a cellule di Langerhans Emocromatosi Ipofisite Ascessi |
||
| Emorragia/ischemia | Apoplessia | ||
| Post-radioterapia | |||
| Post-trauma | |||
| Empty sella | |||
IPOGONADISMI IPERGONADOTROPI vedi capitolo Amenorree
IPOGONADISMO IPOGONADOTROPO DA CAUSA IPOTALAMICA
Può essere definito come una forma di ipogonadismo femminile che coinvolge le strutture ipotalamiche, caratterizzato da una secrezione inadeguatamente bassa di gonadotropine (LH e/o FSH o di entrambe) rispetto ai bassi valori di ormoni steroidei ovarici (ipoestrogenismo). Questo tipo di ipogonadismo è seguito a sua volta da alterazioni funzionali del ciclo mestruale, che talvolta si può accompagnare, nei casi più conclamati, all’assenza di ciclo mestruale spontaneo (amenorrea primaria). Può essere, infatti, caratterizzato dalla mancanza dei caratteri sessuali secondari (se coinvolge la struttura ipotalamica prima dell’adrenarca, pubarca o telarca), mentre può accompagnarsi alla scomparsa del ciclo mestruale, se coinvolge la struttura ipotalamica dopo la pubertà, con regressione dei caratteri sessuali secondari (ipotrofia mammaria, della mucosa vaginale, muscolare, variazione del tono dell’umore, ecc) (2,3).
Ipogonadismo ipogonadotropo idiopatico (vedi anche ipopituitarismo)
Questo tipo di ipogonadismo viene definito anche congenito o non sindromico ed è caratterizzato prevalentemente da un difetto isolato di secrezione, funzione o sensibilità del GnRH. Risulta solitamente accompagnato da ridotti livelli sierici di LH e/o FSH, dalla mancata comparsa della pubertà, dall’assenza dei caratteri sessuali secondari e da una conseguente anomala funzione dell’asse riproduttivo (2). Tali pazienti, nella maggior parte dei casi, non hanno alterazioni staturali, sebbene assumano per la maggior parte un fenotipo enucoide da ipogonadismo, e non presentano il coinvolgimento di altri assi ipofisari. L’incidenza dell’ipogonadismo ipogonadotropo idiopatico (III) può essere considerata di 5-10 nuovi casi su 106 nati e circa il 30% di questi soggetti possono considerarsi da causa genetica nota. A loro volta il 60% dei soggetti affetti da III sono caratterizzati da anosmia (IIIa) e si raccolgono nella Sindrome di Kalmann (KS), patologia caratterizzata da assenza completa o parziale dell’olfatto. Di questi soggetti circa il 15-18% sono portatori di mutazione genetica a carico di alcuni geni responsabili della migrazione delle cellule neuronali del GnRH, come il gene Kalmann 1 (KAL1; KS forma classica), il gene codificante il Fibroblast Growth Factor Receptor-1 (FGF-R1) (4). Tali geni risultano entrambi co-localizzati a livello del bulbo olfattivo e sono i più conosciuti responsabili per la guida assonale nel percorso migratorio delle cellule neuronali del GnRH.
Recentemente si sono riconosciute altre mutazioni, collegate sempre ad anomalie del neurone GnRH, a livello di altri geni responsabili delle codifiche di G-protein, come PROK2 e PROK2-Receptor, che hanno permesso di aggiungere un altro 8-12% ai casi sostenuti da mutazioni genetiche riconosciute.
Il rimanente 40% dei soggetti affetti da III è caratterizzato dalla persistenza della capacità olfattiva, almeno parziale, per cui sono definiti normosmici (IIIn). Il sequenziamento genico ha permesso di identificare alcuni geni coinvolti nella modulazione/espressione della capacità secretiva del GnRH, come i geni codificanti la kisspeptina1 (KISS1) o il suo recettore (KISS1-R), la leptina (LEP) o il suo recettore (LEP-R), la neurokinina B (NKB), codificata da TAC3, appartenente alla famiglia delle tachikinine, o il suo recettore (TAC-R3). Tali soggetti, a seconda della gravità della mutazione o dell’associazione con più mutazioni, possono manifestare una differente alterazione del sistema di regolazione GnRH-dipendente, per cui possono presentarsi con un quadro clinico eterogeneo che può essere compatibile anche con il normale sviluppo dei caratteri sessuali secondari e talvolta della fertilità (4-6).
Le cause genetiche più studiate di III sia anosmico (KS) che normosmico sono riassunte in tabella 2, considerando tuttavia che l’elenco dei geni coinvolti è destinato ad aumentare. È anche da precisare che la frequenza di tali patologie è da 2 a 5 volte inferiore nel sesso femminile rispetto a quello maschile (7).
| Tabella 2 Genetica dell’ipogonadismo ipogonadotropo isolato (da Molecular and Cellular Endocrinology 2010, 324: 30-8) |
|||||
| Gene | Locus | Prodotto del gene | Funzione nell’asse riproduttivo | Trasmissione | Fenotipo |
| Kal 1 | Xp22.3 | Anosmin-1 | Migrazione neuroni GnRH | Legata a X | s. di Kallmann |
| FGF8 FGFR1 |
10q25 8p12 |
FGF-8 e suo recettore | Migrazione neuroni GnRH | AD | s. di Kallmann o o ipogonadismo ipogonadotropo normo-osmico |
| NELF | 9q34.3 | Fattore LHRH nasale embrionario | Migrazione neuroni GnRH | AD (?) | s. di Kallmann |
| PROK2 PROKR2 |
3p21.1 20p12.3 |
Prochineticina-2 e suo recettore | Migrazione neuroni GnRH | AR | s. di Kallmann o o ipogonadismo ipogonadotropo normo-osmico |
| CHD7 | 8q12.1 | Cromodominio elicasi proteina legante DNA | Sviluppo di neuroni GnRH | AD | s. CHARGE, s. di Kallmann o o ipogonadismo ipogonadotropo normo-osmico |
| GnRH GnRH-R |
8p21 4q13.2-3 |
GnRH e suo recettore | Sintesi e segnale GnRH | AR | Ipogonadismo ipogonadotropo normo-osmico |
| KISS1-R | 19p13.3 | Recettore per kisspeptin | Controllo secrezione del GnRH | AR | Ipogonadismo ipogonadotropo normo-osmico |
| TAC3 TAC3-R |
12q13-12 4q25 |
Neurokinina B e suo recettore | Sconosciuta | AR | Ipogonadismo ipogonadotropo normo-osmico |
Sindrome di Kalmann. La tipizzazione del gene KAL1 è avvenuta circa 20 anni fa, mentre negli ultimi 6 anni si sono scoperti altri 5 geni (tab 2), che corrispondono ad altrettante 5 differenti sindromi distinte e numerate in successione (KS da 2 a 6). La sindrome classica (KS1) è sostenuta dalla mutazione/delezione del gene KAL1 contenuto nel cromosoma X, come carattere recessivo, preposto alla codifica dell’anosmina-1, proteina responsabile, nel periodo fetale, della migrazione dei neuroni GnRH dal placode olfattorio all’ipotalamo con la formazione del nucleo “pulse generator” ed anche della formazione del bulbo olfattivo. In circa il 7-10% dei casi si è osservato anche il coinvolgimento del gene FGF-R1, co-espresso nel placode o nel bulbo olfattorio, assieme al KAL1, come espressione monoallelica. Tuttavia, a differenza della forma classica, il differente grado di coinvolgimento dei vari geni riassunti in tabella 2 potrebbe permettere una differente espressione fenotipica, caratterizzata a sua volta da varie espressioni di patologico sviluppo puberale, non escludendo anche la forma fertile. La caratteristica principale della KS classica è l’anosmia, che potrebbe orientare nella diagnosi nel periodo infantile/prepuberale (ad eccezione di casi già noti nel nucleo familiare), sebbene la maggior parte delle diagnosi avvenga nel periodo adolescenziale, soprattutto nelle femmine. Infatti, per il sesso femminile la presenza di amenorrea rappresenta la principale sintomatologia (circa il 90% delle pazienti), accompagnata o meno dallo sviluppo incompleto/assente dei caratteri sessuali secondari. Nella KS, oltre alle note malformazioni a livello cerebrale e cerebellare, si riscontrano malformazioni renali, fino all’agenesia, oculari, del condotto auditivo e cardiache. Tra le forme di KS viene ricordata il tipo 5 (KS 5), dovuto a mutazione del gene chromodomain-helicase-DNA-binding-protein 7 (CHD7), responsabile della sintesi di una proteina localizzata sia a livello del bulbo olfattorio, che nei nuclei produttori di GnRH. Tale proteina sembra coinvolta nella modulazione del sistema di migrazione del neurone GnRH. Questa alterazione genica sostiene la sindrome CHARGE, che rappresenta l’acronimo di Coloboma (dell’iride o della retina), Heart defects (tetralogia di Fallot, pervietà forame ovale, anomalie della valvola aortica), Atresia of Choanae, Retardation of Growth (ritardo di crescita, deficit GH, palatoschisi, fistola esofago-tracheale) and development, Genital anomaly (ipoplasia delle grandi labbra, micropene o criptorchidismo nel maschio) and Ear abnormality (anomalie dell’orecchio esterno, otiti medie ricorrenti, malformazione dei piccoli ossicini, sordità): ha incidenza di 1 su 105 nati ed un numero complessivo di circa 400 soggetti nel nord Europa (4,5).
Sindrome di Laurence–Moon-Biedl. Rappresenta un raro disordine autosomico recessivo, caratterizzato da ritardo mentale, obesità e sindattilia, esadattilia o brachidattilia. Altra caratteristica di tali soggetti è l’alterazione retinica, che vede oltre il 70% dei soggetti divenire ciechi entro la terza decade. In tale quadro sindromico si osserva nel 75% dei casi deficit di secrezione di GnRH, con un quadro di ipogonadismo ipogonadotropo.
Sindrome di Prader-Willi. Alterazione dovuta a delezione del cromosoma 15q, caratterizzata da obesità, iperfagia per alterato controllo dei centri ipotalamici della fame, ipogonadismo da ridotta secrezione di GnRH, ritardo mentale e diabete mellito nell’adulto. Tale disendocrinia si associa inoltre a difetti oculari, scheletrici, auricolari, talvolta associati ad una ridotta funzionalità neuroipofisaria (ADH ed ossitocina), con un quadro spesso di disregolazione del sistema elettrolitico.
Ipogonadismo ipogonadotropo acquisito
Le forme di ipogonadismo ipogonadotropo secondario o acquisito sono caratterizzate da bassi livelli di gonadotropine con livelli di estrogeni ridotti o indosabili. Sono dovute a quadri patologici secondari a patologie espansive, infiammatorie, traumatiche e/o a percorsi terapeutici utilizzati per la cura delle suddette patologie. A differenze delle forme idiopatiche, spesso caratterizzate dal coinvolgimento di un solo asse ormonale, queste possono presentare il contemporaneo coinvolgimento di più sistemi ormonali (oltre alle gonadotropine, anche GH, ACTH, TSH) e/o talvolta da tutti quanti assieme (panipopituitarismo) (8).
Tumori ipotalamici. Sono la causa più frequente in età pediatrica. Solitamente si accompagnano a un differente e/o progressivo interessamento degli assi ormonali: dapprima l’asse somatotropo (con GH deficit), poi quello gonadotropo (LH > di FSH) e infine gli altri.
- Craniofaringioma
- Cisti della tasca di Rathke
- Amartomi e gangliocitomi ipotalamici rappresentano una forma discretamente rara, ma possono essere causa anche di produzione di neuropeptidi (anche GnRH-like) che danno luogo a pubertà precoce. Gli amartomi ipotalamici si sviluppano frequentemente in contiguità con la struttura ipofisaria, rendendone spesso difficile la diagnosi differenziale con un adenoma ipofisario (in alcuni casi solo il riscontro istologico di neuroni ipotalamici rende certa la diagnosi differenziale con gli adenomi ipofisari). Esiste una rara associazione con alterazioni cranio-facciali, ano imperforato, anomalie renali, cardiache e polmonari con ipogonadismo e più raramente con panipopituitarismo che caratterizza la sindrome di Pallister-Hall.
- Altri tumori cerebrali: glioblastomi, gliomi, disgerminomi e teratomi.
- Metastasi cerebrali: possono considerarsi una causa rara di ipogonadismo ipogonadotropo ipotalamico (molto più frequenti sono quelle a livello ipofisario), a partenza da tumori di mammella, polmone, intestino, rene.
Cause infiammatorie/infiltrative. Sono caratterizzate dal coinvolgimento delle strutture ipotalamiche da parte di fenomeni infiammatori di tipo vasculitico o degenerativo/tesaurismosico: granulomatosi di Wegener, sarcoidosi, istiocitosi X o malattia a cellule di Langerhans e tubercolosi.
Cause iatrogene
- Radioterapia per patologie oncologiche cerebrali e/o ipofisarie. La maggiore radiosensibilità delle cellule neuronali ipotalamiche, soprattutto dei neuroni GnRH, rispetto a quelle ipofisarie, giustifica il più frequente coinvolgimento di tale distretto.
- Farmaci. Numerosi farmaci sono in grado di modulare i sistemi ipotalamici di neuroregolazione, come il sistema dopaminergico, noradrenergico, serotoninergico, oppiatergico, gabaergico, ecc, con conseguente disregolazione del sistema ipofisario. Oltre a tutti quelli che interferiscono con il sistema della dopamina e della serotonina (vedi capitolo PRL), sono da segnalare:
- estro-progestinici: l’amenorrea secondaria dopo la sospensione si verifica maggiormente dopo terapie prolungate per oltre 2 anni; il meccanismo è probabilmente la desensibilizzazione del pulse generator del GnRH, ma non vengono esclusi fenomeni ovarici diretti (“torpore ovarico” da insensibilità alle gonadotropine);
- danazolo: agirebbe sia a livello endometriale, inibendo l’attività proliferativa endometriale, sia a livello ipotalamo-ipofisario con attività anti-gonadotropinica (recettoriale?);
- cortisonici: terapie steroidee di lunga durata per patologie infiammatorie croniche (artrite reumatoide, LES, ecc.) esercitano un effetto inibitorio sulla funzionalità ipotalamica.
Ipogonadismo ipogonadotropo funzionale
Rappresenta una forma non organica, spesso reversibile, osservato in soggetti, per lo più molto giovani (seconda, terza decade) con peso corporeo nei limiti di norma (frequentemente appena entro il limite inferiore della norma), ove si siano escluse cause organiche, in cui il sistema ipotalamico preposto al controllo della secrezione del GnRH viene “alterato”. Frequentemente ai bassi livelli di LH, la tropina maggiormente compromessa, e di FSH si associano livelli bassi e/o quasi indeterminabili di 17ß-E2, ma con conservata capacità di risposta ipofisaria al GnRH test. Le alterazioni della secrezione dell’LH in tale forma di ipogonadismo possono variare a seconda dello stato di compromissione del sistema di disregolazione ipotalamico, che a sua volta risulta influenzato principalmente da uno stile di vita non appropriato. Quest’ultimo, infatti, è caratterizzato da eventi stressogeni, come alcune tipologie di lavoro (responsabilità, o che implichino un rischio per lo più emozionale, ecc.), dall’eccessivo controllo alimentare (più frequentemente per la mancata assunzione di carboidrati), dall’eccessiva attività fisica. I meccanismi coinvolti sono ancora discussi, anche se più osservazioni hanno dimostrato il coinvolgimento di più neuro-ormoni, come la leptina (ridotta rispetto alle donne normali e con peso confrontabile), la ghrelina (aumentata), l’allopregnenolone (uno dei più importanti neuro-modulatori femminili legato all’attività steroidea), il sistema oppioide endogeno (attivato con effetto inibitorio sull’attività pulsatile del GnRH ) o NPY. Anche la secrezione del CRH sembra aumentata, con effetto inibitorio secondario sui neuroni ipotalamici GnRH. In alcuni soggetti si sono poi identificati polimorfismi degli stessi geni coinvolti nell’ipogonadismo ipogonadotropo idiopatico normosmico (FGFR1, PROKR2, GnRH-R) (9,10).
Ipogonadismo ipogonadotropo da altre cause
Iperprolattinemie. La PRL a livello ipotalamico inibisce la secrezione dei neuroni GnRH, con meccanismo diretto mediante l’attivazione di recettori specifici o con effetto indiretto, sul turn-over della dopamina (DA). L’aumentato tono dopaminergico prolattina-indotto esercita a sua volta un effetto inibitorio sia a livello ipotalamico (GnRH pulse generator) con riduzione dei picchi, sia a livello ipofisario, frenando secrezione e dismissione di LH. Un altro sistema coinvolto sembra essere quello oppioide endogeno che, stimolato dalla PRL, esercita un ruolo inibitorio/ modulatorio sia ipotalamico (riduce i picchi di GnRH) che ipofisario (riduce secrezione di LH).
Ipercortisolismo. Gli elevati livelli di cortisolo sono in grado, agendo direttamente a livello ipotalamico, di inibire i neuroni GnRH-secernenti, creando una conseguente riduzione della secrezione delle gonadotropine, in particolare dell’LH.
Distiroidismi. Alterati livelli degli ormoni tiroidei possono essere accompagnati da riduzione o blocco dell’attività gonadotropinica ipotalamica. Infatti, nell’ipotiroidismo franco (TSH > 30 mUI/L) si osserva un rallentamento dell’attività pulsatile del GnRH ipotalamico fino al meno frequente blocco (negli ipotiroidismo gravi) con conseguente amenorrea. Diversamente negli ipertiroidismi (TSH < 0.02 mUI/L) si osserva un accorciamento dell’attività ovulatoria, fino alla completa anovulazione. Non è ancora chiarito se in tale quadro ci sia un diretto interessamento dell’attività ipotalamica GnRH-dipendente (3).
IPOGONADISMO IPOGONADOTROPO DA CAUSA IPOFISARIA
La struttura ipofisaria anteriore può essere considerata per cause anatomo-funzionali come all’esterno della barriera emato-encefalica, sebbene ne sia in stretto contatto, e pertanto non protetta dall’effetto farmacologico sistemico di eventuali farmaci distribuiti direttamente dal torrente circolatorio. Anche per gli ipogonadismi ipofisari si riconoscono cause malformativo-idiopatiche (genetiche), tumorali, infiltrativo/infiammatorie, vascolari (ischemico o emorragico), tra le quali possono essere anche incluse le cause post-traumatiche. Vista la distribuzione anatomica topografica dei complessi cellulari ipofisari, che compongono i vari distretti funzionali della stessa ipofisi, si può comprendere come la presenza di una noxa patogena possa corrispondere a elettivi deficit funzionali. Tuttavia, l’effetto funzionale non è sempre coerente con il danno topografico, per le caratteristiche biologiche delle diverse tipologie cellulari ipofisarie. L’incidenza di disfunzione/ipofunzione ipofisaria si assesta tra i 12-45 nuovi casi/anno/106 abitanti, mentre la prevalenza di ipogonadismo da causa ipofisaria si attesta su valori di 300-460 casi/106 di abitanti. Di questi pazienti circa il 43-48% presenta almeno 2-3 sistemi ormonali coinvolti (GH, LH/FSH, ADH) e 3-6% presenta panipopituitarismo (11).
Ipogonadismo ipofisario da causa genetica
Sono forme estremamente rare, spesso caratterizzate dalla compromissione di più settori ipofisari con conseguente panipopituitarismo.
Gene Prop-1: regola la trascrizione di alcune proteine indispensabili per la differenziazione delle cellule ipofisarie anteriori, soprattutto GH, PRL, TSH, LH/FSH e parzialmente ACTH, per cui la mutazione di tale gene si associa ad un deficit ipofisario quasi globale e con ipogonadismo.
Gene HESX1/Rpx: localizzato nel cromosoma 3p212, rappresenta il più precoce elemento trascrizionale per lo sviluppo della ghiandola ipofisaria, la cui proteina interagisce con la proteina del gene Prop-1 per la codifica del DNA. La mutazione è collegata ad una sindrome con panipopituitarismo e displasia setto-ottica, agenesia del corpo calloso ed assenza del setto pellucido (sindrome di De Morsier).
Gene LHX3: gene autosomico recessivo, membro della famiglia di proteine LIM. La sua mutazione è caratterizzata dall’arresto della formazione della tasca di Rathke, con ipoplasia ipofisaria e frequente disfunzione degli assi GH, PRL, TSH e delle gonadotropine.
Gene SOX3:legato al cromosoma X, le mutazioni sono collegate a infertilità per ipopituitarismo e ritardo mentale.
Gene FSHß: gene autosomico recessivo, la cui mutazione sembra essere collegata ad anomala funzionalità del FSH, che non risulta in grado di stimolare il recettore a livello ovarico, per cui nelle forme più gravi si ha amenorrea primitiva (12).
Ipogonadismo ipofisario da tumori o masse
Rappresenta la forma più comune di ipogonadismo da compromissione ipofisaria ed il meccanismo con cui si instaura il danno della funzionalità ipofisaria si può riassumere in un danno:
- meccanico: solitamente associato a macroadenomi ipofisari che comprimono il peduncolo creando una sorta di ischemia meccanica. Possono inoltre essere causa di lesione meccanica altre forme tumorali primitive (craniofaringiomi, meningiomi, cordomi, glioma del nervo ottico e germinomi) e metastatiche (da carcinoma della mammella, polmone, colon e rene);
- endocrino: indotto dal blocco/ inibizione dell’attività ipotalamica per effetto degli elevati livelli di ormoni secreti dall’adenoma stesso: PRLomi (circa il 30%), ACTHomi (circa il 15%), forme a secrezione mista (la maggior parte PRL/GH nel 13%) e gonadotropinomi (generalmente LH) nell’8% (13).
Ipogonadismo ipofisario da infiltrazione/infezione
Come per le strutture ipotalamiche, anche quelle ipofisarie possono essere interessate da patologie infiltrative o granulomatose, come la sarcoidosi o l’istiocitosi-X. Da ricordare che nella forma ipofisaria le forme infiltrative coinvolgono prevalentemente il peduncolo con danno da “disconnessione ipofisaria”, con possibile ipofunzione del sistema GH, LH/FSH, TSH ed ACTH di grado variabile a seconda dell’entità dell’interessamento istologico fino alla completa disconnessione, diabete insipido e iperPRL. L’emocromatosi, caratterizzata dal deposito di ferro nel tessuto cellulare ipofisario, rappresenta una causa relativamente frequente di ipogonadismo da compromissione ipofisaria con un progressivo deficit prevalentemente gonadotropinico (LH > FSH). Sono inoltre da tenere in considerazione ipofisite linfocitica e ascessi.
Ipogonadismo ipofisario da emorragia/ischemia
Apoplessia ipofisaria.
Apoplessia post-partum (sindrome di Sheehan) può essere considerata come una complicanza post-partum per eventi emorragici durante le manovre ostetrico-ginecologiche o per complicanze da ipovolemia (shock ipovolemico). Tale evento sembra una conseguenza della fisiologica ipertrofia ipofisaria gravidica che espone la ghiandola ad un’aumentata sensibilità per gli eventi ipossico-ischemici. Nelle forme più gravi si può osservare un quadro sovrapponibile all’apoplessia ipofisaria, con un aumento della mortalità post-partum, ma le forme più frequenti si accompagnano a deficit parziali (LH/FSH, PRL) che possono giustificare la perdita o l’incapacità dell’allattamento e la successiva amenorrea secondaria. La cefalea di nuova insorgenza, di lieve entità, frontale e/o fronto-temporale, rappresenta un sintomo registrato da oltre il 60% delle pazienti con s. di Sheehan, che nella maggior parte dei casi rimane misconosciuta.
Ipogonadismo post-radioterapico
Può essere considerata la maggior complicanza della radioterapia per patologia oncologica cerebrale e/o ipofisaria.
Ipogonadismo ipofisario post-traumatico
Evento sottostimato fino ad alcuni anni fa, per il quale le pazienti in amenorrea secondaria venivano erroneamente incluse tra le cause ipotalamo-ipofisarie idiopatiche. Emergeva successivamente che tali pazienti erano state esposte ad eventi traumatici cranici (incidenti stradali, cadute accidentali, aggressioni, ecc.). Tale forma è caratterizzata da un deficit ipofisario di vario grado, correlato temporalmente ad un evento traumatico cranico la cui gravità, considerando la scala secondo Glasgow, non risulta tuttavia essere in diretta correlazione con il grado del danno funzionale ipofisario. Si è potuto osservare che nei soggetti dopo un traumatismo cranico circa il 25-30% presenta, dopo 3 mesi dall’evento, un deficit funzionale ipofisario (a carico di GH per il 6-33%, LH/FSH per il 5-20%, ACTH per il 2-16%, TSH per l’1-10% e multiplo per il 4-12%) (14,15).
Sindrome da “Sella Vuota”
Per approfondimenti bibliografici
- The Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea. Fertil Steril 2008, 90 (Suppl 3): S219–25.
- Santoro N. Update in hyper- and hypogonadotropic amenorrhea. J Clin Endocrinol Metab 2011, 96: 3281–8.
- Rothman MS, Wierman ME. Female hypogonadism: evaluation of the hypothalamic–pituitary–ovarian axis. Pituitary 2008, 11: 163–9.
- Semple RK, Topaloglu AKl. The recent genetics of hypogonadotrophic hypogonadism – novel insights and new questions. Clin Endocrinol 2010, 72, 427–35.
- Plant TM. Hypothalamic control of the pituitary-gonadal axis in higher primates: key advances over the last two decades. J Neuroendocrinol 2008, 20, 719–26.
- Ahima RS. No Kiss1ng by leptin during puberty? J Clin Invest 2011, 121: 34–6.
- Bianco DCS, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol 2009, 5: 569–76.
- Brioude F, Bouligand J, Trabado S, et al. Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotype–phenotype relationships. Eur J Endocrinol 2010, 162: 835–51.
- Caronia LM, Martin C, Welt CK, et al. A genetic basis for functional hypothalamic amenorrhea. N Engl J Med 2011, 364: 215–25.
- Miller KK. Endocrine dysregulation in anorexia nervosa update. J Clin Endocrinol Metab 2011, 96: 2939–49.
- McGregor AM. Diagnosis of pituitary disease. Medicine 2005, 37: 393-8.
- Merriam GR Koenig Stress JI. Hypogonadism: not everything that suppresses must converge. Endocrinology 2011, 152: 340–2.
- Golkowski F, Trofimiuk M, Czepko R, et al. Two rare cases of pituitary metastases from breast and kidney cancers. Exp Clin Endocrinol Diabetes 2007, 115: 537–40.
- Tsimaris P, Vrachnis N, Iliodromiti Z, et al. Long-term follow up of adolescent and young adult females with hypergonadotropic hypogonadism. Int J Endocrinol 2012, 10: 1-5.
- Unuane D, Tournaye H, Velkeniers B, Poppe K. Endocrine disorders & female infertility. Best Pract Res Clin Endocrinol Metab 2011, 25: 861–73.
