Epidemiologia dell'ipoparatiroidismo

Massimo Procopio1, Assunta Santonati2 & Antonio Spada2
1SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
2Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
L’ipoparatiroidismo (ipoPTH) è una malattia abbastanza rara causata da un deficit totale o parziale di secrezione di paratormone (PTH) da parte delle ghiandole paratiroidi (ipoPTH vero) (1). La malattia è caratterizzata sul piano biochimico da ipocalcemia, iperfosfatemia e normale funzionalità renale, a fronte di sintomi conclamati o latenti di aumentata eccitabilità neuro-muscolare.
Lo pseudoipoparatiroidismo (pseudoipoPTH) si connota per una resistenza degli organi bersaglio all’azione del PTH, a cui consegue aumentata secrezione compensatoria dell’ormone.
L’ipoPTH è considerata una causa relativamente comune di ipocalcemia, anche se molto meno frequente dell’ipovitaminosi D e dell’insufficienza renale cronica.
Tra le cause di ipoparatiroidismo, la più frequente è la forma post-chirurgica. L'incidenza di tale complicanza, principalmente legata alla chirurgia tiroidea e paratiroidea, varia da casistica a casistica, in relazione all’esperienza del chirurgo e ad altri fattori relativi all'indicazione e all'estensione dell'intervento chirurgico. Casistiche di centri con alti volumi operatori riportano un rischio di ipocalcemia permanente dell'1%, mentre altre casistiche riportano un rischio maggiore (4.4-12.1%) (2). La prevalenza dell'ipoparatiroidismo post-chirurgico è stata stimata intorno a 22/100.000 in uno studio di popolazione danese e a 29/100.000 in uno studio americano della Mayo Clinic (3). Nei pazienti ospedalizzati in Italia nel periodo 2006-2013 la prevalenza riscontrata di ipoparatiroidismo è stata di 3461 pazienti/anno, con una tendenza a decrescere (4); il tasso di ospedalizzazione riscontrato è stato mediamente 5.3/100.000 pazienti. La prevalenza è maggiore nelle donne (71-88%).
In realtà, la reale prevalenza è probabilmente sottostimata per l’assenza di una reale definizione di ipocalcemia, per la variabilità dei range di normalità della calcemia, per la variabilità del tempo di rilevazione della calcemia post-operatoria e dei tempi di follow-up. Nell’ambito dell’ipoparatiroidismo post-chirurgico le condizioni più rischiose sono la tiroidectomia totale (in particolare gli interventi più demolitivi per neoplasie, con svuotamento linfonodale), l’exeresi paratiroidea per iperparatiroidismo e la tiroidectomia totale per m. di Graves (dovuta all’aumentata vascolarizzazione del parenchima) (5). La tiroidectomia sub-totale, con residuo < 1 g per lobo, riduce l’incidenza dell’ipoparatiroidismo post-chirurgico (6). L’ipoparatiroidismo post-chirurgico può essere transitorio nel 4.9-7.3% dei casi: di questi, almeno il 60% si risolve entro 4-6 settimane. L’ipoparatiroidismo diviene permanente nello 0.12-4.6% dei casi ed è definito tale quando si protrae per oltre sei mesi dopo l’intervento chirurgico.
Molto complessa è la definizione di fattori predittivi di ipoparatiroidismo post-chirurgico. Alcuni studi hanno ipotizzato che la rilevazione di livelli di PTH < 10 pg/mL a 4 e 6 ore dall’intervento chirurgico possa predire in modo piuttosto preciso la successiva ipocalcemia (7). Il registro nazionale danese, che ha arruolato 688 pazienti affetti da ipoparatiroidismo post-chirurgico, ha condotto una peculiare valutazione sulle complicanze croniche di questa patologia. In particolare, è stato rilevato un aumentato rischio di complicanze renali (nefrolitiasi, insufficienza renale), crisi comiziali, irritabilità nero-muscolare, disturbi neuro-psichiatrici, infezioni, patologie oftalmiche, cutanee, mentre sembrerebbe essere ridotto il rischio di fratture degli arti superiori e neoplasie gastro-intestinali (3,8). La mortalità cardio-vascolare non sarebbe aumentata nell'ipoparatiroidismo post-chirurgico, mentre aumenterebbe la morbilità, in particolare per cardiopatia ischemica, cardiopatia dilatativa, e scompenso cardiaco (3).
La prevalenza dell'ipoparatiroidismo da causa non chirurgica è stata stimata intorno a 2.3/100.000 (9). Anche in questa forma è stato riportato un aumentato rischio di insufficienza renale, crisi comiziali, disturbi neuro-psichiatrici, infezioni e un ridotto rischio di neoplasie (9), mentre è stato riportato un aumentato rischio di morbilità cardio-vascolare, cataratta e fratture degli arti superiori. La forma acquisita non chirurgica più frequente è quella autoimmune, che può presentarsi in modo isolato o associato ad altre malattie autoimmuni nelle cosiddette sindromi polighiandolari autoimmuni (SPA). L'epidemiologia della forma autoimmune isolata è poco conosciuta e vi sono pochi dati in letteratura, anche per la difficoltà diagnostica dovuta alla mancanza di specifici marcatori immunologici sierici (10,11). La SPA-1 è molto rara, con prevalenza di 1/1.000.000, anche se in alcuni gruppi di popolazione geneticamente isolati, come gli abitanti della Sardegna, la prevalenza può giungere a 1/14.500. Vi sono poi altre forme rare di ipoparatiroidismo acquisito da distruzione ghiandolare secondaria a infiltrazione granulomatosa, metastatica, emosiderinica o danno post-attinico o radio-metabolico. Tra le forme congenite sindromiche, spicca la sindrome di Di George (1:4000-5000 nati vivi). Un solo studio epidemiologico rileva una prevalenza di ipoparatiroidismo ereditario isolato di 7.2 per milione, mentre la forma di ipocalcemia familiare autosomica dominante, legata ad una mutazione iperattivante del gene per il recettore calcio-sensibile (CaSR), può giungere a una prevalenza di 1/70.000.
Bibliografia
- Bilezikian JP, Khan A, Potts JT Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res 2011, 26: 2317-37.
- Balasubramanian SP. Iatrogenic/post-surgical hypoparathyroidism: where do we go from here? Endocrine 2014, 47: 357-9.
- Underbjerg L, Sikjaer T, Mosekilde L, et al. Cardiovascular and renal complications to postsurgical hypoparathyroidism: a Danish nationwide controlled historic follow-up study. J Bone Miner Res 2013, 28: 2277-85.
- Cipriani C, Pepe J, Biamonte F, et al. The epidemiology of hypoparathyroidism in Italy: an 8-year register-based study. Calcif Tissue Int 2016, 100: 278-85.
- Thomusch O, Machens A, Sekulla C, et al. The impact of surgical technique on postoperative hypoparathyroidism in bilateral thyroid surgery. A multivariate analysis of 5846 consecutive patients. Surgery 2003, 133: 180-5.
- Asari R, Passler C, Kaczirek K, et al. Hypoparathyroidism after total thyroidectomy: a prospective study. Arch Surg 2008, 143: 132-7
- Richards ML, Bingener-Casey J, Pierce D, et al. Intraoperative parathyroid hormone assay: an accurate predictor of symptomatic hypocalcemia following thyroidectomy. Arch Surg 2003, 138: 632–5.
- Underbjerg L, Sikjaer T, Mosekilde L, et al. Postsurgical hypoparathyroidism - Risk of fractures, psychiatric diseases, cancer, cataract, and infections. J Bone Miner Res 2014, 29: 2504-10.
- Underbjerg L, Skjaer T, Mosekilde L, et al. The epidemiology of nonsurgical hypoparathyroidism in Denmark: a nationwide case finding study. J Bone Miner Res 2015, 30: 1738-44.
- Betterle C, Garelli S, Presotto F. Diagnosis and classification of autoimmune parathyroid disease. Autoimmunity Rev 2014, 13: 417-22.
- Brandi ML. Genetics of hypoparathyroidism and pseudohypoparathyroidism. J Endocrinol Invest 2011, 34 suppl 7: 27-34.
Classificazione degli ipoparatiroidismi
Massimo Procopio
SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
(aggiornato al 18 dicembre 2019)
L’ipoparatiroidismo può essere distinto in transitorio o permanente, a seconda che la causa sia, rispettivamente, reversibile o no, oppure in totale o parziale a seconda dell’entità della funzionalità paratiroidea residua.
L’ipoparatiroidismo può essere ancora distinto in acuto (sintomi conclamati e importanti) o cronico (sintomi latenti e sfumati o assenti) a seconda della modalità e rapidità di presentazione clinica ed alla durata di malattia.
Sul piano eziologico l’ipoparatiroidismo può essere distinto in acquisito e congenito. Tra le cause acquisite predomina la forma post-chirurgica, conseguente a interventi estesi o re-interventi sul collo (tiroidectomia totale per m. di Basedow, neoplasie tiroidee o della regione cervicale con linfoadenectomia del comparto centrale, gozzo retro-sternale, iperparatiroidismo primario multi-ghiandolare, iperparatiroidismo terziario).
L’altra forma acquisita più comune è l’ipoparatiroidismo autoimmune, in cui vi è generalmente una distruzione su base autoimmune delle paratiroidi. In assenza di altre malattie autoimmuni associate e concomitante assenza di anticorpi specifici, quali anti-NACHT leucine-rich repeat protein 5 (anti-NALP5), anti-interferone omega, anti-triptofano-idrossilasi, anti-aromatico L-aminoacido-decarbossilasi o anti-tirosino-idrossilasi, e di mutazioni del gene regolatore dell'autoimmunità (AIRE), l'eziologia autoimmune rimane ipotetica.
Altre cause acquisite più rare sono: terapia radiometabolica dell'ipertiroidismo, terapia radiante esterna sulla regione cervicale, disordini infiltrativi quali emocromatosi, malattia di Wilson, malattie granulomatose e infiltrazione metastatica, ipomagnesiemia severa conseguente ad alcolismo, malnutrizione e malassorbimento intestinale, ipermagnesiemia.
L’ipoparatiroidismo congenito è generalmente ereditario ed è dovuto a mutazioni di diversi geni o raramente secondario a ipomagnesiemia severa, ipermagnesiemia o iperparatiroidismo primario della madre.
L’ipoparatiroidismo congenito su base genetica può essere isolato o associato ad altri disordini endocrini o nel contesto di quadri sindromici. Nell’ipoparatiroidismo isolato sono interessati geni che sono essenziali per la sintesi, la secrezione del PTH o per lo sviluppo delle ghiandole paratiroidi, come ad esempio il gene del PTH, il gene del recettore calcio-sensibile (CaSR) e il gene glial cell homologue B (GCMB). Si trasmette con ereditarietà autosomica dominante o recessiva, oppure legata al cromosoma X con modalità recessiva.
La mutazione del gene che codifica per il PTH è molto rara e può avere una trasmissione autosomica recessiva o dominante.
La mutazione iperattivante del gene che codifica per il CaSR è ritenuta tra le cause più frequenti di ipoparatiroidismo congenito e interessa generalmente il dominio extra-cellulare del recettore, che è complessato con una proteina G. Ha generalmente una trasmissione autosomica dominante ad alta penetranza e pertanto circa la metà dei parenti di primo grado presenta la malattia, caratterizzata da ipocalcemia e relativa ipercalciuria, con alto rischio di nefrocalcinosi che si sviluppa in corso di terapia suppletiva con calcio e calcitriolo.
Nelle forme legate al cromosoma X con trasmissione recessiva, viene alterato il controllo della trascrizione del gene Sox3, con compromissione dello sviluppo delle ghiandole paratiroidee; solo i maschi sono affetti dalla malattia, con esordio infantile precoce dell’ipocalcemia accompagnata da crisi comiziali.
Tra le cause di ipoparatiroidismo su base genetica associato ad altri disordini è compresa la sindrome polighiandolare autoimmune tipo 1 (SPA-1), caratterizzata dall’associazione di ipoparatiroidismo, morbo di Addison e candidiasi muco-cutanea cronica. Per definire la sindrome, è sufficiente la presenza di ipoparatiroidismo o di un'altra malattia della triade, in presenza di mutazioni del gene AIRE e/o positività di anticorpi specifici quali anti-NALP5, anti-interferone omega, anti-triptofano-idrossilasi, anti-aromatico L-aminoacido-decarbossilasi o anti-tirosino-idrossilasi. L'ipoparatiroidismo può essere associato alla tiroidite autoimmune (in questo caso la condizione è classificata come SPA-3) oppure con altre malattie autoimmuni (si parlerà quindi di SPA-4). L’ipoparatiroidismo, presente in oltre l’80% dei pazienti affetti da SPA-1, si manifesta generalmente nell’infanzia o adolescenza. La comparsa delle altre manifestazioni cliniche può avvenire anche successivamente e pertanto i pazienti devono essere sottoposti a regolare follow-up a lungo termine. La trasmissione avviene generalmente per via autosomica recessiva od occasionalmente autosomica dominante. È causata da una mutazione del gene AIRE, che codifica per un fattore di trascrizione contenente zinco presente nel timo e nei linfonodi. Nella SPA-1 è stato riscontrato frequentemente un anticorpo diretto contro un auto-antigene specifico delle paratiroidi, detto NALP5.
La sindrome di Di George è caratterizzata da: ipoparatiroidismo con ipocalcemia asintomatica (60% dei casi), aplasia o ipoplasia timica con immuno-deficienza, cardiopatia congenita, palatoschisi, facies dismorfica, anomalie renali con ridotta funzionalità renale. È dovuta generalmente a mutazioni de novo, anche se sono state riportate mutazioni ereditate con modalità autosomica dominante. In alcuni pazienti affetti dalla sindrome studi molecolari hanno evidenziato micro-delezioni sulla regione q11.21-q11.23 del cromosoma 22, in cui sono state identificate solo mutazioni puntiformi del gene TBX1. Tale gene codifica per un fattore di trascrizione T-box, che è espresso ampiamente nei tessuti embrionali che danno origine agli organi interessati dalla sindrome. Altri casi indistinguibili dalla sindrome di DiGeorge sono stati riportati in pazienti con delezioni in altre regioni cromosomiche: p13 del cromosoma 10, p13 del cromosoma 17 e q21 del cromosoma 18. Sono state inoltre identificate altre delezioni della regione q11 del cromosoma 22, che possono causare anomalie facciali, cardio-vascolari di tipo cono-truncale e sindrome velo-cardio-facciale, in cui l’ipocalcemia è presente nel 20% dei casi. A causa della variabilità fenotipica, tali condizioni sono state raggruppate sotto l’acronimo CATCH-22, che indica l’associazione di anomalie facciali, ipoplasia timica, palatoschisi, ipocalcemia e delezione della regione q11 del cromosoma 22.