Percorso diagnostico dell'ipogonadismo femminile
Roberto Mioni1 e Vincenzo Toscano2
1 Clinica Medica 3, Dipartimento di Medicina, Università di Padova, Azienda Ospedaliera di Padova
2DAI Scienze Mediche, Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea
I soggetti affetti da ipogonadismo ipogonadotropo, ad eccezione di casi già noti in famiglia o di dismorfismi particolari, generalmente consultano un medico, solitamente il MMG e/o il ginecologo, per la mancanza di caratteri sessuali secondari, nelle forme più complesse, o per l’amenorrea all’età di 12-13 anni.
In presenza della paziente, risultano fondamentali l’adeguata raccolta anamnestica, soprattutto familiare per patologie prepuberali, e uno scrupoloso esame obiettivo. Solitamente negli ipogonadismi ipogonadotropi ipotalamici non si hanno deficit e/o compromissione della funzionalità dell’asse somatotropo e surrenalico, quindi non si osservano difetti di crescita, sebbene manchi solitamente lo spurt gonadale. La relativa e/o totale mancanza di steroidi gonadici non permette il raggiungimento di una struttura ossea epifisaria completa, per cui l’aspetto della paziente può richiamare quello tipico da ipogonadismo (arti di lunghezza maggiore rispetto all’altezza). Deve essere valutato il grado di obesità, quando presente, con l’indice di massa corporea (BMI > 30), escludendo eventualmente le magrezze (BMI < 18). Bisogna eseguire una valutazione diretta dei genitali esterni e quando possibile anche di quelli interni (visita trans-rettale). Importante, se presenti, stabilire il grado di sviluppo adrenarcale, telarcale (BI-V) e puberale (PI-V). Da ricordare che il menarca di solito si accompagna a un telarca di stadio BIV secondo Tanner. Inoltre la presenza di adrenarca spesso si accompagna anche ad iniziale telarca (non oltre BIII), per la presenza di estrogeni provenienti dalla conversione periferica degli androgeni surrenalici. È comunque da tenere in considerazione che le forme idiopatiche a carattere genetico, sia ipotalamico che ipofisario, possono presentare vari gradi di compromissione funzionale, potendo permettere anche il raggiungimento di un livello estrogenico sufficiente all’attivazione endometriale, sebbene quasi sempre non accompagnata da attività ovulatoria (1,2).
Vanno ovviamente ricercati ed esclusi i dismorfismi e/o le anomalie oculari, auricolari, del palato duro, la presenza e/o assenza di falangi (esadattilia o brachidattilia). Deve essere valutata la capacità olfattiva nel riconoscere gli odori (in ambulatorio: caffè, ammoniaca). Nel sospetto si dovrà inviare la paziente ai colleghi ORL per una valutazione olfattometrica formale (eseguibile in pochi centri).
Diagnostica bioumorale
Esami di I livello. Importante è la determinazione dei livelli plasmatici di LH, FSH, prolattina, TSH, 17-β-estradiolo. Nell’ipogonadismo ipogonadotropo, i risultati mostreranno livelli plasmatici bassi di LH ed FSH, talvolta al limite della determinazione del sistema di rilevamento, con livelli di estrogeni bassi e/o molto bassi. Devono essere inoltre esclusi elevati livelli di PRL (se > 200 ng/mL c’è oltre il 90% di possibilità di prolattinoma) ed alterati livelli di funzionalità tiroidea (solitamente gli ipertiroidismi sono accompagnati da una clinica più eclatante) per gli effetti negativi che tali condizioni esercitano sulla funzionalità ipotalamica.
La diagnosi differenziale avviene nei confronti della pubertà ritardata, sebbene questa non risulti facile esecuzione soprattutto quando il ritardo puberale si protrae oltre i 18-20 anni. Alcuni autori individuano la ricerca dei picchi notturni di LH come strumento di utilità clinica per la suddetta diagnosi differenziale, risultando presenti nella fase di pre-attivazione dell’asse ipotalamo-ipofisario nella pubertà ritardata. Nella realtà clinica questo è un esame che ben di rado può essere eseguito (1).
Esami di II livello. Possono essere considerati i test dinamici, come il test con GnRH, che tuttavia non si è dimostrato dirimente, soprattutto nelle forme idiopatiche a espressione parziale. La maggior parte degli autori concorda con la ridotta utilità diagnostica di tale test dinamico per la mancata possibilità di correlare il grado di difetto genetico al grado di difetto funzionale sia ipotalamico che ipofisario, sebbene si sia osservata una correlazione tra entità di risposta allo stimolo e gravità di alterazione morfologica. Neppure l’introduzione del test con l’analogo del GnRH, circa 100 volte più potente del decapeptide nativo ed in grado di esercitare il proprio effetto stimolatorio per un periodo di osservazione di 24 ore, ha permesso di ottenere maggiori informazioni diagnostiche in questo ambito, ma si è rilevato utile nell’identificare le pazienti con iperandrogenismo ovarico.
Per i soggetti con caratteri sessuali secondari, anche iniziali, può risultare utile il test al medrossiprogesterone acetato (MAP-test, con Farlutal 10 mg, 1 cp/die per 5 gg), poichè permette di valutare la sensibilizzazione estrogeno-dipendente delle cellule endometriali. Qualora si sospettino alterazioni o deficit di altri assi, come quello somatotropo, surrenalico e tiroideo, si devono eseguire test di valutazione funzionale per i singoli assi di cui si sospetta il deficit.
In ambienti ultraspecialistici potrebbero essere eseguiti anche ulteriori test. Nelle forme di ipogonadismo funzionale, quadro sempre molto vicino alle forme ipo/anoressiche con disturbi alimentari o per eccessiva attività sportiva, può risultare utile la determinazione della leptina plasmatica. Infatti, a parità di BMI e/o di massa grassa, la leptina risulta significativamente più bassa rispetto ai controlli nella forme da eccessiva attività fisica o da pregresso calo ponderale (disturbi alimentari). In tal senso sembra assumere un significato diagnostico, sempre nell’amenorrea ipotalamica funzionale, la determinazione della proteina legante il retinolo – 4 (RBP-4),di produzione adipocitaria, che è direttamente correlata con lo stato di adiposità (BMI, volume adipocitario, insulino-resistenza) e che risulta significativamente più bassa nei soggetti dimagriti sebbene in attuale normopeso. Da recenti valutazioni sembrerebbe acquistare sempre maggior rilevanza la determinazione dell’ormone anti-mülleriano (AMH), una glicoproteina prodotta dalle cellule della granulosa soprattutto dei follicoli pre-antrali o antrali piccoli. Tale ormone rappresenta fisiologicamente un parametro di reclutamento/maturazione follicolare ovarica, utilizzato come marcatore di riserva follicolare ovarica nei soggetti infertili che si sottopongono alle tecniche di fertilizzazione strumentale, o come marcatore di alterato reclutamento follicolare nella sindrome dell’ovaio policistico o come marcatore oncologico ovarico. L’AMH potrebbe rappresentare un rapido ed utile parametro per differenziare le forme di amenorrea secondaria, poiché nelle amenorree ipotalamiche funzionali i suoi livelli risultano normali e/o più spesso elevati, indice di arresto maturativo follicolare, a differenza di quadri sostenuti da insufficienza ovarica precoce o da fallimento ovarico precoce. È inoltre da ricordare che tale glicoproteina follicolare non subisce significative variazioni durante il ciclo mestruale, rendendola più facilmente interpretabile in ogni momento del ciclo(2,3).
Esami di III livello. Possono essere considerati tali i test di sequenziamento genetico per la ricerca di polimorfismi o di sequenze genetiche non attivate. Tali esami tuttavia avvengono solamente in centri di alta specializzazione e dedicati a studi genetici, per cui la richiesta di tali accertamenti deve essere fortemente indirizzata e motivata con un preciso percorso di screening endocrino-funzionale. Nel sospetto di forme ipofisarie infiammatorie (quadro radiologico compatibile per ipofisite), dopo aver escluso le forme batteriche, micotiche, tubercolari (condizioni generali molto compromesse, indici di flogosi aumentati) o quelle vasculitiche (suggerite dalla positività di anticorpi come ANA, ENA, ANCA), è Importante la determinazione degli anticorpi anti-ipofisi, che permettono di formulare una diagnosi di ipofisite autoimmune. Diversamente, nelle forme da infiltrazione/granulomatose è fondamentale il riscontro istologico da materiale bioptico. Nell’istiocitosi-X si possono osservare tipiche alterazioni del parenchima polmonare, che permettono un accesso bioptico meno invasivo di quello da biopsia ossea del tavolato cranico tecale o addirittura ipofisario trans-sfenoidale(4).
Diagnostica strumentale negli ipogonadismi ipogonadotropi femminili
La RM cerebrale e della struttura ipofisaria è in grado di orientare sulla tipologia e localizzazione della lesione (adenoma, incidentaloma, area ischemica, emorragica, ascesso) e permette di riconoscere le alterazioni del bulbo olfattorio (assenza nel KS) o altre anomalie cerebrali e talvolta cerebellari che caratterizzano il quadro malformativo, frequentemente associato all’anosmia.
L’esame morfologico pelvico (ecografia pelvica standard trans-addominale e se possibile trans-vaginale o trans-rettale) permette di escludere patologie di accompagnamento pelvico ovariche e/o uterine. In tal senso potrebbe essere considerata di aiuto la RM pelvica, in grado di definire meglio alcune patologie dismorfiche polidistrettuali, talvolta presenti nella KS. In caso di accertati dismorfismi, è molto importante l’esecuzione di ecografia, TC e/o RM addominali, per la ricerca di malformazioni a carico degli organi splancnici, come il rene e il pancreas, e di alterazioni cardiache e vascolari.
Nelle forme tumorali o flogistiche possono essere di aiuto le tecniche scintigrafiche isolate o associate a quelle tomografiche, come la scintigrafia con Octreotide marcato (Octreoscan) importante per le patologie tumorali/adenomatose con recettori per la somatostatina, oppure l’uso di leucociti marcati con anticorpi anti-leucociti (Leucoscan) per le forme ascessuali, o l’utilizzo di 18F-FDG PET-TC che permettono di individuare aree cellulari ad elevato turn-over come le masse neoplastiche (primitivo e metastasi) e/o i focolai infiammatori (4,5).
Approfondimenti bibliografici
- Prabhakar VKB, Shalet SM. Aetiology, diagnosis, and management of hypopituitarism in adult life. Postgrad Med J 2006, 82: 259–66.
- Genazzani AD, Ricchieri F, Lanzoni C, et al. Diagnostic and therapeutic approach to hypothalamic amenorrhea. Ann NY Acad Sci 2006, 1092: 103–13.
- Deligeoroglou E, Athanasopoulos N, Tsimaris P, et al. Evaluation and management of adolescent amenorrhea Ann NY Acad Sci 2010, 1205: 23–32.
- Broekmans FJ, Visser JA, Laven JSE, et al. Anti-Mullerian hormone and ovarian dysfunction. Trends Endocrinol Metab 2009, 19: 340-7.
- Committee Opinion no. 502. Primary ovarian insufficiency in adolescent. Obstet Gynecol 2011, 118: 741-5.
Approccio terapeutico all'ipogonadismo femminile
Roberto Mioni1 e Vincenzo Toscano2
1 Clinica Medica 3 , Dipartimento di Medicina, Università di Padova, Azienda Ospedaliera di Padova
2DAI Scienze Mediche, Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea
Il percorso terapeutico che deve essere affrontato in una paziente affetta da ipogonadismo ipogonadotropo, una volta determinata, risolta e/o stabilizzata la causa eziopatogenetica, deve tenere in considerazione alcuni punti fondamentali: il dato anagrafico, la presenza o meno dei caratteri sessuali secondari e di conseguenza il loro grado di evoluzione e la possibilità o meno di ripristinare la fertilità. È anche doveroso precisare che in caso di un deficit multiplo della funzione ipofisaria devono essere stabilizzati in ordine di precedenza l’asse surrenalico, quello tiroideo, quello ovarico/sessuale e infine quello somatotropo. Un’altra attenzione che deve essere esposta alla paziente, soprattutto per le forme ipotalamiche idiopatiche normosmiche è la possibilità di essere fertili, per cui la consapevolezza di tale possibilità nella gestione dell’attività di coppia (1).
Età adolescenziale senza (o con parziale) comparsa dei caratteri sessuali secondari
Secondo le indicazioni dell’ASRM (2), la paziente in queste condizioni dovrebbe avere almeno 13 anni e la terapia più accreditata rimane indubbiamente “l’estrogenizzazione graduale”. Poiché questa andrebbe sicuramente a influire sulla chiusura epifisaria delle ossa lunghe, risulta fondamentale lo stato staturale (quello teorico di riferimento deve essere il punto di arrivo) e la verifica che non ci siano concomitanti deficit dell’asse somatotropo. Una volta appurata la possibilità di proseguire con la terapia, la scelta deve essere fatta sul tipo di estrogeno: c’è molta esperienza con l’uso dell’etinil-estradiolo, sebbene risulti preferibile l’uso dell’estrogeno naturale, come il 17β-estradiolo naturale (il valerato o l’emi-idrato), che risulta meno attivatore degli enzimi epatici, con minor rischio emocoagulativo, e con clearance più completa, soprattutto sui metaboliti glicuronidati. Sempre meno utilizzati risultano gli estrogeni equini coniugati. La forma preferibile in questa fascia d’età risulta quella trans-dermica con estradiolo, che ha il vantaggio di evitare il primo passaggio epatico 17β-estradiolo naturale (Progynova® o Estrofem®): iniziare con 5 µg/Kg/die, aumentare a 10 µg/Kg/die dopo 6 mesi, a 15 µg/Kg/die dopo altri 6 mesi e così via (20, 25, 30 µg/Kg/die), fino ad una dose massima per adulto di 2 mg/die in continuo (3,4);
- Etinil-estradiolo (Etinil-estradiolo IBSA®): iniziare con 0.1 µg/Kg/die, aumentare a 0.2 µg/Kg/die dopo 6 mesi, a 0.3 µg/Kg/die dopo altri 6 mesi e così via fino alla dose media di 20-30 µg/die in continuo nell’età adulta.
All’inizio la terapia dovrebbe prevedere la sola assunzione dell’estrogeno per 12-18 mesi, poi dovrebbe essere controbilanciata dalla somministrazione di progestinici dal 15° al 25° giorno dell’ipotetico ciclo teorico. Ovviamente il percorso viene indicato anche dalla risposta della ghiandola mammaria: raggiunto il grado IV secondo Tanner, all’estrogeno dovrebbe essere associato il progestinico. I preparati suggeriti sono: Farlutal® cp 10 mg o Provera G® cp 10 mg o la forma di progesterone micronizzato (Progeffik® cp 200 mg), che può essere somministrato per os o per via vaginale. Esiste anche la somministrazione come crema vaginale (Esolut®, Crinone®) con apposito dispenser.
Una volta raggiunta la completa estrogenizzazione, con maturazione della mammella in BV secondo Tanner, può essere scelta (solitamente le pazienti la preferiscono soprattutto per non sentirsi diverse dalle altre ragazze che prendono la “pillola”) la somministrazione già completa con pillola monofasica, ben sapendo che la posologia è sicuramente in eccesso rispetto alla necessità funzionale sistemica. La maggior parte degli autori tuttavia caldeggia l’utilizzo del sistema trans-dermico (12).
Terapia trans-dermica. Estradiolo 25 µg/die 1 patch da cambiare ogni 3 giorni secondo lo schema personalizzato dall’autore. Qualora il sistema si staccasse, deve essere reintegrato con uno nuovo, che verrà rimosso secondo lo schema previsto, sebbene ancora attivo. Gli intervalli di incremento posologico possono variare da 6 mesi a 4 mesi, fino al dosaggio di 100 µg/Kg/die seguendo le stesse indicazioni della terapia orale. Una volta raggiunta la maturazione mammaria adeguata, va aggiunto il progestinico. L’autore suggerisce per la dose di progesterone una posologia inferiore (Provera G 5 mg/die o Progeffik 100 mg/die), che permette la riduzione degli effetti emotivi, depressivi e ponderali frequentemente collegati alla somministrazione del progesterone (R. Mioni dati non pubblicati). Esiste anche la confezione trans-dermica settimanale (Femseven® 50 µg/die), tuttavia si osserva con questo schema un maggior numero di complicanze cutanee (irritazione, allergia e/o distacco del sistema), per cui si tende a suggerire quello a cambio ravvicinato (ogni 3 giorni). Lo schema trans-dermico, tuttavia, risulta limitativo nei periodi estivi o per pazienti che svolgano attività sportive acquatiche e/o agonistiche. In questi casi si suggerisce lo schema orale o, quando possibile, la somministrazione vaginale.
I dosaggi certamente sopra-fisiologici prescritti in questa fase di estrogenizzazione si sono rivelati indispensabili per raggiungere il massimo picco di mineralizzazione ossea (massa ossea), parametro che una volta stabilizzato non cresce ulteriormente. Risulta fondamentale per il metabolismo fosfo-calcico l’integrazione con calcio per os (1000-1.200 mg/die) addizionato a vitamina D (almeno 800-1000 U/die) (12,14). Poiché l’assunzione del calcio può dare disturbi gastrici (pirosi, nausea, talvolta vomito), potrebbe risultare utile la dispersione della cp, solitamente effervescente, o della busta di calcio in una bottiglia di acqua da 1.0-1.5 L, da bere durante il giorno, possibilmente lontano dai pasti. La supplementazione con sola vitamina D risulta sufficiente qualora l’apporto alimentare di calcio riuscisse a raggiungere la quantità suggerita: 300-500 cc/die di latte intero + 150-200 g/die di carne (sostituzione 2 uova) + 100 g/die di formaggio (40 g di formaggio grana).
Età adolescenziale con presenza dei caratteri sessuali secondari
Prima di iniziare il percorso terapeutico, risulta importante il grading mammario, secondo la tabella di Tanner, e quello pelvico-uterino, possibilmente con uno studio ecografico con particolare attenzione all’isterometria ed allo spessore endometriale. Inoltre, se non si è già presentato il ciclo mestruale, sapendo che il menarca corrisponde solitamente ad un BIV, risulta utile l’esecuzione del MAP test: se positivo, colloca la paziente a un grado di estrogenizzazione quasi fisiologico, per cui può essere intrapresa la terapia sostitutiva dell’adulto, soprattutto con la necessità di raggiungere o garantire una massa ossea adeguata. Potrebbe essere comunque utile, soprattutto nelle pazienti con amenorrea ipotalamica funzionale, evitare un’eccessiva e troppo rapida estrogenizzazione che potrebbe mettere in difficoltà la compliance della paziente stessa.
Qualora il MAP test risultasse negativo (anche dopo un secondo tentativo ripetuto dopo un intervallo di 2-3 mesi), si dovrebbe prendere in considerazione lo schema a progressiva estrogenizzazione come suggerito per i soggetti a scarsa sensibilizzazione estrogenica. Secondo l’autore, tuttavia, gli intervalli tra una dose e quella successiva, aumentata, si dovrebbero ridurre di un terzo (2 mesi invece di 3), poichè fisiologicamente l’intervallo dallo stadio BIII-IV alla comparsa del menarca è solitamente di 6-12 mesi (4,5,7).
Età adulta
Nell’adulto, dove le cause sono quasi sempre acquisite, la terapia potrà essere scelta a seconda delle caratteristiche e delle esigenze della paziente.
Terapia orale. Una volta raggiunta una normale e fisiologica estrogenizzazione, potrebbe essere suggerita, per comodità, l’assunzione dei preparati già in commercio come le pillole monofasiche, sebbene siano tutte da considerarsi soprafisiologiche. Qualora si volesse personalizzare la terapia sostitutiva, si può valutare l’assunzione degli estrogeni naturali (preferibili) (2.0 mg/die) o di quelli di sintesi come l’etinil-estradiolo (10 µg/die continuato o 20 µg/die per 21-24 giorni). Importante l’associazione con il progestinico (10 mg/die o 200 mg micronizzato) dal 15° al 25° giorno del ciclo teorico. Alcuni gruppi anglosassoni propongono l’associazione del progestinico ogni 30-60 giorni.
Terapia trans-dermica. È un’alternativa terapeutica, consigliabile per i minori effetti collaterali soprattutto a livello epatico e sul metabolismo glucidico e lipidico e per il minor rischio trombofilico. Può essere scelta l’associazione estrogeno e progestinico in sistema monofasico già in commercio (etinil-estradiolo/norelgestromina) con 1 sistema/settimana per 3 settimane e 4° settimana libera. La terapia può comunque essere personalizzata con la scelta di sistemi a solo estrogeno trans-dermico (da 25 µg/die a 50 µg/die - preferibile -, che potrebbero divenire anche 100 µg/die), associando per 10 giorni il progestinico per os o per via trans-vaginale (MAP 10 mg/die o progesterone micronizzato 200 mg/die). L’autore solitamente consiglia l’assunzione di una minor quantità di progesterone (5 mg/die e 100 mg/die, rispettivamente) (6).
Terapia trans-vaginale. È un’altra proposta terapeutica con le stesse caratteristiche della via trans-dermica per quanto riguarda gli effetti sistemici degli estrogeni (fegato e metabolismo glico-lipidico). Esiste già in commercio un sistema ad anello flessibile (etilil-estradiolo e etonogestrel), che viene posizionato in vagina e rimosso dopo 3 settimane per essere riposizionato dopo la 4° settimana riprendendo un nuovo ciclo. Con tale sistema la paziente deve solamente accertarsi che non ci sia l’espulsione accidentale.
La terapia sostitutiva e/o anticoncezionale poneva dei legittimi quesiti per i pazienti portatori di adenomi ipersecernenti (PRL, GH, ACTH) ovviamente in un quadro di stabilizzazione (post-chirurgica o farmacologica) della patologia di base. Sebbene alcuni autori avessero evidenziato un ipotetico peggioramento del volume soprattutto in adenomi PRL- e GH-secernenti, una recente meta-analisi ha dimostrato che tale possibilità sembrerebbe mantenersi solo per i macroadenomi e nei soggetti operati non si sarebbe evidenziata un’aumentata possibilità di recidiva o di una sua facilitazione (7,8).
Terapia per l’induzione della fertilità
Le pazienti affette da ipogonadismo ipogonadotropo da differenti cause possono avere un desiderio di maternità. Da più anni molti gruppi di studio hanno dimostrato la possibilità di raggiungere un normale livello di maturazione dell’asse ipofiso-ovarico in soggetti affetti da ipogonadismo ipogonadotropo, sia con che senza anosmia, e anche in quadri acquisiti, di patologia sia ipotalamica che ipofisaria, ovviamente con l’utilizzo di protocolli terapeutici differenziati.
È importante, prima di iniziare il percorso terapeutico atto al ripristino della fertilità, che ogni paziente abbia raggiunto un grado adeguato di maturità degli organi bersaglio (mammella, ma soprattutto utero e funzionalità endometriale), in quanto, una volta iniziato il percorso della gravidanza, la placenta è in grado di automantenere e assicurare le necessità endocrino-funzionali dell’unità feto-placentare.
Le strategie terapeutiche sono differenti a seconda del livello di lesione (9,10).
Soggetti con deficit ipotalamico. A tali pazienti può essere proposto l’utilizzo di protocolli di induzione con GnRH o con gonadotropine.
- Protocollo con GnRH nativo: sfrutta la capacità di stimolo che il peptide ipotalamico esercita a carico delle cellule gonadotrope ipofisarie. Questo percorso risulta sicuramente più fisiologico e deve assicurare una stimolazione pulsatile con “pulse” ad intervalli di 90-120 minuti. Tale esigenza deriva dalla fisiologica capacità di una down-regulation recettoriale esercitata dal GnRH stesso sul suo recettore a livello ipofisario, con conseguente perdita di attività stimolatoria. Il protocollo pertanto prevede l’applicazione di una pompa di infusione con set per incannulazione ev, che deve essere mantenuta per più giorni e talvolta settimane. Questa tecnica induce un miglior reclutamento follicolare, riduce la possibilità di stimolazioni follicolari multiple e soprattutto riduce il rischio della sindrome da iperstimolazione follicolare. Tuttavia la necessità di mantenere la pompa, con il disagio che ne consegue, e la possibilità di evocare complicanze flebo-trombotiche in corrispondenza del set venoso, hanno fortemente limitato e/o ridotto l’uso di tali programmi di fertilizzazione (8,9).
- Uso delle gonadotropine: esistono più esperienze con relativi protocolli che hanno dimostrato la possibilità di indurre maturazione follicolare:
- somministrazione di solo FSH purificato (da 75 a 225 U) sc: questo protocollo, inizialmente promettente, ha tuttavia evidenziato una minor capacità ovulatoria (circa 20-22%) e quando presente con una peggiore qualità dell’ovocita, con conseguente riduzione della capacità di fertilizzazione. Inoltre con tale protocollo si è osservato un utilizzo finale di maggiori concentrazioni di FSH (fino al 50% in più del numero di fiale);
- somministrazione di FSH combinato con LH: ha dimostrato un minor utilizzo di FSH e migliori risultati (fino al 100% di ovulazione e fino al 60-64% di gravidanza). È stato osservato infatti che esiste un cut-off di concentrazione di LH basale, pre-trattamento, che deve garantire la disponibilità di substrato ormonale androgenico, da parte delle cellule tecali, indispensabile per la costruzione degli estrogeni FSH-dipendente. Il valore soglia di LH è stato identificato a 1.5 UI/L. Alcuni gruppi hanno dimostrato che i risultati migliorano nei soggetti con LH < 1.5 UI/L, utilizzando un pre-trattamento con LH, ad una posologia variabile tra 50 e 75 UI/L/die.
Lo schema per l’induzione del reclutamento del follicolo dominante prevede uno stretto studio ecografico per il monitoraggio follicolare: una volta raggiunto un follicolo di almeno 1.7 cm, viene somministrata una dose di β-hCG (10.000 UI im) per indurre lo scoppio follicolare.
Soggetti con deficit ipofisario. Tali soggetti, a differenza del gruppo delle pazienti ipotalamiche, non hanno la possibilità, maggiormente fisiologica, di utilizzare la terapia con GnRH, ma solo quella con le gonadotropine. Gli schemi pertanto seguono quelli con gonadotropine dei soggetti ipotalamici, che per la maggior parte, presentano valori di LH quasi sempre < 1.0 UI/L. In tali situazioni, pertanto, si presenta la necessità di un periodo di “androgenizzazione tecale” indotta con il pretrattamento con LH ricombinante o con β-hCG, al fine di rendere il milieu androgenico sufficiente per la successiva ed indispensabile aromatizzazione FSH-indotta, per la maturazione ovocitaria (10,11).
Per approfondimenti bibliografici
- Santoro N. Update in hyper- and hypogonadotropic amenorrhea. J Clin Endocrinol Metab 2011, 96: 3281–8.
- Committee Opinion no. 502. Primary ovarian insufficiency in adolescent. 2011, 118: 741-5.
- Prabhakar VKB, Shalet SM. Aetiology, diagnosis, and management of hypopituitarism in adult life. Postgrad Med J 2006, 82: 259–66.
- Genazzani AD, Ricchieri F, Lanzoni C, et al. Diagnostic and therapeutic approach to hypothalamic amenorrhea. Ann NY Acad Sci 2006, 1092: 103–13.
- Deligeoroglou E, Athanasopoulos N, Tsimaris P, et al. Evaluation and management of adolescent amenorrhea. Ann NY Acad Sci 2010, 1205: 23–32.
- Unuane D, Tournaye H, Velkeniers B, et al. Endocrine disorders & female infertility. Best Pract Res Clin Endocrinol Metab 2011, 25: 861–73.
- Kars M, Dekkers OM, Pereira AM, Romijn JA. Update in prolactinomas. Neth J Med 2010, 68: 104-12.
- Shimon I, Barkan A. Estrogen treatment for acromegaly. Pituitary 2012, 15: 601-7.
- Delemarre EM, Felius B, Delemarre-Van de Waal HA. Inducing puberty. Eur J Endocrinol 2008, 159: S9–S15.
- Krause BT, Ohlinger R, Haase A. Lutropin alpha, recombinant human luteinizing hormone, for the stimulation of follicular development in profoundly LH-deficient hypogonadotropic hypogonadal women: a review. Biologics 2009, 3: 337–47.
- Balasch J, Fabregues F, Carmona F, et al. Ovarian luteinizing hormone priming preceding follicle-stimulating hormone stimulation: clinical and endocrine effects in women with long-term hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2009, 94: 2367–73.
Ipogonadismo primario femminile
Agenesia/disgenesia gonadica femminile
Eziopatogenesi e clinica dell'ipogonadismo primario femminile
Cecilia Motta, Laura Proietti Pannunzi, Vincenzo Toscano
Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea, Roma
Definizione ed eziopatogenesi
Una normale funzione ovarica è essenziale per la produzione nella donna degli ormoni sessuali, che sono necessari a un corretto sviluppo del tratto genitale, dei caratteri sessuali secondari, della funzione riproduttiva e al raggiungimento e mantenimento di un’adeguata densità minerale ossea.
Per ipogonadismo primitivo si intende una mancata funzione dell’ovaio associata ad aumentati valori di FSH.
Le cause di ipogonadismo sono estremamente eterogenee: congenite (anomalie cromosomiche, difetti enzimatici) o acquisite, ad esempio a seguito di danni da agenti esterni, natura autoimmune, terapie per patologie neoplastiche (1); nella maggior parte dei casi l’eziologia rimane sconosciuta (2).
Dal punto di vista fisiopatologico, l’insufficienza ovarica può essere dovuta ad accelerato esaurimento del patrimonio follicolare o a ridotta produzione degli ormoni steroidei con perdita degli ovociti; inoltre, un’insufficienza ovarica può anche essere dovuta a mancata migrazione delle cellule germinali o a uno sviluppo ovarico alterato.
| Classificazione degli ipogonadismi primitivi (ipergonadotropi) | ||
| Disgenesia gonadica | con alterazioni del cariotipo | Sindrome di Turner (45, X0) e suoi mosaicismi |
| senza alterazioni del cariotipo | Disgenesia gonadica pura (46, XX e 46, XY o sindrome di Swyer) | |
| Agenesia gonadica | ||
| Difetti enzimatici | Deficit di 17α-idrossilasi Deficit di 17,20 liasi |
|
| Premature Ovarian Failure (POF) | Idiopatica Da agenti esterni: chemioterapia, terapia radiante, ooforite Resistenza ovarica: idiopatica, da mutazioni del recettore per FSH o per LH, premutazione per sindrome dell’X fragile Autoimmune Galattosemia |
|
Clinica generale
I sintomi dell’ipogonadismo primitivo possono variare considerevolmente da paziente a paziente e la patologia può avere un esordio improvviso o può svilupparsi lentamente nel corso di anni. Le forme più severe si presentano prima della pubertà, con mancato sviluppo puberale (assenza di maturazione sessuale e amenorrea primaria) e riduzione della velocità di crescita (2). Circa la metà dei casi di amenorrea primaria è dovuta alla disgenesia ovarica, che consiste nella presenza di streak ovaries (tessuto fibroso) e ipoplasia uterina; nel restante 50% dei casi si può riscontrare la presenza di follicoli all’esame istologico ovarico.
Nella maggior parte dei casi però, l’insufficienza ovarica primitiva si manifesta in età post-puberale e si caratterizza per amenorrea secondaria associata a deplezione prematura dei follicoli o arresto della follicologenesi; clinicamente sono presenti le tipiche manifestazioni climateriche, quali palpitazioni, vampate, intolleranza al caldo, sudorazioni notturne, irritabilità, ansia, depressione, disturbi del sonno, riduzione della libido, secchezza vaginale e astenia.
L’infertilità è una ovvia conseguenza dell’insufficienza ovarica, spesso irreversibile.
La prolungata mancanza di estrogeni può condurre a osteopenia e osteoporosi, oltre ad essere un importante fattore di rischio per malattie neurologiche, metaboliche e cardiovascolari.
Un attento esame obiettivo e un’anamnesi accurata sono indispensabili per guidare il medico nella scelta degli esami da richiedere. Ad esempio, la valutazione dei genitali e dello stadio puberale, nonché la ricerca di caratteristiche peculiari, quali l’assenza del quarto e quinto metacarpo, la presenza di cubito valgo o di petto escavatum, sono elementi da ricercare prima di decidere se richiedere o meno un cariotipo per confermare il sospetto di una patologia cromosomica; invece, la presenza di amenorrea secondaria, precedenti gravidanze, l’esposizione a chemio/radioterapia indirizzerà il clinico verso un’insufficienza ovarica prematura.
Per i dettagli vedi i capitoli relativi a:
Bibliografia
- Santoro N. Mechanisms of premature ovarian failure. Ann Endocrinol 2003, 64: 87-92.
- Beck-Peccoz P, Persani L. Premature ovarian failure. Orphanet J Rare Disease 2006, 1: 9.
Agenesia/disgenesia gonadica femminile
Cecilia Motta, Laura Proietti Pannunzi, Vincenzo Toscano
Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea, Roma
Per disgenesia gonadica si intende una serie di condizioni nelle quali si osserva un anomalo sviluppo gonadico (streak gonads), associato a precoce esaurimento del patrimonio ovocitario e follicolare. Le pazienti affette presentano generalmente amenorrea primaria e sviluppo variabile dei caratteri sessuali secondari.
La disgenesia gonadica è causata da anomalie cromosomiche o genetiche, che determinano un esaurimento prematuro degli ovociti e dei follicoli, con conseguente incremento dei valori di FSH, a causa della riduzione del feed-back negativo esercitato dall’estradiolo e dall’inibina B; se l’insufficienza ovarica è presente prima della pubertà, la paziente non svilupperà i caratteri sessuali secondari.
SINDROME DI TURNER
È una delle malattie cromosomiche più frequenti, dovuta alla mancanza di un cromosoma X, con una prevalenza di 1 su 2500 nati (1) [un numero molto maggiore di embrioni 45,X0 non sopravvive al primo trimestre di gravidanza (2)].
L’età della madre non determina un aumento del rischio e non esistono fattori di rischio ben definiti.
La diagnosi viene posta in pazienti di genere femminile con assenza parziale o completa di un cromosoma X: nell’80% dei casi il cariotipo è 45,X0 e nel 20% mosaicismo. Questo, oltre alla forma più comune (45,X0/46,XX), può presentarsi anche con una linea 46,XY (45, X0/46XY o altre linee con anomalie strutturali della seconda X).
Le manifestazioni cliniche possono essere varie, ma solitamente comprendono bassa statura, torace ampio e piatto con capezzoli molto distanziati (a scudo), cubito valgo, linfedema congenito e mancato sviluppo puberale spontaneo per insufficienza ovarica, con amenorrea primaria, streak gonads, utero ipoplasico e vagina nella norma. In queste pazienti l’ovaio è costituito da tessuto fibroso generalmente privo di follicoli. La presentazione clinica della sindrome di Turner varia nel corso della vita e può essere diagnosticata a qualsiasi età, anche se può essere difficile da riconoscere clinicamente.
- Durante la gravidanza la diagnosi dovrebbe essere sospettata in presenza di un feto femmina con idrope, aumentata translucenza nucale, igroma cistico o linfedema (3).
- Caratteristiche cliniche durante l’infanzia e l’età adulta comprendono pterigio del collo, bassa statura, bassa attaccatura posteriore dei capelli, padiglioni auricolari malformati, torace ampio e piatto, ginocchio varo e cubito valgo, unghie iperconvesse, nevi multi-pigmentati, malformazioni cardiache, malformazioni renali (es. rene “a ferro di cavallo”, doppio distretto renale), perdita uditiva neurosensoriale, otite media ricorrente, tiroidite autoimmune, malattia celiaca, displasia congenita dell’anca e scoliosi.
La presenza di linfedema è la causa più comune per la quale bambine affette da ST giungono all’attenzione del medico, mentre la bassa statura è la causa più frequente durante l’adolescenza. Tuttavia, la paziente può rivolgersi al medico anche in fase successiva, per mancata comparsa del menarca, per infertilità o per amenorrea secondaria.
Nel sospetto di ST l’esame obiettivo non sempre è dirimente, i dosaggi ormonali in genere mostrano elevati livelli di FSH ed LH associati a bassi valori di Estradiolo (E2)(4).
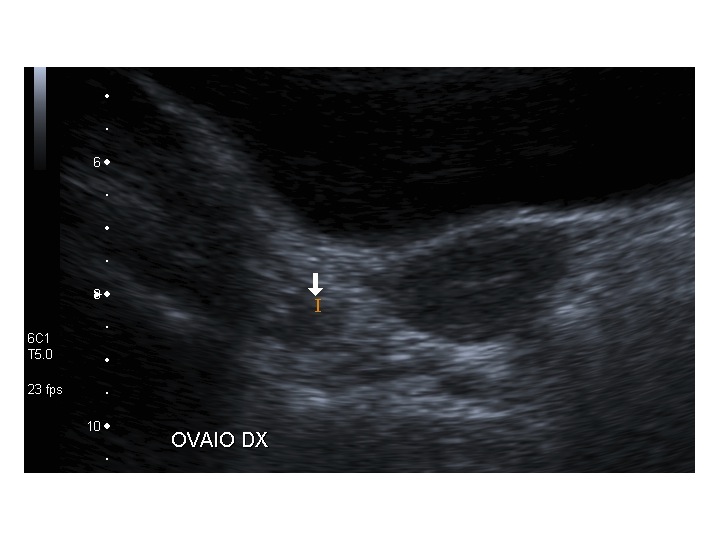
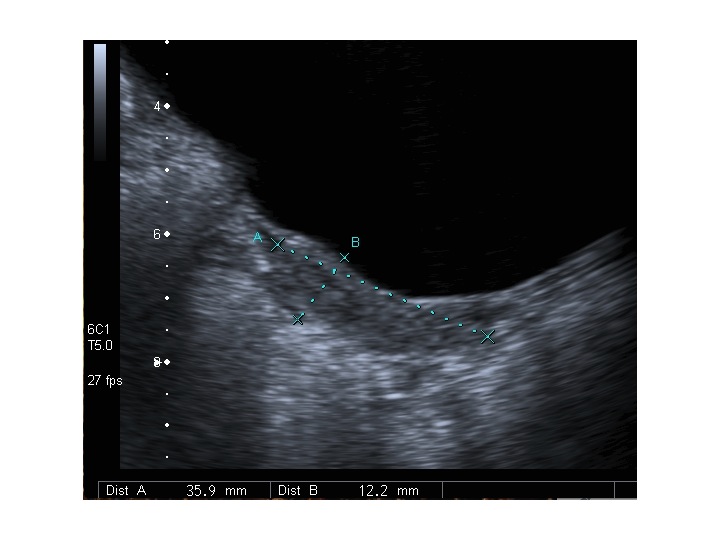
All’ecografia pelvica si osservano le ovaie a “banderella” (figura) e il cariotipo rivela l’assenza di un cromosoma X o la presenza di un mosaicismo, 45 X0/46XX nella forma più comune; nel 5% dei casi di ST può essere presente un frammento di cromosoma Y.
La ST si accompagna ad anomalie congenite cardiovascolari, anatomiche e/o funzionali, nel 50% dei casi: le più frequenti sono la coartazione dell’aorta, la valvola aortica bicuspide, uno sbocco anomalo della vena polmonare. Alterazioni non anatomiche, spesso indipendenti dai difetti anatomici, sono rappresentate da anomalie elettrocardiografiche, come blocco atrioventricolare (presente nel 30% dei casi, associato in modo statisticamente significativo alla presenza di linfedema), deviazione assiale destra, anomalie delle onde T, conduzione atrio-ventricolare accelerata, tratto QTc prolungato (5). La sindrome di Turner non si associa a ritardo mentale.
Sebbene un solo cromosoma X sia sufficiente per la differenziazione della gonade primitiva in tessuto ovarico, per la produzione di ovociti sono necessari due cromosomi X attivi (6); quindi l’aplo-insufficienza di molti geni del cromosoma X nei feti affetti da sindrome di Turner provoca apoptosi degli ovociti, che inizia già dalla 12° settimana di gestazione, con deplezione del patrimonio ovocitario entro i primi 10 anni di vita. Una volta raggiunta l’età dello sviluppo puberale, le pazienti presentano tipicamente mancato sviluppo mammario e amenorrea primaria, associati ad aumento dei valori di FSH. L’amenorrea è causata da ovaie fibrotiche, non in grado di produrre adeguati livelli di estrogeni. I genitali esterni, l’utero e le tube di Falloppio si sviluppano normalmente fino alla pubertà, con successivo arresto di sviluppo, poichè viene a mancare la maturazione estrogeno-indotta. Pazienti con mosaicismo possono presentare un menarca spontaneo con cicli mestruali di ritorno anche per qualche anno.
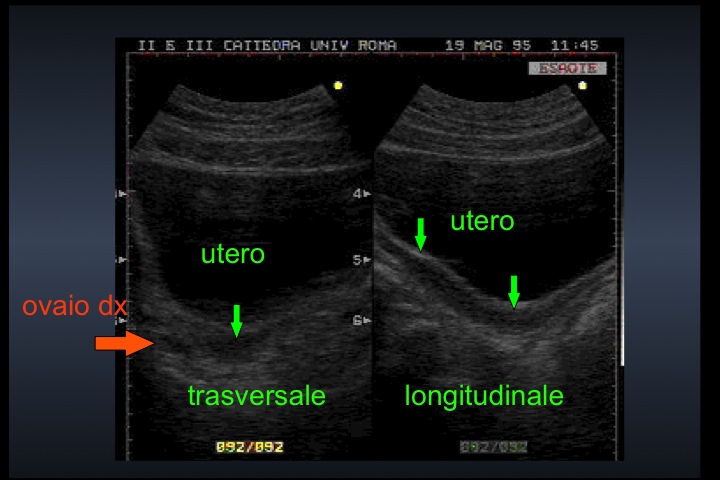
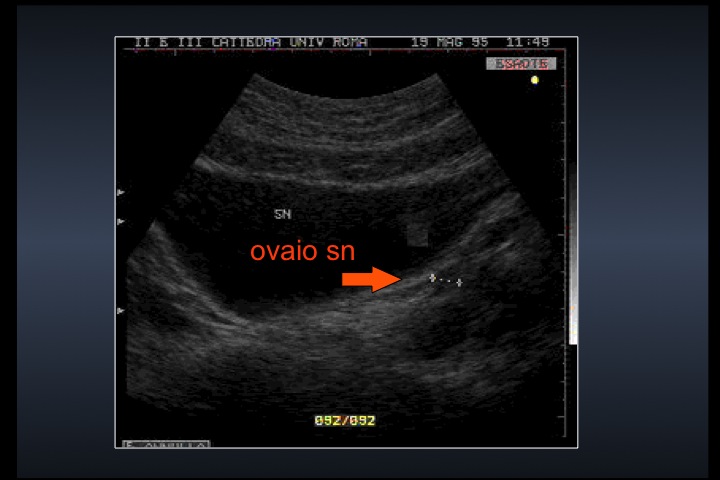
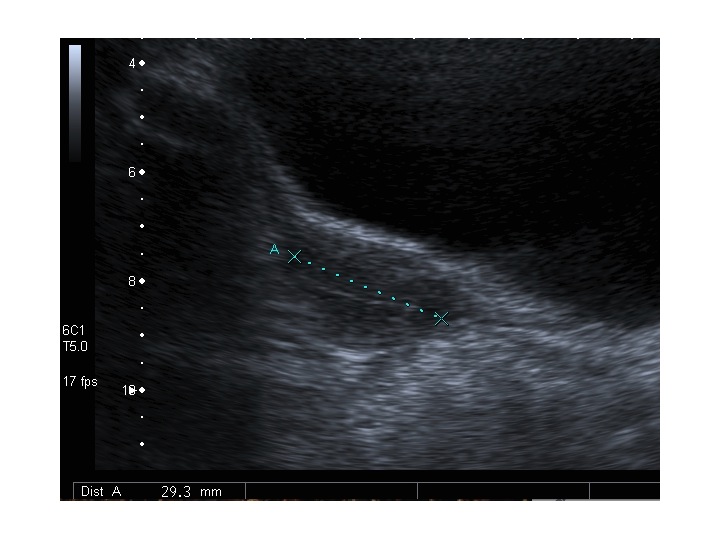
| Modificazioni ecografiche pelviche indotte dalla terapia estrogenica nella s. di Turner | |||
| Pre-terapia con EP | Post-terapia con EP | ||
| Paziente 1 | |||
| Utero: sezione longitudinale |
|
|
|
| Utero: sezione trasversale | 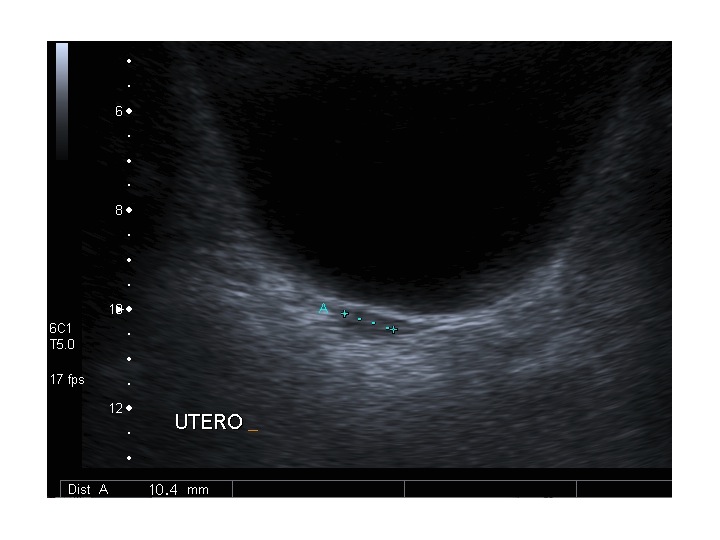 |
 |
|
| Ovaie | 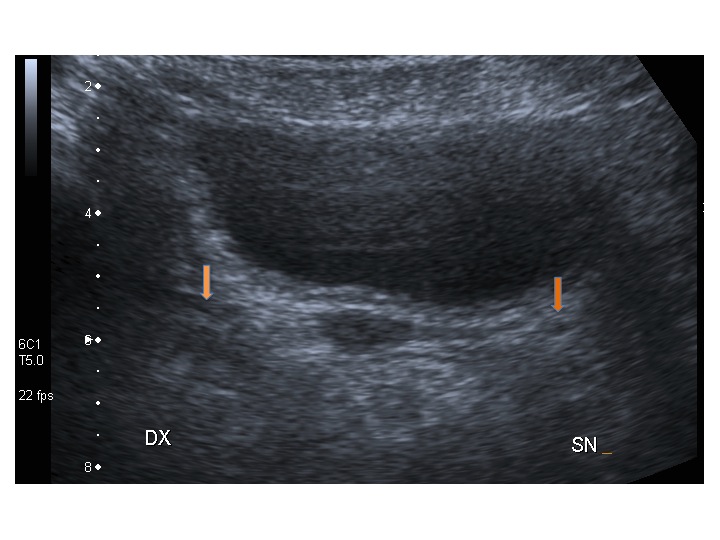 |
|
|
| Paziente 2 | |||
| Utero: sezione longitudinale |
|
|
|
| Utero: sezione trasversale + longitudinale | 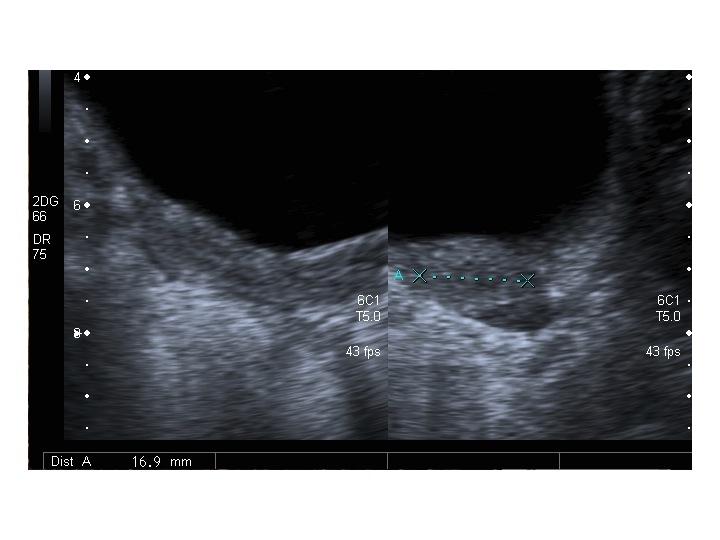 |
 |
|
| Ovaio destro | 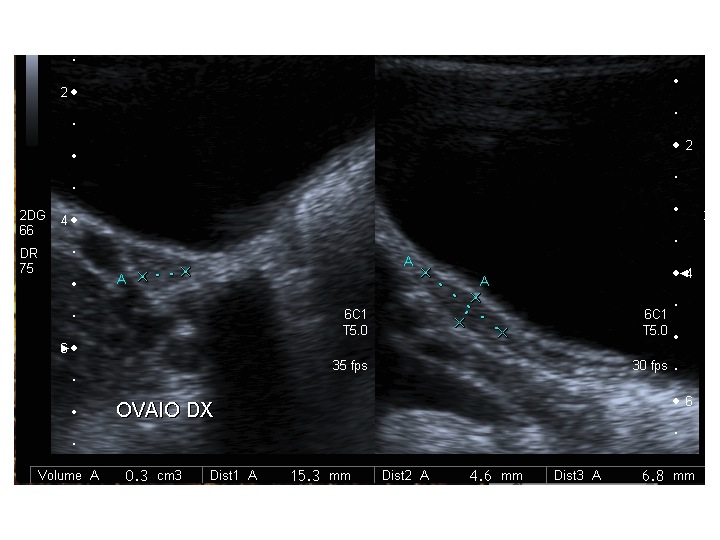 |
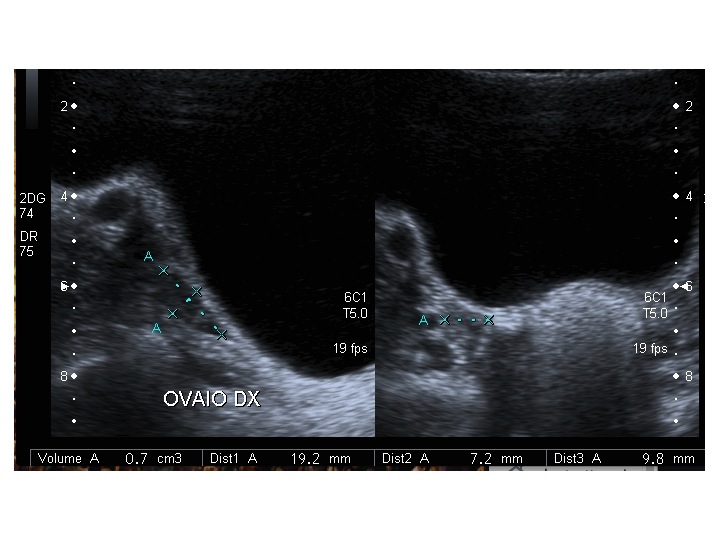 |
|
| Ovaio sinistro | 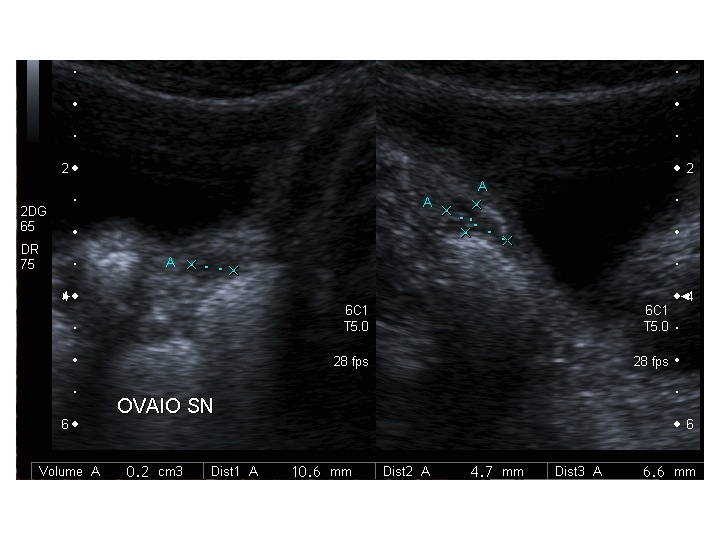 |
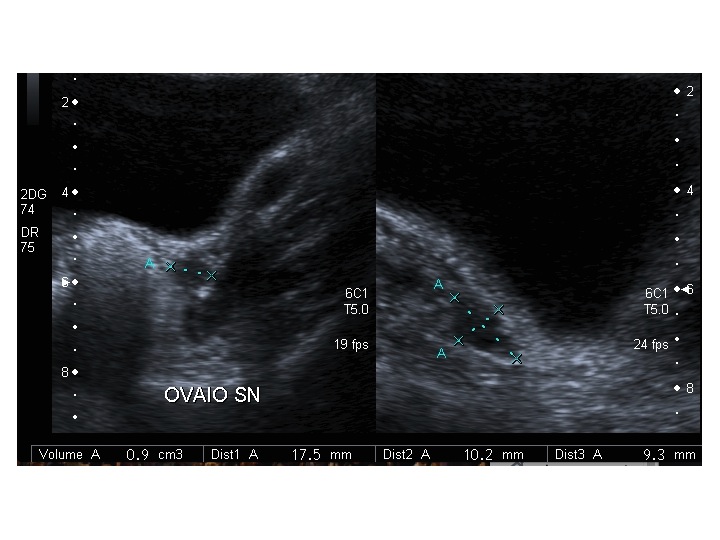 |
|
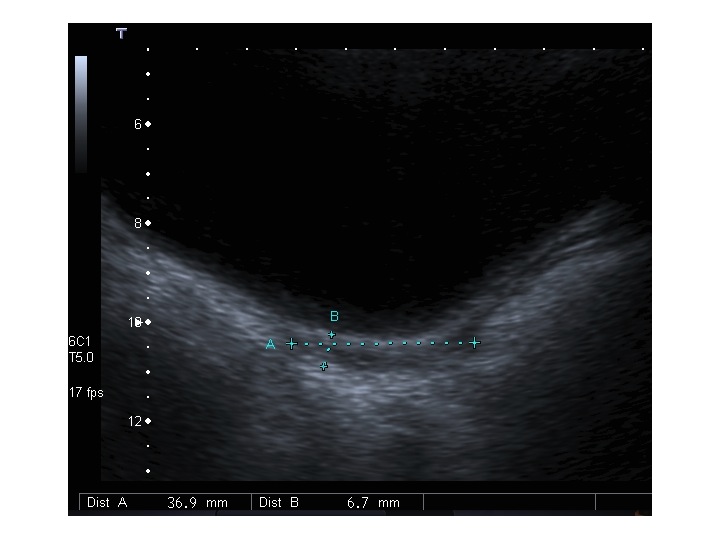
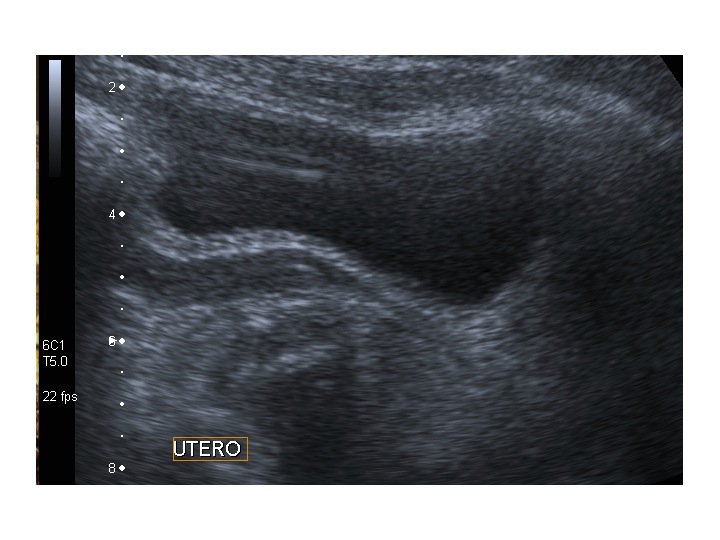
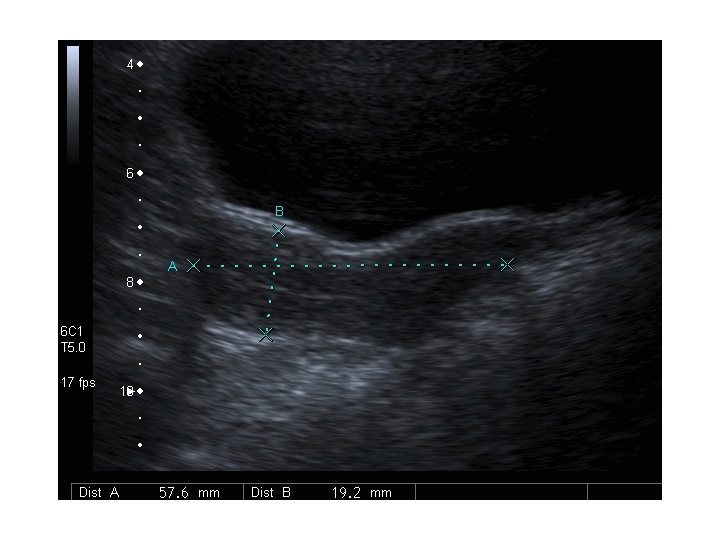
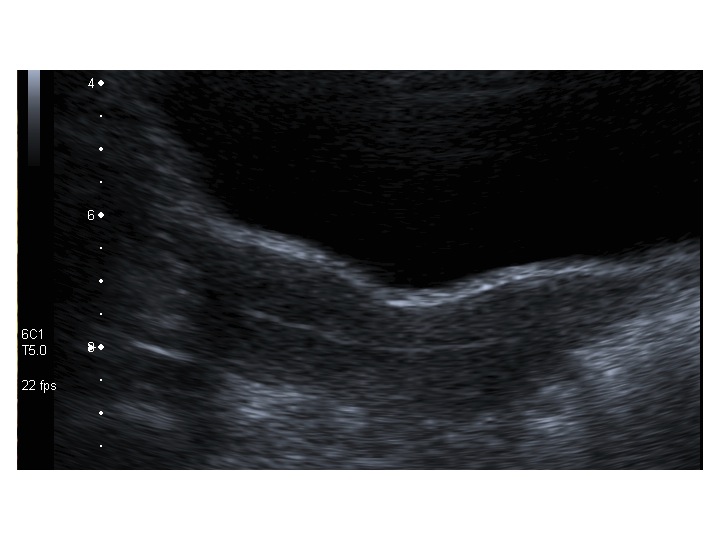
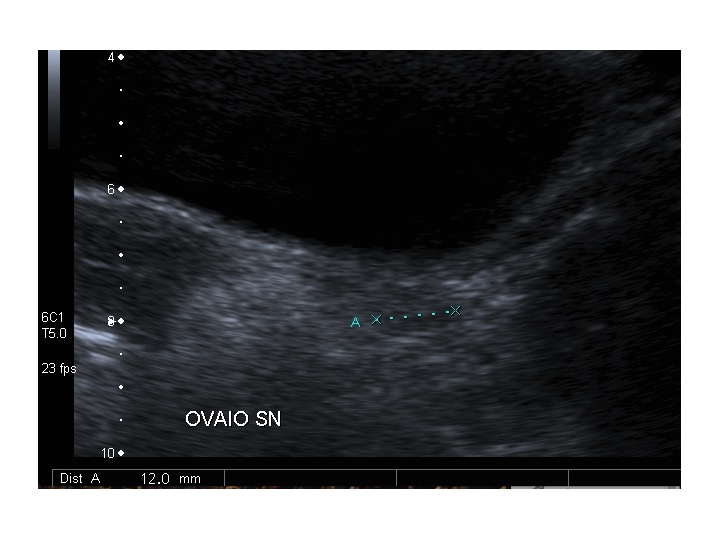
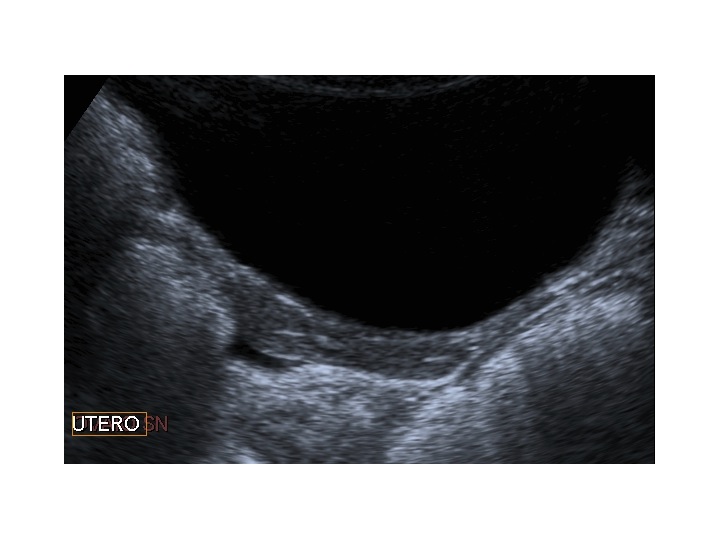
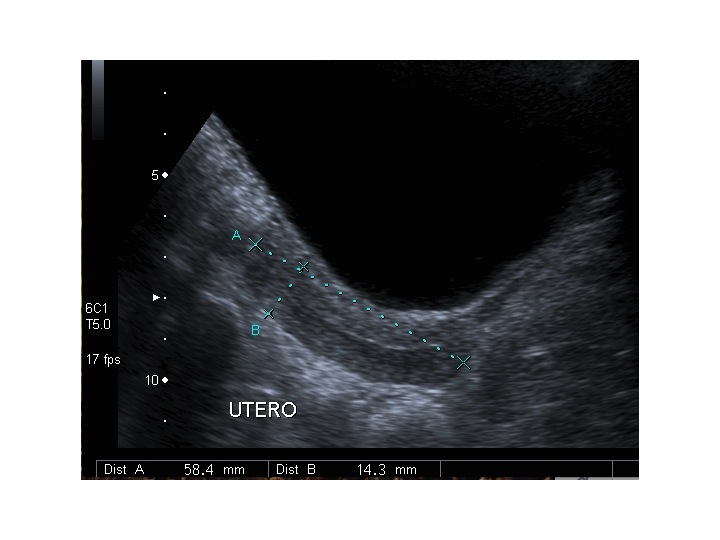
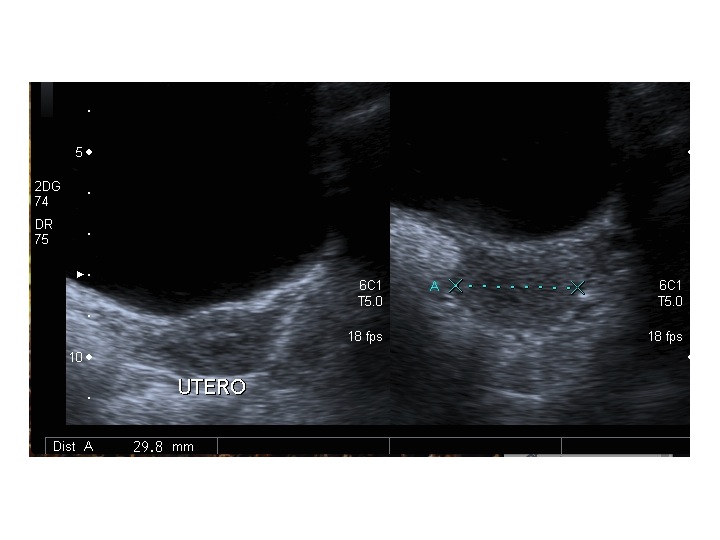
Dopo l'induzione di pubertà è ben visibile la rima endometriale (nella paziente 2 era appena accennata anche nelle immagini pre-terapia), soprattutto nelle sezioni longitudinali, con l'assenza di follicoli a livello ovarico
Altre alterazioni del cromosoma X
Sono dovute a delezioni, inversioni, duplicazioni e traslocazioni bilanciate fra il cromosoma X e gli autosomi, che coinvolgono la regione critica del cromosoma X necessaria allo sviluppo ovarico: Xq13-Xq26 (7).
SINDROMI DA DISGENESIA GONADICA
Il fenotipo della disgenesia gonadica è in genere femminile, tuttavia possono essere presenti o assenti infantilismo sessuale, bassa statura e anomalie somatiche presenti nella sindrome di Turner, in base alla mutazione cromosomica da cui il soggetto risulta affetto (tabella). Può essere presente ritardo mentale (8).
| Alterazioni cromosomiche e manifestazioni cliniche nella disgenesia gonadica | ||||
| Anormalità sesso cromosomico | Cariotipo | Fenotipo | Infantilismo sessuale | Bassa statura |
| Perdita di un X o Y | 45,X | Femminile | + | + |
| Delezione braccio corto X | 46,XXqi | Femminile | + (occ. ±) | + |
| 46,XXp- | Femminile | +, ±, - | + (-) | |
| Delezione braccio lungo X | 46,XXq | Femminile | + | + (-) |
| Delezione delle porzioni terminali di ambedue bracci X | 46,XXr | Femminile | - o + | + |
| Perdita braccio corto Y | 46,XYp- | Femminile | + | + |
Disgenesia gonadica pura
46, XX. Nella maggior parte dei casi ha eziologia sconosciuta, anche se sono stati implicati diversi geni, fra cui il gene BMP15(Bone Morphogenetic Protein-15) localizzato sul braccio corto del cromosoma X (Xp11.2), che codifica per un fattore di crescita della superfamiglia del TGF-ß, prodotto dagli ovociti, implicato nella maturazione follicolare. La diagnosi dovrebbe essere sospettata in soggetti fenotipicamente femmine, con infantilismo sessuale e strutture mulleriane normali, in assenza delle stigmate tipiche della ST. In genere queste pazienti arrivano all’osservazione del pediatra per l’assenza dello sviluppo puberale. Nel 10% dei casi è presente sordità neurosensoriale, che può aiutare il clinico nell’orientamento diagnostico. Clinicamente le pazienti si differenziano dalla Sindrome di Turner perché presentano alta statura (9). Gli esami ormonali rivelano elevati valori di gonadotropine e bassi valori di estrogeni e progesterone. L’esame del cariotipo risulterà normale. Le gonadi di queste pazienti presentano scarso di rischio di degenerazione neoplastica.