Altre rare forme di ipoparatiroidismo su base genetica sono:
- la sindrome caratterizzata da ipoparatiroidismo, ritardo mentale, ritardo di crescita e dismorfismo dovuta a mutazioni del gene TBCE sulla regione q42-43 del cromosoma 1, che codifica per una proteina necessaria per l’assemblaggio micro-tubulare nei tessuti interessati. Nel suo ambito si distinguono:
- la sindrome di Sanjad-Sakati, comune nelle popolazioni arabe, in cui si associano disgenesia paratiroidea, bassa statura, ritardo mentale, microftalmia, microcefalia, piedi e mani corte e anomalie dentarie;
- la sindrome di Kenny-Caffey, in cui si associano ipoparatiroidismo, nanismo, restringimento della cavità midollare delle ossa lunghe e anomalie oculari;
- la sindrome ipoparatiroidismo, sordità, displasia renale, causata da una mutazione del gene GATA3, che codifica per il fattore di trascrizione GATA3 necessario per il normale sviluppo paratiroideo, renale e della vescicola otica.
Infine, devono essere ricordati l’ipoparatiroidismo associato a disfunzione mitocondriale, dovuto a difetti del DNA mitocondriale come nella sindrome di Kearns-Sayre, la sindrome MELAS (encefalopatia mitocondriale, acidosi lattica ed episodi simil-stroke) e la sindrome da deficit della proteina mitocondriale trifunzionale.
| Classificazione eziologica dell'ipoparatiroidismo | ||
| Acquisito | Iatrogeno | Post-chirurgico Post-radioterapia esterna Post-terapia radiometabolica |
| Autoimmune | ||
| Malattie infiltrative | Emocromatosi Malattia di Wilson Malattie granulomatose Infiltrazione metastatica |
|
| Ipomagnesiemia Ipermagnesiemia |
||
| Congenito | Isolato | Gene PTH Gene CaSR Gene GCMB |
| Associato | SPA-1, SPA-3, SPA-4 Sindrome di Di George Sindrome di Sanjad-Sakati Sindrome Kenny-Caffey Sindrome ipoparatiroidismo, sordità, displasia renale Sindrome di Kearns-Sayre Sindrome MELAS Sindrome da deficit della proteina mitocondriale trifunzionale |
|
Bibliografia
- Bilezikian JP, Khan A, Potts JT, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Min Res 2011, 26: 2317-37.
- Brandi ML. Genetics of hypoparathyroidism and pseudohypoparathyroidism. J Endocrinol Invest 2011, 34 (7 suppl): 27-34.
- Betterle C, Garelli S, Presotto F. Diagnosis and classification of autoimmune parathyroid disease. Autoimmunity Rev 2014, 13: 417-22.
Storia naturale dell'ipoparatiroidismo
Massimo Procopio
SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
(aggiornato al 18 dicembre 2019)
La storia naturale di malattia varia a seconda dell’eziologia. L'ipoparatiroidismo può:
- avere un esordio neonatale-infantile;
- manifestarsi nell’età adulta:
- in modo acuto, dovuto al mancato riassorbimento renale di calcio, con conseguente ipocalcemia acuta e sintomatologia neuromuscolare (crampi, tetania), comiziale (convulsioni) o cardiologica (insufficienza cardiaca, aritmie);
- in modo cronico, quando decade anche l’effetto fosfaturico, con conseguente iperfosfatemia e deposizione di cristalli di fosfato di calcio e sintomatologia neuromuscolare sfumata (parestesie) nonché manifestazioni peculiari di danno degli organi bersaglio, quali cataratta, calcificazione dei nuclei della base o altre calcificazioni ectopiche (1).
In studi di coorte nell'ipoparatiroidismo post-chirurgico è stato riportato un aumentato rischio di complicanze renali (nefrolitiasi, insufficienza renale), crisi comiziali, disturbi neuro-psichiatrici, infezioni, mentre sembra essere ridotto il rischio di fratture degli arti superiori e delle neoplasie gastro-intestinali (2-4). Nella forma da causa non chirurgica è stato riscontrato che il rischio di tali comorbilità è aumentato o ridotto in maniera simile, salvo il rilievo di un aumentato rischio di morbilità cardio-vascolare, cataratta e fratture degli arti superiori (5).
La terapia suppletiva convenzionale con calcio e calcitriolo è spesso causa di ipercalciuria ed è chiamata in gioco nello sviluppo di temibili complicanze, quali la nefrocalcinosi, la nefro-urolitiasi e l’insufficienza renale cronica. In uno studio di coorte, il 31% dei pazienti sottoposti a "renal imaging" mediante ecografia e/o TC presentava nefrolitiasi, mentre la frequenza di insufficienza renale cronica stadio 3 o più avanzato era da 2 a 17 volte maggiore rispetto ai controlli di pari età (4).
Bibliografia
- Bilezikian JP, Khan A, Potts JT Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res 2011, 26: 2317-37.
- Underbjerg L, Sikjaer T, Mosekilde L, et al. Cardiovascular and renal complications to postsurgical hypoparathyroidism: a Danish nationwide controlled historic follow-up study. J Bone Miner Res 2013, 28: 2277-85.
- Underbjerg L, Sikjaer T, Mosekilde L, et al. Postsurgical hypoparathyroidism - Risk of fractures, psychiatric diseases, cancer, cataract, and infections. J Bone Miner Res 2014, 29: 2504-10.
- Mitchell DM, Regan S, Cooley MR, et al. Long-term follow up of patients with hypoparathyroidism. J Clin Endocrinol Metab 2012, 97: 4507-14.
- Underbjerg L, Skjaer T, Mosekilde L, et al. The epidemiology of nonsurgical hypoparathyroidism in Denmark: a nationwide case finding study. J Bone Miner Res 2015, 30: 1738-44.
Manifestazioni acute di ipoparatiroidismo
Rossella Antonelli
UOC Endocrinologia e Diabetologia, Polo Ospedaliero Integrato Ospedale S. Eugenio & Ospedale CTO, Università Tor Vergata, Roma
(aggiornato al 18 dicembre 2019)
Il calcio, sotto forma di ione libero (Ca++) è di fondamentale importanza per numerose funzioni cellulari: regola la secrezione endocrina ed esocrina di numerose ghiandole, la trasmissione neuromuscolare e alcune attività enzimatiche.
L’ipoparatiroidismo causa ipocalcemia, in quanto la secrezione di paratormone è inadeguata nel mobilizzare calcio dall’osso, nel riassorbire calcio dalla parte distale del nefrone e nello stimolare l’attività dell’1α-idrossilasi renale; come risultato, viene prodotta 1,25-vitamina D in maniera insufficiente ad assicurare un adeguato assorbimento intestinale di calcio.
La maggior parte delle manifestazioni cliniche dell’ipocalcemia è conseguenza dello stato di ipereccitabilià neuromuscolare da essa indotta, in quanto viene ridotta la differenza tra potenziale a riposo e potenziale soglia della membrana cellulare, viene ridotto l’inotropismo cardiaco e ridotte le resistenze vascolari periferiche (1,2).
Un’ipocalcemia stabile e presente da tempo solitamente non si associa a manifestazioni cliniche importanti, mentre il rapido decremento dei valori plasmatici di calcio ionizzato, spesso determinato da cause favorenti, caratterizza la crisi ipocalcemica, la cui manifestazione più importante è la tetania (3). Le cause favorenti più frequenti sono:
- la concomitante presenza di iperfosfatemia (l’aumento del fosfato circolante lega il calcio libero);
- l’alcalosi, sia respiratoria sia metabolica (l’aumento di HCO3- riduce la quota di calcio ionizzato, l’aumento del pH aumenta l’affinità delle proteine leganti il calcio, riducendone la quota attiva). Inoltre, alcalosi e diminuzione dei livelli di calcio ione influenzano l’eccitabilità neuromuscolare in maniera sinergica; la cronica riduzione di calcio ionizzato nel liquor sensibilizza il centro respiratorio e predispone il paziente all’iperventilazione e all’alcalosi respiratoria. I livelli di calcio ionizzato si abbassano di circa il 3-8% per ogni incremento di pH pari a 0.1;
- l’ipo e l’ipermagnesiemia possono entrambe indurre un ipoparatiroidismo funzionale;
- l’ipo- e l’iperpotassiemia, attraverso la variazione dell’eccitabilità neuromuscolare;
- la gravidanza e l'allattamento.
Le manifestazioni cliniche generalmente compaiono quando la calcemia totale scende sotto i 7-7.5 mg/dL e il calcio ionizzato è ≤ 1.15 mmol/L.
- Sintomi neuromuscolari: parestesie e formicolio prevalentemente a carico di regione peri-orale, lingua e dita, iperestesie cutanee e per gli stimoli luminosi e acustici, crampi muscolari, tetania, contrazione della muscolatura liscia intestinale con vomito, diarrea e pollachiuria.
- Sintomi neurologici: cefalea, compromissione di grado variabile delle funzioni cognitive e della memoria a breve e lungo termine, crisi epilettiche ad espressione variabile (dalle non tipiche, confinate ad episodi di perdita di coscienza, alle convulsive tipiche, quali episodi di grande male e alle crisi focali).
- Sintomi cardio-vascolari: ipotensione, deficit di contrattilità miocardica con riduzione della gittata cardiaca, causato principalmente da inibizione del rilasciamento in fase diastolica, bradicardia, aritmie, diminuzione della sensibilità alla digitale, insufficienza cardiaca refrattaria, aumentata incidenza di eventi cardio-vascolari (principalmente cardiopatia ischemica), senza peraltro incremento della mortalità cardio-vascolare, arresto cardiaco (4). La disfunzione cardiaca risponde brillantemente alla correzione dell’ipocalcemia, con risoluzione nel 98% dei pazienti (5).
La crisi tetanica è caratterizzata da:
- spasmi della muscolatura del volto e degli arti;
- adduzione delle braccia al tronco, flessione dell’avambraccio sul braccio;
- spasmo carpo-pedale (flessione del gomito, polso e articolazioni metacarpo-falangee, estensione delle articolazioni inter-falangee e adduzione del pollice – mano da ostetrico – con inarcamento della superficie plantare dei piedi);
- trisma – risus sardonicus;
- opistotono;
- faringo-spasmo, laringo-spasmo, bronco-spasmo.
Sintomi neuromuscolari senza crisi tetanica classica, prevalenti nel sesso femminile, correlati prevalentemente a iperventilazione o ipomagnesemia, sono caratteristici della spasmofilia (6).
Segni di ipocalcemia
- Segno di Trousseau: si evidenzia gonfiando il bracciale dello sfigmomanometro intorno al braccio, per almeno tre minuti, appena al di sopra della pressione sistolica; la conseguente ischemia dei nervi motori determina dopo pochi minuti uno spasmo dei muscoli carpali, generalmente a dita estese. In caso di esito negativo della manovra di Trousseau, si può eseguire una prova di iperventilazione (30 atti respiratori in un minuto), che in alcuni casi favorisce la comparsa del crampo tetanico (test di Trousseau – von Bonsdorff) (fig. 1).
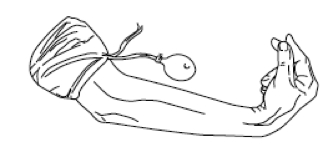
Figura 1. Segno di Trousseau, mano da ostetrico.
- Segno di Chvostek (fig. 2): contrazione dei muscoli facciali, includendo i muscoli peri-orali e l’orbicularis oculi, ottenuta percuotendo il decorso del nervo facciale in due punti specifici:
- da 0.5 a 1 cm sotto il processo zigomatico dell’osso temporale, 2 cm anteriormente al lobo dell’orecchio, in linea con l’angolo della mandibola (Chvostek 1);
- su una linea che unisce lo zigomo con l’angolo superiore della bocca, ad un terzo di distanza dallo zigomo (Chvostek 2).
È presente in circa il 25% dei pazienti normali; d’altra parte, circa il 30% dei pazienti con ipocalcemia ha uno Chvostek negativo.
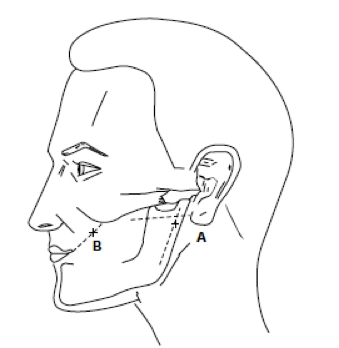
Figura 2. A: Chvostek 1; B: Chvostek 2
Sebbene i due segni non siano mai stati direttamente comparati, il segno di Trousseau viene ritenuto un indice più specifico di ipocalcemia che il segno di Chvostek. In un unico studio, il 94% dei pazienti con ipocalcemia confermata aveva un Trousseau positivo, mentre solo l’1% dei sani presentava un segno positivo.
- Alterazioni ECG:
- allungamento dell’intervallo QT corretto per la frequenza cardiaca (è dovuto all’allungamento dell’ST, che si verifica senza variazione di durata dell’onda T, piuttosto che ad un’alterazione del complesso QRS). Questa caratteristica, oltre che dell’ipocalcemia, è caratteristica anche dell’ipotermia;
- occasionale appiattimento, appuntimento o inversione dell’onda T;
- tachicardia sopra-ventricolare con BAV 2:1 e BAV completi.
- Papilledema: l’ipocalcemia grave si può inoltre associare a una riduzione del riassorbimento del liquor. L’ipertensione endocranica che ne deriva - pseudotumor cerebri, definito benigno, e presente in altre endocrinopatie - può essere evidenziata all’esame del fundus oculi con il rilievo di papilledema (turgore delle vene, emorragie a fiamma, stella maculare, papilla pallida a margini indistinti). Il papilledema migliora con la correzione dell’ipocalcemia. La complicanza più temibile del papilledema è la perdita del visus; il meccanismo principale per il danno permanente del nervo ottico è la stasi del flusso assoplasmico e la conseguente ischemia intra-neuronale (7).
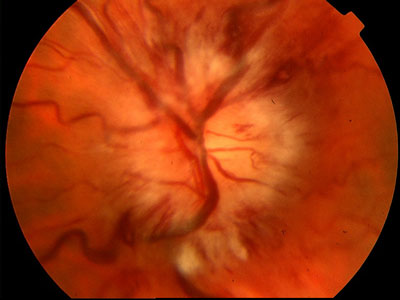
Figura 3. Papilledema
Bibliografia
- Bilezikian JP, Khan A, Potts JT Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res 2011, 26: 2317-37.
- Shoback D. Hypoparathyroidism. N Engl J Med 2008, 359: 391-402.
- Williams A, et al. Tetany: a diagnostic dilemma. J Anaesthesiol Clin Pharmacol 2011, 27: 393-4.
- Behagel A, Donal E. Hypocalcemia–induced dilated cardiomyopathy: a case report. Eur J Echocard 2011, 10: E 38.
- Newman BD, et al. Reversible cardiac dysfunction associated with hypocalcemia: a systematic review and metanalysis of individual patient data. Hearth Fail Rev 2014, 19: 199-205.
- Bazzichi L, et al. Spasmophilia comorbidity in fibromyalgia syndrome. Clin Exp Rheumatol 2010, 28 (6 suppl 63): S94-9.
- Lee AG, Wall M. Papilledema: are we nearer to a Consensus on pathogenesis and treatment? Curr Neurol Neurosci Rep 2012, 12: 334-9.
Manifestazioni specifiche di ipoparatiroidismo
Assunta Santonati & Antonio Spada
Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
La sintomatologia dell’ipoparatiroidismo dipende dalla severità e dalla cronicità dell’ipocalcemia. Nelle forme croniche di ipoparatiroidismo, l’ipocalcemia si instaura lentamente ed è di entità minore. L'ipocalcemia cronica è frequentemente asintomatica. La presenza di ipoparatiroidismo è spesso suggerita dalle manifestazioni cliniche della malattia di base (es. cataratta, calcificazioni dei gangli della base e candidosi cronica nell'ipoparatiroidismo idiopatico) (1-3).
Nell’ipoparatiroidismo cronico si instaurano lesioni causate dalla deposizione di fosfato di calcio a livello dei tessuti, in particolare a livello cutaneo, del cristallino e nel sistema nervoso centrale. Inoltre, l’ipocalcemia provoca disturbi al trofismo dei tessuti di origine ectodermica (in particolare cute ed annessi, denti).
Manifestazioni cutanee
L'ipocalcemia cronica determina disturbi del trofismo cutaneo: la cute si presenta in genere secca e facilmente desquamante. Può inoltre essere presente una forma peculiare di dermatite, nota come impetigo erpetiforme. I peli sono diradati al pube e alle ascelle, mentre le unghie sono fragili e deformate, con striature trasversali.
C’è una maggiore esposizione alle micosi. Occasionalmente nell'ipocalcemia si verificano infezioni da Candida, ma esse sono più comuni nei pazienti affetti da ipoparatiroidismo idiopatico.
Alterazioni dentarie
Risultano frequenti. Nell’infanzia l’ipocalcemia può determinare ipoplasia dentaria o ipoplasia dello smalto, che appare fissurato con striature orizzontali giallastre. Nei bambini con quadro di ipoparatiroidismo è inoltre possibile osservare ritardo o mancata eruzione della dentizione permanente, aumento di frequenza della carie dentaria, iposviluppo delle radici dei molari e, in alcuni casi, perdita dei denti.
Alterazioni neurologiche
A causa della deposizione di fosfato tricalcico a livello dei gangli della base, evento che può verificarsi in tutte le forme di ipocalcemia cronica e che può essere rilevato mediante studio TC, si osservano deficit cognitivi significativi, anomalie neuro-psichiatriche e sintomi extra-piramidali, dal parkinsonismo (bradicinesia, amimia facciale, tremori, impaccio della deambulazione), ai più rari movimenti coreici o emiballismo. Possono essere rilevate anche manifestazioni cognitive e psichiatriche: difficoltà nella concentrazione, riduzione dell’acuità mentale, depressione, ansia, somatizzazioni, ostilità, tendenza psicotica. Le cause di tali complicanze rimangono indefinite, ma sono chiamate in causa le calcificazioni del sistema nervoso centrale (nuclei della base, aree cerebrali e cerebellari), l’ipocalcemia (che può esacerbare le disfunzioni indotte dalle calcificazioni), il recettore PTH2 e il suo ligando tubero-infundibolare TIP39, che possono influenzare diverse funzioni neuro-endocrine (percezione del dolore, ansia, depressione, funzioni ipofisarie) (4).
Alterazioni oculari
Nelle ipocalcemie di vecchia data viene osservata cataratta sub-capsulare, legata alla deposizione di fosfato tricalcico. Ha una prevalenza di circa il 50% ed è correlata all’età del paziente e alla durata e gravità dell’ipocalcemia. Tale manifestazione non è reversibile in seguito alla correzione della calcemia, sebbene questa possa arrestarne la progressione (5). Più raramente si osservano casi di blefarospasmo, intensa fotofobia e cherato–congiuntivite cronica.
Alterazioni cardiache
Nei pazienti con ipocalcemia grave possono occasionalmente svilupparsi aritmie o arresto cardiaco. L'ECG nell'ipocalcemia mostra tipicamente un prolungamento degli intervalli QTc e ST. Si possono anche osservare alterazioni della ripolarizzazione, come una forma appuntita dell'onda T o la sua inversione.
Nei quadri di ipoparatiroidismo è possibile osservare scompenso cardiaco congestizio da cardiopatia dilatativa (resistente alla digitale), che regredisce con la correzione dell’ipocalcemia, ed eventi cardio-vascolari ischemici maggiori senza incremento della mortalità cardio-vascolare.
Neuropatia autonomica cardio-vascolare
È una compromissione del sistema cardio-vascolare autonomo, causa di aumento della mortalità e associato ad aumento dell’affaticabilità. Alcuni studi ne stanno valutando la correlazione con ipoparatiroidismo, ipocalcemia, livelli di PTH e iperfosfatemia (6). Da questi primi studi emergerebbe la prevalenza di questo tipo di neuropatia nel 23% nel gruppo di controllo versus il 78% nel gruppo dei pazienti con ipoparatiroidismo.
Alterazioni ossee
Se si va a valutare la massa ossea, con la DXA si riscontra un incremento densitometrico, associato però a bassi livelli di turnover osseo, come dimostrato dai ridotti livelli dei marcatori biochimici e dall’istomorfometria dinamica. L’esame di biopsie ossee alla tomografia microcomputerizzata evidenzia un aumento del volume, dello spessore e del numero di trabecole ossee, nonché della densità connettivale, con associata riduzione degli indici dinamici di neoformazione ossea e della superficie di mineralizzazione (studiati dopo marcatura con tetraciclina marcata) (5,7). La rigidità e la durezza dell’osso ipoparatiroideo, indotto dall’alto contenuto minerale, compromette la capacità dell’osso di deformarsi, assorbire e dissipare energia. Non ci sono però dati definitivi sulle fratture, mentre non si sono rilevate fratture atipiche.
Alterazioni renali
Nefrolitiasi e nefrocalcinosi, con coliche renali e insufficienza renale cronica, sono complicanze dell’ipoparatiroidismo per la necessità di supplementare calcio e vitamina D in presenza di alterato assorbimento a causa della carenza di PTH. La nefrolitiasi (2-15% dei pazienti) ha frequenza quattro volte maggiore rispetto ai controlli; la nefrocalcinosi è particolarmente frequente nei casi di mutazione del recettore CaSR, per l’aumentata predisposizione ad espellere calcio per via renale. Complessivamente, predittori di ridotta filtrazione glomerulare sono l’età del paziente, la durata di malattia, l’ipercalcemia relativa (8).
Bibliografia
- Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bras Endocrinol Metab 2006, 50: 664-73.
- Bilezikian JP, Khan A, Potts JT Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Min Res 2011, 26: 2317-37.
- Cooper MS. Disorders of calcium metabolism and parathyroid disease. Best Pract Res Clin Endocrinol Metab 2011, 25: 975-83.
- Usdin TB, et al. New members of parathyroid hormone/parathyroid hormone receptor family: the parathyroid receptor 2 and tuberoinfundibolar peptide of 39 residues. Front Neuroendocrinol 2000, 21: 349-83.
- Underbjerg L, Sikjaer T, Mosekilde L, Rejnmark L. Postsurgical hypoparathyroidism - Risk of fractures, psychiatric diseases, cancer, cataract, and infections. J Bone Miner Res 2014, 29: 2504-10.
- Tabacco G, et al. Cardiovascular autonomic neuropathy as a new complication of post-surgical hypoparathyroidism. J Bone Mineral Res 2019, 34: 475-81.
- Rubin MR, et al. Three dimensional cancellous bone structure in hypoparathyroidism. Bone 2010, 46: 190-5.
- Levey AS, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009, 150: 604-12.
Diagnostica dell'ipoparatiroidismo
Marco Gallo
SC Endocrinologia Oncologica DU, AOU Città della Salute e della Scienza – Molinette (Torino)
Diagnosi di laboratorio e clinica
Si basa sul riscontro di ipocalcemia associata a livelli circolanti di PTH indosabili o inappropriatamente normal-bassi, che consentono di distinguere l’ipoparatiroidismo da altre cause d’ipocalcemia (1).
In presenza di alterazioni della protidemia o dell’equilibrio acido-base, può essere preferibile la determinazione del calcio ionizzato o la correzione della calcemia totale sulla base dei livelli in eccesso o in difetto di albuminemia, utilizzando la formula:
calcemia corretta = calcemia misurata (in mg/dL) + [0.8 x (4 - albuminemia (in g/dL)].
È generalmente presente ipocalciuria (pur in presenza di un’escrezione relativa aumentata), tranne nella forma genetica a trasmissione autosomica dominante legata a mutazione iperattivante del recettore del calcio [CaSR, calcium-sensing receptor], nella quale l’escrezione urinaria di calcio/creatinina risulta sovrapponibile a quella dei soggetti sani.
Ulteriori caratteristiche bioumorali sono costituite dalla presenza di iperfosfatemia e ipofosfaturia (nel caso di un apporto dietetico di fosfati normale), con valori normali o ridotti degli indici di turn-over osseo (come la fosfatasi alcalina ossea) e riduzione dei livelli plasmatici di 1,25(OH)2 vitamina D, prodotta a livello renale con il contributo fondamentale del PTH. Quando disponibile, la determinazione dell’escrezione urinaria di AMP ciclico (cAMP nefrogeno) mostra valori caratteristicamente ridotti.
Nella diagnosi di laboratorio degli ipoparatiroidismi e delle ipocalcemie deve essere compresa anche la valutazione della magnesiemia, dal momento che il deficit di tale elemento (per es in alcolismo, denutrizione, malassorbimento) sopprime il rilascio del PTH e determina un certo grado di resistenza periferica all’ormone. Anche un’ipermagnesiemia (spesso iatrogena) può indurre ipoparatiroidismo funzionale.
Anticorpi isolati rivolti contro le paratiroidi (per es, anticorpi anti-NALP5, NACHT leucine-rich protein 5) sono stati rilevati nel 40% circa dei soggetti con ipoparatiroidismo autoimmune (isolato o associato ad altre manifestazioni patologiche). In questi soggetti, inoltre, è stata spesso riscontrata la positività anticorpale contro il dominio extra-cellulare del CaSR; non è tuttavia chiaro se tali anticorpi svolgano un ruolo causale nella patogenesi della malattia, o fungano semplicemente da marcatori di danno tissutale (2-4). Pochi centri sono attualmente attrezzati per la loro determinazione. Nelle forme di (verosimile) ipoparatiroidismo autoimmune, inoltre, è appropriato escludere la presenza di altri deficit ormonali (valutazione della funzionalità surrenalica, gonadica e tiroidea).
Ormai in disuso sono le valutazioni dinamiche, come il test con EDTA (etilen-diamino-tetracetato) per la valutazione della riserva paratiroidea (che induce o accentua l’ipocalcemia) e quello di Ellsworth-Howard per la diagnostica differenziale tra ipofunzione paratiroidea e resistenza al PTH (misurazione del cAMP urinario dopo somministrazione parenterale di PTH).
Nella diagnostica dell’ipoparatiroidismo rivestono peraltro un’importanza fondamentale l’anamnesi e la valutazione clinica. Con l’anamnesi familiare si potrà valutare l’esistenza di altri membri con storia clinica nota o suggestiva di ipoparatiroidismo su base genetica (per es ipocalcemia di ndd).
L’anamnesi patologica dovrà indagare la pregressa esecuzione d’interventi chirurgici in regione cervicale, l’esposizione a trattamenti radianti o radiometabolici (131I), la coesistenza di patologie autoimmuni (per es nell’ambito della sindrome polighiandolare autoimmune di tipo 1), di malattie d’organo o sistemiche (talassemia, emocromatosi, malattia di Wilson, patologie granulomatose), o ancora l’assunzione di farmaci in grado di slatentizzare ipocalcemie in presenza di quadri d’insufficienza paratiroidea (chemioterapici, ketoconazolo, bisfosfonati, prazolici).
La valutazione clinica potrà evidenziare l’esistenza di cicatrici cervicali, ipostaturismi, vitiligine o candidosi muco-cutanee; rilevare la presenza di parestesie, crampi, spasmofilia/tetania o le manifestazioni psichiatriche e neurologiche (irritabilità, depressione, ipereccitabilità neuromuscolare, convulsioni, deficit uditivo, ritardo mentale), così come i segni cutaneo-annessiali di malattia (cute ruvida e secca, diradazione pilifera, fragilità ungueale) o documentare le caratteristiche cranio-facciali suggestive delle forme sindromiche.
La semeiotica dell’ipoparatiroidismo comprende il segno di Trousseau (spasmo carpale evocabile gonfiando il bracciale dello sfigmomanometro 20 mmHg sopra il valore della pressione sistolica e tenendolo gonfio per 1-3 minuti, interrompendo in tal modo l’afflusso di sangue all’arto superiore - cosiddetta “mano da ostetrico”) e quello di Chvostek, meno sensibile e specifico (contrazione della muscolatura facciale omolaterale indotta dalla percussione della regione pre-auricolare, lungo il tragitto del nervo facciale).
Figura 1. Sopra segno di Chvostek, sotto segno di Trousseau
Diagnostica genetica
Rare forme ereditarie di ipoparatiroidismo potranno richiedere conferma genetica mediante la ricerca delle mutazioni potenzialmente responsabili, come (5):
- gene CaSR (locus 3q21.1), gene GCM2 [glial cell missing-2 gene](locus 6p24.2) e gene PTH (locus 11p15): forme di ipoparatiroidismo isolato a tramissione autosomica dominante o recessiva;
- gene SOX3 (locus Xq26-27): forme legate al cromosoma X [X-linked];
- microdelezioni gene TBX1 (locus 22q11.2): responsabili della sindrome di DiGeorge;
- aploinsufficienza del cromosoma 10p e del gene GATA3 (locus 10p14-10): sindrome di Barakat/HDR;
- gene TBCE (locus 1q42-43): sindromi di Kenny-Caffey e di Sanjad-Sakati;
- deficit geni mitocondriali: sindromi di Kearns-Sayre e di Pearson, encefalomiopatia mitocondriale, deficit LCHAD (long-chain 3-hydroxyacyl CoA dehydrogenase);
- mutazioni responsabili di patologie ereditarie da accumulo: emocromatosi, malattia di Wilson;
- gene AIRE (locus 21q22.23; oltre 200 mutazioni note): responsabili di ipoparatiroidismi autoimmuni.
Al momento, è controverso quando dover ricorrere alle indagini genetiche nei casi di ipoparatiroidismo idiopatico.
Indagini strumentali
Un ipoparatiroidismo di lunga durata può inoltre accompagnarsi a tipiche alterazioni di alcune indagini strumentali legate all’ipocalcemia o al quadro clinico, peraltro non necessarie per la diagnosi:
- ECG: allungamento del tratto QT corretto per la frequenza cardiaca (QTc), inversione dell’onda T (fig. 2);
- EEG: anomalie simil-comiziali, salve parossistiche di onde lente ad alto voltaggio;
- EMG: spike ad alta frequenza;
- Ecocardiogramma: compromissione della funzione sistolica;
- Esami radiologici: calcificazioni dei tessuti molli o intra-craniche (nuclei della base; fig. 3), nefrocalcinosi;
- Densitometria ossea DEXA: aumento della massa ossea per riduzione del turn-over scheletrico (pur in presenza di alterazioni microstrutturali, rilevabili all’istomorfometria ossea);
- Valutazione oculistica (oftalmoscopia, lampada a fessura): cheratopatia a banda, opacamento del cristallino, cataratta lenticolare posteriore, papilledema da ipertensione endocranica.