46, XY o sindrome di Swyer. Si manifesta in soggetti con fenotipo di tipo femminile, genitali esterni e interni di tipo femminile e cariotipo XY. Nel 15-30% dei casi è causata da una mutazione di SRY (sex determining region of Y chromosome) o altre alterazioni del cromosoma Y. Si caratterizza per la mancata produzione dell’Anti Mullerian Hormone (AMH) e del testosterone, necessari per la corretta differenziazione del tratto genito-urinario in senso maschile (10). Esclusi i casi diagnosticati con diagnosi prenatale, le pazienti giungono all’osservazione del clinico per il mancato sviluppo puberale, nonostante sia presente l’adrenarca. In genere la statura è normale e non si osservano stigmate della ST. In tale epoca presentano genitali esterni femminili normali. L’esame del cariotipo risulterà XY. Sono presenti gonadi disgenetiche non del tutto sviluppate, che si associano a un aumento del rischio di sviluppare tumori addominali (più comunemente disgerminomi). La diagnosi differenziale si pone con la disgenesia ovarica ipergonadotropa (disgenesia gonadica tipo 46,XX) e con tutte le forme di disgenesia gonadica completa (CGD) 46,XY sindromica [ad esempio, la sindrome di Frasier, la displasia campomelica e la DSD (disordine di sviluppo sessuale) 46,XY con insufficienza surrenalica]. Sebbene alcuni casi di CGD 46,XY siano sporadici, alle famiglie affette può essere proposta la consulenza genetica.
È possibile la diagnosi prenatale nelle famiglie nelle quali è stata precedentemente confermata un'anomalia genetica, anche se viene consigliata solo nei casi sindromici.
La presa in carico consiste nella rimozione del tessuto gonadico disgenetico, in quanto è elevato il rischio di trasformazione neoplastica. A seconda della diagnosi genetica, devono essere presi in considerazione diversi disturbi, ad esempio l’insufficienza renale nella sindrome di Frasier e le malformazioni correlate.
AGENESIA GONADICA
È una malattia molto rara, che comprende uno spettro di anomalie genitali, secondarie alla regressione dello sviluppo del testicolo tra l'8° e la 14° settimana di gravidanza.
I pazienti affetti presentano cariotipo XY; il fenotipo dipende dall'epoca della regressione del testicolo fetale e varia da quello femminile al maschio anorchide:
- se la regressione testicolare fetale si manifesta tra l'8° e la 10° settimana di gestazione, i pazienti presenteranno genitali esterni femminili, talora ambigui, in assenza delle gonadi, utero ipoplasico e dotti genitali rudimentali;
- se invece la regressione testicolare si manifesta dopo le 12-14 settimane di gestazione, i pazienti presenteranno fenotipo maschile, con anorchia o gonadi atrofiche (testicoli rudimentali).
I fenotipi intermedi si manifestano con ambiguità sessuale, associata a dotti genitali, seno urogenitale e differenziazione dei genitali esterni variabile. Tutti i pazienti mostrano agenesia o atrofia delle gonadi.
La causa non è nota. Alcuni casi di consanguineità tra i genitori e la ricorrenza tra fratelli hanno suggerito un difetto genetico autosomico recessivo. Si sospetta che siano coinvolte mutazioni in uno o più geni autosomici implicati nello sviluppo gonadico, che mappano nella regione distale all'SRY.
BIBLIOGRAFIA
- Nielsen J, Wohlert M. Chromosome abnormalities found among 34.910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum Genet 1991, 87: 81-3.
- Menasha J, Levy B, Hirschhorn K, Kardon NB. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: new insights from a 12-year study. Genet Med 2005, 7: 251-63.
- Sanders RC, Blackmon LR. Structural fetal abnormalities: the total picture. 2nd ed. Mosby, St. Louis, MO, 2002: 15-6.
- Pinsker JE. Turner Syndrome: updating the paradigm of clinical care. J Clin Endocrinol Metab 2012, 97: E994-E1003.
- Gardner G, Shoback D. Greenspan’s basic & clinical endocrinology. Mc Graw Hill 9th Edition: 495-7.
- De Vos M, Devroey P, Fauser BCJM. Primary ovarian insufficiency. Lancet 2010, 376: 911-21.
- Schlessinger D, Herrera L, Crisponi L, et al. Genes and translocations involved in POF. Am J Med Genet 2002, 111: 328-33.
- Gardner G, Shoback D. Greenspan’s basic & clinical endocrinology. Mc Graw Hill 9th Edition, 2011: 497-9.
- Persani L, Rossetti R, Cacciatore C, Bonomi M. Primary Ovarian Insufficiency: X chromosome defects and autoimmunity. J Autoimmun 2009, 33: 35-41.
- Cools M, Pleskacova J, Stoop H, et al. Gonadal pathology and tumor risk in relation to clinical characteristics in patients with 45,X/46,XY mosaicism. J Clin Endocrinol Metab 2011, 96: E1171-80.
POF (Premature Ovarian Failure)
Cecilia Motta, Laura Proietti Pannunzi, Vincenzo Toscano
Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea, Roma
Definizione ed eziopatogenesi
Vari termini sono stati utilizzati per indicare la premature ovarian failure (POF), fra cui menopausa precoce e insufficienza ovarica primitiva (POI). Ultimamente la definizione più diffusa è insufficienza ovarica primitiva. Si caratterizza per la presenza di amenorrea, primaria o secondaria, elevati livelli plasmatici di FSH e ridotti livelli plasmatici di estrogeni in donne di età < 40 anni.
La prevalenza della POI è all’incirca di 1 su 10.000 donne di età < 20 anni, 1 su 1000 di età < 30 anni e 1 su 100 di età < 40 anni (1).
La POI è caratterizzata dalla perdita di ovociti, dalla mancanza della follicolo-genesi e della produzione di estrogeni ovarici e dall’infertilità. In alcuni casi è stata descritta la transitoria o parziale ripresa dell’attività ovarica (2).
Può essere dovuta a malattie autoimmuni, a danni ovarici da agenti esterni, a resistenza del tessuto ovarico all’azione delle gonadotropine, anche se, nella maggior parte dei casi, la causa è sconosciuta.
| Geni implicati nella POF (modificata da 8) | |||
| Categorie | Cromosoma | Gene | Locus genico |
| Mutazioni identificate | Geni sul cromosoma X | FMR1 | Xq27.3 |
| FMR2 | Xq28 | ||
| BMP15 | Xp11.2 | ||
| Geni su autosomi | FOXL2 | 3q22-q23 | |
| FSH-R | 2p21-p16 | ||
| LH-R | 2p21 | ||
| Variante ß-FSH | 11p13 | ||
| LH ß | 19q13.32 | ||
| Inibina A | 2q33-q36 | ||
| GALT | 9p13 | ||
| AIRE | 21q22.3 | ||
| EIF2b2,-4,-5 | 14q24.3, 2p23.3, 3q27 | ||
| NOGGIN | 17q22 | ||
| POLG | 15q25 | ||
| Geni candidati | Sul cromosoma X | DIAPH2 | Xq22 |
| DFFRX | Xp11.4 | ||
| XPNPEP2 | Xq25 | ||
| ZFX | Xq22.3-p21.3 | ||
| FSHPRH1 | Xq22 | ||
| XIST | Xq13.2 | ||
| Su autosomi | WT1 | 11p13 | |
| ATM | 11q22.3 | ||
| Mutazioni non identificate | Sul cromosoma X | AT2 | Xq22-23 |
| c-kit | 4q12 | ||
| SOX3 | Xq26-q27 | ||
| Su autosomi | MTS | 19p13.3-13.2 | |
Una delle alterazioni genetiche implicate nella POI è la condizione di portatore per la premutazione della sindrome della X fragile, un disordine X-linked che si associa a ritardo mentale. Il gene FMR1, localizzato sul braccio lungo del cromosoma X (Xq27) presenta ripetizioni della tripletta CGG nella regione 5’ UTR (untranslated): alleli con meno di 40 ripetizioni sono normali, alleli con ripetizioni fra 40 e 55 sono intermedi, alleli con ripetizioni fra 55 e 200 sono considerati “premutati”, i soggetti affetti dalla sindrome presentano più di 200 ripetizioni. Donne portatrici della “premutazione” presentano aumentato rischio di sviluppare insufficienza ovarica primitiva, probabilmente perché le aumentate concentrazioni intracellulari di mRNA possono sequestrare le proteine leganti CGG, importanti nell’elaborazione dell’RNA. Sembra che il rischio di sviluppare POI sia associato col numero di ripetizioni, e sia massimo sopra le 80 ripetizioni, fino ad un plateau di 100 ripetizioni, per poi decrescere con ripetizioni di numero superiore (3,4).
Altre alterazioni genetiche descritte sono le mutazioni di FMR2, non distante dal gene FMR1, e mutazioni inattivanti il recettore per l’FSH o il recettore per l’LH (3,5).
Esistono inoltre patologie sindromiche con difetti a carico degli autosomi, che possono associarsi a POI:
- Galattosemia: malattia genetica dovuta al deficit dell’enzima galattosio-1-fosfato uridil-trasferasi, con conseguente accumulo di galattosio. Ha un’incidenza di 1 su 60.000 nati ed è ereditata per via autosomica recessiva. A questa patologia si associata quasi sempre insufficienza ovarica, probabilmente dovuta all’apoptosi degli ovociti o delle cellule stromali a causa dell’effetto tossico dei metaboliti del galattosio (6).
- Atassia tele-angectasia: dovuta a mutazioni del gene ATM 3), è l'associazione tra un'immunodeficienza combinata grave (che interessa in particolare la risposta immuno-umorale) e un'atassia cerebellare progressiva. È caratterizzata da segni neurologici, tele-angectasie, suscettibilità alle infezioni e rischio elevato di sviluppare un tumore; si associa a menopausa precoce.
- Sindrome BPES tipo 1 (blefarofimosi-ptosi-epicanto-situs inversus): dovuta a mutazioni del gene FOXL2, che ha un ruolo importante nei primi stadi di differenziazione ovarica e nel mantenimento della funzione ovarica, oltre ad un presunto ruolo nel metabolismo del colesterolo e nella steroidogenesi (3).
- La POI può essere di natura autoimmune, causata da una disregolazione generalizzata del sistema immunitario, con associazione ad altremalattie autoimmuni:
- sindrome polighiandolare autoimmune (SPA) tipo I: è causata dalla mutazione del gene AIRE, localizzato sul cromosoma 21, che regola la funzione dei linfociti T, con manifestazioni cliniche sin dall’infanzia (candidiasi muco-cutanea, ipoparatiroidismo, insufficienza surrenalica, POI nel 60% dei casi) (7);
- SPA tipo II: associazione di insufficienza surrenalica, ipotiroidismo e POI (presente nel 10% dei casi) (7);
- SPA tipo III, caratterizzata da tireopatia autoimmune associata ad altre malattie autoimmuni (esclusi morbo di Addison e ipoparatiroidismo).
Il processo autoimmune può essere diretto contro antigeni specifici delle cellule germinali ovariche o dei fattori regolatori; sono stati inoltre descritti anticorpi circolanti diretti contro cellule steroidogeniche (3). Nelle ooforiti autoimmuni la fertilità può essere parzialmente conservata e può osservarsi un'insufficienza ovarica intermittente; in questi casi la secrezione di inibina B è conservata, perché l’infiltrato infiammatorio coinvolge le cellule della teca (7).
La POI può derivare, oltre che da ovariectomia, da insulti esterni, quali agenti virali e agenti tossici come il fumo, e, più frequentemente, chemio- o radioterapia eseguite per patologie neoplastiche; inoltre anche altri farmaci, tra cui alcuni utilizzati nel trattamento delle patologie autoimmuni o per prevenire il rigetto di organi trapiantati, possono portare a danno ovarico.
La chemioterapia può indurre un’alterazione degli stadi di maturazione dei follicoli e/o deplezione dei follicoli primordiali. Gli agenti alchilanti, come quelli utilizzati nel trattamento del linfoma di Hodgkin e nelle malattie autoimmuni, sono i farmaci più frequentemente associati a danno ovarico; l’azione citotossica di questi agenti, infatti, non richiede la presenza di proliferazione cellulare, pertanto essi possono distruggere gli ovociti quiescenti e probabilmente anche le cellule della granulosa del follicolo primordiale. Al contrario, gli anti-metaboliti, farmaci che solitamente si utilizzano nel trattamento delle neoplasie mammarie, causano danni minori in quanto agiscono su cellule in proliferazione (3).
Il tessuto ovarico è inoltre estremamente sensibile al danno indotto da radiazioni. Fattori di rischio per lo sviluppo di POI da radiazione sono l’età della paziente, nonché l’estensione ed il tipo di radioterapia eseguita: una dose singola elevata è più dannosa per gli ovociti rispetto a multiple dosi frazionate, e ovviamente l’irradiazione addominale e pelvica si associa più frequentemente a POI, anche se l’irradiazione scatter può causare danni anche quando il tessuto ovarico non è nel campo d’irradiazione (3).
Clinica e diagnostica
La presentazione tipica della POF è amenorrea secondaria o oligomenorrea in donna con meno di 40 anni, accompagnata o meno da vampate.Nella maggior parte dei casi non ci sono segni o sintomi evidenti che precedono la scomparsa deicicli mestruali(9); in genere le pazienti riferiscono normale sviluppo puberale, con menarca e adrenarca in età fisiologica e con cicli successivi regolari, fino all'inizio dei sintomi dell’insufficienza ovarica. Spesso le pazienti si rivolgono al medico per la mancata ripresa del ritmo mestruale dopo una gravidanza o dopo l’assunzione di un estroprogestinico orale.
La diagnosi di tale condizione può essere difficoltosa, considerando che non esistono ancora criteri standardizzati per quanto riguarda la durata dell’amenorrea e il grado di elevazione delle gonadotropine (10). L'assenza di criteri standardizzati rappresenta un problema di non poco conto per il medico, che deve decidere quanto attendere prima di richiedere accertamenti in donne che si rivolgono a lui per assenza del flusso mestruale. Raccomandazione generale è che le giovani donne che manifestano amenorrea per un periodo di 3 o più mesi consecutivi dovrebbero avere una valutazione adeguata già al momento della loro prima visita (11), mediante anamnesi dettagliata, senza tralasciare la possibile storia familiare di POF. La maggior parte degli autori suggerisce che per porre diagnosi sia necessaria una condizione di amenorrea di almeno 3-6 mesi e livelli di gonadotropine compresi fra 10 e 20 UI/L. Tuttavia, Tibiletti et al suggeriscono di utilizzare i seguenti criteri: amenorrea da almeno 6 mesi ed FSH > 40 UI/L in due misurazioni effettuate ad almeno 4-6 settimane l'una dall'altra (12). I livelli di E2 sono in genere bassi, < 50 pg/mL, e si associano a follicoli assenti o non funzionanti (13); vanno comunque escluse altre possibili cause di amenorrea, quali gravidanza, sindrome dell’ovaio policistico, iperprolattinemia e distiroidismi. Valori di FSH maggiori dell’LH aiutano a distinguere la POF dal picco pre-ovulatorio dell’LH.