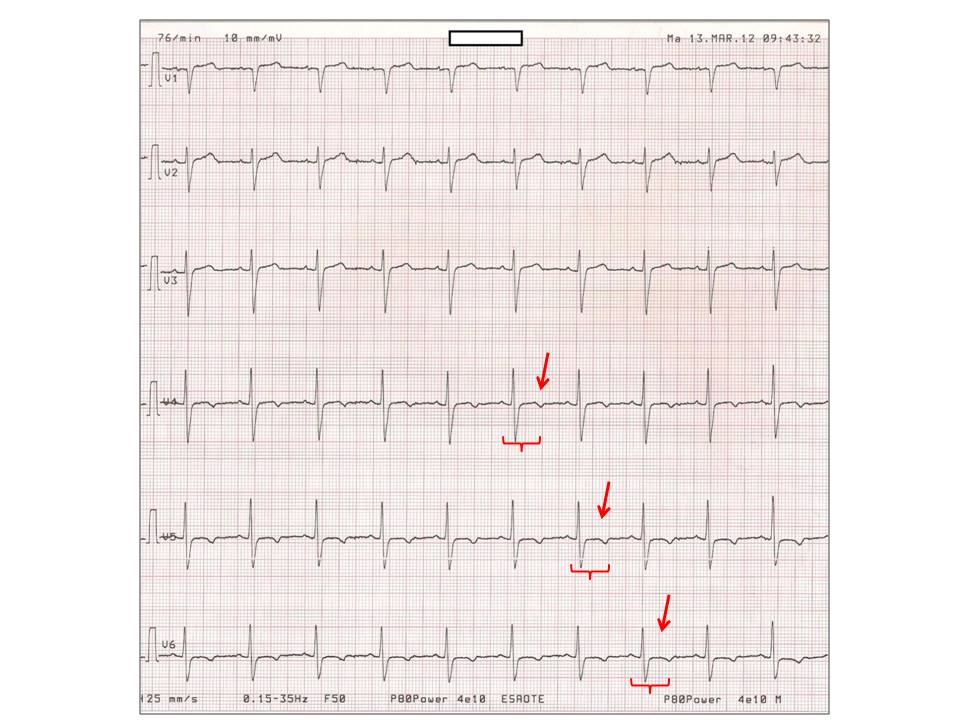
Figura 2. Ritmo sinusale (76 bpm) con inversione delle onde T in V4-V6 (frecce) e prolungamento del QTc (0.615 sec.) caratteristico delle ipocalcemie severe, a carico prevalentemente del segmento ST, più che delle onde T (invertite, ma di ampiezza relativamente normale); la paziente, al momento dell’ECG, aveva un calcio ionizzato di 0.71 mmol/l.
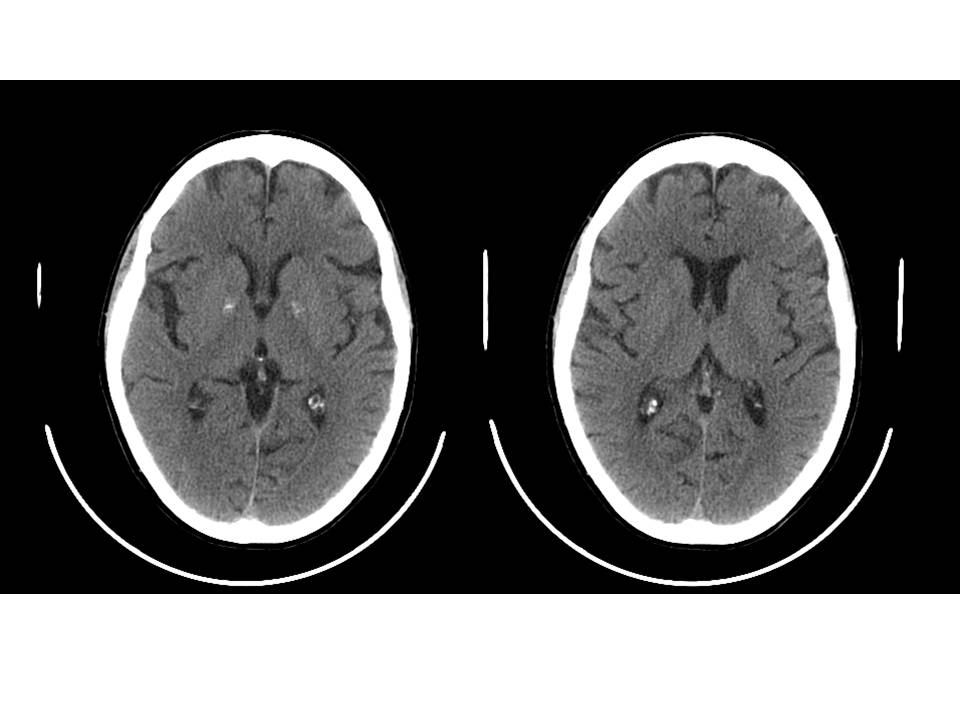
Figura 3. TC encefalo: minute calcificazioni a livello dei nuclei della base
Bibliografia
- Shoback D. Hypoparathyroidism. N Engl J Med 2008, 359: 391-402.
- Bilezikian JP, Khan A, Potts JT Jr, et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res 2011, 26: 2317-37.
- Brown EM. Anti-parathyroid and anti-calcium sensing receptor antibodies in autoimmune hypoparathyroidism. Endocrinol Metab Clin North Am 2009, 38: 437-45.
- Betterle C, Garelli S, Presotto F. Diagnosis and classification of autoimmune parathyroid disease. Autoimmun Rev 2014, 13: 417-22.
- Brandi ML. Genetics of hypoparathyroidism and pseudohypoparathyroidism. J Endocrinol Invest 2011, 34 (7 suppl): 27-34.
Overview terapia ipoparatiroidismo
Massimo Procopio1, Assunta Santonati2 & Antonio Spada2
1SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
2Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
La terapia dell’ipoparatiroidismo deve avere questi obiettivi:
- migliorare i sintomi dell’ipocalcemia;
- mantenere la calcemia nei valori bassi della normalità;
- mantenere la fosfatemia entro i limiti della norma;
- evitare l’ipercalciuria;
- mantenere un rapporto Ca/P < 55 (in mg/dL);
- evitare le calcificazioni ectopiche (soprattutto i danni renali);
- migliorare la qualità della vita.
| Terapia dell’ipoparatiroidismo: obiettivi biochimici | |
| Calcemia | Limite inferiore di normalità (8-8.5 mg/dL) |
| Fosfatemia | Range di normalità (chelanti se > 6.5 mg/dL) |
| Magnesio | Range di normalità (supplementazione se livelli ridotti) |
| Calciuria | < 4 mg/kg peso corporeo |
| 25-OH-vitamina D | > 20 ng/mL |
Nell’ipoparatiroidismo cronico la terapia si basa sull’utilizzo di sali di calcio (in aggiunta all’apporto alimentare) e calcitriolo.
In alcuni casi è necessario ricorrere a farmaci che aumentano il riassorbimento di calcio in sede renale per ridurre l’ipercalciuria, quali i diuretici tiazidici, e che riducono l’assorbimento intestinale di fosfati per ridurre l’iperfosfatemia, come il magnesio idrossido + algedrato (Maalox).
Nell’ipoparatiroidismo acuto l’obiettivo terapeutico primario è il controllo dei sintomi dell’ipocalcemia. In una situazione di emergenza quando vi è il sospetto di ipocalcemia e compaiono manifestazioni di tetania muscolare, convulsioni, ipotensione e aritmie cardiache, vi è indicazione alla somministrazione endovenosa di calcio ancor prima di conoscere il valore del Ca++, passando appena possibile alla somministrazione di calcio e calcitriolo per via orale. In caso di ipomagnesiemia, deve essere somministrato solfato di magnesio per via endovenosa.
Una concomitante condizione di ipovitaminosi D dovrebbe essere trattata con colecalciferolo allo scopo di mantenere livelli di 25OH vitamina D ≥ 30 ng/mL, per consentire gli effetti pleiotropici di tale ormone.
Il follow-up terapeutico prevede il controllo semestrale di calcemia, fosfatemia, creatininemia e calciuria 24 ore. Se clinicamente indicato, ogni 5 anni eseguire ecografia renale, esame oftalmologico per cataratta, eventualmente imaging del SNC per escludere calcificazioni.
Bibliografia
- Shoback D. Hypoparathyroidism. N Engl J Med 2008, 359: 391-402.
- Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bras Endocrinol Metab 2006, 50: 664-73.
- Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab 2012, 26: 517-22.
Calcio nella terapia dell'ipoparatiroidismo
Massimo Procopio1, Assunta Santonati2 & Antonio Spada2
1SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
2Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
Nel trattamento dell’ipoparatiroidismo cronico si usano i sali di calcio per os, in aggiunta a un adeguato apporto alimentare di calcio, che va sempre raccomandato, essendo il calcio alimentare meglio tollerato e meglio assorbito. Fonti alimentari di calcio sono le acque, il latte e i suoi derivati (rispetto a qualsiasi altro alimento forniscono più calcio, proteine, magnesio, potassio, zinco, fosforo in rapporto al contenuto calorico), soia, mandorle, rucola, quinoa, broccoli, semi di chia e di lino. Si utilizzano 800–2000 mg/die di calcio elementare in 3-4 dosi. Dopo intenso esercizio fisico è necessario un supplemento di calcio.
Il calcio carbonato è il più economico, ha il più elevato contenuto di calcio elementare (40%) ed è disponibile in compresse da 500 e 1000 mg. Bisogna sempre considerare che la capacità intestinale di assorbimento di un bolo alimentare di calcio non supera i 500 mg, quindi evitare la somministrazione di compresse il cui contenuto in sali di calcio ecceda i 600 mg, per prevenire sovraccarichi renali. Il calcio è più tollerabile se viene assunto ai pasti e necessita di una normale acidità gastrica per un’efficiente solubilizzazione e assorbimento. La somministrazione di calcio carbonato lontano dal pasto funziona per lo più come chelante del fosforo, non essendo assorbito, e può pertanto essere utilizzato nel controllo dell’iperfosfatemia. Sono comuni gli effetti collaterali gastro-intestinali (sensazione di peso gastrico), pertanto nei pazienti con acloridria e in terapia con inibitori della pompa protonica, l’assorbimento è ridotto e può essere favorito dall’assunzione di spremute di agrumi.
Il calcio citrato (contenuto di calcio elementare 21%) in generale presenta la migliore tollerabilità. Non risente dell’acidità gastrica, quindi è parimenti assorbito se assunto a digiuno o dopo il pasto, in pazienti affetti da acloridria o che assumono inibitori di pompa protonica. È meglio assorbibile nei pazienti sottoposti a by-pass gastrico, condizione in cui il passaggio del duodeno limita la superficie di assorbimento del calcio. L'anione citrato riduce l'effetto litogenico del calcio urinario: la tendenziale riduzione della saturazione dell'ossalato di Ca lo dimostra. L'aumento dell'escrezione urinaria di citrato compensa l'ipercalciuria, creando un ambiente meno propenso alla formazione di calcoli. Questa azione, che si esplica attraverso l’inibizione della nucleazione e della crescita dei cristalli, oltre ad essere legata alla riduzione della concentrazione ionica del calcio nelle urine, sembra legata a un effetto diretto del citrato sulla superficie dei cristalli; a tale proposito la letteratura anglosassone ha coniato per gli inibitori il termine di “stone poisons” (veleni dei calcoli), per definire l’importanza degli inibitori in generale e di questo anione in particolare, nel contrastare i processi di formazione, crescita ed aggregazione dei cristalli.
Altri sali di calcio sono il calcio lattato (13%) ed il calcio gluconato (9.3%). Infine, il calcio fosfato (38.3%) è da evitare nell’ipoparatiroidismo perché peggiora l’iperfosfatemia.
La terapia dell’ipocalcemia acuta si basa sull’impiego di sali di calcio per uso endovenoso, tra i quali troviamo il calcio gluconato (contiene circa 94 mg di calcio elementare per grammo di sale, 9.4%), e il calcio cloruro (272 mg di calcio elementare per grammo di sale, 27.2%). Il calcio cloruro è meno usato per il suo effetto irritante e la scarsa tollerabilità in caso di stravaso ematico (necrosi tessutale).
In pratica si somministrano 1-2 fiale di calcio gluconato da 1 g in 100 cc NaCl 0.9% o glucosata 5% in 2-5 min. La prima infusione di calcio consente solitamente di ottenere un aumento transitorio (circa 2 ore) del Ca++ ed è pertanto necessario instaurare un’infusione continua ev di calcio gluconato nel modo seguente: calcio gluconato 6 fiale da 1 g (60 mL) in 500 cc di soluzione glucosata 5% o NaCl 0.9% (pari a 1 mg/mL circa di calcio elementare), da infondere a una velocità iniziale di 0.5 mL/kg/h (pari a 0.5 mg/kg/h di calcio elementare), aumentando fino a 2.0 mg/kg/h. Per ottenere una velocità di infusione di 0.5 mg/kg/h:
- paziente di 50 kg: velocità di infusione 25 mL/h;
- paziente di 75 kg: velocità di infusione 37 mL/h;
- paziente di 90 kg: velocità di infusione 45 mL/h.
Durante l’infusione è necessario monitorare sia il Ca++ ogni 4-6 ore, titolando di conseguenza la velocità di infusione, sia la potassiemia (che tende a scendere). Nei pazienti digitalizzati monitorare l’ECG e controllare più spesso la calcemia. L’infusione va sospesa alla scomparsa dei sintomi e al raggiungimento di valori stabili di Ca++ > 0.9 mmol/L o calcemia totale > 1.88 mmol/L (7.5 mg/dL), proseguendo con la terapia orale.
Bibliografia
- Tartaglino B. Sali di Calcio. In Farmaci e Procedure in Medicina d’Urgenza, 2° Edizione, Edizioni Medico-Scientifiche srl, 2007: 114-120.
- Recker RR. Calcium absorption and achlorhydria. N Engl J Med 1985, 313: 70–3.
- Levine BS, et al. Effect of calcium citrate supplementation on urinary calcium oxalate saturation in female stone formers: implications for prevention of osteoporosis. Am J Clin Nutr 1994, 60: 592-6.
- Caudarella R, Vescini F, Buffa A, Stefoni S. Citrate and mineral metabolism: kidney stones and bone disease. Front Biosci 2003, 8: s1084-106.
Scheda sali di calcio
Fabio Vescini
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria S. Maria della Misericordia, Udine
Meccanismo d’azione
Il calcio è il principale costituente, insieme al fosfato, della parte minerale dell’osso. Esso è anche essenziale per lo svolgimento di importanti funzioni, quali la contrazione muscolare, la trasmissione nervosa, la permeabilità capillare, l’attività coenzimatica, la produzione di energia nei mitocondri.
Preparazioni
Calcio Carbonato
- cp 500 mg: Cacit
- cp effervescenti 1000 mg: Cacit, calcio carbonato EG, Calciodie, Calcium Sandoz, Idracal, Lubical
- cp masticabili 1250 mg (pari a 500 mg di calcio elementare): Metocal
- bustine 1000 mg: Rex
- bustine 2500 mg: calcio Savio, Carbosint
Calcio carbonato e lattogluconato
- cp 500 mg: Calcium Sandoz
- bustine 500 mg: Calcium Sandoz
- cp 1000 mg: Calcium Sandoz
- bustine da 1000 mg: Calcium Sandoz
Calcio acetato
cp 667 mg: Phoslo
Calcio Carbonato + Colecalciferolo
- cp 500 mg + 440 U: Eurocal D3, Ideos, Orotre
- bustine 500 mg + 440 U: Cacit D3
- cp 600 mg + 400 U: Metocal D3, Natecal D3, Tonacal D3
- cp 600 mg + 1000 U: Demilos, Natemille, Riliscal
- cp 1000 mg + 880 U: Biocalcium D3, Calcicol D3, Calcimed, Calcium D3 Sandoz
- bustine 1000+ 880 U: Cacit D3, Cadtre, calcio carbonato + vitamina D3 ABC, calcio carbonato + vitamina D3 Actavis, calcio carbonato + vitamina D3 Almus, calcio carbonato + vitamina D3 DOC, calcio carbonato + vitamina D3 EG, calcio carbonato + vitamina D3 Germed, calcio carbonato + vitamina D3 Pensa, calcio carbonato + vitamina D3 Sofar, calcio carbonato + vitamina D3 Tecnigen, calcio carbonato + vitamina D3 Union Health, Ditrost, Donicil, Eurocal D3
Calcio Fosfato + Colecalciferolo
- fl os 1290 mg (pari a calcio ione mg 500) + 200 U: Calisvit
- bustine 1200 mg + 800 U: Foscal D3, Osteofos D3
Calcio acetato + magnesio carbonato
435 mg + 235 mg: Osvaren
Preparati per uso ev
Calcio cloruro: sono in commercio:
- fiale al 10%, contenenti 1 g/10 mL = 100 mg/mL: calcio cloruro Bioindustria, calcio cloruro Galenica Senese, calcio cloruro Monico, calcio cloruro SALF
- fiale al 5%, contenenti 500 mg/10 mL = 50 mg/mL: calcio cloruro Galenica Senese, calcio cloruro Monico, calcio cloruro SALF
Calcio gluconato: sono in commercio fiale al 10% da 5 e 10 mL, contenenti 100 mg/mL: calcio gluconato Bioindustria, calcio gluconato Braun, calcio gluconato Galenica Senese, calcio gluconato Monico, calcio gluconato SALF
Indicazioni
Trattamento e prevenzione del deficit di calcio.
In associazione alla vitamina D come aggiunta a terapie specifiche per il trattamento dell’osteoporosi in soggetti a rischio di carenza concomitante di vitamina D e calcio.
Contro-indicazioni
Ipercalcemia o ipercalciuria
Calcolosi renale attiva (nefrolitiasi, nefrocalcinosi)
Immobilizzazione prolungata (specie se accompagnata da ipercalciuria e/o da ipercalcemia)
Insufficienza renale (necessità di calibrare la dose in base al metabolismo calcio-fosforo)
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti
Precauzioni d’uso
Somministrare con cautela in concomitanza a diuretici tiazidici (rischio di ipercalcemia), digitale (rischio di aritmie)
Somministrare a 2 ore di distanza dall’assunzione di bisfosfonati, sodio floruro e tetracicline
Limitazioni prescrittive
Nessuna
Magnesio nella terapia dell'ipoparatiroidismo
Massimo Procopio1, Assunta Santonati2 & Antonio Spada2
1SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
2Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
La più comune formulazione di magnesio per via endovenosa è il solfato di magnesio (MgSO4) in fiale da 1-2-2.5 g in 10 mL; 1 grammo di solfato di magnesio contiene circa 100 mg di magnesio, equivalenti a 4 mmol o 8 mEq di magnesio elementare.
Dovrebbe essere diluito in soluzione fisiologica di cloruro di sodio allo 0.9% o glucosio al 5%, al fine di ottenere una concentrazione di magnesio elementare non superiore al 20%.
In caso di ipomagnesiemia severa (Mg++< 0.5 mmol/L), infondere solfato magnesio 1 g/h per le prime 6 ore, seguiti da 1 g ogni 4-6 ore fino a correzione. Si consiglia di non superare 8-12 g di solfato di magnesio nelle prime 24 ore, seguite da 4-6 g/die per 3-4 giorni, al fine di saturare i depositi.
In caso di ipomagnesiemia moderata, si consiglia di infondere 1 g ogni 6 ore per il primo giorno e 1 g ogni 12 ore nei giorni successivi.
Anche nell’ipoparatiroidismo cronico può essere indicata la supplementazione orale di magnesio. Una tipica dose di magnesio in pazienti con normale funzionalità renale varia da 240 a 1000 mg/die in dosi refratte. Un effetto collaterale frequente può essere la diarrea. Se la supplementazione orale di sali di magnesio non è tollerata dal paziente, si può utilizzare l’amiloride, grazie alla sua azione Mg-risparmiatrice renale.
Bibliografia
- Tartaglino B. Magnesio (solfato). In Farmaci e Procedure in Medicina d’Urgenza, 2° Edizione, Edizioni Medico-Scientifiche srl, 2007: 241-7.
- Ayuk J, Gittoes NJL. How should hypomagnesaemia be investigated and treated? Clin Endocrinol 2011, 75: 743–6.
Scheda sali di magnesio
Massimo Procopio
SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
Meccanismo d'azione
Il magnesio è il secondo principale catione intracellulare dopo il potassio e svolge un ruolo critico in numerosi processi cellulari:
- funzionamento di molti enzimi, quali creatin-chinasi, protein-chinasi, Na-K-ATPasi, esochinasi, Ca-ATPasi, adenilato-ciclasi, guanilato-ciclasi, fosfofrutto-chinasi, 5-fosforibosil-pirofosfato-sintetasi, miosin-chinasi;
- stabilizzazione delle membrane cellulari, modulando la conduzione nervosa, l’attività del canale del calcio e il trasporto del potassio
- antagonista del calcio nel processo di contrazione/rilascio muscolare, secrezione di neurotrasmetittori, conduzione del potenziale d’azione del tessuto nodale cardiaco;
- secrezione di PTH
- sensibilità insulinica
- funzione strutturale (proteine, poliribosomi, acidi nucleici, enzimi, mitocondri).
Quando somministrato in infusione endovenosa ha un’azione stabilizzante di membrana, rallenta la trasmissione dell’impulso neuromuscolare (riduce la quantità di acetil-colina liberata nella placca neuromuscolare) e induce rilassamento del muscolo scheletrico e liscio (attività miorilassante, anti-convulsivante, tocolitica, vasodilatatrice); rallenta la conduzione dell’impulso elettrico cardiaco seno-atriale ed atrio-ventricolare e aumenta la refrattarietà del nodo atrio-ventricolare (attività anti-aritmica); possiede attività anti-aggregante.
Preparazioni, via di somministrazione, posologia
La forma più comunemente usata di magnesio è il solfato di magnesio, disponibile in fiale da 1-2-2.5 g/10 mL (galenico).
Può essere somministrato per via endovenosa, intramuscolare (in soluzioni al 25-50%) e orale. La velocità di somministrazione ev non dovrebbe superare 150 mg/min; in infusione 2 g/h, tranne che nelle emergenze.
La posologia varia a seconda dell’indicazione clinica:
- ipomagnesiemia moderata: infusione ev di 1 g ogni 6 ore per il primo giorno, 1 g ogni 12 ore nei giorni successivi
- ipomagnesiemia severa: infusione ev di 1g/h per le prime 6 ore, seguiti da 1 g/4-6 h fino a correzione
- pre-eclampsia ed eclampsia:
- 4-6 g ev in 5-15 min seguiti da 2-3 g/h in infusione continua
- 5 g im in ciascun gluteo, seguiti da 5 g im ogni 4 ore.
- torsione di punta:
- adulti: 1-2 g ev in 2-3 min, il bolo si può ripetere dopo 10-15 min; ai boli si può fare seguire un’infusione di 0.25-2 g/h fino a 48 h
- bambini: 25-50 mg/kg ev
- asma severo
- adulti: 2 g ev, in 5-20 min
- bambini: 25-100 mg/kg ev
Altre formulazioni:
- magnesio pidolato:
Indicazioni
Ipomagnesiemia, ipokaliemia severa e/o refrattaria alla sola somministrazione di potassio, pre-eclampsia ed eclampsia, torsione di punta, asma riacutizzato severo.
Altre indicazioni meno certe sono la fibrillazione o tachicardia ventricolare, la tachicardia atriale multifocale, la tossicità digitalica, la fibrillazione atriale, il tetano ed il brivido da adattamento termoregolatorio.
Contro-indicazioni
Ipocalcemia non dovuta ad ipomagnesiemia, grave insufficienza renale, blocco atrio-ventricolare di grado avanzato, blocco bi-fascicolare, bradicardia sinusale.
Effetti collaterali
In caso di somministrazione troppo rapida si possono avere vasodilatazione, ipotensione ed asistolia.
Gli effetti collaterali dipendono dalla magnesiemia (valori normali 1.5-2.5 mg/dL):
- > 3 mg/dL: depressione del sistema nervoso centrale, depressione della trasmissione neuromuscolare e dei riflessi tendinei profondi;
- > 5 mg/dL: flushing, sonnolenza;
- > 10 mg/dL: scomparsa dei riflessi tendinei, blocchi cardiaci, depressione respiratoria;
- > 12.5 mg/dL blocco cardiaco completo ed arresto respiratorio.
Altri effetti collaterali sono rappresentati dalla comparsa di rash cutanei, sudorazione, nausea, vomito, ipotermia, ipocalcemia.
Limitazioni prescrittive
No
Bibliografia
- Tartaglino B. Magnesio (solfato) In: Farmaci e Procedure in Medicina d’Urgenza, 2° Edizione, Edizioni Medico-Scientifiche srl, 2007: 241-7.
Vitamina D e suoi metaboliti nella terapia dell'ipoparatiroidismo
Massimo Procopio1, Assunta Santonati2 & Antonio Spada2
1SCDU Endocrinologia, Diabetologia e Metabolismo, Azienda Ospedaliera S. Giovanni Battista, Torino
2Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
Nella terapia dell’ipoparatiroidismo è indispensabile associare analoghi attivi della vitamina D, sotto forma di calcitriolo (1,25OH2D) o altri analoghi 1-alfa idrossilati come 1-alfacalcidolo, per os alla dose di 0.25–2 µg/die, per permettere l’assorbimento intestinale di calcio. Infatti, i composti 1-alfa idrossilati sono gli analoghi della vitamina D più potenti, che consentono di usare dosaggi ridotti, raggiungono in breve tempo (3-6 giorni) il massimo effetto, che in altrettanto breve tempo svanisce dopo sospensione della terapia (3-6 giorni). Il ricorso al calcitriolo nell’ipoparatiroidismo è chiaramente correlato all’assenza del PTH, che, insieme al frequente riscontro di iperfosfatemia, impedisce l’idrossilazione in posizione 1, quindi l’attivazione, della 25-OH-vitamina D. Titolare verso l’alto la dose di calcitriolo può consentire di ridurre la supplementazione orale di calcio. Queste caratteristiche rendono questi composti di più facile e sicuro impiego, con minor rischio di grave tossicità rispetto agli analoghi non idrossilati in posizione 1-alfa, quali l’ergocalciferolo, il colecalciferolo, il calcifediolo e il diidro-tachisterolo. Il razionale dell’uso delle altre formulazioni di vitamina D non attivate in alcune particolari situazioni, risiede nel fatto che molti tessuti formano autonomamente 1-25-OH-vitamina D, con potenziali effetti benefici extra-scheletrici.
Il dosaggio farmacologico di ergo- e cole-calciferolo, dotati di minore potenza, può variare tra 0.625-5 mg/die. Affinchè possano espletare l’effetto farmacologico e legarsi, sia pure con minore affinità, al recettore della vitamina D, necessitano di essere idrossilati in posizione 25 in sede epatica. Il tempo di raggiungimento del massimo effetto in questo caso varia tra circa 3 e 10 settimane, mentre quello di effetto residuo dopo sospensione varia tra 6 e 17 settimane.
Il calcifediolo non deve subire alcuna conversione e può legarsi al recettore della vitamina D sia pure con minore affinità. Il dosaggio giornaliero può variare tra 20 e 250 µg/die, il tempo di raggiungimento del massimo effetto varia tra 2 e 4 settimane e quello di scomparsa dopo sospensione tra 3 e 7 settimane.
Infine, il diidro-tachisterolo, che è un prodotto semi-sintetico e non necessita di alcuna conversione, viene somministrato a un dosaggio giornaliero variabile tra 250-1000 µg/die. Il tempo di raggiungimento del massimo effetto è di 2-4 settimane e il tempo di effetto residuo dopo sospensione è di 1-3 settimane.
Bibliografia
- Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bras Endocrinol Metab 2006, 50: 664-73.
- Brandi L, et al. Management of hypoparathyroidism: summary statement and guidelines. J Clin Endocrinol Metab 2016, 101: 2273-83.
Scheda vitamina D
Fabio Vescini
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria S. Maria della Misericordia, Udine
VITAMINA D naturale e 25-idrossilata
Meccanismo d’azione
La somministrazione di vitamina D incrementa l’assorbimento intestinale di calcio e fosfato, favorendo la mineralizzazione ossea.
Preparazioni, via di somministrazione, posologia
Colecalciferolo:
- gocce orali 10.000 U.I. (Annister, Balavita, Briogen D3, Colcad, colecalciferolo DOC, colecalciferolo EG, colecalciferolo Ipso Pharma, colecalciferolo Mylan, colecalciferolo Pensa, colecalciferolo Sandoz, colecalciferolo Teva, colecalciferolo Zentiva, Dibase, Dicolev, Dipront, Disteomin, Divod, Ibitred, Lampard, Medivid, Tomaino, Tredimin, Trevid, Xarenel, Zibenak): 20 gtt alla settimana (salvo diverso parere medico)
- gocce orali 20.000 U.I. (colecalciferolo Dompè)
- flaconcino da 2.5 mL 25.000 U.I. (Annister, Balavita, Briogen D3, Colcad, colecalciferolo DOC, colecalciferolo EG, colecalciferolo Ipso Pharma, colecalciferolo Mylan, colecalciferolo Pensa, colecalciferolo Sandoz, colecalciferolo Teva, colecalciferolo Zentiva, Dibase, Dicolev, Dipront, Disteomin, Divod, Ibitred, Lampard, Medivid, Tomaino, Tredimin, Trevid, Xarenel, Zibenak): 1 fl al mese (salvo diverso parere medico)
- flaconcino da 2.5 mL 50.000 U.I. (Annister, Balavita, Briogen D3, Colcad, colecalciferolo DOC, colecalciferolo EG, colecalciferolo Ipso Pharma, colecalciferolo Mylan, colecalciferolo Pensa, colecalciferolo Sandoz, colecalciferolo Teva, colecalciferolo Zentiva, Dibase, Dicolev, Dipront, Disteomin, Divod, Lampard, Medivid, Tomaino, Tredimin, Trevid, Xarenel, Zibenak)
- fiale da 100.000 U.I. (colecalciferolo DOC, Dibase, Xarenel): 1 fl im/os ogni 4-6 mesi (salvo diverso parere medico)
- fiale da 300.000 U.I. (Dibase, Xarenel): 1 fl im/os ogni 12 mesi (salvo diverso parere medico)
- compresse 800 U.I. (Annova)
- compresse 1000 U.I. (Annova)
- compresse 7000 U.I. (Annova)
- compresse 30.000 U.I. (Annova)
- capsule molli 1000 U.I. (Fedivelle, Nodigap)
- capsule molli 10.000 U.I. (Nodigap)
- capsule molli 20.000 U.I. (Fedivelle, Nodigap)
- capsule molli 25.000 U.I. (colecalciferolo Invos)
- capsule molli 50.000 U.I. (Nodigap)
- capsule rigide 1000 U.I. (Dibaselab, Xarenel, Xarexen)
- capsule rigide 2000 U.I. (Dibase, Xarenel)
- capsule rigide 6000 U.I. (Dibase, Xarenel)
- capsule rigide 25.000 U.I. (Dibase, Vitenson, Xarenel)
- capsule rigide 50.000 U.I. (Xarenel)
- capsule rigide 100.000 U.I. (Xarenel)
Ergocalciferolo:
- fiale da 400.000 U.I. (Ostelin): 1 fl im/os ogni 3 mesi (salvo diverso parere medico) non più disponibile
Alfacalcidolo
- capsule molli 0.25 mg: Alpha D3, Dediol, Diseon, Diserinal, Ostidil D3
- capsule molli 1 mg: Alpha D3, Diseon, Deril, Diserinal, Ostidil D3, Sefal
- gocce orali, soluzione 10 mL 2mg/mL: Alpha D3, Dediol, Diseon, Geniad.