Una pratica comune è quella di eseguire un test di stimolo con progestinico. Si somministrano 10 mg di medrossiprogesterone acetato per 5 giorni consecutivi: in presenza di adeguati livelli di estrogeni endogeni, comparirà un sanguinamento nei 2-5 giorni successivi. Nel caso di POF di solito il test è negativo, a causa della carenza degli steroidi sessuali. Bisogna però tener conto che il test può essere fuorviante, sia in condizioni di funzione ovarica intermittente, sia poiché il 50% delle donne con POF può avere una condizione di ipo-estrogenismo, soprattutto nelle fasi iniziali della malattia; questo test potrebbe quindi portare a un ritardo della diagnosi (10).
Quando si sospetta una POF può essere utile la valutazione della riserva follicolare con il dosaggio di altri peptidi di origine ovarica, quali l’inibina B e l’ormone anti-mülleriano (AMH). Bassi livelli di inibina B possono predire la deplezione follicolare prima che si verifichi l’aumento dell’FSH. L’AMH sembra essere il marcatore più affidabile della riserva ovarica ed è il meno influenzato dalla fase del ciclo mestruale (14).
Dal punto di vista metabolico, le pazienti affette da POF spesso presentano un profilo lipidico pro-aterogeno e una maggiore prevalenza di alterata glicemia a digiuno (con progressione al diabete mellito tipo 2). L’ipertensione arteriosa, più comunemente sistolica e spesso notturna, è presente nel 25% delle adolescenti e nel 50% delle adulte affette da POF.
Nonostante la terapia sia la stessa per le varie forme di POF, è importante capire quale sia l’eziologia dell’insufficienza ovarica e per tale motivo l’anamnesi rimane un punto fondamentale. Qualora si sospetti una base autoimmunitaria, per la presenza di altre patologie autoimmuni, è importante eseguire la valutazione degli anticorpi anti-ovaio. Anche se questi sono stati trovati nel 10-69% delle donne con POF, questo dato non è di particolare utilità, in quanto la positività si trova anche in un numero significativo di controlli (15). La forte associazione di POF con la sindrome polighiandolare autoimmune (SPA) rende essenziale lo screening per questa condizione.
Bibliografia
- Coulam CB, Adamson SC, Annegers JF. Incidence of premature ovarian failure. Obstet Gynecol 1986, 67: 604-6.
- Taylor AE, Adams JM, Mulder JE, et al. A randomized, controlled trial of estradiol replacement therapy in women with hypergonadotropic amenorrhea. J Clin Endocrinol Metab 1996, 81: 3615-21.
- De Vos M, Devroey P, Fauser BCJM. Primary ovarian insufficiency. Lancet 2010, 376: 911-21.
- Wittenberger M, Hagerman R, Sherman S, et al. The FMR1 premutation and reproduction. Fertil Steril 2007, 87: 456-65.
- Laml T, Preyer O, Umek W, et al. Genetic disorders in premature ovarian failure. Hum Reprod Update 2002, 8: 483-91.
- Berry G. Galactosemia and amenorrhea in the adolescent. Ann NY Acad Sci 2008, 1135: 112-7.
- Persani L, Rossetti R, Cacciatore C, Bonomi M. Primary ovarian insufficiency: X chromosome defects and autoimmunity. J Autoimmun 2009, 33: 35-41.
- Goswami D, Conway GS. Premature ovarian failure. Hum Reprod Update 2005, 11: 391-410.
- Woad KJ, Watkins WJ, Prendergast D, Shelling AN. The genetic basis of premature ovarian failure. Aust N Z J Obstet Gynaecol 2006, 46: 242-4.
- Panay N, Kalu E. Management of premature ovarian failure. Best Pract Res Clin Obstet Gynaecol 2009, 23: 129-40.
- Van Karseren YM, Schoemaker J. Premature ovarian failure: a systematic review on therapeutic interventions to restore ovarian function and achieve pregnancy. Hum Reprod Update 1999, 5: 483–92.
- Tibiletti MG, Testa G, Vegetti W, et al. The idiopathic forms of premature menopause and early menopause show the same genetic pattern. Hum Reprod 1999, 14: 2731-4.
- Rebar RW. Premature ovarian failure. Obstet Gynecol 2009, 113: 1355-63.
- Broekmans FJ, Kwee J, Hendriks DJ, et al. A systematic review of tests predicting ovarian reserve and IVF outcome. Hum Reprod Update 2006, 12: 685–718.
- Wheatcroft NJ, Salt C, Milford-Ward A, et al. Identification of ovarian antibodies by immunofluorescence, enzyme-linked immunosorbent assay or immunoblotting in premature ovarian failure. Hum Reprod 1997, 12: 2617–22.
Approccio terapeutico all’ipogonadismo primario femminile
Cecilia Motta, Laura Proietti Pannunzi, Vincenzo Toscano
Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea, Roma
L’ipogonadismo ipergonadotropo si caratterizza per la presenza di bassi livelli di estrogeni in fasi della vita in cui questi dovrebbero invece essere presenti. Tale carenza espone le pazienti affette a un incrementato rischio di morbilità e mortalità: se non adeguatamente trattate, possono incorrere in osteoporosi, patologie cardiovascolari, decadimento cognitivo, parkinsonismi (1).
Come già detto per l’ipogonadismo ipogonadotropo, determinata la causa eziopatogenetica, è fondamentale tener conto di alcuni aspetti: l’età della paziente, la presenza/assenza dei caratteri sessuali secondari e il loro grado di sviluppo, la possibilità o meno di ripristinare la fertilità (2).
È inoltre necessaria una corretta informazione circa la condizione clinica e un’attività di counseling, che aiuti la paziente nell’accettazione della propria patologia.
Pazienti in età fertile che devono esser sottoposte a chemio/radioterapia o terapie anti-rigetto andrebbero indirizzate a centri specialistici dove poter ricevere le informazioni adeguate circa la possibilità di eseguire una crioconservazione del tessuto ovarico. Questa è una tecnica in grado di permettere una gravidanza futura a donne affette da malattie prevalentemente oncologiche e in cui la malattia stessa, o le terapie messe in atto per curarla, compromettono in modo grave il patrimonio ovocitario (3). La crioconservazione del tessuto ovarico può essere presa in considerazione in tutte le pazienti affette da neoplasie maligne, purché non presentino nessuna delle condizioni seguenti:
- età > 38 anni (perché, dopo questa età, la quantità e la qualità degli ovociti presenti nella corteccia ovarica sono, di massima, così basse da ridurre in modo critico il successo delle procedure di successivo utilizzo del tessuto ovarico prelevato);
- positività alle infezioni da HBV, HCV, HIV, sifilide (per i problemi di sicurezza nella conservazione del tessuto ovarico);
- condizioni cliniche che comportino la presenza di rischio chirurgico o anestesiologico superiore a quello generico (a causa del tempo chirurgico laparoscopico).
Per la terapia in età adolescenziale cfr ipogonadismi ipogonadotropi.
Per età adulta cfr terapia ipogonadismi ipogonadotropi, fatta eccezione della terapia per l’induzione della fertilità.
Bibliografia
- Vujovic S, Brincat M, Erel T, et al; European Menopause and Andropause Society. EMAS position statement: managing women with premature ovarian failure. Maturitas 2010, 67: 91-3.
- De Vos M, Devroey P, Fauser BCJM. Primary ovarian insufficiency. Lancet 2010, 376: 911-21.
- Goswami D, Conway GS. Premature ovarian failure. Hum Reprod Update 2005, 11: 391-410.
Acne
Roberto Castello1 & Cecilia Motta2
1Medicina/Endocrinologia, AOUI Verona
2Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea, Roma
(aggiornato al 31 dicembre 2015)
L'acne presenta una prevalenza di circa l’85% fra i 12 e i 24 anni; inoltre, può anche persistere nell'età adulta nonostante precedenti trattamenti, influendo negativamente sulla qualità della vita.
Nelle linee guida canadesi riguardanti la terapia dell’acne, redatte principalmente da dermatologi, si distinguono tre categorie di acne:
- acne comedonica: piccole papule bianche (comedoni chiusi) oppure grigio-bianche (comedoni aperti) causate dall'occlusione completa o parziale dei dotti con accumulo di sebo;
- acne papulo-pustolosa di grado lieve-moderato: lesioni infiammatorie per lo più superficiali;
- acne di grado severo: pustole profonde o noduli, talora dolorosi, talora estesi, fino a distruggere il tessuto cutaneo; un sottotipo dell'acne di grado severo, molto raro, è l’acne conglobata, consistente in noduli infiammatori e cisti che possono residuare in cicatrici.
Le linee guida hanno preso in considerazione il trattamento dell'acne volgare in età adulta e adolescenziale, escludendo le forme a patogenesi diversa (neonatale, infantile, late-onset, fulminante, follicoliti, rosacea, ecc). Poichè non esistono studi clinici riguardanti l'acne comedonica, le raccomandazioni sul trattamento di questa forma sono state estrapolate dai dati sulle lesioni acneiche non infiammatorie presenti negli studi eseguiti sull’acne di grado lieve-moderato.
La terapia di prima linea più appropriata dovrebbe essere scelta sulla base di eventuali precedenti terapie, avendo informazioni sulla loro efficacia e su come sono state tollerate, sulla base del tipo di pelle del paziente, del veicolo utilizzato, e della praticità di applicazione/somministrazione (localizzazione dell’acne, frequenza di applicazione), oltre ai costi del farmaco. L'efficacia del trattamento andrebbe valutata dopo almeno 2-3 mesi di terapia.
Acne comedonica
Come prima linea di terapia vengono raccomandate terapie topiche:
- monoterapie a base di retinoidi (tretinoina, adapalene e tazarotene) o di benzoil-perossido (raccomandazione di media forza con dubbi di grado moderato sull’efficacia);
- combinazioni a dosi fisse di benzoil-perossido con retinoidi o clindamicina (raccomandazione di media forza con dubbi di grado moderato sull’efficacia).
I pazienti con pelle secca o molto sensibile potrebbero preferire creme o lozioni, mentre quelli con pelle grassa potrebbero preferire gel.
Il benzoil-perossido in gel, sia al 2.5% che al 5%, si è dimostrato più efficace del veicolo (placebo) e ha una rapida insorgenza d'azione.
Adapalene e tazarotene, ugualmente efficaci, sono risultati superiori nel trattamento dell’acne comedonica rispetto alla tretinoina; il tazarotene può essere più irritante.
La combinazione fra adapalene 0.1% e benzoil-perossido 2.5% è risultata di uguale o superiore efficacia rispetto alla monoterapia con adapalene o con benzoil-perossido; allo stesso modo la combinazione fra clindamicina 1% e benzoil-perossido 5% è risultata superiore rispetto alla sola clindamicina ed equivalente al solo benzoil-perossido.
Se la combinazione a dosi fisse non porta un miglioramento dopo 2-3 mesi di applicazioni, si consiglia di aggiungere un retinoide topico (soprattutto tazarotene o adapalene).
Se la risposta alla monoterapia con retinoide topico o benzoil-perossido oppure a una delle combinazioni a dosi fisse già discusse fosse inadeguata, le LG consigliano di prendere in considerazione la terapia di combinazione fra clindamicina 1.2% e tretinoina 0.025% (gel) e, nella donna, la terapia con contraccettivi orali (raccomandazione di scarsa forza con dubbi di grado elevato sull’efficacia).
Acne papulo-pustolosa di grado lieve-moderato, localizzata
Anche per questo tipo di acne le terapie topiche sono una terapia ragionevole. Per il trattamento delle lesioni infiammatorie sono fortemente raccomandate tutte e tre le opzioni terapeutiche:
- monoterapie a base di retinoidi o di benzoil-perossido (raccomandazione di elevata forza con dubbi di grado lieve sull’efficacia);
- combinazioni a dosi fisse di clindamicina 1% con benzoil-perossido 5% oppure di adapalene 0.1% con benzoil-perossido 2.5% in gel (raccomandazione di elevata forza con dubbi di grado lieve sull’efficacia) oppure clindamicina 1.2% con tretinoina 0.025% (raccomandazione di scarsa forza con dubbi di grado elevato sull’efficacia).
Per acne più estesa o localizzata in aree corporee non facilmente raggiungibili, si raccomanda di utilizzare terapie sistemiche in aggiunta alla terapia topica.
Il benzoil-perossido 2%, 5% e 10% si è dimostrato superiore rispetto al placebo e il suo effetto sembra essere più rapido rispetto a quello della tretinoina.
In prima linea possono essere utilizzati anche retinoidi topici: adapalene 0.1% e 0.3%, tazarotene, trenitoina 0.025%, 0.04% e 0.05% sono più efficaci del placebo sulle lesioni infiammatorie; mentre il tazarotene si è dimostrato superiore rispetto alla tretinoina sulle lesioni comedoniche, esso appare equivalente alla tretinoina sulle lesioni infiammatorie.
Entrambe le combinazioni si sono dimostrate superiori rispetto al placebo e la combinazione contenente clindamicina si è anche dimostrata superiore rispetto ai due principi attivi in monoterapia.
Acne papulo-pustolosa di grado lieve-moderato, estesa
Oltre alle terapie topiche già raccomandate sopra, le LG raccomandano l'aggiunta di:
- un antibiotico per via sistemica (raccomandazione di media forza con dubbi di grado moderato sull’efficacia);
- un contraccettivo orale (raccomandazione di media forza con dubbi di grado moderato sull’efficacia).
Tetraciclina, doxiciclina e minociclina si sono dimostrate superiori rispetto al placebo nel ridurre le lesioni di tipo infiammatorio; l’utilizzo di questi agenti in monoterapia è sconsigliato per il rischio di selezione di batteri antibiotico-resistenti. Poiché la minociclina potrebbe essere associata a un aumentato rischio di lupus ed epatite, si consiglia di preferire tetraciclina o doxiciclina.
È stato dimostrato che le combinazioni di etinil-estradiolo con levonorgestrel, con drospirenone, con norgestimate e con ciproterone acetato sono tutte similmente efficaci nel trattamento dell'acne.
Acne di grado severo
Le LG raccomandano fortemente l'utilizzo di:
- isotretinoina per via orale (raccomandazione di elevata forza con dubbi di grado lieve sull’efficacia);
- antibiotici per via sistemica in combinazione con benzoil-perossido con o senza retinoidi topici (raccomandazione di media forza con dubbi di grado moderato sull’efficacia).
L’isotretinoina per via orale è fortemente raccomandata nonostante vi sia un unico trial controllato con placebo eseguito sull'utilizzo di isotretinoina nell'acne conglobata di grado severo. Vista la potenzialità di effetti collaterali e di teratogenicità, tale terapia va maneggiata da specialisti esperti e va integrata con misure contraccettive.
La terapia combinata è consigliata in pazienti che non possono fare uso della isotretinoina.
Inoltre nelle donne potrebbe essere considerata anche la terapia con contraccettivi orali.
Punti rimasti insoluti:
- mancano standard universali per quantificare la severità dell’acne;
- non vi sono alti livelli di evidenza per i trattamenti che sono utilizzati più frequentemente;
- non è nota l’efficacia minima delle terapie, tale da diventare rilevante sulla qualità di vita dei pazienti;
- non vi sono certezze circa la durata dell’utilizzo degli antibiotici per via orale, anche per minimizzare lo sviluppo di batteri resistenti;
- non vi sono adeguate informazioni relative all'efficacia di queste terapie sull’acne localizzata al dorso, infatti quasi tutti gli studi hanno valutato l’acne localizzata al volto.
| Preparati a disposizione in commercio | ||
| Principio attivo | Nome commerciale | Veicolo |
| Benzoil Perossido | Benzac 5%-10%, Benzac clean 5%, | gel |
| Panoxil 4% | crema | |
| Adapalene | Differin 0.1% | gel, crema |
| Tretinoina | Airol 0.05% | soluzione cutanea, crema |
| Tretinoina Same 0.05% | crema | |
| Vesanoid 10 mg | cps molli | |
| Tazarotene | Zorac 0.05%, 0.1% | gel acquoso |
| Adapalene + Benzoil Perossido | Epiduo 1%, 2.5% | gel |
| Clindamicina 5% + Benzoil Perossido 1% | Duac | gel |
| Isotretinoina | Aisoskin 10 mg, 20 mg | cps molli |
| Isdiben 5 mg, 10 mg, 20 mg, 40 mg | cps molli | |
| Isoriac 10 mg, 20 mg | cps molli | |
| Isotretinoina Difa Cooper 10 mg, 20 mg, 40 mg | cps molli | |
| Isotretinoina Stiefel 0.05% | crema | |
| Isotrex 0.05% gel | gel | |
Asai Y, Baibergenova A, Dutil M, et al. Management of acne: Canadian clinical practice guideline. CMAJ 2015, DOI:10.1503 /cmaj.140665
Clinica e diagnostica iperandrogenismi e irsutismi
Roberto Castello, Medicina/Endocrinologia, AOUI Verona
Francesca Zambotti, Endocrinologia e Malattie del Metabolismo, AOUI Verona
DEFINIZIONE, EPIDEMIOLOGIA, EZIOLOGIA
Iperandrogenismo è un termine usato per descrivere i più comuni segni clinici presenti nelle donne con iperandrogenemia, ossia irsutismo, acne, alopecia androgenetica e virilizzazione, ma indica anche lo stato iperandrogenico sottostante che ne è l’origine (1).
E’ una patologia endocrina frequente, con prevalenza del 5-10% nella popolazione generale delle donne in età fertile (2). L’espressione clinica dell’iperandrogenismo coinvolge da un lato l’unità pilo-sebacea con irsutimo, acne e alopecia, e, dall’altro, la funzione riproduttiva, con irregolarità mestruali e infertilità.
L’iperandrogenismo può essere una spia di patologie cardiovascolari, come ipertensione arteriosa, microangiopatia, dislipidemia ed altri disordini metabolici come il diabete mellito.
L’iperandrogenismo è di solito dovuto a iperproduzione di androgeni, che può derivare dall’ovaio, dal surrene o da entrambi, o dipendere da un’alterazione del recettore per gli androgeni sui tessuti bersaglio o da alterazioni della conversione periferica di precursori steroidei in androgeni.
| Distribuzione di frequenza delle diverse cause di iperandrogenismo | ||
| Cause | Patologia | Frequenza |
| S. dell’ovaio policistico (PCOS) | 70-80% | |
| Irsutismo idiopatico | 10-15% | |
| Surrenaliche |
Iperplasie surrenaliche congenite a insorgenza tardiva |
1-5% |
| Ovariche |
Difetti della steroidogenesi ovarica: tumori a cellule di Leydig, tumori cistici benigni, tumori a cellule steroidee, tumori a cellule ilari |
|
| Insulino-resistenza | HAIRAN syndrome Difetti genetici di azione insulinica (leprecaunismo, s. di Rabson Mendenhall, lipodistrofie) |
|
| Ipofisarie | Acromegalia Iperprolattinemia |
|
| Tiroidee | Ipotiroidismo Ipertiroidismo |
|
| Esogene | Assunzione di androgeni | |
Nel recente consensus statement dell’AE-PCOS Society si propongono cinque principali patologie da eccesso di androgeni (3):
- PCOS, se è presente iperandrogenismo clinico e/o biochimico associato a disfunzione ovulatoria e/o ad ovaio policistico;
- iperandrogenismo idiopatico, se è presente iperandrogenismo clinico e/o biochimico con regolari cicli ovulatori e normale morfologia ovarica;
- irsutismo idiopatico, che è la presenza d’irsutismo in donne con normali livelli ematici di androgeni e regolari cicli ovulatori in assenza di ovaio micropolicistico;
- iperplasia surrenalica congenita a insorgenza tardiva;
- neoplasie secernenti androgeni.
Un prolattinoma può stimolare la produzione ovarica di androgeni (4).
Il termine ipertecosi è utilizzato per indicare un quadro anatomo-patologico caratterizzato dalla presenza di gruppi di cellule tecali luteinizzate distribuiti in tutto lo stroma ovarico (5). Alcuni autori includono tale quadro nella PCOS, sulla base del fatto che aspetti istologici di questo tipo possono essere talora riscontrati in donne con tale condizione. Altri autori utilizzano il termine solo per i quadri clinicamente eclatanti, che sono inusuali, e considerano ipertecosi e PCOS come due entità distinte. Il quadro clinico tipico è quello di un’intensa androgenizzazione che può arrivare alla virilizzazione, con livelli di androgeni circolanti chiaramente elevati, ovaie ingrandite e di consistenza aumentata, in genere senza follicoli. I livelli di LH tendono ad essere meno elevati rispetto a quanto si riscontra nella PCOS. In questi casi spesso è presente anche una severa insulino-resistenza con alterazioni metaboliche. In molti casi vi è obesità centrale e talora acanthosis nigricans. Questo quadro si può osservare anche in epoca post-menopausale e va differenziato dai tumori ovarici androgeno-secernenti. La diagnosi definitiva è anatomo-patologica (6).
HAIRAN Syndrome (Hyperandrogenic-Insulin Resistant-Acanthosis Nigricans) raggruppa alcune differenti sindromi, di solito ereditarie, che si associano a gravi alterazioni dell’azione insulinica per difetti recettoriali o post-recettoriali del segnale insulinico. Il compensatorio aumento dell’insulina circolante assieme all’LH stimola la secrezione di un eccesso di androgeni dall’ovaio, simile a quella indotta da neoplasie secernenti androgeni. Molto frequentemente si riscontra la presenza di acanthosis nigricans. I pazienti hanno un’elevata incidenza di diabete mellito, ipertensione e patologie cardiovascolari (7).
MANIFESTAZIONI CLINICHE DI IPERANDROGENISMO
Irsutismo
Acne
L’acne è comune nelle adolescenti, tanto da essere presente in quasi il 50% di esse, ma la persistenza oltre i venti anni pone il sospetto d’iperandrogenismo, specialmente se resistente alle terapie dermatologiche o se associata con irsutismo ed irregolarità mestruali. Poiché la suscettibilità dell’unità pilo-sebacea alla stimolazione androgenica è variabile, risulta variabile anche l’espressione clinica dell’iperandrogenismo, senza che si possa stabilire alcuna correlazione fra l’entità dell’acne ed i livelli di testosterone sierico (1).
Alopecia androgenetica
È la perdita di capelli mediata dagli androgeni, i quali riducono la durata dell’anagen, causando così il progressivo assottigliamento del follicolo pilifero fino alla caduta del pelo. Tipicamente la perdita di capelli avviene al vertice cranico, ma può interessare la corona laterale e infine produrre una perdita diffusa.
Le donne con alopecia non mostrano in genere elevati livelli circolanti di androgeni, ma un’aumentata espressione della 5α-reduttasi e dei recettori per gli androgeni. Sono inoltre ridotti i livelli di citocromo P450 necessario alla conversione degli androgeni in estrogeni.
Altre possibili cause di alopecia sono la predisposizione genetica, carenze nutrizionali, cali ponderali rapidi, anemia, disfunzioni tiroidee, alcuni farmaci come danazolo, anabolizzanti e isotriteonina, infezioni, malattie autoimmuni, traumi, neoplasie.
Per una classificazione clinica dell’entità del problema si utilizza la scala di Ludwig (8, 9).

Virilizzazione
E’ una manifestazione clinica rara, caratterizzata da ipertrofia clitoridea (prodotto del diametro traverso e sagittale del glande del clitoride > 35 mm2), approfondimento del tono di voce in senso maschile, ipertrofia muscolare, atrofia mammaria, irsutismo severo, alopecia androgenetica, fisionomia androgina, amenorrea (2). Tipicamente si associa a livelli di androgeni molto elevati, attribuibili a tumori secernenti androgeni surrenalici o ovarici, ipertecosi o iperplasia congenita surrenalica. Il sospetto si pone per la rapida progressione delle manifestazioni d’iperandrogenismo (1, 10).
DIAGNOSI
Anamnesi:
- anamnesi familiare: calvizie precoce (< 35 anni) nei parenti maschi;
- sviluppo puberale: età al telarca, adrenarca e menarca;
- storia mestruale: frequenza e durata dei cicli mestruali;
- anamnesi ginecologica ed ostetrica;
- storia delle manifestazioni di iperandrogenismo: età e modalità d’insorgenza e di progressione nel tempo di irsutismo, acne, seborrea, alopecia;
- andamento del peso;
- anamnesi farmacologica;
- anamnesi patologica, in particolare in merito a galattorrea e disfunzioni tiroidee.
Disfunzione ovulatoria
Ci sono vari gradi di disfunzioni ovulatoria che possono portare a infertilità e clinicamente si manifestano con irregolarità mestruali, come oligo-amenorrea, menorragia, metrorragia. Un certo numero di pazienti con iperandrogenismo può presentarsi con cicli regolari, ma avere cicli oligo-ovulatori e allungato tempo di concepimento (1).
Impatto psicologico
L’iperandrogenismo può avere effetti psico-sociali profondi in adolescenti e giovani donne in età fertile, portando ansia e depressione. La compresenza di obesità può avere un effetto additivo negativo sull’autostima e sull’immagine di sé. La paura del giudizio altrui può portare alcune donne all’auto-isolamento e ritardare lo sviluppo delle capacità sociali (1).
Esame obiettivo (11):
- parametri vitali e antropometrici: BMI, circonferenza vita, circonferenza fianchi;
- irsutismo: score di Ferriman-Gallwey, frequenza delle depilazioni;
- seborrea ed acne: sedi e entità;
- alopecia: tipo ed entità;
- virilizzazione: visita ginecologica;
- segni di iperinsulinismo o di altre patologie endocrine: acanthosis nigricans, obiettività tiroidea, galattorrea, strie rubrae.
Valutazione laboratoristica
È volta ad individuare l’origine dell’iperproduzione di androgeni. I dosaggi ormonali devono essere sempre fatti in fase follicolare precoce o dopo tre mesi di amenorrea.
Indagini di secondo livello:
- test di soppressione con desametasone ad alte dosi;
- test di stimolo con ACTH: se 17OH-progesterone basale > 2 ng/mL (6 nmol/L);
| Test di stimolo con ACTH | |
| 17-OH-progesterone a 60 minuti dallo stimolo | > 9 ng/mL: iperplasia surrenalica non classica (late onset 21-hydroxylase deficiency) |
| 3.5 - 9 ng/mL: forma eterozigote del difetto di 21-idrossilasi | |
- GnRH test: utile solo negli ambienti di ricerca nel quadro di PCOS (1);
- OGTT per glicemia ed insulinemia;
- Clamp euglicemico iperinsulinemico: utile solo negli ambienti di ricerca.
Esami strumentali:
- ecografia pelvica ed eventuale TC ovarica;
- ecografia surrenalica ed eventuale TC surrenalica;
- cateterismo delle vene surrenaliche nei casi dubbi.
Negli esseri umani si distinguono tre tipi di pelo (2):
- lanugo: peluria che ricopre il feto;
- vello: pelo sottile, di solito non più lungo di 2 mm, scarsamente o non pigmentato, che ricopre le aree cutanee apparentemente prive di peli;
- pelo terminale: lungo, duro, pigmentato, si trova sia in aree non steroido-dipendenti (cuoio capelluto, ciglia e sopracciglia), sia in aree dipendenti dagli steroidi sessuali.