Calcifediolo:
- gocce orali 10 mL/1.5 mg (Didrogyl): 10 gtt x 2 volte alla settimana (salvo diverso parere medico)
- capsule molli 30 µg (Rayaldee)
- capsule molli 0.266 mg (Neodidro): 1 cps al mese
Indicazioni
Prevenzione e trattamento della carenza di vitamina D.
Contro-indicazioni
Ipercalcemia, ipercalciuria di severa entità (nell’iperparatiroidismo primitivo è consigliata una somministrazione di colecalciferolo al fine di mantenere la concentrazione plasmatica di 25(OH)-vitamina D fra 20 e 30 ng/mL).
Calcolosi renale attiva (nefrolitiasi, nefrocalcinosi).
Insufficienza renale.
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti.
Precauzioni d’uso
Somministrare con cautela in:
- concomitanza con diuretici tiazidici (rischio di ipercalcemia)
- malattie granulomatose (rischio di aumentata conversione in calcitriolo)
Utilizzo in gravidanza
Nel feto la vitamina D può avere effetti tossici: esiste una correlazione tra eccesso di assunzione o estrema sensibilità materna alla vitamina durante la gravidanza e ritardo dello sviluppo fisico e mentale del bambino, stenosi aortica sopra-valvolare e retinopatia. L'ipercalcemia materna può anche portare alla soppressione della funzione paratiroidea nei neonati con conseguente ipocalcemia, tetania e convulsioni. Il farmaco deve essere utilizzato a dosi adeguate, sotto stretto controllo e solo se i benefici per la madre superano i potenziali rischi per il nascituro.
Utilizzo durante l'allattamento
La vitamina D e i suoi metaboliti passano nel latte materno; a fronte dei potenziali rischi per il neonato, si sconsiglia la somministrazione del farmaco nel corso dell'allattamento.
Limitazioni prescrittive
Nota 96.
CALCITRIOLO
Meccanismo d’azione
Il calcitriolo è il metabolita attivo della vitamina D che si forma a livello renale a partire dal suo precursore, il 25(OH)-colecalciferolo. Il calcitriolo favorisce l'assorbimento intestinale del calcio e regola la mineralizzazione ossea. Nei pazienti con grave insufficienza renale la formazione di calcitriolo endogeno si riduce progressivamente, partecipando all’instaurarsi dell'osteodistrofia renale. In questi pazienti il calcitriolo esogeno è in grado di normalizzare l'assorbimento intestinale del calcio, correggendo l'ipocalcemia e riducendo la biosintesi endogena di PTH.
Preparazioni
Formulazioni orali
- capsule molli 0.25 µg: Calcitriolo DOC, Calcitriolo EG, Calcitriolo Mylan, Calcitriolo TEVA, Rocaltrol
- capsule molli 0.50 µg: Calcitriolo DOC, Calcitriolo EG, Calcitriolo Mylan, Calcitriolo TEVA, Rocaltrol
Formulazioni endovenose
- 1 µg/mL: Calcijex
Posologia
La dose deve essere determinata sulla base della risposta individuale. Le dosi possono essere aumentate di 0.25 µg ad intervalli di 2-4 settimane se non è stata conseguita una soddisfacente risposta biochimica o clinica. Durante tale periodo la calcemia va controllata almeno due volte alla settimana: se i livelli di calcio nel siero aumentano di 1 mg/dL (250 micromoli/L) oltre l'intervallo di normalità (9-11 mg/dL - 2250-2750 micromoli/L), il trattamento deve essere sospeso sino a quando la calcemia rientra nei limiti di norma. Generalmente una dose giornaliera di 0.5-1.0 µg è sufficiente per ottenere la risposta appropriata. Possono essere richieste dosi maggiori se contemporaneamente viene fatto uso di barbiturici o anti-convulsivanti.
Indicazioni
Osteodistrofia renale in pazienti con insufficienza renale cronica, in particolare in quelli sottoposti ad emodialisi.
Ipoparatiroidismo idiopatico o chirurgico.
Pseudoipoparatiroidismo.
Rachitismo ipofosfatemico vitamina D-resistente e rachitismo familiare vitamina D pseudo-dipendente.
Osteoporosi post-menopausale.
Contro-indicazioni ed effetti collaterali
Ipersensibilità al calcitriolo (o a farmaci della stessa classe) o ad uno qualsiasi degli eccipienti
Ipercalcemia, ipercalciuria di severa entità
Calcolosi renale attiva (nefrolitiasi, nefrocalcinosi)
I principali effetti collaterali sono ipercalcemia e ipercalciuria: a causa della breve emivita del calcitriolo, l'ipercalcemia tenderà a diminuire nell'arco di 2-7 giorni successivi alla sospensione del trattamento
È stato segnalato un leggero aumento, non progressivo, dei livelli di enzimi epatici (AST, ALT).
Precauzioni d’uso
Somministrare con cautela in concomitanza a diuretici tiazidici (rischio di ipercalcemia) o digitale (rischio di aritmie cardiache)
Durante il trattamento con calcitriolo non debbono essere somministrati farmaci contenenti magnesio, in quanto si può verificare la comparsa di ipermagnesiemia.
Limitazioni prescrittive
Nessuna
Aspetti specifici nella terapia dell'ipoparatiroidismo
Assunta Santonati & Antonio Spada
Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
Prevenzione dell'ipercalciuria
Poiché la terapia calcio-vitaminica su cui si basa il trattamento dell’ipoparatiroidismo non corregge il ridotto riassorbimento renale di calcio, che è un difetto specifico della carenza di PTH, è necessario porre attenzione, sin dalle fasi iniziali della terapia, nell’evitare una calciuria eccessiva, che può predisporre a nefrocalcinosi e nefro-urolitiasi.
Per ridurre la calciuria è talvolta necessario l’impiego di diuretici tiazidici, che favoriscono il riassorbimento tubulare del calcio. Il riassorbimento del calcio è incrementato dai tiazidici con un duplice meccanismo:
- azione diretta sul trasporto di questo ione nel tubulo distale;
- stimolo indiretto al riassorbimento lungo tutto il nefrone, conseguente alla deplezione del volume extra-cellulare.
A ciò consegue riduzione della calciuria e aumento della calcemia. L’aggiunta di amiloride al tiazidico può ulteriormente ridurre la perdita di calcio, ridurre il rischio di ipokaliemia e la perdita renale di magnesio.
Anche una dieta iposodica sembra avere un effetto benefico sulla riduzione della calciuria, probabilmente sempre attraverso uno stimolo indiretto al riassorbimento del calcio escreto a livello urinario, mediato da una deplezione del volume extra-cellulare.
PTH ricombinante
La terapia ormonale sostitutiva con PTH è stata da tempo invocata come potenziale terapia ottimale nel trattamento dell’ipoparatiroidismo (1), poiché l’omeostasi del calcio non è completamente garantita con l’utilizzo solo di calcio e metaboliti attivi della vitamina D. Il PTH gioca un ruolo centrale nell’omeostasi minerale, attraverso il riassorbimento renale di calcio, la promozione dell’escrezione renale di fosfati, la conversione della 25-idrossivitamina D a 1,25-diidrossivitamina D (1-25OH₂D), la forma prontamente attiva, con un ruolo peculiare nel consentire l’assorbimento di calcio e fosforo a livello intestinale. Inoltre, il PTH è un potente regolatore del turn-over osseo, che nell’ipoparatiroidismo risulta essere soppresso. La terapia convenzionale con calcio e vitamina D, pur potendo correggere l’ipocalcemia, non è in grado di supplire alle sovra-riportate azioni peculiari del PTH. Per questo, in condizioni di grave ipoparatiroidismo, quando la terapia convenzionale, pur ad alti dosaggi di supplementazione, non è in grado di indurre eucalcemia, ma è gravata da ipercalciuria non risolvibile con la terapia tiazidica, dalla presenza di calcificazioni extra-scheletriche (come nefrolitiasi, nefrocalcinosi e/o insufficienza renale) o da palesi alterazioni della qualità della vita (QoL), è necessario valutare la terapia con rhPTH come unico rimedio fisiologico sostitutivo.
Molti studi hanno valutato l’efficacia del teriparatide (PTH 1-34) (2-4) e del PTH intatto (1-84) (5,6). Già studi preliminari avevano evidenziato come l’ipercalciuria fosse un problema meno frequente nei pazienti in terapia con PTH 1-34 sottocute per 2 volte al giorno. Infatti, in soggetti ipoparatiroidei adulti, la somministrazione di teriparatide e PTH 1-84 ha reso possibile la riduzione della dose quotidiana di supplementazione di calcio e calcitriolo e tali risultati sono stati confermati anche per l’età pediatrica.
Anche se sia il PTH 1-34 che il PTH 1-84 hanno dimostrato buona tollerabilità e studi recenti di trattamento fino a 5 anni non hanno rilevato casi di tumore osseo, la black box sulla potenzialità oncogena (derivante dalla comparsa di osteosarcoma in un topo sottoposto ad esposizione sovra-fisiologica a lungo termine) grava sulla somministrazione di questi farmaci e ne controindica l’uso in alcune situazioni. In particolare, la terapia non può essere somministrata in pazienti: “attualmente in trattamento o precedentemente trattati con radioterapia allo scheletro, con neoplasie maligne a carico dello scheletro o metastasi ossee, ad aumentato rischio basale di osteosarcoma come i pazienti con malattia ossea di Paget o patologie ereditarie” (7). Sempre maggiori sono le evidenze per le quali la terapia ormonale sostitutiva a base di PTH ha dimostrato di essere sicura e di rappresentare una valida opzione in pazienti selezionati.
Con la determina del 27 maggio 2013, l'ente regolatorio nazionale (AIFA) ha autorizzato in Italia l'uso di PTH ricombinante quale terapia sostitutiva ormonale per la cura dell’ipoparatiroidismo cronico grave, inserendo il teriparatide nell’elenco dei medicinali erogabili a totale carico, ai sensi della legge 23 dicembre 1996, n° 648. In particolare, l’AIFA ha stabilito che possono usufruire della terapia con teriparatide (per un periodo di tempo comunque non superiore a 36 mesi, limitazione eliminata nell'ottobre 2021) pazienti affetti da ipoparatiroidismo cronico grave, di età > 18 anni che rientrino in una o più delle seguenti categorie:
- pazienti divenuti ipoparatiroidei permanenti dopo tiroidectomia totale e che sviluppano durante l’utilizzo della terapia convenzionale con calcio e vitamina D la formazione di calcoli o la deposizione di calcio a livello del parenchima renale (nefrocalcinosi);
- pazienti che già in partenza presentano un difetto della funzionalità renale, che potrebbe aggravarsi con l’uso del calcio e della vitamina D;
- pazienti non rispondenti alla vitamina D, che dopo interventi di tiroidectomia totale complicatasi con ipoparatiroidismo grave hanno manifestato gravi episodi di ipocalcemia e ipomagnesiemia, risoltisi solo con la somministrazione di PTH;
- pazienti affetti da mutazioni attivanti del calcium-sensing receptor (CaR), che, in quanto affetti da questa mutazione, non sono adeguatamente responsivi alla terapia con calcio e vitamina D;
- forme congenite di insufficienza delle ghiandole paratiroidee, come esemplificato dalla sindrome di DiGeorge, o forme di ipoparatiroidismo autoimmune nell’ambito delle sindromi polighiandolari autoimmuni, condizioni di ipoparatiroidismo da distruzione ghiandolare per invasione da parte di cellule tumorali o per accumuli intra-ghiandolari di ferro (emocromatosi) o rame (morbo di Wilson).
Costituiscono criteri di esclusione i pazienti:
- con età < 18 anni;
- ben compensati dalla terapia con calcio e vitamina D, che non manifestano alterazioni della funzionalità renale e che non hanno mai presentato sviluppo di calcolosi renale o nefrocalcinosi;
- con insufficienza renale grave (nell'insufficienza di grado moderato è possibile la prescrizione, ma è raccomandata cautela);
- in gravidanza o in allattamento;
- con insufficienza epatica grave, non essendo disponibili dati a riguardo.
Inoltre, il farmaco deve essere utilizzato con cautela nei pazienti con urolitiasi in fase attiva o recente, perché può potenzialmente peggiorare questa condizione.
La posologia che ha dimostrato di ottenere i migliori risultati in termine di correzione dei livelli di calcemia, fosfatemia, magnesiemia e calciuria, a fronte di scarsi e ben tollerabili effetti collaterali, è di 0.5-0.7 µg/kg/die (circa 20-80 µg/die), in doppia somministrazione sottocutanea. Tale posologia iniziale andrà poi calibrata in base alla risposta del paziente in corso di follow-up. Considerando che attualmente il dispositivo di somministrazione del teriparatide eroga una quantità fissa e non modificabile di prodotto (pari a 20 µg/dose), la dose iniziale da somministrare potrebbe essere aggiustata lievemente, in eccesso o in difetto, rispetto alla dose calcolata, per ottenere la precisa erogazione prevista (tra 1 e 4 dosi).
È infine da ricordare la necessità di monitorare il paziente in corso di terapia, secondo le modalità e i tempi riassunti in tabella, la necessità di acquisire il consenso informato per la prescrizione di tale trattamento e la necessità di rilevare e trasmettere i dati di spesa.
| Monitoraggio in corso di terapia con PTH (mesi) | ||||||||||
| Parametro | 0 | 0.5 | 1 | 2 | 3 | 4 | 6 | 12 | 18 | 24 |
| Calcemia | x | x | x | x | x | x | x | x | x | x |
| Fosfatemia | x | x | x | x | x | x | x | x | x | x |
| Magnesiemia | x | x | x | x | x | x | x | x | x | x |
| ALP ossea | x | x | x | x | x | x | x | x | x | x |
| Calciuria e fosfaturia/24 h | x | x | x | x | x | x | x | x | x | |
| AST/ALT | x | x | x | x | x | x | x | x | ||
| Creatininemia | x | x | x | x | x | x | x | x | ||
| Azotemia | x | x | x | x | x | x | x | x | ||
| Sodiemia | x | x | x | x | x | x | ||||
| Potassiemia | x | x | x | x | x | x | ||||
| Emocromo | x | x | x | x | x | x | ||||
| Elettroforesi sieroproteine | x | x | x | x | x | x | ||||
| Uricemia | x | x | x | x | x | |||||
| Colesterolemia | x | x | x | x | x | |||||
| 25OH-D | x | x | x | x | ||||||
| BMD | x | x | x | |||||||
| Test di gravidanza | x | |||||||||
Due recenti studi multicentrici italiani hanno dimostrato che la terapia con teriparatide (PTH 1-34) in duplice somministrazione sc giornaliera in pazienti ipoparatiroidei post-chirurgici è in grado di ripristinare i normali livelli di calcemia e di ridurre il fabbisogno di sali di calcio e vitamina D (8). L’incremento della calcemia avviene rapidamente (già entro i primi 15 giorni) e si mantiene stabile. Contemporaneamente si assiste alla riduzione dei valori di fosfatemia. La riduzione dell’apporto farmacologico di calcio e vitamina D permette la progressiva riduzione della calciuria, endpoint peculiare nella protezione dei danni renali indotti dalla terapia convenzionale. Un recente lavoro ha inoltre dimostrato che i livelli sierici di fosfato diminuiscono rapidamente, già alla 1° settimana di terapia e rimangono bassi per tutta la durata della terapia.
Un altro risultato rilevante di questi studi è la dimostrazione, attraverso questionari, del significativo miglioramento della QoL, sul piano fisico e mentale (9). La stabilità della calcemia, la netta riduzione della supplementazione di calcio e vitamina D, i possibili effetti trofici diretti del PTH sul SNC sono la possibile spiegazione.
Analoghi risultati sono stati raggiunti in studi che hanno utilizzato il PTH 1-84 in mono-somministrazione giornaliera nei pazienti ipoparatiroidei (10-13). Un recente lavoro (10), che ha valutato i risultati del trattamento con rhPTH 1-84 a sei anni, ha dimostrato la riduzione del 53% della supplementazione di calcio e del 67% di vitamina D; il 48% dei pazienti ha completamente sospeso la terapia con calcio e vitamina D. Significativo anche il miglioramento della QoL, indipendente dalla calcemia basale o dalle successive modificazioni, correlato ai valori di QoL all’inizio della terapia: tanto più bassi i valori di QoL prima della terapia con PTH, tanto maggiori i miglioramenti indotti dal rhPTH 1-84.
Nel novembre 2019 sono stati pubblicati i risultati dell’estensione dello studio REPLACE (13), che ha valutato a 5 anni i pazienti in trattamento con rhPTH: sono stati ulteriormente confermati i risultati già noti in termini di riduzione della necessità di supplementazione di calcio e calcitriolo (rispettivamente, -53.4% e -75.7%), nonchè i benefici in termine di sicurezza renale. Infatti, nell’arco dei 5 anni è stata dimostrata la progressiva riduzione della calciuria e la stabilità della creatinina e della GFR. Sono stati valutati i marcatori di turnover osseo (BTM) che, come noto, sono inappropriatamente bassi nei pazienti ipoparatiroidei. La loro concentrazione sierica raggiunge un plateau dai 6 ai 16 mesi dall’inizio del trattamento, poi declina, rimanendo nei limiti di normalità per CTx e BAP e leggermente al di sopra per P1NP. Non sono state osservate modificazioni di BMD per colonna e femore, mentre è stato rilevato un miglioramento a livello del terzo distale del radio.
I numerosi lavori pubblicati sui risultati di tali trattamenti in termine di sicurezza ed efficacia sostengono con forza l’indicazione alla terapia con rhPTH 1-84 in pazienti selezionati affetti da ipoparatiroidismo grave, disponibile per ora negli USA e in alcuni paesi europei e speriamo a breve anche in Italia.
Bibliografia
- Sikjaer T, Rejnmark L, Mosekilde L. PTH treatment in hypoparathyroidism. Curr Drug Saf 2011, 6: 89-99.
- Winer KK, Ko CW, Reynolds JC, et al. Long-term treatment of hypoparathyroidism: a randomized controlled study comparing parathyroid hormone-(1-34) versus calcitriol and calcium. J Clin Endocrinol Metab 2003, 88: 4214-20.
- Winer KK, Sinaii N, Peterson D, et al. Effects of once versus twice-daily parathyroid hormone 1-34 therapy in children with hypoparathyroidism. J Clin Endocrinol Metab 2008, 93: 3389-95.
- Winer KK, Sinaii N, Reynolds J, et al. Long-term treatment of 12 children with chronic hypoparathyroidism: a randomized trial comparing synthetic human parathyroid hormone 1-34 versus calcitriol and calcium. J Clin Endocrinol Metab 2010, 95: 2680-8.
- Rubin MR, Sliney J Jr, McMahon DJ, et al. Therapy of hypoparathyroidism with intact parathyroid hormone. Osteoporos Int 2010, 21: 1927-34.
- Rubin M, Dempster D, Sliney J, et al. PTH(1-84) administration reverses abnormal bone remodeling dynamics and structure in hypoparathyroidism. J Bone Miner Res 2011, 26: 2727-36.
- Jolette J, Wilker CE, Smith SY, et al. Defining a noncarcinogenic dose of recombinant human parathyroid hormone 1-84 in a 2-year study in Fischer 344 rats. Toxicol Pathol 2006, 34: 929-40.
- Santonati A, et al. PTH (1-34) for surgical hypoparathyroidism: a prospective open label investigation of efficacy and quality of life. J Clin Endocrinol Metab 2015, 100: 3590-7.
- Vokes TJ, et al. Recombinant human parathyroid hormone effect on health-related quality of life in adults with chronic hypoparathyroidism. J Clin Endocrinol Metab 2018, 103: 722-31.
- Rubin MR, et al. Therapy of hypoparathyroidism with PTH (1-84): a prospective six year investigation of efficacy and safety. J Clin Endocrinol Metab 2016, 101: 2742-50.
- Clarke BL, et al. Effects of parathyroid hormone rhPTH(1–84) on phosphate homeostasis and vitamin D metabolism in hypoparathyroidism: REPLACE phase 3 study. Endocrine 2017, 55: 273-82.
- Sikjaer T, et al. Effects of PTH(1–84) therapy on muscle function and quality of life in hypoparathyroidism: results from a randomized controlled trial. Osteoporos Int 2014, 25: 1717-26.
- Mannstadt M, et al. Safety and efficacy of 5 years of treatment with recombinant human parathyroid hormone in adults with hypoparathyroidism. J Clin Endocrinol Metab 2019, 104: 5136-47.
Marco Gallo
SC Endocrinologia Oncologica DU, AOU San Giovanni Battista – Molinette (Torino)
Forme transitorie
L’ipoparatiroidismo transitorio è una complicanza frequente degli interventi di tiroidectomia totale e di chirurgia paratiroidea, ed è legato generalmente al danno provocato dalla legatura dei vasi paratiroidei, con conseguente ischemia o infarcimento emorragico. Solo una minoranza di questi pazienti è infatti destinata a sviluppare un ipoparatiroidismo permanente, completo o parziale (cosiddetta “insufficienza paratiroidea”), con persistenza del quadro a distanza di oltre 6 mesi dall’intervento.
Molte forme di ipoparatiroidismo transitorio sono lievi e asintomatiche, determinando modeste riduzioni della calcemia e non imponendo quindi un trattamento. Ipocalcemie severe e prolungate, per quanto transitorie, riguardano spesso pazienti sottoposti a interventi di paratiroidectomia per iperparatiroidismo primario o di tiroidectomia per morbo di Basedow, per la “sindrome dell’osso affamato” conseguente alla risoluzione della patologia e all’ipoparatiroidismo funzionale delle ghiandole sane.
Un’altra forma di ipoparatiroidismo transitorio è quello neonatale: in alcuni casi, nei primi giorni dopo la nascita, è osservabile un’accentuazione della fisiologica caduta dei livelli sierici del calcio, dovuta per lo più a ritardata maturazione delle paratiroidi. La severità di queste forme è limitata, ma talvolta è necessaria l’infusione di calcio ev; talvolta le manifestazioni sono più severe, per soppressione dell’attività paratiroidea fetale legata a stati di iperparatiroidismo o ipercalcemici nella madre.
Vari studi hanno valutato l’ipotesi che il dosaggio intra-operatorio o post-operatorio precoce (nelle prime 4-24 ore dall’intervento) dei livelli di calcio e PTH possa risultare predittivo della successiva comparsa d’ipocalcemia e ipoparatiroidismo, per anticipare il trattamento integrativo e contrastare l’insorgenza dei sintomi, ma non esistono al momento raccomandazioni univoche in questo senso (1,2). D’altra parte, il trattamento generalizzato con calcio e calcitriolo di tutti i pazienti può indurre ipercalcemia in alcuni. A seconda della patologia di fondo, del tipo d’intervento e dell’organizzazione locale, l’ipocalcemia può essere gestita con atteggiamenti differenziati, in maniera preventiva o reattiva.
Gestione pre-operatoria. Dal momento che un deficit concomitante di vitamina D può contribuire alla comparsa di ipocalcemia nel post-operatorio, secondo vari autori è indicata la supplementazione di vitamina D nelle settimane precedenti l’intervento, ottenendo livelli sierici > 30 ng/mL (1). La presenza di fattori di rischio per l’insorgenza di ipoparatiroidismo post-chirurgico può indurre a trattare preventivamente con calcio e calcitriolo individui selezionati, a prescindere dall’andamento della calcemia dopo l’intervento.
Gestione peri-operatoria. La determinazione precoce dei livelli post-operatori di PTH, ove possibile, sembra avere una buona capacità predittiva del successivo sviluppo di ipocalcemia. È stato proposto che una riduzione dell’ormone alla 6° ora dopo l’intervento del 90% rispetto ai valori pre-operatori costituisca indicazione ad avviare il trattamento con calcio e calcitriolo, senza attendere la riduzione della calcemia e/o la comparsa di sintomi (3,4). In uno studio randomizzato e controllato, la supplementazione nel peri-operatorio con solo alfa-calcidiolo (2 µg/die il giorno prima e il giorno dopo l'intervento) si è dimostrata in grado di ridurre in maniera significativa l'incidenza di ipocalcemia transitoria e dei suoi sintomi (5).
Gestione post-operatoria. Il trattamento post-operatorio dell’ipoparatiroidismo si basa sulla supplementazione di calcio (dapprima mediante infusione ev [2-8 g/die di calcio gluconato in soluzione fisiologica o glucosata al 5%, da infondere nelle 24 ore], passando poi a formulazioni per os [1-3 g/die di calcio elementare] alla ripresa dell’alimentazione) e di calcitriolo (0.5-1.5 μg/die), irrinunciabile in caso d’ipocalcemia significativa (< 8.0 mg/dL). Tale trattamento deve essere eventualmente associato a integrazione di magnesio (2-8 g/die), in caso di deficit.
Una revisione sistematica e metanalisi della letteratura (pubblicata nel 2015) ha identificato la supplementazione post-operatoria con calcio e vitamina D (oltre alla tiroidectomia bilaterale subtotale) come misura efficace nella prevenzione dell’ipocalcemia transitoria (6).
È in fase di valutazione l’utilizzo di brevi periodi di trattamento sostitutivo con rhPTH, nella gestione iniziale dell’ipoparatiroidismo post-chirurgico transitorio.
Al momento della dimissione ospedaliera, i pazienti dovranno ricevere informazioni relative ai segni e ai sintomi d’ipocalcemia, programmando (in maniera integrata con il Medico di Medicina Generale) controlli settimanali per la calcemia (fino alla sua stabilizzazione) e dopo 1 mese dall’intervento per il PTH. Nella maggior parte dei casi sarà possibile una progressiva riduzione di calcio e vitamina D (per esempio, dimezzando le dosi in maniera alternata dell’uno e dell’altra), fino alla sospensione completa.
Bibliografia
- Khan MI, Waguespack SG, Hu MI. Medical management of postsurgical hypoparathyroidism. Endocr Pract 2011, 17 suppl 1: 18-25.
- Edafe O, Antakia R, Laskar N, et al. Systematic review and meta-analysis of predictors of post-thyroidectomy hypocalcaemia. Br J Surg 2014, 101: 307–20.
- Noordzij JP, Lee SL, Bernet VJ, et al. Early prediction of hypocalcemia after thyroidectomy using parathyroid hormone: an analysis of pooled individual patient data from nine observational studies. J Am Coll Surg 2007, 205: 748-54.
- Grodski S, Lundgren CI, Sidhu S, et al. Postoperative PTH measurement facilitates day 1 discharge after total thyroidectomy. Clin Endocrinol 2009, 70: 322-5.
- Genser L, Trésallet C, Godiris-Petit G, et al. Randomized controlled trial of alfacalcidol supplementation for the reduction of hypocalcemia after total thyroidectomy. Am J Surg 2014, 207: 39-45.
- Antakia R, Edafe O, Uttley L, et al. Effectiveness of preventative and other surgical measures on hypocalcemia following bilateral thyroid surgery: a systematic review and meta-analysis. Thyroid 2015, 25: 95-106.
Ipoparatiroidismo in gravidanza
Assunta Santonati & Antonio Spada
Endocrinologia, Ospedale San Giovanni Addolorata, Roma
(aggiornato al 18 dicembre 2019)
La gravidanza e l’allattamento si associano a rilevanti modificazioni dell’omeostasi calcio-fosforica, provocate dalle alterazioni nella sintesi, nel metabolismo e nell’escrezione del calcio e degli ormoni calciotropi tipiche di queste fasi della vita della donna. Per soddisfare lo sviluppo scheletrico del feto durante la gravidanza, aumenta il fabbisogno di calcio e circa l'80% del contenuto di calcio, fosforo e magnesio presente nel feto viene reclutato entro il 3° trimestre di gravidanza. Nella seconda metà della gravidanza si osserva un fisiologico incremento dei livelli di 1,25(OH)2 vitamina D, per la sintesi placentare e l’effetto stimolante degli estrogeni e del lattogeno placentare. Nelle madri normo-paratiroidee la calcemia rimane invariata, grazie all’incremento dalla 3° alla 13° settimana di gestazione della proteina correlata al PTH (PTHrP), proveniente da placenta, mammella e utero, che sarà triplicata al termine della gravidanza. La PTHrP migliora il riassorbimento renale di calcio e fosfato, stimola la CYP27b1, aumentando la produzione di calcitriolo, che a sua volta migliora l’assorbimento intestinale di calcio e fosforo. Durante l’allattamento, invece, è abitualmente osservabile un incremento dei livelli plasmatici materni di fosforo e calcio ionizzato, con progressiva normalizzazione della 1,25(OH)2 vitamina D (1).