Il pelo ha un proprio ciclo vitale, suddivisibile nelle fasi:
- anagen: fase di crescita che per i peli corporei può durare 3-6 mesi, per i capelli 2-6 anni;
- catagen: fase stazionaria di progressivo rallentamento della crescita;
- telogen: fase di quiescenza;
- caduta del pelo.
Si definisce irsutismo l’eccessiva presenza nella donna di pelo terminale a distribuzione maschile. Si sviluppa quando i follicoli piliferi presenti in aree cutanee androgeno-sensibili iniziano a produrre pelo terminale al posto del vello normalmente presente nella donna in tali aree (12). L’irsutismo deve essere distinto dall’ipertricosi, che si definisce come eccessiva crescita di vello su tutta la superficie corporea, la cui presenza è influenzata soprattutto da fattori ereditari o è indotta da fattori iatrogeni (fenitoina, diazossido, minoxidil, ciclosporina, glucocorticoidi), squilibri metabolici (anoressia nervosa) o irritazioni cutanee.
La prevalenza dell’irsutismo è di circa 4.3-10.8% in neri e caucasici, inferiore negli asiatici (3). Poiché il 70-80% delle donne con iperandrogenismo presenta irsutismo (13), questo è il più affidabile criterio clinico per la diagnosi d’iperandrogenismo.
Gli androgeni sono importanti nel determinare sia il tipo, sia la distribuzione del pelo sulla superficie corporea (2). Sono essenziali nell’indurre la trasformazione del vello in pelo terminale e prolungano la fase anagen (11). Nell’irsutismo l’attività della 5alfa-reduttasi e la produzione di DHT dal testosterone sono aumentate (11), ma l’irsutismo non dipende solo dalle concentrazioni di androgeni circolanti, bensì anche dalle concentrazioni locali di androgeni e dalla sensibilità del follicolo pilifero agli androgeni, che è diversa nelle varie aree cutanee. Ascelle e pube richiedono basse concentrazioni di androgeni per la trasformazione da vello a pelo terminale, altre aree di pilificazione richiedono concentrazioni maggiori (2).
Non esiste quindi una diretta correlazione fra l’entità dell’irsutimo e le concentrazioni di androgeni circolanti. Infatti, pazienti con grave iperandrogenemia possono non avere irsutismo e, al contrario, donne con grave irsutismo possono avere livelli di androgeni circolanti assolutamente nella norma (3). Anche altri ormoni, fra cui GH e ormoni tiroidei, influenzano la crescita dei peli (2).
Eziopatogenesi
Varie sono le possibili cause (vedi tabella eziologia dell’iperandrogenismo) (7,14-16).
| Distribuzione del pelo terminale in diverse condizioni endocrine | ||||||
| Situazione | Tipo di pelo terminale | |||||
| Palpebre | Scalpo | Dorsale | Pubico | Addomino-toracico | Facciale | |
| Bambino normale | + | + | - | - | - | - |
| Femmina adulta normale | + | + | + | + | - | - |
| Iperandrogenismo femminile | + | + | + | + | ± | ± |
| Maschio adulto | + | + | + | + | + | + |
| Deficit 5-alfa-reduttasi tipo II | + | + | + | + | - | - |
| Deficit del recettore androgenico | + | + | - | - | - | - |
Stanczyk FZ. Diagnosis of hyperandrogenism: biochemical criteria. Best Pract Res Clin Endocrinol Metab 2006, 20: 177-91
Approccio alla donna con irsutismo
L’approccio diagnostico nella paziente con irsutismo prende in considerazione cinque principali patologie da eccesso di androgeni, cfr. consensus statement dell’AE-PCOS Society (3), come già riportato precedentemente.
Diagnosi clinica
Molte donne che si lamentano di un eccesso di peli non soffrono in realtà di vero irsutismo. Si può considerare nella norma la presenza di pochi peli terminali visibili sul volto, attorno alle areole mammarie, sulla parte inferiore del dorso e dell’addome, mentre è indice d’iperandrogenismo la presenza di peli terminali sulla parte superiore del dorso e sul torace (2). Il metodo più diffuso di valutazione clinica dell’entità e della distribuzione dell’irsutismo è il punteggio di Ferriman-Gallwey (17,18) nella sua versione modificata, che prevede la valutazione di 9 aree delle 11 originariamente proposte da Ferriman e Gallwey, assegnando ad ognuna un punteggio da 0 (assente crescita) a 4 (estesa presenza). Gamba ed avambraccio sono esclusi dalla valutazione. Nonostante sia il metodo più usato in clinica, ha molte limitazioni, fra le quali il fatto di essere un metodo soggettivo, semi-quantitativo, con notevole variabilità inter-individuale e che non esiste ancora esplicito accordo sul valore da considerare patologico anche in considerazione delle notevoli differenze etniche nella pilificazione corporea. Attualmente, la maggior parte dei clinici indica come cut-off per la definizione di irsutismo un punteggio ≥ 8, valore che risulta valido per caucasici e neri, ma che andrebbe forse ridotto a 3 negli asiatici basandosi sul punteggio corrispondente al 95° centile di una popolazione di donne in età fertile (3).
L’irsutismo si definisce:
- lieve per punteggio fino a 15
- moderato fra 16 e 25
- severo fra 25 e 36.
Conseguenze metaboliche e cardiovascolari dell’iperandrogenismo e dell’irsutismo
Il rischio di avere alterazioni metaboliche e dei marcatori di rischio cardiovascolare nella donna con irsutismo correla con la severità dell’iperandrogenemia (19) (vedi capitolo PCOS). E’ stato suggerito di indagare il profilo di rischio cardiovascolare nelle donne con patologie da lieve iperandrogenismo solo attraverso la valutazione di parametri come BMI, circonferenza vita e fianchi e pressione arteriosa, riservando solo alle donne obese o che presentano anche altri fattori di rischio cardiovascolare approfondimenti più specifici, come profilo lipidico, indici di tolleranza glucidica e di insulino-resistenza (3).
BIBLIOGRAFIA
- Goodman NF. AACE Medical Guidelines for the Diagnosis and Treatment of Hyperandrogenic Disorders. Endocr Pract 2001, 7: 120-34.
- Yildiz BO. Diagnosis of hyperandrogenism: clinical criteria. Best Pract Res Clin Endocrinol Metab 2006, 20: 167-76.
- Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 2012, 18: 146-70.
- Legro RS. Evaluation and Treatment of Polycystic Ovary Syndrome. 19 September 2009, Endotext http://www.endotext.org/female/female6/femaleframe6.htm
- Culiner A, Shippel S. Virilism and theca cell hyperplasia of the ovary: a syndrome. J Obstet Gynaecol Br Commonw 1949, 56: 439-45.
- Castello R, Moghetti P. Manuale per valutazione e inquadramento delle patologie gonadiche. AME 2009
- The Practice Committee of the American Society for Reproductive Medicine. The evaluation and treatment of androgen excess. Fertil Steril 2006, 86 (S 4): S241-7.
- Rebora A. Pathogenesis of androgenetic alopecia. J Am Acad Dermatol 2004, 50: 777-9.
- Ludwig E. Classification of the types of androgenetic alopecia (common baldness) occurring in the female sex. Br J Dermatol 1977, 97: 247-54.
- Marshburn PB, Carr BR. Hirsutism and virilization. A systematic approach to benign and potentially serious causes. Postgrad Med 1995, 97: 99-102.
- Stanczyk FZ. Diagnosis of hyperandrogenism: biochemical criteria. Best Pract Res Clin Endocrinol Metab 2006, 20: 177-91.
- Rosenfield RL. Hirsutism. N Engl J Med 2005, 353: 2578-88.
- Azziz R, Carmina E, Sawaya ME. Idiopathic hirsutism. Endocr Rev 2000, 21: 347-62.
- Randall B, Barnes MD, et al. Hyperandrogenism, hirsutism and polycystic ovary syndrome. 2003 Endotext http://www.endotext.org/female/female6/femaleframe6.htm
- O’Driscoll JB, Mamtora H, Higginson J, et al. A prospective study of the prevalence of clear-cut endocrine disorders and polycystic ovaries in 350 patients presenting with hirsutism or androgenic alopecia. Clin Endocrinol (Oxf) 1994, 41: 231-6.
- Azziz R, Sanchez LA, et al. Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab 2004, 89: 453-62.
- Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab 1961, 21: 1440-7.
- Hatch R, Rosenfield RL, et al. Hirsutism: implications, etiology and management. Am J Obstet Gynecol 1981, 140: 815-30.
- Wild RA, Carmina E, Diamanti-Kandarakis E, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab 2010, 95: 2038–49.
Terapia di iperandrogenismi/irsutismo
Cecilia Motta1 & Roberto Castello2
1Cattedra e UOC Endocrinologia, Dipartimento Medicina Clinica e Molecolare, La Sapienza Università di Roma, Azienda Ospedaliera Sant'Andrea, Roma
2Medicina/Endocrinologia, AOUI Verona
(aggiornato all'8 marzo 2016)
Considerazioni generali sulla terapia farmacologica dell’iperandrogenismo
L’irsutismo è un segno clinico e non una malattia, per cui non sempre richiede trattamento, una volta escluse cause maggiori, e molto dipende dalla percezione del problema da parte della donna affetta più che dalla reale entità dell’irsutismo.
Se la donna esprime il desiderio di una gravidanza a breve, il trattamento del solo sintomo irsutismo va rimandato a dopo la gravidanza.
Solo uno è il farmaco approvato per il trattamento dell’iperandrogenismo nella donna (ciproterone acetato 2 mg), mentre gli anti-androgeni (il cui impiego è off-label) migliorano l’irsutismo rispetto al placebo, ma non esistono studi di buona qualità, su larga scala e di lunga durata tali da consentire una classifica di efficacia di questi farmaci (1). Sono sempre necessari 6 mesi di terapia prima di vedere benefici ed il trattamento farmacologico è sintomatico: alla sospensione della terapia il problema si ripresenta (2).
Vanno sempre associate misure cosmetiche che permettano di gestire il problema in attesa del manifestarsi dell’effetto dei farmaci. Le misure cosmetiche possono essere utilizzate da sole per il trattamento dell’irsutismo lieve e localizzato ad aree ristrette (3).
Opzioni terapeutiche possibili (4):
- soppressione degli androgeni surrenalici con glucocorticoidi: desametasone;
- soppressione degli androgeni ovarici con estro-progestinici o GnRH agonisti;
- anti-androgeni: spironolattone, flutamide, bicalutamide;
- inibitori della 5α-reduttasi: finasteride;
- insulino-sensibilizzanti: metformina;
- modificazioni dello stile di vita: calo ponderale e attivitàfisica;
- trattamenti cosmetici.
La scelta del trattamento dipende da (4):
- origine dell’iperandrogenismo
- problema percepito dalla paziente e suoi obiettivi (volontàdi una gravidanza);
- valutazione dei rischi e benefici a breve e a lungo termine dei vari trattamenti.
Terapia con glucocorticoidi (vedi capitolo surrene).
Terapia con contraccettivi ormonali
Ampiamente usata. Nell’evoluzione delle formulazioni si è via via diminuito il contenuto di estrogeni per ridurre il rischio di eventi avversi, in particolare trombo-embolici. Vanno preferiti quelli in cui il progestinico non è di derivazione androgenica o è dotato di attività anti-androgenica (ciproterone, drospirenone, clormadinone). Oltre alla formulazione orale classica, sono disponibili anche in forma di cerotto settimanale e anello vaginale mensile. La contraccezione ormonale non va prescritta in caso di patologie cardio-vascolari e trombosi venose. Si possono associare ad altri farmaci anti-androgeni.
Terapia con anti-androgeni e inibitore della 5α-reduttasi
Gruppo di farmaci utilizzati per ridurre l’effetto degli androgeni circolanti. Ad eccezione del ciproterone acetato, sono usati off-label, perché nessuno ha l’indicazione d’uso nella donna. La possibilità di ambiguità genitale nel feto maschio ne controindica l’uso in gravidanza e vanno sempre associati ad un’efficace contraccezione.
Ciproterone acetato: compete con il recettore per androgeni, ha debole attività corticosteroidea, ha effetto anti-gonadotropo e progestinico, permette anche la riduzione della funzionalità delle ghiandole sebacee. Poiché è dotato di effetto progestinico, in presenza di utero il ciproterone va associato ad estrogeni. La formulazione di associazione fissa di etinil-estradiolo con 2 mg di ciproterone acetato è l’unica con la specifica indicazione nella donna al trattamento di malattie dermatologiche androgeno-dipendenti (4). Altri regimi di trattamento prevedono 12.5-100 mg/die di ciproterone acetato nei primi 10 giorni di un trattamento con estrogeni o estro-progestinici (a questi dosaggi il suo impiego è da considerare off-label). Può causare alterazioni metaboliche, aumento di peso, edema, perdita della libido, alterazioni del tono dell’umore e cefalea. Ad alte dosi può dare tossicità epatica (2). L’acne e la seborrea solitamente rispondono in tempi più brevi rispetto all'irsutismo o all'alopecia.
Spironolattone: diuretico, anti-aldosteronico e anti-androgeno utilizzato, off-label, nel trattamento dell’irsutismo. Compete con il testosterone e il di-idrotestosterone per il legame al recettore per androgeni anche a livello del follicolo pilifero ed inibisce l’attività della 5alfa-reduttasi. È dotato di bassa attività progestinica; ad alte dosi inibisce la steroidogenesi ovarica e surrenale. È usato alla posologia di 50-100 mg/die, bilanciando benefici ed effetti collaterali. Fra questi ultimi si annoverano irregolarità mestruali dose-dipendenti, specialmente polimenorrea, ma spesso transitoria. Se persistente, va aggiunto un estro-progestinico o va ridotta la posologia. Poliuria, tensione mammaria, disturbi gastro-intestinali sono comuni, ma spesso lievi e transitori. Rari sono ipotensione e iperpotassiemia. L’associazione con i contraccettivi orali minimizza gli effetti collaterali e ha un’efficacia maggiore sull’iperandrogenismo rispetto al solo spironolattone (1,2,4). Va usato con attenzione nei pazienti con ipotensione e richiede regolari controlli dei livelli sierici di sodio, potassio e creatinina.
Flutamide: farmaco non steroideo inibitore competitivo del legame degli androgeni al loro recettore; inibisce la sintesi o il metabolismo degli androgeni causando riduzione dei livelli circolanti di androgeni. È considerato un anti-androgeno puro, perché non si lega ad altri recettori steroidei. Ha indicazione d’uso solo per il carcinoma prostatico ed è utilizzata off-label nella donna per l’iperandrogenismo. L’abituale posologia è di 125-250 mg/die. L’effetto collaterale più frequente è una modesta secchezza cutanea dovuta alla riduzione della produzione di sebo; rara ma potenzialmente grave è la possibilità di tossicità epatica fatale, che si può manifestare anche a distanza di molti mesi dall’avvio del trattamento. L’uso della flutamide richiede perciò l’esecuzione di controlli frequenti della funzionalità epatica. Va associata ad adeguata contraccezione. Va quindi riservata ai casi più severi d’iperandrogenismo o resistenti ad altri farmaci (1,2). Analogo comportamento ha un suo derivato, la bicalutamide.
Chetoconazolo: anti-micotico con la capacità di inibire la sintesi steroidea. La possibilità di tossicità epatica anche grave e d’iposurrenalismo (4) ne hanno reso l’uso poco diffuso.
Finasteride: inibitore della 5α-reduttasi responsabile della conversione del testosterone in diidro-testosterone. Agisce sull'isoforma 2 dell’enzima prevalentemente espressa nella prostata e nel tratto genito-urinario, ma anche nei follicoli piliferi. È disponibile in formulazione da 5 mg per il trattamento dell’iperplasia prostatica benigna e da 1 mg per il trattamento dell’alopecia androgenetica nei maschi. Nella donna è utilizzato, off-label, nel trattamento dell’irsutismo. La posologia abituale è di 2.5-5 mg/die. È meglio tollerata degli altri anti-androgeni. Fra gli effetti collaterali comuni è descritta solo la riduzione della libido. La sua assunzione va associata a un’adeguata contraccezione (1,2). Per la sua modalità d'azione, in corso di terapia si riscontra un aumento dei livelli di testosteronemia (4).
Eflornitina (Vaniqa)
Inibisce irreversibilmente l'ornitin-decarbossilasi, enzima coinvolto nella produzione dello stelo pilifero da parte del follicolo. L’eflornitina in forma di crema è indicata per il trattamento dell'irsutismo facciale nella donna e va applicata 2 volte al giorno. L'efficacia è stata dimostrata esclusivamente per le aree interessate del viso e sotto il mento. Si è notato un miglioramento della condizione entro 8 settimane dall'inizio del trattamento ed il 32% delle pazienti aveva un marcato miglioramento dopo 24 settimane rispetto al placebo. È necessario un trattamento continuo per mantenere gli effetti benefici. Se non si notano effetti entro 4 mesi dall'inizio della terapia, l'uso deve essere interrotto. Di solito ben tollerata, può causare irritazione cutanea di lieve intensità, di solito risolta senza l'interruzione del farmaco, acne e secchezza della cute. Nel caso di sviluppo d’irritazione cutanea o d’intolleranza, la frequenza delle applicazioni deve essere ridotta a una volta al giorno. Non è stata studiata nelle pazienti con grave insufficienza renale e si deve quindi essere cauti nel prescriverla a tali pazienti. I dati in gravidanza sono limitati ed è stata evidenziata tossicità riproduttiva negli animali, pertanto non va utilizzata in gravidanza e in allattamento (1). In genere è da associare alla terapia cosmetica.
Terapia cosmetica
Ha un ruolo centrale nel trattamento dell’irsutismo. Nei casi d’irsutismo lieve può essere da sola sufficiente per controllare il problema, nei casi più gravi vale come terapia adiuvante per rimuovere i peli terminali già presenti, su cui i farmaci non hanno capacità di azione, e in attesa del manifestarsi dell’efficacia degli anti-androgeni.
Possibili opzioni sono decolorazione, rasatura, creme depilatorie, pinzette, cerette, elettrolisi, luce pulsata e laser.
Pinzette e cerette sono efficaci, ma possono comportare irritazione cutanea locale, la quale a sua volta può paradossalmente aumentare la crescita pilifera.
L’elettrolisi consiste nell’inserire un sottile ago elettrificato nel follicolo pilifero di ogni singolo pelo causandone la distruzione. Non è pertanto applicabile su aree vaste. Può causare dolore e irritazione cutanea transitori e cicatrici se l’operatore non è esperto.
Il laser e la luce pulsata si basano sulla foto-termolisi selettiva del pelo, che, essendo pigmentato, assorbe la luce laser o pulsata in maniera maggiore della cute circostante, fino a riscaldarsi tanto da bruciare il follicolo senza danneggiare la cute. Sono pertanto più efficaci quando è maggiore il contrasto di pigmentazione fra cute e pelo, ossia in donne di pelle chiara con peli scuri. Può causare una transitoria irritazione cutanea e più raramente, vesciche, croste e alterazioni della pigmentazione cutanea. La foto-termolisi laser sembra il metodo più efficace, ma è costosa e richiede multiple sedute senza ottenere un risultato davvero permanente (2).
Terapia con insulino-sensibilizzanti (vedi capitolo PCOS).
Terapia con GnRH agonisti
È il trattamento più efficace in caso di severo iperandrogenismo di origine ovarica. Induce ipoestrogenismo marcato, con conseguente sintomatologia tipica, vaso-motoria principalmente.
Terapia con bromocriptina o cabergolina
Utile solo in donne con iperprolattinemia.
Modificazione dello stile di vita (vedi capitolo PCOS).
Durata ed efficacia del trattamento
La terapia ormonale con desametasone deve essere proseguita indefinitamente e la posologia va adattata mantenendo il dosaggio minimo efficace (5). La terapia con estro-progestinici e anti-androgeni dovrebbe essere proseguita per almeno due anni, comunque sempre valutandone la risposta clinica. In casi particolari potrebbe essere proseguita per 4-5 anni.
Circa il 20% delle pazienti con iperandrogenismo non risponde all’iniziale trattamento ormonale entro 6-8 mesi (5).
Bibliografia
- Legro RS. Evaluation and Treatment of Polycystic Ovary Syndrome. 19 September 2009 Endotext.
- Moghetti P, Toscano V. Treatment of hirsutism and acne in hyperandrogenism. Best Pract Res Clin Endocrinol Metab 2006, 20: 221–34.
- Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 2012, 18: 146-70.
- Goodman NF. AACE Medical Guidelines for the Diagnosis and Treatment of Hyperandrogenic Disorders. Endocr Pract 2001, 7: 120-34.
- The Practice Committee of the American Society for Reproductive Medicine. The evaluation and treatment of androgen excess. Fertil Steril 2006, 86 (S 4): S241-7.
Aggiornamento 2016
Nel 2015 è stata pubblicata una revisione Cochrane sulle diverse terapie disponibili farmacologiche per trattare tale patologia (non sono stati considerati laser e luce pulsata) (1).
Gli autori hanno valutato 157 trial controllati randomizzati (RCT), pubblicati fino a giugno 2014, condotti su pazienti irsute e affette da PCOS, irsutismo idiopatico e iperandrogenismo idiopatico. In totale sono state incluse 10.550 donne con un'età media di 25 anni. L’effetto delle terapie è stato studiato per 6 o 12 mesi.
La maggior parte degli RCT è risultato ad alto rischio di bias: fra questi, il più frequente era la mancanza di valutazione in cieco. In 48 studi vi erano dati non utilizzabili; solo in pochi casi erano stati considerati come outcome primario il miglioramento dell’irsutismo riportato dalle pazienti o le modificazioni nella qualità della vita; solo in metà dei casi venivano riportati gli eventi avversi. Nella quasi totalità degli RCT gli outcome consistevano nelle modifiche del grado di irsutismo, valutate dal clinico, e dei valori di androgeni; in alcuni casi sono state prese in considerazione anche le modifiche del BMI e di altri segni clinici di iperandrogenismo. La qualità delle evidenze è risultata da moderata a molto bassa.
I dati ottenuti sul confronto fra due contraccettivi orali (CO) (etinil-estradiolo 35 µg e ciproterone acetato 2 mg vs etinil-estradiolo 30 µg e desogestrel 0.15 mg) hanno mostrato come entrambi siano associati con una riduzione nello score di Ferriman e Gallwey (FG score) in caso di irsutismo di grado medio, con nessuna differenza di efficacia fra i due preparati (evidenza di qualità bassa).
L’assunzione di flutamide 250 mg per due volte al giorno è associata con un significativo decremento dello FG score quando comparata con placebo (2 studi per un totale di 60 pazienti; evidenza di qualità molto bassa).
Lo spironolattone 100 mg/die (dati ottenuti da un unico studio, eseguito su 20 pazienti) risulta più efficace rispetto al placebo nel trattamento dell’irsutismo; è risultato avere una efficacia simile alla flutamide (evidenza di qualità molto bassa) e alla finasteride (evidenza di qualità bassa), in altri due studi.
Sia la finasteride che gli analoghi del GnRH hanno mostrato, nella maggior parte dei lavori, risultati inconsistenti.
La metformina non ha dimostrato alcun beneficio sul FG score (7 studi su 264 pazienti, evidenza di bassa qualità).
Non è stato possibile eseguire l’analisi quando in aggiunta ai CO era somministrato ciproterone acetato (20-100 mg/die), a causa dell’eterogeneità clinica e metodologica fra gli studi; comunque, sembrerebbe che l’aggiunta del ciproterone acetato ai CO produca una riduzione maggiore in termini FG score.
La dieta ipocalorica si è ovviamente dimostrata associata a una riduzione dell'indice di massa corporea (BMI), ma non alla riduzione del grado di irsutismo.
Non sono stati trovati studi che valutassero gli effetti delle misure cosmetiche (elettrolisi, cera), nonostante queste siano ampiamente utilizzate.
Gli eventi avversi descritti sono stati: tensione mammaria e cute secca per finasteride e flutamide, metrorragia per spironolattone, vampate e cefalea per analoghi del GnRH.
In sintesi:
- i CO sono efficaci nel trattamento dell’irsutismo di grado moderato (evidenze di bassa qualità);
- la terapia con flutamide 250 mg x 2/die o con spironolattone 100 mg/die sembra essere efficace e sicura nell’irsutismo di grado più severo, anche se la qualità delle evidenze varia dal basso al molto basso;
- i dati riguardanti la finasteride 5 mg/die sono inconsistenti, non si possono dare indicazioni;
- la metformina non appare essere efficace sull' irsutismo (evidenze di bassa qualità);
- i dati riguardanti gli analoghi del GnRH sono inconsistenti e sicuramente presentano effetti collaterali abbastanza significativi.
Dalla pubblicazione di questa review fino ad agosto 2015 sono stati pubblicati altri 5 studi sull'argomento ma nessuno ha modificato alcuna di queste conclusioni (2). La maggior parte degli studi presentava un alto rischio di bias a causa della mancanza dell’esecuzione degli stessi “in cieco” e di un’alta quota di dropout; inoltre molti trial sono stati eseguiti su poche pazienti, e solo in una minima parte (29 studi) si è valutata la qualità della vita.
I risultati ottenuti da questa review concordano con le indicazioni riportate nelle due Linee Guida (LG) dell’Endocrine Society (3,4), riguardanti diagnosi e terapia dell’irsutismo e della PCOS. Le LG raccomandano come prima linea di trattamento l'utilizzo di CO, a cui aggiungere, dopo sei mesi di trattamento se la risposta ai CO non è stata “ottimale”, un anti-androgeno o la finasteride; viene inoltre riportato che i farmaci insulino-sensibilizzanti presentano pochi o nulli benefici nel trattamento dell'irsutismo. Durante la terapia, in attesa dei risultati che non si evidenziano prima di 6-12 mesi, è consigliato utilizzare procedure cosmetiche.
Sono comunque necessari ulteriori studi che vadano a comparare l’efficacia dei CO in combinazione con anti-androgeni o finasteride vs le altre terapie. Mancano inoltre dati che confrontino fra loro i diversi anti-androgeni o gli anti-androgeni e la finasteride. Tutti i trial dovrebbero sempre analizzare l’impatto di questi trattamenti sulla qualità della vita e fornire rapporti dettagliati e completi sugli eventi avversi.
Bibliografia
- Zuuren EJ, Fedorowicz Z, Carter B, Pandis N. Interventions for hirsutism (excluding laser and photoepilation therapy alone). Cochrane Database Syst Rev 2015, 4: CD010334.
- Zuuren EJ, Fedorowicz Z. Interventions for hirsutism. JAMA 2015, 314: 1863-4.
- Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2008, 93: 1105-20.
- Legro RS, Arslanian SA, Ehrmann DA, et al. Diagnosis and treatment of polycystic ovary syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2013, 98: 4565-92.
Epidemiologia della PCOS
Paolo Moghetti
Sezione di Endocrinologia e Metabolismo, Dipartimento di Medicina, Università di Verona, e UO di Endocrinologia e Malattie Metaboliche, Azienda Ospedaliera Universitaria Integrata, Verona
La sindrome dell’ovaio policistico (PCOS) è una condizione assai comune, anche se le sue precise dimensioni epidemiologiche restano mal definite. Questa incertezza è legata a diversi fattori: l’eterogeneità dei criteri di diagnosi utilizzati, la scarsa riproducibilità di molte delle misure utilizzate ai fini diagnostici, la mancanza di un “gold standard” per la formulazione di una diagnosi certa, l’afferenza di queste pazienti a figure mediche diverse, la frequente incompletezza dell’iter diagnostico abitualmente utilizzato in queste donne. Questi ultimi aspetti sono legati alla marcata eterogeneità di presentazione clinica della PCOS e alla scarsa percezione delle sue variegate implicazioni da parte di molti medici, che in genere si concentrano sugli aspetti fenotipici individuali che hanno portato la paziente all’attenzione di un determinato specialista.
Utilizzando i criteri NIH (vedi oltre per gli aspetti concernenti le modalità di diagnosi della PCOS), alcuni limitati studi hanno riscontrato una prevalenza della PCOS, nelle donne in età riproduttiva, intorno al 6-8% (1-4). Con i criteri definiti successivamente nella Consensus di Rotterdam, attualmente i più largamente utilizzati, la prevalenza è mal precisata ma certamente più elevata: alcuni dati preliminari suggeriscono possa raggiungere valori compresi fra il 12 e il 18% (4). Infine, anche con i criteri AE-PCOS non vi sono informazioni precise, ma dati preliminari suggeriscono che, come atteso sulla base del confronto fra i diversi criteri, con questa modalità di diagnosi i valori di prevalenza possano avere dimensioni intermedie fra quelle ottenibili con i criteri NIH e con i criteri della Consensus di Rotterdam, intorno al 10-12% (4).
La formulazione attuale dei criteri di diagnosi di questa sindrome non consente di porre la diagnosi in età prepuberale o in età post-menopausale, anche se le manifestazioni tipiche della PCOS possono avere dei segni anticipatori prima dell’età riproduttiva e delle sequele in età climaterica. In queste epoche della vita non è quindi possibile fare stime di prevalenza della patologia, anche se le implicazioni fisiopatologiche e terapeutiche di questi segni precoci e tardivi sono potenzialmente rilevanti.
Bibliografia
- Asuncion M, Calvo RM, San Millan JL, et al. A prospective study of the prevalence of the polycystic ovary syndrome in unselected Caucasian women from Spain. J Clin Endocrinol Metab 2000, 85: 2434–8.
- Azziz R, Woods KS, Reyna R, et al. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab 2004, 89: 2745–9.
- Diamanti-Kandarakis E, Kouli CR, Bergiele AT, et al. A survey of the polycystic ovary syndrome in the Greek island of Lesbos: hormonal and metabolic profile. J Clin Endocrinol Metab 1999, 84: 4006–11.
- March WA, Moore VM, Willson KJ, et al. The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod 2010, 25: 544-51.
Clinica e diagnostica PCOS
Paolo Moghetti
Sezione di Endocrinologia e Metabolismo, Dipartimento di Medicina, Università di Verona, e UO di Endocrinologia e Malattie Metaboliche, Azienda Ospedaliera Universitaria Integrata, Verona
Anche gli aspetti clinici sono inevitabilmente legati, almeno in parte, ai criteri di diagnosi utilizzati. È quindi opportuno premettere quali sono questi criteri allo stato attuale e come sono stati individuati. Sfortunatamente c’è ancora discussione su come la diagnosi di PCOS debba essere formulata e, come già ricordato, manca un riferimento certo che consenta di saggiare la validità effettiva dei criteri utilizzati. Pertanto questi criteri sono legati a opinioni autorevoli basate su elementi indiretti, ma non a evidenze inequivocabili.
La diagnosi di PCOS è stata a lungo considerata equivalente a quella della sindrome descritta da Stein & Leventhal nel 1935, in epoca pre-ecografica (1). Questi autori riportarono alcuni casi caratterizzati da ovaie ingrandite e policistiche, alla laparotomia, in donne che presentavano amenorrea secondaria, infertilità, obesità e talora irsutismo. Questa descrizione viene in genere considerata la prima relativa alla PCOS, anche se un quadro simile era già stato riportato da Antonio Vallisneri nel 1723, in un case-report su una singola paziente (2), e poi da altri autori nel secolo successivo.
Una rivisitazione del problema, con definizione per la prima volta di precisi criteri di diagnosi, è stata effettuata nel 1990, in occasione di una conferenza di consenso sul tema organizzata negli USA dal National Institute of Child Health and Human Development, uno dei National Institutes of Health (NIH), le cui conclusioni furono pubblicate due anni dopo. In quell’occasione il consenso fra i partecipanti alla conferenza era comunque stato assai relativo e il documento anticipava l’opportunità di una rivalutazione a distanza delle conclusioni faticosamente raggiunte. I criteri definiti in quella occasione, chiamati successivamente criteri NIH (3), stabilivano che la diagnosi poggiava sulla presenza contemporanea di oligoanovulazione cronica (spesso documentata indirettamente dalle irregolarità mestruali) e iperandrogenismo (documentato indifferentemente sia da evidenze cliniche, come irsutismo, acne o alopecia androgenetica, che biochimiche, rappresentate dall’aumento dei livelli circolanti di un qualsiasi androgeno), in assenza di altre patologie che potessero dar origine a queste manifestazioni (in primo luogo iperplasia surrenalica congenita, tumori androgeno-secernenti o iperprolattinemia). Questi criteri stabilivano quindi che la diagnosi di PCOS è comunque una diagnosi di esclusione, data la mancanza di qualsiasi elemento patognomonico. La consensus NIH giunse alla conclusione che il quadro ecografico ovarico e le frequenti alterazioni metaboliche di queste donne non erano elementi da utilizzare nella diagnosi. Alla base di queste decisioni stavano l’aspecificità delle alterazioni ecografiche, spesso presenti anche in donne senza altre anomalie, e la difficoltà di misurare in maniera semplice e riproducibile l’insulino-resistenza. L’esclusione degli aspetti desumibili dall’ecografia era comunque anche legata alla scarsa diffusione che all’epoca questa tecnica aveva sul territorio americano.
Le decisioni di questa conferenza hanno goduto di largo credito, anche perché rappresentavano il primo tentativo di codificare la diagnosi di PCOS, ma non sono mai state accettate completamente dalla scuola britannica, fortemente legata per motivi storici agli aspetti ecografici. Questo ha prodotto l’inconveniente della pubblicazione di lavori difficilmente confrontabili, in quanto condotti su pazienti in cui la diagnosi era stata posta con criteri differenti. Per superare questa situazione di stallo, nel 2003 è stata convocata a Rotterdam una nuova conferenza di consenso, ad opera delle società della riproduzione europea e americana (ESHRE e ASRM). Le conclusioni di questa conferenza furono pubblicate l’anno seguente (4,5) e i criteri che ne sono derivati prendono il nome di criteri di Rotterdam. In sostanza, queste società hanno fatto proprie le conclusioni della conferenza NIH, reintroducendo però l’aspetto ecografico come elemento diagnostico e stabilendo che la diagnosi di PCOS poteva essere posta in presenza di almeno due elementi fra oligoanovulazione cronica, iperandrogenismo (definito come stabilito dai criteri NIH) e quadro ecografico ovarico, sempre in assenza di possibili cause secondarie. Per quanto concerne l’aspetto ecografico, sulla base della pur limitata letteratura sull’argomento, è stato stabilito che le caratteristiche da usare per formulare una diagnosi di ovaio micropolicistico sono un volume > 10 mL e/o un numero di follicoli di almeno 12, in almeno un ovaio. Questa decisione, che permetteva in sostanza alla scuola americana e a quella britannica di formulare la diagnosi formalmente sulla base dei medesimi criteri, ma conservando di fatto la possibilità di usare modalità differenti, ha avuto come conseguenza più rilevante il fatto che sotto l’etichetta PCOS possono oggi essere comprese situazioni caratterizzate da aspetti diversi, i cosiddetti fenotipi della PCOS. In particolare, accanto ai quadri in cui sono presenti tutti e tre gli elementi diagnostici (oligoanovulazione, iperandrogenismo e ovaio micropolicistico), ci possono essere situazioni in cui sono presenti solo oligoanovulazione e iperandrogenismo (fenotipo classico o NIH), ovvero solo oligoanovulazione e ovaio micropolicistico (fenotipo normoandrogenico) o infine iperandrogenismo e ovaio micropolicistico (fenotipo ovulatorio). Anche questa consensus è stata comunque oggetto di vivace dibattito, con posizioni autorevoli sostanzialmente diverse (6,7). In ogni caso questi criteri sono quelli su cui oggi c’è il maggior consenso, pur tutt’altro che unanime. Anche l’Endocrine Society, nella sua linea guida sulla diagnosi e terapia della PCOS pubblicata nel dicembre 2013, ha deciso di attenersi a queste raccomandazioni(8).
Una posizione critica nei confronti dei criteri di Rotterdam è in particolare quella della Androgen Excess and PCOS (AE-PCOS) Society, una società scientifica internazionale cui aderiscono molti studiosi di questa patologia. Questa società ha pubblicato una propria revisione dei criteri di diagnosi nel 2006 (9) e ha ribadito questa posizione, in un documento più ampio, nel 2009 (10). I criteri AE-PCOS riprendono nella sostanza molte delle conclusioni raggiunte nella consensus di Rotterdam, ma se ne distinguono nella sostanza, in quanto individuano l’iperandrogenismo come elemento centrale, indispensabile alla diagnosi, e lasciano un ruolo gerarchicamente secondario agli aspetti ovarici, funzionali (oligoanovulazione) e morfologici (aspetto micropolicistico). Nei criteri AE-PCOS, infatti, l’iperandrogenismo è un elemento comunque necessario, mentre è sufficiente uno qualsiasi fra oligoanovulazione e aspetto ecografico ovarico come ulteriore requisito per poter porre la diagnosi, sempre in assenza di cause secondarie. Questi criteri stabiliscono inoltre una gerarchia anche nelle modalità di diagnosi dell’iperandrogenismo, definendo come elementi preferenziali l’irsutismo, sul piano clinico, e il testosterone libero, misurato o calcolato in modo adeguato e non ricorrendo al dosaggio diretto routinario di questo parametro, su quello biochimico.
L’AE-PCOS ha successivamente rivisto anche le modalità di valutazione del criterio ecografico, su cui va notato che in generale vi è una certa confusione. In particolare, molti non sono consapevoli del fatto che la consensus di Rotterdam, nel definire il cut-off del numero dei follicoli da utilizzare nella diagnosi di PCOS, si era rifatta ai risultati di un singolo lavoro in cui questo calcolo era stato effettuato sull’intero ovaio e non su una singola sezione massima, come spesso avviene invece nella pratica clinica. La revisione AE-PCOS è stata stimolata dal fatto che l’uso di strumentazioni ecografiche moderne permette oggi la visualizzazione di un numero di follicoli maggiore e che se il conteggio viene effettuato effettivamente sull’intero ovaio il risultato è in molte donne superiore ai limiti stabiliti dai criteri di Rotterdam, con ulteriore e artificiosa amplificazione delle dimensioni epidemiologiche della PCOS. L’AE-PCOS ha individuato ora in 25 il cut-off diagnostico del numero dei follicoli sull’intero ovaio, lasciando invariato quello dei 10 mL in relazione al volume ovarico (11). Va peraltro sottolineato che questa procedura di valutazione è piuttosto indaginosa, se non si dispone di un ecografo con uno specifico software, e richiede comunque uno strumento tecnologicamente avanzato, con una sonda suggerita da almeno 8 MHz. Se questa non è disponibile, viene suggerito di utilizzare il solo criterio del volume, che peraltro ha una performance diagnostica minore di quella del conteggio dei follicoli. Va notato che se si effettua il conteggio dei follicoli su una singola scansione ovarica la soglia diagnostica è ovviamente più bassa, individuata in uno studio di confronto in 9 follicoli, ma questa modalità di valutazione non è ufficializzata dalle attuali linee guida. Nel documento AE-PCOS è stato anche valutato l’utilizzo del dosaggio dell’AMH come possibile alternativa ai criteri ecografici. Le attuali importanti problematiche di standardizzazione nel dosaggio di questo ormone hanno però suggerito di non ricorrere per il momento a questa strategia, che in prospettiva appare molto interessante.
La tabella 1 riassume e confronta i diversi criteri utilizzati per la diagnosi di PCOS nella donna adulta.
| Tabella 1 Confronto dei diversi criteri utilizzati per la diagnosi di PCOS nella donna adulta |
|
| Criteri NIH 1990 | Necessari tutti i seguenti tre elementi:
|
| Criteri di Rotterdam 2003 |
Necessari almeno due (qualsiasi) fra gli elementi 1-3 + l’elemento 4:
|
| Criteri AE-PCOS 2006/2013 |
Necessario l’elemento 1 + almeno uno (qualsiasi) fra gli elementi 2-3 + l’elemento 4:
|
† Il volume ovarico può essere calcolato, utilizzando la formula dell’ellissoide, a partire dai tre diametri dell’ovaio: 0.52·(D1·D2·D3)
* Il testosterone libero può essere misurato con metodo diretto valido (dialisi all’equilibrio) o calcolato, a partire da un dosaggio di buona qualità del testosterone totale e dell’SHBG, ad esempio utilizzando il calcolatore online dell’International Society for the Study of the Aging Male (ISSAM)
Va ricordato che questi criteri sono applicabili con molta difficoltà nei primi anni dopo il menarca. Infatti, in adolescenza è estremamente comune osservare cicli irregolari e anovulatori e l’aspetto ovarico è spesso micropolicistico. Anche per la diagnosi di iperandrogenismo clinico vi sono delle difficoltà, in quanto l’acne è un fenomeno assai frequente in questa età, mentre l’irsutismo è spesso ancora poco espresso. La problematica della diagnosi di PCOS in adolescenza è stato uno dei temi trattati in un’altra conferenza di consenso, tenutasi ad Amsterdam nel 2010 sotto l’egida di ESHRE e ASRM, che ha affrontato vari aspetti concernenti la salute della donna con PCOS. Le conclusioni di questa consensus sono state pubblicate nel 2012 (12). Per quanto riguarda la diagnosi di PCOS in adolescenza, la consensus di Amsterdam ha concluso che è necessaria la presenza di tutti e tre gli elementi usati per la diagnosi nella donna adulta e non solo di due fra questi. Inoltre, è necessario che i disturbi mestruali siano presenti per almeno due anni dopo il menarca o che vi sia un’amenorrea primaria all’età di 16 anni e che sia presente l’aumento del volume ovarico oltre 10 mL. Infine, è stata sottolineata l’importanza di documentare un aumento dei livelli degli androgeni rispetto all’evidenza clinica di iperandrogenismo (12). Anche la linea guida dell’Endocrine Society ha affrontato questa problematica, dando particolare enfasi ai fini della diagnosi di PCOS in adolescenza alla persistente presenza di alterazioni mestruali e di segni clinici e/o biochimici di iperandrogenismo. Per quanto riguarda quest’ultimo aspetto, consapevole dei limiti di questa valutazione, ha suggerito di utilizzare i cut-off della donna adulta in relazione ai livelli degli androgeni e di non valorizzare ai fini diagnostici l’acne isolata. Ha suggerito inoltre di non utilizzare oligo-anovulazione e alterazioni ecografiche ovariche a fini diagnostici in questa età (8).
La linea guida dell’Endocrine Society ha analizzato anche la possibilità di una diagnosi di PCOS dopo la menopausa, concludendo che non vi sono criteri utilizzabili a questo scopo, ma che una diagnosi presuntiva può essere comunque posta in presenza di una storia clinica suggestiva, specie in presenza di un quadro ecografico ovarico tipico (8).
Da quanto già discusso si può intuire come la presentazione clinica della PCOS sia alquanto variabile e come l’utilizzo di un criterio diagnostico piuttosto di un altro possa condizionare questa eterogeneità intrinseca alla sindrome, per come intendiamo oggi questa entità nosologica. Data la mancanza di adeguati studi di popolazione, tutte le casistiche risentono inoltre di un bias potenziale, legato alla motivazione che ha portato la paziente all’osservazione del medico. Nei centri endocrinologici ci potrà quindi spesso essere una selezione preferenziale di casi caratterizzati da iperandrogenismo od obesità, mentre nei centri ginecologici la casistica risulterà arricchita di casi con alterazioni morfologiche dell’ovaio o con infertilità. Con queste limitazioni in mente, è utile prendere visione di alcune ampie casistiche pubblicate, che descrivono le caratteristiche delle pazienti iperandrogeniche afferenti a singoli centri, in gran parte con PCOS (13,14), e far riferimento alla metanalisi condotta dalla task force della AE-PCOS Society, che ha redatto le raccomandazioni diagnostiche e terapeutiche di questa società (10).
L’analisi della letteratura condotta dalla AE-PCOS (10) conclude, in particolare, che l’irsutismo è un aspetto più specifico della PCOS rispetto ad altre manifestazioni cliniche di iperandrogenismo. Infatti, il 75-80% delle donne irsute ha questa patologia, contro il 20-40% di quelle che presentano solo acne e appena il 10% di quelle con alopecia isolata. Anche in termini assoluti l’irsutismo è di gran lunga la più comune presentazione clinica di iperandrogenismo nelle donne con PCOS, eventualmente associato ad altre manifestazioni sostenute da un’eccessiva azione androgenica. Complessivamente si stima che l’irsutismo ricorra nel 66-75% delle donne con PCOS, mentre l’acne è stata riportata nel 12-24% dei casi e l’alopecia è stata raramente valutata in maniera sistematica, con conseguente grande discrepanza di dati fra i pochi e limitati studi sull’argomento (5-50%). Va ricordato che la crescita del pelo presenta sensibili differenze etniche e quindi la frequenza dell’irsutismo e i parametri di riferimento usati per definire i limiti “normali” di questo fenomeno possono essere sensibilmente diversi a seconda del gruppo etnico cui appartiene la paziente (15). Questi aspetti sono descritti in maggior dettaglio nella sezione di Endowiki sull’iperandrogenismo in generale. Nei gruppi etnici in cui la pilificazione è scarsa e l’irsutismo è quindi infrequente o comunque difficilmente percepibile, come accade in alcune popolazioni dell’estremo oriente, la presenza di iperandrogenismo clinico può di conseguenza facilmente sfuggire.
Le alterazioni del ciclo mestruale, più spesso oligoamenorrea ma talora polimenorrea, sono un altro aspetto molto comune in queste pazienti. La frequenza di questo fenomeno viene riportata infatti in almeno tre quarti delle donne con PCOS e spesso riflette la presenza di oligoanovulazione. Tuttavia, le due cose non devono essere semplicisticamente considerate equivalenti. Ci sono infatti donne con irregolarità lievi che hanno comunque cicli ovulatori e soprattutto donne con cicli sostanzialmente regolari ma anovulatori. In particolare, quest’ultima evenienza si osserva più spesso nelle donne con manifestazioni cutanee di iperandrogenismo. In queste donne è stato rilevato che l’anovulazione può ricorrere nel 20-30% dei casi. Pertanto in una donna irsuta con cicli regolari è sempre necessario valutare la funzione ovulatoria. Le irregolarità mestruali possono avere d’altra parte cause diverse, che vanno tenute presenti nella diagnosi differenziale. Le alterazioni mestruali delle donne con PCOS tendono ad attenuarsi con l’aumentare dell’età, come altri aspetti di questa sindrome. È interessante notare che recentemente alcuni studi hanno suggerito che le pazienti con PCOS possano essere caratterizzate da una vita fertile più prolungata rispetto a quella delle altre donne e che quindi possano recuperare nel tempo almeno in parte in termini procreativi (16).
Ancora più aspecifico è il quadro ecografico ovarico (per le immagini vedi capitolo ecografia). Questo aspetto è presente in molte donne per il resto normali e non va quindi considerato di per sè patologico. È stato infatti stimato che il 20-30% delle donne della popolazione generale abbia un aspetto “micropolicistico” dell’ovaio (17-19), che non va assolutamente considerato equivalente alla diagnosi di PCOS. La valutazione ecografica è influenzata dalle modalità con cui l’esame viene effettuato e dalle caratteristiche antropometriche della paziente. La via trans-vaginale è quella ideale, ma non è sempre applicabile o accettata. Va ricordato che nella popolazione generale la frequenza dell’aspetto micropolicistico dell’ovaio si riduce con l’età e scende sotto il 10% oltre i 35 anni. Quindi aumenta parallelamente il valore predittivo positivo del suo riscontro. D’altra parte i cut-off del volume e soprattutto del numero di follicoli definiti dalla consensus di Rotterdam sono anch’essi oggetto di discussione. Alcuni dati suggeriscono che il limite superiore della volumetria ovarica normale possa essere più basso (fino a 7-8 mL) e che la sensibilità delle strumentazioni ecografiche più moderne possa permettere di visualizzare un maggior numero di follicoli, con conseguente possibile sovrastima del numero di donne caratterizzate da un eccessivo numero di follicoli. Di questo aspetto si è già parlato sopra. Discusso è anche l’utilizzo di indicatori ecografici diversi, come il rapporto fra area stromale e area ovarica totale (20).
L’obesità è un aspetto frequente nelle donne con PCOS e che contribuisce a rendere elevato il rischio metabolico di queste pazienti e probabilmente anche l’entità delle loro alterazioni riproduttive ed endocrine. La prevalenza di sovrappeso e obesità in queste pazienti resta comunque mal definita e appare diversa in aree geografiche differenti. Mal definite sono anche le basi fisiopatologiche di questa associazione (21). Vi sono solide evidenze che l’insulino-resistenza, verosimilmente attraverso l’iperinsulinemia che l’accompagna, svolga un ruolo patogenetico nella PCOS, soprattutto attraverso l’aumento degli androgeni liberi che consegue agli incrementati livelli degli androgeni e ai ridotti livelli della SHBG. È quindi ragionevole ipotizzare che la comparsa della PCOS sia favorita dall’eccesso ponderale, anche se i dati in proposito non sono univoci. D’altra parte è anche possibile che l’eccesso di androgeni possa favorire l’accumulo di grasso a livello tronculare. Queste due ipotesi non sono mutuamente esclusive. Va tenuto presente che nelle donne con PCOS l’eccesso di grasso centrale può realizzarsi anche con un BMI normale (22). Nella valutazione degli aspetti antropometrici di queste pazienti è quindi importante rilevare gli indici di adiposità centrale, come la circonferenza vita, e non limitarsi alla misura del peso.
Nella diagnostica della PCOS vanno distinte le indagini necessarie ai fini della diagnosi e quelle utili a definire le problematiche complessive potenzialmente associate a questa sindrome. Le prime sono ovviamente legate a quanto stabilito dai criteri utilizzati. Adoperando i criteri della consensus di Rotterdam o della AE-PCOS, oltre agli elementi clinici (valutazione dell’irsutismo in primo luogo), la diagnostica poggia quindi su quattro aspetti: il dosaggio degli androgeni, la valutazione della funzione ovulatoria, lo studio degli aspetti ecografici ovarici, l’esclusione delle cause secondarie. Per quanto riguarda la diagnostica relativa alle altre problematiche di queste pazienti, l’aspetto più rilevante è certamente quello metabolico.
Il dosaggio degli androgeni rappresenta un punto controverso in questa valutazione, in relazione soprattutto ai gravi limiti dei dosaggi routinari di questi ormoni. Questi aspetti sono discussi in un’altra sezione di endowiki.. Sulla base dei criteri correnti, ai fini diagnostici questo dosaggio può non essere indispensabile in presenza di chiaro irsutismo. D’altra parte, va ricordato che anche la valutazione clinica dell’iperandrogenismo soffre di limitazioni importanti, data la soggettività del punteggio di Ferriman-Gallwey, con cui abitualmente si misura l’irsutismo nella pratica clinica, e date le differenze individuali ed etniche nella risposta del follicolo pilifero agli androgeni. Qualsiasi opzione rischia quindi di determinare falsi positivi e falsi negativi. In presenza di irsutismo, la Endocrine Society suggerisce di ricorrere al dosaggio del testosterone solo quando il punteggio di Ferriman-Gallwey supera il valore di 15 ovvero vi siano altri elementi, quali disturbi mestruali, infertilità, obesità centrale o acanthosis nigricans, che suggeriscano la possibilità di un quadro di PCOS (23). La consensus ESHRE/ASRM di Amsterdam e lo statement della AE-PCOS Society sul tema concludono invece che è sempre indicata una quantificazione degli androgeni e in particolare del testosterone libero in presenza di un irsutismo di qualsiasi entità (12,15). In ogni caso il dosaggio degli androgeni richiede accuratezza e deve essere interpretato alla luce del quadro clinico. Attualmente è in corso uno sforzo per mettere a punto metodiche affidabili di dosaggio del testosterone, soprattutto attraverso la spettrometria di massa. Vi sono comunque metodiche RIA con cui sono stati osservati risultati sovrapponibili a quelli della spettrometria di massa (24). Gli altri parametri ormonali, in particolare le gonadotropine, sono oggi considerati di limitata utilità nella diagnosi di PCOS, se non per escludere altre patologie in situazioni clinicamente dubbie.
Per i motivi già ricordati, la funzione ovulatoria deve essere verificata in tutte le donne con sospetta PCOS e cicli regolari o quasi regolari, ad esempio attraverso il dosaggio del progesterone nella presunta fase luteinica (circa una settimana prima del giorno in cui è attesa la mestruazione successiva). In presenza di lievi alterazioni del ciclo può essere considerato il ricorso a 2-3 prelievi seriati, per essere certi di effettuare l’esame nella finestra temporale corretta. Il risultato dovrebbe essere idealmente confermato da un secondo dosaggio in un ciclo successivo. Il valore di progesterone sopra il quale si può parlare di avvenuta ovulazione varia in rapporto al singolo laboratorio ma è comunque intorno a 3-4 ng/mL (9.5-12.7 nmol/L). In alternativa a questo dosaggio, si può ricorrere al monitoraggio ecografico o della temperatura basale, tenendo presenti i limiti intrinseci a quest’ultima metodica.
Gli aspetti ecografici ovarici sono già stati discussi. È necessario che l’esame venga effettuato nella presunta fase follicolare, possibilmente per via trans-vaginale, e che l’operatore abbia cura di misurare i tre diametri dell’ovaio, per poterne calcolare il volume, oltre al numero e alle dimensioni dei follicoli. Va specificato che l’esame non è diagnostico e deve essere ripetuto in caso di riscontro di un follicolo dominante (oltre 10 mm), di un corpo luteo (che comunque depone per avvenuta ovulazione) o di una formazione cistica. Le linee guida internazionali (4,5,10) raccomandano unanimemente di non tener conto dei referti ecografici che non quantifichino il volume ovarico e il numero dei follicoli.
Le cause secondarie da escludere per poter porre la diagnosi di PCOS possono essere diverse, ma non appare razionale ed economica una ricerca sistematica di ognuna di queste. Il documento della AE-PCOS (10) suggerisce di effettuare sempre il dosaggio del 17-idrossiprogesterone, per lo screening dell’iperplasia surrenalica congenita late-onset, con eventuale test all’ACTH di verifica in caso di valori basali superiori a 2 ng/mL (6 nmol/L). L’opportunità di un dosaggio sistematico del TSH e della prolattina resta invece controversa, anche se può essere raccomandabile. La linea guida dell’Endocrine Society (8) suggerisce invece, a questo proposito, il dosaggio sistematico di 17-idrossiprogesterone, prolattina e TSH. Discusso è anche il dosaggio sistematico dell’insulinemia come indicatore di una condizione di severa insulino-resistenza, mentre la ricerca di una possibile neoplasia androgeno-secernente o di una sindrome di Cushing va ragionevolmente limitata ai casi con quadro clinico sospetto (in particolare comparsa recente o brusca accentuazione delle manifestazioni di iperandrogenismo nel caso delle neoplasie che producono androgeni).