Anche il decorso clinico delle patologie paratiroidee può quindi risentirne, con sintomi e alterazioni bioumorali, che possono confondersi con quelli ascrivibili alla gestazione e all’allattamento. Durante tali periodi è comune osservare, nelle donne con ipoparatiroidismo, una riduzione del fabbisogno di calcio e calcitriolo; talvolta, invece, può rendersi necessario un incremento della dose di calcitriolo (slatentizzazione di ipoparatiroidismi parziali per infusione di magnesio, come terapia tocolitica) (2-4). Una gestione terapeutica scorretta dell’ipoparatiroidismo può avere conseguenze sfavorevoli per la madre e il feto, con aumento della morbilità e della mortalità materna e perinatale. L’ipocalcemia aumenta l’eccitabilità della muscolatura uterina, riducendo il potenziale a riposo e amplificando la frequenza degli spike delle fibre muscolari, con incremento del rischio di aborto spontaneo.
Sia in gravidanza sia durante l’allattamento, l’obiettivo terapeutico è il mantenimento dei livelli di calcemia sierica nella parte bassa del range di normalità e dei livelli di 25-idrossivitamina D e calciuria nell'intervallo di riferimento normale. È quindi fondamentale, in gravidanza, un attento e frequente monitoraggio (ogni 2-4 settimane, da ravvicinare se si modifica il fabbisogno di supplementazione) dei livelli materni di calcio ionizzato, fosfato, GFR, magnesio e 1,25(OH)2 vitamina D, per garantirne una supplementazione adeguata:
- un trattamento insufficiente potrà determinare nel feto iperparatiroidismo compensatorio con ipercalcemia transitoria;
- un trattamento eccessivo, per contro, determinerà l’ipotrofia delle paratiroidi fetali con conseguente ipocalcemia (5,6).
Durante la gravidanza evitare l’idroclorotiazide, perchè è classificata dalla FDA come farmaco rischioso, e il rhPTH perchè non è stato adeguatamente valutato.
Durante l’allattamento, è altrettanto importante proseguire il monitoraggio di calcemia e vitamina D (ogni 1-3 mesi); la supplementazione di quest’ultima dovrà avvenire con la forma attiva (calcitriolo), per il passaggio nel latte della 25OH-vitamina D e il potenziale rischio conseguente di ipervitaminosi nel neonato (1,2).
Bibliografia
- Cooper MS. Disorders of calcium metabolism and parathyroid disease. Best Pract Res Clin Endocrinol Metab 2011, 25: 975-83.
- Krysiak R, Kobielusz-Gembala I, Okopien B. Hypoparathyroidism in pregnancy. Gynecol Endocrinol 2011, 27: 529-32.
- Kovacs CS. Calcium and bone metabolism disorders during pregnancy and lactation. Endocrinol Metab Clin North Am 2011, 40: 795-826.
- Khan AA, et al. Standards of care for hypoparathyroidism in adults: a Canadian and International Consensus. Eur J Endocrinol 2019, 180: P1-22.
- Hsu SC, Levine MA. Perinatal calcium metabolism: physiology and pathophysiology. Semin Neonatol 2004, 9: 23-36.
- Steffensrud S. Parathyroids: the forgotten glands. Neonatal Netw 2000, 19: 9-16.
Nefrolitiasi
Gregorio Guabello
Ambulatorio di Patologia Osteo-Metabolica, UO Reumatologia, Istituto Ortopedico Galeazzi, Milano
(aggiornato al 28 aprile 2015)
EPIDEMIOLOGIA ED EZIOLOGIA
La calcolosi renale interessa il 7.5% della popolazione generale (M:F = 2-4:1), soprattutto i bianchi non ispanici.
È principalmente di 2 tipi:
- calcolosi calcica: calcoli di calcio ossalato mono e bi-idrato (pH acido) e di calcio fosfato (apatite, pH alcalino): 70-75% casi;
- calcolosi urica: calcoli di acido urico e di urato monosodico e di ammonio (pH acido): 5-10% casi.
Casi più rari sono quelli su base genetica, infettiva e quelli su base iatrogena (1) (tabella 1).
| Tabella 1 Classificazione eziologica dell’urolitiasi |
||
| Tipo | Frequenza | |
| Metabolica | Calcica | 70-75% |
| Urica | 5-10% | |
| Genetica | Calcoli di cistina nella cistinuria, pH acido | 1% |
| Infettiva | Calcoli di struvite-magnesio ammonio fosfato da batteri del genere Proteus ureasi-produttori (per scissione dell’urea urinaria in anidride carbonica e ammoniaca, quest’ultima idrolizzata a ione ammonio, con conseguente incremento del pH e diminuzione della solubilità degli ioni fosfato e magnesio) | 10-15% |
| Iatrogena | Calcoli formati da farmaci cristallizzati: triamterene, indinavir, nelfinavir, amprenavir, atazanavir, acyclovir, sulfamidici | |
FISIOPATOLOGIA DELLA CALCOLOSI CALCICA (calcoli di calcio ossalato e fosfato)
La formazione di questo tipo di calcoli non dipende tanto dalla saturazione di sali di calcio nelle urine, quanto da alterazione dell’equilibrio fra agenti promotori e inibitori della litogenesi (tabella 2): un aumento dei promotori e/o una diminuzione degli inibitori rappresenta il “primum movens” per la formazione di piccoli cristalli urinari che si aggregano, formando nel tempo un calcolo più o meno grande (processo di cristallizzazione).
| Tabella 2 Equilibrio fattori litogenetici urinari |
||
| Promotori | Calcio Ossalato Urato Sodio Basso volume urinario pH acido |
|
| Inibitori | Inorganici | Magnesio Citrato |
| Organici | Nefrocalcina Frammento-1 della protrombina urinaria Osteopontina |
|
I principali fattori eziopatogenetici (2) sono elencati in tabella 3.
| Tabella 3 Classificazione eziopatogenetica litiasi calcica |
||
| Ipercalciuria (3)(40-50% dei casi) | Idiopatica | |
| Secondaria | Acidosi tubulare renale distale di tipo I Altre |
|
| Iperossaluria | Ereditaria: aumentata conversione di gliossilato in ossalato | |
| Secondaria (enterica): gli acidi grassi non digeriti nel colon saponificano con il calcio, creando un eccesso di ossalato libero per l’assorbimento intestinale | Dieta ipocalcica Morbo di Crohn Chirurgia bariatrica Sindrome dell’intestino corto Diarrea cronica Insufficienza pancreatica |
|
| Ipocitraturia | Ereditaria | |
| Secondaria | Dieta acidogena Ipokaliemia Diarrea Infezioni Acidosirenale tubulare distale Acetazolamide |
|
| Ipomagnesiuria | ||
| Iperuricosuria | ||
Acidosi tubulare renale distale di tipo I
Tale condizione, che merita un approfondimento a parte,si definisce come l’incapacità del tubulo renale di acidificare le urine per difetto della rigenerazione distale di HCO3- a livello del tubulo contorto distale.
Può dipendere da diverse cause (tabella 4).
| Tabella 4 Cause di acidosi tubulare tipo I |
|
| Genetiche | Autosomica dominante o recessiva |
| Secondarie | Malattie autoimmuni: cirrosi biliare primitiva, LES, sindrome di Sjögren, artrite reumatoide |
| Malattie granulomatose: sarcoidosi | |
| Malattie endocrine: ipertiroidismo, iperparatiroidismo primario | |
| Nefro-uropatie: nefrocalcinosi, infezioni croniche o ostruzione cronica delle vie urinarie | |
| Trapianto di rene e rigetto di trapianto di rene | |
| Malattie epatiche: epatite cronica attiva | |
| Emopatie: anemia a cellule falciformi | |
| Farmaci: amfotericina B, FANS, toluene, litio | |
Dal punto di vista clinico, si presenta nel bambino con ritardo crescita, rachitismo, vomito, ipotonia, sordità e nell’adulto con osteomalacia e nefrolitiasi/nefrocalcinosi.
Il sospetto diagnostico si pone in presenza di acidosi metabolica (bicarbonati ematici ≤ 20 mEq/L), non altrimenti spiegabile, con gap anionico normale, in associazione con ipercloremia, ipopotassiemia, ipercalciuria, ipofosfatemia, ipocitraturia. La diagnosi viene confermata dalla valutazione del pH urinario (> 5.3-5.5) e dalla ridotta escrezione urinaria di ammonio delle 24 ore.
La terapia consiste nella correzione dell’acidosi con sodio bicarbonato o sodio citrato per os e nella supplementazione di potassio con potassio citrato, che possiede anche effetto ipocalciurico (4).
Casi particolari di ipercalciuria
In ambito strettamente endocrinologico, si verificano nei pazienti affetti da iperaldosteronismo primario (5) (per ridotto riassorbimento di sodio e calcio a livello del tubulo contorto prossimale) e nei pazienti affetti da ipoparatiroidismo cronico trattati con dosi eccessive di calcio carbonato e calcitriolo (per assente azione del PTH a livello del tubulo renale).
FISIOPATOLOGIA DELLA CALCOLOSI URICA (calcoli di acido urico)
I calcoli di acido urico si possono formare in quelle condizioni di iperuricemia da iperproduzione di acido urico con iperuricosuria (> 750 mg/24 ore nella donna, > 800 mg/24 ore nel maschio) (tabella 5).
| Tabella 5 Cause di iperuricemia (6,7) |
||
| Primitiva | Poligenica | |
| Da difetto biochimico noto | Deficit di ipoxantina-guanina fosforibosil-transferasi (s. di sindrome di Lesch-Nyhan) Iperattività di PRPP-sintetasi Deficit di adenina-fosforibosil-transferasi |
|
|
Secondaria |
Dieta ricca di purine Diabete mellito di tipo 2 Obesità Sindrome metabolica Accelerato turn-over degli acidi nucleici in caso di malattie mielo-linfoproliferative, anemia emolitica cronica, radio-chemioterapia |
|
DIAGNOSTICA DI LABORATORIO E IMAGING DELLA CALCOLOSI RENALE
Lo screening metabolico:
- deve essere eseguito nei pazienti affetti da calcolosi multipla o recidivante;
- può essere preso in considerazione nei pazienti con calcoli non calcarei, funzione renale ridotta, singolo rene funzionante, storia familiare di calcolosi, aumentato rischio di ossaluria enterica, anomalie anatomiche del tratto urinario, infezioni recidivanti del tratto urinario.
Gli esami di laboratorio possono essere così schematizzati:
- esami ematici: calcemia, fosforemia, magnesiemia, creatininemia, 25OH-vitamina D, sodiemia, potassiemia, cloremia,bicarbonatemia, uricemia, PTH;
- esami urinari: esame urine completo + urinocoltura, esami sulle urine 24 ore(calciuria, fosfaturia, citraturia, ossaluria, creatininuria,azoturia, magnesiuria, sodiuria, potassiuria, cloruria, uricosuria e se disponibilisolfato e ammonio urinario 24 ore).
Alcuni laboratori specializzati forniscono un’elaborazione grafica a colori del rischio litogeno (8), che guida medico e paziente nella comprensione dei fattori che inducono un'elevata sovra-saturazione urinaria, consentendo di monitorare nel tempo, in modo chiaro e intuitivo, l'evoluzione della malattia e i risultati del programma di trattamento e di prevenzione.
Gli esami strumentali utili per la diagnosi di sede della calcolosi sono Rx addome diretto (i calcoli di calcio, struvite e cistina sono radio-opachi, mentre i calcoli di acido urico sono radio-trasparenti), ecografia dell’apparato urinario e TC addome senza mdc.
Il gold standard per lo studio della composizione del calcolo (espulso) è rappresentato dalla spettro-fotometria agli infrarossi (apatite e brushite per i calcoli di fosfato di calcio; forma COD da ipercalciuria e forma COM da iperossaluria per i calcoli di ossalato di calcio); altri metodi sono la cristallografia/difrattometria a raggi X e la microscopia a luce polarizzata (1).
TERAPIA DELLA CALCOLOSI RENALE
Calcolosi calcica
Si basa su: dieta, diuretici calcio-ritentivi, alkali del potassio e del magnesio (vedi capitolo Ipercalciurie) (3).
Calcolosi urica
La terapia di I linea comprende: terapia idropinica (diuresi > 2-3 L/die), alkalinizzanti per innalzare il pH urinario a 6-6.5 (citrato di potassio 4-9 g in 3 somministrazioni/die), dieta ipoproteica e prevalentemente vegetariana (cibi da evitare: crostacei, carne rossa, lievito).
La terapia II linea è rappresentata dai farmaci inibitori della xantino-ossidasi, enzima chiave nella sintesi dell’acido urico nell’ambito del catabolismo delle purine: allopurinolo (inibitore non specifico della xantino-ossidasi, alla dose media di 300 mg/die) e febuxostat (inibitore specifico della xantino-ossidasi, più efficace, alla dose media di 80 mg/die). In entrambi i casi i dosaggi vanno modulati in base alla funzione renale (1).
Calcolosi cistinica
La terapia ha essenzialmente due obiettivi:
- ridurre la concentrazione urinaria della cistina: terapia idropinica (diuresi > 2-3 L/die) e dieta povera di sodio e metionina (carne, pesce, uova, latticini) precursore della cistina;
- aumentare la solubilità urinaria della cistina: alkalinizzanti(citrato di potassio 4-9 g in 3 somministrazioni/die con target pHurinario > 7) e, se la cistinuria è > 300 µmol/mmol creatinina, tiopronina (forma un complesso farmaco-cisteinapiù solubile della cistina), alla dose nel bambino di 20-50 mg/kg/die in 3 somministrazioni (max 750 mg/die) e nell’adulto di 20 mg/kg/die in 3 somministrazioni. Tra gli effetti collaterali di tiopronina c’è la glomerulopatia proteinurica, per cui è utile controllo periodico della proteinuria (1).
Calcolosi infetta
La terapia prevede: terapia idropinica (diuresi > 3 L/die), antibiotici mirati, acidificanti (L-metionina), inibitori dell’ureasi (acido aceto-idroxamico) (1).
Terapia urologica della calcolosi renale
Prevede terapia chirurgica, rimozione endoscopica, litotripsia (1).
BIBLIOGRAFIA
- Rosen CJ. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. John Wiley & Sons, 8th Edition, 2013.
- Aggarwal KP, Narula S, Kakkar M, Tandon C. Nephrolithiasis: molecular mechanism of renal stone formation and the critical role played by modulators. Biomed Res Int 2013, 2013: 292953.
- Guabello G. Le ipercalciurie. Endowiki.
- Seidowsky A, Moulonguet-Doleris L, Hanslik T, et al. Tubular renal acidosis. Rev Med Interne 2014, 35: 45-55.
- Ceccoli L, Ronconi V, Giovannini L, et al. Bone health and aldosterone excess. Osteoporos Int 2013, 24: 2801-7.
- Faglia G. Malattie del sistema endocrino e del metabolismo. 4° Ed, McGraw-Hill, 2006.
- Billiet L, Doaty S, Katz JD, Velasquez MT. Review of hyperuricemia as new marker for metabolic syndrome. ISRN Rheumatol 2014, 2014: 852954.
- www.lithocenter.it.