Come ricordato, l’insulino-resistenza è un'anomalia metabolica frequente nella PCOS, soprattutto ma non esclusivamente nelle pazienti obese. Una verifica di questo aspetto resta però problematica. La sua valutazione richiede infatti metodiche, come il clamp euglicemico iperinsulinemico, non applicabili nella pratica clinica comune. In molti casi si ricorre pertanto a indici surrogati, come l’indice HOMA, derivato dai valori di glicemia e insulinemia a digiuno. Nell’eventuale utilizzo di questo parametro, come di altri indici surrogati simili, va tenuto presente che il dosaggio dell’insulina non è standardizzato e che pertanto gli intervalli di riferimento da utilizzare sono condizionati dal metodo specificamente impiegato. Queste difficoltà hanno portato in passato alla proposta di ricorrere a indici indiretti di insulino-resistenza, come quelli usati per la diagnosi di sindrome metabolica (25). Va notato che l’eterogeneità fra i diversi fenotipi clinici della PCOS si esprime anche in termini di anomalie metaboliche e in particolare di insulino-resistenza. Questa infatti appare essere una caratteristica del fenotipo classico della sindrome e in misura minore di quello ovulatorio, ma non di quello normo-androgenico (26). La caratterizzazione accurata di queste pazienti, al di là dell’assegnazione dell’etichetta comune di PCOS, può quindi essere utile anche nel definire quali pazienti sottoporre ad approfondimenti in questa direzione.
La frequente presenza di alterazioni del profilo lipidico nelle donne con PCOS richiede in ogni caso che la diagnostica di queste pazienti includa una misura di colesterolo totale, LDL, HDL e non-HDL e trigliceridi (27). Il colesterolo HDL ridotto è l’alterazione metabolica più frequente in queste donne, probabilmente per l’azione sinergica di insulino-resistenza e iperandrogenismo sui livelli di questo parametro.
Il rischio di alterazioni della tolleranza ai carboidrati è aumentato nelle donne con PCOS, anche indipendentemente dalla presenza di obesità (28). L’opportunità di effettuare un carico orale di glucosio in tutte le donne con PCOS, con misura della glicemia a due ore, è però un altro aspetto discusso. La AE-PCOS aveva inizialmente proposto un impiego sistematico di questa indagine (29), alla luce delle evidenze che la glicemia a digiuno non ha sufficiente sensibilità nell’individuare le donne con PCOS che presentano un'alterata tolleranza al glucosio (IGT) (30). Più recentemente un documento della stessa società, concernente valutazione e approccio terapeutico al rischio cardiovascolare di queste pazienti, ha comunque optato per un utilizzo più mirato dell’indagine (27), limitandone l’indicazione ai soggetti con alcuni fattori di rischio: obesità, età oltre 40 anni, storia personale di diabete gestazionale o storia familiare di diabete tipo 2. La AE-PCOS suggerisce anche di ripetere il test, se negativo, ogni due anni.
La linea guida dell’Endocrine Society discute una serie di altre problematiche che sono state associate alla PCOS, suggerendo uno screening sistematico di depressione e ansia, attraverso un breve questionario e con successivo eventuale invio allo specialista, e della sindrome delle apnee notturne, in particolare nelle pazienti in sovrappeso, con eventuale successiva polisonnografia (8). Suggerisce inoltre di tenere presenti i rischi potenziali di neoplasia endometriale e NAFLD (nonalcoholic fatty liver disease), senza però suggerire uno screening sistematico di queste patologie. Altri autori suggeriscono uno screening relativamente anche ad altre aree della sfera psichica, quali i disturbi del comportamento alimentare, il vissuto sessuale, l’immagine di sé.
La tabella 2 riassume le indagini da considerare nella diagnostica di una donna con sospetta PCOS, ai fini della diagnosi di questa patologia e per l’inquadramento complessivo.
| Tabella 2 Valutazioni e indagini principali da considerare nella paziente con sospetta PCOS (pannello A) e ulteriori valutazioni da effettuare dopo la diagnosi (pannello B) |
||
| A. Valutazioni per la diagnosi di PCOS | Esame clinico | valutazione di grado di irsutismo, acne, alopecia androgenetica segni clinici di possibili cause secondarie |
| Esami strumentali | ecografia pelvica (in fase follicolare se è presente ciclo mestruale sufficientemente regolare, possibilmente per via trans-vaginale; misura dei tre diametri ovarici e del numero e delle dimensione dei follicoli in ciascun ovaio) | |
| Dosaggi di laboratorio basali |
in fase follicolare (in qualsiasi momento se il ciclo è molto irregolare o assente):
In fase luteinica (se il ciclo è presente e sufficientemente regolare):
|
|
| Test dinamici | ACTH-test (limitatamente ai casi con 17OH-P basale ≥ 2 ng/mL) Soppressione con desametasone a basse dosi - test di Nugent (limitatamente ai casi di sospetta s. di Cushing, eventuali ulteriori indagini successive) |
|
| B. Altre valutazioni utili all’inquadramento complessivo | Esame clinico | misura di BMI, circonferenza vita, valori pressori ricerca acanthosis nigricans valutare possibile sindrome delle apnee notturne |
| Dosaggi di laboratorio basali | glicemia profilo lipidico insulinemia (opzionale) |
|
| Test dinamici | OGTT (in presenza di fattori di rischio†, opzionale negli altri casi) con misura della glicemia a due ore (dosaggio dell’insulinemia dopo OGTT opzionale) | |
| Valutazione psicologica | questionario di screening | |
† Fattori di rischio principali per alterazioni della tolleranza ai carboidrati sono: presenza di obesità, età > 40 anni, storia personale di diabete gestazionale, storia familiare di diabete tipo 2.
Bibliografia
- Stein I, Leventhal M. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol 1935, 29: 181–5.
- Vallisneri A. Istoria della generazione dell'uomo e degli animali, se sia da' vermicelli spermatici o dalle uova, con un trattato nel fine della sterilità, e dei suoi rimedj. Hertz GG, Venezia, 1721.
- Zawadski JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine FP, Merriam GR, eds. Polycystic ovary syndrome. Blackwell Scientific Publications, Boston, MA, 1992: 377–84.
- The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 2004, 19: 41–7.
- The Rotterdam ESHRE/ASRM-sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004, 81: 19–25.
- Azziz R. Controversy in clinical endocrinology. Diagnosis of polycystic ovarian syndrome: the Rotterdam criteria are premature. J Clin Endocrinol Metab 2006, 91: 781-5.
- Franks S. Controversy in clinical endocrinology. Diagnosis of polycystic ovarian syndrome: in defense of the Rotterdam criteria. J Clin Endocrinol Metab 2006, 91: 786-9.
- Legro RS, Arslanian SA, Ehrmann DA, et al. Diagnosis and treatment of polycystic ovary syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2013, 98: 4565-92.
- Azziz R, Carmina E, Dewailly D, et al; Androgen Excess Society. Position statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab 2006, 91: 4237-45.
- Azziz R, Carmina E, Dewailly D, et al; Task Force on the Phenotype of the Polycystic Ovary Syndrome of The Androgen Excess and PCOS Society. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril 2009, 91: 456-88.
- Dewailly D, Lujan ME, Carmina E, et al. Definition and significance of polycystic ovarian morphology: a task force report from the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update E-pub 2013 Dec 16.
- Fauser BC, Tarlatzis BC, Rebar RW, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril 2012, 97: 28-38.
- Azziz R, Sanchez LA, Knochenhauer ES, et al. Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab 2004, 89: 453-62.
- Carmina E, Rosato F, Jannì A, et al. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab 2006, 91: 2-6.
- Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 2012, 18: 146-70.
- Kalra SK, Ratcliffe SJ, Dokras A. Is the fertile window extended in women with polycystic ovary syndrome? Utilizing the Society for Assisted Reproductive Technology registry to assess the impact of reproductive aging on live-birth rate. Fertil Steril 2013, 100: 208-13.
- Clayton RN, Ogden V, Hodgkinson J, et al. How common are polycystic ovaries in normal women and what is their significance for the fertility of the population? Clin Endocrinol (Oxf) 1992, 37: 127–34.
- Michelmore KF, Balen AH, Dunger DB, et al. Polycystic ovaries and associated clinical and biochemical features in young women. Clin Endocrinol (Oxf) 1999, 51: 779–86.
- Koivunen R, Laatikainen T, Tomas C, et al. The prevalence of polycystic ovaries in healthy women. Acta Obstet Gynecol Scand 1999, 78: 137–41.
- Fulghesu AM, Angioni S, Frau E, et al. Ultrasound in polycystic ovary syndrome--the measuring of ovarian stroma and relationship with circulating androgens: results of a multicentric study. Hum Reprod 2007, 22: 2501-8.
- Hoeger KM, Oberfield SE. Do women with PCOS have a unique predisposition to obesity? Fertil Steril 2012, 97: 13-7.
- Carmina E, Bucchieri S, Esposito A, et al. Abdominal fat quantity and distribution in women with polycystic ovary syndrome and extent of its relation to insulin resistance. J Clin Endocrinol Metab 2007, 92: 2500-5.
- Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2008, 93: 1105-20.
- Legro RS, Schlaff WD, Diamond MP, et al; Reproductive Medicine Network. Total testosterone assays in women with polycystic ovary syndrome: precision and correlation with hirsutism. J Clin Endocrinol Metab 2010, 95: 5305-13.
- Alberti KG, Eckel RH, Grundy SM, et al; International Diabetes Federation Task Force on Epidemiology and Prevention; Hational Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; International Association for the Study of Obesity. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120: 1640-5.
- Moghetti P, Tosi F, Bonin C, et al. Divergences in insulin resistance between the different phenotypes of the polycystic ovary syndrome. J Clin Endocrinol Metab 2013, 98: E628-37.
- Wild RA, Carmina E, Diamanti-Kandarakis E, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab 2010, 95: 2038-49.
- Moran LJ, Misso ML, Wild RA, et al. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update 2010, 16: 347-63.
- Salley KE, Wickham EP, Cheang KI, et al. Glucose intolerance in polycystic ovary syndrome--a position statement of the Androgen Excess Society. J Clin Endocrinol Metab 2007, 92: 4546-56.
- Legro RS, Kunselman AR, Dodson WC, et al. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab 1999, 84: 165-9.
Valutazione ecografica nella policistosi ovarica
Ricciarda Raffaelli & Elena Lavarini
UO Ginecologia e Ostetricia, Policlinico GB Rossi, Università di Verona
(aggiornato al 14 aprile 2017)
La sindrome dell’ovaio policistico (PCOS) è stata definita nel 1990 durante la consensus conference statunitense tenutasi presso i National Institutes of Health (NIH). In questa occasione è stata stilata la prima linea guida sulla PCOS, la quale non considerava l’aspetto ecografico delle ovaie tra i criteri proposti per la diagnosi. L’ecografia ovarica viene menzionata per la diagnosi di PCOS nel 2003, durante la consensus conference di Rotterdam organizzata dalla Società Europea per la Riproduzione e l’Embriologia umana (ESHRE) e dalla Società Americana per la Medicina della Riproduzione (ASRM), che revisionava le linee guida del NIH. Le conclusioni di questo convegno prendono il nome di 'criteri di Rotterdam' e sostengono che la PCOS è una sindrome che deve essere valutata dopo avere escluso altri disordini secondari e non è sufficiente un singolo criterio diagnostico per definirla. Deve essere caratterizzata da almeno due dei tre parametri seguenti: oligo o anovulazione cronica, segni clinici o biochimici di iperandrogenismo e riscontro ecografico di ovaio policistico (1). La maggior parte dei partecipanti alla conferenza riteneva, infatti, che fosse necessario aggiungere la valutazione ecografica delle gonadi femminili ai criteri stabiliti dal NIH (anovulazione cronica ed iperandrogenismo). In accordo con la letteratura del tempo, venivano definite le caratteristiche ecografiche necessarie a identificare con maggior sensibilità e specificità la morfologia dell’ovaio policistico.
Gli elementi da valutare sono il numero di follicoli antrali misurati durante la fase follicolare, che deve essere di 12 o più follicoli in almeno un ovaio, con diametro compreso tra 2 e 9 mm e/o presenza di volume ovarico aumentato (> 10 mL) (2), calcolato usando la formula: lunghezza x larghezza x profondità x 0.5 (3). La distribuzione dei follicoli può essere omessa, mentre l’aumento del volume ovarico è una caratteristica tipica dell’ovaio policistico. Viene specificato inoltre che nel caso in cui l’ecografia venga eseguita in presenza di un follicolo dominante > 10 mm o di una cisti ovarica, l'esame deve essere ripetuto nel corso del ciclo mestruale seguente.
Nel 2009 la Androgen Excess-PCOS Society (AE-PCOS) ha proposto la propria revisione dei criteri diagnostici, puntando sull’iperandrogenismo (clinico o/e biochimico) e sulle disfunzioni ovariche (oligo-anovulazione e/o PCO). Il quadro ecografico secondo AE-PCOS non è un fattore indispensabile nella diagnosi (4). Storicamente, infatti, l’ecografia ovarica veniva effettuata quando la PCOS era già diagnosticata, in qualità di ulteriore accertamento e non per stabilire una diagnosi de novo (5). Questo è dovuto al fatto che in passato non c’era un consenso unanime sulle caratteristiche ecografiche necessarie per definire un ovaio policistico, né tale unanimità è stata raggiunta attualmente. Con il tempo, tuttavia, si è ottenuto il miglioramento delle tecnologie, dei software e l’introduzione di nuove sonde ecografiche, come quella da 8 MHz, migliorando sia la capacità di visualizzazione dei dettagli dell’ovaio, che la precisione delle misurazioni volumetriche dello stesso, permettendo così di revisionare i parametri precedentemente stabiliti.
Nel 2013 una revisione della letteratura ad opera dell’AE-PCOS (6) propone come criterio ecografico una soglia più alta del numero di follicoli (≥ 25 per ovaio), poiché, utilizzando la nuova sonda da 8 MHz, i 12 follicoli menzionati dai precedenti criteri di Rotterdam rappresentano un cut-off troppo basso, che indurrebbe a includere donne sane nel gruppo affetto da ovaio policistico. Per chi non è dotato di tale sonda ad alta frequenza invece, rimangono validi i precedenti criteri di Rotterdam.
Per la valutazione dell’ovaio policistico, la letteratura specifica propone altri parametri ecografici, che non hanno ancora ottenuto un consenso tale da essere inseriti tra i criteri diagnostici, quali la distribuzione dei follicoli, il rapporto tra area stromale e area totale dell’ovaio e la vascolarizzazione ovarica. Questi parametri sarebbero utili soprattutto nelle adolescenti non sessualmente attive, dove l’ecografia pelvica trans-addominale è preferibile all'approccio trans-vaginale, nonostante essa fornisca delle immagini a risoluzione minore soprattutto in caso di obesità.
Silfen et al (7) riportano che la distribuzione dei follicoli in pazienti con PCOS è periferica nel 100% delle obese e nel 75% delle non obese, rispetto al 31% dei controlli. Secondo Panchal et al (8), gli androgeni provocherebbero una proliferazione delle cellule stromali e della teca, provocando un aumento dello stroma, con conseguente spostamento dei follicoli in periferia. Il verificarsi di tale evento richiede del tempo, per cui il riscontro di una morfologia ovarica a distribuzione periferica dei follicoli è suggestivo di una malattia più severa.
Lo stroma ovarico viene maggiormente studiato con l’introduzione delle sonde 3D, che permettono di effettuare il calcolo del rapporto tra area dello stroma e area totale dell’ovaio, significativamente aumentato nella PCOS per il fenomeno spiegato in precedenza. L’aumento di questo rapporto è presente nel 20% delle donne nella popolazione generale ed è significativamente correlato con irsutismo e alti livelli di androgeni. Anche questo parametro rimane controverso in letteratura per la diagnosi di PCOS.
Un altro parametro che viene discusso in letteratura è la vascolarizzazione del parenchima ovarico. Alcuni studi, infatti, hanno dimostrato come sia comune un aumento del flusso sanguigno nelle pazienti con PCOS (10). Per la valutazione della vascolarizzazione vengono utilizzati la metodologia Doppler e la sonda 3D. Attualmente sono pochi gli studi che valutano i valori di riferimento dei parametri Doppler dell’arteria ovarica, per definire una morfologia ovarica policistica. Si è visto che gli indici di flusso e la vascolarizzazione sono significativamente più alti nelle pazienti con PCOS normopeso, rispetto a quelle in sovrappeso. Inoltre, tali parametri sembrano avere molta importanza nella risposta clinica alle terapie per l’infertilità. I risultati dell’indagine Doppler-flussimetrica di pazienti con PCOS potrebbero essere utili per comprendere l'eziologia della malattia e attuarne il follow-up clinico.
Bibliografia
- Rotterdam ESHRE/ASRM Sponsored PCOS Consensus Worshop Group. Revised 2003 consensus on diagnostic criteria and long-term healt risks related to polycystic ovary syndrome. Fertil Steril 2004, 81: 19-25.
- Balen AH, Laven JSE, Tan SL, Dewailly D. Ultrasound assessment of polycystyc ovary: international consensus definition. Hum Reprod Update 2003, 9: 505-14.
- Swanson M, Sauerbrei EE, Cooperberg PL. Medical implication of ultrasonically detected polycystic ovaries. J Clin Ultrasound 1981, 9: 219-22.
- Azziz R, Carmina E, Dewailly D, et al. The Androgen Excess and PCOS Society criteria for polycystic ovary sindrome: the complete task force report. Fertil Steril 2009, 91: 456-8
- Senaldi L, Gopi RP, Milla S, Shan B. Is ultrasound useful in the diagnosis of adolescents with polycystic ovary sindrome? J Pediatr Endocr Met 2015, 28: 608-12.
- Dewailly D, Lujan ME, Carmina E, et al. Definition and significance of polycystic ovarian morphology: a task force report from the Androgen Excess and Polycystyc Ovary Syndrome Society. Hum Reprod Update 2014, 20: 334-52.
- Silfen ME, Denburg MR, Manibo AM, et al. Early endocrine, metabolic, and sonographic characteristics of polycystic ovary syndrome (PCOS): comparison between non obese and obese adolescents. J Clin Endocrinol Metab 2003, 88: 4682-8.
- Panchal S, et al. Baseline scan and ultrasound diagnosis of PCOS. J Ultrasound Obstet Gynecol 2012, 6: 290-9.
- Pan HA, Wu MH, Cheng YC, et al. Quantification of Doppler signal in polycystic ovarian syndrome using 3D power Doppler ultrasonography. Hum Reprod 2002, 17: 2484.
- Shresta SM, Costello MF, Sjoblom P, et al. Power Doppler ultrasound assessment of follicular phase and its relationship with outcome of in vitro fertilisation. J Assist Reprod Genet 2006, 23: 161-9.
Terapia PCOs
Paolo Moghetti
Sezione di Endocrinologia e Metabolismo, Dipartimento di Medicina, Università di Verona, e UO di Endocrinologia e Malattie Metaboliche, Azienda Ospedaliera Universitaria Integrata, Verona
Il trattamento della PCOS è ancora in larga parte sintomatico e frequente è il ricorso a farmaci non approvati per questo utilizzo. E’ opportuno puntualizzare alcune differenze di approccio legate all’età della paziente.
Terapia nell'adolescente
Terapia in età fertile
Terapia dopo la menopausa
| Come PCOS? Trucco mnemonico per la valutazione e il trattamento della PCOS (modificato da Setji TL, et al. Polycystic ovary syndrome: update on diagnosis and treatment. Amer J Med 2014, 127: 912-9) |
||
| Valutazione | Trattamento | |
| Cicli | Indaga le mestruazioni: la lunghezza normale del ciclo è di 28 giorni (range 21-35) |
Se amenorrea ≥ 3 mesi (e gravindex negativo): MAP test
Metformina (come seconda linea) |
| Metabolismo | OGTT 75 g con misurazione glicemia a 0 e 120 min Profilo lipidico Test epatici (se altri fattori di rischio per steatosi non alcolica) |
Modifiche dello stile di vita: dieta, esercizio fisico e calo ponderale (se sovrappeso o obesità) Metformina per intolleranza glucidica non controllata dalle modifiche dello stile di vita Statine se indicate secondo linee guida (ATP-III/ACC/AHA) |
| Psicosociale | Valuta depressione e disturbi del comportamento alimentare Sottolinea l’importanza del problema medico PCOS e dai sostegno senza giudicare Discuti la gestione dello stress Rinforza i comportamenti di autocura |
Se si identificano depressione o disturbi del comportamento alimentare, consulenza psichiatrica o terapia anti-depressiva |
| Cosmetica | Valuta l’irsutismo con la scala di Ferriman-Gallwey Valuta acne e stempiatura Dosa gli androgeni se hai dubbi sul grado di irsutismo o in caso di sintomi atipici |
Contraccettivi ormonali contenenti estrogeni Anti-androgeni come spironolattone o finasteride (teratogeni, da usare solo con contraccezione efficace) Ciproterone acetato |
| Ovulazione e fertilità | Informa sulla diminuzione (ma non assenza) di fertilità Valuta gli obiettivi relativi alla fertilità |
Se desiderio di gravidanza, invia a un centro specializzato per eventuale terapia con clomifene Metformina ha ruolo limitato |
| Sleep apnea | Esegui uno screening: sonnolenza diurna, cefalea mattutina, sintomi da reflusso, russamento, pause respiratorie | Studio del sonno Se diagnosi di sleep apnea, CPAP |
Terapia della PCOS nell'adolescente
Paolo Moghetti
Sezione di Endocrinologia e Metabolismo, Dipartimento di Medicina, Università di Verona, e UO di Endocrinologia e Malattie Metaboliche, Azienda Ospedaliera Universitaria Integrata, Verona
Come già ricordato, la diagnosi di PCOS poggia su elementi mal definibili nei primi anni dopo il menarca e richiede quindi cautela. Nondimeno vi sono situazioni in cui l’espressione clinica della sindrome è chiara precocemente e si ritiene che queste manifestazioni meritino un trattamento (1,2), pur in assenza di studi longitudinali che permettano di valutarne gli effetti a distanza.
Va tenuto presente che l’eccesso ponderale sembra giocare un ruolo importante nel mantenimento nel tempo delle alterazioni mestruali comuni nei primi anni dell’adolescenza (3,4). Di conseguenza, deve essere posta particolare energia nel cercare di intervenire su questo aspetto, laddove presente, con interventi basati innanzitutto sulla dieta e sull’attività fisica.
È interessante notare che le figlie delle donne con PCOS presentano precocemente iperinsulinemia e successivamente un aumento degli androgeni nel corso della pubertà (5,6), suggerendo che questi aspetti possano essere valorizzati come possibili manifestazioni precoci della sindrome. Le modalità con cui intervenire in adolescenza sulle manifestazioni cliniche di PCOS restano comunque controverse (7).
Gli estroprogestinici vengono considerati uno strumento di prima linea, in aggiunta alle modifiche dello stile di vita, anche nell’adolescente con PCOS, sia per contrastare le manifestazioni di iperandrogenismo che a scopo contraccettivo (2,8). Gli studi di confronto fra contraccettivi diversi sono estremamente scarsi e limitati e quindi la scelta di un preparato piuttosto che di un altro non è basata su solide evidenze. L’Endocrine Society suggerisce l’utilizzo dei preparati ormonali anche in epoca pre-menarcale, se i segni clinici e biochimici di iperandrogenismo sono chiari e lo sviluppo puberale è avanzato (almeno stadio puberale Breast 4 sec Tanner) (2).
Anche la metformina viene spesso utilizzata in questi casi, sia per i suoi benefici metabolici che per facilitare il controllo del peso (2,8), ma va ricordato che è un utilizzo off label. Dati limitati suggeriscono che la combinazione modifiche dello stile di vita/metformina/estroprogestinico possa permettere di attenuare gli effetti metabolici sfavorevoli dei contraccettivi orali nelle adolescenti obese con PCOS (9).
Alcuni autori propongono nelle adolescenti con PCOS anche l’utilizzo di piccole dosi di flutamide (62.5 mg/die), eventualmente in combinazione con la metformina. Gli studi che suggeriscono benefici con questa strategia sono però limitati e in gran parte non controllati. Inoltre, va tenuta presente la potenziale epatotossicità, non frequente ma talora grave, di questo farmaco anti-androgeno. Al momento non vi sono segnalazioni di epatotossicità severa con 62.5 mg/die di flutamide, ma l’ipotesi che questo effetto avverso sia dose-dipendente non è documentata con certezza. Va segnalato che reazioni molto gravi sono state riportate anche con dosi di 125 mg/die e che il fenomeno si può presentare anche dopo diversi mesi di trattamento (10). Va anche ricordato che l’utilizzo degli anti-androgeni richiede particolare cautela nell’adolescente, per il maggior rischio di una compliance inadeguata nei confronti della copertura contraccettiva (2).
Bibliografia
- Fauser BC, Tarlatzis BC, Rebar RW, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril 2012, 97: 28-38.
- Legro RS, Arslanian SA, Ehrmann DA, et al. Diagnosis and treatment of polycystic ovary syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2013, 98: 4565-92.
- Metcalf MG, Skidmore DS, Lowry GF, et al. Incidence of ovulation in the years after the menarche. J Endocrinol 1983, 97: 213–9.
- van Hooff MH, Voorhorst FJ, Kaptein MB, et al. Predictive value of menstrual cycle pattern, body mass index, hormone levels and polycystic ovaries at age 15 years for oligo-amenorrhoea at age 18 years. Hum Reprod 2004, 19: 383-92.
- Kent SC, Gnatuk CL, Kunselman AR, et al. Hyperandrogenism and hyperinsulinism in children of women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab 2008, 93: 1662-9.
- Sir-Petermann T, Codner E, Pérez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 2009, 94: 1923-30.
- Warren-Ulanch J, Arslanian S. Treatment of PCOS in adolescence. Best Pract Res Clin Endocrinol Metab 2006, 20: 311-30.
- Geller DH, Pacaud D, Gordon CM, Misra M; of the Drug and Therapeutics Committee of the Pediatric Endocrine Society. State of the Art Review. Emerging Therapies: The Use of Insulin Sensitizers in the Treatment of Adolescents with Polycystic Ovary Syndrome (PCOS). Int J Pediatr Endocrinol 2011, 2011: 9.
- Hoeger K, Davidson K, Kochman L, et al. The impact of metformin, oral contraceptives, and lifestyle modification on polycystic ovary syndrome in obese adolescent women in two randomized, placebo-controlled clinical trials. J Clin Endocrinol Metab 2008, 93: 4299-306.
- Brahm J, Brahm M, Segovia R, et al. Acute and fulminant hepatitis induced by flutamide: case series report and review of the literature. Ann Hepatol 2011, 10: 93-8.